
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePostsynthetic ionization of an imidazole-containing metal–organic framework for the cycloaddition of carbon dioxide and epoxides†
Jun
Liang
ab,
Rui-Ping
Chen
a,
Xiu-Yun
Wang
a,
Tao-Tao
Liu
a,
Xu-Sheng
Wang
a,
Yuan-Biao
Huang
*a and
Rong
Cao
*ab
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, University of Chinese Academy of Sciences Fujian, Fuzhou, 350002, P. R. China. E-mail: rcao@fjirsm.ac.cn; Fax: +86 591 8379 6710
bDepartment of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China
First published on 2nd November 2016
Abstract
A bifunctional imidazolium functionalized zirconium metal–organic framework (Zr-MOF), (I−)Meim-UiO-66 (2), was successfully prepared from the imidazole-containing Zr-MOF Im-UiO-66 (1) by a post-synthetic modification (PSM) method. It was found that the crystal size and pore features of the imidazole-containing 1 could be tuned at the nanoscale. The bifunctional MOF 2, containing Brønsted acid sites and iodide ions, was shown to be an efficient and recyclable heterogeneous catalyst for the cycloaddition of carbon dioxide (CO2) with epoxides, without the use of any co-catalyst, at ambient pressure. The solvent-free synthesis of the cyclic carbonate from CO2 and an epoxide was monitored by in situ Fourier transform infrared spectroscopy (FT-IR) and an acid/base synergistic catalysis mechanism was proposed. We hope that our strategy provides an effective approach for the introduction of functional N-heterocyclic groups into MOFs for potential applications.
Introduction
The conversion of greenhouse carbon dioxide (CO2), as a renewable C1 synthon, into value-added chemicals such as cyclic carbonates, is one of the hot topics in the field of catalysis.1 However, due to the inert nature of CO2, finding a chemical fixation process with a high efficiency is still a great challenge.2 Up to now, various homogeneous catalysts including quaternary ammonium complexes,3 alkali metal salts,4 ionic liquids,5 and metal–organic complexes2a,6 have been applied to the cycloaddition of CO2 with epoxides into cyclic carbonates. However, homogeneous catalysts often suffer from laborious and expensive purification processes, whereas the use of porous heterogeneous catalysts could overcome the product–catalyst separation problem.7Recently, metal–organic frameworks (MOFs), as a class of porous crystalline compounds composed of metal nodes and organic linkers, have shown workable applications in gas adsorption and catalysis due to their chemical and composition tunabilities as well as their permanent porosities and large surface areas.8,9 Although a few MOF catalysts have been utilized for the coupling reactions of CO2 with epoxides, most of them are efficient only under high pressure conditions or with co-catalysts, such as organic bases and organic salts, which are uneconomical and complicated.10 Thus the synthesis of porous MOF materials with efficient catalytic sites, which can be used for the conversion of CO2 without a co-catalyst at atmospheric pressure is highly desirable. As is known, imidazoliums have been employed as efficient catalysts for the cycloaddition reaction of CO2.5e,11 Despite that, a handful of imidazolium-based MOFs have been synthesized via pre-synthetic strategies, although none of them have been utilized for the chemical fixation of CO2.12 Furthermore, N-heterocyclic functional groups such as imidazole (im) and imidazolium could enhance the “CO2 capture” capability due to the weak interactions between the CO2 molecule and the nitrogen atom.6,13 However, it remains a challenge to obtain porous and stable MOFs with free imidazole groups through avoiding undesirable coordination with metal nodes in direct synthesis.14 In particular, UiO-66,15b as an archetype of a 12-connected Zr-based MOF, is among the most stable and tunable MOFs due to its high valence metal-oxide clusters (Zr6) and its variable connectivity.15 UiO-66 derivatives are typically constructed from Zr6 clusters and linear dicarboxylic linkers, and the produced open Brønsted-acid sites (Zr–OH/Zr–OH2) could enhance the acidic catalysis by decreasing the number of linkers.16 Thus, we hope that the isoreticular synthesis of imidazole-containing UiO-66 could pave the way towards imidazolium functionalized MOFs, which can be employed as bifunctional catalysts containing Brønsted-acid sites and halogen anions for carbon dioxide capture and synergistic conversion.17
Herein, we first synthesized a bifunctional imidazolium decorated Zr-MOF, (I−)Meim-UiO-66 (2), via isoreticular synthesis and a post-synthetic modification (PSM) method.18 Interestingly, 2 is shown to be an efficient and recyclable heterogeneous catalyst for the cycloaddition of CO2 with epoxides, without the use of any co-catalyst, at ambient pressure. As shown in Scheme 1, a delicate linear linker, 2-(imidazol-1-yl)terephthalic acid (Im-H2BDC), was selected in order to construct imidazolium-Zr-MOF efficiently (for details, see Fig. S1†).
