DOI:
10.1039/C6SC04477H
(Edge Article)
Chem. Sci., 2017,
8, 1876-1882
A Lewis acid β-diiminato-zinc-complex as all-rounder for co- and terpolymerisation of various epoxides with carbon dioxide†
Received
7th October 2016
, Accepted 12th November 2016
First published on 17th November 2016
Abstract
A β-diiminato–zinc–N(SiMe3)2 complex (1) was synthesised and fully characterised, including an X-ray diffraction study. The activity of catalyst 1 towards the coupling reaction of CO2 and various epoxides, including propylene oxide (PO), cyclohexene oxide (CHO), styrene oxide (SO), limonene oxide (LO), octene oxide (OO) and epichlorohydrin (ECH), was investigated. Terpolymerisation of CO2, PO and LO, as well as CO2, CHO and PO, was successfully realised, resulting in polymers with adjustable glass transition temperatures and transparencies. Reaction conditions such as temperature, pressure and catalyst concentration were varied to find the optimal reaction values, especially regarding LO/CO2. In situ IR experiments hinted that at 60 °C and a critical LO concentration, polymerisation and depolymerisation are in an equilibrium (ceiling effect). Pressurising catalyst 1 with carbon dioxide resulted in a dimeric catalyst (2) with a OSiMe3 group as a new initiator. Homopolymerisation of different epoxides was carried out in order to explain the reactivity concerning copolymerisation reaction of CO2 and epoxides.
Introduction
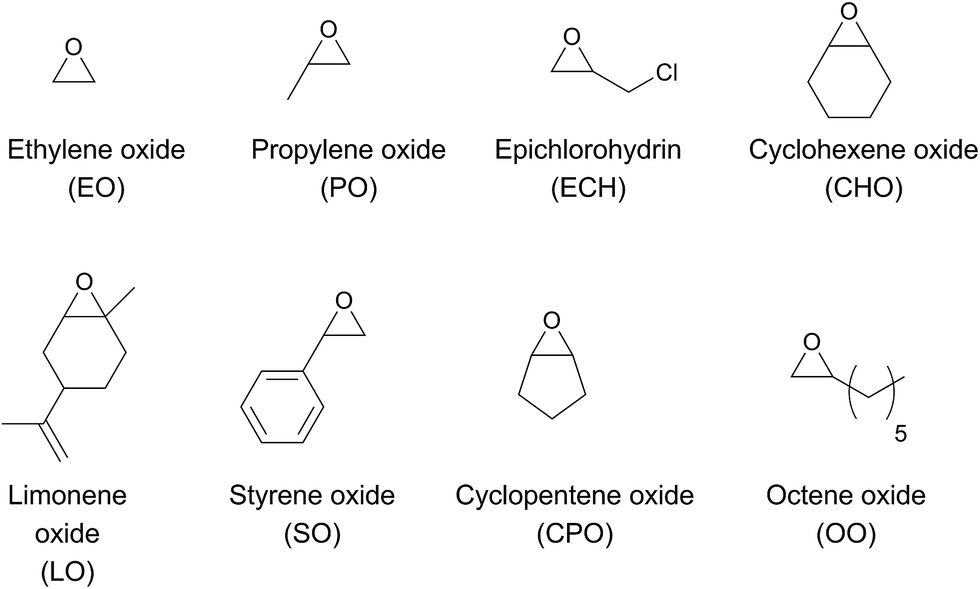
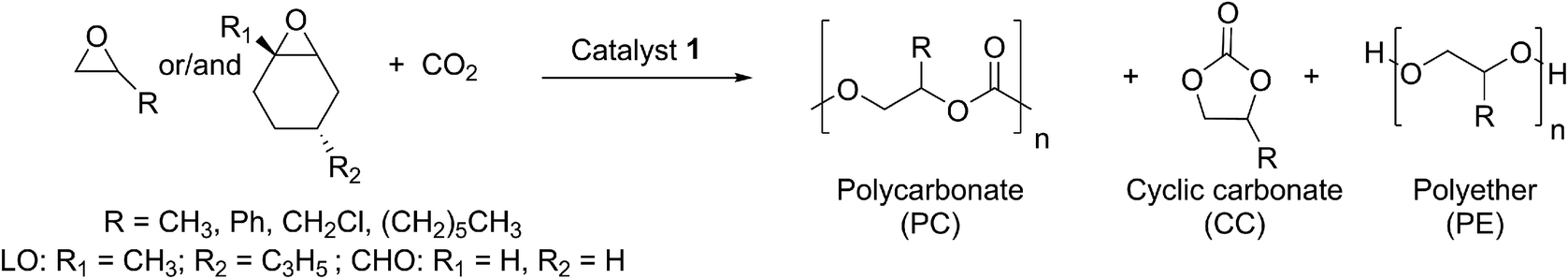
The chemical fixation of carbon dioxide for the preparation of cyclic- or polycarbonates has been targeted by many research groups to date.1–6 Polycarbonates from renewable sources have the potential to replace petroleum-based plastics such as poly(propylene), poly(ethylene), polystyrene or bisphenol A carbonate. Until now, none of the current polycarbonates derived from carbon dioxide and epoxides (Fig. 1) display all of the desired polymer properties. Poly(propylene carbonate) (PPC), which is already produced in industry, is biocompatible and biodegradable. It can be used as a film due to its good barrier for O2 and water, it burns in air without emitting toxic materials and it has a long elongation at break. However, its glass transition temperature (Tg) of 25–40 °C (body temperature) limits the range of its industrial applications.7,8 In contrast, poly(cyclohexene carbonate) (PCHC) has a high Tg of ca. 120 °C, but its brittleness causes unfavourable mechanical properties. Terpolymerisation of different epoxides and CO2 is a promising strategy towards altering disadvantageous homopolymer properties.9–11 It is still challenging, as most complexes are specific for only one monomer. Besides, the majority of catalysts that are able to polymerise different epoxides are based on the toxic metals cobalt and chromium.9,11,12 Apart from the production of typical polycarbonates like PPC or PCHC, fabrication of bio-based poly(limonene carbonate) (PLimC) has attracted much attention recently.13–20 Limonene oxide (LO) is based on limonene, which is one of the most common terpenes in nature, found in the peel of many citrus fruits.21 Elastic properties, water solubility, antibacterial characteristics or hydrophobicity can be introduced by post-modification of the double bond of the PLimC.19 However, there are only a few catalysts reported in the literature which are active for this copolymerisation, as LO exhibits an internal, tri-substituted motif with a high kinetic barrier.14,15,17 So far, turnover frequencies (TOFs) of the complexes are still low, and the reported catalysts are sensitive towards high epoxide-to-catalyst ratios.14,15 β-diiminato–zinc (BDI–zinc) complexes exhibit the best activities, but are only able to copolymerise trans-LO.15,17 Recently, a revised strategy for the synthesis of trans-enriched LO (84% trans) was published to avoid redundant cis-LO.13 There is already an easy synthetic approach for a successful kinetic resolution with up to 98% de for both diastereomers.22 We recently reported an activity enhancement of BDI-based zinc complexes regarding copolymerisation of CO2 and cyclohexene oxide (CHO) by introducing electron-withdrawing groups.23,24 Herein, we present the utilisation of a newly developed Lewis acid BDI–zinc complex for co- and terpolymerisation of CO2 and an expanded set of epoxides, including the sterically demanding monomer LO.
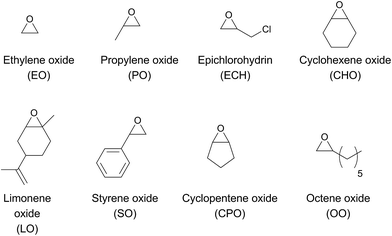 |
| | Fig. 1 Various epoxides for copolymerisation with carbon dioxide. | |
Results and discussion
Synthesis of β-diiminato-zinc-NTMS2 (1)
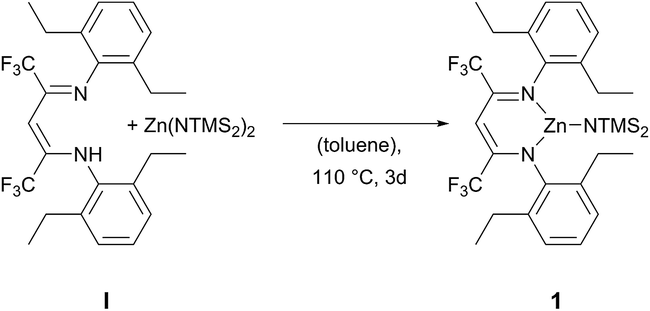
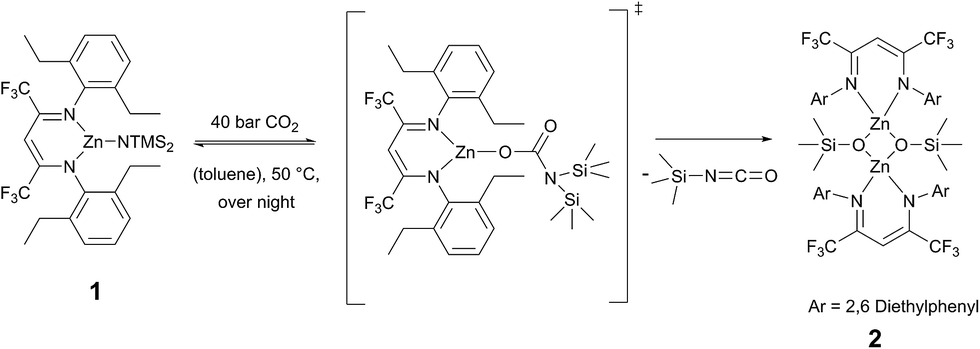
The ligand BDICF3–H(I) can be synthesised by a literature procedure in a condensation reaction with TiCl4, 2,6-diethylamine and 1,1,1,5,5,5-hexafluroroacetylacetone.25 In order to obtain the corresponding complex (1), ligand I was stirred at 110 °C with 1.2 equivalents of bis[bis(trimethylsilyl)-amido]zinc in dry toluene for three days under argon atmosphere (Scheme 1).
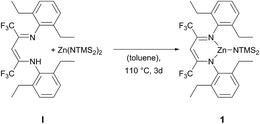 |
| | Scheme 1 Synthesis of β-diiminato–zinc–N(SiMe3)21 by reaction of BDICF3 ligand I and the zinc precursor. | |
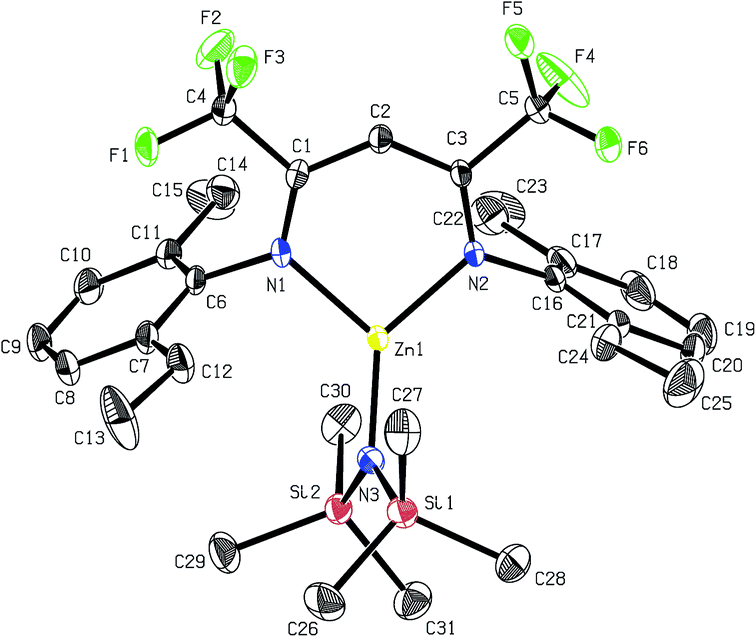
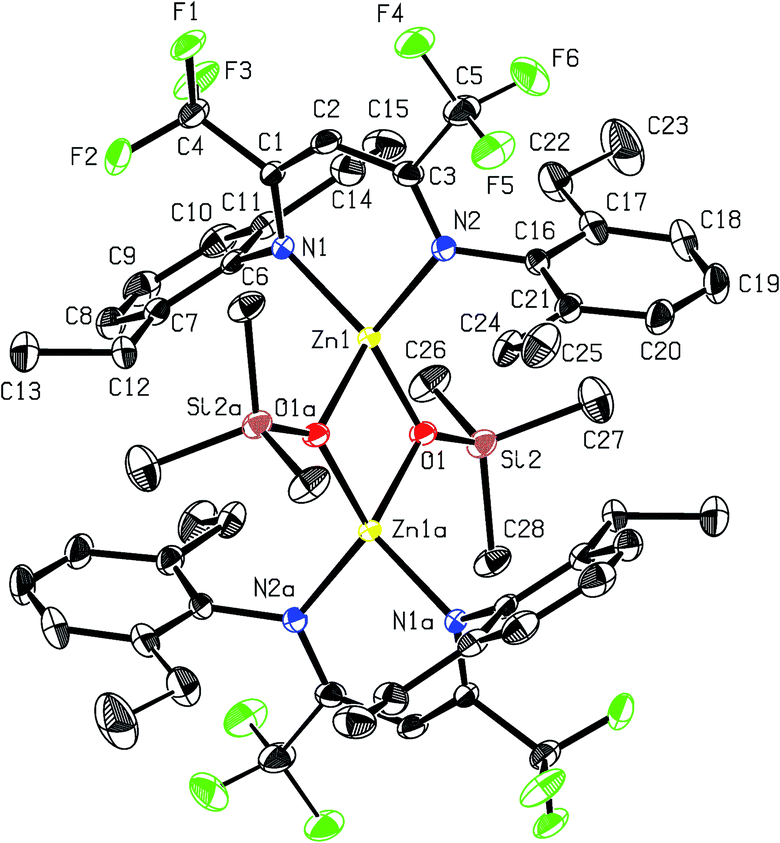
Recrystallisation from toluene at −35 °C afforded yellow crystals, which were suitable for single-crystal X-ray diffraction (SC-XRD). The molecular structure of catalyst 1 in the solid state is depicted in Fig. 2. The metal core adopts a trigonal-planar geometry with a Zn–N(SiMe3)2 bond length of 1.869(2) Å. Compound 1 can be compared with a literature known iPr-BDICH3–Zn–N(SiMe3)2 complex (4), which bears methyl instead of trifluoromethyl groups in the pentane backbone and iso-propyl groups instead of ethyl groups at the aniline (Zn–N(SiMe3)2, 1.896(2) Å).26 The Zn1–N1 (1.986(2) Å) and Zn1–N2 (1.967(2) Å) bonds differ slightly in their length, as the N(SiMe3)2 ligand is marginally shifted to one side (C2, Zn1 and N3 are not strictly linear). Pressurising a solution of the catalyst in toluene with 40 bar CO2 resulted in precipitation of a new dimeric catalyst [BDICF3–Zn-μ-OSiMe3]2 (2). SC-XRD revealed the resulting structure of this compound, which is presented in Fig. 3. During this reaction (Scheme 2), CO2 is inserted into the Zn1–N3 bond, and after migration of the trimethylsilyl group, trimethylsilyl isocyanate is released (the 1H-NMR spectrum of the mother liquor is shown in Fig. S1†).27
