
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step electric-field driven methane and formaldehyde synthesis from liquid methanol†
Giuseppe
Cassone
 *a,
Fabio
Pietrucci
b,
Franz
Saija
c,
François
Guyot
b and
A. Marco
Saitta
b
*a,
Fabio
Pietrucci
b,
Franz
Saija
c,
François
Guyot
b and
A. Marco
Saitta
b
aInstitute of Biophysics – Czech Academy of Sciences, Královopolská 135, 61265 Brno, Czech Republic. E-mail: giuseppe.cassone@impmc.upmc.fr
bSorbonne Universités, Université Pierre et Marie Curie Paris 06, Institut de Minéralogie, de Physique des Matériaux et de Cosmochimie, CNRS, Muséum national d'Histoire naturelle, Institut de Recherche pour le Développement, Unité Mixte de Recherche 7590, F-75005 Paris, France. E-mail: fabio.pietrucci@impmc.upmc.fr; fguyot@mnhn.fr; marco.saitta@impmc.upmc.fr
cCNR-IPCF, Viale Ferdinando Stagno d'Alcontres 37, 98158 Messina, Italy. E-mail: saija@ipcf.cnr.it
First published on 5th December 2016
Abstract
The reaction pathways connecting methanol to methane and formaldehyde are among the most emblematic in chemistry because of their outstanding interest in the fields of energy, synthesis, and bio- and geo-chemistry. Despite of its fundamental nature, the one-pot synthesis of formaldehyde and methane stemming from methanol has never been reported before. Here we present a study, based on ab initio molecular dynamics and free-energy methods, in which the simultaneous oxidation and reduction (i.e., the disproportionation) of liquid methanol into methane and formaldehyde has been achieved at ambient temperature through the application of a static electric field. Because strong electric fields can be generated in the proximity of field emitter tips, this finding shows that the challenge of experimentally disproportionating methanol into formaldehyde and methane could be attempted. We show that the methanol “solvent” molecules play a major role in this process and that the chemical pathway connecting methanol to the detected products in the bulk liquid phase is very different from its reproduced gas-phase counterpart. Finally, we demonstrate that switching on an external electric field drastically modifies the reaction network of methanol, lowering some activation barriers, stabilizing the methane and formaldehyde products, and opening otherwise difficult-to-achieve chemical routes.
I. Introduction
Albeit the reaction network characterizing the chemistry of methanol (CH3OH) is highly multifaceted,1–6 the industrially relevant reaction routes starting from the simplest alcohol are well-established.7 There exists a huge variety of chemical transformations in which methanol can be employed as a primary reactant and that can give rise to the formation of hydrocarbons,1,4 olefins,2 di-hydrogen,3 dimethyl ether,5 and so on. Due to this evidence, in addition to the fact that methanol can be used as an energy storage system, the possibility to base the world economy on this simple compound has been envisaged.7While methane (CH4) is typically found as a by-product of the hydrocarbon synthesis from methanol1 and, for commercial reasons, the reverse reaction – i.e., from methane to methanol – is strongly encouraged,8,9 formaldehyde (H2CO) synthesis holds a privileged place among the chemical pathways that can be erected from methanol. Indeed, due to its industrial and economic importance,10 great effort has been made in order to control and enhance the production yields of the simplest aldehyde stemming from the simplest alcohol.
The conversion of methanol to formaldehyde is traditionally achieved in the gas phase and in the presence of very specific catalysts11–15 (i.e., molybdenum oxide based catalysts,11 silver,16 copper surfaces,17,18etc.). Also in plasma-phase chemistry, some chemical routes that branch out from methanol decomposition show the presence of formaldehyde19 or of the latter and methane20 among a plethora of other gas-phase by-products.
Although the production of formaldehyde stemming from methanol can be also obtained under supercritical aqueous conditions (i.e., T ∼ 500 °C and P ∼ 24 MPa),21 the unique renowned way to synthesise the simplest aldehyde in the condensed phase under standard conditions occurs by means of alcohol dehydrogenase (ADH) enzymes in biological systems (e.g., in the human liver).
Recently, methanol chemistry has received attention in astrochemistry and in the fast-growing field of prebiotic chemistry. Indeed, newly proposed mechanisms of hydrocarbon production from the simplest alcohol in interstellar space have just been suggested by Olah et al.22 Incidentally, a steady growing consensus on (proto)sugar synthesis, starting from methanol as the unique carbon source, has been recorded:23,24 it is plausible that methanol-based chemistry may play an extremely relevant role in synthesising sugars through formaldehyde.
Electric fields can operate as promoters of specific reaction channels. The first experimental evidence that an electric field can control chemical reactions has been recently provided.25 On the other hand, recent ab initio molecular dynamics (AIMD) studies have succeeded in describing complex chemical reactions of small organic molecules under extreme conditions of confinement26 and pressure,27,28 and, more specifically, in the case of strong electric fields in water,29 quantitatively confirmed by experiments,30,31 including a study mimicking in silico the historical Miller experiment.32
Here we present a so far unreported chemical reaction which occurs when a sample of neat liquid methanol is exposed to a static and homogeneous electric field. The formation of formaldehyde, with the consequent release of methane and water, has been detected during our computational experiment. In order to assess the energetic contribution of the field to the reaction and the field-induced changes in the thermodynamic paths undertaken by the newly revealed reaction, we have employed our own recent method33 of unbiased exploration of chemical reaction networks, capable of revealing unexpected pathways and microscopic mechanisms and, at the same time to provide the free-energy landscape of those reactions, fully including the effect of the explicit solvent and of the thermodynamic conditions.