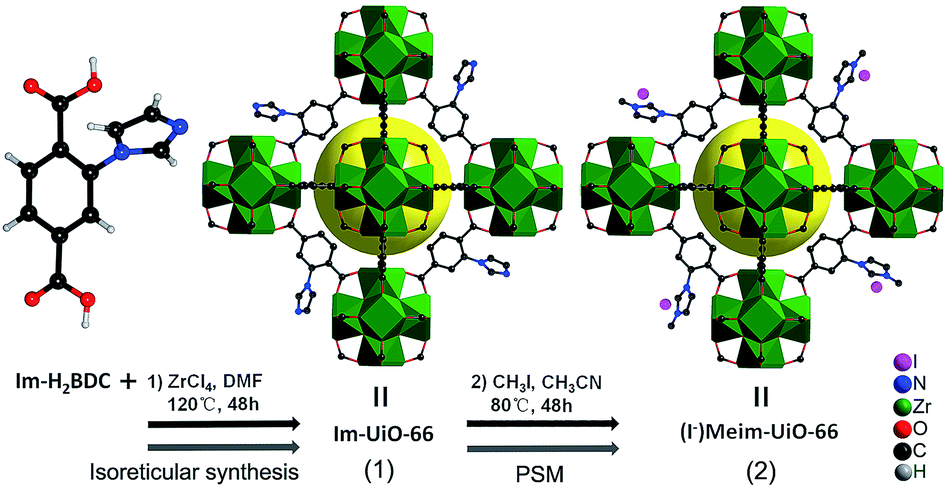 | ||
| Scheme 1 Syntheses of 1 and 2via reticular chemistry and by a PSM method, respectively. Green polyhedra: Zr6 clusters. Yellow ball: micropores. Only part of the linkers are shown for clarity. | ||
Results and discussion
Syntheses and characterizations of Im-UiO-66 (1) and (I−)Meim-UiO-66 (2)
Initially, Im-UiO-66 (1) was readily synthesized in 75% yield by the solvothermal reaction of an equimolar mixture of Im-H2BDC·HCl and ZrCl4 in DMF at 120 °C for 48 h. The powder X-ray diffraction (PXRD) patterns of the resultant solid matched well with the calculated pattern of UiO-66 (Fig. 1a), which indicated that the two compounds had similar topological structures (Fig. S5†).15b Interestingly, the obtained uniform small nanocrystals (20–30 nm) of Im-UiO-66 (1) were able to facilitate contact with the substrate and enhance the catalysis (Fig. 1c and S6†).9c The framework of 1 was chemically stable in aqueous sodium hydroxide and hydrochloric acid conditions (pH = 1–14), as confirmed by the PXRD patterns collected for each sample (Fig. S7†). Furthermore, 1 had a Brunauer–Emmett–Teller (BET) surface area of 538 m2 g−1 (Fig. 1e and Table S1†). Notably, 1, which contained imidazole groups, indeed showed a higher CO2 adsorption uptake (5.85 wt%) than UiO-66 (3.39 wt%) at room temperature (Fig. 1f). In order to further investigate the structural features and active components of this framework, 1 was measured by CO2-TPD and NH3-TPD, which revealed the existence of a large amount of medium Lewis basic sites (free imidazole groups) and acid sites (probably Brønsted-acid (Zr–OH/Zr–OH2) sites of defect Zr6 nodes)18 in 1 (for details, see Fig. S8†).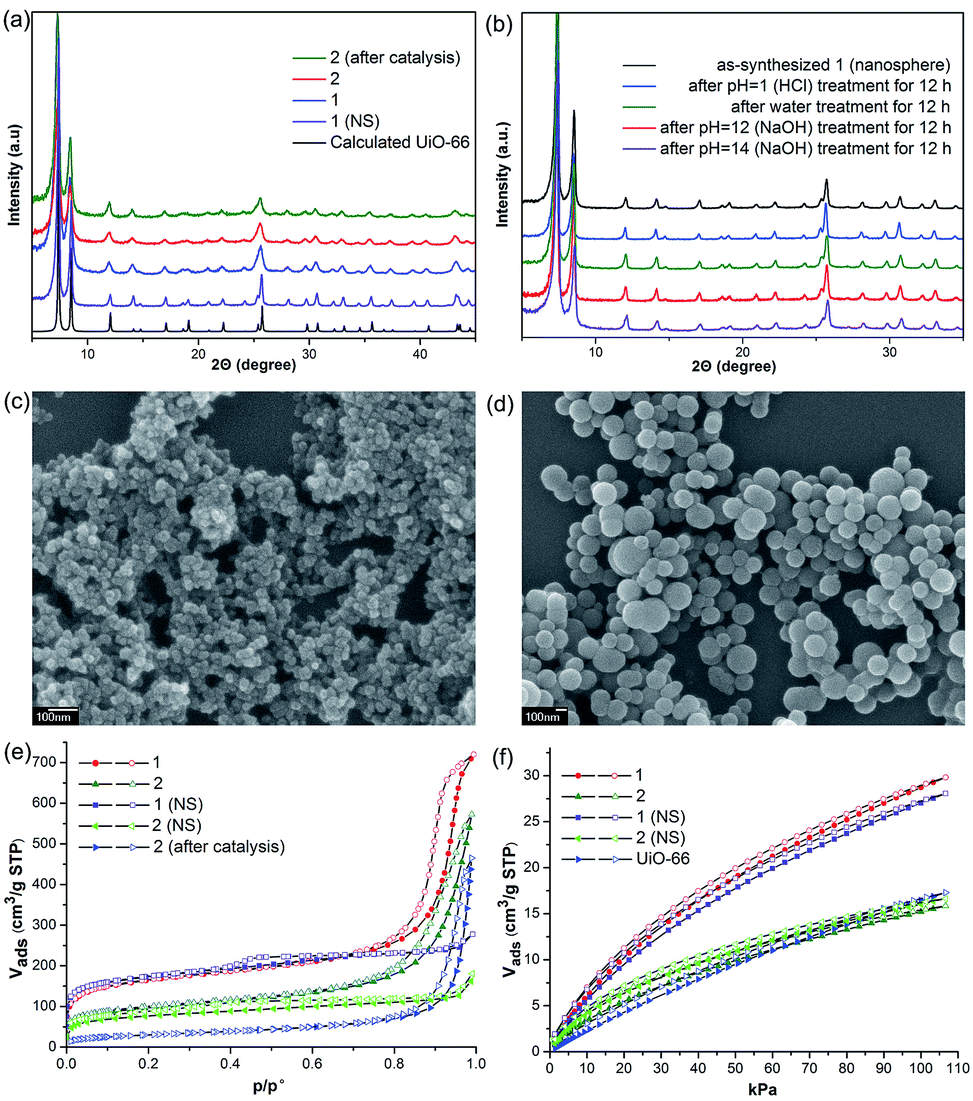 | ||
| Fig. 1 (a) PXRD of 1, 1 (nanosphere, NS), 2 and 2 after catalysis. (b) The PXRD patterns of 1 (NS) after treatment with different aqueous solutions. The SEM images of (c) as-synthesized nanoparticles of 1 and (d) nanospheres of 1 (NS). Scale bar: 100 nm. (e) N2 sorption isotherms for 1, 2, 1 (nanosphere, NS) and 2 (NS) at 77 K. (f) CO2 sorption isotherms for 1, 2, 1 (NS), 2 (NS) and UiO-66 at 298 K. Solid symbols denote adsorption, open symbols denote desorption (P/Po = partial pressure). | ||
The successful synthesis of robust 1 with uncoordinated imidazole groups prompted us to implement a post-synthetic functionalization of 1 with methyl iodide (Scheme 1). The PXRD pattern indicated that the targeted 2 retained its original framework integrity (Fig. 1a). To determine the degree of methyl-modification of 1, 1H NMR analyses were performed after the digestion of 2 in DMSO-d6 and HF (Fig. S9†), and indicated that ca. 75% of the imidazole groups were converted to imidazolium, based on the molar ratio of the (I−)Meim-BDC2− and Im-BDC2− linkers. The formation of 2 was further confirmed by mass spectra, FT-IR and solid 13C NMR (Fig. S10 and S11†). Compared with the 13C NMR spectrum of 1, the 13C NMR spectrum of 2 showed a unique peak at 36.0 ppm, which was ascribed to the methyl carbon in the framework (Fig. 2).19 Energy dispersive spectroscopy (EDS) images (Fig. S12†) confirmed the homogeneous distribution of the iodide throughout the interior of the 2 crystals. After incorporation of the methyl groups, 2 still exhibited a high BET surface area of 328 m2 g−1 (Fig. 1e) and a good CO2 adsorption uptake of 3.1 wt% at room temperature (Fig. 1f). The zero-coverage adsorption enthalpies for CO2 at 298 K were 27.4 and 44.2 kJ mol−1 for 1 and 2, respectively (Fig. S13†). The Qst value of 1 was higher than that of most metal azolate frameworks (MAFs) but slightly lower than that of nitrogen rich IFMC-1 (30.7 kJ mol−1).13b On the other hand, the Qst value of 2 containing imidazolium groups was moderate, which surpassed that of most reported MOFs, such as MOF-5 (34 kJ mol−1) and HKUST-1 (35 kJ mol−1), but which was lower than that of MOFs with CO2-philic sites, including MOF-74-Mg (47 kJ mol−1) and TBA@bio-MOF-1 (55 kJ mol−1) (Table S2†).8e,13c
 | ||
| Fig. 2 Solid-state 13C NMR spectra of Im-UiO-66 and (I−)Meim-UiO-66. | ||
It is very interesting that the well-defined hierarchically porous nanospheres (100–300 nm) of Im-UiO-66, 1 (NS), can also be obtained under a high concentration of acetic acid by a solvothermal method, as evidenced by the PXRD patterns (Fig. 1a) and the SEM images (Fig. 1d). Although 1 (NS) had similar structures to those of 1, the porosity features were quite different. As shown in Fig. 1e, 1 (NS) showed combined isotherms of types I and IV.
