
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring secondary-sphere interactions in Fe–NxHy complexes relevant to N2 fixation†
Sidney E.
Creutz
and
Jonas C.
Peters
 *
*
California Institute of Technology, Division, of Chemistry and Chemical Engineering, Pasadena, California 91125, USA. E-mail: jpeters@caltech.edu
First published on 8th December 2016
Abstract
Hydrogen bonding and other types of secondary-sphere interactions are ubiquitous in metalloenzyme active sites and are critical to the transformations they mediate. Exploiting secondary sphere interactions in synthetic catalysts to study the role(s) they might play in biological systems, and to develop increasingly efficient catalysts, is an important challenge. Whereas model studies in this broad context are increasingly abundant, as yet there has been relatively little progress in the area of synthetic catalysts for nitrogen fixation that incorporate secondary sphere design elements. Herein we present our first study of Fe–NxHy complexes supported by new tris(phosphine)silyl ligands, abbreviated as [SiPNMe3] and [SiPiPr2PNMe], that incorporate remote tertiary amine hydrogen-bond acceptors within a tertiary phosphine/amine 6-membered ring. These remote amine sites facilitate hydrogen-bonding interactions via a boat conformation of the 6-membered ring when certain nitrogenous substrates (e.g., NH3 and N2H4) are coordinated to the apical site of a trigonal bipyramidal iron complex, and adopt a chair conformation when no H-bonding is possible (e.g., N2). Countercation binding at the cyclic amine is also observed for anionic {Fe–N2}− complexes. Reactivity studies in the presence of proton/electron sources show that the incorporated amine functionality leads to rapid generation of catalytically inactive Fe–H species, thereby substantiating a hydride termination pathway that we have previously proposed deactivates catalysts of the type [EPR3]FeN2 (E = Si, C).
Introduction
Whereas there has been sustained interest in modeling the structures and functions of metalloenzymes using synthetic, small-molecule systems,1,2 models of metalloenzyme active sites that reproduce only the primary coordination sphere rarely capture the catalytic activity of interest. In the vast majority of cases, model complexes lack important peripheral secondary sphere interactions commonly present in biological systems. Structural, computational, and mechanistic studies on metalloenzymes indicate that critical secondary sphere interactions, such as hydrogen bonding, commonly facilitate the enzyme's catalytic activity, mediated at one or more active site metal centers; these interactions can both stabilize intermediates and orchestrate the necessary arrangement of reactants.3Synthetic inorganic chemists have increasingly taken up the challenge of preparing coordination complexes that incorporate secondary sphere interactions, especially via hydrogen bonding, with the hope of realizing both structurally and functionally faithful models of metalloenzyme active sites. In a number of cases, especially in the context of proton reduction and oxygen activation, these approaches have been notably successful, both with respect to engendering favorable catalytic reactivity and in stabilizing reactive species such as terminal metal oxos.2
A role for secondary sphere interactions in nitrogen fixation is less well established. Biological nitrogen fixation is a fascinatingly complex process that is catalyzed by several iron-containing active sites, of which the most well-studied is the Fe7MoS9C cluster in the iron–molybdenum cofactor (FeMoco). There is great interest in understanding the mechanism(s) by which nitrogenases mediate nitrogen fixation, but much uncertainty remains, including the potential presence or importance of hydrogen bonding interactions near the active site.4–7
In the nitrogenase enzyme of Azotobacter vinelandii, a highly conserved histidine residue (His-195) is poised above the central irons (Fe2 and Fe6) on one face of the cluster. Based on computations and biological studies of mutant enzymes, it has been suggested that one or more of these iron centers may be the binding site for N2 and the locus of catalytic reduction to ammonia (Fig. 1).4,5 Removing this histidine residue via site-directed mutagenesis shuts down N2 fixation,8 suggesting that it may participate in the reaction, perhaps via hydrogen bonding or by acting as a proton shuttle to the active site. A recent crystal structure of a CO-bound form of FeMoco appears consistent with the presence of a hydrogen bond between the terminal oxygen atom of a bridging CO ligand and the N–H moiety of this histidine residue.9
