
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mapping a protein recognition centre with chiral photoactive ligands. An integrated approach combining photophysics, reactivity, proteomics and molecular dynamics simulation studies†
Daniel
Limones-Herrero
 a,
Raúl
Pérez-Ruiz
a,
Emilio
Lence
b,
Concepción
González-Bello
b,
Miguel A.
Miranda
*a and
M. Consuelo
Jiménez
*a
a,
Raúl
Pérez-Ruiz
a,
Emilio
Lence
b,
Concepción
González-Bello
b,
Miguel A.
Miranda
*a and
M. Consuelo
Jiménez
*a
aDepartamento de Química/Instituto de Tecnología Química UPV-CSIC, Universitat Politècnica de València, Camino de Vera s/n, 46022, Valencia, Spain. E-mail: mmiranda@qim.upv.es; mcjimene@qim.upv.es
bCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Departamento de Química Orgánica, Universidade de Santiago de Compostela, C/ Jenaro de la Fuente s/n, 15782 Santiago de Compostela, Spain
First published on 5th January 2017
Abstract
A multidisciplinary strategy to obtain structural information on the intraprotein region is described here. As probe ligands, (S)- and (R)-CPFMe (the methyl esters of the chiral drug carprofen) have been selected, while bovine α1-acid glycoprotein (BAAG) has been chosen as a biological host. The procedure involves the separate irradiation of the BAAG/(S)-CPFMe and BAAG/(R)-CPFMe complexes, coupled with fluorescence spectroscopy, laser flash photolysis, proteomic analysis, docking and molecular dynamics simulations. Thus, irradiation of the BAAG/CPFMe complexes at λ = 320 nm was followed by fluorescence spectroscopy. The intensity of the emission band obtained after irradiation indicated photodehalogenation, whereas its structureless shape suggested covalent binding of the resulting radical CBZMe˙ to the biopolymer. After gel filtration chromatography, the spectra still displayed emission, in agreement with covalent attachment of CBZMe˙ to BAAG. Stereodifferentiation was observed in this process. After trypsin digestion and ESI-MS/MS, the incorporation of CBZMe was detected at Phe68. Docking and molecular dynamics simulation studies, which were carried out using a homology model of BAAG, reveal that the closer proximity of the aromatic moiety of the (S)-enantiomer to the phenyl group of Phe68 would be responsible for the experimentally observed, more effective chemical modification of the protein. The proposed tridimensional structure of BAAG covalently modified by the two enantiomers is also provided. In principle, this approach can be extended to a variety of protein/ligand complexes.
Introduction
The intraprotein regions of proteins are not passive receptacles for substrate binding, but very complex microenvironments that employ a wide variety of physicochemical interactions (van der Waals forces, hydrogen bonding, etc.) for the complexation of substrates. The sites are formed by side chains of specific amino acid residues in a particular three-dimensional arrangement that play an important role in the accommodation of ligands. Besides the favourable structural requirements, stabilising ligand–protein interactions are necessary to form complexes. These interactions form the basis of molecular recognition, which is essential for most of the biological functions of these biomacromolecules, such as biodistribution (transport proteins), biotransformation (enzymes) or activation/deactivation (receptors).1Experimentally, precise structural information can be obtained from techniques such as cryo-EM or X-ray crystallography, when suitable crystals of the ligand–protein complex are obtained with the appropriate diffracting properties.2 However, the development of alternative and structurally informative methodologies is still highly desirable. In principle, a wide variety of ligands contain photoreactive chromophores whose excitation could give rise to very short-lived excited states or reactive intermediates and are capable of undergoing covalent photobinding to amino acid residues.3 As these species have negligible capability of diffusing away from the place where they are generated, photoreaction has to take place in close proximity to them. Thus, identification of the molecular recognition centre would be achieved by photoinduced covalent modification of key amino acid residues by the ligand. This is reminiscent of photoaffinity labelling, but without the need to carry out time consuming chemical syntheses of reactive derivatives, which in addition may present binding affinities different to those of the unaltered ligands.4
Among the ligands containing photoreactive chromophores, a number of xenobiotics (drugs, cosmetics, etc.) have been reported, which possess the ability to induce changes in the sensitivity of a subject with respect to UV-Vis radiation, whether of solar or artificial origin. Such a phenomenon, called photosensitivity, occurs when the photoexcited xenobiotic is able to interact with cellular components of an organism, especially in the skin.
Drug-induced photosensitivity includes photoallergic reactions, which are thought to involve covalent drug–protein photobinding, leading to the formation of a complete antigen that may trigger a hypersensitivity reaction due to a cell-mediated immune response.5 Hence, transport proteins have been employed as models to investigate the photobinding process. Most of the work that has been done uses serum albumins,6 but very little is known on other relevant transport proteins, such as α1-acid glycoproteins (AAGs). In general, the existing studies are limited to detecting the irreversible incorporation of the drug chromophore to the protein by means of gel filtration chromatography followed by absorption or emission spectroscopy, but they do not provide structural information.7
The goal of the present work is to obtain structural information on the recognition centre of AAGs by exploiting the photobinding of a ligand. Several activities of physiological significance have been described for AAGs, such as their ability to bind and transport small hydrophobic molecules and to act as immuno-modulating agents. The AAGs’ serum concentrations remain constant under normal physiological conditions but increase markedly during acute-phase reactions, in response to systemic tissue injury, inflammation or infection. Thus, AAGs are considered as major members of the positive acute phase protein family. Specifically, bovine α1-acid glycoprotein (BAAG) has been chosen as the biological host due to its ability to bind and transport endogenous or exogenous ligands, with basic or neutral character, mainly in one large and flexible binding site. The protein consists of one polypeptide chain and is highly glycosylated; the BAAG molecular weight has been determined as 33.8 kDa by MALDI-TOF mass spectrometry analysis.8
With regard to the probe ligand, CPFMe, the two enantiomers of the methyl ester of the chiral non-steroidal anti-inflammatory drug carprofen (CPF) have been selected (see chemical structures in Fig. 1). Introduction of the methyl group in arylpropionic acids, such as in CPFMe, leads to substrates that should bind to BAAG more efficiently than the parent drug.9 It has been reported that CPF is able to induce photoallergy, manifested as photocontact dermatitis.10 The effects were initially observed in patients under medical treatment and then, after introduction for veterinary purposes, in factory workers producing the drug and in pet owners. The main photoreaction of CPF in aqueous solution is dehalogenation, a process that generates highly reactive aryl radicals (CBZ˙, Fig. 1) capable of causing irreversible covalent chemical modification of proteins. Alternatively, in the presence of the appropriate hydrogen donors, the product resulting from dehalogenation is CBZ (Fig. 1).11
 | ||
| Fig. 1 Chemical structures of CPF and derivatives. | ||
Specifically, the strategy employed in this work involves the separate irradiation of the two enantiomers of CPFMe encapsulated within BAAG, to obtain their respective covalent adducts, coupled with a multidisciplinary methodology that comprises fluorescence and transient absorption spectroscopies, proteomic analysis and docking and molecular dynamics simulation studies. The results clearly show that after the dehalogenation of CPFMe, in order to afford CBZMe˙, irreversible covalent modification of BAAG occurs to Phe68. Moreover, the process is stereoselective, with higher chemical modification of the protein for the (S)-enantiomer.