II. Methods
Based on the Car–Parrinello (CP) approach,34 we performed a series of AIMD simulations of a sample of liquid methanol at ambient temperature under the action of intense electric fields. Our sample contained 32 CH3OH molecules (i.e., 192 atoms) arranged in a cubic cell with side parameter a = 12.93 Å, so as to reproduce the experimental density of 0.79 g cm−3 at about 300 K. As usual, the structure was replicated in space by using periodic boundary conditions. We gradually increased the intensity of the electric field with a step increment of about 0.05 V Å−1 from zero up to a maximum of 0.60 V Å−1, cumulating a total simulation time of about 35 ps. The fictitious electronic mass was set to a value of 300 a.u., with a cutoff energy of 35 Rydberg (Ry) and a cutoff energy for the charge density of 280 Ry, which allowed us to adopt a timestep of 0.12 fs. With such cutoff values the sample is described in a realistic way since the core electronic interaction is being depicted through ultrasoft pseudopotentials (USPP). As for exchange and correlation effects, we adopted the Perdew–Burke–Ernzerhof (PBE) functional.35 The dynamics of the ions were simulated classically within a constant number, volume, and temperature (NVT) ensemble.The free-energy landscape reconstruction has been executed in two steps. The first step had the aim of exploring the relevant basins and the chemical pathways in the Collective-Variables (CV) space. This has been achieved by employing the method presented in ref. 33. The second step aimed at the effective sampling of the known pathways by means of the umbrella sampling36 approach, which has been exploited for the first time in a complementary fashion to the previous method. This protocol was revealed to be completely reliable both for gas phase and condensed phase chemical reactions. See the ESI† for further details on all the performed calculations and for the full treatment of the gas-phase counterpart of the reaction here analysed.
III. Results and discussion
A. One-pot synthesis of formaldehyde and methane
By applying a static electric field of intensity equal to or greater than 0.30 V Å−1 on a sample of liquid methanol, it is possible to induce the cleavage of the OH alcohol covalent bond which results in the concept of generalization of pH in methanol.37 The dissociated proton can then migrate via a Grotthuss-like mechanism along the H-bond network.37 This process is assisted by a certain fraction of ionic species, such as the methanol cation CH3OH2+ (methyloxonium) and anion CH3O− (methoxide), which transiently exist and are responsible for the ionic charge flow in the system. Hence, if the field strength is increased above this threshold, the field effects will be manifested also as a consequence of the presence of several ions. This leads to locally enhanced contributions to the external electrostatic potential which, in turn, produce an increase in molecular reactivity. If in neat water, under ambient conditions, fields of ∼0.50 V Å−1 can give rise only to improved proton transfer,29 the reactivity of carbon atoms in methanol leads to more complex chemical transformations. In fact, we observe that a field strength equal to 0.55 V Å−1, assisted by local molecular advantageous circumstances (i.e., nearest neighbors arrangements), is able to break one of the covalent bonds in some methyl groups of methoxide molecules and to create formaldehyde. The formation of this species is naturally accompanied by the release of water and methane according to the following reaction: | (1) |