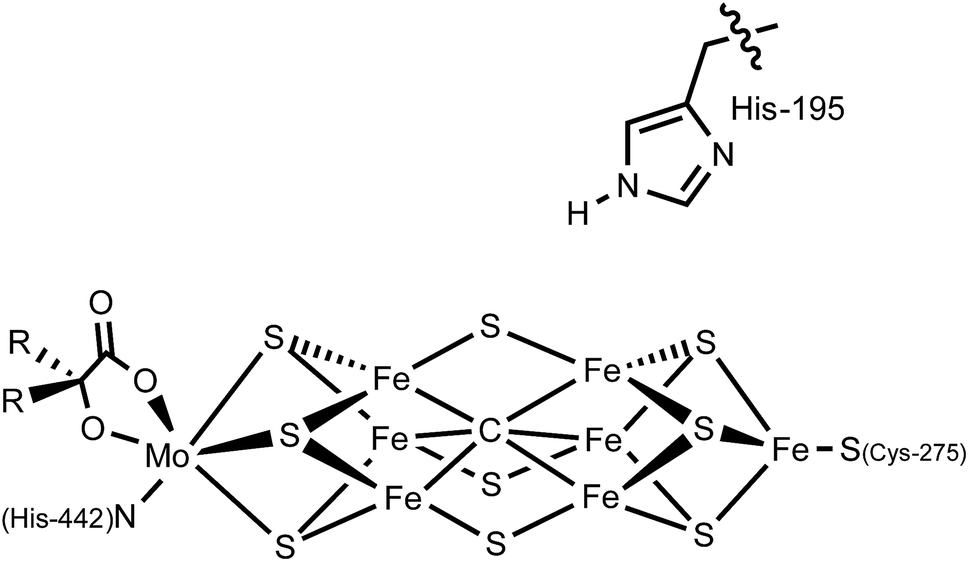 | ||
| Fig. 1 Schematic of the nitrogenase FeMo cofactor; His-195 may interact via hydrogen-bonding with the active site. | ||
While there has been considerable effort invested in the development of functional inorganic Mo and Fe systems as limiting models of biological nitrogen fixation,10,11 there has been correspondingly less investment of effort towards incorporating secondary sphere interactions into such models. There are nonetheless several studies that have probed for secondary sphere interactions in M–NxHy species (Fig. 2). For instance, within group 6, the influence of pendant amine groups on the protonation of Mo– and W–N2 complexes was examined,12 and Cr–N2 complexes supported by ligands featuring pendant tertiary amines were shown to undergo protonation to liberate some NH3 (Fig. 2a);13 it remains as yet unclear what role, if any, these amines play in the N2 protonation in this Cr system. In relation to Fe–NxHy systems, secondary sphere participation has been highlighted in an iron catalyst for hydrazine disproportionation; ligand –NH groups were proposed to aid in the delivery of protons to the substrate (Fig. 2b).14 Recent work from our laboratory furnished a structurally characterized Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NNH2+ species derived from protonation of Fe–N2; this reactive species was stabilized in the solid state by hydrogen bonding interactions to exogenous anion and ethereal solvent.15 Finally, Fe(II)–NH3 and Fe(III)–NH3 complexes bearing secondary-sphere hydrogen-bonding interactions were recently characterized by the Borovik group (Fig. 2c).16 Examples of secondary sphere interactions in M–NxHy species are known for other transition metals as well.17
NNH2+ species derived from protonation of Fe–N2; this reactive species was stabilized in the solid state by hydrogen bonding interactions to exogenous anion and ethereal solvent.15 Finally, Fe(II)–NH3 and Fe(III)–NH3 complexes bearing secondary-sphere hydrogen-bonding interactions were recently characterized by the Borovik group (Fig. 2c).16 Examples of secondary sphere interactions in M–NxHy species are known for other transition metals as well.17
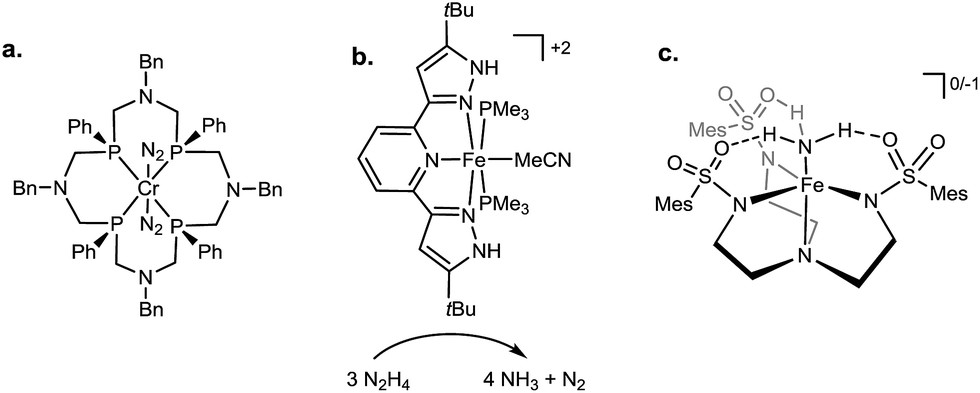 | ||
| Fig. 2 Select previously reported systems incorporating hydrogen-bonding or proton-responsive ligands for the binding and/or conversion of nitrogenous substrates. (a) Treatment of a Cr(0)–(N2)2 complex within a scaffold bearing tertiary amines with acid produces ammonia and hydrazine.13a (b) The participation of a proton-responsive ligand is invoked in the disproportionation of hydrazine by Fe(II).14 (c) Fe(II) and Fe(III) complexes of ammonia show multiple hydrogen-bonding interactions with ligand.16 | ||
Our lab has extensively studied a series of tetradentate trisphosphine ligands with apical N, Si, B, or C donors in the context of N2 activation and reduction, including catalytic N2-to-NH3 conversion.11a,b,18 We sought to redesign these ligands to enable the examination of intramolecular secondary sphere H-bonding interactions within Fe–NxHy species in systems that mediate N2-to-NH3 conversion, and to probe the resulting consequences on reactivity.
Towards this end we report here our first foray in this effort via the synthesis of tetradentate tris(phosphine)silyl ligands that incorporate tertiary amines as hydrogen bond acceptors. We have successfully elucidated the presence of intramolecular hydrogen-bonding interactions with several Fe–NxHy species of relevance to catalytic N2 reduction. Additionally, we have shown that the presence of the pendant tertiary amine dramatically alters the outcome of Fe–N2 protonation reactions in these scaffolds.
Results and discussion
Ligand design and synthesis