These results indicated that the pores of this MOF were hierarchically constructed from both micropores and mesopores. 1 (NS) had mesopores with a diameter of 34 Å and micropores with diameters of 6 Å and 11 Å, as determined by the NLDFT-based method (Fig. S14†). The mesopore features of 1 (NS) were further confirmed by high-resolution transmission electron microscopy (HRTEM; Fig. S15†). Furthermore, 1 (NS) exhibited a slightly higher BET surface area (560 m2 g−1) than 1 (538 m2 g−1), but a slightly lower CO2 adsorption uptake (5.5 wt%). 1 (NS) retained its full crystallinity after treatment with different aqueous solutions (pH = 1–14), as demonstrated by the PXRD patterns of the collected samples (Fig. 1b). Porous 2 (nanosphere, NS) was prepared by the same PSM method. A higher degree of ionization (85%) for 2 (NS) was observed due to its higher crystallinity and the presence of mesopores (Fig. S16†). As a result, the BET surface area (255 m2 g−1) of 2 (NS) was much lower than that of 2 (328 m2 g−1), but the CO2 adsorption uptake (3.26 wt%) was comparable.
Catalytic activity
Considering the coexistence of the basic/acidic sites and the good CO2 adsorption uptake of 2, we examined the catalytic performance of the nanoparticles of 2 towards the cycloaddition of CO2 with epoxides to produce cyclocarbonates (Table 1). As shown in Table S3,† without any co-catalyst, 2 exhibits a highly efficient catalytic performance for the cycloaddition of epichlorohydrin with CO2 into cyclocarbonate at 120 °C under 1 atm CO2 pressure, with a yield of 93% (Table S3,† entry 6). In contrast, nanosphere 2 (NS) exhibited a lower activity, with a yield of 59% under similar conditions (Fig. 1a and d and Table S4,† entry 2). The enhanced catalytic activity of 2 indicated that the small nanoparticles with larger surface areas could provide many more active sites, thus improving the efficiency of heterogeneous reactions. Taking into account the conversion and TON, the optimal conditions were set as: epoxide (10 mmol), catalyst (0.050 g, 0.745 mol%), PCO2 (0.1 MPa), temperature (120 °C) and time (24 h).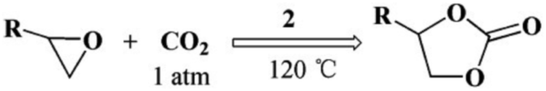
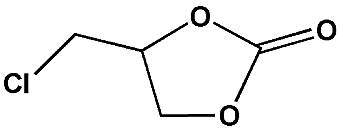


In order to perform control experiments, Zr-MOFs without imidazole groups, such as UiO-66 and NH2-UiO-66, were synthesized according to the literature (Fig. S17†).15d It was observed that UiO-66 was almost ineffective for this reaction, while NH2-UiO-66 showed little activity with a very low conversion (11%) at ambient pressure (Table S4,† entry 3, 4). Additionally, the combination of Im-H2BDC and ZrCl4 led to a low yield (Table S4,† entry 6). These results highlighted the catalytic efficiency of the targeted porous framework 2, which possesses a Brønsted-acid site and an iodide ion, towards the cycloaddition of CO2 with epoxides. It should be noted that the catalytic performance of bifunctional 2 was much better than that of other MOF systems where co-catalysts are generally required for this reaction.10
To shed light on the mechanism of the heterogeneous catalytic CO2 fixation, in situ FT-IR measurements were performed to monitor the reaction process at 1 atm CO2 pressure (Fig. 3). The absorption intensities of the carbonyl group characteristic peaks (ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)), centered at 1735 cm−1 and 1800 cm−1, rapidly increased with the reaction time, indicating the formation of chloropropene carbonate.4c In addition to the typical asymmetric vibrations (2361 cm−1 and 2342 cm−1) of CO2, one new peak centered at 2329 cm−1 was observed after the addition of epichlorohydrin during the reaction and disappeared at the end of the reaction. This signal might be tentatively ascribed to that of the CO2 molecules being attacked by the anionic intermediate species formed by epoxide and iodide (Scheme 2, stage III).