Although the intensity of these fields appears somewhat prohibitive, they are so far outweighed in disparate electrode tip experiments,38–41 and the dissociation of simple molecules has been reported for field strengths of ∼0.3 V Å−1.30 Moreover, the spontaneous presence of even stronger fields in the proximity of certain clean and apolar mineral surfaces42 and in regions surrounding solvated ionic species43–45 is well established.
For the sake of a clearer description, albeit in pure liquid methanol there is no difference between solute and solvent molecules, we will indicate as solvent all the other methanol molecules surrounding the ones involved in the chemical reactions.
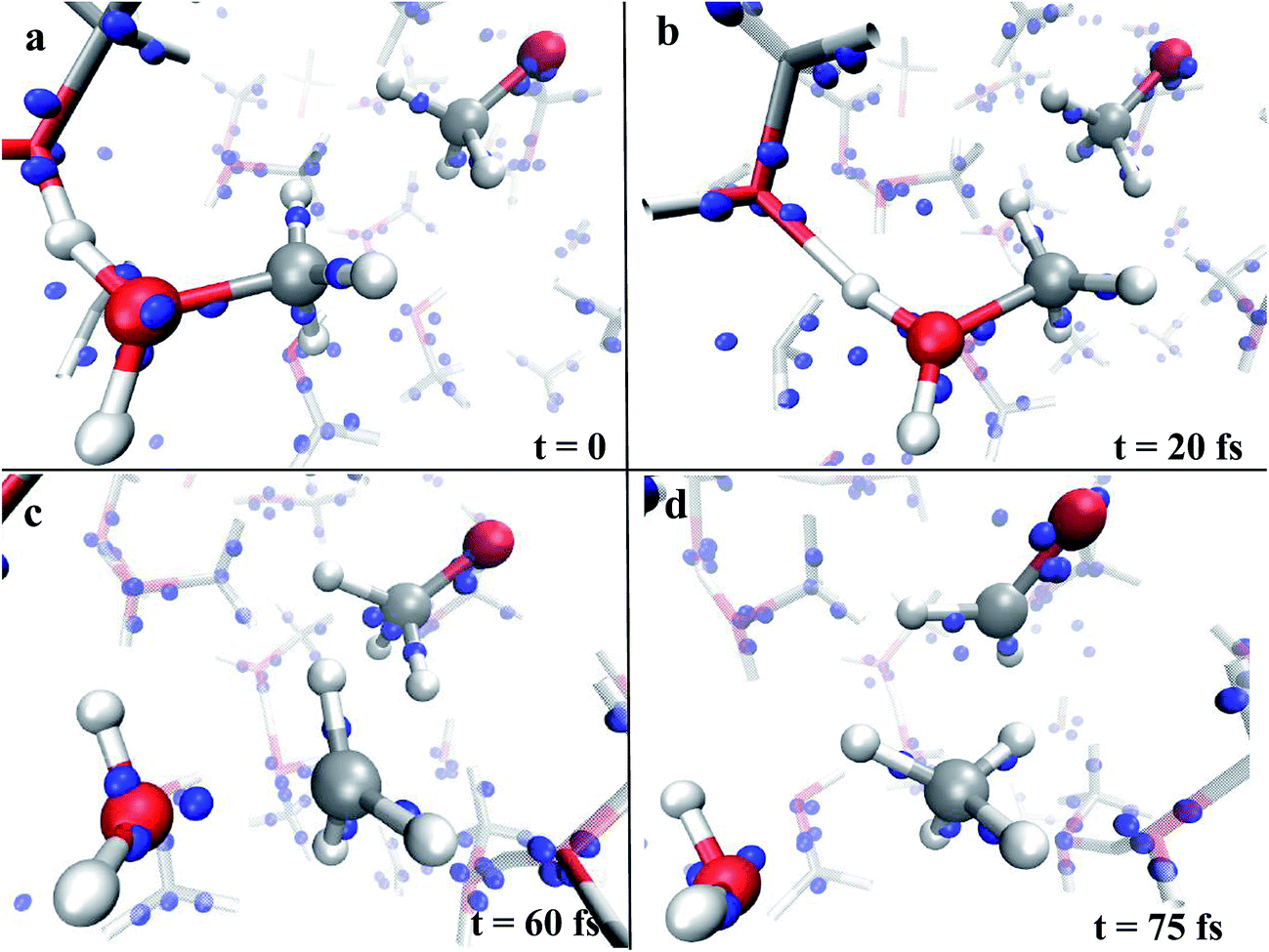
In the condensed phase, at a given instant, several molecules are arranged in such a way that the most electronegative “pole” is oriented towards the field direction (e.g., see the methoxide CH3O− in Fig. 1a). Moreover, also as a consequence of the activated proton transfer, some methyloxonium cations may be oriented with the excess proton in opposition to the electric field direction, as shown in Fig. 1a and e. In particular, during the proton migration process, an oxygen to oxygen intermolecular distance of ∼2.3 Å with an evenly shared proton can be transiently observed. This represents the formation of a transient Zundel-like ion,37 shown in Fig. 1a and e for two field intensities (i.e., the [C2H6O2H3]+ complex), which readily leads to the release of a proton from one donor methanol molecule to an acceptor one. This way, the typical CO bond length of the just formed methyloxonium cation (Fig. 1b and f) becomes sensibly higher than the one characterizing the neutral molecular state in which this species lay before accepting the proton from the solvent. The stretch of the CO bond, if assisted by the local presence of a methoxide anion with its methyl group as its first neighbor, leads to a visible decrease of the CO bond strength (see Fig. 1b, c, f and g). Indeed in few dozens of fs the CO covalent bond of CH3OH2+ is broken whereas the distance between the carbon atom of the newly formed methenium ion CH3+ (see Fig. 1c and g) and the closest methyl hydrogen atom of CH3O− readily approaches a very small value. The final step of this concerted reaction is represented by the ultrafast release of a hydride H− stemming from the methoxide anion, which recombines with the methenium just after the “umbrella inversion” of the latter (Fig. 1d and h). The transformation of methoxide into formaldehyde is characterized by a drastic reduction of the relative CO bond length (i.e., ∼1.2 Å), which is a manifestation of its peculiar double bond formation. A short movie in mpg format of the whole reaction mechanism is available.† The mechanistic pictures described in Fig. 1 are in practice stackable and a recurrent pattern can be recognized for the whole process: the proton transfer that triggers the reaction and the subsequent electrostatic approach of the two counterions, which are arranged in a specific orientation with respect to each other and with respect to the field. Although the observed methanol dismutation appears to be highly unlikely under standard conditions and at high temperatures (calculations not shown), the electric field is able to orient the system towards the formaldehyde, methane, and water synthesis. Moreover, by switching off the external field at a strength of 0.55 V Å−1, once the first formaldehyde, water, and methane molecules have been formed in the system, it has been observed that they remain stable.