We targeted modification of the [SiP3]Fe system we have studied previously (SiP3 = tris(phosphine)silyl)18 by inclusion of tertiary amines within the phosphine donor groups as conduits of secondary-sphere functionality, anticipating the stability of such motifs to the presence of strong reductants and acids, reagents we have employed in Fe-mediated N2-to-NH3 conversion.11 Incorporation of the pendant amine groups within six-membered heterocyclic phosphine/amine rings was expected to provide sufficient rigidity to position the hydrogen-bonding groups around the substrate binding cavity while allowing sufficient flexibility for the ligand to adjust to the presence of different substrates in the metal binding pocket.Synthesis of the required donor arm (L0) involved generating o-bromophenyldivinylphosphine oxide followed by cyclization with methylamine via a double Michael-type addition.19 The resulting phosphine oxide azacycle was then reduced to the desired phosphine (Scheme 1), followed by lithiation of the donor arm and addition to an appropriate electrophile; unsymmetric ([SiPiPr2PNMe]H, L1) and symmetric ([SiPNMe3]H, L2) tris(phosphine)silyl ligands were assembled in this fashion (Scheme 2).
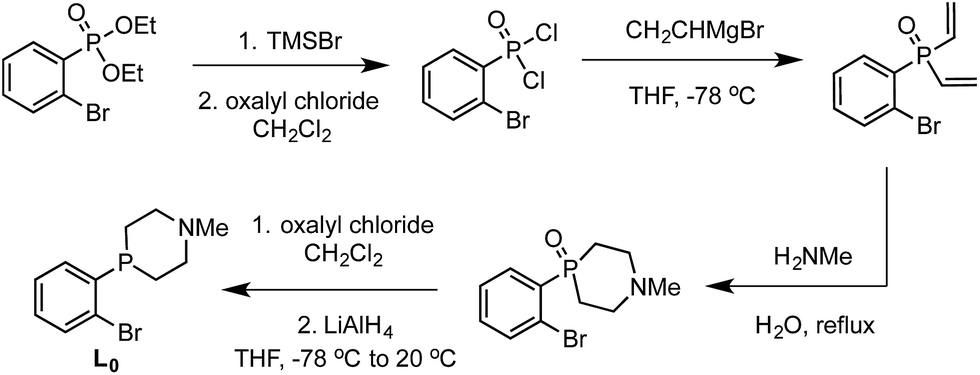 | ||
| Scheme 1 Synthesis of ligand arm L0. | ||
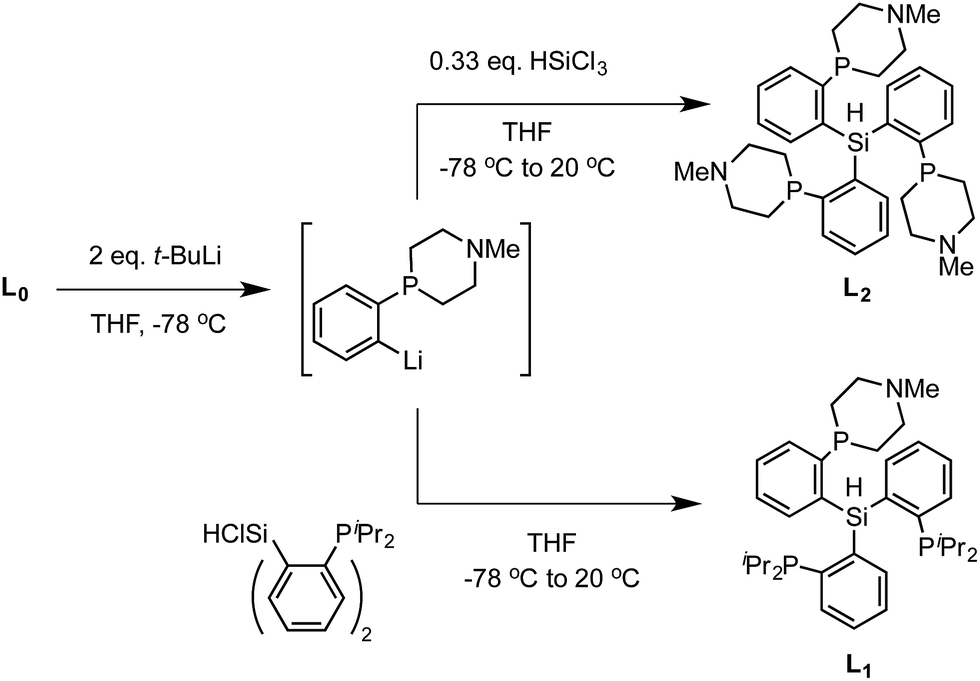 | ||
| Scheme 2 Synthesis of ligands L1 and L2. | ||
Synthesis of precursor iron complexes of L1 and L2
Metallation of the new ligands L1 and L2 on iron followed an approach similar to that used previously for the preparation of complexes of the parent ligand [SiPiPr3]H;18b,c initial complexation of the phosphines L1 or L2 with FeCl2, followed by treatment with methylmagnesium chloride, putatively generates a transient Fe–Me species that loses methane via Si–H activation to afford trigonal bipyramidal Fe(II) chloride complexes. Sodium amalgam (Na/Hg) reduction then generates the corresponding Fe(I) complexes with loss of sodium chloride and uptake of N2 (Scheme 3 and Fig. 3). Comparison of the spin states and IR parameters for these complexes to the parent [SiPiPr3]Fe system establishes that the electronic properties of the azacyclic phosphine donor are very similar to the parent donor—2, 2′, and [SiPiPr3]FeN2 are all low-spin, S = 1/2 complexes with similar N–N vibrational frequencies (2005, 2007, and 2008 cm−1, respectively). This fact should facilitate interpretation of the effects of secondary sphere interactions on the properties of these respective complexes.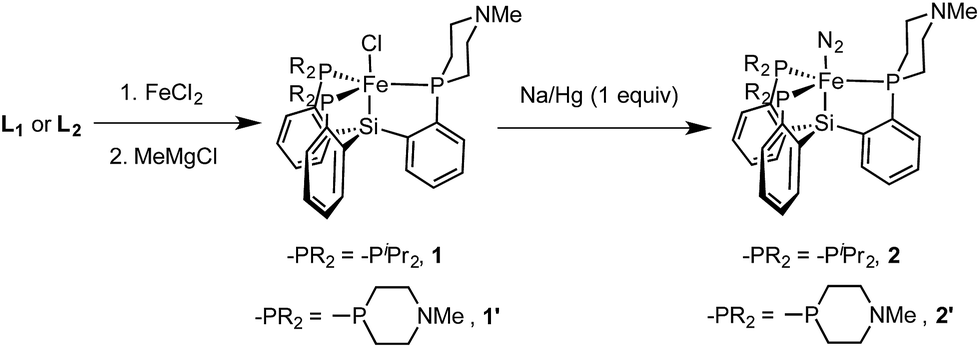 | ||
| Scheme 3 Synthesis of Fe(II) and Fe(I) precursor complexes. | ||
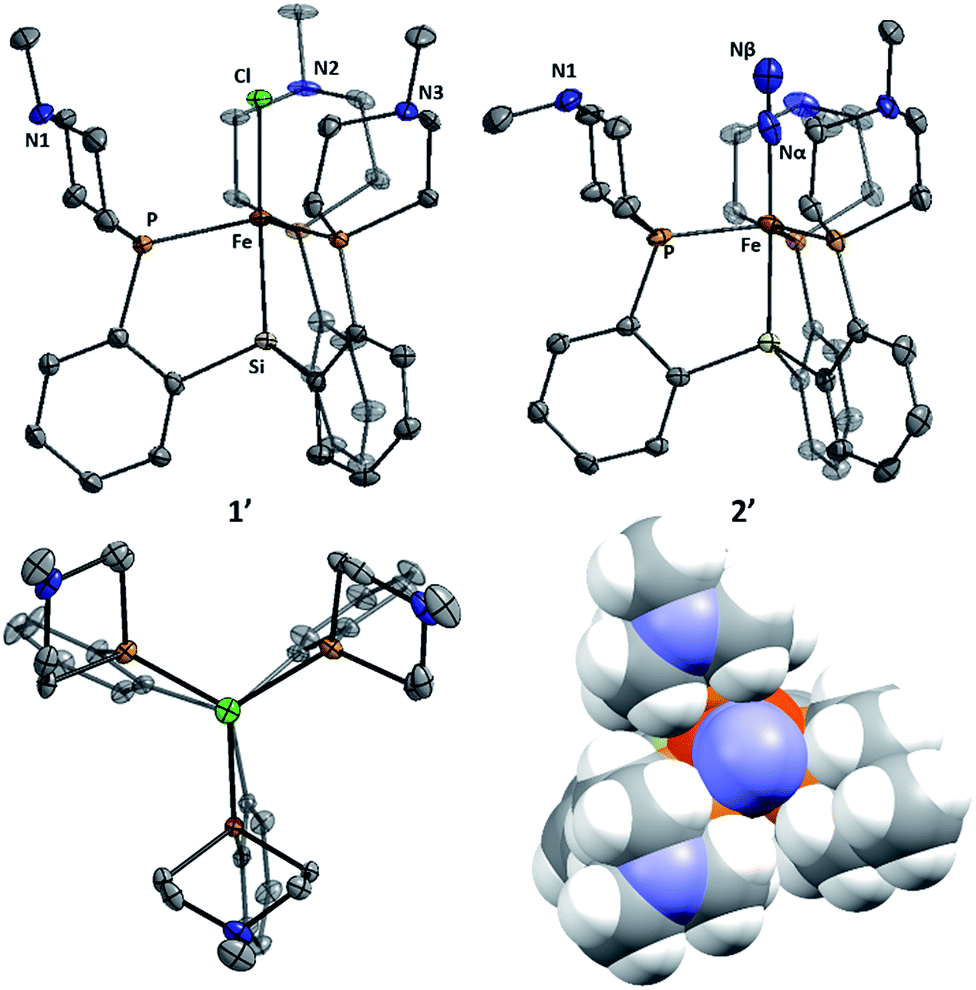 | ||
| Fig. 3 Structures of 1′ (left, two views) and 2′ (right, with space-filling view down N–N–Fe axis). Solvent molecules and hydrogen atoms omitted for clarity. Thermal ellipsoids are shown at 50% probability. | ||
The azaphosphacycles in these complexes, in which no hydrogen bonding is present, adopt a chair conformation, with the N–Me group residing in either a pseudo-axial or pseudo-equatorial position. This is to be expected for a saturated six-membered ring,20 and in this conformation the tertiary amine is not well positioned for hydrogen bonding to an NxHy substrate coordinated to iron in the axial site. As described below, hydrogen bonding requires the ring to adopt the energetically less favorable boat or twist-boat conformation. This feature provides a useful structural diagnostic for the presence of hydrogen bonding interactions.
Generation of Fe–NxHy complexes
We have synthesized and structurally characterized iron complexes coordinated by the NxHy ligands NH3, N2H4, and NH2 using L1 and L2. These compounds demonstrate the ability of these new auxiliary ligands to engage in hydrogen bonds with reduced NxHy substrates.Access to the hydrazine and ammonia adducts proceeds via oxidation of 2 or 2′ with [Fc][BArF4] (ArF = 3,5-bistrifluoromethylphenyl; Fc = ferrocenium) to generate in situ an L1[Fe(II)] or L2[Fe(II)] complex which is presumably solvated by THF (Scheme 4), followed by treatment with hydrazine or gaseous ammonia to afford the desired complex. The resulting Fe–NH3 adducts 3 and 3′, and the Fe–N2H4 adduct 4 are stable to vacuum and are readily isolated in pure form; they have been crystallographically characterized. Complex 4′ is not sufficiently stable to be isolated (vide infra).