O)), centered at 1735 cm−1 and 1800 cm−1, rapidly increased with the reaction time, indicating the formation of chloropropene carbonate.4c In addition to the typical asymmetric vibrations (2361 cm−1 and 2342 cm−1) of CO2, one new peak centered at 2329 cm−1 was observed after the addition of epichlorohydrin during the reaction and disappeared at the end of the reaction. This signal might be tentatively ascribed to that of the CO2 molecules being attacked by the anionic intermediate species formed by epoxide and iodide (Scheme 2, stage III).
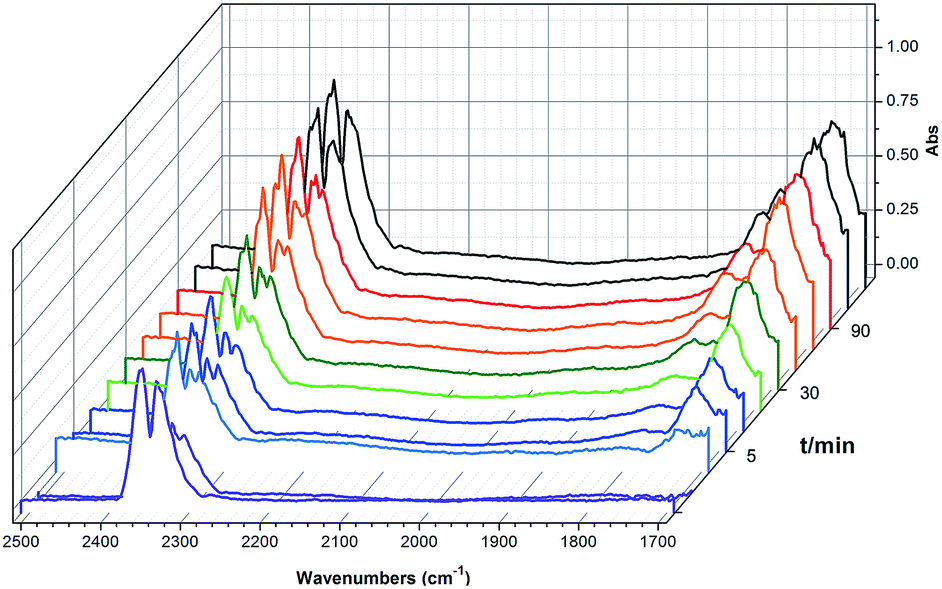 | ||
| Fig. 3 In situ FT-IR spectra of the synthesis of chloropropene carbonate catalyzed by 2 at 100 °C under an atmospheric dilute CO2/N2 gas mixture. IR absorption of 2 is subtracted for clarity. | ||
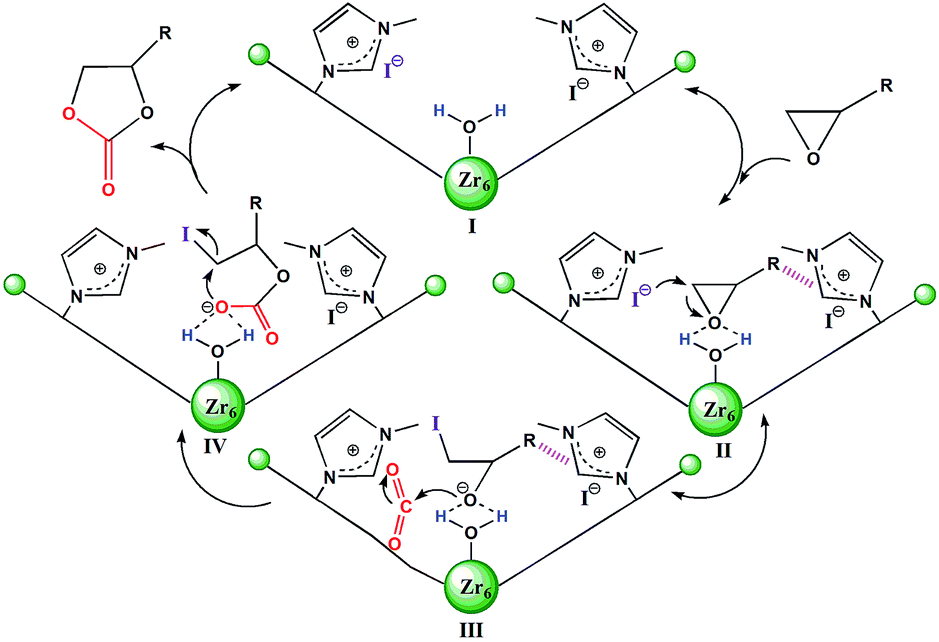 | ||
| Scheme 2 Plausible mechanism of 2 catalyzed cycloaddition reaction of CO2 with epoxides. The hashed bonds represent π⋯π, cationic⋯π or hydrogen bonding interactions. | ||
Based on the structural features and catalytic performances of 2 as well as the in situ FT-IR results, a plausible catalytic mechanism is proposed as shown in Scheme 2. Firstly, the epoxide ring is adsorbed onto 2 and stabilised by non-covalent interactions, such as hydrogen bonding and π⋯π interactions (stage II). Then the ring-opening step occurs due to the synergistic activation effect of the Brønsted-acid (Zr–OH/Zr–OH2) and the nucleophilic Lewis base I− (stage III). Immediately, CO2 inserts into this anionic intermediate species to form an acyclic ester (stage IV), which is transformed to a carbonate by intramolecular cyclization, followed by the return of the original catalyst for the new catalytic cycle (stage I).7e On the other hand, the catalytic activities of 1 for the coupling reactions of 1,2-epoxyhexane or styrene oxide and CO2 were investigated, which further confirmed the existence of Brønsted-acid (Zr–OH/Zr–OH2) sites in 1 (Table S5†).16 Moreover, it was noted that halohydrocarbons, such as epichlorohydrin, could react with the imidazole-based framework to form a complex ionic catalyst that self-catalyzes the reaction based on a similar mechanism.18b
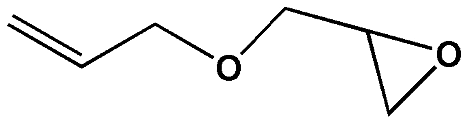
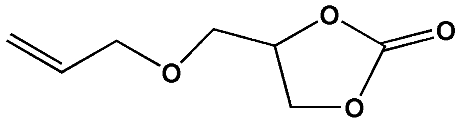
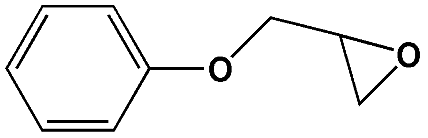
To examine the scope of the substrates for the cycloaddition reaction of CO2, other epoxides were studied under optimal conditions. As shown in Table 1, the epoxide allyl glycidyl ether gave a high conversion (92%) and a satisfactory carbonate selectivity (>81%) (Table 1, entry 2). However, phenyl glycidyl ether afforded a moderate conversion (76%) and a high selectivity (>92%) (Table 1, entry 3). In contrast, the chain epoxides propylene oxide, 1,2-epoxyhexane and 1,2-epoxyoctane gave a moderate to low epoxide conversion and selectivity (Table 1, entries 5–7). We ascribed the higher carbonate selectivity of the aromatic epoxides over those of the chain epoxides to the possible π⋯π, cationic⋯π and hydrogen bonding interactions between the aromatic substrates and the catalyst frameworks (Table 1, entries 2–4).
Separation of the catalyst from the reaction mixture is easily carried out by centrifugation. This allows the catalyst to be recycled six times for use in the cycloaddition reaction of epichlorohydrin with CO2, without a noticeable loss in activity (Table S6 and Fig. S21†), despite the fact that the BET surface area of 2 decreases to 109 m2 g−1 after catalysis (Fig. 1e). The decreased surface area without sacrifice of the activity of 2 indicated that the active sites on the external surfaces could effectively promote the catalytic reaction. Additionally, the PXRD pattern of 2 after catalysis is in agreement with that of synthesized 2 (Fig. 1a), which demonstrates that the catalyst framework is retained.