 | ||
| Fig. 1 Formaldehyde and methane formation mechanism in the presence of a static electric field oriented along the positive z-axis (i.e., the blue Cartesian axis direction) with strengths of 0.55 V Å−1 (left panel) and 0.60 V Å−1 (right panel). Red, silver, and white coloring refers to oxygen, carbon, and hydrogen atoms, respectively. Four typical distances (shown in Å) have been chosen in order to better follow the reaction: the oxygen to oxygen separation between one solvent molecule and the (future, in panel (a)) CH3OH2+, the CO bond length of the latter, the distance between the carbon of methyloxonium and the closest methyl hydrogen atom of CH3O−, and the CO bond length of the latter. A proton transfer event (a and e) leads to the elongation of the CO bond of the newly formed methyloxonium cation (b and f), i.e., from 1.47 Å to 1.59 Å (left panel) and from 1.48 Å to 1.57 Å (right panel). Simultaneously, electrostatic effects further shorten the CH distance between the carbon of CH3OH2+ and its closest extramolecular hydrogen (b and f). This way the methyloxonium species undergoes the cleavage of its characteristic CO bond (i.e., CO distance of 1.96 Å (left panel) and 1.79 Å (right panel)) and a methenium ion CH3+ is thus transiently formed (c and g). The ultrafast recombination of the latter with a hydride H− stemming from the methoxide ion leads to the formation of water, methane, and formaldehyde (d and h). | ||
A useful method that characterizes the electronic ground state properties of a condensed system is represented by the Wannier functions46,47 and, in particular, by the Maximally Localised Wannier Functions (MLWF).48–50 One of the key factors that can be extracted from the MLWF are their charge centers which are a sort of quantum equivalent of the classical concept of the location of an electron pair and thus allow for direct visualization of the bond’s behaviour. The Wannier charge centers characterizing the dynamical progress of the observed reaction at 0.55 V Å−1 are displayed in Fig. 2. In particular, Fig. 2a and b show in the foreground the proton transfer process between two methanol molecules and, in the right part of the relative panels, a methoxide molecule. Since methanol is by itself a polar molecule and, secondly, because of the field-induced polarization effects, the charge centers identifying the CO bonds are slightly shifted towards the oxygen atoms of the shown species. A magnification of the progress of the reaction is shown in Fig. 2c and d. In particular, in Fig. 2c, the cleavage of the methyloxonium CO bond and the further approach of the respective Wannier center to the oxygen atom of the forming water molecule should be noted. This leads to the release of the methenium cation CH3+ as a transient species. In the meantime, a dislocation of the Wannier centers from their standard positions of the methyl group of methoxide has been observed. Indeed, the methyl hydrogen closest to the methenium cation is preparing to bring an electron pair and thus becoming a hydride H−. The further neutralization between the two transient species (i.e., CH3+ and H−) is shown in Fig. 2d. In the latter, besides trivial distortions due to the re-arrangement of the local electronic structure, the formation of the typical formaldehyde CO double bond can be observed.
 | ||
| Fig. 2 Wannier charge centers (blue small spheres) characterizing the dynamical evolution of the reaction shown in the left panel of Fig. 1, which leads to the formation of water, methane, and formaldehyde. The electric field orientation is exactly the same as that shown in Fig. 1 (i.e., from left to right). | ||
In order to better visualize and quantify the solvent effects in screening the bare action of the applied electric field on the reactant atoms of the reaction, a Löwdin population analysis has also been performed both in the condensed and gas phase (see the ESI†). This investigation suggests that the solvent is able to locally screen the field effect on the sampled atomic configurations. The microscopic evidence of the assistance of the solvent in guiding the reaction (i.e., via proton transfers) and the feeble field-induced changes of the atomic charge distributions suggest that the correlations between the interacting molecules play an important role in the evolution of reaction (1).
