
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of acceptor nucleophilicity on the glycosylation reaction mechanism†
S.
van der Vorm
,
T.
Hansen
,
H. S.
Overkleeft
,
G. A.
van der Marel
and
J. D. C.
Codée
*
Leiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: jcodee@chem.leidenuniv.nl
First published on 9th November 2016
Abstract
A set of model nucleophiles of gradually changing nucleophilicity is used to probe the glycosylation reaction mechanism. Glycosylations of ethanol-based acceptors, bearing varying amounts of fluorine atoms, report on the dependency of the stereochemistry in condensation reactions on the nucleophilicity of the acceptor. Three different glycosylation systems were scrutinized, that differ in the reaction mechanism, that – putatively – prevails during the coupling reaction. It is revealed that the stereoselectivity in glycosylations of benzylidene protected glucose donors are very susceptible to acceptor nucleophilicity whereas condensations of benzylidene mannose and mannuronic acid donors represent more robust glycosylation systems in terms of diastereoselectivity. The change in stereoselectivity with decreasing acceptor nucleophilicity is related to a change in reaction mechanism shifting from the SN2 side to the SN1 side of the reactivity spectrum. Carbohydrate acceptors are examined and the reactivity–selectivity profile of these nucleophiles mirrored those of the model acceptors studied. The set of model ethanol acceptors thus provides a simple and effective “toolbox” to investigate glycosylation reaction mechanisms and report on the robustness of glycosylation protocols.
Introduction
The connection of two carbohydrate building blocks to construct a glycosidic linkage in a glycosylation reaction is one of the most important and one of the most difficult steps in the assembly of an oligosaccharide.1–3 The stereoselective formation of 1,2-cis glycosidic linkages remains a major synthetic challenge and often requires careful tuning of reaction conditions for a profitable outcome.4 The variation in stereochemical outcome of a chemical glycosylation reaction originates from the different mechanistic pathways that can be followed for the union of an activated donor glycoside and an acceptor. Fig. 1 depicts the current understanding of the continuum of mechanisms operational during a glycosylation reaction. The activation of a donor glycoside leads to an array of reactive intermediates, formed from the donor glycoside and the activator derived counterion. α- and β-configured covalent reactive intermediates can be formed and these are in equilibrium with less stable and more reactive oxocarbenium ion based species. These can be either closely associated with the counterion providing close (or contact) ion pairs (CIPs), or further separated from their counterion in solvent separated ion pairs (SSIPs). These reactive intermediates can be attacked by an incoming nucleophile following a reaction mechanism with both SN1 and SN2 features. The covalent species are displaced in a reaction mechanism having an associative SN2-character, while the oxocarbenium ion-like intermediates are engaged in an SN1-like reaction. The exact position(s) on the continuum where a given glycosylation reaction takes place, and hence the stereoselectivity of the process, depends critically on the reactivity of both reaction partners: the donor and acceptor glycoside. The impact of the reactivity of the donor glycoside on the stereochemical outcome has been studied extensively, and the effect of functional and protecting groups on glycosyl donor reactivity is well documented.5–10 In contrast, the influence of the reactivity of the nucleophile (the acceptor) on the outcome of a glycosylation reaction remains poorly understood.11–18 We here present a systematic study to determine the effect of acceptor nucleophilicity on the stereochemical course of a glycosylation reaction. We show how a simple “toolset” of partially fluorinated alcohols13 can be used to dissect reaction mechanisms that are operational during a glycosylation reaction. It is revealed that the stereoselectivity of some glycosylation systems varies more with changing acceptor nucleophilicity than others and we relate these differences to changes in reaction pathways that are followed. A panel of model carbohydrate acceptors is scrutinized to place the reactivity of these building blocks in the context of the nucleophilicity scale set by the series of fluorinated ethanols.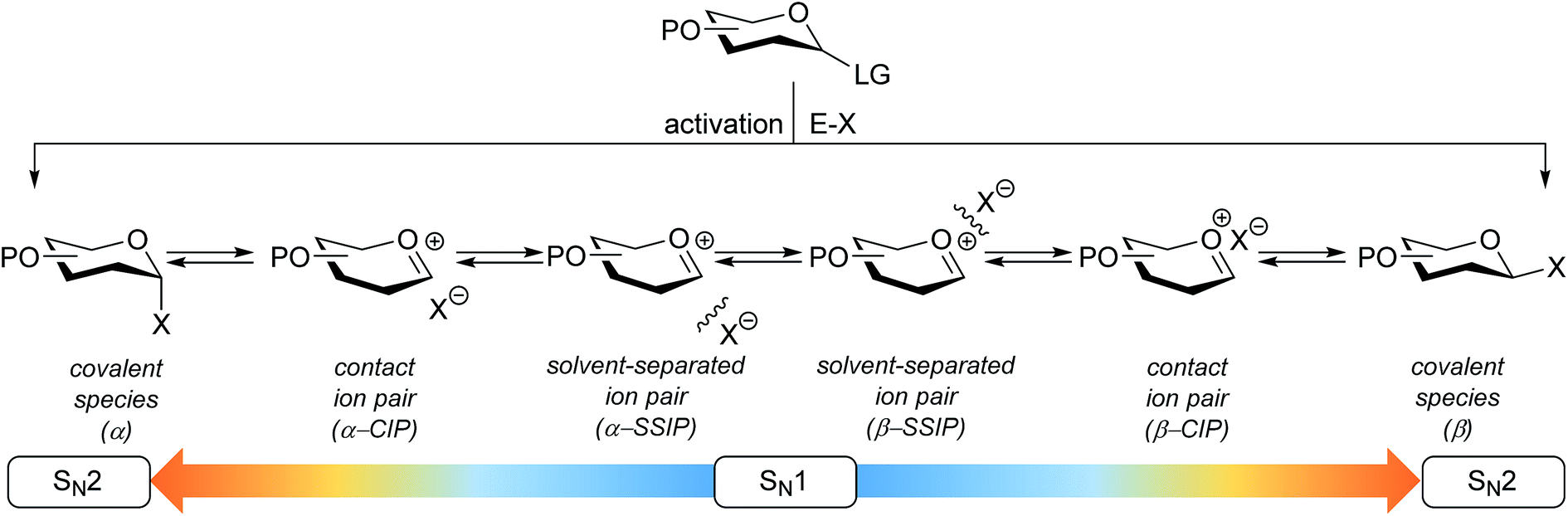 | ||
| Fig. 1 The reaction mechanism manifold operational during glycosylation reactions. | ||
Results and discussion
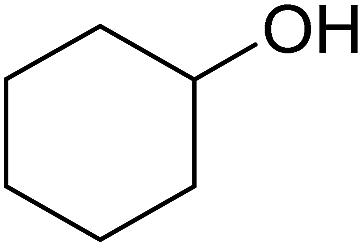
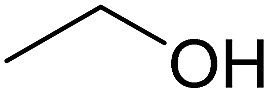
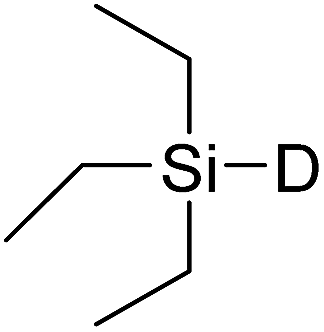
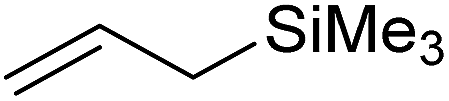
In this study the effect of acceptor nucleophilicity on the glycosylation selectivity is systematically investigated by the hand of a set of model O-nucleophiles, encompassing ethanol, monofluoroethanol (MFE), difluoroethanol (DFE), trifluoroethanol (TFE), hexafluoro-iso-propanol (HFIP) and cyclohexanol, as well as a C-nucleophile, allyltrimethylsilane (allyl-TMS), and a deuterium nucleophile, deuterated triethylsilane (TES-D).12,13 Next a series of carbohydrate acceptors is used to place the reactivity of these alcohols in the context of the reactivity of the ethanol model acceptors (see Fig. 2B and C). Three glycosylation systems have been investigated with these acceptors: the benzylidene mannose and analogous benzylidene glucose system as well as the mannuronic acid system (see Fig. 2A). These systems have been selected because they have previously been studied in depth to provide insight into the major reaction pathways that operate during glycosylation reactions of these donors (vide infra). Although these three glycosylation systems all selectively provide 1,2-cis-products, the major product-forming pathways significantly differ.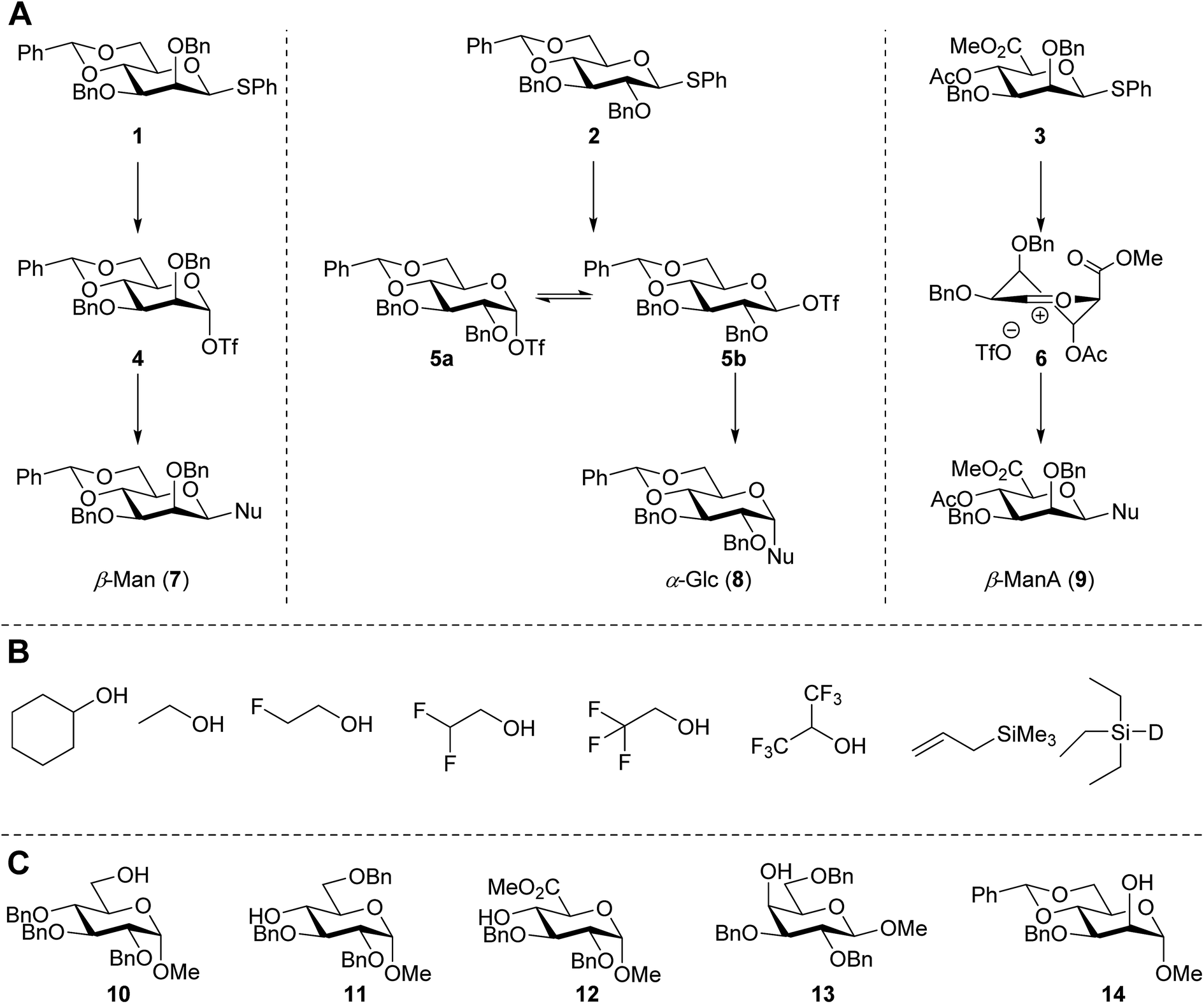 | ||
| Fig. 2 (A) The benzylidene mannose, benzylidene glucose and mannuronic acid glycosylation systems studied and the major glycosylation pathways of these donors. (B) Set of model nucleophiles used in this study. (C) Set of carbohydrate alcohols used. | ||
The benzylidene mannose system, introduced by Crich and co-workers for the stereoselective construction of β-mannosidc linkages, represents the best studied glycosylation system to date.19,20 It has been found that benzylidene mannose donors can be transformed into the corresponding α-anomeric triflate 4 upon activation. These triflates have been extensively characterized in variable temperature NMR studies.21–24 A significant body of evidence has been gathered through a vast amount of glycosylation reactions,19–23,25–33 the establishment of kinetic isotope effects in combination with computational methods,34,35 and the application of cation clock methodology,36–38 to indicate that these triflates can be substituted in an SN2-manner to provide β-mannosides. However, an alternative hypothesis to account for the β-selectivity of benzylidene mannose glycosylations has also been forwarded. This hypothesis is based on a B2,5-oxocarbenium ion as product forming intermediate.39–42
The closely related benzylidene glucose system provides α-selective glycosylation reactions.21,22,29,40,43–47 It has been proposed that this selectivity originates from an in situ anomerization kinetic scheme, in which the initially formed α-triflate 5α anomerizes into its more reactive β-couterpart 5β.21 Substitution of this species provides the α-glucosyl products. Mechanistic studies, amongst others kinetic isotope effect and cation clock experiments, using the reactive nucleophile iso-propanol have provided support for this pathway.34,37,38
Glycosylations of mannuronic acids have been shown to proceed in a highly selective manner to provide β-mannuronic acid products. Based on the conformational behavior of the donors and the intermediate α-triflates 18α, adopting an 1C4 conformation,48,49 the high reactivity of these donors50,51 and a large variety of glycosylation reactions, both in solution,50,52–55 and on fluorous56 and solid supports,57 it has been postulated that the selectivity in these glycosylation reactions can be related to the intermediacy of an 4H3 oxocarbenium ion-like intermediate.53,54,58
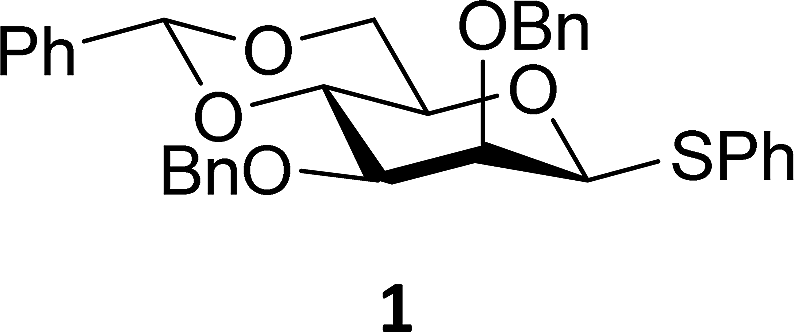
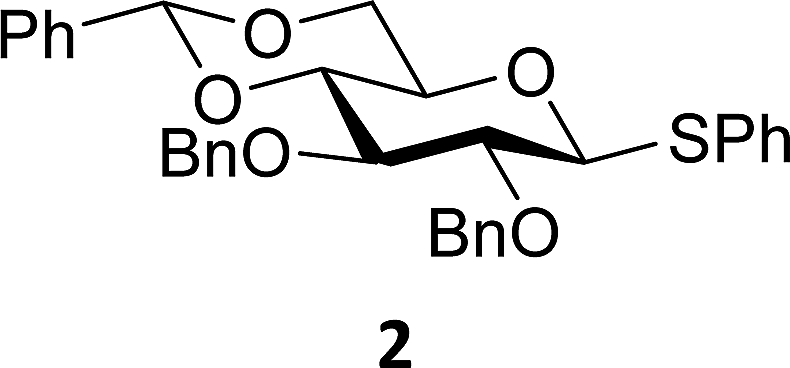
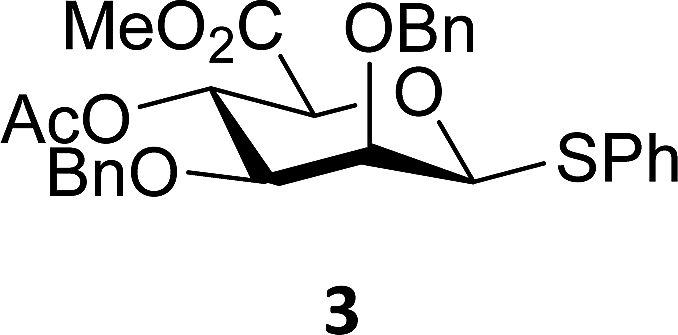
The experimental setup that we used in this study is based on pre-activation of the thioglycoside donors 1,59221 and 3 using a slight excess of diphenyl sulfoxide and triflic anhydride (Ph2SO/Tf2O) at low temperature. This transforms all three donors into the corresponding anomeric triflates,21–24,48,60 prior to addition of the acceptor nucleophiles. The pre-activation set-up generates a pool of reactive intermediates in the absence of the acceptor, thereby eliminating product forming pathways that originate from direct displacement reactions on the activated parent donor species. Table 1 summarizes the results obtained with the three donor systems and the set of model acceptors. As a measure for the reactivity of the used acceptors, Mayr's nucleophilicity parameters have been tabularized where available.61–63 The field inductive parameters for the –CH3, –CH2F, –CHF2 and –CF3 groups have also been shown to indicate the gradual increase of electron withdrawing character of these groups.64
| Acceptor | N | F |
|
|
|
|---|---|---|---|---|---|
Product, α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β (yield)c :β (yield)c |
Product, α:β (yield)c |
Product, α:β (yield)c |
|||
| a Mayr's nucleophilicity parameters. b Field inductive parameters. c α/β-Ratios were established by NMR spectroscopy of the crude and purified reaction mixtures. d Both anomers of donor glycoside were also found after the glycosylation reaction. Literature yields of 1H40: 57% and 2H40: 56%. | |||||
|
— | — | 1A | 2A | 3A |
| 1:6 |
1:5 |
1:8 |
|||
| (96%) | (71%) | (83%) | |||
|
7.44 | 0.01 | 1B | 2B | 3B |
| 1:5 |
1:10 |
1:8 |
|||
| (70%) | (68%) | (95%) | |||
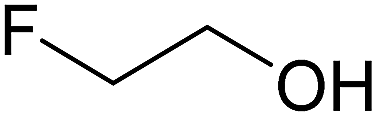
|
— | 0.15 | 1C | 2C | 3C |
| 1:5 |
1:3 |
1:6 |
|||
| (86%) | (70%) | (70%) | |||
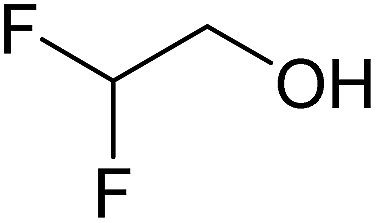
|
— | 0.29 | 1D | 2D | 3D |
| 1:5 |
5:1 |
1:5 |
|||
| (90%) | (70%) | (87%) | |||
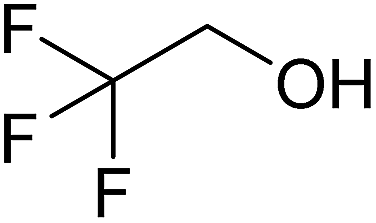
|
1.11 | 0.38 | 1E | 2E | 3E |
| 1:4 |
>20:1 |
1:2.5 |
|||
| (78%) | (64%) | (85%) | |||
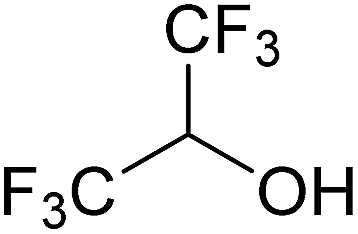
|
−1.93 | — | 1F | 2F | 3F |
| 3:1 |
>20:1 |
1:1 |
|||
| (56%) | (65%) | (52%) | |||
|
3.58 | — | 1G | 2G | 3G |
| <1:20 |
>20:1 |
<1:20 |
|||
| (60%) | (79%) | (95%) | |||
|
1.68 | — | 1H | 2H | 3H |
| <1:20 |
>20:1 |
<1:20 |
|||
| (44%)d | (42%)d | (40%)d | |||
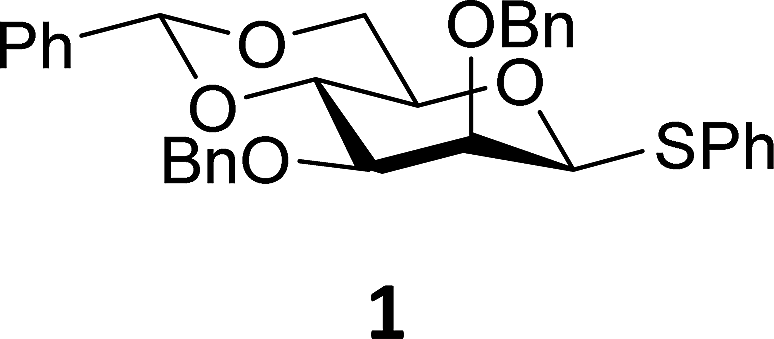
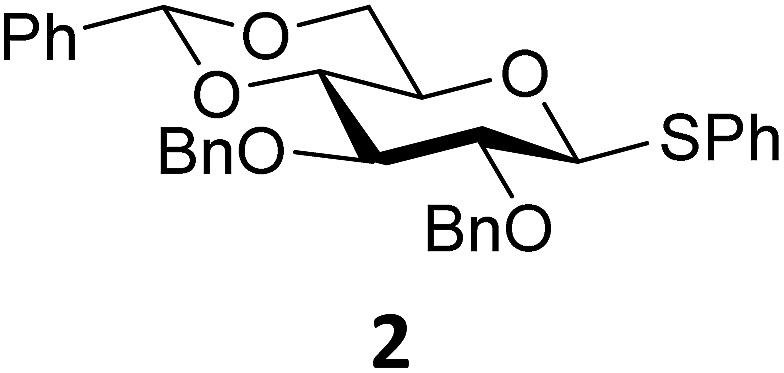
From the results depicted in Table 1 it becomes immediately apparent that the stereoselectivity of the benzylidene mannose and mannuronic acid systems shows relatively little variation with changing nucleophilicity, where the stereoselectivity of the glycosylations involving the benzylidene glucose donor changes significantly depending on the reactivity of the used nucleophile. Reactive nucleophiles such as ethanol, cyclohexanol and MFE predominantly provide β-linked products (2A, 2B and 2C), where the use of less reactive nucleophiles such as DFE, TFE, HFIP, TES-D and allyl-TMS leads to the preferential formation of the α-glucosyl products (2D–2H). A clear trend becomes apparent between the reactivity of the non-fluorinated and partially fluorinated ethanols and the stereoselectivity of the glucosylations involving these acceptors. The formation of the β-linked products 2A,652B and 2C can be explained to originate from an SN2-like substitution on the intermediate α-triflate 5α (see Fig. 3). The α-products in these glucosylations (α-2A, α-2B, α-2C) may be formed from the corresponding β-glucosyl triflate 5β, as postulated by Crich and co-workers and as supported by kinetic isotope effect and cation clock studies.34,35,37,66 It is however less likely that the unreactive O-nucleophiles, such as TFE and HFIP, and the weak C- and D-nucleophiles, are capable of displacing the anomeric triflate 5 in an SN2-manner. Woerpel and co-workers have previously shown that TFE requires a glycosylating agent bearing significant oxocarbenium ion character.13 An explanation for the observed α-selectivity in the glucosylations of these nucleophiles may be found in the SN1-like substitution on the benzylidene glucose oxocarbenium ion 15. This ion preferentially adopts a 4H3/4E-structure, as verified by several computational studies,67,68 that is attacked in a diastereoselective fashion from the bottom face, leading via a chair-like transition state to the α-linked products. As the reactivity of the nucleophile diminishes, it is likely that the amount of SN2-character in the substitution of the β-triflate 5β gradually decreases and the amount of SN1-character with the intermediacy of the corresponding CIP and SSIP (15) increases.13 The least reactive nucleophiles require the most “naked” oxocarbenium ions, with the triflate counterions significantly, if not completely, dissociated from the carbohydrate ring.
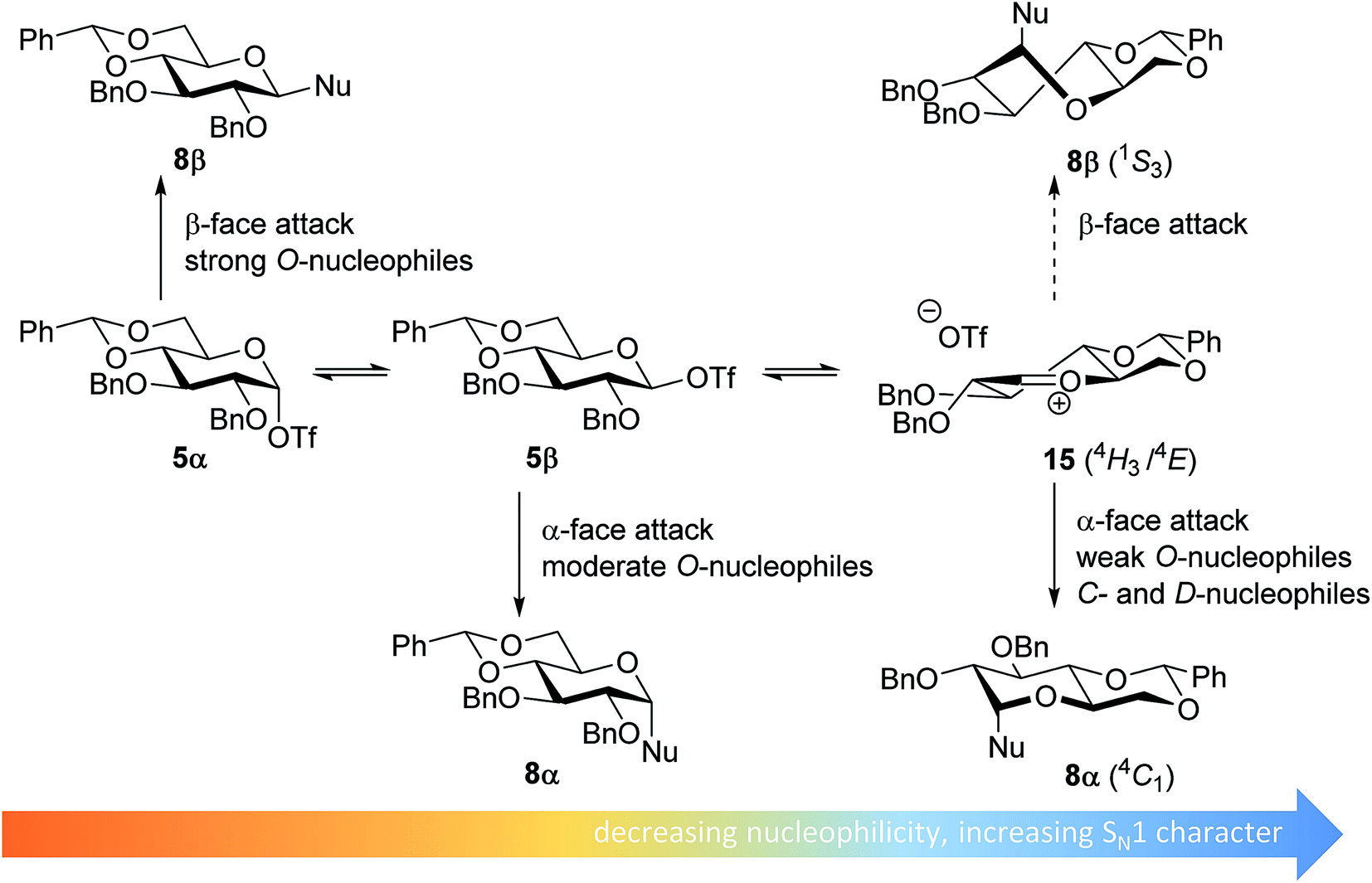 | ||
| Fig. 3 Mechanistic pathways to account for the selectivity in glycosylations of benzylidene glucose donors. | ||
The stereoselectivity of the benzylidene mannose systems seems to be less sensitive to variation in nucleophilicity of the acceptor. Donor 1 provides β-selective glycosylations with the range of acceptors studied. There is a slight decrease in selectivity going from the reactive O-nucleophiles to the weak O-nucleophiles and the condensation of benzylidene mannose 1 with HFIP proceeds with moderate α-selectivity. The most likely explanation for the β-selectivity observed with the reactive O-nucleophiles is an associative SN2-type substitution of the intermediate α-triflate 4 (see Fig. 4). As discussed above, it is unlikely that unreactive acceptors such as TFE and HFIP react in an SN2-type reaction, directly displacing the α-mannosyl triflate 4. Formation of the β-linked products formed from the unreactive acceptors and donor 1 may be better explained with an oxocarbenium ion-like product forming intermediate. Various theoretical studies have indicated that the B2,5-oxocarbenium ion 16 is the most stable benzylidene mannose oxocarbenium ion conformer.67,68 This oxocarbenium ion is preferentially attacked from the convex top-face, as attack from the bottom face would lead to unfavorable interactions with the pseudo-axial H-2 and to an eclipsed C-1–C-2 configuration upon rehybridization.36,40,69,70
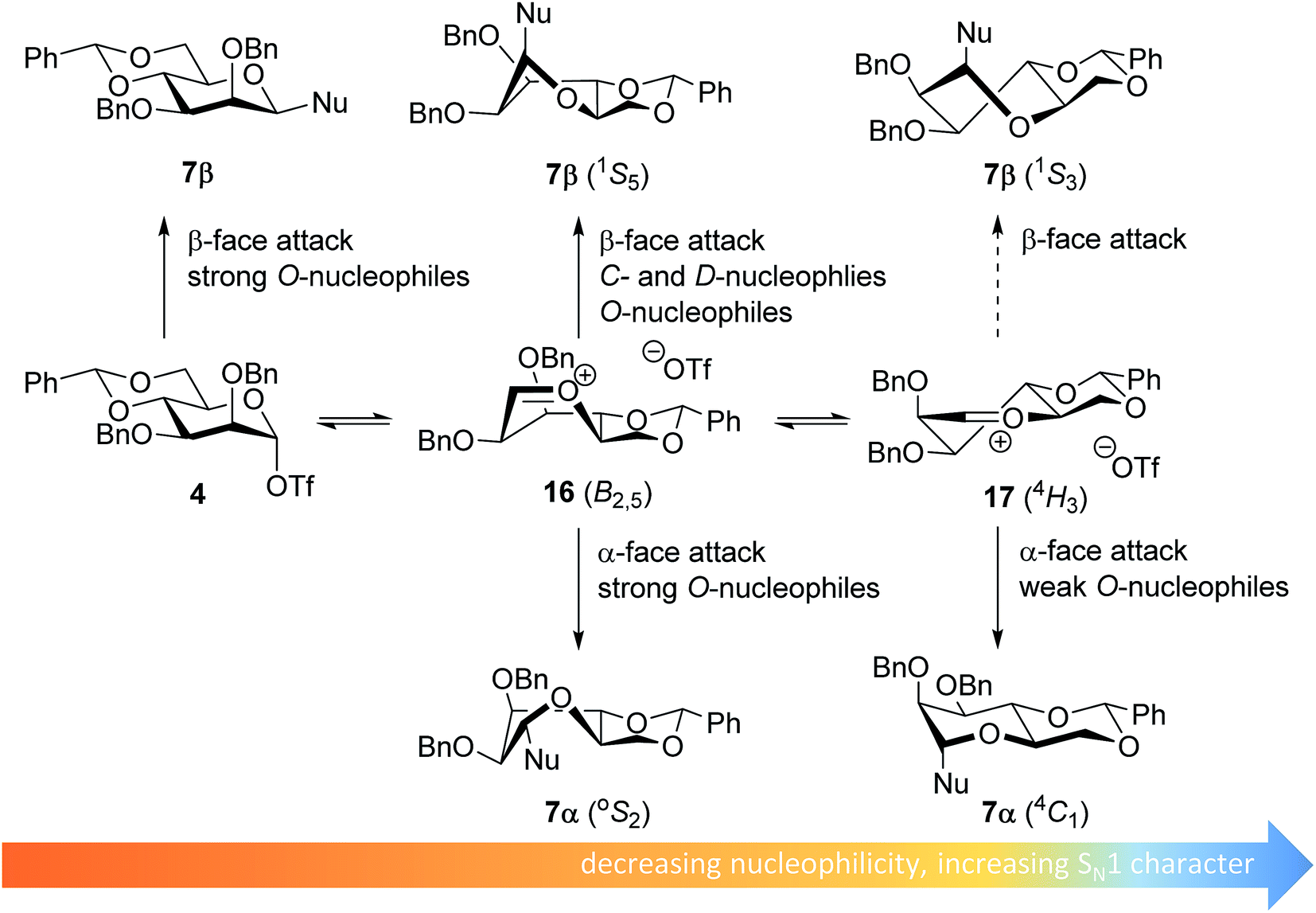 | ||
| Fig. 4 Mechanistic pathways to account for the selectivity in glycosylations of benzylidene mannose donors. | ||
The α-products formed in the condensations of donors 1 likely originate from an oxocarbenium ion intermediate. Reactive O-nucleophiles may react with an oxocarbenium ion in a relatively indiscriminative manner leading to the formation of both α- and β-products.11–13 Because unreactive O-nucleophiles are expected to react in a more diastereoselective fashion with an oxocarbenium ion, it is unlikely that the α-products derived from the weak O-nucleophiles, such as TFE and HFIP, originate from the B2,5-oxocarbenium ion 15. Instead, α-face attack on the 4H3 half chair conformer 17 may be a plausible reaction pathway to account for the α-products of the less reactive O-nucleophiles. In a later transition state, product development control plays a more important role and the developing anomeric effect and the low energy chair conformation that results from the α-face attack on the 4H3 half chair 17, make this trajectory favorable.71 For the weak C- and D-nucleophiles, which react in a highly selective β-manner, this latter pathway does not play a major role, and these nucleophiles attack the B2,5-oxocarbenium ion 16 selectively from the top face.40,72
In line with the benzylidene mannose system, the mannuronic acid donor provides β-selective condensations with all acceptors explored, except with the very unreactive O-nucleophile HFIP where both anomers were formed in equal amounts. Where reactions with nucleophilic O-nucleophiles can be expected to form from the α-triflate 18α,34–37 the weaker O-nucleophiles and allyl-TMS and TES-D will react preferentially with an oxocarbenium ion (Fig. 5). We have previously postulated that the 3H4 half chair mannuronic acid oxocarbenium ion 6 is the most stable oxocarbenium ion conformer.51,54,55 To substantiate this hypothesis, we have calculated the energy associated with a range of mannuronic acid oxocarbenium ion conformers (see Fig. 5 and ESI†) using DFT-calculations at the B3LYP/6-311G level.73 From these calculations the 3H4 conformer 6 appears to be significantly more stable (by >5 kcal mol−1) than other conformers such as the alternative 4H3 half chair 19 and the B2,5 boat conformers. The relative stability of the 3H4 half chair oxocarbenium ion can be explained by favorable interaction of the ring substituents with the electron depleted carbocation. Hyperconjugative stabilization of the C-2–H-2 bond and through space stabilization of the pseudo-axial C-3, C-4 oxygen atoms and the axial C-5 carboxylate each contribute to the stability of the half chair oxocarbenium ion.51,54,74–76 This oxocarbenium ion is preferentially attacked from the top face to provide the β-linked products via a chair-like transition state. For the weaker O-nucleophiles, a later transition state leads to significant steric interactions with the axial substituents in the 3H4 half chair oxocarbenium 6 and a reaction pathway, involving attack of the nucleophiles on the higher energy 4H3 half chair oxocarbenium ion 19 becomes relevant. In line with the discussion above, product development control is favorable for the formation of α-O-mannuronic acids.
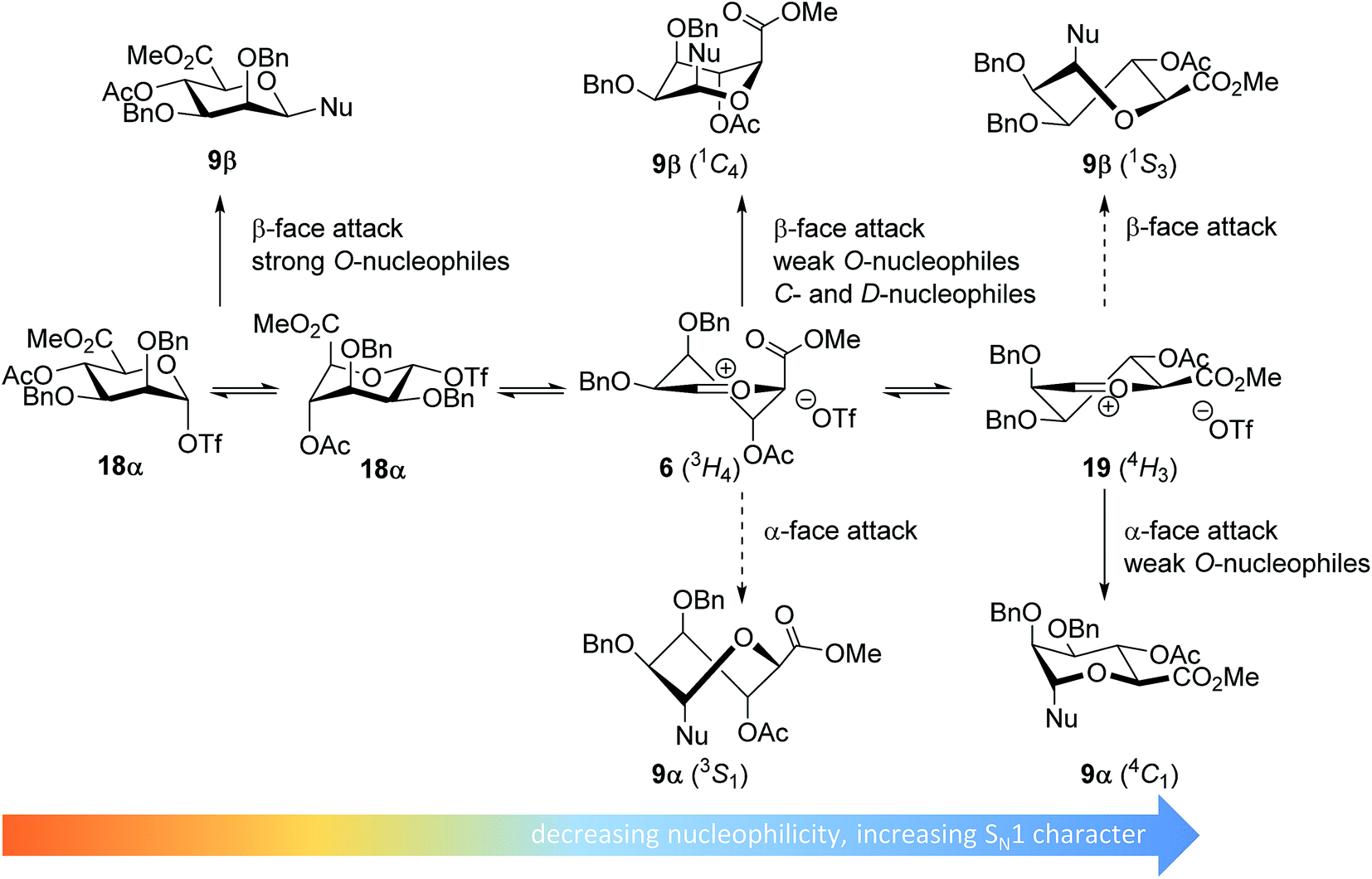 | ||
| Fig. 5 Mechanistic pathways to account for the selectivity in glycosylations of mannuronic acid donors. | ||
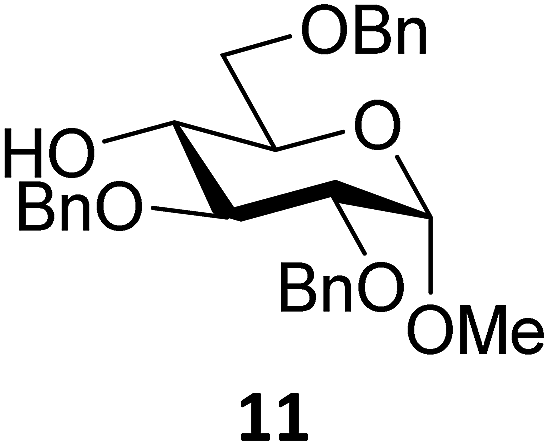
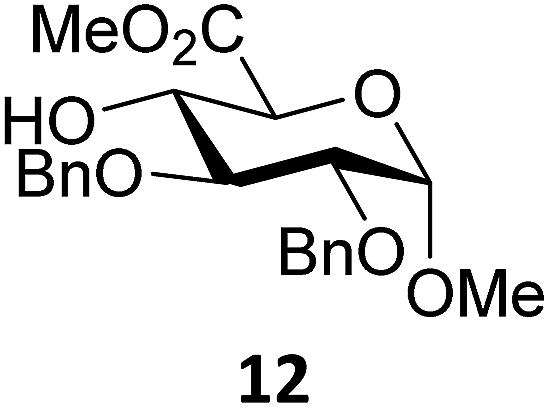
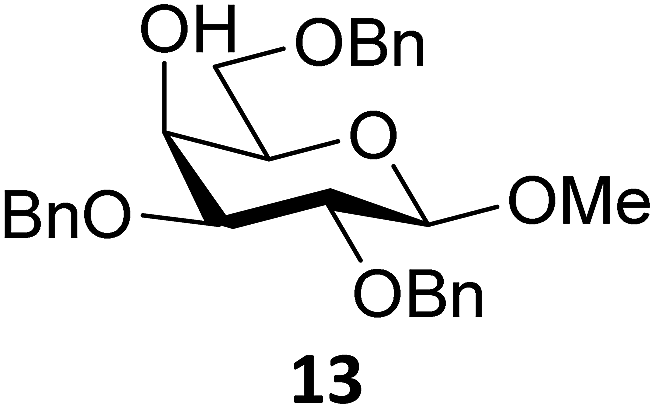
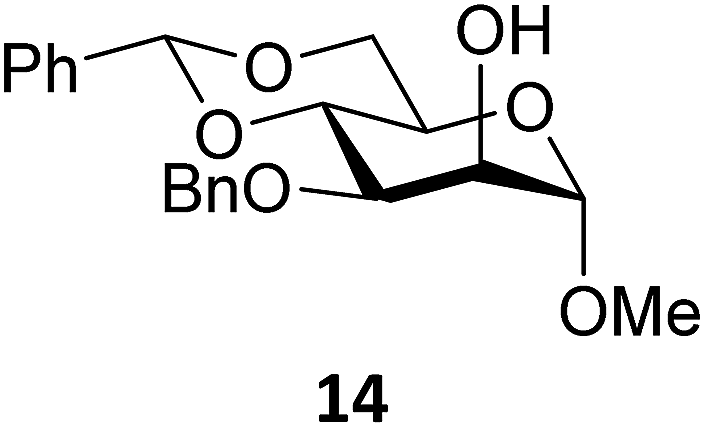
Next, we explored the set of carbohydrate acceptors depicted in Fig. 2C. The results of these condensation reactions are summarized in Table 2. Where it can be reasoned that the secondary carbohydrate acceptors 11,7712,781377 and 1479 electronically resemble DFE and TFE, because of the amount of electron withdrawing β- and/or γ- and δ-substituents, the size of the carbohydrate acceptors obviously differs significantly from the small ethanol based acceptors. The picture that emerges from Table 2 follows in broad lines the results described in Table 1 and corroborates this analysis. The benzylidene glucose donor system 2 shows most variation in stereoselectivity, where both the benzylidene mannose and mannuronic acid donors 1 and 3 provide β-selective reactions with all carbohydrate acceptors studied. The series of benzylidene glucose condensations again reveals that reactive O-nucleophiles can provide β-selective glycosylations, while less reactive O-nucleophiles give the α-linked products. The electron withdrawing effect of the C-5 carboxylate in acceptor 12, makes this acceptor less reactive and more α-selective than its C-5-benzyloxymethylene counterpart 11. In line with the discussion above, formation of the β-linked products can be explained with triflate 5α as product forming intermediate. Less reactive acceptors require a glycosylating species that is more electrophilic and react in a more dissociative substitution reaction, with a substantial amount of oxocarbenium ion character and the glucose ring taking up a 4H3-like structure (15).
The benzylidene mannose and mannuronic acid donors 1 and 3 provide very β-selective condensation reactions, in line with the vast amount of previously reported glycosylations of these two donors. Based on the results presented here and in previous work the following picture emerges. Reactive carbohydrate acceptors react in a reaction with significant SN2-character, displacing the anomeric α-triflate (4 and 18α). Weaker nucleophiles, such as most secondary carbohydrate acceptors, will react with a species that bears more carbocation character. For the benzylidene mannose donor, this species will resemble B2,5 boat oxocarbenium ion 16, where the reactive mannuronic acid reactive intermediate will be structurally close to 3H4 oxocarbenium ion 6. The minor α-products in these condensations likely arise from a higher energy 4H3 oxocarbenium ion 19, through a transition state that benefits from a developing anomeric effect and favorable conformational properties.
Conclusions
The influence of structural changes in a glycosyl donor on the outcome of a glycosylation reaction, in terms of yield and stereoselectivity, has received considerable attention over the years and many ingenious donor systems have been developed for the stereoselective construction of glycosidic bonds. The influence of the reactivity of the acceptor in glycosylation reactions, on the other hand, is less well understood. Here we have investigated in a systematic manner how the outcome of a glycosylation system can change depending on the gradually changing reactivity of the nucleophile. We have shown that a series of partially fluorinated alcohols of gradually decreasing nucleophilicity, can be used to map how the stereoselectivity of a glycosylation system varies with changing acceptor reactivity. The simple “toolset” of partially fluorinated ethanols represents a rapid and easy means to dissect SN2-type (for ethanol) and SN1-type (for trifluoroethanol and hexafluoro-iso-propanol) glycosylation reaction mechanisms.80 It is expected that application of this set of model nucleophiles to newly developed glycosylation methodology or re-investigation of already established methods will bring detailed insight into the complex and intriguing glycosylation reaction mechanism. This will allow for more directed optimization of glycosylation reactions, taking away the trial and error component and ill-understood reaction protocols that have plagued carbohydrate chemistry for so long.References
- T. J. Boltje, T. Buskas and G.-J. Boons, Nat. Chem., 2009, 1, 611–622 Search PubMed.
- M. Sinnott, Carbohydrate Chemistry and Biochemistry: Structure and Mechanism, The Royal Society of Chemistry, 2007 Search PubMed.
- A. V. Demchenko, Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- C. S. Bennett, Selective Glycosylations: Synthetic Methods and Catalysts, Wiley VCH Verlag GmbH, 2017 Search PubMed.
- M.-K. Cheung, N. Douglas, B. Hinzen, S. Ley and X. Pannecoucke, Synlett, 1997, 257–260 CrossRef CAS.
- N. L. Douglas, S. V. Ley, U. Lücking and S. L. Warriner, J. Chem. Soc., Perkin Trans. 1, 1998, 51–66 Search PubMed.
- D. K. Baeschlin, L. G. Green, M. G. Hahn, B. Hinzen, S. J. Ince and S. V. Ley, Tetrahedron: Asymmetry, 2000, 11, 173–197 CrossRef CAS.
- Z. Zhang, I. R. Ollmann, X.-S. Ye, R. Wischnat, T. Baasov and C.-H. Wong, J. Am. Chem. Soc., 1999, 121, 734–753 Search PubMed.
- J.-C. Lee, W. A. Greenberg and C.-H. Wong, Nat. Protoc., 2007, 1, 3143–3152 Search PubMed.
- T. K. Ritter, K.-K. T. Mong, H. Liu, T. Nakatani and C.-H. Wong, Angew. Chem., Int. Ed., 2003, 42, 4657–4660 Search PubMed.
- J. R. Krumper, W. A. Salamant and K. A. Woerpel, Org. Lett., 2008, 10, 4907–4910 Search PubMed.
- J. R. Krumper, W. A. Salamant and K. A. Woerpel, J. Org. Chem., 2009, 74, 8039–8050 Search PubMed.
- M. G. Beaver and K. A. Woerpel, J. Org. Chem., 2010, 75, 1107–1118 Search PubMed.
- J. Kalikanda and Z. Li, Carbohydr. Res., 2011, 346, 2380–2383 Search PubMed.
- M. Heuckendorff and H. H. Jensen, Eur. J. Org. Chem., 2016, 5136–5145 Search PubMed.
- M. L. Bohn, M. I. Colombo, C. A. Stortz and E. A. Rúveda, Carbohydr. Res., 2006, 341, 1096–1104 Search PubMed.
- M. I. Colombo, C. A. Stortz and E. A. Rúveda, Carbohydr. Res., 2011, 346, 569–576 Search PubMed.
- B. Schumann, S. G. Parameswarappa, M. P. Lisboa, N. Kottari, F. Guidetti, C. L. Pereira and P. H. Seeberger, Angew. Chem., Int. Ed., 2016, 55, 14431–14434 Search PubMed.
- D. Crich and S. Sun, J. Org. Chem., 1996, 61, 4506–4507 Search PubMed.
- D. Crich and S. Sun, J. Org. Chem., 1997, 62, 1198–1199 Search PubMed.
- D. Crich and W. Cai, J. Org. Chem., 1999, 64, 4926–4930 Search PubMed.
- D. Crich and M. Smith, Org. Lett., 2000, 2, 4067–4069 Search PubMed.
- D. Crich and M. Smith, J. Am. Chem. Soc., 2001, 123, 9015–9020 Search PubMed.
- T. G. Frihed, M. Bols and C. M. Pedersen, Chem. Rev., 2015, 115, 4963–5013 Search PubMed.
- D. Crich and S. Sun, J. Am. Chem. Soc., 1998, 120, 435–436 Search PubMed.
- D. Crich and V. Dudkin, J. Am. Chem. Soc., 2001, 123, 6819–6825 Search PubMed.
- D. Crich and M. Smith, J. Am. Chem. Soc., 2002, 124, 8867–8869 Search PubMed.
- K. Ohara, C.-C. Lin, P.-J. Yang, W.-T. Hung, W.-B. Yang, T.-J. R. Cheng, J.-M. Fang and C.-H. Wong, J. Org. Chem., 2013, 78, 6390–6411 Search PubMed.
- D. Crich and A. U. Vinod, Org. Lett., 2003, 5, 1297–1300 Search PubMed.
- D. Crich, W. Li and H. Li, J. Am. Chem. Soc., 2004, 126, 15081–15086 Search PubMed.
- C. Mukherjee, K. Ranta, J. Savolainen and R. Leino, Eur. J. Org. Chem., 2012, 2957–2968 Search PubMed.
- M. Poláková, M. U. Roslund, F. S. Ekholm, T. Saloranta and R. Leino, Eur. J. Org. Chem., 2009, 870–888 Search PubMed.
- D. Crich and S. Sun, J. Am. Chem. Soc., 1997, 119, 11217–11223 Search PubMed.
- M. Huang, G. E. Garrett, N. Birlirakis, L. Bohé, D. A. Pratt and D. Crich, Nat. Chem., 2012, 4, 663–667 Search PubMed.
- D. Crich and N. S. Chandrasekera, Angew. Chem., Int. Ed., 2004, 43, 5386–5389 Search PubMed.
- M. Huang, P. Retailleau, L. Bohé and D. Crich, J. Am. Chem. Soc., 2012, 134, 14746–14749 Search PubMed.
- P. O. Adero, T. Furukawa, M. Huang, D. Mukherjee, P. Retailleau, L. Bohé and D. Crich, J. Am. Chem. Soc., 2015, 137, 10336–10345 Search PubMed.
- M. Huang, T. Furukawa, P. Retailleau, D. Crich and L. Bohé, Carbohydr. Res., 2016, 427, 21–28 Search PubMed.
- L. Bohé and D. Crich, Carbohydr. Res., 2015, 403, 48–59 Search PubMed.
- M. Moumé-Pymbock and D. Crich, J. Org. Chem., 2012, 77, 8905–8912 Search PubMed.
- M. Heuckendorff, P. S. Bols, C. B. Barry, T. G. Frihed, C. M. Pedersen and M. Bols, Chem. Commun., 2015, 51, 13283–13285 Search PubMed.
- M. Heuckendorff, J. Bendix, C. M. Pedersen and M. Bols, Org. Lett., 2014, 16, 1116–1119 Search PubMed.
- D. Crich, M. de la Mora and A. U. Vinod, J. Org. Chem., 2003, 68, 8142–8148 Search PubMed.
- W. F. J. Hogendorf, L. J. V. den Bos, H. S. Overkleeft, J. D. C. Codée and G. A. V. der Marel, Bioorg. Med. Chem., 2010, 18, 3668–3678 Search PubMed.
- E. C. Lourenço, C. D. Maycock and M. Rita Ventura, Carbohydr. Res., 2009, 344, 2073–2078 Search PubMed.
- D. Crich and I. Sharma, J. Org. Chem., 2010, 75, 8383–8391 CrossRef CAS PubMed.
- E. Bousquet, M. Khitri, L. Lay, F. Nicotra, L. Panza and G. Russo, Carbohydr. Res., 1998, 311, 171–181 Search PubMed.
- M. T. C. Walvoort, G. Lodder, J. Mazurek, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, J. Am. Chem. Soc., 2009, 131, 12080–12081 Search PubMed.
- J. Rönnols, M. T. C. Walvoort, G. A. van der Marel, J. D. C. Codée and G. Widmalm, Org. Biomol. Chem., 2013, 11, 8127–8134 Search PubMed.
- M. T. C. Walvoort, W. de Witte, J. van Dijk, J. Dinkelaar, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, Org. Lett., 2011, 13, 4360–4363 Search PubMed.
- J. D. C. Codée, L. J. van den Bos, A.-R. de Jong, J. Dinkelaar, G. Lodder, H. S. Overkleeft and G. A. van der Marel, J. Org. Chem., 2009, 74, 38–47 Search PubMed.
- L. J. van den Bos, J. Dinkelaar, H. S. Overkleeft and G. A. van der Marel, J. Am. Chem. Soc., 2006, 128, 13066–13067 Search PubMed.
- L. J. van den Bos, J. D. C. Codée, R. E. J. N. Litjens, J. Dinkelaar, H. S. Overkleeft and G. A. van der Marel, Eur. J. Org. Chem., 2007, 3963–3976 Search PubMed.
- J. D. C. Codée, M. T. C. Walvoort, A.-R. de Jong, G. Lodder, H. S. Overkleeft and G. A. van der Marel, J. Carbohydr. Chem., 2011, 30, 438–457 Search PubMed.
- J. Dinkelaar, A. R. de Jong, R. van Meer, M. Somers, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, J. Org. Chem., 2009, 74, 4982–4991 Search PubMed.
- S.-L. Tang and N. L. B. Pohl, Org. Lett., 2015, 17, 2642–2645 Search PubMed.
- M. T. C. Walvoort, H. van den Elst, O. J. Plante, L. Kröck, P. H. Seeberger, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Angew. Chem., Int. Ed., 2012, 51, 4393–4396 Search PubMed.
- M. T. C. Walvoort, J. Dinkelaar, L. J. van den Bos, G. Lodder, H. S. Overkleeft, J. D. C. Codée and G. A. van der Marel, Carbohydr. Res., 2010, 345, 1252–1263 Search PubMed.
- Y. J. Lee, J. Y. Baek, B.-Y. Lee, S. S. Kang, H.-S. Park, H. B. Jeon and K. S. Kim, Carbohydr. Res., 2006, 341, 1708–1716 Search PubMed.
- T. G. Frihed, M. T. C. Walvoort, J. D. C. Codée, G. A. van der Marel, M. Bols and C. M. Pedersen, J. Org. Chem., 2013, 78, 2191–2205 Search PubMed.
- S. Minegishi, S. Kobayashi and H. Mayr, J. Am. Chem. Soc., 2004, 126, 5174–5181 Search PubMed.
- J. Ammer, C. Nolte and H. Mayr, J. Am. Chem. Soc., 2012, 134, 13902–13911 Search PubMed.
- J. Ammer and H. Mayr, J. Phys. Org. Chem., 2013, 26, 59–63 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 Search PubMed.
- K. Worm-Leonhard, K. Larsen and K. J. Jensen, J. Carbohydr. Chem., 2007, 26, 349–368 Search PubMed.
- Acid catalyzed anomerization reactions do not play a major role in the studied glycosylation reactions as the released acid is scavenged by tri-tert-butyl methyl pyrimidine (TTBP) and executed at low temperature. See ref. 42.
- D. M. Whitfield, Carbohydr. Res., 2007, 342, 1726–1740 Search PubMed.
- T. Hosoya, P. Kosma and T. Rosenau, Carbohydr. Res., 2015, 411, 64–69 Search PubMed.
- H. Wang and K. N. Houk, Chem. Sci., 2014, 5, 462–470 Search PubMed.
- D. Crich, Acc. Chem. Res., 2010, 43, 1144–1153 Search PubMed.
- J. I. Seeman, Chem. Rev., 1983, 83, 83–134 Search PubMed.
- D. Crich and I. Sharma, Org. Lett., 2008, 10, 4731–4734 Search PubMed.
- Calculated energies (kcal mol−1) of the methyl 2,3,4-tri-O-methyl mannuronate oxocarbenium ion (B3LYP/6-311G, with a PCM model to correct for solvation in dichloromethane): 3H4 = 0.0, 4H3 = +5.0, B2,5 = +6.1.
- J. A. C. Romero, S. A. Tabacco and K. A. Woerpel, J. Am. Chem. Soc., 2000, 122, 168–169 Search PubMed.
- L. Ayala, C. G. Lucero, J. A. C. Romero, S. A. Tabacco and K. A. Woerpel, J. Am. Chem. Soc., 2003, 125, 15521–15528 Search PubMed.
- C. G. Lucero and K. A. Woerpel, J. Org. Chem., 2006, 71, 2641–2647 CrossRef CAS PubMed.
- E. I. Balmond, D. M. Coe, M. C. Galan and E. M. McGarrigle, Angew. Chem., Int. Ed., 2012, 51, 9152–9155 Search PubMed.
- A. B. Ingle, C.-S. Chao, W.-C. Hung and K.-K. T. Mong, Org. Lett., 2013, 15, 5290–5293 Search PubMed.
- M. Giordano and A. Iadonisi, J. Org. Chem., 2014, 79, 213–222 Search PubMed.
- The concentration dependence of SN2-type reactions indicates that this parameter should be carefully considered in optimizing these glycosylations. See for example ref. 36 and T. Fang, Y. Gu, W. Huang and G.-J. Boons, J. Am. Chem. Soc., 2016, 138, 3002–3011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04638j |
| This journal is © The Royal Society of Chemistry 2017 |