Conclusion
In summary, we have successfully synthesized a bifunctional porous imidazolium functionalized Zr-MOF, (I−)Meim-UiO-66, via isoreticular synthesis and a PSM method. This new MOF can serve as an efficient heterogeneous catalyst towards the capture and coupling of CO2 with epoxides at ambient pressure, without a co-catalyst, under mild conditions. It was found that the smaller particles of 2 led to a higher catalytic activity towards CO2 fixation than that of nanosphere 2 (NS). Considering the high stabilities, hierarchical pores and modifiable features of 1 and 1 (NS), it is expected that more functional materials based on Im-UiO-66 will be explored in the future.20Experimental section
Imidazole-containing Zr-MOFs: nanocrystals of Im-UiO-66 (1) were prepared by adding Im-H2BDC·HCl·H2O (57 mg), ZrCl4 (47 mg) and acetic acid (1.2 mL) successively into 5 mL of DMF. The mixture was sonicated sufficiently before being transferred into an autoclave. The autoclave was heated at 393 K for 48 h. The resultant gel was centrifuged and washed repeatedly with DMF and methanol to yield 1. Nanospheres of Im-UiO-66 were synthesized in a similar way: Im-H2BDC·HCl·H2O (57 mg), Zr(NO3)4·5H2O (86 mg) and acetic acid (2 mL) were successively added into 3 mL of DMF. The mixture was sonicated sufficiently before being transferred into an autoclave. Imidazolium functionalized Zr-MOFs: Im-UiO-66 (0.346 g) was stirred and refluxed in a CH3CN (15 mL) suspension of methyl iodide (0.5 mL) for 48 h. The precipitate was collected and washed with CH3CN, MeOH and ether before drying under vacuum to yield (I−)Meim-UiO-66 (2).Acknowledgements
We acknowledge the financial support from the 973 Program (2014CB845605 and 2013CB933200), NSFC (21671188, 21273238, 21521061, and 21331006), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), Youth Innovation Promotion Association, CAS (2014265), and Chunmiao Project of the Haixi Institute of the Chinese Academy of Sciences (CMZX-2014-004).Notes and references
- (a) I. Omae, Coord. Chem. Rev., 2012, 256, 1384 CrossRef CAS; (b) A. A. Olajire, J. CO2 Util., 2013, 3–4, 74 CrossRef CAS; (c) J. Klankermayerf, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296 CrossRef PubMed; (d) A. Schoedel, Z. Ji and O. M. Yaghi, Nat. Energy, 2016, 1, 16034 CrossRef.
- (a) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed; (b) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC; (c) S. Das, F. D. Bobbink, A. Gopakumar and P. J. Dyson, Chimia, 2015, 69, 765–768 CrossRef CAS PubMed; (d) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347 RSC.
- (a) J. Xu, F. Wu, Q. Jiang and Y. X. Li, Catal. Sci. Technol., 2015, 5, 447 RSC; (b) C. C. Rocha, T. Onfroy, J. Pilmé, A. Denicourt-Nowicki, A. Roucoux and F. J. Launay, J. Catal., 2016, 333, 29 CrossRef.
- (a) K. Kasuga and N. Kabata, Inorg. Chim. Acta, 1997, 257, 277 CrossRef CAS; (b) S. Kumar and S. L. Jain, Ind. Eng. Chem. Res., 2014, 53, 541 CrossRef CAS; (c) X. Liu, S. Zhang, Q. W. Song, X. F. Liu, R. Ma and L. N. He, Green Chem., 2016, 18, 2871 RSC.
- (a) J. Peng and Y. Deng, New J. Chem., 2001, 25, 639 RSC; (b) J. Sun, W. Cheng, W. Fan, Y. Wang, Z. Meng and S. Zhang, Catal. Today, 2009, 148, 361 CrossRef CAS; (c) A. L. Girard, N. Simon, M. Zanatta, S. Marmitt, P. Gonçalves and J. Dupont, Green Chem., 2014, 16, 2815 RSC; (d) C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134 CrossRef CAS PubMed; (e) V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615 CrossRef PubMed.
- (a) L. Zhang and Z. M. Hou, Chem. Sci., 2013, 4, 3395 RSC; (b) X. Jiang, F. Gou, F. J. Chen and H. W. Jing, Green Chem., 2016, 18, 3567 RSC.
- (a) X. C. Wang, Y. Zhou, Z. Guo, G. J. Chen, J. Li, Y. M. Shi, Y. Liu and J. Wang, Chem. Sci., 2015, 6, 6916 RSC; (b) D. Yu and Y. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20184 CrossRef CAS PubMed; (c) Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960 Search PubMed; (d) H. C. Cho, H. S. Lee, J. Chun, S. M. Lee, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 917 RSC; (e) G. Fiorani, W. S. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375 RSC; (f) T.-T. Liu, J. Liang, Y.-B. Huang and R. Cao, Chem. Commun., 2016, 52, 13228–13291 Search PubMed.
- (a) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed; (b) Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586 CrossRef CAS PubMed; (c) K.-J. Chen, D. G. Madden, T. Pham, K. A. Forrest, A. Kumar, Q.-Y. Yang, W. Xie, B. Spce, J. J. Perry IV, J.-P. Zhang, X.-M. Cheng and M. J. Zaworotko, Angew. Chem., Int. Ed., 2016, 55, 10268 CrossRef CAS PubMed; (d) S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348–352 CrossRef CAS PubMed; (e) Q. Yang, S. Vaesen, F. Ragon, A. D. Wiersum, D. Wu, A. Lago, T. Devic, C. Matineau, F. Taulelle, P. L. Llewellyn, H. Jobic, C. Zhong, C. Serre, G. D. Weireld and G. Maurin, Angew. Chem., Int. Ed., 2013, 52, 1 CrossRef.
- (a) A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606 CrossRef CAS PubMed; (b) A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804 RSC; (c) W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677 RSC; (d) H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- (a) C. M. Miralda, E. E. Macias, M. I. Zhu, P. Ratnasamy and M. A. Carreon, ACS Catal., 2012, 2, 180 CrossRef CAS; (b) J. Kim, S. N. Kim, H. G. Jang, G. Seo and W. S. Ahn, Appl. Catal., A, 2013, 453, 175 CrossRef CAS; (c) S. Wang and X. C. Wang, Angew. Chem., Int. Ed., 2016, 55, 2308 CrossRef CAS PubMed; (d) D. X. Ma, B. Y. Li, K. Liu, X. Zhang, W. J. Zou, Y. Q. Yang, G. H. Li, Z. Shi and S. H. Feng, J. Mater. Chem. A, 2015, 3, 23136 RSC; (e) J. Zheng, M. Y. Wu, F. L. Jiang, W. P. Su and M. C. Hong, Chem. Sci., 2015, 6, 3466 RSC; (f) P. Z. Li, X. J. Wang, J. Liu, J. S. Lim, R. Zou and Y. J. Zhao, J. Am. Chem. Soc., 2016, 138, 2142 CrossRef CAS PubMed.
- (a) A. Barbarini, R. Maggi, A. Mazzacani, G. Mori, G. Sartori and R. Sartorio, Tetrahedron Lett., 2003, 44, 2931 CrossRef CAS; (b) T. Werner, N. Büttner and H. Tenhumberg, ChemCatChem, 2014, 6, 3493 CrossRef CAS.
- (a) C. I. Ezugwu, N. A. Kabir, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 307, 188 CrossRef CAS; (b) K. Fujie and H. Kitagawa, Coord. Chem. Rev., 2016, 307, 382 CrossRef CAS.
- (a) Q. Lin, T. Wu, S. T. Zheng, X. H. Bu and P. Y. Feng, J. Am. Chem. Soc., 2012, 134, 784 CrossRef CAS PubMed; (b) J. S. Qin, D. Y. Du, W. L. Li, J. P. Zhang, S. L. Li, Z. M. Su, X. L. Wang, Q. Xu, K. Z. Shao and Y. Q. Lan, Chem. Sci., 2012, 3, 2114 RSC; (c) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724 CrossRef CAS PubMed.
- J. C. Wang, J. P. Ma, Q. K. Liu, Y. H. Hu and Y. B. Dong, Chem. Commun., 2016, 52, 6989 RSC.
- (a) Y. Bai, Y. B. Dou, L. H. Xie, W. Rutledge, J. R. Li and H.-C. Zhou, Chem. Soc. Rev., 2016, 45, 2327 RSC; (b) J. H. Cacka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed; (c) S. Yuan, L. F. Zou, H. X. Li, Y. P. Chen, J. S. Qin, Q. Zhang, W. G. Lu, M. B. Hall and H.-C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef; (d) F. Zhang, S. Zheng, Q. Xiao, Y. J. Zhong, W. D. Zhu, A. Lin and M. S. El-Shall, Green Chem., 2016, 18, 2900 RSC; (e) G. Ferey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS PubMed; (f) G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749 CrossRef.
- (a) Y. Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Chem. Commun., 2016, 52, 7806 RSC; (b) M. J. Cliffe, W. Wan, X. D. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CAS.
- (a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed; (b) N. W. Ockwig, O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS PubMed; (c) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (d) Y. Zhang and D. S. W. Lim, ChemSusChem, 2015, 8, 2606 CrossRef CAS PubMed.
- (a) S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS PubMed; (b) P. L. Arnold, A. C. Scarisbrick, A. J. Blake and C. Wilson, Chem. Commun., 2001, 2340 RSC . The epichlorohydrin substrate reacted with imidazole groups on 1 to form complex ionic catalyst (Cl−)Rim-UiO-66. This is confirmed by mass and FT-IR spectra (Fig. S19 and S20†).
- J. A. Stewart, R. Drexel, B. Arstad, E. Reubsaet, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2016, 18, 1605 RSC.
- (a) K. Fujie and H. Kitagawa, Coord. Chem. Rev., 2016, 307, 382 CrossRef CAS; (b) S. Zhang, K. Dokko and M. Watanabe, Chem. Sci., 2015, 6, 3684 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General comments, syntheses and characterizations of the MOFs mentioned in the manuscript, details of the single crystal diffraction experiments, PXRD, TG and additional figures. CCDC 1499016. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04357g |
| This journal is © The Royal Society of Chemistry 2017 |