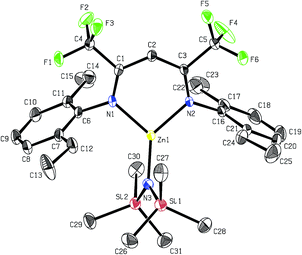 |
| | Fig. 2 ORTEP style representation of 1 (hydrogen atoms omitted for clarity) with ellipsoids drawn at the 50% probability level. Selected bond lengths (Å) and bond angles (deg): Zn1–N1 1.986(2), Zn1–N2 1.967(2), Zn1–N3 1.869(2), N1–Zn1–N2 97.11(8), N3–Zn1–N2 134.61(9), N3–Zn1–N1 128.11(9). | |
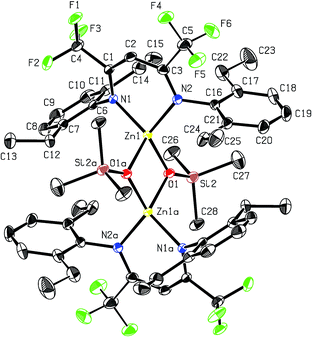 |
| | Fig. 3 ORTEP style representation of 2 (hydrogen atoms omitted for clarity) with ellipsoids drawn at the 50% probability level. The complex features an intramolecular centre of inversion. Disordered groups are indicated by thin bonds. Selected bond lengths (Å) and bond angles (deg): Zn1–Zn2 2.9486(4), Zn1–N1 2.035(1), Zn1–N2 2.029(1), Zn1–O1 1.966(1), Zn1–O1A 2.019(1), N1–Zn1–N2 94.36(5), O1–Zn1–N2 120.97(5), O1–Zn1–N1 126.35(5), O1–Zn1–O1A 84.55(4). | |
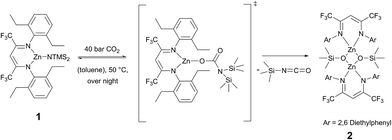 |
| | Scheme 2 Catalyst 2 is synthesized via pressurizing 1 with 40 bar CO2 in toluene. | |
Complex 2 is a dimeric catalyst having four electron withdrawing CF3 groups in the ligand backbones. The solid state structure shows a siloxide-bridged dimer with a distorted tetrahedral geometry around each zinc centre. In comparison to 1 the Zn–N bond lengths of 2 are significantly elongated (Zn1– N1 = 2.035(1) Å, Zn1–N2 = 2.029(1) Å). The Zn–Zn distance of 2.9486(4) Å is very short, similar to a reported [BDICH3–Zn-μ-OSiMe3]2 complex.27 In contrast, pressurising a previously reported catalyst BDICF3–Zn–ethyl (3) with CO2 at 50 °C overnight did not lead to the insertion of CO2 (no reaction at all was observed).