As stated before, field intensities of the order or greater than 1 V Å−1 are currently found (locally) in disparate condensed systems in which simple solvated ionic species are present,43–45 or even at the surface of clean and apolar oxides, such as the (001) MgO surface.42 In addition, external field strengths of the order of 0.5 V Å−1 have been also employed in order to reproduce in silico the historical Miller experiment.32 In field emitter tip experiments, field strengths of 1–3 V Å−1 are recorded38–40 and it has been proven that intensities of ∼0.3 V Å−1 are necessary in order to induce the cleavage of some covalent bonds (i.e., water dissociation).29,30 Since a very similar computational setup29 has been exploited in order to accurately predict the experimental dissociation threshold of the water molecule,31 thus confirming some preliminary30 and extremely recent51 results, there is no reason to think that the same reliability is not preserved for the present chemical reaction. Hence, all the evidence suggests the concrete experimental feasibility of reproducing the proposed reaction by exploiting, as an example, the high field capability of field emitter tips.
B. Free-energy landscapes in the liquid phase
Despite the importance of elucidating the microscopic process that characterizes the above discussed chemical reaction, more fundamental thermodynamic considerations are in order, beyond its mechanistic description. Enhanced sampling techniques52 allow for the evaluation of the free energy surface (FES) of disparate processes and, recently, a path-Collective-Variables (path-CV) metadynamics method has been developed,33 which is particularly useful for condensed phase reactions. Within this technique, the portions of the phase space close to the reactants (i.e., 2 neutral methanol molecules in a bath of methanol) and the products (i.e., a formaldehyde, a methane, and a water molecule surrounded by methanol molecules) basins have been at first explored and consequently sampled in solution (and in the gas phase, see the ESI†). This way, the FES of reaction (1) has been reconstructed in the zero-field regime and under the action of a field strength of 0.30 V Å−1. The choice of sampling the system under a lower field intensity than that which renders the reaction barrierless (0.55 V Å−1) is dictated by the necessity of monitoring and quantifying the field-induced changes on the free energy landscape for field intensities that are not large enough to induce the barrierless formation of formaldehyde, methane, and water. In Fig. 3, the free energy landscape of reaction (1) in the absence of the electric field is shown. The chemical transformation is inhibited by the presence of a high free-energy barrier of 45 kcal mol−1. The catalysis effects of the external electric field are manifested from the induced changes on the FES of reaction (1), as shown in Fig. 4. Even in the presence of a field strength lower than that capable of rendering the reaction barrierless, the products are stabilized: a comparable free energy estimate characterizes the initial and the final states. Another important aspect concerns the field-induced changes in the transition region (S ∼ 1.45). By comparing the FES evaluated in the zero-field regime (Fig. 3) with that in the presence of a field strength of 0.30 V Å−1 (Fig. 4), an evident modification of the shape can be recognized. In fact, a field strength that is roughly half (0.30 V Å−1) of that which is able to overcome the reaction barrier (0.55 V Å−1), definitely lowers a portion of the barrier of approximately a third with respect to that of the fieldless case, i.e., ∼30 kcal mol−1vs. ∼45 kcal mol−1, respectively. This finding is not so surprising if one takes into account that the solvent is fundamental in assisting this kind of reaction. Indeed, the field strength of 0.30 V Å−1 represents the known methanol dissociation threshold37 and the formation of ionic species such as CH3OH2+ and CH3O− produces intense local fields, as is usual when dealing with solvated ions.43–45 Moreover, the region of the free energy barrier that is significantly modified by the field action corresponds to the effectively recorded passage in our unbiased AIMD simulation (i.e., it is the natural reaction pathway when a strong electric field is applied). Hence the external electric field is able to open reaction pathways that were avoided in the zero-field regime. Indeed, the two observed reaction mechanisms in absence of the electric field and the one observed at 0.55 V Å−1 during our theoretical experiment are substantially different as shown in Fig. 5. | ||
| Fig. 3 FES of reaction (1) in the zero-field regime. The energy scale (depth) is in kcal mol−1 whereas the S-axis and the Z-axis represent the progress along the reaction and a sort of distance from its ideal path, respectively. Low values of S characterize a system of pure liquid methanol whereas high values of this parameter describe a sample composed of a formaldehyde, a methane, and a water molecule in a bath of methanol molecules. In the picture frames are shown the local structures of reactants, transition states, and products corresponding to their relative location on the FES in the space spanned by the CV. | ||
 | ||
| Fig. 4 FES of reaction (1) under a relatively moderate field strength of 0.30 V Å−1. The energy scale (depth) is in kcal mol−1 whereas the S-axis and the Z-axis represent the progress along the reaction and a sort of distance from its ideal path, respectively. Low values of S characterize a system of pure liquid methanol whereas high values of this parameter describe a sample composed of a formaldehyde, a methane, and a water molecule in a bath of methanol molecules and its ionic equivalents. In the picture frames are shown the local structures of reactants, transition states, and products corresponding to their relative location on the FES in the space spanned by the CV. For (S; Z) ∼ (1.45; 0.3–0.4), where is located the observed transition state in presence of a field of 0.55 V Å−1, the free energy is ∼30–35 kcal mol−1. This value is not negligibly lower than that observed for the fieldless case (i.e., ∼45 kcal mol−1) (see Fig. 3). | ||
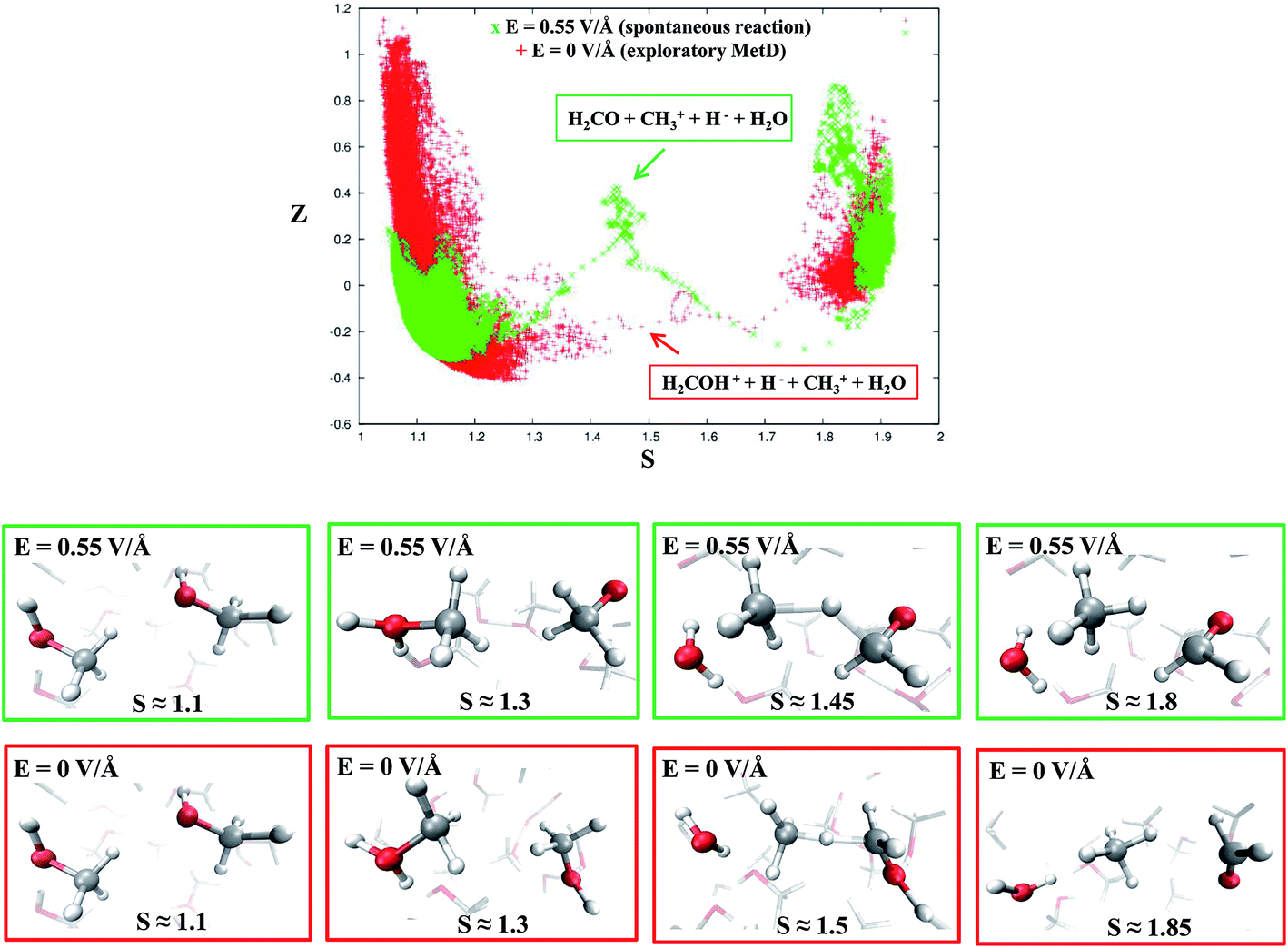 | ||
| Fig. 5 Upper panel: reaction pathways for reaction (1) in the CV space at the field strength that renders the reaction barrierless (green crosses) and in the zero-field regime, i.e., metadynamics-driven reaction (red crosses). Middle panel (green frames): dynamical evolution of the reaction in the presence of a field strength of 0.55 V Å−1 in which reactants, intermediates, transition states, and products (from left to right) are shown, respectively. Lower panel (red frames): dynamical evolution of the reaction in the biased metadynamics simulation in the zero-field regime in which reactants, intermediates, transition states, and products (from left to right) are shown, respectively. | ||
C. Transition state detection
With the aim of fully characterizing the transition states (TSs) of all the observed reactions, at zero or finite fields, committor analysis53 has been performed, by shooting many unbiased trajectories, each one starting from a structure thought to be a plausible transition state. In the middle panel of Fig. 6 the most likely TS of the reaction in the zero-field regime is shown. Launching 40 independent simulations from this specific configuration, in the absence of an electric field, it has been observed that 60% and 40% of them fell in the reactant and in the product basin, respectively (upper part of Fig. 6). We thus define this initial molecular state as the likely TS. Moreover, from a similar kind of statistical analysis, additional powerful information can be gained if one performs the committor analysis on the same configuration TS in the presence of a field with an intensity of 0.30 V Å−1, once oriented along the positive z-axis (right part of Fig. 6) and once towards the opposite direction (left part of Fig. 6). The change of the topological arrangement of the molecules with respect to the field direction induces dramatically different paths. In one case the reaction proceeds always to the products, whereas in the other case the atomic configuration always and suddenly evolves to the typical reactants configuration. This means that the orientation of the field qualitatively changes some features of the evolution of a given chemical reaction. The committor analysis has also been conducted in order to identify the TS of the reaction at 0.30 V Å−1, which is shown in the middle panel of Fig. 7. Again, 50 ± 10% of the trajectories starting from the shown TS structure under the effect of a field intensity equal to 0.30 V Å−1 fell either in the reactant or in the product basin. This TS is different from that recorded in the zero-field regime, suggesting that the electric field strength effectively modifies the accessible reaction pathways by changing the TS. The latter, in this case, resembles more the reactant state with respect to that of the zero-field regime, where the TS shows a molecular configuration towards the product state. Of course, it is not just the field intensity that plays a crucial role in shaping the TSs, but field-induced differences may be ascribed also to the peculiar field direction. As shown in the lower part of Fig. 6, if the field orientation is reversed, the trajectories evolve either always towards the reactants or always towards the products. This latter finding echoes the recent discovery that an electric field can promote or hinder a chemical reaction;25 moreover, our molecular-level study indicates that the electric field is capable of finely shaping the pathways of a chemical transformation depending on its strength and on its direction.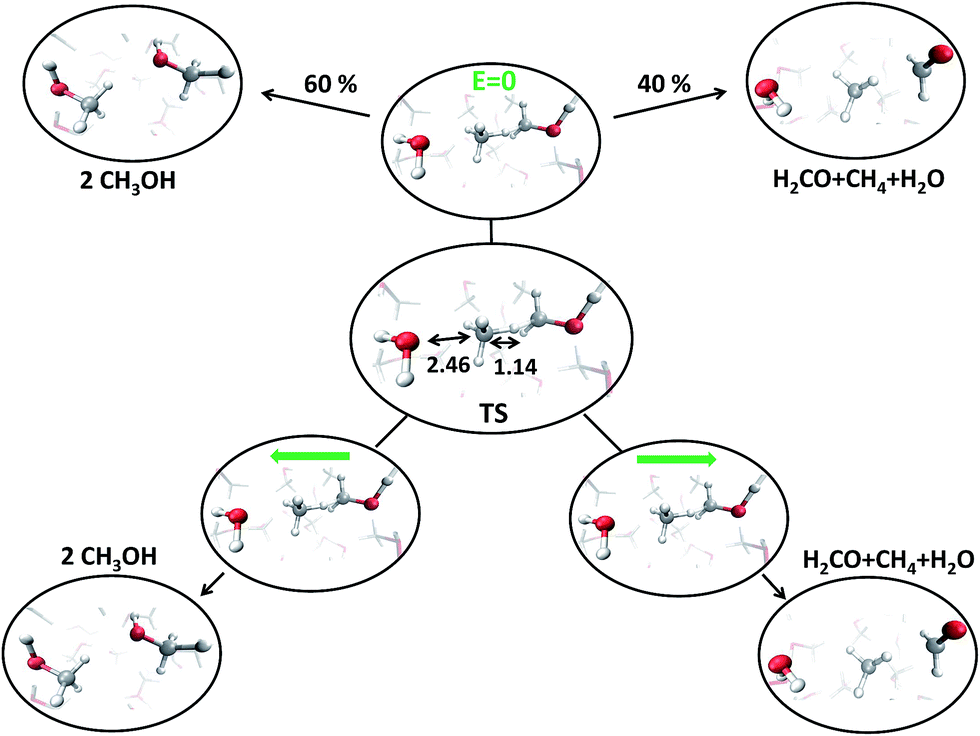 | ||
| Fig. 6 Transition state (TS) (center) of the metadynamics-driven reaction, as confirmed by the committor analysis shown in the upper part; some relevant distances (in Å), such as the CO and the CH lengths are highlighted. By launching 40 independent AIMD simulations from the TS, 60% and 40% of them fall in the reactant and product basin, respectively. From the center to the lower part, the same analysis is shown by applying, independently, opposite electric field directions. In the left part, the reaction is always inhibited (i.e., 100% of the trajectories fall into the reactant state) whereas, in the right part, it is always promoted (i.e., 100% of the trajectories fall into the product state). | ||
