
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO/O2 assisted oxidative carbon–carbon and carbon–heteroatom bond cleavage for the synthesis of oxosulfonates from DMSO and olefins†
Ailong
Shao‡
ab,
Meng
Gao‡
*a,
Songtao
Chen
a,
Tao
Wang
*a and
Aiwen
Lei
 *ab
*ab
aNational Research Center for Carbohydrate Synthesis, Key Laboratory of Chemical Biology, Jiangxi Normal University, Nanchang 330022, Jiangxi, PR China. E-mail: drmenggao@163.com; wangtao@jxnu.edu.cn
bCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, P. R. China. E-mail: aiwenlei@whu.edu.cn
First published on 7th December 2016
Abstract
Selective carbon–carbon and carbon–heteroatom bond cleavage was achieved in a one reaction system. With this strategy a novel Pd/Cu-catalyzed aerobic oxidative oxosulfonation of olefins with DMSO has been developed. Preliminary mechanistic investigations indicated that CO/O2 assisted the bond cleavage and the leaving groups from the starting materials were trapped by O2 and underwent a hydroxylation process.
In the past few years, transition-metal-catalyzed carbon–carbon and carbon–heteroatom bond activation (cleavage) has attracted much attention, due not only to its fundamental scientific interest but also its potential usage in organic synthesis.1 Methods for these strategies mainly involve strain-release, aromatization, and chelation-assistance, and the research topics mainly focus on stoichiometric reactions such as transition-metal insertion into carbon–carbon bonds via stable metallacycle formation.1e,2 Although significant progress has been made in this emerging field, the reactivity, selectivity, and efficiencies in these strategies are still far from satisfactory due to the thermodynamic stability of the unstrained C–C and C–heteroatom bonds. Furthermore, few examples are compatible with a variety of bond cleavages in one transformation.3 Hence, the development of an efficient catalytic system towards unstrained C–C and C–heteroatom bond cleavage is always highly attractive. Due to continuous research interests into C–C and C–heteroatom bond cleavage, we discovered a transformation in which C–C, C–S, C–O, C–Br etc. bond cleavage was efficiently achieved in a one reaction system. The leaving groups could be phenyl, methyl, cyclohexyl, carboxyl, phosphate or bromine (Scheme 1).
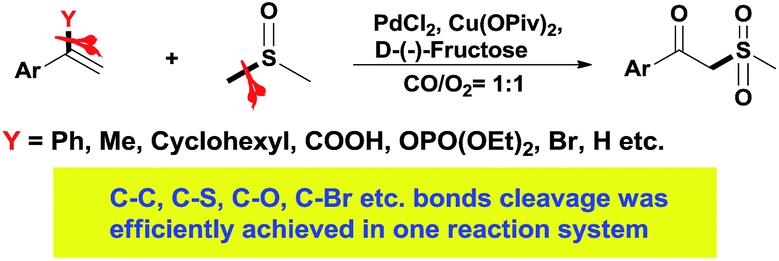 | ||
| Scheme 1 The synthesis of oxosulfonates via C–C/C–S bond cleavage. | ||
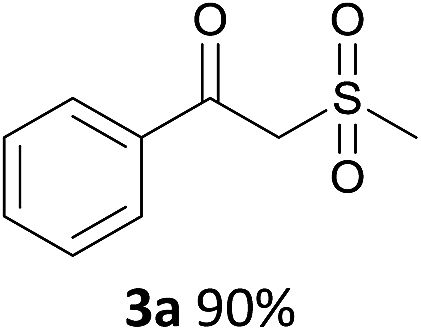
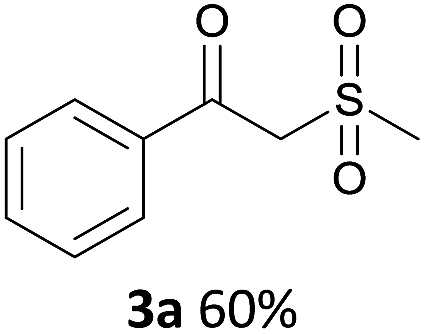
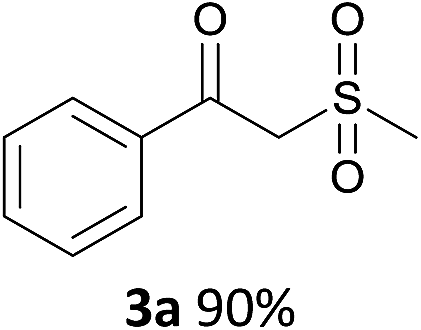
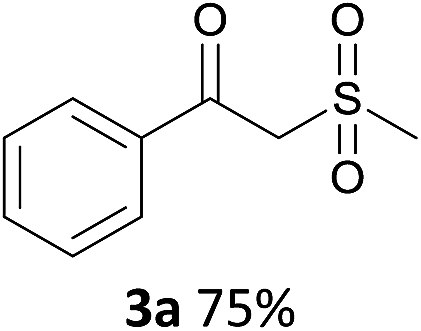
With the combination of ethene-1,1-diyldibenzene, PdCl2, Cu(OAc)2 and ethylene glycol in the presence of 1 atm CO/O2 at 80 °C, the unexpected β-oxo sulfone compound 3a and phenol were isolated and identified (Scheme 2 and Table 1), which indicated that C–S/C–C bond cleavage and an O2 fixation process were involved. To the best of our knowledge, the selective cleavage of unactivated C–S/C–C bonds is still a challenging topic to date.4 Furthermore, β-oxo sulfone derivatives are important chemical feedstocks which are commonly used precursors in the synthesis of a series of useful biologically active molecules and basic scaffolds of numerous pharmaceutically important molecules.5 No examples have been described in the literature of β-oxo methyl sulfones being prepared using a Pd/Cu catalyst from the direct cleavage of C–C and C–S bonds. Therefore, we decided to further investigate this transformation by utilizing commercially available dimethyl sulfoxide to realize the synthesis of β-oxo methyl sulfone derivatives.
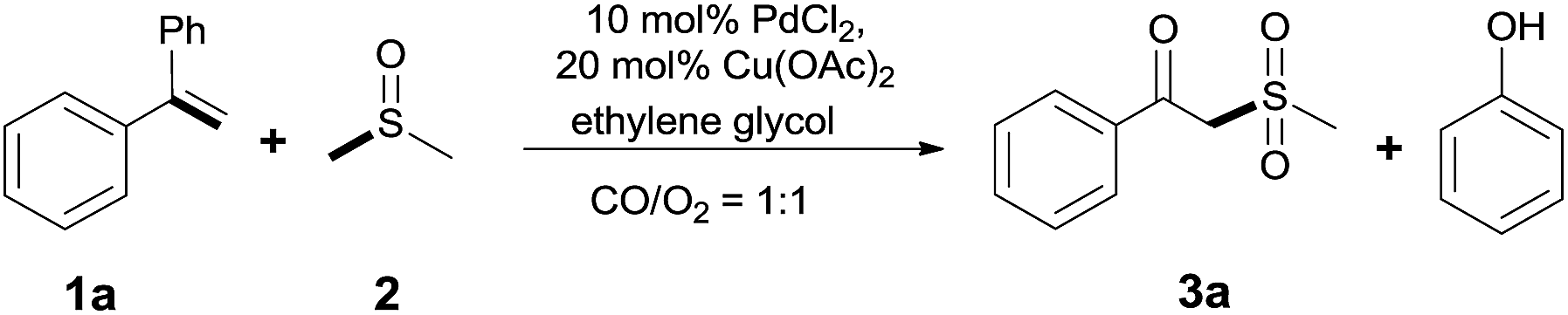 | ||
| Scheme 2 The reaction between ethene-1,1-diyldibenzene and dimethyl sulfoxide. | ||
|
|
||||
|---|---|---|---|---|
| Entry | [Pd] | [Cu] | Additive | Yieldb [%] |
a Reactions were carried out on a scale of 0.20 mmol of 1a and 1 mL of 2a in the presence of 10 mol% [Pd], 20 mol% [Cu] and 40 mol% additive in 1 atm CO/O2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 15 h.
b Isolated yields.
c Without CO. :1) for 15 h.
b Isolated yields.
c Without CO.
|
||||
| 1 | PdCl2 | Cu(OAc)2− | — | N.R |
| 2 | PdCl2 | Cu(OAc)2 | Glycol | 30 |
| 3 | PdCl2 | Cu(OAc)2 | Glycerol | 44 |
| 4 | PdCl2 | Cu(OAc)2 | 1,10-Phen | Trace |
| 5 | PdCl2 | Cu(OAc)2 | PPh3 | N.R |
| 6 | PdCl2 | Cu(OAc)2 | L-Proline | Trace |
| 7 | PdCl2 | Cu(OAc)2 | Sucrose | 41 |
| 8 | PdCl2 | Cu(OAc)2 | D-(−)-Glucose | 77 |
| 9 | PdCl2 | Cu(OAc)2 | D-(−)-Fructose | 80 |
| 10 | PdCl2 | — | D-(−)-Fructose | Trace |
| 11 | PdCl2 | CuCl | D-(−)-Fructose | 15 |
| 12 | PdCl2 | CuOAc | D-(−)-Fructose | 35 |
| 13 | PdCl 2 | Cu(OPiv) 2 | D -(−)-Fructose | 90 |
| 14 | — | Cu(OPiv)2 | D-(−)-Fructose | N.R |
| 15 | Pd(OAc)2 | Cu(OPiv)2 | D-(−)-Fructose | 70 |
| 16 | Pd/C | Cu(OPiv)2 | D-(−)-Fructose | 40 |
| 17c | PdCl2 | Cu(OPiv)2 | D-(−)-Fructose | N.R |
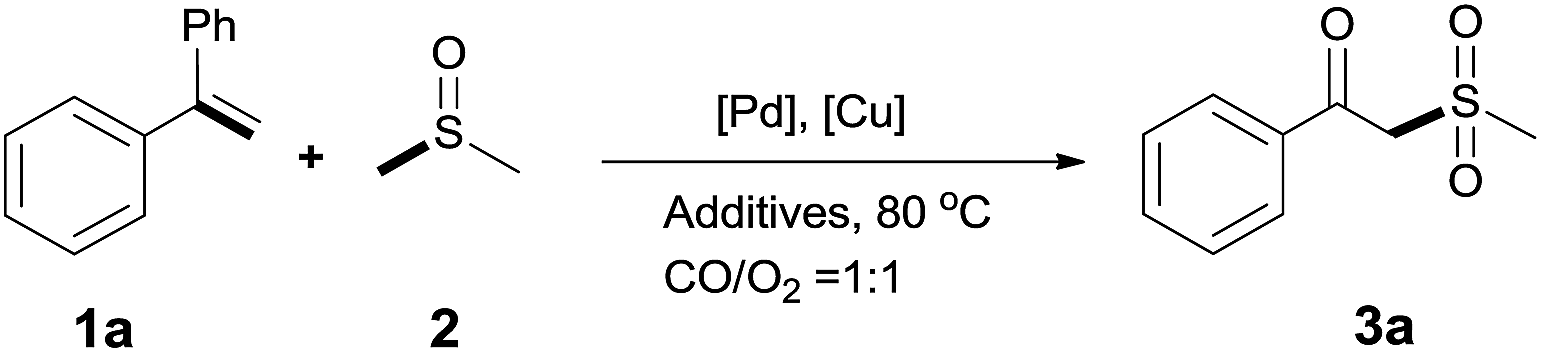
Our efforts started with treating ethene-1,1-diyldibenzene with DMSO in the presence of catalytic Pd/Cu salts and polyhydroxy additives. Notably, the use of polyhydroxy compounds, such as ethylene glycol and glycerol, is essential for the reaction and CO is also indispensable (Table 1, entry 1–3 and 17). Removing or replacing the polyhydroxy compounds with other ligands, such as 1,10-phen, PPh3, or L-proline, both turned out to be ineffective (Table 1, entry 4–6). It is well known that carbohydrates are commonly available and exist widely in nature so could also be used in organic synthesis because of the polyhydroxy functional groups.6 Hence, we next screened a series of carbohydrates as the additives. To our delight, D-(−)-fructose could afford the product in 80% yield and D-(−)-glucose could also afford the product in 77% yield (Table 1, entry 8 and 9). When sucrose was used, a lower yield was obtained (Table 1, entry 7). Pd and Cu salts were also essential in this transformation. After screening a series of conditions, we found that Cu(OPiv)2 and PdCl2 were the best catalysts in this reaction (Table 1, entries 10–16). After considerable efforts, the combination of PdCl2 (0.1 eq.), Cu(OPiv)2 (0.2 eq.) and D-(−)-fructose (0.4 eq.) in the presence of 1 atm CO/O2 (1:1) at 80 °C was found to be the best reaction conditions for this aerobic oxidative cross-coupling (Table 1, entry 13).
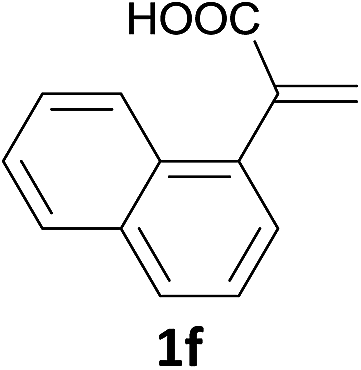
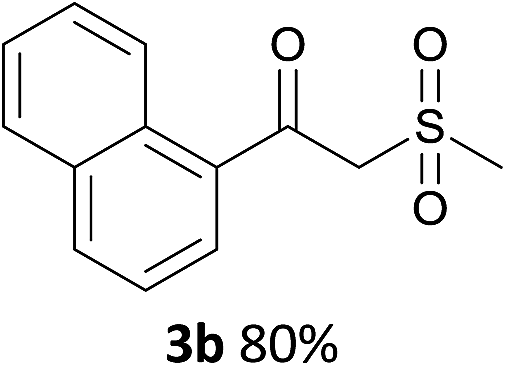
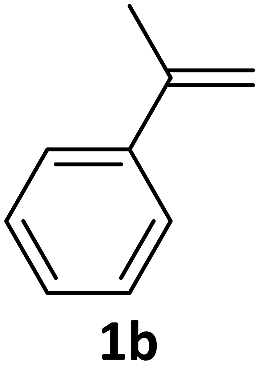
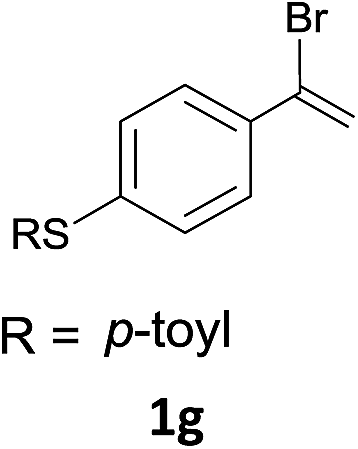
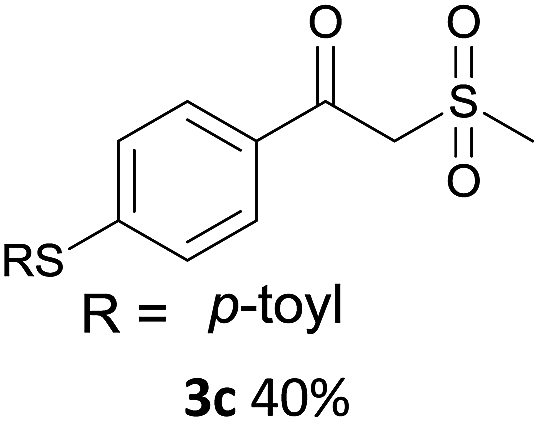
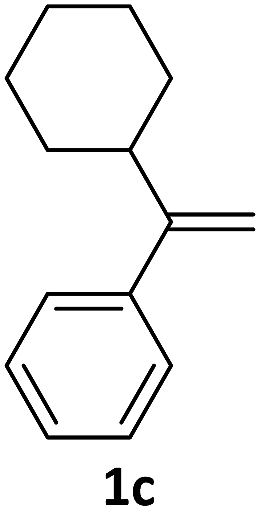
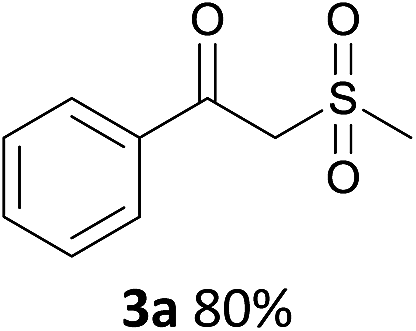
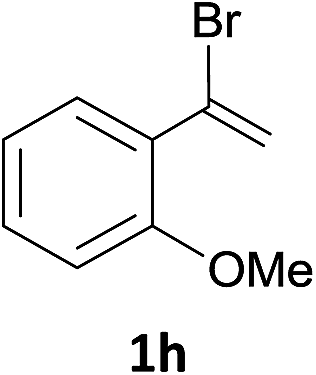
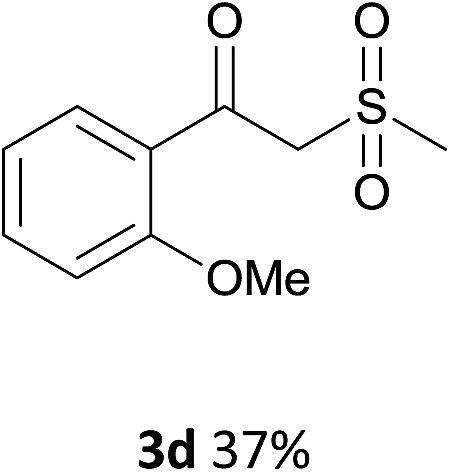
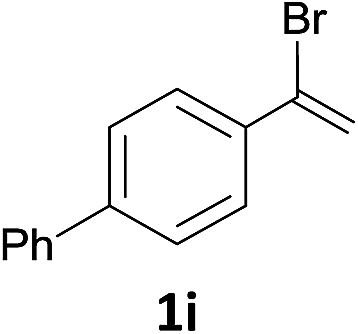
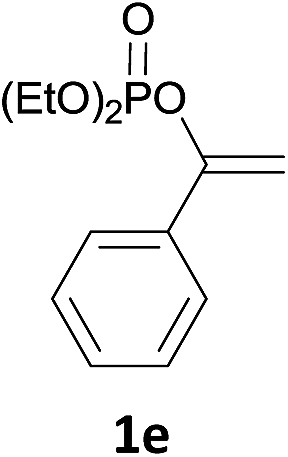
With the optimized conditions in hand, various α-substituted phenylethylenes, 1, were tested to react with dimethyl sulfoxide (Table 2). To our delight, the reaction was readily extended to a variety of α-substituted phenylethylenes, 1, and methyl, phenyl, cyclohexyl or carboxyl substituted phenylethylene could provide 2-(methylsulfonyl)-1-phenylethanone 3a in good yields, in which C–C and C–S bond cleavage was involved (Table 2, 1a, 1b, 1c, 1d and 1f). In addition, diethyl(1-phenylvinyl)phosphate 1e was also found to be a suitable reaction partner with dimethyl sulfoxide in the reaction, in which C–O and C–S bond cleavage was involved. It was noteworthy that various α-bromostyrene derivatives, such as (4-(1-bromovinyl)phenyl)(p-tolyl)sulfane 1g, 4-(1-bromovinyl)-1,1′-biphenyl 1i and 1-(1-bromovinyl)-2-methoxybenzene 1h, could also be added to the β-oxo sulfone without any difficulties, in which C–Br and C–S bond cleavage was involved.
|
|
|||
|---|---|---|---|
| Substrate | Product | Substrate | Product |
|
a Reaction conditions: 1 (0.25 mmol), 2 (1 mL), PdCl2 (10 mol%), Cu(OPiv)2 (20 mol%), D-(−)-fructose (40 mol%) and 1 atm CO/O2 (1:1) at 80 °C for 15 h. Isolated yields.
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
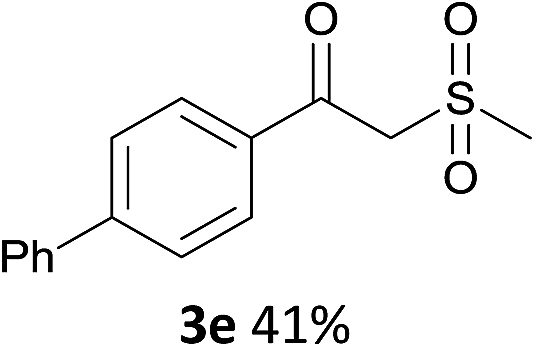
Since the products are β-oxo sulfones, an indisputable advancement is the application of simple olefins in this reaction. To our delight, the reaction was readily extended to a variety of simple aryl alkenes, either electron-withdrawing or electron-donating substituted groups or halogen groups at the aromatic ring could be introduced into the desired product under the standard reaction conditions (Scheme 3, 3f, 3g–3r). Styrenes bearing alkyl substituents, such as methyl and tert-butyl groups, afforded the desired products in moderate to good yields (Scheme 3, 3f, 3g–3i). It was noteworthy that the position of the substituent seems to have an influence on the product yield (Scheme 3, 3f, 3h and 3i). Several typical functional groups such as sulfhydryl, trifluoromethyl and cyano groups were well tolerated (Scheme 3, 3o, 3p, 3r). Meanwhile, 2-vinylnaphthalene 4q could also be transformed into the target product in 60% yield (Scheme 3, 3q).
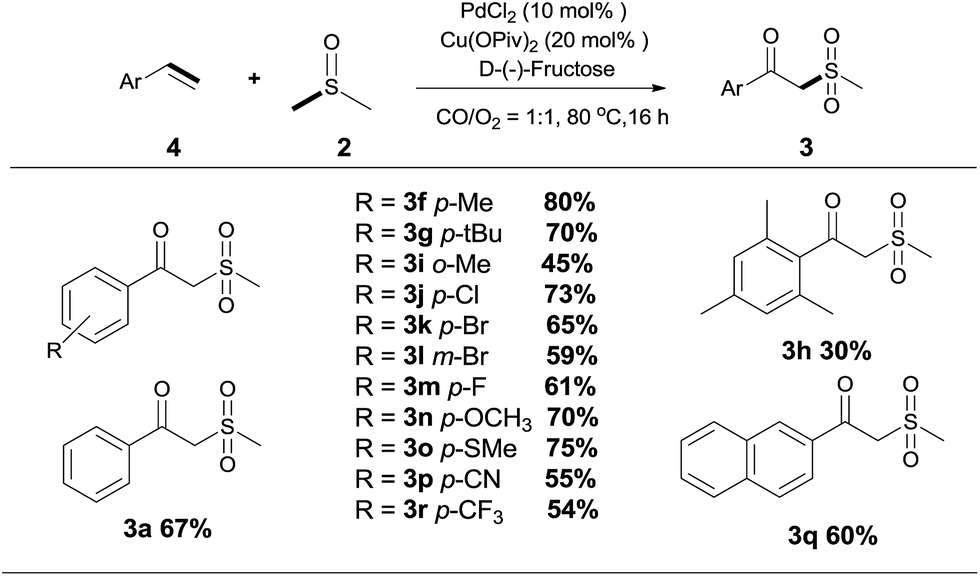 | ||
| Scheme 3 Reactions of alkenes 4 and dimethyl sulfoxide 2. Reaction conditions: 4 (0.20 mmol), 2 (1 mL), PdCl2 (10 mol%), Cu(OPiv)2 (20 mol%), D-(−)-fructose (40 mol%) and 1 atm CO/O2 (1:1) at 80 °C for 15 h. Isolated yields. | ||
To gain preliminary mechanistic information about this transformation, the styrene was reacted with deuterated DMSO to generate the desired deuterium labeled product in 88% yield under the standard reaction conditions (Scheme 4a). From the 1H NMR spectrum, the resonance at δ = 2.0–4.0 ppm was not observed. However, the resonance at δ = 4.61 ppm still existed, which demonstrated that the methylene of the product was from the styrene. The leaving Ph group was transformed into phenol (GC yield 82%). Meanwhile, (1-cyclohexylvinyl)benzene was also tested in the reaction, and the leaving cyclohexyl group was also transformed into cyclohexanol (Scheme 4b). Furthermore, the by-product of diethyl (1-phenylvinyl)phosphate 1e was also identified by LC-MS (see more details in the ESI†). Isotopic labeling experiments with 18O2 were also performed (Scheme 4c), and the results demonstrated that the two additional oxygen atoms present in the product and the oxygen present in phenol all came from 18O2.
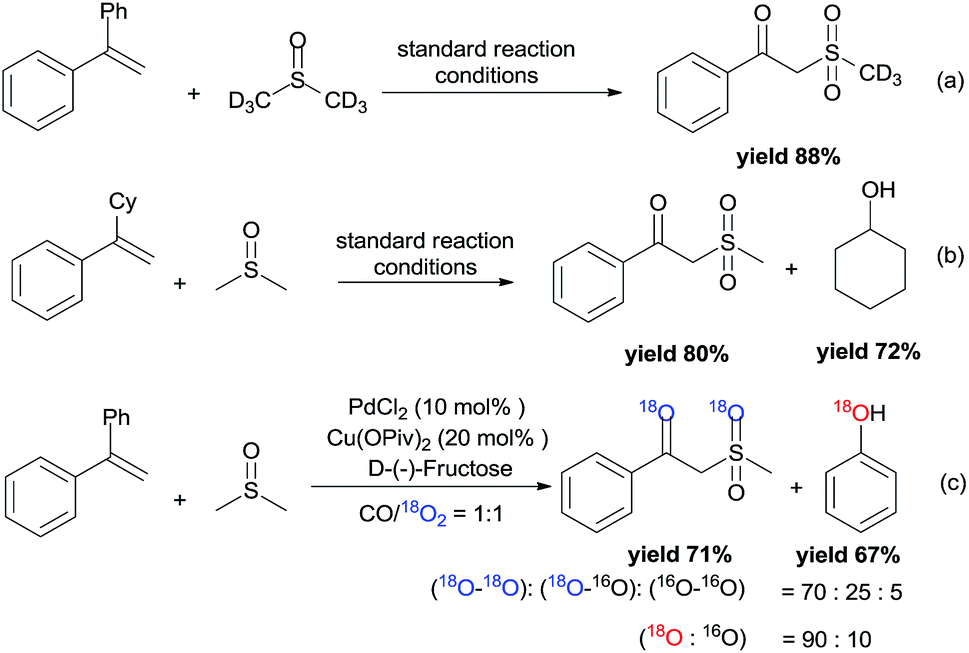 | ||
| Scheme 4 Labelling and trapping experiments. | ||
Radical-trapping experiments were carried out under the standard conditions. The addition of the radical scavengers TEMPO and BHT only reduced the reaction yields slightly, which suggests that radical intermediates might not be the active species in this oxidative transformation (Scheme 5).
 | ||
| Scheme 5 Radical inhibiting experiment. | ||
By employing dimethyl sulfone as the substrate instead of DMSO to react with 1a under the standard conditions, no corresponding β-oxo sulfone product was produced (Scheme 6a). This indicates that the lone pair of electrons on DMSO is essential to the C–S bond cleavage. Furthermore, 2-(methylsulfonyl)-1,1-diphenylethanol did not lead to the desired product under our reaction conditions (Scheme 6b), which suggests that the formation of the carbonyl and the C–C bond cleavage might go through a synergistic process.
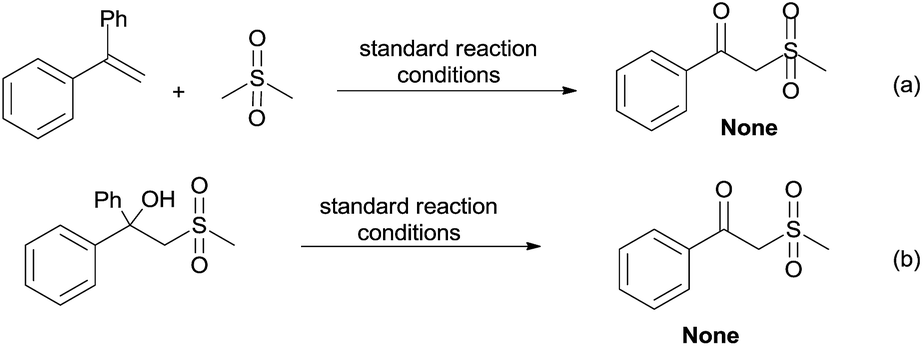 | ||
| Scheme 6 Control experiment. | ||
On the basis of the above results and previous studies,7 we proposed a mechanism for this oxidative transformation (Scheme 7): (1) the deprotonation of DMSO by Pd(II) affords the intermediate 5 (DFT calculations were also performed, see more details in the ESI†); (2) the insertion of CO leads to the intermediate 6; (3) the β-heteroatom (SOCH3) elimination via7 produces intermediate 8 and the ketene; (4) the insertion of 1a into 8 gives the alkylpalladium intermediate 9; (5) although the detailed mechanism is not clear yet, via intermediate 10, the oxidative C–C or C–X bond cleavage and the C–O bond formation takes place in the presence of copper, oxygen and the carbohydrate.
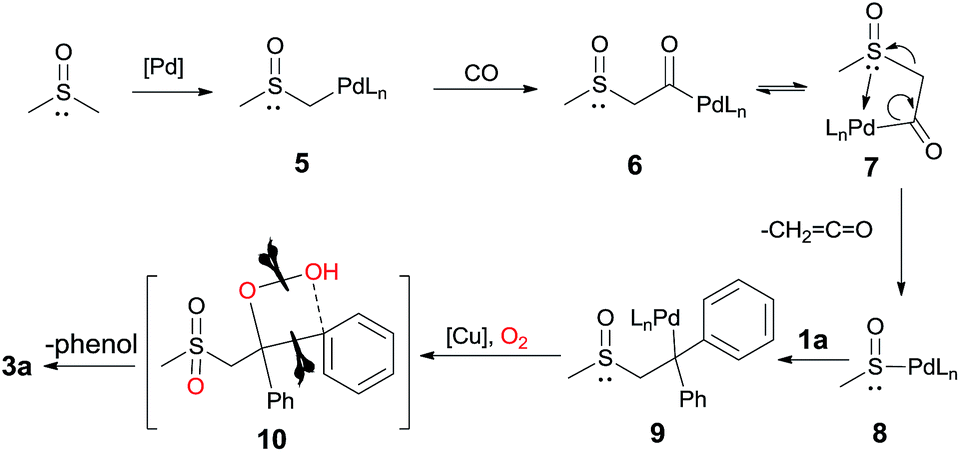 | ||
| Scheme 7 Proposed mechanism for C–S and C–C bond cleavage. | ||
As shown in the mechanism, the ketene was from the C–S bond cleavage and the insertion of CO. The ketene easily reacted with H2O to form acetic acid. Hence, we performed an acetic acid trapping experiment (Scheme 8). 2-(Bromomethyl)naphthalene was added into the solution after the reaction was completed, and deuterium labeled naphthalen-2-ylmethyl acetate was produced, which suggests the formation of ketene.
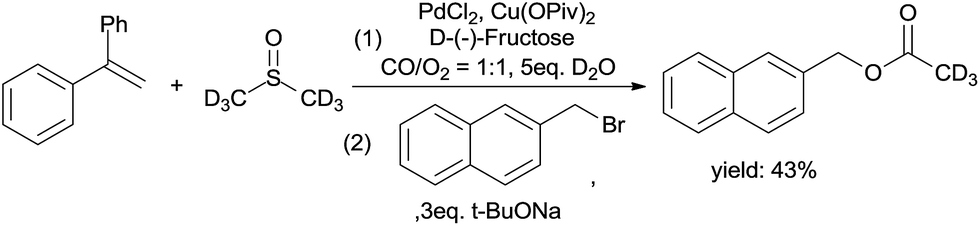 | ||
| Scheme 8 Acetic acid trapping experiment. | ||
In summary, we have demonstrated the efficient and attractive aerobic oxidative oxosulfonation of olefins with DMSO, in which C–C, C–S, C–O, C–Br etc. bond cleavage is involved. A diverse collection of valuable β-oxo sulfones was easily synthesized by this protocol. Preliminary mechanistic investigation was also performed, indicating that CO and O2 assisted this oxidative carbon–carbon and carbon–heteroatom bond cleavage and the leaving groups from the starting materials were trapped by O2 and underwent a hydroxylation process.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21562026 and 21262018) and JiangXi Provincial Natural Science Foundation of China (20161BAB203085).Notes and references
- (a) R. H. Crabtree, Chem. Rev., 1985, 85, 245–269 CrossRef CAS; (b) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed; (c) P. Steenwinkel, R. A. Gossage and G. van Koten, Chem.–Eur. J., 1998, 4, 759–762 CrossRef CAS; (d) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870–883 CrossRef; (e) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC; (f) T. Jia, A. Bellomo, K. E. L. Baina, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 3740–3743 CrossRef CAS PubMed.
- (a) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed; (b) H. Liu, M. Feng and X. Jiang, Chem.–Asian J., 2014, 9, 3360–3389 CrossRef CAS PubMed; (c) C. He, S. Guo, L. Huang and A. Lei, J. Am. Chem. Soc., 2010, 132, 8273–8275 CrossRef CAS PubMed; (d) Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222–234 CrossRef CAS PubMed; (e) A. de Meijere, Chem. Rev., 2003, 103, 931–932 CrossRef CAS; (f) O. G. Kulinkovich, Chem. Rev., 2003, 103, 2597–2632 CrossRef CAS PubMed; (g) K. Chen, H. Li, Z.-Q. Lei, Y. Li, W.-H. Ye, L.-S. Zhang, J. Sun and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 9851–9855 CrossRef CAS PubMed; (h) S. K. Hanson, R. Wu and L. A. P. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS PubMed; (i) A. Dermenci, J. W. Coe and G. Dong, Org. Chem. Front., 2014, 1, 567–581 RSC; (j) T. Wang and N. Jiao, J. Am. Chem. Soc., 2013, 135, 11692–11695 CrossRef CAS PubMed.
- (a) S. Hanessian, G. Yang-Chung, P. Lavallee and A. G. Pernet, J. Am. Chem. Soc., 1972, 94, 8929–8931 CrossRef CAS; (b) Y. Xia, Z. Liu, Z. Liu, R. Ge, F. Ye, M. Hossain, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2014, 136, 3013–3015 CrossRef CAS PubMed; (c) L. Zhang, X. Bi, X. Guan, X. Li, Q. Liu, B.-D. Barry and P. Liao, Angew. Chem., Int. Ed., 2013, 52, 11303–11307 CrossRef CAS PubMed; (d) C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257–15262 CrossRef CAS PubMed.
- (a) F. Luo, C. Pan, L. Li, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 5304–5306 RSC; (b) Y. Lv, Y. Li, T. Xiong, W. Pu, H. Zhang, K. Sun, Q. Liu and Q. Zhang, Chem. Commun., 2013, 49, 6439–6441 RSC; (c) G. Yuan, J. Zheng, X. Gao, X. Li, L. Huang, H. Chen and H. Jiang, Chem. Commun., 2012, 48, 7513–7515 RSC; (d) L. Chu, X. Yue and F.-L. Qing, Org. Lett., 2010, 12, 1644–1647 CrossRef CAS PubMed; (e) Y. Jiang and T.-P. Loh, Chem. Sci., 2014, 5, 4939–4943 RSC; (f) X. Shi, X. Ren, Z. Ren, J. Li, Y. Wang, S. Yang, J. Gu, Q. Gao and G. Huang, Eur. J. Org. Chem., 2014, 2014, 5083–5088 CrossRef CAS; (g) J. Wen, H. Cheng, S. Dong and C. Bolm, Chem.–Eur. J., 2016, 22, 5547–5550 CrossRef CAS PubMed; (h) X. Shen, Q. Liu, W. Zhang and J. Hu, Eur. J. Org. Chem., 2016, 2016, 906–909 CrossRef CAS; (i) K.-J. Wei, Z.-j. Quan, Z. Zhang, Y.-x. Da and X.-c. Wang, RSC Adv., 2016, 6, 78059–78063 RSC; (j) Y. Uetake, T. Niwa and T. Hosoya, Org. Lett., 2016, 18, 2758–2761 CrossRef CAS PubMed; (k) L. Wang, W. He and Z. Yu, Chem. Soc. Rev., 2013, 42, 599–621 RSC; (l) F. Pan, H. Wang, P.-X. Shen, J. Zhao and Z.-J. Shi, Chem. Sci., 2013, 4, 1573–1577 RSC; (m) S. G. Modha, V. P. Mehta and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 5042–5055 RSC; (n) X. Jiang, C. Wang, Y. Wei, D. Xue, Z. Liu and J. Xiao, Chem.–Eur. J., 2014, 20, 58–63 CrossRef CAS PubMed; (o) E. Baciocchi, E. Fasella, O. Lanzalunga and M. Mattioli, Angew. Chem., Int. Ed. Engl., 1993, 32, 1071–1073 CrossRef; (p) H. Huang, J. Li, C. Lescop and Z. Duan, Org. Lett., 2011, 13, 5252–5255 CrossRef CAS PubMed.
- (a) O. Pestovsky, S. Stoian, E. L. Bominaar, X. Shan, E. Münck, L. Que and A. Bakac, Angew. Chem., Int. Ed., 2005, 44, 6871–6874 CrossRef CAS PubMed; (b) Q. Lu, J. Zhang, F. Wei, Y. Qi, H. Wang, Z. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156–7159 CrossRef CAS PubMed; (c) O. Lanzalunga, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 322–330 CrossRef CAS.
- (a) D. Cheng, F. Gan, W. Qian and W. Bao, Green Chem., 2008, 10, 171–173 RSC; (b) A. Gual, C. Godard, C. Claver and S. Castillon, Eur. J. Org. Chem., 2009, 1191–1201 CrossRef CAS; (c) M. M. K. Boysen, Chem.–Eur. J., 2007, 13, 8648–8659 CrossRef CAS PubMed.
- (a) L. L. Shiu, C. C. Yu, K. T. Wong, B. L. Chen, W. L. Cheng, T. M. Yuan and T. Y. Luh, Organometallics, 1993, 12, 1018–1020 CrossRef CAS; (b) H. Zhao, A. Ariafard and Z. Lin, Organometallics, 2006, 25, 812–819 CrossRef CAS; (c) G. Zhu and X. Lu, Organometallics, 1995, 14, 4899–4904 CrossRef CAS; (d) S. R. Samanta, R. Choudhury and V. Ramamurthy, J. Phys. Chem. A, 2014, 118, 10554–10562 CrossRef CAS PubMed; (e) E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed; (f) S. Itoh, Acc. Chem. Res., 2015, 48, 2066–2074 CrossRef CAS PubMed; (g) S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 2004, 304, 864–867 CrossRef CAS PubMed; (h) S. A. M. Dowling-Mitchell, W. Henderson, E. R. T. Tiekink, J. Petrakopoulou and M. P. Sigalas, Organometallics, 2012, 31, 4662–4669 CrossRef CAS; (i) R. Usón, J. Forniés, M. Tomás, B. Menjón and A. J. Welch, J. Organomet. Chem., 1986, 304, C24–C26 CrossRef; (j) W. Henderson, R. D. W. Kemmitt, J. Fawcett, L. J. S. Prouse and D. R. Russell, J. Chem. Soc., Chem. Commun., 1986, 1791–1793 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04480h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |