
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadically promoted formation of a molecular lasso†
Yuping
Wang
 a,
Junling
Sun
a,
Zhichang
Liu
a,
Majed S.
Nassar
b,
Youssry Y.
Botros
bc and
J. Fraser
Stoddart
*a
a,
Junling
Sun
a,
Zhichang
Liu
a,
Majed S.
Nassar
b,
Youssry Y.
Botros
bc and
J. Fraser
Stoddart
*a
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: stoddart@northwestern.edu; Tel: +1-847-491-3793
bJoint Center of Excellence in Integrated Nano-Systems (JCIN), King Abdul-Aziz City for Science and Technology (KACST), P. O. Box 6086, Riyadh 11442, Saudi Arabia
cPanaceaNano, Inc., 2265 East Foothill Boulevard, Pasadena, California 91107, USA
First published on 16th January 2017
Abstract
Two potential viologen-based molecular lasso precursors—both composed of a 4,4′-bipyridinium (BIPY2+) unit as part of a rope appended to a cyclobis(paraquat-p-phenylene) (CBPQT4+) loop—that have been designed to mimic the threading/unthreading motion of lasso peptides, have been synthesised and characterised. Solution and solid-state experiments reveal that, when the BIPY2+ unit in the rope and the CBPQT4+ loop are connected by a bulky linker, no lasso-like conformational transformation is observed between the different redox states on account of steric effects. In sharp contrast, when the linker size is small, the molecule can be switched between (i) a free rope-like conformation in its fully oxidised state and (ii) a self-entangled lasso-like conformation under reducing conditions employing either chemical or electrochemical stimuli: the BIPY˙+ unit in the rope resides inside the cavity of the CBPQT2(˙+) loop, forming a pseudo[1]rotaxane. The switching process is reversible and stereochemically unambiguous. This research shows how tiny structural differences can induce significantly different self-complexing properties and sheds light on designing functional artificial actuators.
Introduction
As chemists, we often claim that we draw inspiration from Mother Nature. Lasso peptides,1,2 which contain 16–21 amino acid residues,3 constitute members of the ribosomally assembled and post-translationally modified peptides (RiPPs) superfamily.4 The understanding of their biofunctions5,6 has attracted the attention of chemists, principally on account of their remarkable stabilities, resulting from their self-entangled conformations. Specifically, composed of macrocycles and axles that thread the macrocycles, lasso peptides exhibit their biological activities by adopting compact—in comparison with their unthreaded analogues—lasso-like conformations. Designing and synthesising abiotic counterparts, not only provides us with wholly synthetic compounds that can mimic the threading-unthreading behaviour7 of lasso peptides, but it also enriches the family of artificial molecular actuators displaying controllable intramolecular motions. So far, among all the attempts that have been made, (pseudo)[1]rotaxanes,8–13 which bear (Fig. 1) macrocycles (loops) encircling or otherwise14 tails that are connected covalently to the loops, have been shown3 to be promising synthetic targets. Usually, the formation of the mechanically interlocked compounds in the form of (pseudo)[1]rotaxanes are driven by weak interactions such as (i) hydrogen bonds between crown ether15 and ammonium or triazolium ions,13,16–18 (ii) hydrophobic forces in water between the cavities of cyclodextrins and aromatic guest substrates,11,19–21 (iii) metal–ligand coordinative interactions22 and (iv) donor–acceptor interactions10,23 between cyclobis(paraquat-p-phenylene) (CBPQT4+) host and electron-rich tetrathiafulvalene (TTF) and 1,5-dioxynaphthalene (DNP) units. Amongst all the reported examples,13,22 however, pseudo[1]rotaxanes that are able to switch reversibly between self-entangled and disentangled states remain a rare phenomenon. Moreover, the actuation processes are usually triggered exclusively by chemical stimuli, which inevitably introduces additional species onto the scene and so complicates the situation. In order to avoid all these disadvantages, we have designed a new class of pseudo[1]rotaxane actuated by a different type of noncovalent bonding interaction. Not so long ago, we discovered24,25 that under reducing conditions, the radical cationic state of the bipyridinium unit, namely BIPY˙+, interacts strongly with the bisradical dicationic state CBPQT2(˙+). Since this radical-mediated recognition has been demonstrated26,27 to be reversible in solution, upon varying the external applied electro-potential, we decided to take advantage of this recognition to construct pseudo[1]rotaxane-based lassos. It is noteworthy that radical-pairing recognition has also been shown28 to drive the self-sorting behavior of numerous other organic radicals, e.g., thiazyl radicals. | ||
| Fig. 1 Illustrations of the transformation of a lasso precursor between its rope-like and lasso-like conformations employing (a) a rope and (b) a graphical representation of 13(˙+) and 23(˙+). The terms we chose for the description in this paper are shown and the two pathways, (i) threading and (ii) self-entanglement approach, are denoted by the red dot lines. It should be noted that in (b), the plane labelled with “+” sign faces up after process (i) and faces down after process (ii). | ||
Results and discussion
When designing the structures of the pseudo[1]rotaxanes, two general mechanisms of actuation come to mind. They involve (Fig. 1) (i) a threading29 approach where the end of the tail is small enough to go through the cavity of the loop, forming the pseudo[1]rotaxane and (ii) a self-entanglement approach, in which the tail is pulled into the cavity of the loop forming a pseudo[1]rotaxane, as a result of one portion of the loop—i.e., the portion connected to the tail—rotating like a revolving door (Fig. 1b) with respect to the mean plane of the ring.3,13,22 In both approaches (i) and (ii), the rope-like conformations are converted into the lasso-like conformations, while forming (Fig. 1) nooses at the same time. Although the second one—namely the revolving-door mechanism—is less obvious, it can be considered as a viable pathway since it is not necessary for the entire tail portion to pass through the cavity of the loop during the revolving process. As a result, the tail can be substituted by a bulky group with some desired function, such that, not only will it stand a good chance of introducing new properties into the pseudo[1]rotaxane, but it will also exclude the possibility of intermolecular complexation, e.g., daisy chain-style oligomer formation.12,30–32 On the basis of these considerations, we report (Scheme 1) herein how 16+ and 26+ were prepared by Cu(I)-catalysed alkyne-azide cycloadditions (CuAAC) between (i) a BIPY2+-based half-dumbbell component 52+—in which one end is functionalised with a 2,6-diisopropylphenyl group to act as a stopper—and (ii) a couple of alkyne-functionalised CBPQT4+ derivatives 34+ and 44+, respectively. The resulting molecular-lasso precursors 16+ and 26+ were readily purified by column chromatography (SiO2: 2% w/v MeCN solution of NH4PF6 as the eluent), resulting in yields of 60 and 80%, respectively. It should be emphasised that, despite the fact that both 16+ and 26+ are composed of similar loops and tail components, we were interested as to whether the differences in the constitutions of their linkers, connecting the loops to their tails, would lead to differences in their conformational behaviour under reducing conditions. Specifically, in the case of 16+, the linker contains a relatively bulky imide group, whereas in the case of 26+, it is replaced by a much smaller ester function, increasing the freedom of motion of the substituted p-phenylene linker of the loop components, e.g., the motion about its para-substituted axis. In the fully oxidised states, no interaction is present between the tail and the loop components in either 16+ or 26+, an observation which can be ascribed to the strong coulombic repulsion that surely exists between the BIPY2+ units. By contrast, upon reduction, both 16+ and 26+ can accept three electrons readily to generate their corresponding trisradical tricationic states—namely 13(˙+) and 23(˙+)—where it is possible, in principle at least, for them to undergo self-interlacing as a result of intramolecular radical-pairing interactions involving all three BIPY˙+ units.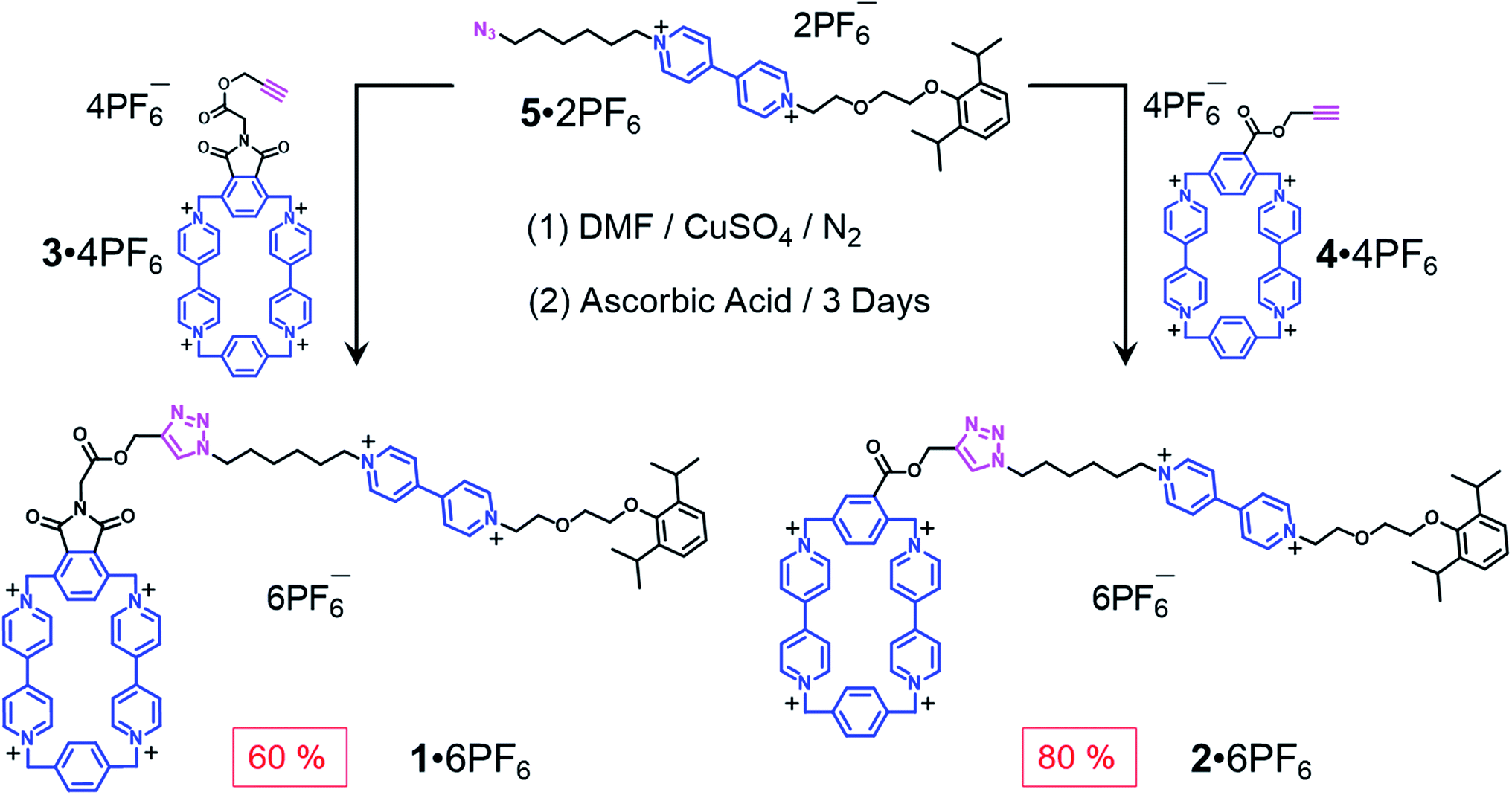 | ||
| Scheme 1 Syntheses of the two potential viologen-based molecular lassos 16+ and 26+. | ||
The self-complexing properties of 13(˙+) and 23(˙+) were first of all investigated by UV/Vis/NIR spectrophotometry. Following the reduction of 16+ to its radical state by Zn dust, the absorption spectra of a series of MeCN solutions, containing different concentrations of 13(˙+) ranging from 10–80 μM, were recorded. It transpires (Fig. 2a) that at all concentrations, strong absorption bands centered upon 600 nm emerge, an observation which is a characteristic feature of the BIPY˙+ radical cation. In addition, absorption bands centered on 900 nm grow in their intensities as well, suggesting that the radical-pairing interactions drive the formation of the (BIPY˙+)2 radical dimer33,34 rather than an intramolecular trisradical tricationic “complex”. Moreover, the absorption intensities of these bands correlate (Fig. 2b) linearly with the concentration of 13(˙+), an observation which indicates that pimerization33 takes place intramolecularly in a side-on fashion, i.e., the BIPY˙+ unit of the tail folds back to interact in an alongside manner with one of the two BIPY˙+ units in the loop component.
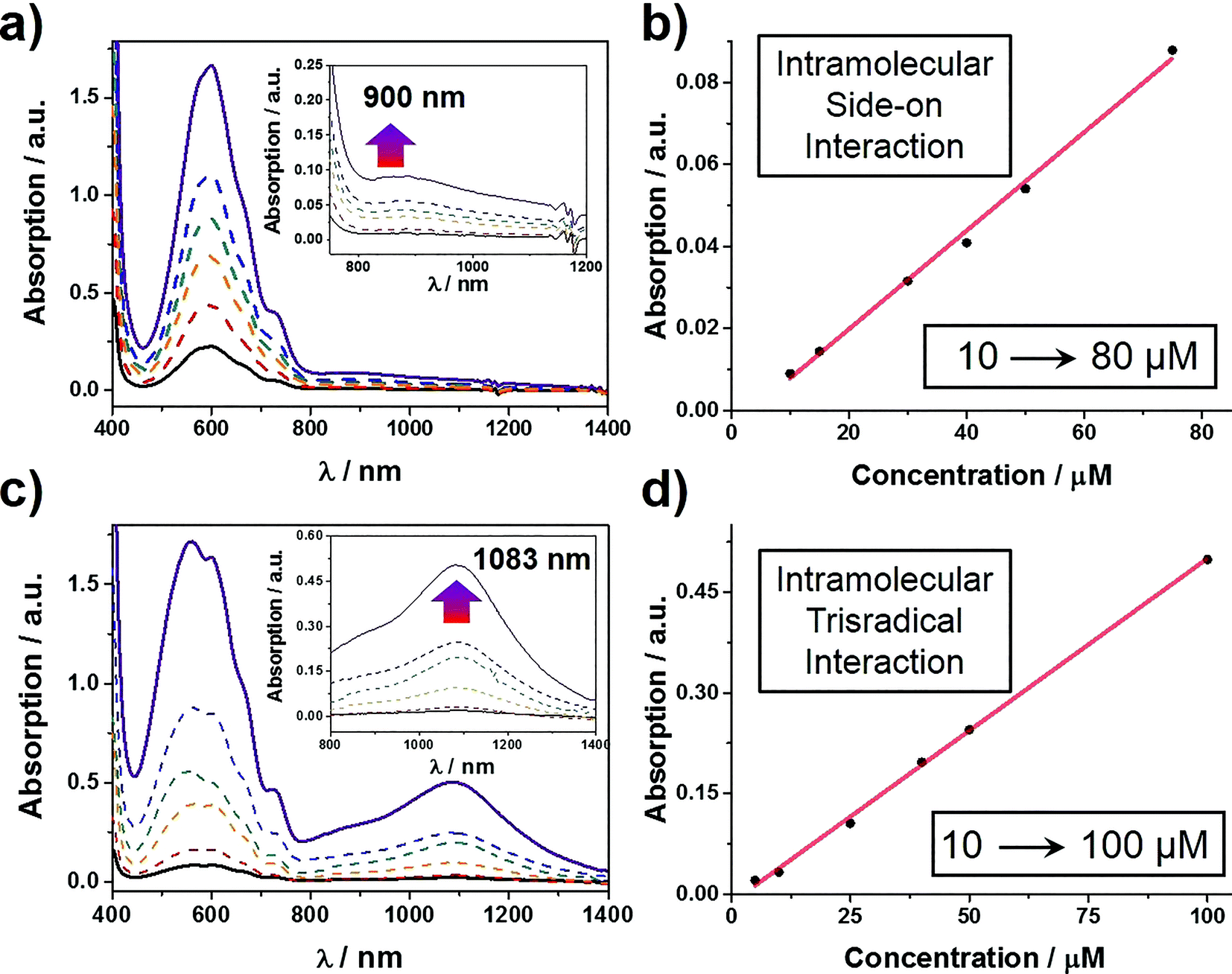 | ||
| Fig. 2 UV/Vis/NIR absorption spectra of (a) 13(˙+) and (c) 23(˙+) recorded over a range of different concentrations. Dependence of the intensity of the band at (b) at 900 nm for 13(˙+) and (d) 1083 nm for 23(˙+) on concentration. In both cases, the linear relationships point to the fact that absorption bands result from intramolecular radical-pairing interactions. | ||
By contrast, the MeCN solutions of 23(˙+) show (Fig. 2c) very different spectrophotometric behaviour. On treating the MeCN solutions of 26+ with an excess of Zn dust, the solution turned dark purple within a minute. In comparison with the weak absorption bands observed around 900 nm in the case of 13(˙+), the spectra of 23(˙+) display strong absorption bands centered on 1083 nm, demonstrating unequivocally the formation of a trisradical trication inclusion “complex”. This process, which is more favoured than the side-on pimerization,33 is also driven by intramolecular radical-pairing interactions. This conclusion is supported by the fact that the absorption intensities show (Fig. 2d) a linear dependency on concentration. Because of the existence of the bulky 2,6-diisopropylphenyl group at the terminus of the tail component, it follows that this interlaced pseudo[1]rotaxane-like conformation of 23(˙+) can only be accessed by an approach wherein the p-phenylene linker of the loop component experiences a revolving-door transformation, allowing the BIPY˙+ unit in the tail to enter the cavity of the loop component, forming a molecular lasso. This mechanism also explains why the self-interlacing phenomenon is not observed in the case of 13(˙+), since it is hampered from occurring by the much bulkier imide function, i.e., the door is prevented by its very size from revolving. Furthermore, in order to test the reversibility of these reduction-induced self-complexing processes, we exposed CD3CN solutions containing 13(˙+) and 23(˙+), respectively, to oxygen in the air. 1H NMR spectra were recorded during the time when their purple CD3CN solutions turned colorless. The results show (see Section 9 in the ESI†) that, after the re-oxidation, the 1H NMR spectra are almost identical to those recorded before reduction, indicating that the initial disentangled rope-like conformations of 16+ and 26+ are restored, thus confirming that these self-complexing processes can be driven reversibly by redox stimuli. It is also noteworthy that, when undergoing oxidation by the oxygen in the air, the CD3CN solution of 23(˙+) demonstrates (see Section 10 in the ESI†) a higher resistance than that of 13(˙+) to the oxidation on account of its self-interlaced lasso-like conformation, a phenomenon which is reminiscent of the fact that lasso peptides exhibit6 stability towards degradation by compounds in their vicinity.
In an attempt to elucidate the mechanism of electron transfer during the self-complexing processes of 13(˙+) and 23(˙+), cyclic voltammetry (CV) was performed. While the CV profile (Fig. 3, blue trace) of an equimolar mixture of 52+ and 34+ in MeCN indicates (see Section 4 in the ESI†) the formation of an inclusion complex, namely 5˙+ ⊂ 32(˙+), upon scanning the potential in a negative direction, this scenario is not observed (Fig. 3, red trace) in the case of 16+. The first broad peak at −434 mV, which is a combination of a two-electron, followed by a single-electron, reduction process corresponding to 16+ → 13(˙+), experiences significant cathodic shifts of 53 and 138 mV, respectively, compared with those (−381 and −296 mV) of the equimolar mixture of 52+ and 34+, demonstrating that the radical species are less stable compared with the formation of the trisradical inclusion complex, an observation which is consistent with the hypothesis that two of the BIPY˙+ units in 13(˙+) interact in a side-on fashion, giving rise to weaker radical-pairing interactions. Since 34+ has a higher reduction potential (Fig. S7†) than 52+, we believe that the ring component in 16+ first of all receives two electrons, followed by the single-electron reduction of the half-dumbbell component, resulting in all the BIPY2+ units being present in their radical cationic states. Next, the trisradical trication 13(˙+) accepts one electron at −705 mV and another two electrons at −862 mV to give the fully neutral state 1(0).
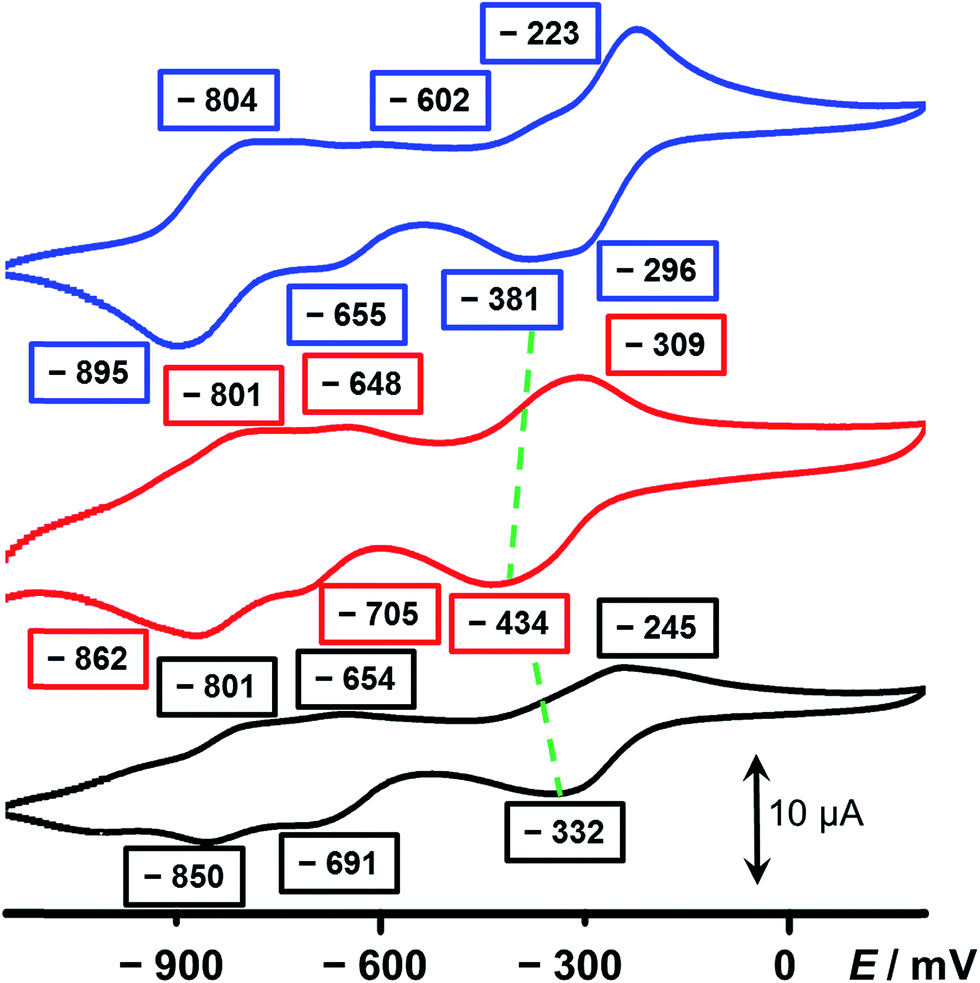 | ||
| Fig. 3 Cyclic voltammograms of (i) an equimolar mixture of the half-dumbbell 52+ and 34+ (blue), (ii) 16+ (red) and (iii) 26+ (black). The green dashed lines denote the shifts in potentials between the complex and the two components. A glassy carbon working electrode, a platinum counter electrode, and a Ag/AgCl reference electrode were used in the characterisation of the MeCN solutions (0.1 mM for each component) at 298 K with 0.1 M TBAPF6 serving as the electrolyte. A scan rate of 200 mV s−1 was used in all the analyses. | ||
The CV of 26+ also reveals three sets of reduction peaks. Two of them, which emerge at −691 and −850 mV—corresponding to the 23(˙+)/22(˙+) and the 22(˙+)/20 redox couples, respectively—have values close to those of 16+. The other reduction peak which emerges at −332 mV is also a combination of two successive reduction processes, involving 26+ → 22(˙+)2+ and 22(˙+)2+ → 23(˙+), and showing a significant anodic shift compared with that of 16+ and even the equimolar mixture. This observation can be ascribed to the fact that, once 26+ is reduced to 23(˙+), the smaller-sized linker between the loop and the tail of the rope allow the trisradical “complexation” to take place immediately, consequently stabilising the radical species. On the other hand, since this process is an intramolecular one, the close proximity between the loop and the half-dumbbell component of the rope, compared with the equimolar mixture of 52+ and 34+, facilitates the occupation by the BIPY˙+ unit inside the cavity of the loop and gives rise to an even higher potential value than that of the mixture. The numbers of electrons involved in the reduction processes have been confirmed (see Section 8 in the ESI†) by differential pulse voltammetric (DPV) experiments. In all, the conclusions drawn from the CV are in a good agreement with the observations made from the UV/Vis/NIR spectra, illustrating the fact that subtle structural differences between 16+ and 26+ lead to their having very different molecular-level properties in their radical states. While 26+ can form a self-interlaced lasso-like conformation by means of a revolving-door mechanism, a similar change in conformation is precluded by the bulkier linker in its analogue 16+, leaving an unthreaded conformation with a side-on interaction between the loop and the tail of the rope as the only alternative.
Further insight into how the structural differences affect the binding modes of 16+ under reducing conditions comes from single-crystal X-ray crystallography. Single crystals of 34+, suitable for X-ray crystallographic analysis, were obtained from slow vapor diffusion of iPr2O into an MeCN solution (1 mM) of 34+ during one week at room temperature. The solid-state structure reveals that the loop retains its rectangular shape in the presence of the imide functional group, while the alkyne-terminated tail is orientated (Fig. 4a) inwards toward the centre of the loop. The centroid-to-centroid distance between the two BIPY2+ units is (Fig. 4b) 6.93 Å, while the span between the two oxygen atoms of the imide group is 4.55 Å. It has been shown25,35,36 that the dimensions of the CBPQT4+ loop are barely affected at all by its redox states, making it reasonable to assume that these distance parameters in 34+ remain unchanged when it is reduced to 32(˙+). As a consequence, in the case of 13(˙+), when the imide-attached p-phenylene function attempts to undergo the revolving-door route to form the lasso-like pseudo[1]rotaxane, a high-energy conformation where only ca. 1.20 Å separates the oxygen atom and the plane of the BIPY2+ units in the CBPQT4+ loop is generated, disfavouring self-entanglement. In addition, a close contact (2.38 Å) between the oxygen atom on the imide and the Hα proton of the BIPY2+ unit is observed (Fig. 4c), further disfavouring the revolving-door mechanism since (i) the hydrogen bonding interactions between the oxygen atom and the Hα proton in this conformation have to be broken, and (ii) it is apparent that the imide group will be hindered sterically first of all by the Hα proton before reaching into the centre of the loop as a result of the revolving-door mechanism. Taken together, these geometric parameters observed in the solid-state structures of 34+ confirm that the lasso-like pseudo[1]rotaxane conformation in the case of 13(˙+) is not accessible.
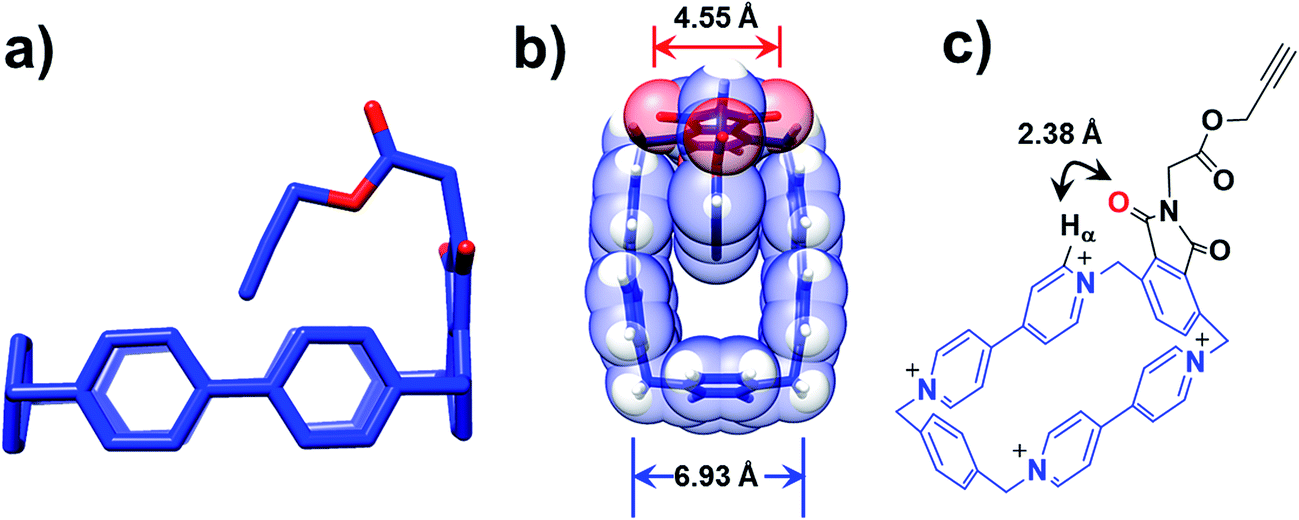 | ||
| Fig. 4 Solid-state structure of the CBPQT4+ derivative 34+ obtained by single-crystal X-ray crystallography. (a) Wireframe representation of 34+ from a side-on perspective, which shows the alkyne group pointing inwards towards the centre of the loop. (b) Wireframe and space-filing representation from a plan perspective, where the distances between (i) the two oxygen atoms of the imide group and (ii) two centroids of the BIPY2+ units are shown. (c) A short distance contact between the Hα on the BIPY2+ unit and O atom of the imide is shown. The PF6− counterions are omitted for the sake of clarity. | ||
Conclusions
In summary, two potential molecular lassos, 16+ and 26+, which respond to both chemical and electrochemical stimuli as a consequence of the recognition between the BIPY˙+ radical cations, have been prepared. Their self-complexing properties under reducing conditions, designed to mimic those of the lasso peptides, have been investigated in considerable detail. UV/Vis/NIR spectrophotometric and electrochemical studies, along with single crystal X-ray crystallography, reveal that, in the case of 13(˙+), the bulky imide group linking the loop and the half-dumbbell component of the rope only allows (Fig. 5a) the BIPY˙+ groups in the loop and the tail of the rope to “dimerise” in a side-on interacting fashion. In comparison, the smaller-size linker in 23(˙+) renders it possible to form (Fig. 5b) a mechanically interlocked lasso-like pseudo[1]rotaxane conformation, employing a revolving-door mechanism on the part of the p-phenylene ring. More importantly, this self-interlacing behaviour, driven by intramolecular trisradical tricationic interactions, is reversible upon changing the external potentials, as evidenced by both cyclic voltammetry and 1H NMR spectroscopy. In conclusion, we have made two potential molecular lassos, one of which can switch reversibly between disentangled and self-entangled states, and in so doing form a noose, reminiscent of lasso peptides. Besides, by comparing the different actuating mechanisms of these two potential viologen-based molecular lassos, we demonstrate how the molecular-level properties of analogous compounds can be affected drastically by subtle structural differences, while shedding light on the synthesis of a molecular lasso with a well-designed and finely tuned means of actuating it.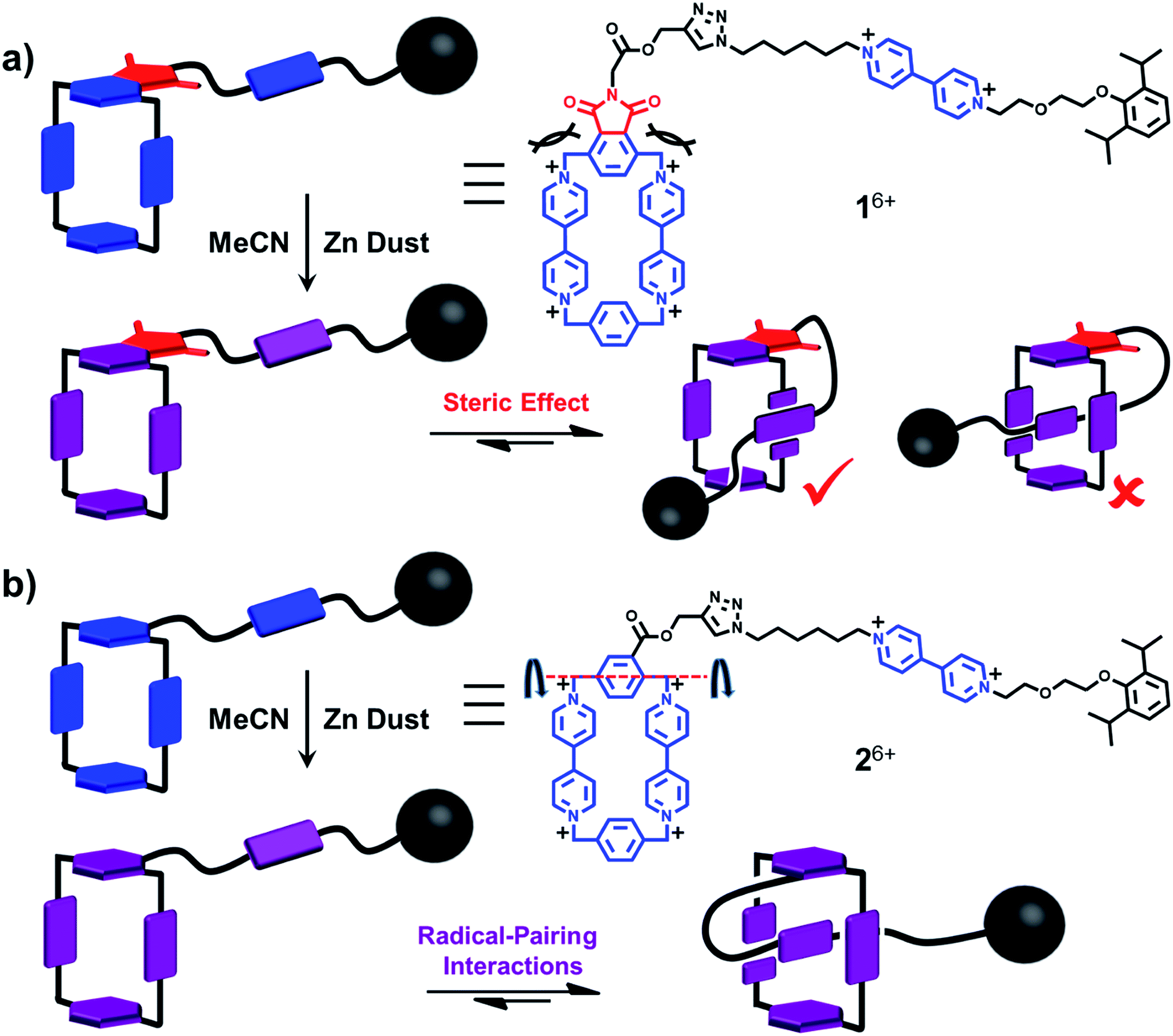 | ||
| Fig. 5 A schematic summary of the reduction-induced motions in the potential viologen-based molecular lassos 16+ and 26+. (a) Conversion of 16+ to its reduced state, 13(˙+), by treating with Zn dust, where the self-entanglement is prohibited because of the disfavoured steric repulsion and only a side-on interacting mode is allowed. (b) Conversion of 26+ to its reduced state, 23(˙+), by treating with Zn dust, which triggered a self-entanglement process to form a lasso-like pseudo[1]rotaxane with a noose-like conformation, as a result of strong intramolecular radical-pairing interactions. | ||
Acknowledgements
This research is part of the Joint Center of Excellence in Integrated Nano-Systems (JCIN) at King Abdulaziz City for Science and Technology (KACST) and Northwestern University (NU). The authors would like to thank both KACST and NU for their continued support of this research.References
- M. O. Maksimov, S. J. Pan and A. J. Link, Nat. Prod. Rep., 2012, 29, 996 RSC.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909 CrossRef CAS PubMed.
- C. Clavel, K. Fournel-Marotte and F. Coutrot, Molecules, 2013, 18, 11553 CrossRef CAS PubMed.
- J. E. Velásquez and W. van der Donk, Curr. Opin. Chem. Biol., 2011, 15, 11 CrossRef PubMed.
- K. Kuznedelov, E. Semenova, T. A. Knappe, D. Mukhamedyarov, A. Srivastava, S. Chatterjee, R. H. Ebright, M. A. Marahiel and K. Severinov, J. Mol. Biol., 2011, 412, 842 CrossRef CAS PubMed.
- I. Mathavan, S. Zirah, S. Mehmood, H. G. Choudhury, C. Goulard, Y. Li, C. V. Robinson, S. Rebuffat and K. Beis, Nat. Chem. Biol., 2014, 10, 340 CrossRef CAS PubMed.
- M. O. Maksimov and A. J. Link, J. Am. Chem. Soc., 2013, 135, 12038 CrossRef CAS PubMed.
- C. Reuter, A. Mohry, A. Sobanski and F. Vögtle, Chem.–Eur. J., 2000, 6, 1674 CAS.
- K. Hiratani, M. Kaneyama, Y. Nagawa, E. Koyama and M. Kanesato, J. Am. Chem. Soc., 2004, 126, 13568 CrossRef CAS PubMed.
- Y. Liu, A. H. Flood, R. M. Moskowitz and J. F. Stoddart, Chem.–Eur. J., 2005, 11, 369 CrossRef PubMed.
- X. Ma, D. Qu, F. Ji, Q. Wang, L. Zhu, Y. Xu and H. Tian, Chem. Commun., 2007, 1409 RSC.
- N. L. Strutt, H. Zhang, M. A. Giesener, J. Lei and J. F. Stoddart, Chem. Commun., 2012, 48, 1647 RSC.
- C. Clavel, C. Romuald, E. Brabet and F. Coutrot, Chem.–Eur. J., 2013, 19, 2982 CrossRef CAS PubMed.
- G. W. Gokel, Chem. Soc. Rev., 1992, 21, 39 RSC.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723 CrossRef CAS PubMed.
- Q. Zhou, P. Wei, Y. Zhang, Y. Yu and X. Yan, Org. Lett., 2013, 15, 5350 CrossRef CAS PubMed.
- H. Li, X. Li, H. Agren and D.-H. Qu, Org. Lett., 2014, 16, 4940 CrossRef CAS PubMed.
- P. W. C. Clavel, K. Fournel-Marotte and F. Coutrot, Chem. Sci., 2015, 6, 4828 RSC.
- A. Miyawaki, P. Kuad, Y. Takashima, H. Yamaguchi and A. Harada, J. Am. Chem. Soc., 2008, 130, 17062 CrossRef CAS PubMed.
- L. Zhu, H. Yan and Y. Zhao, Int. J. Mol. Sci., 2012, 13, 10132 CrossRef CAS PubMed.
- S. Di Motta, T. Avellini, S. Silvi, M. Venturi, X. Ma, H. Tian, A. Credi and F. Negri, Chem.–Eur. J., 2013, 19, 3131 CrossRef CAS PubMed.
- Z. Xue and M. F. Mayer, J. Am. Chem. Soc., 2010, 132, 3274 CrossRef CAS PubMed.
- Y. Liu, S. Saha, S. A. Vignon, A. H. Flood and J. F. Stoddart, Synthesis, 2005, 18, 3437 Search PubMed.
- A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Cotí, D. Benítez, E. Tkatchouk, J.-C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, Nat. Chem., 2010, 2, 42 CrossRef CAS PubMed.
- A. C. Fahrenbach, J. C. Barnes, D. A. Lanfranchi, H. Li, A. Coskun, J. J. Gassensmith, Z. Liu, D. Benítez, A. Trabolsi, W. A. Goddard III, M. Elhabiri and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 3061 CrossRef CAS PubMed.
- C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714 CrossRef CAS PubMed.
- Y. Wang, M. Frasconi, W.-G. Liu, J. Sun, Y. Wu, M. S. Nassar, Y. Y. Botros, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, ACS Cent. Sci., 2016, 2, 89 CrossRef CAS PubMed.
- J. M. Rawson, A. Alberola and A. Whalley, J. Mater. Chem., 2006, 16, 2560 RSC.
- Y. Liu, C. Chipot, X. Shao and W. Cai, J. Phys. Chem. C, 2014, 118, 19380 CAS.
- C. Romuald, A. Ardá, C. Clavel, J. Jiménez-Barbero and F. Coutrot, Chem. Sci., 2012, 3, 1851 RSC.
- C. Romuald, G. Cazals, C. Enjalbal and F. Coutrot, Org. Lett., 2013, 15, 184 CrossRef CAS PubMed.
- J.-C. Chang, S.-H. Tseng, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Nat. Chem., 2017, 9, 128 CrossRef.
- W. Geuder, S. Hünig and A. Suchy, Tetrahedron, 1986, 42, 1665 CrossRef CAS.
- T. M. Bockman and J. Kochi, J. Org. Chem., 1990, 55, 4127 CrossRef CAS.
- B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1547 CrossRef.
- M. Frasconi, I. R. Fernando, Y. Wu, Z. Liu, W.-G. Liu, S. M. Dyar, G. Barin, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 11057 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1517068. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc05035b |
| This journal is © The Royal Society of Chemistry 2017 |