 | ||
| Fig. 7 Transition state (TS) (center) for a field strength of 0.30 V Å−1, as confirmed by the committor analysis shown in the upper part; some relevant distances (in Å), such as the CO and the CH lengths are highlighted. Comparing them with the zero-field case: in the presence of the field the reaction is “anticipated”, with the TS more resembling the reactant configuration. By launching 40 independent AIMD simulations from the TS, 40% and 60% of them fall in the reactant and product basin, respectively. From the center to the lower part, the same analysis is shown by applying in one case a reversed electric field direction, whereas in the other case the zero-field has been also tested for a one-to-one comparison with the zero-field case of Fig. 6. In both cases, the reaction is always inhibited (i.e., 100% of the trajectories fall into the reactant state). | ||
IV. Conclusions
In this paper we studied the effects of the application of intense electric fields on bulk liquid methanol by means of ab initio molecular dynamics and free-energy techniques. Our results revealed that field strengths beyond 0.50 V Å−1 are able to trigger the one-pot formation of formaldehyde, methane, and water from neat liquid methanol. This disproportionation reaction can be seen as a new basic chemistry “textbook” example and it may open new chemical pathways for formaldehyde synthesis from methanol. Indeed, electric fields of this order of magnitude (and even higher) are currently generated in the proximity of field emitter tips and the proposed reaction may be experimentally reproduced.We show the way in which the electric field plays a major role both in modifying the free-energy activation barriers and in reshaping the methanol reaction networks. This is due to the key role of the surrounding “solvent” methanol molecules in triggering and promoting the field-induced chemical pathways. This reaction happens each time these microscopic conditions are met and it is clear that strong electric fields help it dramatically. The importance of the “solvent” molecules is highlighted, inter alia, by the fact that the gas-phase variant of the disproportionation reaction of methanol is strongly discouraged, being characterized by a very high activation barrier even in the presence of the electric field. However, in the reproduced gas-phase reaction, an evident field-induced increase of the selectivity of the reaction has been recorded.
On one hand, we provide a powerful and efficient method for exploration of chemical reactions in the condensed phase, and on the other hand, our results show the effects that external electric fields have on orienting and modifying chemical reactions and, moreover, they reveal several microscopic and thermodynamic aspects through which the field acts. All the presented findings suggest the concrete experimental feasibility of synthesising formaldehyde and methane from liquid methanol by means of the high field capability of field emitter tip apparatus.
Acknowledgements
We acknowledge the GENCI-IDRIS French National Supercomputing Facility for CPU time under projects x2015091387 and x2016091387. G. C. thanks M. S. Cassone for graphical assistance and G. Mondello for helpful discussions. G. C. thanks S. Savasta and P. V. Giaquinta for encouragements and support.References
- C. D. Chang, Catal. Rev.: Sci. Eng., 1983, 25, 1–118 Search PubMed.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1939 CrossRef.
- P. J. de Wild and M. J. F. M. Verhaak, Catal. Today, 2000, 60, 3–10 CrossRef.
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149, 458–464 CrossRef.
- M. Xu, J. H. Lunsford, D. N. Goodman and A. Battacharyya, Appl. Catal., A, 1997, 149, 289–301 CrossRef.
- W. Li, C. Pan, Q. Zhang, Z. Liu, J. Peng, P. Chen and H. Lou, Bioresour. Technol., 2011, 102, 4884–4889 CrossRef PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, Winheim, Germany, 2009 Search PubMed.
- H. D. Gesser, N. R. Hunter and C. B. Prakash, Chem. Rev., 1985, 85, 235–244 CrossRef.
- D. Wolf, Angew. Chem., Int. Ed., 1998, 37, 3351–3353 CrossRef.
- H. R. Gerberich and G. C. Seaman, Formaldehyde in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley and Sons, New York, USA, 1994 Search PubMed.
- T. Waters, R. A. J. O'Hair and A. G. Wedd, J. Am. Chem. Soc., 2002, 125, 3384–3396 CrossRef PubMed.
- J. Döbler, M. Pritzsche and J. Sauer, J. Am. Chem. Soc., 2005, 127, 10861–10868 CrossRef PubMed.
- M. House, A. F. Carley and M. Bowker, J. Catal., 2007, 252, 88–96 CrossRef.
- K. Yoshizawa and Y. Kagawa, J. Phys. Chem. A, 2000, 104, 9347–9355 CrossRef.