Results and discussion
The required (S)- and (R)-enantiomers of CPFMe were prepared from their corresponding enantiopure free acids, (R)- and (S)-CPF, which were separated from the commercial racemic mixture by chiral HPLC, and then subjected to esterification with methanol to afford (R)- and (S)-CPFMe (see details in the ESI†).Photophysical studies
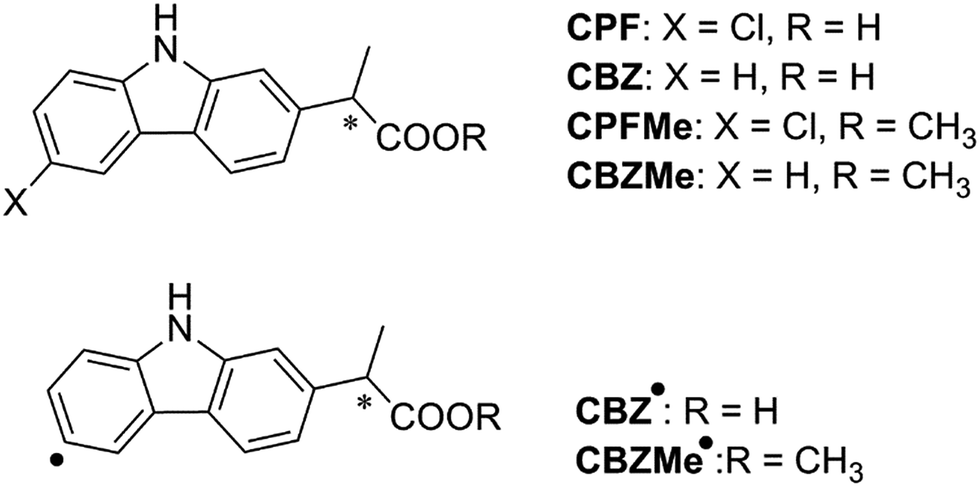
Since the photophysical properties of a given chromophore may be strongly dependent on the experienced microenvironment, a thorough photophysical study including fluorescence spectroscopy and laser flash photolysis was performed on the BAAG/CPFMe complexes.Solutions of (S)- and (R)-CPFMe (3.3 × 10−5 M) were prepared in the presence of 2 equivalents of BAAG, in phosphate-buffered saline (PBS). Fig. 2A shows the absorption spectra of BAAG/(S)-CPFMe and BAAG/(R)-CPFMe; the maxima in the UVB-UVA region appear at 300, 334 and 348 nm. The spectra of the isolated CPFMe (in MeCN, due to its poor solubility in PBS) and BAAG are shown as controls. The fluorescence spectra of the BAAG/CPFMe complexes at λexc = 320 nm consist of a band with two maxima at 360 and 375 nm (Fig. 2B).
 | ||
| Fig. 2 (A) Absorption spectra of CPFMe (in MeCN), BAAG, BAAG/(R)-CPFMe and BAAG/(S)-CPFMe in PBS. (B) Fluorescence spectra of BAAG/(R)-CPFMe and BAAG/(S)-CPFMe in PBS/air at λexc = 320 nm. | ||
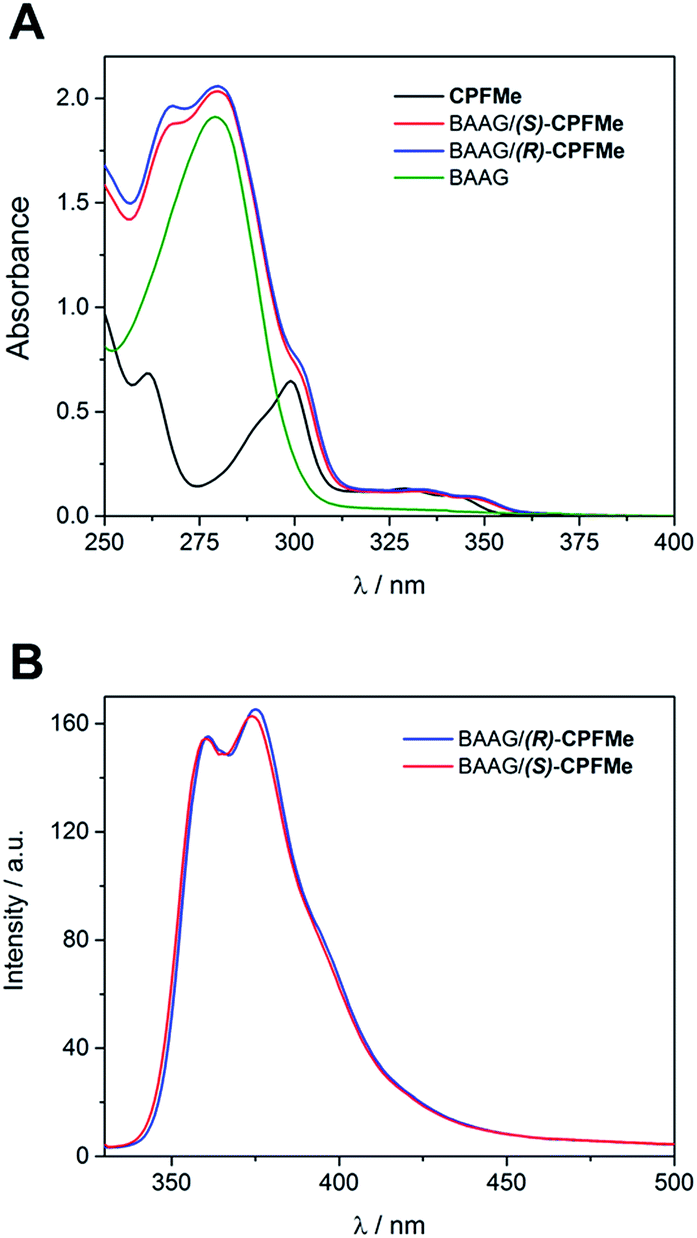
Laser flash photolysis of BAAG/(S)-CPFMe and BAAG/(R)-CPFMe was performed at λexc = 355 nm. The results in different time windows for the (R)-enantiomer are shown in Fig. 3A. Thus, a broad band with maximum at 450 nm was observed, attributed to the triplet excited state (3CPFMe*), confirmed by the literature data for CPF in MeCN.7c
 | ||
| Fig. 3 Laser flash photolysis of BAAG/CPFMe at λexc = 355 nm, in PBS/air. (A) Transient absorption spectra in different time windows for (R)-CPFMe. (B) Normalised decay kinetic traces at λmon = 450 nm for both enantiomers. [CPFMe] = 3.3 × 10−4 M, [BAAG] = 6.6 × 10−4 M. | ||
For the (S)-enantiomer, the shapes of the spectra were similar. However, stereodifferentiation was observed in the decay kinetics of BAAG/(R)-CPFMe and BAAG/(S)-CPFMe monitored at λ = 450 nm (Fig. 3B). Thus the triplet lifetime (τT) value for the (S)-enantiomer was much shorter (13 μs) than that of the (R)-analog (18 μs). This could be related to the more efficient quenching of 3CPFMe* by electron transfer from Trp in the case of (S)-CPFMe, which is in agreement with a closer distance from (S)-CPFMe to a Trp residue than in the case of (R)-CPFMe, within the binding site pocket.12 Stereodifferentiation in the triplet lifetimes of ligands within transport proteins has previously been observed in other cases, such as carprofen, flurbiprofen, flurbiprofen methyl ester or 2-anthracene propionic acid incorporated into serum albumins.7c,14
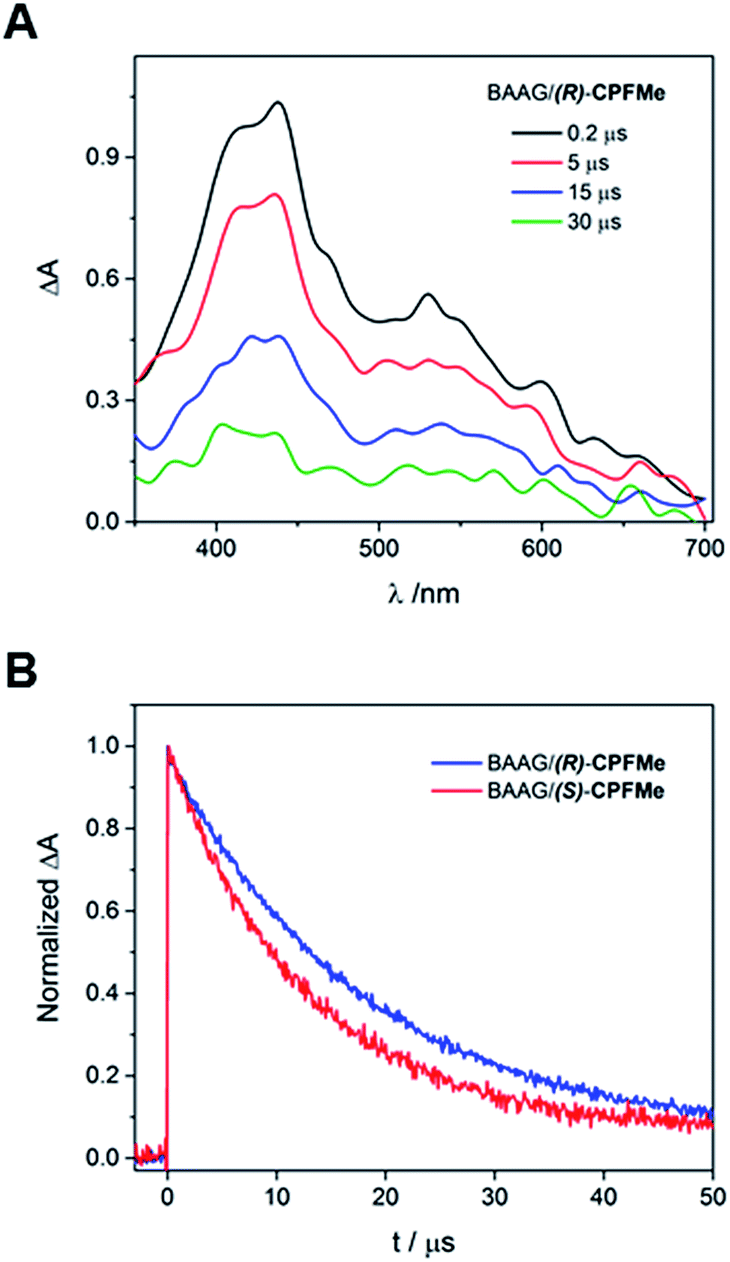
Solutions of BAAG/(S)-CPFMe and BAAG/(R)-CPFMe, again at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 protein/CPFMe molar ratio, were irradiated separately at λ = 320 nm, and the process was followed by fluorescence spectroscopy. The spectra recorded before and after 15 minutes of irradiation are shown in Fig. 4A. For both CPFMe enantiomers, the emission bands of the irradiated samples did not exhibit the characteristic fine structures with two well-defined maxima of CPFMe, and were both more intense. This is in agreement with the occurrence of the photodehalogenation reaction leading to products with fluorescence quantum yields much higher than that of CPFMe.13
:1 protein/CPFMe molar ratio, were irradiated separately at λ = 320 nm, and the process was followed by fluorescence spectroscopy. The spectra recorded before and after 15 minutes of irradiation are shown in Fig. 4A. For both CPFMe enantiomers, the emission bands of the irradiated samples did not exhibit the characteristic fine structures with two well-defined maxima of CPFMe, and were both more intense. This is in agreement with the occurrence of the photodehalogenation reaction leading to products with fluorescence quantum yields much higher than that of CPFMe.13
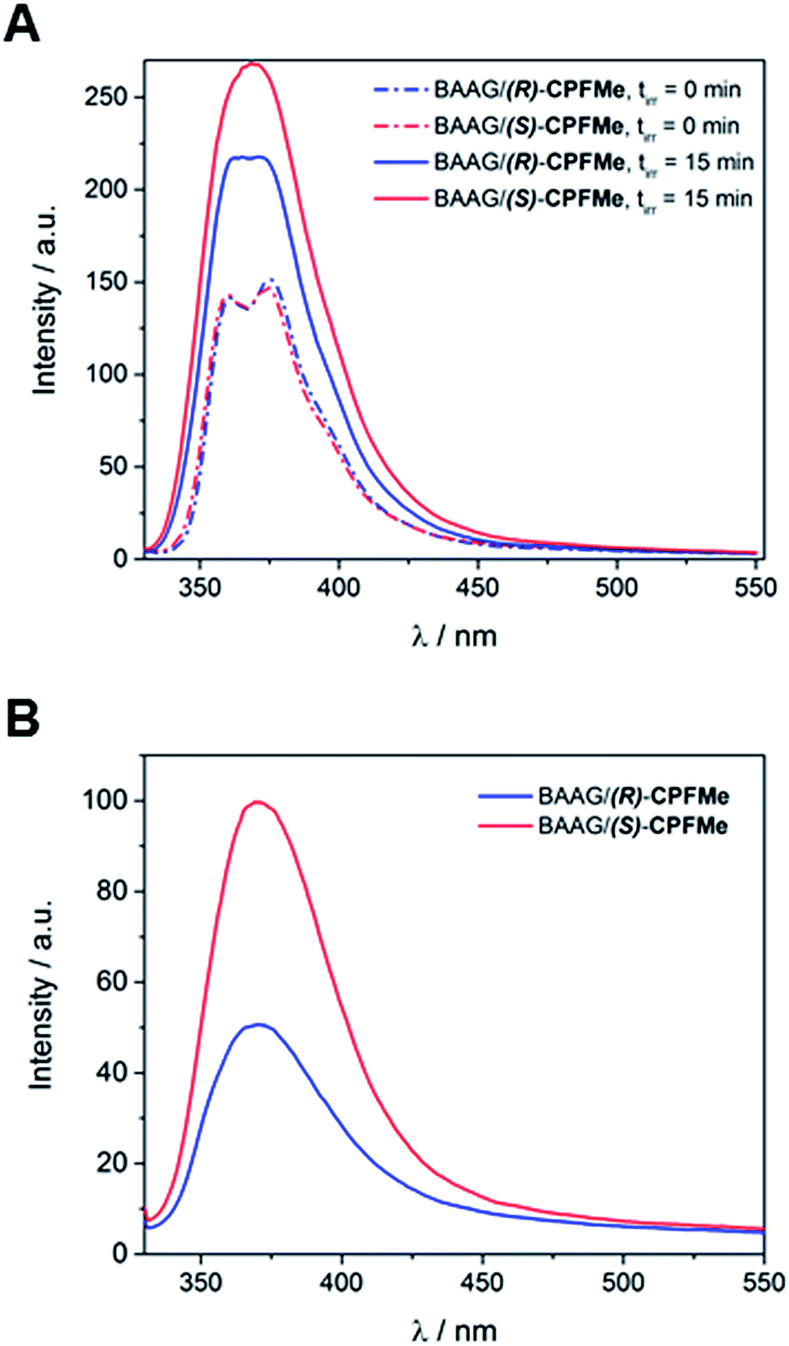 | ||
| Fig. 4 (A) Fluorescence spectra of non- and 15 min-irradiated samples of BAAG/CPFMe. (B) Fluorescence spectra of 15 min-irradiated samples of BAAG/CPFMe after GndCl treatment and filtration through Sephadex. [CPFMe] = 3.3 × 10−5 M, λexc = 320 nm, [BAAG] = 6.6 × 10−5 M. Experiment errors were lower than 5% of the obtained values. | ||
To check whether covalent modification of the protein was occurring, the photolysates were treated with 6 M of guanidinium chloride and filtered through Sephadex (details in ESI†), a process that allows the removal of the species with the lower molecular weight. The obtained spectra are shown in Fig. 4B. Clearly, emission is still observed, indicating that this CBZMe-derived species is covalently attached to BAAG. In parallel, non-irradiated samples of BAAG/CPFMe were subjected to the same work up as control experiments. As expected for the non-protein bound complexes, no emission was observed in these cases.
Interestingly, the CPFMe binding to BAAG resulted in being stereoselective, which would be in agreement with the relative lifetime values obtained by LFP.
Proteomic analysis
Having demonstrated the photobinding of CPFMe to BAAG by fluorescence measurements, the formation of covalent photoadducts was investigated in more detail by proteomic analysis. These studies would provide precise information of which amino acid(s) were covalently modified. Thus, the photoreactivity of the two enantiomers of CPFMe with BAAG was addressed by HPLC-nanoESI analysis. The irradiated BAAG/(S)-CPFMe and BAAG/(R)-CPFMe mixtures were filtered to remove excess ligand; then underwent trypsin digestion, which cleaves the peptide chain mainly at the carboxyl side of Lys or Arg residues (unless they neighbour a Pro residue), and finally HPLC-MS/MS analysis was performed. This was intended to obtain information on the modified peptide sequence and to characterize the adduct. Full scan and fragmentation data files were analysed using the Mascot® database search engine (Matrix Science, Boston, MA, USA) and by entering variable modifications that take into account the main possible residues (FWY) able to react with the carbazolyl radical CBZMe˙ obtained after dehalogenation of CPFMe. The sequence coverage was 90% for BAAG/(R)-CPFMe and 93% for BAAG/(S)-CPFMe. The results for the (R)-enantiomer are shown in Fig. 5A, with the non-matched peptides indicated in violet.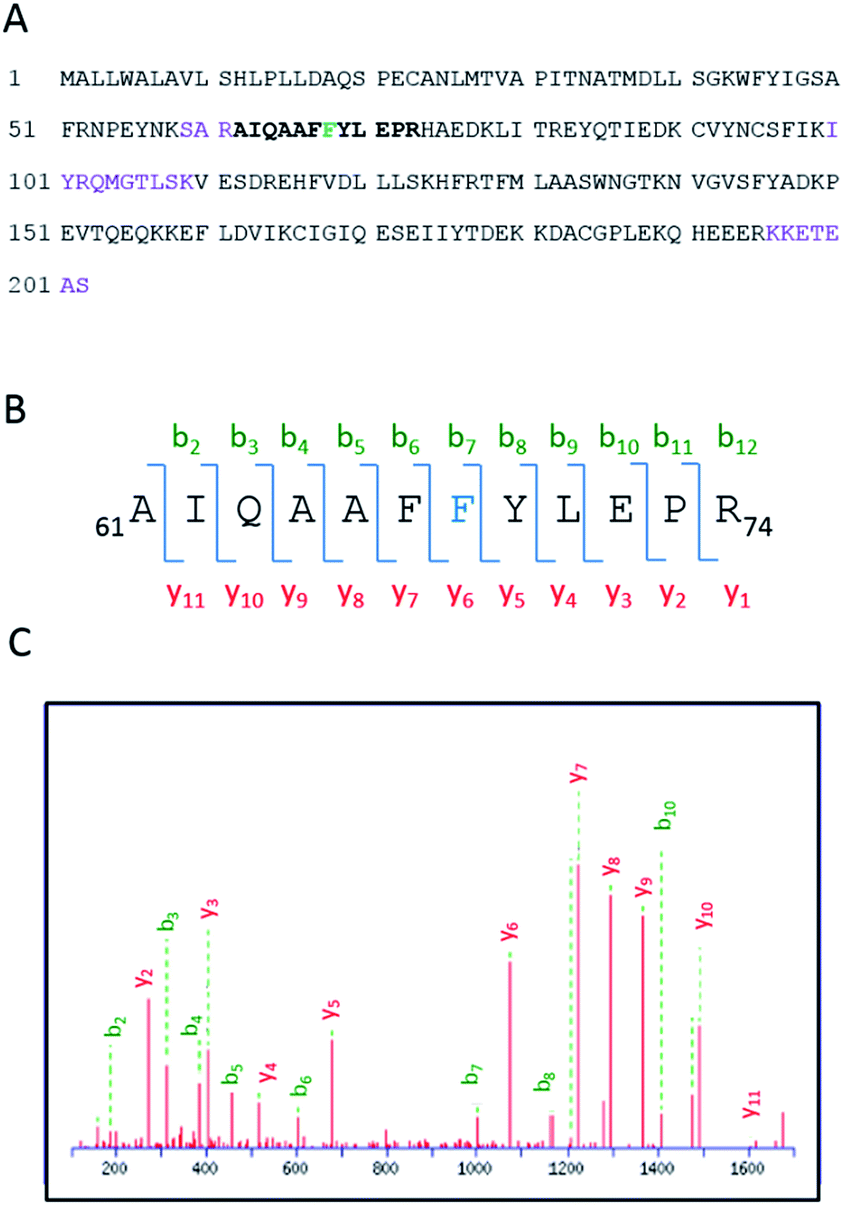 | ||
| Fig. 5 (A) Amino acid sequence of BAAG. The modified peptide is emboldened, and the altered amino acid residue is indicated in blue. (B) Modified peptide and fragmentation keys. (C) ESI-MS/MS spectra and fragmentation pathway of the CBZMe-modified peptide. | ||
The main result confirmed the identification of only one CBZMe–BAAG derived peptide adduct, 61AIQAAFFYLEPR74, for both CPFMe enantiomers (Fig. 5B).
The modification site of the peptide was assessed by tandem mass experiments on the trypsin digests. The ESI-MS/MS spectra and fragmentation pattern are shown in Fig. 5C. Their related data are summarised in Table 1. The peptide sequence agreed well with the “y” and “b” ion series. The MS/MS fragment ions showed an unmodified y ion series from y1 to y5, whereas an increment of m/z 398 was detected at Phe68 between y5 and y6 corresponding to CBZMe˙–Phe˙(–H2O). Accordingly, the b ion series suffered the same increase from b6 to b7. Thus, the modified amino acid is Phe68.
| Amino acid | y | m/z | b | m/z |
|---|---|---|---|---|
| A | b1 | 72.044 | ||
| I | y11 | 1605.8100 | b2 | 185.1285 |
| Q | y10 | 1492.7260 | b3 | 313.1870 |
| A | y9 | 1364.6674 | b4 | 384.2241 |
| A | y8 | 1293.6303 | b5 | 455.2263 |
| F | y7 | 1222.5932 | b6 | 602.3297 |
| F | y6 | 1075.5247 | b7 | 1000.4927 |
| Y | y5 | 677.3617 | b8 | 1163.5560 |
| L | y4 | 514.2984 | b9 | 1276.6401 |
| E | y3 | 401.2143 | b10 | 1405.6827 |
| P | y2 | 272.1717 | b11 | 1502.7355 |
| R | y1 | 175.1190 |
In order to ascertain whether Phe can react directly with 3CPFMe*, a triplet quenching experiment was performed. Thus, LFP of CPFMe in acetonitrile at λexc = 355 nm was run in the presence and also in the absence of Phe methyl ester, but no differences were found in the triplet lifetimes. In contrast, it has been previously established that quenching by Trp occurs at diffusion controlled rates.12 Combining this information with the proteomic results, a plausible mechanism is proposed (Scheme 1). It involves the reaction of 3CPFMe* with Trp to afford the corresponding radical ion pairs; the CBZMe˙ radical formed after the loss of Cl−would react with Phe, resulting in covalent binding to this residue.