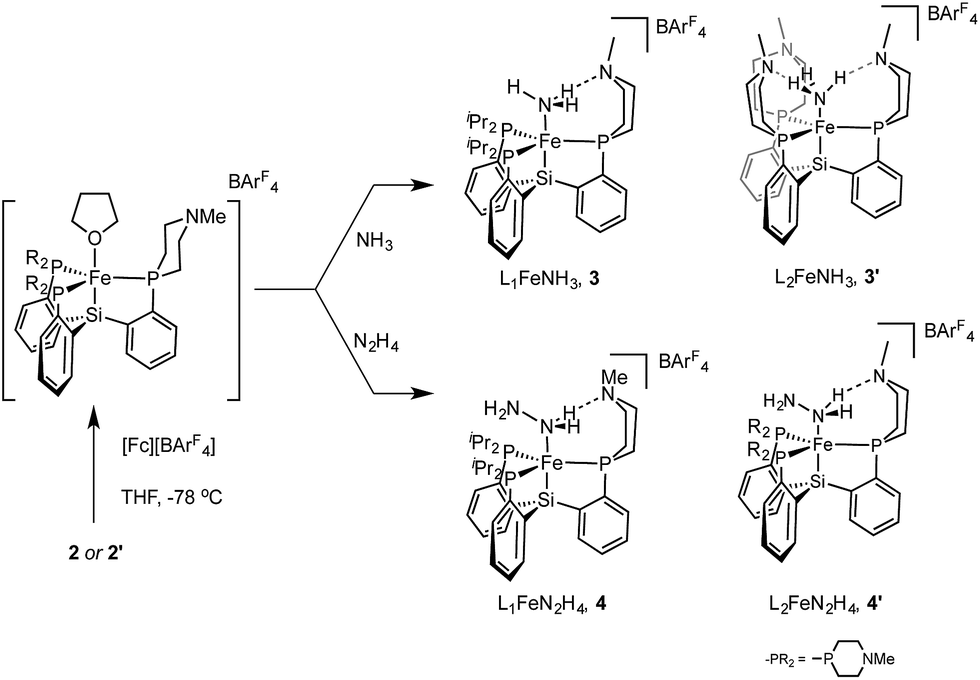 | ||
| Scheme 4 Synthesis of cationic NH3 and N2H4 adducts of LFe(II). | ||
The X-ray structures of 3, 3′, and 4 are gratifying in that they clearly illustrate the presence of hydrogen bonding interactions between the tertiary amines of the tris(phosphine)silyl ligands and the N–H bonds of the respective Fe–NxHy species (Fig. 4). The boat-type conformations of the six-membered azaphosphine rings in 3, 3′, and 4 are diagnostic for the presence of at least moderately strong hydrogen bonds; the energetic benefit of the hydrogen bonds must be sufficient to overcome the energetic penalty of adopting the disfavored boat (vs. chair) conformation (vide infra). Accordingly, chair conformations of the azaphosphine rings are instead observed in all structures we have examined that lack hydrogen bond interactions with an apically coordinated substrate.
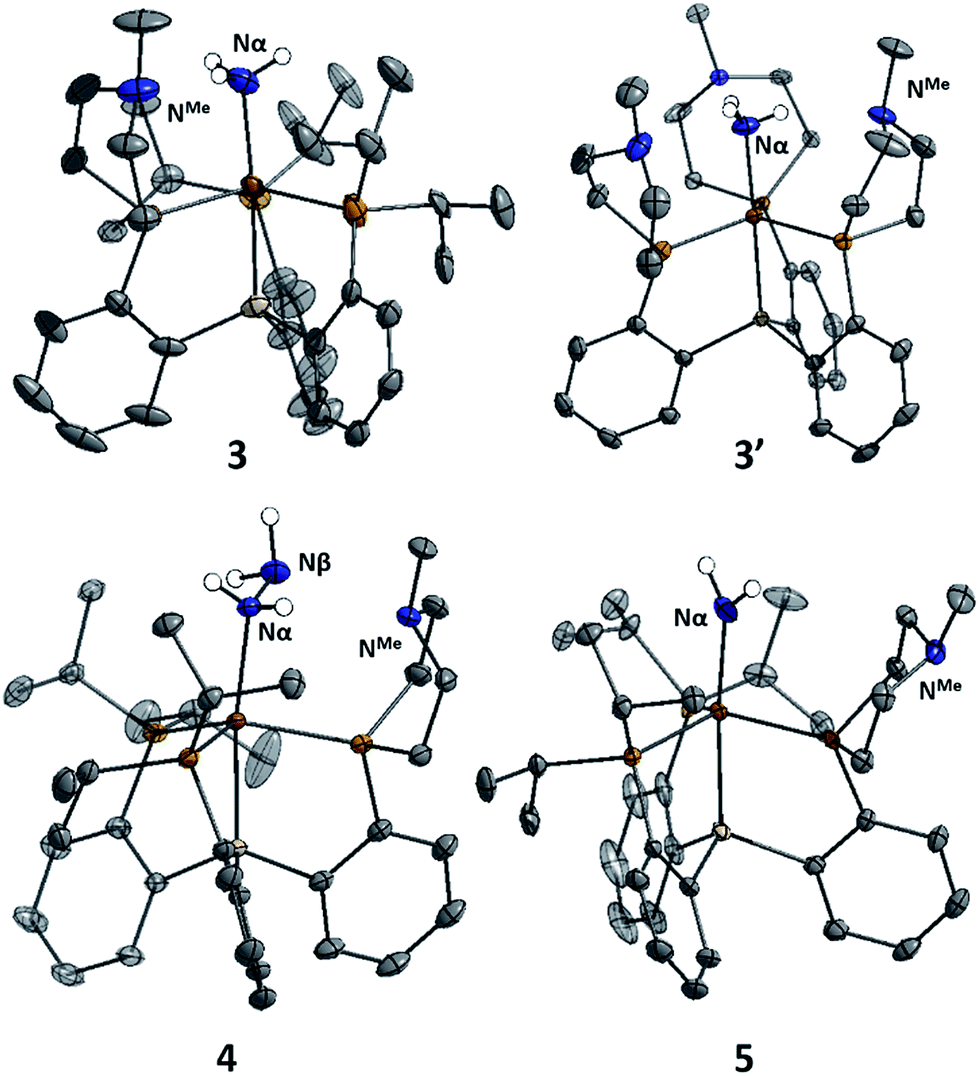 | ||
| Fig. 4 Structure of ammonia, hydrazine, and amide complexes 3, 3′, 4, and 5. BArF4 counteranions, solvent molecules, and carbon-bound hydrogen atoms have been omitted for clarity. Thermal ellipsoids are shown at 50% probability. | ||
In the structures of complexes 3, 3′, and 4, the N-bound hydrogen atoms were located in the density difference map and their positions were allowed to refine freely. In all cases where hydrogen bonds were apparent from the conformation of the azaphosphacycles, hydrogen atoms were located in the expected positions, bound to the Fe-coordinated nitrogen atom and appropriately oriented for interaction with the tertiary amine acceptors. In the case of ammonia complex 3′, two independent molecules were found to be present in the asymmetric unit. In one of these molecules all three ligand azaphosphacycles occupy a boat conformation indicative of hydrogen bonding interactions with the three ammonia hydrogen atoms; in the second molecule, two of the ligand arms are engaged in hydrogen bonding, while the third arm is disordered (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) between boat and chair conformations, perhaps suggestive of a weaker interaction. The shortest donor–acceptor bond lengths between Nα and NMe are 2.994 Å, 2.984 Å, and 3.074 Å in 3, 3′, and 4, respectively. The donor–acceptor distances for the hydrogen bonds are in the regime of what has been classified as a “moderate” strength hydrogen bond.21
:2) between boat and chair conformations, perhaps suggestive of a weaker interaction. The shortest donor–acceptor bond lengths between Nα and NMe are 2.994 Å, 2.984 Å, and 3.074 Å in 3, 3′, and 4, respectively. The donor–acceptor distances for the hydrogen bonds are in the regime of what has been classified as a “moderate” strength hydrogen bond.21
Since engaging in hydrogen bonds with a coordinated substrate requires the six-membered rings of the ligand to adopt a higher-energy conformation (boat rather than chair), a lower limit for the strength of the hydrogen bonds can be obtained if the difference in energy between these two conformations is known. For cyclohexane, the twist-boat conformation lies approximately 5.5 kcal mol−1 higher in energy than the chair conformation.20 To approximate the energy difference for our system, DFT optimizations were carried out on the uncoordinated ligand L1 with the azaphosphine ring in the chair (as in the structure of 1′ or 5, vide infra) or boat (as in the structure of 3) conformation, with ligand isopropyl groups truncated to methyls (see ESI† for details). The energy difference between the two conformers was calculated to be approximately 4.1 kcal mol−1; this value corresponds to the minimum stabilization energy imparted by each hydrogen bond, again consistent with a hydrogen bond of moderate strength.
Both 4 and 4′ are unstable in solution, decomposing to give the ammonia complexes 3 and 3′, respectively. While 4 decomposes over the course of several hours at 60 °C, 4′ decomposes so quickly in solution that it has not been possible to isolate it in pure form; full conversion to 3′ is observed within several minutes at room temperature. The parent complex, {[SiPiPr3]FeN2H4}{BArF4}, is also susceptible to a similar decomposition process; however, in this case full conversion to the ammonia complex requires heating at 60 °C for several days (see ESI†). The kinetics of these decomposition reactions show complex behavior that we suspect are indicative of autocatalytic reactions, similar to that reported for the metalloboratrane complex {[TPBFe]N2H4}{BArF4}.22 The nature of the autocatalyst has not, however, been identified in either case and complicates quantitative kinetic analysis. Qualitatively, the rate of the decomposition reaction appears to be increased in the presence of hydrogen bond acceptors within the secondary coordination sphere. Similar observations have been made in another iron system that is capable of catalytically disproportionating hydrazine.14
Terminal parent amide (–NH2) complexes of iron are relatively rare despite their relevance as a possible late stage intermediate in Fe-mediated N2-to-NH3 conversion;4 only two examples of terminal Fe–NH2 species have been previously structurally characterised.22,23 The Fe(II) amide complex L1Fe–NH2 (5) is readily synthesized by treating L1FeCl (1) with excess NaNH2 in 1:1 THF/NH3 solvent (Scheme 5). The identity of the –NH2 ligand was confirmed by digesting the complex with HCl and then analyzing the resulting solution for ammonia using the indophenol method;24 quantitative generation of ammonia was established. Complex 5 has been structurally characterized and does not show any hydrogen bonding to the ligand in the solid state (Fig. 4). Consistent with this, the azaphosphine ring adopts a chair conformation. The Fe–NH2 ligand in 5 is expected to be less acidic than the Fe–NH3+ ligand in the cationic complexes 3 and 3′; further stabilization of the smaller degree of partial positive charge on the Fe–NH2 hydrogens via hydrogen bonding is hence not sufficiently favorable to overcome the energetic cost of conformationally flipping the azaphosphacycle to the boat conformation.
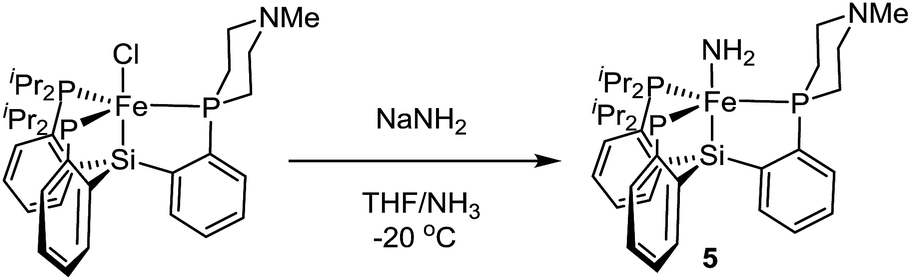 | ||
| Scheme 5 Synthesis of a parent amide complex (5). | ||
Oxidation of 5 with [Fc][BArF4] in Et2O in an attempt to generate L1Fe–NH2+ instead afforded the ammonia complex 3 as the primary detectable product. We had hoped that generation of L1Fe–NH2+ might afford a viable precursor to a terminal L1Fe(NH) species. However, the instability of L1Fe–NH2+ (or a corresponding L1Fe(NH) species in the event a proton is transferred to the cyclic amine) suggests that reaction with solvent (perhaps via HAT) and/or disproportionation is too rapid. Carrying out the reaction in thawing 2-MeTHF does allow for the detection of a new S = 1/2 species by EPR at 77 K (see ESI†), but this species decays upon warming. Attempts to trap or further characterize this intermediate have not yet been successful.
Synthesis and reactivity of reduced Fe–N2 complexes
The Fe systems we have studied previously that generate significant or catalytic amounts of NH3 from N2 use anionic (pre)catalysts of the type LnFeN2−.11 Therefore, we synthesized and characterized L1FeN2− and L2FeN2− as their sodium salts by reduction of 1 and 1′ with excess sodium amalgam in THF (Scheme 6). The solid-state structure of 6′ (ν(NN) = 1878 cm−1) illustrates that, in addition to acting as a hydrogen-bond acceptor, the tertiary amine in the secondary coordination sphere can serve as a Lewis base and interact with a Lewis acidic countercation such as [Na(THF)3]+ (Fig. 5). In this case, the N–Na distances are sufficiently long that the six-membered azacycle maintains a distorted chair conformation to accommodate this interaction. In the solid state the Fe–N–N angle, which is typically very close to 180° in terminal Fe–N2 complexes,11,18,25 is distorted to 171.7° due to interaction with the Na cation; the cation position is constrained by coordination to the pendant amine in the ligand. In contrast, the crystal structure of 6 (ν(NN) = 1874 cm−1) shows intermolecular coordination of the sodium cation to the amine of a neighboring molecule, forming an infinite chain structure; in this case the azacycle again adopts a chair conformation and the Fe–N–N angle is nearly linear (Fig. 5).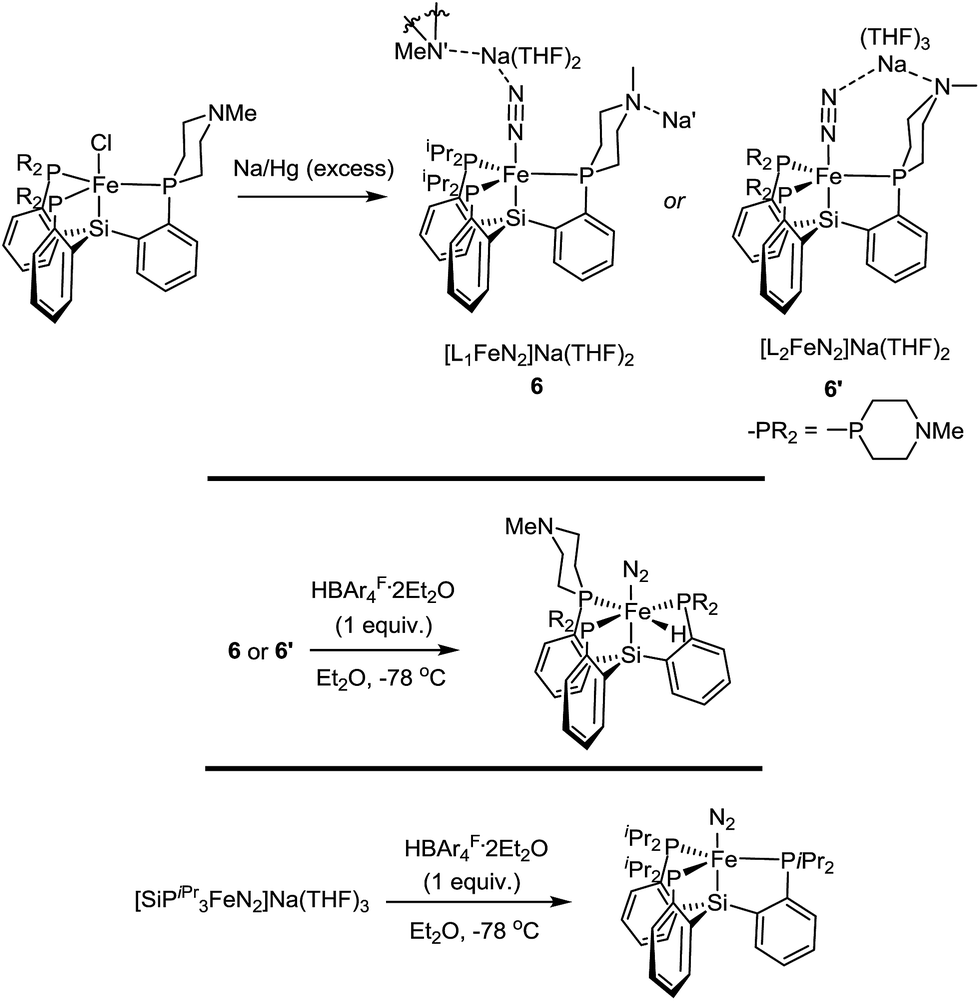 | ||
| Scheme 6 Synthesis of Fe(0)–N2 complexes, and their reaction profiles with HBArF4·2Et2O. | ||
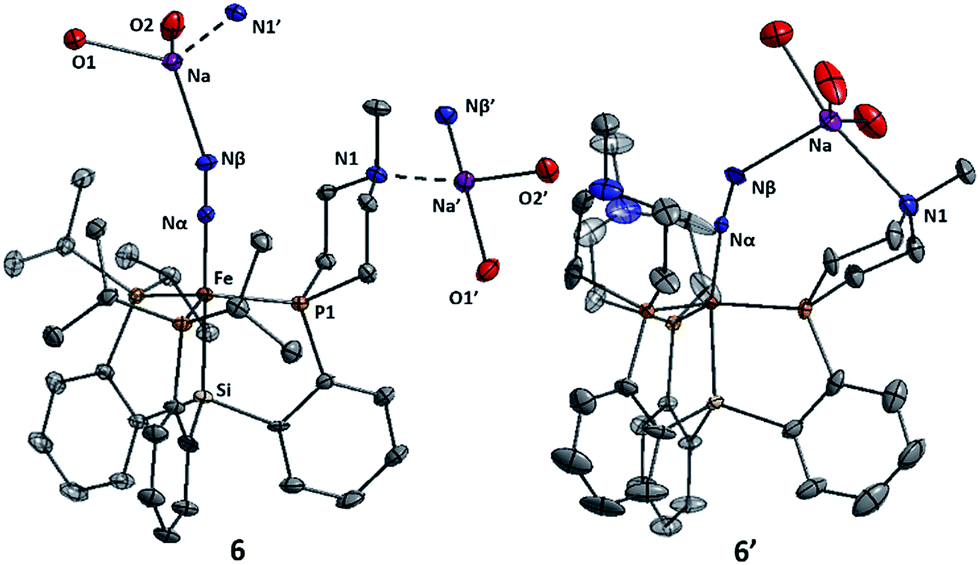 | ||
| Fig. 5 Structures of complexes 6 and 6′. For 6, the intermolecular interaction (dashed lines) between the sodium cation and the tertiary amine group of a neighboring molecule (N1′) is shown, as well as the interaction with the sodium countercation of another neighbor (Na′). Thermal ellipsoids are shown at 50% probability, and hydrogen atoms and uncoordinated solvent are omitted for clarity. Coordinated THF molecules are truncated to show only the oxygen atom bound to Na. | ||
{[SiPiPr3]FeN2}− can catalyze N2-to-NH3 conversion with poor efficiency at very high acid/reductant loading,26 and at lower loadings of 48 equiv. HBArF4·2Et2O and 50 equiv. KC8 generates 0.8 ± 0.4 equivalents of NH3 per Fe (Et2O, −78 °C, 1 atm N2).11a In contrast, exposure of L1FeN2− and L2FeN2− complexes 6 and 6′ to analogous reaction conditions affords no detectable NH3. This dichotomy in reaction profile is interesting; the stoichiometric reactions of these respective species with acid provide some clues as to the reason for the disparate reactivity.
In the case of {[SiPiPr3]FeN2}−, treatment with one equivalent of either HBArF4·2Et2O or the tertiary ammonium acid [HNiPr2Et][BArF4] at −78 °C results in clean oxidation to [SiPiPr3]FeN2, a reaction which is believed to proceed with formal loss of ½H2 from a transient Fe–NNH intermediate.15 However, with either 6 or 6′, the same reaction conditions instead result in immediate generation of the hydride complexes L1Fe(N2)(H) and L2Fe(N2)(H) (Scheme 6). Given that formation of a transient Fe–NNH species is believed to be a necessary first step in the transformation of N2 to NH3 on complexes of these types,15,26,27 it seems plausible that 6 and 6′ do not give rise to Fe–NNH species on exposure to acid.
The difference in product profiles between {[SiPiPr3]FeN2}− and L1FeN2− or L2FeN2− upon treatment with acid can be rationalized by considering the kinetically and thermodynamically preferred sites of protonation in these complexes. The metal center is the thermodynamically preferred site of protonation in each system (i.e., to afford an LFe(N2)(H) product); for 6, DFT computations suggest the proton affinity of the iron center is at least 20 kcal mol−1 higher than for protonation at N2 or at the donor arm amine position (see ESI† for details). However, in the absence of an exposed basic site on the ligand, the kinetic site of protonation is most likely the terminal N-atom of the N2 ligand due to steric crowding at the iron center. If we presume protonation at the N2 transiently generates an Fe–NNH species with {[SiPiPr3]FeN2}− we can conclude that there is thereafter no kinetically facile pathway for the proton to then migrate to Fe; therefore the Fe–NNH species instead decomposes via other (presumed bimolecular) pathways that release H2. When a tertiary amine is instead present in a phosphine donor arm, the amine can serve as the kinetic site of protonation, and can likely act as a proton shuttle to transfer a proton from N2 to the iron center, leading to stable iron hydride complexes (Fig. 6). A similar observation has been made in the case of protonation of anionic tungsten N2 complexes with and without pendant amines in the ligand.12b,c
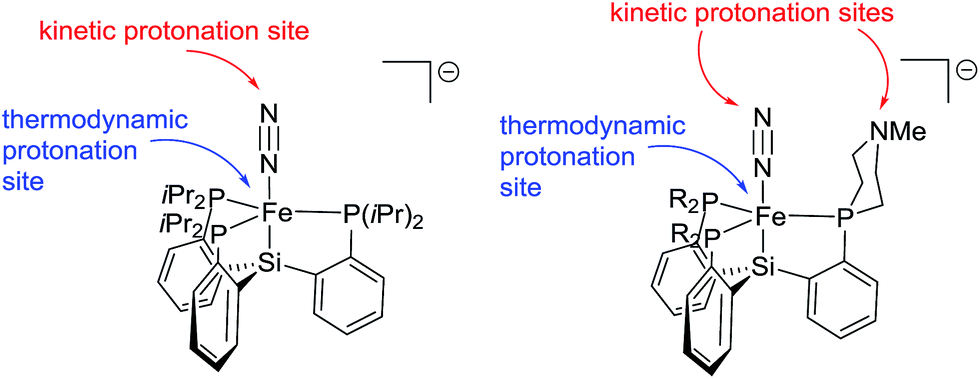 | ||
| Fig. 6 Kinetic and thermodynamic protonation sites of {[SiPR3]FeN2}− anions. | ||
We have previously observed the formation of iron hydride species during the course of catalytic N2-to-NH3 conversion. In the case of NH3 production catalyzed by the tris(phosphine)borane complex {[TPiPrB]FeN2}−, we have demonstrated that such a hydride species most likely serves as an off-path catalyst resting state.26 While the mechanisms of N2-to-NH3 conversion on the analogous alkyl- and silyl-supported complexes, {[CPiPr3]FeN2}− and {[SiPiPr3]FeN2}−, have not yet been as thoroughly studied, we have proposed that the [EPiPr3]Fe(N2)(H) (E = C, Si) hydride complexes may be thermodynamic sinks that irreversibly deactivate the catalyst when formed under the reaction conditions.11b The evidence presented here corroborates this, and supports the idea that efficient molecular catalysts for N2-to-NH3 conversion must either avoid the formation of metal hydrides, or facilitate kinetically competent pathways for these hydride complexes to reenter the catalytic cycle, likely through the evolution of H2.26
Conclusions
We have demonstrated the incorporation of secondary-sphere interactions into iron–phosphine scaffolds relevant to synthetic nitrogen fixation. The presence of modestly strong hydrogen bonds between pendant ligand functionalities and possible N2 reduction products, ammonia and hydrazine, has been demonstrated; the ligand donor arms respond to the presence of favorable H-bonding interactions with a coordinated NxHy substrate via conformational flipping of the cyclic phosphine/amine donor arm from a chair to a boat confirmation. While these new phosphine/amine ligands demonstrate an approach to tuning the secondary coordination sphere in Fe–N2 systems, the present systems do not show improved catalysis for N2 fixation but instead completely shut down such function. Kinetically facile formation of iron hydride products occurs instead due to the presence of the pendant amine proton shuttle. As the modified complexes are not competent catalysts, caution must be exercised in extrapolating these results to the catalytic systems, including the nitrogenase cofactor itself. Nevertheless, further tuning of the scaffold, such as additional steric protection of the metal center to hinder protonation, better pKa-tuning of the pendant base, or design of a system where hydrogen-bonding is less conformationally disfavored, may improve the reactivity. Work to further explore structure–function relationships in systems of these types is underway.Acknowledgements
This work was supported by the NIH (GM 070757) and the Gordon and Betty Moore Foundation. Larry Henling and Mike Takase are thanked for crystallographic assistance.Notes and references
- Selected reviews and examples: (a) R. H. Holm and E. I. Solomon, Chem. Rev., 2004, 104, 347 CrossRef CAS PubMed; (b) S. A. Cook and A. S. Borovik, Acc. Chem. Res., 2015, 48, 2407 CrossRef CAS PubMed; (c) C. J. Cramer and W. B. Tolman, Acc. Chem. Res., 2007, 40, 601 CrossRef CAS PubMed; (d) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Jr., Chem. Rev., 2004, 104, 939 CrossRef CAS PubMed; (e) S. Ohta, Y. Ohki, T. Hashimoto, R. E. Cramer and K. Tatsumi, Inorg. Chem., 2012, 51, 11217 CrossRef CAS PubMed; (f) J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561 CrossRef CAS PubMed; (g) E. Y. Tsui, J. S. Kanady and T. Agapie, Inorg. Chem., 2013, 52, 13833 CrossRef CAS PubMed; (h) J. Chatt and G. J. Leigh, Chem. Soc. Rev., 1972, 1, 121 RSC; (i) J. A. Kovacs and L. M. Brines, Acc. Chem. Res., 2007, 40, 501 CrossRef CAS PubMed; (j) S. Itoh, Acc. Chem. Res., 2015, 48, 2066 CrossRef CAS PubMed; (k) S. Hematian, I. Garcia-Bosch and K. D. Karlin, Acc. Chem. Res., 2015, 48, 2462 CrossRef CAS PubMed; (l) C. T. Lyons and T. D. P. Stack, Coord. Chem. Rev., 2013, 257, 528 CrossRef CAS PubMed; (m) C. Citek, S. Herres-Pawlis and T. D. P. Stack, Acc. Chem. Res., 2015, 48, 2424 CrossRef CAS PubMed; (n) J. Serrano-Plana, I. Garcia-Bosch, A. Company and M. Costas, Acc. Chem. Res., 2015, 48, 2397 CrossRef CAS PubMed; (o) S. Friedle, E. Reisner and S. J. Lippard, Chem. Soc. Rev., 2010, 39, 2768 RSC.
- Selected reviews and examples: (a) A. Dutta, J. A. S. Roberts and W. J. Shaw, Angew. Chem., Int. Ed., 2014, 53, 6487 CrossRef CAS PubMed; (b) R. M. Henry, R. K. Shoemaker, D. L. DuBois and M. Rakowski DuBois, J. Am. Chem. Soc., 2006, 128, 3002 CrossRef CAS PubMed; (c) M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62 RSC; (d) T. B. Rauchfuss, Acc. Chem. Res., 2015, 48, 2107 CrossRef CAS PubMed; (e) R. L. Shook and A. S. Borovik, Inorg. Chem., 2010, 49, 3646 CrossRef CAS PubMed; (f) C. E. MacBeth, A. P. Golombek, V. G. Young, C. Yang, K. Kuczera, M. P. Hendrich and A. S. Borovik, Science, 2000, 289, 938 CrossRef CAS PubMed; (g) C. E. MacBeth, R. Gupta, K. R. Mitchell-Koch, V. G. Young, G. H. Lushington, W. H. Thompson, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2004, 126, 2556 CrossRef CAS PubMed; (h) D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2010, 132, 12188 CrossRef CAS PubMed; (i) C. T. Carver, B. D. Matson and J. M. Mayer, J. Am. Chem. Soc., 2012, 134, 5444 CrossRef CAS PubMed; (j) C. Constentin, S. Drouet, M. Robert and J.-M. Saveant, Science, 2012, 338, 90 CrossRef PubMed; (k) C. J. Chang, L. L. Chng and D. G. Nocera, J. Am. Chem. Soc., 2003, 125, 1866 CrossRef CAS PubMed; (l) C. H. Lee, D. K. Dogutan and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 8775 CrossRef CAS PubMed; (m) H. S. Soo, A. C. Komor, A. T. Iavarone and C. J. Chang, Inorg. Chem., 2009, 48, 10024 CrossRef CAS PubMed; (n) E. M. Matson, J. A. Bertke and A. R. Fout, Inorg. Chem., 2014, 53, 4450 CrossRef CAS PubMed; (o) E. M. Matson, Y. J. Park and A. R. Fout, J. Am. Chem. Soc., 2014, 136, 17398 CrossRef CAS PubMed; (p) C. M. Wallen, L. Palatinus, J. Bacsa and C. C. Scarborough, Angew. Chem., Int. Ed., 2016, 55, 11902 CrossRef CAS PubMed.
- Selected reviews and examples: (a) R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239 CrossRef CAS PubMed; (b) Y. Lu and J. S. Valentine, Curr. Opin. Struct. Biol., 1997, 7, 495 CrossRef CAS PubMed; (c) L. L. Kiefer, S. A. Paterno and C. A. Fierke, J. Am. Chem. Soc., 1995, 117, 6831 CrossRef CAS; (d) L. Vogt, D. J. Vinyard, S. Khan and G. W. Brudvig, Curr. Opin. Chem. Biol., 2015, 25, 152 CrossRef CAS PubMed; (e) Y. Kung and C. L. Drennan, Curr. Opin. Chem. Biol., 2011, 15, 276 CrossRef CAS PubMed; (f) C. M. Moore, E. W. Dahl and N. K. Szymczak, Curr. Opin. Chem. Biol., 2015, 25, 9 CrossRef CAS PubMed; (g) J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273 CrossRef CAS PubMed; (h) D. Fernandez, I. Pallares, J. Vendrell and F. X. Aviles, Biochimie, 2010, 92, 1484 CrossRef CAS PubMed; (i) M. G. I. Galinato, T. Spolitak, D. P. Ballou and N. Lehnert, Biochemistry, 2011, 50, 1053 CrossRef CAS PubMed; (j) S. A. Cook, E. A. Hill and A. S. Borovik, Biochemistry, 2015, 54, 4167 CrossRef CAS PubMed.
- (a) B. M. Hoffman, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2009, 42, 609 CrossRef CAS PubMed; (b) B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2013, 46, 587 CrossRef CAS PubMed; (c) P. C. Dos Santos, R. Y. Igarashi, H.-I. Lee, B. M. Hoffman, L. C. Seefeldt and D. R. Dean, Acc. Chem. Res., 2005, 38, 208 CrossRef CAS PubMed.
- (a) B. M. Barney, R. Y. Igarashi, P. C. Dos Santos, D. R. Dean and L. C. Seefeldt, J. Biol. Chem., 2004, 279, 53621 CrossRef CAS PubMed; (b) L. C. Seefeldt, I. G. Dance and D. R. Dean, Biochemistry, 2004, 43, 1401 CrossRef CAS PubMed; (c) P. E. M. Siegbahn, J. Am. Chem. Soc., 2016, 138, 10485 CrossRef CAS PubMed; (d) J. Schimpl, H. M. Petrilli and P. E. Blochl, J. Am. Chem. Soc., 2003, 125, 15772 CrossRef CAS PubMed; (e) P. P. Hallmen and J. Kastner, Z. Anorg. Allg. Chem., 2015, 641, 118 CrossRef CAS; (f) J. Kastner, S. Hemmen and P. E. Blochl, J. Chem. Phys., 2005, 123, 074306 CrossRef PubMed.
- J. B. Howard and D. C. Rees, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17088 CrossRef CAS PubMed.
- I. Dance, Dalton Trans., 2012, 41, 7647 RSC.
- C.-H. Kim, W. E. Newton and D. R. Dean, Biochemistry, 1995, 34, 2798 CrossRef CAS PubMed.
- T. Spatzal, K. A. Perez, O. Einsle, J. B. Howard and D. C. Rees, Science, 2014, 345, 1620 CrossRef CAS PubMed.
- (a) D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76 CrossRef CAS PubMed; (b) K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120 CrossRef CAS PubMed; (c) S. Kuriyama, K. Arashiba, K. Nakajima, H. Tanaka, N. Kamaru, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2014, 136, 9719 CrossRef CAS PubMed.
- (a) J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef CAS PubMed; (b) S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 1105 CrossRef CAS PubMed; (c) G. Ung and J. C. Peters, Angew. Chem., Int. Ed., 2015, 54, 532 CAS; (d) P. J. Hill, L. R. Doyle, A. D. Crawford, W. K. Myers and A. E. Ashley, J. Am. Chem. Soc., 2016, 138, 13521 CrossRef CAS PubMed.
- (a) L. A. Labios, Z. M. Heiden and M. T. Mock, Inorg. Chem., 2015, 54, 4409 CrossRef CAS PubMed; (b) C. J. Weiss, J. D. Egbert, S. Chen, M. L. Helm, R. M. Bullock and M. T. Mock, Organometallics, 2014, 33, 2189 CrossRef CAS; (c) P. Bhattacharya, D. E. Prokopchuk and M. T. Mock, Coord. Chem. Rev., 2016 DOI:10.1016/j.ccr.2016.07.005.
- (a) M. T. Mock, S. Chen, M. O'Hagan, R. Rousseau, W. G. Dougherty, W. S. Kassel and R. M. Bullock, J. Am. Chem. Soc., 2013, 135, 11493 CrossRef CAS PubMed; (b) J. D. Egbert, M. O'Hagan, E. S. Wiedner, R. M. Bullock, N. A. Piro, W. S. Kassel and M. T. Mock, Chem. Commun., 2016, 52, 9343 RSC.
- K. Umehara, S. Kuwata and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 6754 CrossRef CAS PubMed.
- J. Rittle and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 4243 CrossRef CAS PubMed.
- N. S. Sickerman, S. M. Peterson, J. W. Ziller and A. S. Borovik, Chem. Commun., 2014, 50, 2515 RSC.
- (a) D. Sellmann, A. Hille, A. Rosler, F. W. Heinemann, M. Moll, G. Brehm, S. Schneider, M. Reiher, B. A. Hess and W. Bauer, Chem.–Eur. J., 2004, 10, 819 CrossRef CAS PubMed; (b) M. Reiher, O. Salomon, D. Sellmann and B. A. Hess, Chem.–Eur. J., 2001, 7, 5195 CrossRef CAS PubMed; (c) O. Tutusaus, C. Ni and N. K. Szymczak, J. Am. Chem. Soc., 2013, 135, 3403 CrossRef CAS PubMed; (d) H.-F. Hsu, W.-C. Chu, C.-H. Hung and J.-H. Liao, Inorg. Chem., 2003, 42, 7369 CrossRef CAS PubMed.
- (a) C. E. MacBeth, S. B. Harkins and J. C. Peters, Can. J. Chem., 2005, 83, 332 CrossRef CAS; (b) N. P. Mankad, M. T. Whited and J. C. Peters, Angew. Chem., Int. Ed., 2007, 46, 5768 CrossRef CAS PubMed; (c) M. T. Whited, N. P. Mankad, Y. Lee, P. F. Oblad and J. C. Peters, Inorg. Chem., 2009, 48, 2507 CrossRef CAS PubMed; (d) Y. Lee, N. P. Mankad and J. C. Peters, Nat. Chem., 2010, 2, 558 CrossRef CAS PubMed; (e) M.-E. Moret and J. C. Peters, Angew. Chem., Int. Ed., 2011, 50, 2063 CrossRef CAS PubMed; (f) M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118 CrossRef CAS PubMed; (g) J. Rittle and J. C. Peters, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15898 CrossRef CAS PubMed.
- D. J. Collins, L. E. Rowley and J. M. Swan, Aust. J. Chem., 1974, 27, 841 CrossRef CAS.
- (a) F. A. Carey and R. J. Sundberg, Conformations of Cyclohexane Derivatives, in Advanced Organic Chemistry, Part A: Structure and Mechanisms, Springer, New York, 4th edn, 2004, pp. 135–136 Search PubMed; (b) M. Squillacote, R. S. Sheridan, O. L. Chapman and F. A. L. Anet, J. Am. Chem. Soc., 1975, 97, 3244 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- J. S. Anderson, M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 534 CrossRef CAS PubMed.
- D. J. Fox and R. G. Bergman, J. Am. Chem. Soc., 2003, 125, 8984 CrossRef CAS PubMed.
- M. W. Weatherburn, Anal. Chem., 1967, 39, 971 CrossRef CAS.
- B. Peigne and G. Aullon, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 369 CAS.
- T. Del Castillo, N. Thompson and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 5341 CrossRef CAS PubMed.
- J. S. Anderson, G. E. Cutsail, J. Rittle, B. A. Connor, W. A. Gunderson, L. Zhang, B. M. Hoffman and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 7803 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic data, reactivity studies, computational studies, crystallographic information. CCDC 1511362 (1′), 1511363 (2′), 1511369 (3), 1511371 (3′), 1511365 (4), 1511368 (5), 1511367 (6), 1511364 (6′), 1511366 and 1511370 contain supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04805f |
| This journal is © The Royal Society of Chemistry 2017 |