- A. M. Beale, S. D. M Jacques, E. Sacaliuc-Parvalescu, M. G. O'Brien, P. Barnes and B. M. Weckhuysen, Appl. Catal., A, 2009, 363, 143–152 CrossRef.
- A. Nagy, G. Mestl, T. Rühle, G. Weinberg and R. Schlögl, J. Catal., 1998, 179, 548–559 CrossRef.
- A. K. Chen and R. Masel, Surf. Sci., 1995, 343, 17–23 CrossRef.
- A. F. Carley, P. R. Davies, G. G. Mariotti and S. Read, Surf. Sci., 1996, 364, L525–L529 CrossRef.
- G. Chuah, N. Kruse, J. Block and G. Abend, J. Phys., Colloq., 1988, 49, 215–220 CrossRef.
- H. Zhang, X. Li, F. Zhu, Z. Bo, K. Cen and X. Tu, Int. J. Hydrogen Energy, 2015, 40, 15901–15912 CrossRef.
- S. F. Rice, T. B. Hunter, Å. C. Rydén and R. G. Hanush, Ind. Eng. Chem. Res., 1996, 35, 2161–2171 CrossRef.
- G. A. Olah, T. Mathew, G. K. Surya Prakash and G. Rasul, J. Am. Chem. Soc., 2016, 138, 1717–1722 CrossRef PubMed.
- P. de Marcellus, C. Meinert, I. Myrgorodska, L. Nahon, T. Buhse, L. Le Sergent d’Hendecourt and U. J. Meierhenrich, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 965–970 CrossRef PubMed.
- C. Meinert, I. Myrgorodska, P. de Marcellus, T. Buhse, L. Nahon, S. V. Hoffmann, L. Le Sergent d’Hendecourt and U. J. Meierhenrich, Science, 2016, 352, 208–212 CrossRef PubMed.
- A. C. Aragones, N. L. Haworth, N. Darwish, S. Ciampi, N. J. Bloomfield, G. G. Wallace, I. Diez-Perez and M. L. Coote, Nature, 2016, 531, 88–91 CrossRef PubMed.
- E. Schreiner, N. N. Nair and D. Marx, J. Am. Chem. Soc., 2008, 130, 2768–2770 CrossRef PubMed.
- N. Goldman, J. E. Reed, L. E. Fried, I.-F. W. Kuo and A. Maiti, Nat. Chem., 2010, 2, 949–954 CrossRef PubMed.
- M.-S. Lee and S. Scandolo, Nat. Commun., 2011, 2, 185 CrossRef PubMed.
- A. M. Saitta, F. Saija and P. V. Giaquinta, Phys. Rev. Lett., 2012, 108, 207801 CrossRef PubMed.
- E. M. Stuve, Chem. Phys. Lett., 2012, 519–520, 1–17 CrossRef.
- Z. Hammadi, M. Descoins, E. Salanon and R. Morin, Appl. Phys. Lett., 2012, 101, 243110 CrossRef.
- A. M. Saitta and F. Saija, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13768–13773 CrossRef CAS PubMed.
- F. Pietrucci and A. M. Saitta, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15030–15035 CrossRef CAS PubMed.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 ( Phys. Rev. Lett. , 1997 , 78 , 1396 ) CrossRef CAS PubMed.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- G. Cassone, P. V. Giaquinta, F. Saija and A. M. Saitta, J. Chem. Phys., 2015, 142, 054502 CrossRef PubMed.
- D. Price and J. W. Halley, J. Electroanal. Chem., 1983, 159, 347 CrossRef.
- J. Kreuzer, Surf. Sci., 1991, 246, 336–347 CrossRef.
- W. Schmickler, Surf. Sci., 1995, 335, 416–421 CrossRef CAS.
- W.-K. Lee, et al. , Nano Res., 2013, 6, 767–774 CrossRef CAS.
- S. Laporte, F. Finocchi, L. Paulatto, M. Blanchard, E. Balan, F. Guyot and A. M. Saitta, Phys. Chem. Chem. Phys., 2015, 17, 20382–20390 RSC.
- B. Sellner, M. Valiev and S. M. Kathman, J. Phys. Chem. B, 2013, 117, 10869–10882 CrossRef CAS PubMed.
- B. Reischl, J. Köfinger and C. Dellago, Mol. Phys., 2009, 107, 495–502 CrossRef CAS.
- Y. Bronstein, P. Depondt, L. E. Bove, R. Gaal, A. M. Saitta and F. Finocchi, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 024104 CrossRef.
- G. H. Wannier, Phys. Rev., 1937, 52, 191–197 CrossRef CAS.
- W. Kohn, Phys. Rev., 1959, 115, 809–821 CrossRef.
- N. Marzari and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 12847–12865 CrossRef CAS.
- N. Marzari, A. A. Mostofi, J. R. Yates, I. Souza and D. Vanderbilt, Rev. Mod. Phys., 2012, 84, 1419–1475 CrossRef CAS.
- A. A. Mostofi, J. R. Yates, Y.-S. Lee, I. Souza, D. Vanderbilt and N. Marzari, Comput. Phys. Commun., 2008, 178, 685–699 CrossRef CAS.
- J. R. Choudhuri, D. Vanzo, P. A. Madden, M. Salanne, D. Bratko and A. Luzar, ACS Nano, 2016, 10, 8536–8544 CrossRef CAS PubMed.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2001, 99, 12562–12566 CrossRef PubMed.
- P. G. Bolhuis, D. Chandler, C. Dellago and P. L. Geissler, Annu. Rev. Phys. Chem., 2002, 53, 291–318 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04269d |
| This journal is © The Royal Society of Chemistry 2017 |