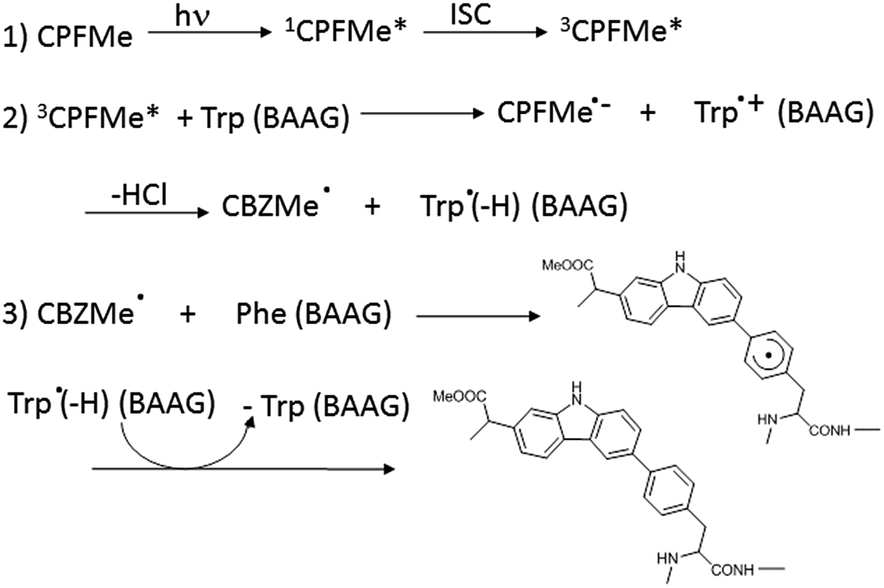 | ||
| Scheme 1 | ||
Molecular modelling
Due to (i) the specific covalent modification of Phe68 in BAAG by (S)- and (R)-CPFMe and (ii) reasoning that the differences observed between the two enantiomers would be due to the binding mode of these ligands in the recognition centre of the protein, computational studies were performed. To this end, molecular docking using GOLD 5.2 (ref. 15) was carried out using a homology model of BAAG. As no three-dimensional structure of BAAG is available, a homology model was constructed using the Phyre2 homology modelling web server16 (see a detailed description in the ESI, pp. S9 and S10†). A comparison of the crystal structure of the HAAG/(R)-1-glycerol acetate complex (PDB code 3KQ0 (ref. 17)) and the constructed three-dimensional structure of apo-BAAG reveals that its recognition centre is a large apolar pocket (Fig. S1A and B†). It mainly involves Leu70, Ile98, Leu107, Val118, Leu131, Phe68, Phe129, and Trp44. As for the HAAG protein, the pocket is closed by an electrostatic interaction between two polar residues, which for BAAG are Lys109 and Glu83. In addition, the recognition centre includes another aromatic residue, specifically, Tyr146. More importantly, the experimentally observed covalently modified Phe68 residue is located in close contact with Trp44, which would trigger the photocleavage reaction. A second phenylalanine residue, Phe129, is also located in the vicinity of Trp44.With the BAAG homology model in hand, docking studies were then performed. The proposed binding modes were further analysed by Molecular Dynamics (MD) simulation studies in order to assess the stability and therefore the reliability of the postulated binding. The results of these studies are discussed below.
Docking and MD simulation studies were carried out using GOLD 5.2 (ref. 15), with the protein geometries found in the apo-BAAG homology model. The position of (R)-1-glycerol acetate in PDB code 3KQ0 was used to define the active-site and the radius was set to 8 Å. The proposed binding modes of (S)- and (R)-CPFMe were further analysed by MD simulation studies. As expected, at the beginning of the simulation (the first ∼25 ns) a small displacement of the two ligands was observed resulting from the initial adjustment of the bulky ligands with flexible side chains (ester) into apo-structures (Fig. S2†). However, eventually the complexes revealed to be stable during most of the simulation (∼75 ns) with the aromatic moiety located in close proximity to Phe68 (Fig. 6).
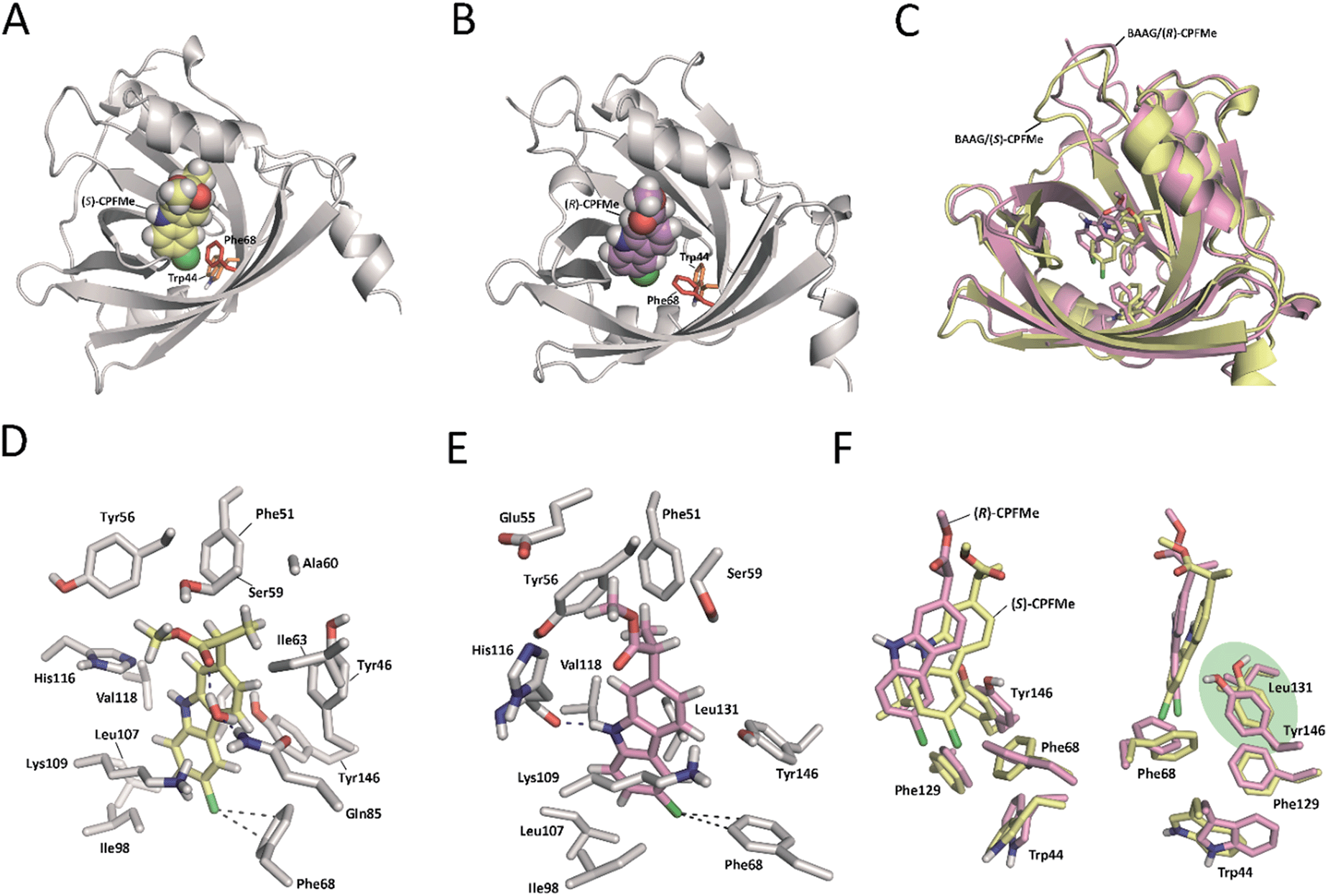 | ||
| Fig. 6 Binding modes of (S)- (A and D) and (R)-CPFMe (B and E) obtained by docking and MD simulation studies of the recognition centre of the BAAG protein (grey). The viewpoints showed for the ligands (S)-CPFMe (yellow) and (R)-CPFMe (pink) correspond to snapshots after 50 ns of MD simulation. (C and F) Comparisons of the binding modes of (S)- (yellow) and (R)-CPFMe (pink) in the recognition centre of the BAAG protein. Relevant side chain residues are shown and labelled. Polar (blue) contacts and distances (grey) to Phe68 are shown as dashed lines. Note how the C6 atom of the two isomers is located close to Phe68 and how Tyr146 and Leu131 side chains prevent the covalent modification of Phe129. | ||
Our computational studies revealed that the binding mode of the two enantiomers would be in general quite similar in terms of the type of amino acids involved and the kind of interactions with them. The binding free energies of each ligand were calculated using the MM/PBSA18 approach in implicit water (generalised Born, GB) as implemented in Amber. (S)-CPFMe has a binding free energy that is −0.9 kcal lower than that of (R)-CPFMe (Table S1†). Similar results were obtained using the water-swap application.19 In particular, the aromatic moiety of the ligands would establish lipophilic interactions with the side chains of residues Ile98, Leu107, Val118, Ile63, and Lys109 (carbon chain) and CH–π interactions with the side chain of Leu114 (Fig. 6D and E). Besides this similarity involving the aromatic ring of the ligands, the stereochemistry of the flexible side chain proved to be a key element in the proximity of the C6 carbon atom of the two enantiomers to the phenyl group of the experimentally observed covalently modified Phe68, which would explain its different behaviour (Fig. 6C and F). Whereas for (S)-CPFMe, the position of the ester chain is anchored in the recognition centre by hydrogen bonding with the side chain of Gln85 via a water molecule, however, for (R)-CPFMe the chain is quite flexible. Gln85 is located in the vicinity of Phe68. In addition, the NH group of the aromatic moiety in (R)-CPFMe would interact by a strong hydrogen bonding interaction with the main carbonyl group of His116 and the methyl group of the side chain would be located in the apolar pocket, involving Phe34 and Tyr39. These residues are located in the opposite site of Phe68. As a consequence of these attractive interactions, (R)-CPFMe and therefore its C6 carbon atom would be located further from the phenyl group of Phe68 than in the case of (S)-CPFMe. This displacement of (R)-CPFMe towards the opposite site of the recognition centre would explain the less efficient covalent modification of the Phe68 residue (Fig. 6F). Analysis of the contribution of each residue to the binding free energy carried out with the water-swap19 application also revealed the interaction of the (S)-enantiomer with Phe68 would be 8-fold stronger than its enantiomer (Fig. S3†). A study of the relative distances between the C6 atom in the two ligands and the CZ (para) and CE1 and CE2 (meta) carbon atoms in Phe68 during the simulation is shown in Fig. 7. For (S)-CPFMe in the last 60 ns of simulation, the average distances between the C6 atom and the CZ, CE1 and CE2 carbon atoms in Phe68 were found to be 5.4, 5.4 and 6.0 Å, respectively (Fig. 7C and E). For (R)-CPFMe, the average distances were 6.0, 6.0 and 6.6 Å, respectively (Fig. 7D and F). It is important to highlight that once the C–Cl bond cleavage occurs, the generated radical would be located closer to the phenyl ring of Phe68 as this would reduce steric hindrance.
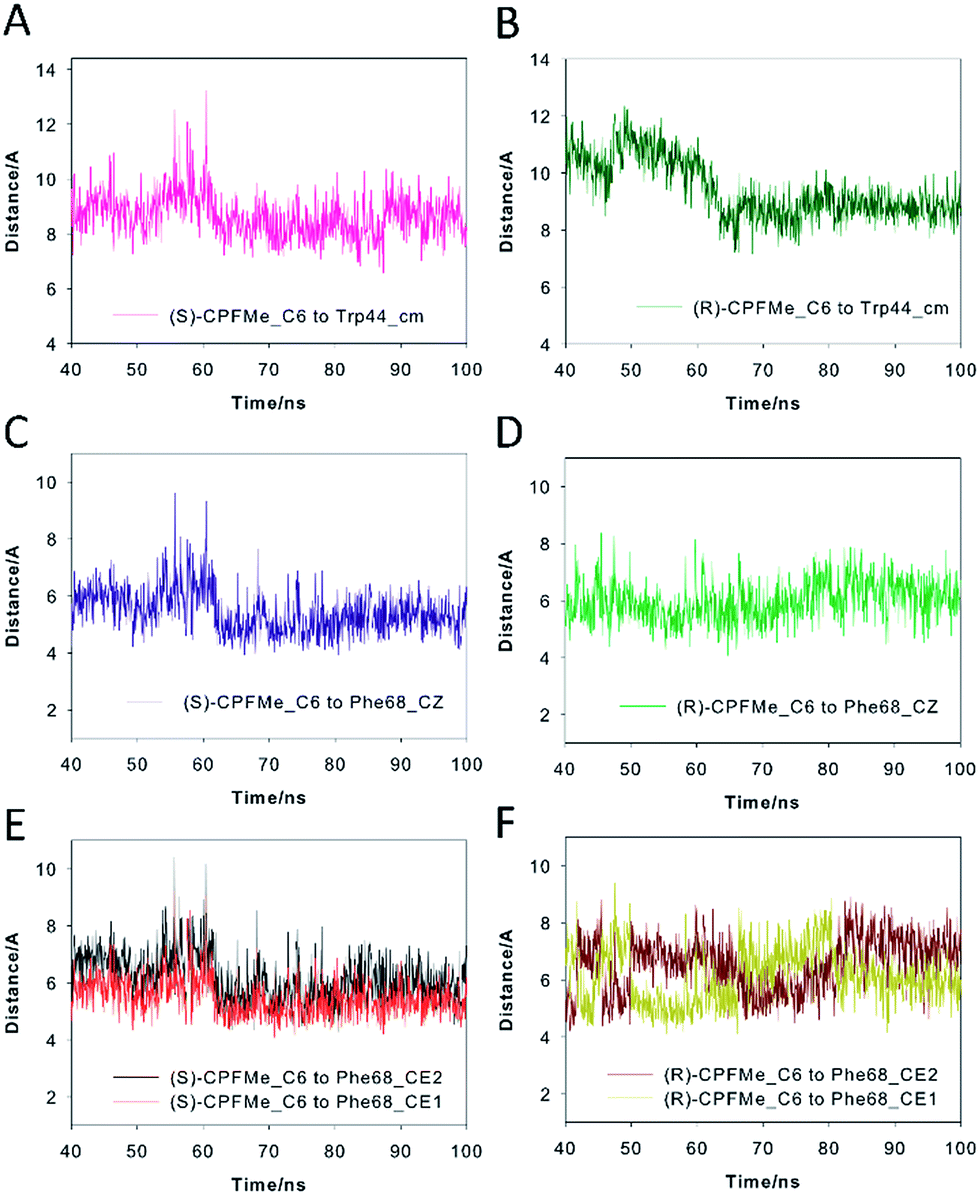 | ||
| Fig. 7 Variation of the relative distances between the C6 atom of (S)-CPFMe (A, C and E) and (R)-CPFMe (B, D and F) and the CZ atom (para-), the CE1 and CE2 atoms (meta-) of Phe68 and the centre of mass of Trp44 in the corresponding BAAG/CPFMe protein complexes during the last 60 ns of the simulation. Note how the C6 atom of (S)-CPFMe remains closer to Phe68 than its enantiomer during the simulation. | ||
The results of our computational studies also explain why the covalent modification of Phe68 is only observed in spite of the presence of another phenylalanine in the vicinity of the ligands, specifically, Phe129. The proximity and higher accessibility of Phe68 to the ligands would explain this fact. Thus, an analysis of the relative distance between the C6 carbon atom in the two ligands and the CZ (para) and CE1 and CE2 (meta) carbon atoms of Phe68 and Phe129 revealed that the ligands are closer to Phe68 than Phe129 (Fig. S4†). In addition, as shown in Fig. 7F, the proximity of Tyr146 and Leu131 to the aromatic rings of the two ligands would prevent reaction with Phe129, which is located behind those residues. Moreover, as expected, the covalently modified Phe68 is located in the vicinity of a tryptophan residue, specifically, Trp44 (Fig. 7B). Our simulation studies showed that this residue is also located closer to the C6 carbon atom of (S)-CPFMe than its enantiomer during the simulation with an average distance of 8.6 Å vs. 9.2 Å for (R)-CPFMe during the last 60 ns of the simulation (Fig. 7A and B). The centre of mass of Trp44 was considered for these calculations.
In an effort to obtain further details of the covalent modification of Phe68 by the two enantiomers, the possible insertion at its para and meta positions were then explored by MD simulation studies (Fig. 8 and S5†). For (S)-CPFMe, the analysis of the relative distances between the C6 carbon atom of the ligand and the CZ, CE1 and CE2 carbon atoms in Phe68 revealed that the two positions are equally accessible (Fig. 7C and E). In addition, no significant binding differences were observed in the two possible adducts (Fig. 8C). The two would be well embedded in the recognition centre. These findings suggest that, from a computational point of view, both para and meta adducts would be possible. On the contrary, the most plausible covalent modification of the Phe68 residue by the R enantiomer would be at its para position (Fig. 8B). In this case: (i) the CZ carbon atom of Phe68 would be located closer to the C6 carbon atom of the ligand than the CE1 and CE2 atoms (Fig. 7Dvs.F); and (ii) the corresponding meta adduct would open the recognition centre up to 5.4 Å to accommodate the ester side chain in this arrangement (Fig. 8D).
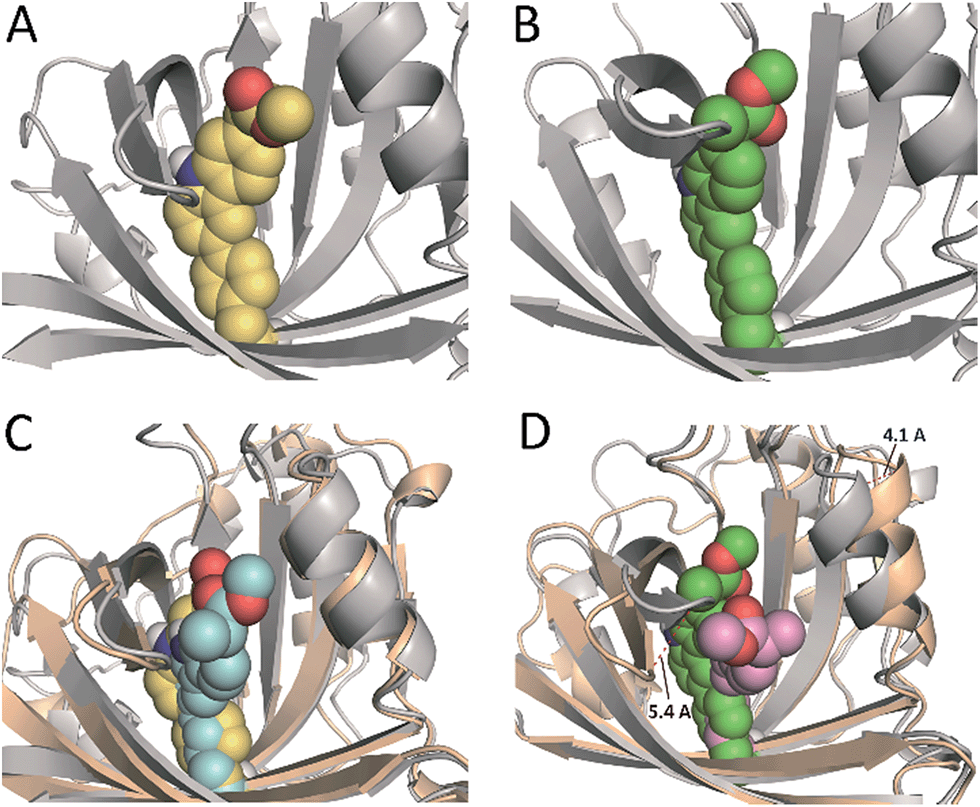 | ||
| Fig. 8 (A and B) Proposed tridimensional structure of BAAG covalently modified by (S)-CPFMe (A, yellow) and (R)-CPFMe (B, green) at the para-position of Phe68, obtained by MD simulation studies. (C and D) Comparison of the resulting para- [protein (grey)] and meta- [protein (wheat)] BAAG/(S)-CPFMe (C) and BAAG/(R)-CPFMe (D) adducts. meta-Covalently modified Phe68 residues by (S)- and (R)-CPFMe are shown as blue and pink spheres, respectively. The viewpoints shown correspond to snapshots after 70 ns of MD simulation. Note how for (R)-CPFMe, the meta-adduct causes a large opening of the recognition centre of up to 5.4 Å. | ||
Conclusions
A multidisciplinary strategy has been employed here to obtain structural information on the intraprotein region. The principle has been proven by separate irradiation of BAAG/(S)-CPFMe and BAAG/(R)-CPFMe complexes, coupled with photophysical studies, proteomic analysis, and docking and molecular dynamics simulations. Overall, the obtained results indicate that the ligand is located in a large apolar pocket that mainly involves Leu70, Ile98, Leu107, Val118, Leu131, Phe68, Phe129, and Trp44. Steady-state irradiation of the complexes leads to the dehalogenation of CPFMe, ultimately resulting in the covalent photobinding of the intermediate aryl radical CBZMe˙ to Phe68. This process, which is more efficient for the (S)-enantiomer, could be triggered by the neighbouring Trp44. Accordingly, the triplet excited state lifetime of the BAAG/(S)-CPFMe complex is shorter than that of BAAG/(R)-CPFMe. Thus, this strategy allows the identification of the molecular recognition centre of BAAG through the photoinduced modification of key amino acid residues by CPFMe and could, in principle, be extended to a variety of protein/ligand complexes. We believe that the studies reported here would be helpful for studying the recognition centre of diverse proteins with photoactive ligands. The only requirement for the methodology to be applicable is the presence of an active chromophore in the ligand that, after light absorption, can generate reactive species capable of attacking one or several amino acid residues, leading to the formation of covalent, irreversible adducts.Acknowledgements
Financial support from the Spanish Ministry of Economy and Competitiveness (CTQ2013-47872-C2-1-P, SAF2013-42899-R, BES-2011-043706), Generalitat Valenciana (PROMETEOII/2013/005), Instituto de Salud Carlos III (RD12/0013/0009), Xunta de Galicia (GRC2013-041), the Consellería de Cultura, Educación e Ordenación Universitaria (Centro singular de investigación de Galicia accreditation 2016-2019, ED431G/09) and the European Regional Development Fund (ERDF) is gratefully acknowledged. E. L. thanks the Xunta de Galicia for a postdoctoral fellowship. We are grateful to the Centro de Supercomputación de Galicia (CESGA) for use of the Finis Terrae II supercomputer. The proteomic analysis was performed in the proteomics facility of SCSIE University of Valencia that belongs to ProteoRed PRB2-ISCIII and is supported by grant PT13/0001, of the PE I+D+i 2013–2016, funded by ISCIII and FEDER.Notes and references
- (a) J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111 RSC; (b) R. E. Babine and S. L. Bender, Chem. Rev., 1997, 97, 1359 CrossRef CAS PubMed; (c) M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479 CrossRef CAS PubMed; (d) K. N. Houk, A. G. Leach, S. P. Kim and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872 CrossRef CAS PubMed; (e) R. D. Smith, A. L. Engdahl, J. B. Dunbar and H. A. Carlson, J. Chem. Inf. Model., 2012, 52, 2098 CrossRef CAS PubMed; (f) A. J. T. Smith, X. Zhang, A. G. Leach and K. N. Houk, J. Med. Chem., 2009, 52, 225 CrossRef CAS PubMed; (g) H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924 CrossRef CAS PubMed; (h) X. Zhang and K. N. Houk, Acc. Chem. Res., 2005, 38, 379 CrossRef CAS PubMed; (i) D. H. Williams, E. Stephens, D. P. O'Brien and M. Zhou, Angew. Chem., Int. Ed., 2004, 43, 6596 CrossRef CAS PubMed; (j) A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775 CrossRef CAS PubMed.
- (a) R. F. Thompson, M. Walker, C. A. Siebert, S. P. Muench and N. A. Ranson, Methods, 2016, 100, 3 CrossRef CAS PubMed; (b) A. M. Hassell, G. An, R. K. Bledsoe, J. M. Bynum, H. L. Carter III, S.-J. J. Deng, R. T. Gampe, T. E. Grisard, K. P. Madauss, R. T. Nolte, W. J. Rocque, L. Wang, K. L. Weaver, S. P. Williams, G. B. Wisely, R. Xua and L. M. Shewchuka, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 72 CrossRef CAS PubMed; (c) P. Nollert, M. D. Feese and B. L. Staker, H. Kim, Protein X-Ray Crystallography in Drug Discovery, Pharmaceutical Sciences Encyclopedia, John Wiley & Sons, 2010 Search PubMed; (d) D. R. Cooper, P. J. Porebski, M. Chruszcz and W. Minor, Expert Opin. Drug Discovery, 2011, 6, 771 CrossRef CAS PubMed; (e) A. L. Carvalho, J. Trincão and M. J. Romão, Methods Mol. Biol., 2009, 572, 31 CrossRef PubMed.
- (a) S. Montanaro, V. Lhiaubet-Vallet, M. C. Jiménez, M. Blanca and M. A. Miranda, ChemMedChem, 2009, 4, 1196 CrossRef CAS PubMed; (b) E. Nuin, D. Pérez-Sala, V. Lhiaubet-Vallet, I. Andreu and M. A. Miranda, Front. Pharmacol., 2016, 7, 277 Search PubMed.
- (a) Y. Hatanaka and Y. Sadakane, Curr. Top. Med. Chem., 2002, 2, 271 CrossRef CAS PubMed; (b) F. Kotzyba-Hibert, I. Kapfer and M. Goeldner, Angew. Chem., Int. Ed., 1995, 34, 1296 CrossRef CAS.
- (a) I. Andreu, C. Mayorga and M. A. Miranda, Curr. Opin. Allergy Clin. Immunol., 2010, 10, 303 CrossRef CAS PubMed; (b) M. Gonçalo, Phototoxic and Photoallergic Reactions, in Contact Dermatitis, ed. J. D. Johansen, P. J. Frosch and J.-P. Lepoittevin, Springer-Verlag, Berlin, 2011, p. 361 Search PubMed; (c) J. Ferguson, Drug and Chemical Photosensitivity, in Photodermatology, ed. J. L. M. Hawk, Arnold, London, 1999, p. 155 Search PubMed.
- P. Jones, In vitro phototoxicity assays, in Principles and Practice of Skin Toxicology, ed. R. Chilcott and S. Price, John Wiley & Sons, 2008, p. 169 Search PubMed.
- (a) J. Moser, A. Hye, W. W. Lovell, L. K. Earl, J. V. Castell and M. A. Miranda, Toxicol. In Vitro, 2001, 15, 333 CrossRef CAS PubMed; (b) J. Moser, F. Boscá, W. W. Lovell, J. V. Castell, M. A. Miranda and A. Hye, J. Photochem. Photobiol., B, 2000, 58, 13 CrossRef CAS; (c) V. Lhiaubet-Vallet, Z. Sarabia, F. Boscá and M. A. Miranda, J. Am. Chem. Soc., 2004, 126, 9538 CrossRef CAS PubMed.
- (a) K. Matsumoto, K. Sukimoto, K. Nishi, T. Maruyama, A. Suenaga and M. Otagiri, Drug Metab. Pharmacokinet., 2002, 17, 300 CrossRef CAS PubMed; (b) T. Fournier, N. Medjoubi-N and D. Porquet, Biochim. Biophys. Acta, 2000, 1482, 157 CrossRef CAS PubMed; (c) K. Tamura, T. Yatsu, H. Itoh and Y. Motoi, Nippon Juigaku Zasshi, 1989, 51, 987 CrossRef CAS PubMed; (d) M. Nakano, K. Kakehi, M. Tsai and Y. C. Lee, Glycobiology, 2004, 14, 431 CrossRef CAS PubMed; (e) F. Ceciliani, V. Pocacqua, E. Provasi, C. Comunian, A. Bertolini, V. Bronzo, P. Moroni and P. Sartorelli, Vet. Res., 2005, 36, 735 CrossRef CAS PubMed.
- M. H. Rahman, K. Yamasaki, Y.-H. Shin, C. C. Lin and M. Otagiri, Biol. Pharm. Bull., 1993, 16, 1169 CAS.
- (a) V. Lhiaubet-Vallet and M. A. Miranda, in Handbook of Organic Photochemistry and Photobiology, 3rd edn, 2012, vol. 2, p. 1541 Search PubMed; (b) A. C. Kerr, F. Muller, J. Ferguson and R. S. Dawe, Br. J. Dermatol., 2008, 159, 1303 CrossRef CAS PubMed.
- F. Boscá, S. Encinas, P. F. Heelis and M. A. Miranda, Chem. Res. Toxicol., 1997, 10, 820 CrossRef PubMed.
- V. Lhiaubet-Vallet, M. A. Miranda and F. Boscá, J. Phys. Chem. B, 2007, 111, 423 CrossRef CAS PubMed.
- D. Limones-Herrero, R. Pérez-Ruiz, M. C. Jiménez and M. A. Miranda, Org. Lett., 2013, 15, 1314 CrossRef CAS PubMed.
- (a) M. C. Jiménez, M. A. Miranda and I. Vayá, J. Am. Chem. Soc., 2005, 127, 10134 CrossRef PubMed; (b) R. Alonso, M. C. Jiménez and M. A. Miranda, Org. Lett., 2011, 13, 3860 CrossRef CAS PubMed; (c) I. Vayá, C. J. Bueno, M. C. Jiménez and M. A. Miranda, ChemMedChem, 2006, 1, 1015 CrossRef PubMed; (d) I. Vayá, C. J. Bueno, M. C. Jimenez and M. A. Miranda, Chem.–Eur. J., 2008, 14, 11284 CrossRef PubMed; (e) I. Vayá, V. Lhiaubet-Vallet, M. C. Jiménez and M. A. Miranda, Chem. Soc. Rev., 2014, 43, 4102 RSC.
- http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ .
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845 CrossRef CAS PubMed.
- D. L. Schonfeld, R. B. Ravelli, U. Mueller and A. Skerra, J. Mol. Biol., 2008, 384, 393 CrossRef PubMed.
- B. R. Miller III, T. D. McGee Jr, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314 CrossRef PubMed.
- (a) C. J. Woods, M. Malaisree, S. Hannongbua and A. J. Mulholland, J. Chem. Phys., 2011, 134, 054114 CrossRef PubMed; (b) C. J. Woods, M. Malaisree, J. Michel, B. Long, S. McIntosh-Smith and A. J. Mulholland, Faraday Discuss., 2014, 169, 477 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, additional computational calculations, figures and references. See DOI: 10.1039/c6sc04900a |
| This journal is © The Royal Society of Chemistry 2017 |