Copolymerisation of epoxides and CO2 with catalyst 1
After full characterisation of 1 by NMR spectroscopy, elemental analysis and SC-XRD, the compound was employed as a catalyst for copolymerisation experiments of CO2 and epoxides (Table 1). There is a huge variety of epoxides and only selected examples were used within this work in order to show the catalyst's capacity. Previously, we reported about the activity of a BDICF3–Zn–ethyl catalyst (3) towards copolymerisation of CHO/CO2. PCHC with a TOF of 4800 h−1, 91% polycarbonate linkages and a molecular weight of 102 kg mol−1 (PDI 1.3, bimodal) was achieved.24 Catalyst 1 yielded 57% PCHC with good selectivity for carbonate linkages (93%) within 12.5 min (TOF 5520 h−1) under the same reaction conditions ([Cat]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CHO] = 1:2000, 50 °C and 40 bar CO2). The resulting polymer was analysed by gel permeation chromatography (GPC), which showed a bimodal distribution curve and a molecular weight of 134 kDa with a PDI of 1.7. Especially regarding PCHC, this bimodality is often seen in the literature and can be explained by the presence of residual water.28–30 Another interesting monomer is propylene oxide (PO), as it is very cheap and can also be produced from CO2. It has one main drawback, as owing to a facile depolymerisation (backbiting), the formation of cyclic propylene carbonate is often observed. Catalyst 3 is not able to produce PPC (see further discussion below). In contrast, experiments (Table 1, entries 3–5) demonstrate that with catalyst 1, copolymerisation of PO and CO2 is possible. At room temperature, a selectivity for PPC of up to 78% was achieved, whereas no formation of ether linkages was observed. PPC with a high molecular weight of 98 kg mol−1 and a narrow PDI of 1.4 could be achieved using 0.1 mol% catalyst. Raising the temperature to 50 °C led to better activity, but almost exclusively, cyclic propylene carbonate was isolated. Furthermore, the catalyst was tested for styrene oxide (SO), epichlorohydrin (ECH), LO and octene oxide (OO). The coupling reaction of CO2 and ECH at 40 °C led solely to the formation of cyclic ECH carbonate. At 20 °C, no reaction was observed. With regard to SO, there are only a few reports in the literature dealing with the coupling reaction of SO/CO2, and they are again mostly based on Co(III)–salen systems.31–34 Complex 1 is indeed able to copolymerise SO and CO2, with a high epoxide-to-catalyst ratio, but only a moderate selectivity for polycarbonate of 66% (34% cyclic styrene carbonate). Furthermore, an Mn of 19.6 kDa with a narrow PDI (1.3) was measured via GPC in tetrahydrofuran. Finally, even LO/CO2 copolymerisation was realised with good selectivity for PLimC and moderate TOF (variation of the reaction conditions is summarised in Table 3). The excellent versatility of catalyst 1 was shown in terpolymerisation reactions with CHO/PO/CO2 or LO/PO/CO2. High-molecular-weight polymers were produced, whereby the glass transition temperature could be varied from 39 °C (PPC/PLimC, Table 1, entry 10, Fig. S8†) to 50 °C (PPC/PCHC, Table 1, entry 11, Fig. S8†). UV-vis measurements showed that films of LO/PO/CO2 terpolymer (0.70 mm thick) have a very good transmission of 95–99% in the range 400–800 nm (Fig. S9†). A film out of a CHO/PO/CO2 terpolymer (0.13 mm thick) exhibits transmission of 80–90% in the range 400–800 nm (Fig. S9†). Further investigations are ongoing to examine the influence of different epoxide ratios on glass transition temperatures, transmissions and strain–stress behaviour.
:[CHO] = 1:2000, 50 °C and 40 bar CO2). The resulting polymer was analysed by gel permeation chromatography (GPC), which showed a bimodal distribution curve and a molecular weight of 134 kDa with a PDI of 1.7. Especially regarding PCHC, this bimodality is often seen in the literature and can be explained by the presence of residual water.28–30 Another interesting monomer is propylene oxide (PO), as it is very cheap and can also be produced from CO2. It has one main drawback, as owing to a facile depolymerisation (backbiting), the formation of cyclic propylene carbonate is often observed. Catalyst 3 is not able to produce PPC (see further discussion below). In contrast, experiments (Table 1, entries 3–5) demonstrate that with catalyst 1, copolymerisation of PO and CO2 is possible. At room temperature, a selectivity for PPC of up to 78% was achieved, whereas no formation of ether linkages was observed. PPC with a high molecular weight of 98 kg mol−1 and a narrow PDI of 1.4 could be achieved using 0.1 mol% catalyst. Raising the temperature to 50 °C led to better activity, but almost exclusively, cyclic propylene carbonate was isolated. Furthermore, the catalyst was tested for styrene oxide (SO), epichlorohydrin (ECH), LO and octene oxide (OO). The coupling reaction of CO2 and ECH at 40 °C led solely to the formation of cyclic ECH carbonate. At 20 °C, no reaction was observed. With regard to SO, there are only a few reports in the literature dealing with the coupling reaction of SO/CO2, and they are again mostly based on Co(III)–salen systems.31–34 Complex 1 is indeed able to copolymerise SO and CO2, with a high epoxide-to-catalyst ratio, but only a moderate selectivity for polycarbonate of 66% (34% cyclic styrene carbonate). Furthermore, an Mn of 19.6 kDa with a narrow PDI (1.3) was measured via GPC in tetrahydrofuran. Finally, even LO/CO2 copolymerisation was realised with good selectivity for PLimC and moderate TOF (variation of the reaction conditions is summarised in Table 3). The excellent versatility of catalyst 1 was shown in terpolymerisation reactions with CHO/PO/CO2 or LO/PO/CO2. High-molecular-weight polymers were produced, whereby the glass transition temperature could be varied from 39 °C (PPC/PLimC, Table 1, entry 10, Fig. S8†) to 50 °C (PPC/PCHC, Table 1, entry 11, Fig. S8†). UV-vis measurements showed that films of LO/PO/CO2 terpolymer (0.70 mm thick) have a very good transmission of 95–99% in the range 400–800 nm (Fig. S9†). A film out of a CHO/PO/CO2 terpolymer (0.13 mm thick) exhibits transmission of 80–90% in the range 400–800 nm (Fig. S9†). Further investigations are ongoing to examine the influence of different epoxide ratios on glass transition temperatures, transmissions and strain–stress behaviour.
Table 1 Coupling reaction of CO2 and epoxides with catalyst 1a
|
|
| Entry |
Epoxide |
[Epoxide]: [(1)] |
p [bar] |
T [°C] |
Time [h] |
Yield [%] |
TONb |
TOFc [h−1] |
Selectivity (NMR)d [PC]:[CC]:[PE] |
Terpolymer composition (NMR)e [PC]:[PPC]:[CC] |
M
n (PDI)f,g [kg mol−1] |
|
Reaction scheme refers to copolymerisation of PO, SO, OO, ECH with CO2 (PLimC, PCHC and terpolymers look different).
The turnover number (TON) is the ratio of the number of moles of epoxide consumed to the number of moles of catalyst.
TOF is TON/time.
[PC]:[CC]:[PE], assigned by the relative integrals of the signals for polycarbonate (PC), cyclic carbonate (CC) and polyether (PE).
[PC]:[PPC]:[CC], assigned by the relative signals for (a) entry 10: PLimC:PPC:cPC (b) entry 11: PCHC:PPC:cPC before precipitation.
Determined by GPC, calibrated with polystyrene standards in chloroform.
Determined by GPC, calibrated with polystyrene standards in thf.
5.0 mL toluene.
|
| 1 |
CHO |
2000 |
40 |
50 |
0.2 |
57 |
1150 |
5520 |
93:0:7 |
— |
134 (1.7)g |
| 2 |
CHO |
4000 |
40 |
50 |
0.58 |
50 |
2010 |
3440 |
91:0:9 |
— |
135 (1.4)g |
| 3 |
POh |
1000 |
30 |
25 |
20 |
56 |
560 |
30 |
62:38:0 |
— |
98 (1.4)f |
| 4 |
POh |
1000 |
30 |
50 |
1 |
12 |
120 |
120 |
3:94:3 |
— |
— |
| 5 |
PO |
500 |
30 |
25 |
3 |
32 |
160 |
53 |
78:22:0 |
— |
21 (1.2)f |
| 6 |
SO |
1000 |
30 |
25 |
20 |
51 |
510 |
26 |
66:34:0 |
— |
20 (1.3)g |
| 7 |
OO |
1000 |
40 |
40 |
24 |
92 |
920 |
38 |
33:66:0 |
— |
107 (1.7)f |
| 8 |
ECH |
1000 |
30 |
40 |
16 |
44 |
440 |
28 |
0:100:0 |
— |
— |
| 9 |
LO |
250 |
30 |
40 |
16 |
73 |
90 |
180 |
100:0:0 |
— |
33 (1.9)f |
| 10 |
LO/PO |
250/600 |
30 |
25 |
6 |
40 |
330 |
55 |
98:2:0 |
51:47:2 |
95 (1.4)f |
| 11 |
CHO/PO |
500/500 |
30 |
25 |
14 |
65 |
650 |
46 |
99:1:0 |
59:40:1 |
272 (1.3)f |
Ring-opening homopolymerisation of PO, CHO and LO
The copolymerisation experiments with different epoxides and CO2 showed that catalyst 1 is very active for all tested monomers. On the other hand, catalyst 3 with a different initiator (ethyl) is only able to copolymerise CHO/CO2 and is not active for PO/CO2 or LO/CO2. SC-XRD revealed that complex 1 inserts CO2 into the Zn–N bond in order to form the dinuclear complex 2, whereas complex 3 did not show any reaction with CO2. However, the synthesis of 2 is slow, and only low conversion is observed.
To get more insight into the reaction mechanism, ring-opening homopolymerisation experiments with catalysts 1–3 and PO, CHO and LO were performed (Table 2). It was therefore found that CHO can be ring opened by complexes 1, 2 and 3 (Table 2, entries 1, 4 and 7). In this case, there is no need for CO2 in order to start the polymerisation. However, catalyst 3 shows only low conversion for formation of poly(cyclohexene oxide) due to a low initiator efficiency (Table 2, entry 3). In contrast to CHO, PO can only be slowly homopolymerised by catalyst 2. The 1H-NMR spectra showed that coordination of the epoxide to the respective catalyst occurs in all cases, as the signal of the initiation group is marginally shifted, but only catalyst 2 is able to ring open PO (Fig. S2–S4†). LO is also coordinated (1H-NMR spectra, Fig. S2–S4†), but apparently none of the initiation groups are able to attack the epoxide. All results indicate that the true active species during copolymerisation of CO2 and epoxides with complex 1 is formed in situ in the reaction with CO2 (Scheme 2, intermediate). Furthermore, in literature, it was shown that acetate is a good initiator for the coupling reaction of CO2/LO, which resembles the in situ formed –OCONTMS2 group.15 As catalyst 3 is not able to insert CO2, PO and LO cannot be ring-opened, and no copolymerisation occurs.
Table 2 Ring-opening homopolymerisation of PO, CHO and LO with complexes 1–3a
| Entry |
Cat |
Epoxide |
[Epoxide]:[Cat] |
Yield [%] |
TONb |
|
All experiments were performed at room temperature (∼23 °C) over 16 hours in neat epoxide.
The turnover number (TON) is the ratio of the number of moles of epoxide consumed to the number of moles of catalyst.
|
| 1 |
1
|
CHO |
500 |
52 |
260 |
| 2 |
1
|
PO |
500 |
0 |
0 |
| 3 |
1
|
LO |
250 |
0 |
0 |
| 4 |
2
|
CHO |
500 |
56 |
280 |
| 5 |
2
|
PO |
500 |
5 |
25 |
| 6 |
2
|
LO |
250 |
0 |
0 |
| 7 |
3
|
CHO |
500 |
5 |
25 |
| 8 |
3
|
PO |
500 |
0 |
0 |
| 9 |
3
|
LO |
250 |
0 |
0 |
Variation of the reaction conditions of the coupling reaction of CO2/LO
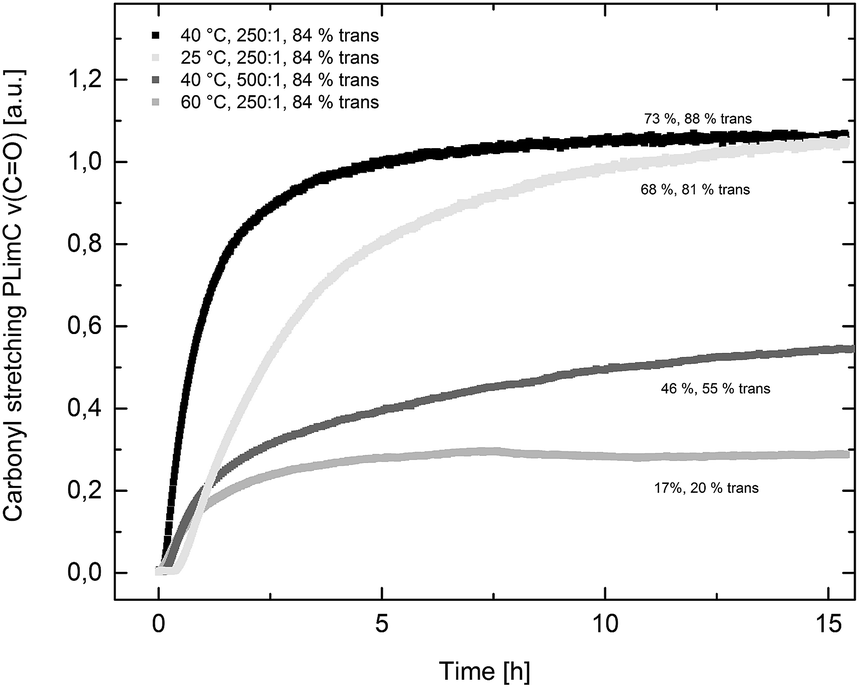
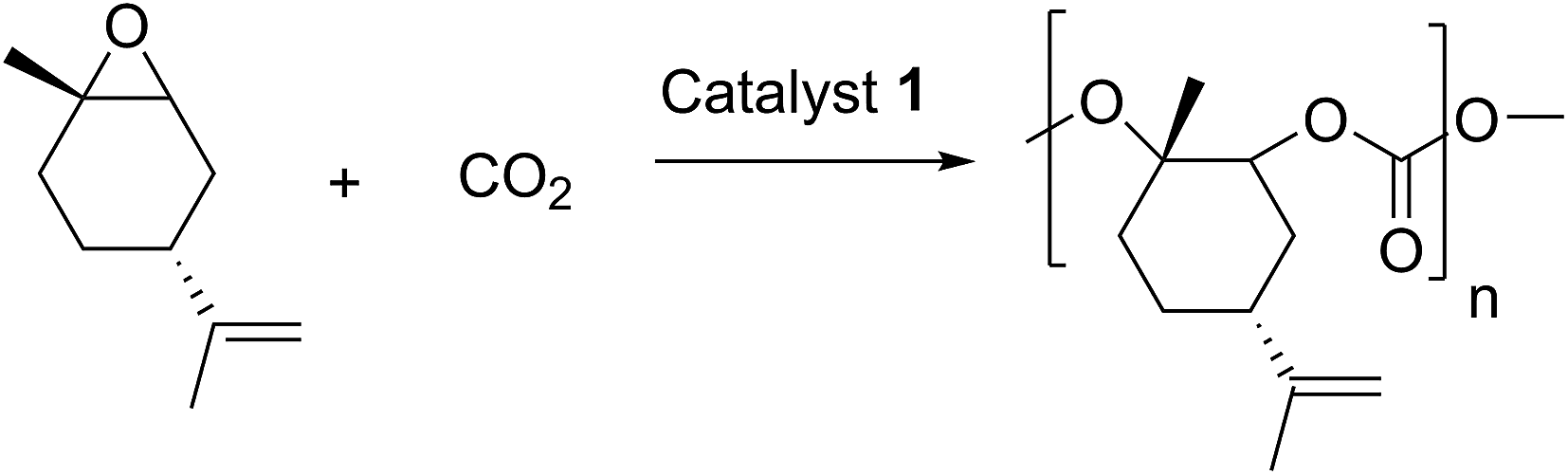
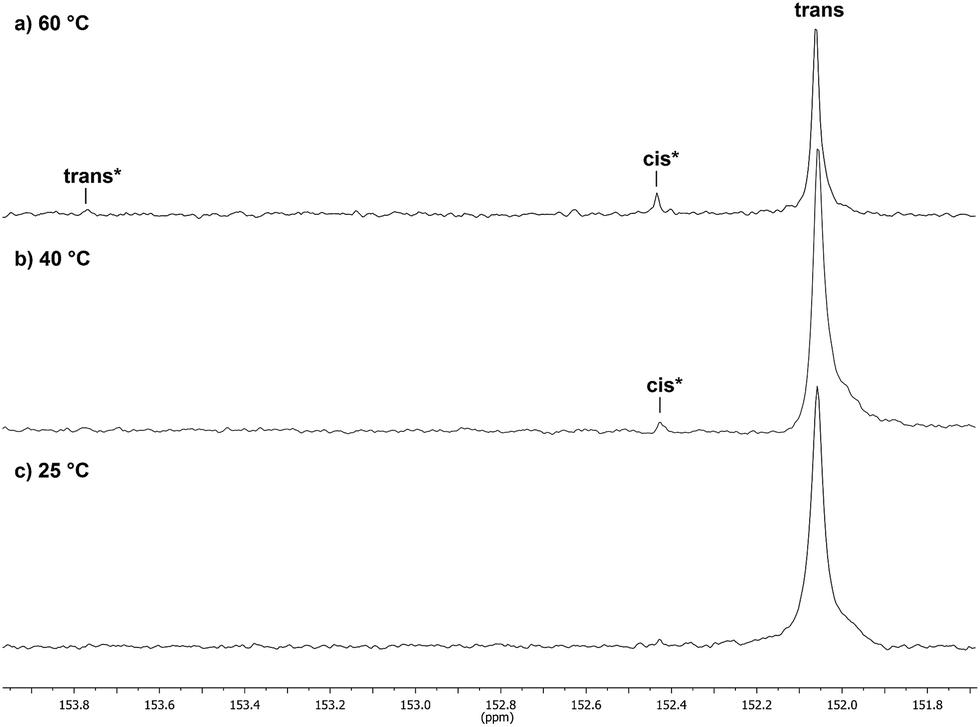
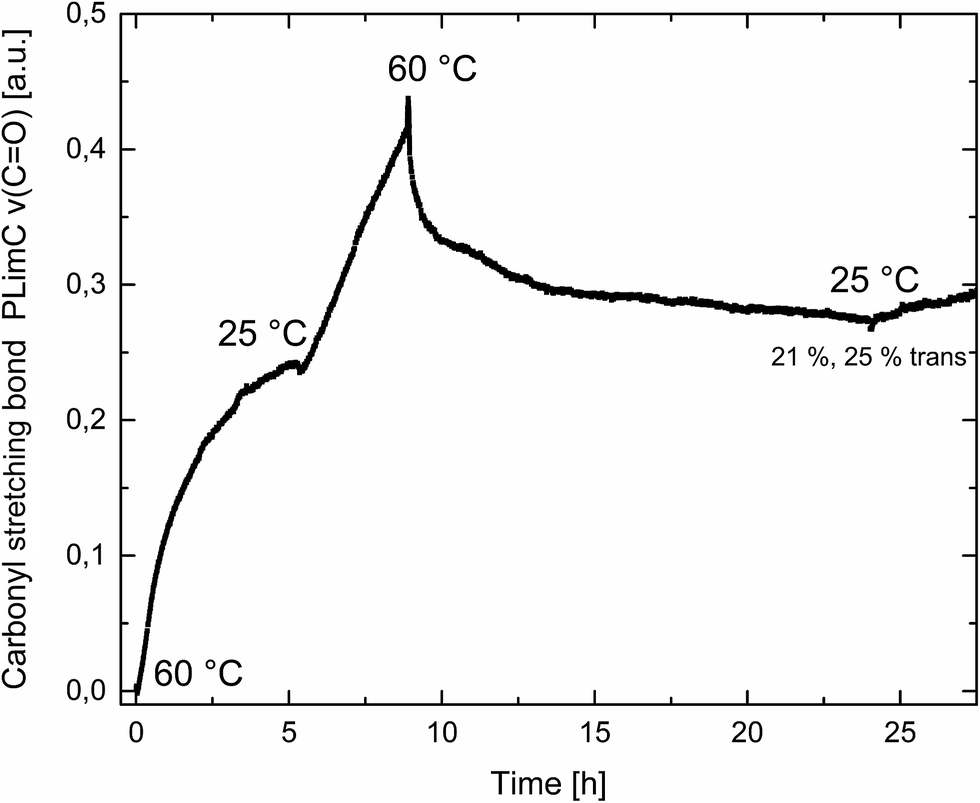
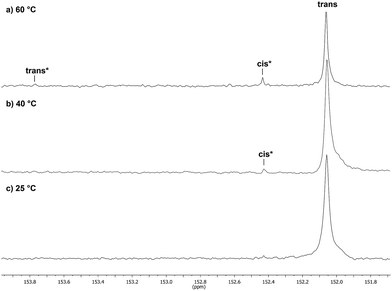
Catalyst 1 was also tested for LO/CO2 copolymerisation. Firstly, the reaction temperature was varied between 25 °C, 40 °C and 60 °C at an epoxide-to-catalyst ratio of 250:1 (0.4 mol%) and 30 bar. All experiments were conducted in an autoclave with in situ IR monitoring to follow the reaction progress (Fig. 4). Raising the temperature from 25 °C to 40 °C resulted in a higher TOF, but similar overall yields (full conversion was not achieved perhaps owing to the high viscosity of the polymer mixture). At 60 °C, catalyst 1 only yielded 17% PLimC. The in situ IR spectrum shows that after about 4 h, the carbonyl-stretching band of PLimC is not increasing anymore. Coates et al. reported very low conversions at 50 °C for their BDI–Zn catalyst owing to an axial attack at the stereogenic carbon centre of (1R,4R)-1-methyl-4-(prop-1-en-2-yl)-7-oxabicyclo[4.1.0]heptane (cis-LO) at this temperature (formation of trans*-PLimC, slow reaction due to steric hindrance).15 The 1H and 13C-NMR spectra of our polymers showed that (1S,4R)-1-methyl-4-(prop-1-en-2-yl)-7-oxabicyclo-[4.1.0]heptane (trans-LO) is primarily attacked by the catalyst at the sterically less hindered C-atom (trans-PLimC) at all temperatures. If the trans-LO is opened at the most substituted carbon centre of the oxirane unit, inversion of the configuration leads to the formation of cis*-PLimC. At higher temperatures, the attack at the stereogenic centre of trans-LO and cis-LO is more likely than at 25 °C (Table 3, entry 3, up to 5% cis*-PLimC, 1% trans*-PLimC). Decreasing the catalyst concentration to 0.2 mol% leads to a slow reaction after 4 h and a lower overall conversion (Table 3, entry 8, 46% yield). A yield of 3% cis*-PLimC was determined, accompanied by low molecular masses and a broad PDI. Interestingly, the TOF could be increased in a solvent-free polymerisation using 50 bar CO2 at 0.2 mol% catalyst and 60 °C (Table 3, entry 14). A maximum TOF of 310 h−1 was achieved, which is the highest reported TOF for the formation of PLimC so far.13 However, once again, only up to 40% yield was reached after 16 h (Table 3, entry 14). We considered cis-LO as a catalyst quencher, as this monomer stays untouched at low temperatures (13C-NMR spectrum, Fig. 5c), whereas at 60 °C, the formation of approximately 1% trans*-PLimC was observed (Table 3, entry 3). Therefore, the amount of cis-LO was first varied from <1% to 10% and 46% at 40 °C (30.5 mmol, 5.0 mL LO with different composition, 2 mL toluene, 30 bar). In these cases, no deactivation of the catalyst was observed (Fig. S7†). With a higher amount of cis-LO, the overall yield for trans-PLimC was even increased owing to a good solubility of the polymer in the residual cis-LO (lower viscosity) and a higher catalyst concentration with regard to trans-LO. The highest reported molecular masses of 145 kg mol−1 with a small PDI of 1.3 were achieved using >99% trans-LO at 40 °C and 30 bar CO2. Applying the same conditions at 60 °C, the catalyst yielded again only 11–26% PLimC (Table 3, entries 3, 5 and 7). Using >99% trans-LO, the IR monitoring of the reaction showed that the intensity of the carbonyl-stretching band of PLimC increases only very slightly after 2 h (Fig. S6†). Thus, cis-LO could be excluded as a catalyst poison. Another possibility is deactivation of complex 1via the double bond of cis-/trans-LO. Zn–π interactions are rare, but are reported in the literature.35,36 In order to verify the influence of the double bond, LO (54% trans) was hydrated with PtO2/H2, and the resulting menthene oxide was copolymerised with CO2 at 30 bar, 0.4 mol% catalyst 1 and 25 °C or 60 °C (ESI Table S1, Fig. S10 and S11†). An initiation time of about 40 min was observed at 25 °C, and high-molecular-weight trans-polymenthene carbonate (PMenC) (Mn = 149 kDa, PDI 1.2) was produced. At 60 °C and over 16 h, only 4% regioirregular PMenC (Fig. S11†) was generated (Mn = 5.6 kDa, PDI 1.3), which indicates that even in the absence of a double bond, complex 1 is deactivated. In summary, cis-LO as well as the double bond of LO could be excluded as catalyst poisons. A third possible explanation for all the aforementioned observations is that the ceiling temperature of PLimC is around 60 °C at an LO concentration of about 3.5 mol L−1. Every polymerisation is an equilibrium reaction, whereby the balance can be shifted towards the formation of polymer or the reverse by varying temperature, pressure and concentration of the monomer.37 At 60 °C, a critical concentration of LO is achieved during the polymerisation, where the reverse reaction is just as fast as the polymer formation itself. We do not detect the formation of cyclic limonene carbonate by in situ IR spectroscopy and 1H-NMR, so the reverse reaction is the release of CO2 and LO (and not the usual backbiting). An experiment in the autoclave with in situ IR monitoring was performed at 60 °C, 30 bar and a catalyst loading of 0.4 mol% (Fig. 6). After the signal of the carbonyl stretching of PLimC approached a plateau, the temperature was decreased to 25 °C. The polymer signal started to increase linearly as soon as the right temperature was achieved. After a few hours, a temperature of 60 °C was applied again, and the carbonyl stretching of PLimC decreased to the original level from the beginning of the cycle. That means that depolymerisation occurred until the critical LO concentration (ca. 3.5 mol L−1) was achieved. In a last step, the temperature was set to 25 °C, which led again to an increasing polymer signal, though with a clearly decreased slope. Therefore, besides this thermodynamic control, there should be, in addition, kinetic aspects which have to be considered, e.g. deactivation of the catalyst by traces of contaminants in LO.
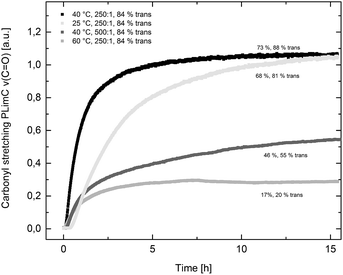 |
| | Fig. 4 The formation of PLimC against time (carbonyl stretching ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in a.u. against time in h). Yields are given concerning 30.5 mmol cis-/trans LO and regarding the actual amount of trans-LO (x% trans). O) in a.u. against time in h). Yields are given concerning 30.5 mmol cis-/trans LO and regarding the actual amount of trans-LO (x% trans). | |
Table 3 Copolymerisation of limonene oxide and carbon dioxide with catalyst 1 under different reaction conditionsa
 |
| | Fig. 5
13C-NMR spectra (expansion 153.9–151.6 ppm) of PLimC, produced at different polymerisation temperatures: (a) 60 °C (Table 3, entry 3), (b) 40 °C (Table 3, entry 2) and (c) 25 °C (Table 3, entry 1). | |
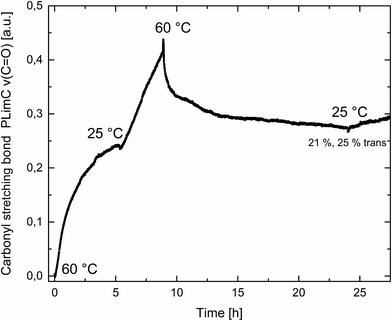 |
| | Fig. 6 Formation of the carbonyl stretching of PLimC in a.u. against time in h at different temperatures. Overall conversion was 21%, and regarding trans-LO, 25%. | |
Conclusion
In summary, we introduced a versatile Lewis acid BDI-based zinc catalyst with an N(SiMe3)2 initiation group for copolymerisation of many different epoxides and CO2. We found out that pressurising the catalyst with CO2 leads to the insertion of the latter and a subsequent release of trimethylsilyl isocyanate. Ring-opening studies of PO, CHO and LO indicate that the real active species is primarily the in situ generated carbonato-intermediate. Even terpolymerisation experiments with different monomers were successful, as they resulted in polymers with tuneable properties such as Tg or transparency. In addition, copolymerisation reactions with LO/CO2 were performed, which showed that catalyst 1 is the most active catalyst for the formation of PLimC reported so far. However, at a temperature of 60 °C, conversions were much lower than expected. cis-LO and the double bond of LO could be excluded as catalyst poisons. In situ IR experiments showed that at 60 °C, depolymerisation is in an equilibrium with polymer formation at a critical LO concentration.
Acknowledgements
The authors would like to thank Dr Richard O. Reithmeier and Dr Peter T. Altenbuchner for proofreading the manuscript. We would like to thank Rike Adams, Martin Machat, Markus Pschenitza and Christina Schwarzenböck for the valuable discussions. We would like to thank Dr A. Pöthig for crystallographic advice.
Notes and references
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 Search PubMed.
- G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc. London, Ser. A, 2016, 374, 20150085 Search PubMed.
-
B. Rieger, A. Künkel and G. W. Coates, Synthetic Biodegradable Polymers, Springer, Berlin, Heidelberg, Germany, 2012 Search PubMed.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 Search PubMed.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 Search PubMed.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 Search PubMed.
- G. A. Luinstra and E. Borchardt, Adv. Polym. Sci., 2012, 245, 29–48 Search PubMed.
- G. A. Luinstra and E. Borchardt, Polym. Rev., 2008, 48, 192–219 Search PubMed.
- L. Shi, X.-B. Lu, R. Zhang, X.-J. Peng, C.-Q. Zhang, J.-F. Li and X.-M. Peng, Macromolecules, 2006, 39, 5679–5685 Search PubMed.
- D. J. Darensbourg, R. R. Poland and A. L. Strickland, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 127–133 Search PubMed.
- W.-M. Ren, X. Zhang, Y. Liu, J.-F. Li, H. Wang and X.-B. Lu, Macromolecules, 2010, 43, 1396–1402 Search PubMed.
- J. E. Seong, S. J. Na, A. Cyriac, B.-W. Kim and B. Y. Lee, Macromolecules, 2010, 43, 903–908 Search PubMed.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 Search PubMed.
- L. Peña Carrodeguas, J. González-Fabra, F. Castro-Gómez, C. Bo and A. W. Kleij, Chem.–Eur. J., 2015, 21, 6115–6122 Search PubMed.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 Search PubMed.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo and G. W. Coates, Macromolecules, 2015, 48, 2534–2550 Search PubMed.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 1215–1218 Search PubMed.
- C. Li, R. J. Sablong and C. E. Koning, Angew. Chem., Int. Ed., 2016, 55, 11572–11576 Search PubMed.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 Search PubMed.
- C. Martín and A. W. Kleij, Macromolecules, 2016, 49, 6285–6295 Search PubMed.
- R. Ciriminna, M. Lomeli-Rodriguez, P. Demma Cara, J. A. Lopez-Sanchez and M. Pagliaro, Chem. Commun., 2014, 50, 15288–15296 Search PubMed.
- D. Steiner, L. Ivison, C. T. Goralski, R. B. Appell, J. R. Gojkovic and B. Singaram, Tetrahedron: Asymmetry, 2002, 13, 2359–2363 Search PubMed.
- S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579–4582 RSC.
- M. Reiter, A. Kronast, S. Kissling and B. Rieger, ACS Macro Lett., 2016, 5, 419–423 Search PubMed.
- A. Kronast, M. Reiter, P. T. Altenbuchner, C. Jandl, A. Pöthig and B. Rieger, Organometallics, 2016, 35, 681–685 Search PubMed.
- M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 Search PubMed.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 Search PubMed.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 Search PubMed.
- K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 Search PubMed.
- H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 Search PubMed.
- G.-P. Wu, S.-H. Wei, X.-B. Lu, W.-M. Ren and D. J. Darensbourg, Macromolecules, 2010, 43, 9202–9204 Search PubMed.
- R.-J. Wei, X.-H. Zhang, B.-Y. Du, X.-K. Sun, Z.-Q. Fan and G.-R. Qi, Macromolecules, 2013, 46, 3693–3697 Search PubMed.
- Y. Dienes, W. Leitner, M. G. J. Muller, W. K. Offermans, T. Reier, A. Reinholdt, T. E. Weirich and T. E. Muller, Green Chem., 2012, 14, 1168–1177 Search PubMed.
- G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, B. Li, Y.-P. Zu and D. J. Darensbourg, Energy Environ. Sci., 2011, 4, 5084–5092 Search PubMed.
- A. Wooten, P. J. Carroll, A. G. Maestri and P. J. Walsh, J. Am. Chem. Soc., 2006, 128, 4624–4631 Search PubMed.
- R. Benn, H. Grondey, H. Lehmkuhl, H. Nehl, K. Angermund and C. Krüger, Angew. Chem., Int. Ed. Engl., 1987, 26, 1279–1280 Search PubMed.
-
G. Odian, Principles of Polymerization, John Wiley & Sons, New York, 1981 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, GPC data, NMR spectra and crystallographic data. CCDC 1508589–1508590. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04477h |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence