
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold(III)–arene complexes by insertion of olefins into gold–aryl bonds†
Feriel
Rekhroukh
ab,
Charlie
Blons
ab,
Laura
Estévez‡
c,
Sonia
Mallet-Ladeira
d,
Karinne
Miqueu
*c,
Abderrahmane
Amgoune
 *ab and
Didier
Bourissou
*ab
*ab and
Didier
Bourissou
*ab
aUniversité de Toulouse, UPS, 118 route de Narbonne, 31062 Toulouse, France. E-mail: amgoune@chimie.ups-tlse.fr; dbouriss@chimie.ups-tlse.fr
bCNRS, LHFA, UMR 5069, F-31062 Toulouse, France
cCNRS, Univ Pau & Pays Adour, Institut des Sciences Analytiques et de Physico-Chimie pour l'environnement et les Matériaux, UMR 5254, Pau, 64000, France. E-mail: karinne.miqueu@univ-pau.fr
dUniversité Paul Sabatier, Institut de Chimie de Toulouse (FR 2599), 118 route de Narbonne, 31062 Toulouse Cedex 9, France
First published on 19th April 2017
Abstract
The synthesis and characterization of the first gold(III)–arene complexes are described. Well-defined (P,C)-cyclometalated gold(III)–aryl complexes were prepared and characterized by NMR spectroscopy. These complexes swiftly and cleanly reacted with norbornene and ethylene to provide cationic gold(III)–alkyl complexes, in which the remote phenyl ring was η2-coordinated to gold. The interaction between the aromatic ring and the gold(III) center was thoroughly analyzed by NMR spectroscopy, X-ray diffraction, and DFT calculations. The π–arene coordination was found to significantly influence the stability and reactivity of low coordinated gold(III) alkyl species.
Introduction
Transition metal π-complexes are key intermediates in many catalytic transformations involving alkenes, alkynes, allenes, and arenes1 as substrates. They represent an important class of organometallic compounds, with Zeise's salt [KPtCl3(H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)] standing as the pioneering archetype.2 Nowadays, π-complexes are well-documented with almost all transition metals and their bonding situations being well understood (cf. the Dewar–Chatt–Duncanson model). However, this general picture does not apply to gold. Gold(I) π-complexes have been known for more than four decades and have recently witnessed an upsurge of interest owing to their relevance to the catalytic functionalisation of π-substrates.3 However, in stark contrast with their Pd(II) and Pt(II) congeners, gold(III) π-complexes are extremely rare.4 To date, only a few gold(III) alkene complexes have been authenticated,5–8 following the landmark work of Bochmann and Tilset in 2013.5,6 Moreover, despite the considerable development of gold(III) chemistry over the last few years,4,9–11 other types of gold(III) π-complexes are hitherto unknown.
CH2)] standing as the pioneering archetype.2 Nowadays, π-complexes are well-documented with almost all transition metals and their bonding situations being well understood (cf. the Dewar–Chatt–Duncanson model). However, this general picture does not apply to gold. Gold(I) π-complexes have been known for more than four decades and have recently witnessed an upsurge of interest owing to their relevance to the catalytic functionalisation of π-substrates.3 However, in stark contrast with their Pd(II) and Pt(II) congeners, gold(III) π-complexes are extremely rare.4 To date, only a few gold(III) alkene complexes have been authenticated,5–8 following the landmark work of Bochmann and Tilset in 2013.5,6 Moreover, despite the considerable development of gold(III) chemistry over the last few years,4,9–11 other types of gold(III) π-complexes are hitherto unknown.
In particular, no gold(III)–arene complex has been characterized to date. This is all the more striking and unfortunate as gold(III) complexes have the remarkable ability to activate aromatic C–H bonds, and π–arene complexes likely represent the initial stage of such processes.12–14 This view is supported by a recent computational study by Swang and co-workers.15 Accordingly, benzene was predicted to coordinate in an η1-fashion to a dicationic gold(III) fragment [(bpy)AuIII(C6H5)]2+, and low activation barriers were computed for benzene rotation as well as proton jumping between the coordinated phenyl and benzene ligands. However, attempts to synthesize relevant gold(III) complexes were unsuccessful.15 While gold(III)–arene complexes remain unprecedented, it must be noted that a few gold(I) complexes featuring intra and intermolecular Au(I)–arene interactions have been structurally characterized.16–19 In these complexes, the arene acts as a weakly coordinating ligand and it can be easily displaced by substrates such as alkynes.
The key bottlenecks in the quest for gold(III)–arene complexes lie in the design and access of low coordinated cationic gold(III) complexes. We have recently developed a simple synthetic route to cyclometalated [(P,C)Au(III)–Me]+ complexes and have shown their ability to readily insert olefins into the gold(III)–carbon bond.8,20 The resulting low coordinated gold(III) alkyl complexes showed a high tendency for β-hydride elimination21 and enabled the first authentication of a C–H agostic interaction with gold.22 These results motivated us to explore the synthesis of gold(III)–arene complexes by olefin insertion into a gold(III)–aryl bond (Chart 1).
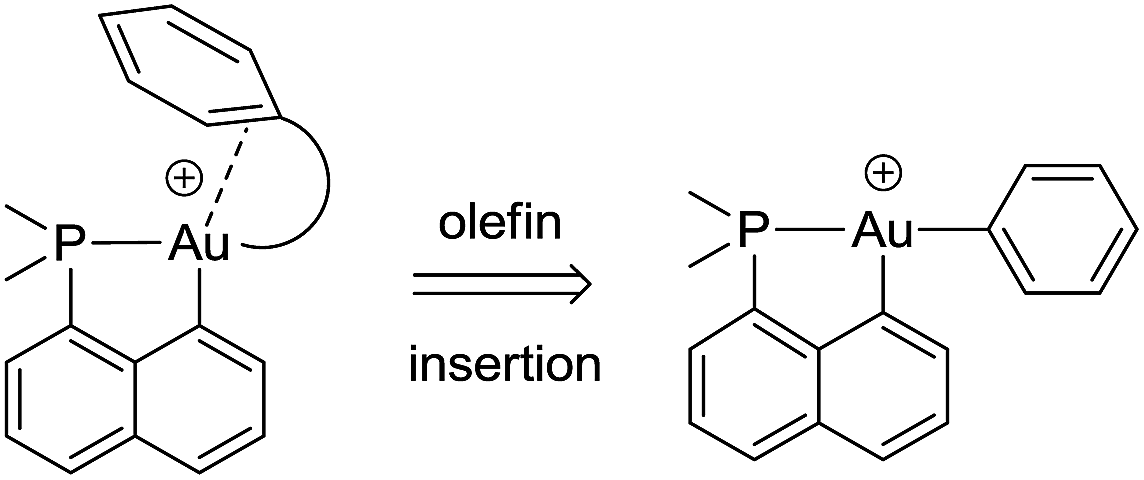 | ||
| Chart 1 Schematic representation of the targeted gold(III)–arene complexes and their generation by olefin insertion into a Au–aryl bond. | ||
As reported hereafter, this strategy proved fruitful and empowered us to characterize gold(III)–arene complexes. Migratory insertion of norbornene and ethylene into the Au(III)–Ph bonds is shown to be a very facile process that leads to gold(III) alkyl complexes stabilized by intramolecular coordination of the phenyl ring. The mechanism of formation of the gold(III)–arene complexes and their bonding situation have been thoroughly investigated spectroscopically, crystallographically, and computationally. The influence of arene coordination on the fate of low coordinated gold(III) alkyl species is also discussed.
Results and discussion
Synthesis and characterization of the cationic (P,C) gold(III)–phenyl complex 3
The cationic [(P,C)Au(III)Ph]+ complex 3 was prepared in two steps (Scheme 1) from the [(P,C)AuI2] precursor 1 resulting from chelation-assisted oxidative addition to gold.23 First, treatment of 1 with PhMgBr (2.2 equiv.) enabled selective monoarylation, and complex 2 was isolated as a pale yellow powder (67% yield) after flash chromatography. Its molecular structure was ascertained by multinuclear NMR spectroscopy, high-resolution mass spectrometry (HRMS), elemental analysis, and X-ray diffraction.24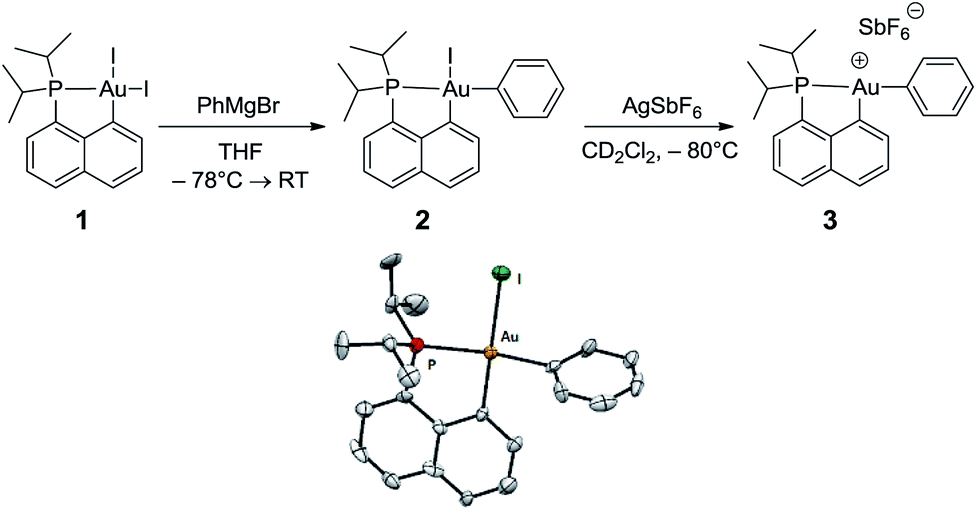 | ||
| Scheme 1 Preparation of the cationic gold(III)–phenyl complex 3 and molecular view of its neutral precursor 2 with thermal ellipsoids drawn at the 50% probability level. | ||
The iodide at the gold was then abstracted by adding complex 2 to AgSbF6 in dichloromethane. According to 31P NMR monitoring, the reaction was instantaneous at −80 °C and afforded a mixture of two cationic species in a 70/30 ratio (δ31P: 77.4 and 80.8 ppm, respectively). The 1H and 13C NMR spectra also display two sets of similar signals, suggesting that the two species correspond to closely related forms of complex 3.25 Variable-temperature 31P NMR studies indicated that the ratio between the two forms does not vary significantly from −80 to −20 °C (decomposition into several unidentified species occurs above −20 °C). However, the existence of a dynamic exchange between the two species was clearly apparent from the cross-peaks observed in the 2D 31P{1H} EXSY NMR spectrum. The 13C NMR signal for the phenyl ipso carbon atom Cipso bound to gold is deshielded by about 10 ppm upon cationization (δ from 168.4 ppm for 2 to 177.2 ppm for the major form of 3). The large 2JPC coupling constant is retained (134 Hz for 2, 120 Hz for 3), indicating that the phenyl ring still occupies the position trans to the phosphine moiety, in line with the dissymmetric trans influence of the (P,C) chelate. The reaction of complex 3 with olefins was then investigated by carrying out the iodide abstraction of 2 in the presence of norbornene or ethylene.
Reaction of the cationic (P,C) gold(III)–phenyl complex 3 with norbornene
First, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of complex 2 and norbornene (NB) was treated with AgSbF6 at −80 °C and the reaction mixture was warmed to room temperature. The 31P NMR spectrum showed the immediate formation of a single compound at δ 73.6 ppm. The 1H NMR spectrum revealed the complete consumption of NB (the corresponding olefin signal disappeared) and the presence of new aliphatic signals in line with the formation of the gold(III) norbornyl complex 4. Mono-insertion of NB into the Au–Ph bond of 3 was apparent from the integrals of the respective 1H NMR signals and was unambiguously confirmed by HRMS analysis. The cationic species 4 is too sensitive to be isolated in pure form, but NMR monitoring indicates that it is stable at room temperature (decomposition is observed after 48 h in solution at RT). The behavior of the gold(III) phenyl complex 3 towards NB markedly differs from that we reported previously for the corresponding Au(III) methyl derivative.8,20 In the latter case, double insertion of NB was observed, and the resulting product decomposed rapidly at low temperature despite its stabilization by γ-CH agostic interaction.22 To gain more insight into the stability of 4 and assess the possible π-coordination of the phenyl ring to gold, we assigned all the 1H and 13C NMR signals of 4via 2D HSQC and HMBC experiments.24 It is important to note that NMR authentication of such π-interactions is per se challenging due to their weak and fluxional character. Accordingly, no diagnostic NMR data have been reported for the few characterized gold(I)–arene complexes. High fluxionality has also been noticed upon NMR characterization of Au(III)–olefin.5–8 In the case of complex 4, it is considered that the pendant phenyl ring is ideally positioned to occupy the vacant coordination site at the gold, with the norbornyl spacer inducing some rigidity (the gold atom and phenyl group are located on the exo face and are mutually cis). The 1H and 13C NMR spectra of 4 display broad resonance signals for the phenyl ring at room temperature, indicating some dynamic behavior. However, lowering the temperature to −20 °C resulted in well-resolved 1H and 13C NMR spectra.
:1 mixture of complex 2 and norbornene (NB) was treated with AgSbF6 at −80 °C and the reaction mixture was warmed to room temperature. The 31P NMR spectrum showed the immediate formation of a single compound at δ 73.6 ppm. The 1H NMR spectrum revealed the complete consumption of NB (the corresponding olefin signal disappeared) and the presence of new aliphatic signals in line with the formation of the gold(III) norbornyl complex 4. Mono-insertion of NB into the Au–Ph bond of 3 was apparent from the integrals of the respective 1H NMR signals and was unambiguously confirmed by HRMS analysis. The cationic species 4 is too sensitive to be isolated in pure form, but NMR monitoring indicates that it is stable at room temperature (decomposition is observed after 48 h in solution at RT). The behavior of the gold(III) phenyl complex 3 towards NB markedly differs from that we reported previously for the corresponding Au(III) methyl derivative.8,20 In the latter case, double insertion of NB was observed, and the resulting product decomposed rapidly at low temperature despite its stabilization by γ-CH agostic interaction.22 To gain more insight into the stability of 4 and assess the possible π-coordination of the phenyl ring to gold, we assigned all the 1H and 13C NMR signals of 4via 2D HSQC and HMBC experiments.24 It is important to note that NMR authentication of such π-interactions is per se challenging due to their weak and fluxional character. Accordingly, no diagnostic NMR data have been reported for the few characterized gold(I)–arene complexes. High fluxionality has also been noticed upon NMR characterization of Au(III)–olefin.5–8 In the case of complex 4, it is considered that the pendant phenyl ring is ideally positioned to occupy the vacant coordination site at the gold, with the norbornyl spacer inducing some rigidity (the gold atom and phenyl group are located on the exo face and are mutually cis). The 1H and 13C NMR spectra of 4 display broad resonance signals for the phenyl ring at room temperature, indicating some dynamic behavior. However, lowering the temperature to −20 °C resulted in well-resolved 1H and 13C NMR spectra.
The CH of the norbornyl moiety bound to gold resonates in 13C NMR as a doublet at 57.0 ppm with a large 2JPC coupling constant of 72.5 Hz, indicating a trans arrangement of the phosphorus atom and norbornyl group (Fig. 1a). In addition, six 13C NMR signals are found for the phenyl ring, suggesting hindered rotation around the Csp3–Csp2 bond and possibly some interaction between the phenyl ring and the gold center. The chemical shift of the ipso carbon is shifted to high field compared to that of 2-phenylbicycloheptane (PBH)26 (δ 111.8 ppm for 4vs. 147.6 ppm for PBH) and appears as a doublet due to coupling with phosphorus (JPC = 9.1 Hz). The presence of the gold(III) center also affects the ortho-CH signals, one of them being shifted to high field by about 10 ppm (δ 124.1 and 135.6 ppm). These spectroscopic data are strongly reminiscent of those observed by Cheng and Catellani for related neutral Pd(II)–arene complexes27–29 and suggest η2-coordination of the phenyl ring to gold(III). Some of these Pd(II)–arene complexes have been crystallographically characterized and their bonding situation has been thoroughly investigated.27–29
 | ||
| Fig. 1 Complexes 4 and 4′: (a) diagnostic NMR data determined experimentally at −20 °C, (b) molecular structure of 4′ in the solid state with ellipsoids set at 50% probability and hydrogen atoms expected on Cortho omitted for clarity, (c) superposition of the donor π(CipsoCortho) and acceptor σ*(AuCnaphthyl) NBOs accounting for the Au(III)–arene π-interaction in 4. | ||
With the aim of obtaining structural confirmation of the gold(III)–arene interaction, we prepared another gold(III)–aryl complex 2′ with an OMe group in the para position of the phenyl ring (Scheme 2). This electron-rich arene ring was envisioned to impart greater stability to the gold(III) π-complex and hopefully higher crystallinity. Starting from 2′, the insertion of norbornene also works well. The ensuing gold(III)–arene complex 4′ was isolated and fully characterized. To our delight, crystals suitable for X-ray diffraction analysis were obtained in this case. Accordingly, complex 4′ (Fig. 1b) adopts a 3-coordinated T-shaped structure (PAuCNB and PAuCnaphthyl bond angles of 177.29(7)° and 84.15(7)°, respectively). As expected, the insertion of norbornene into the Au–Ph bond places the gold atom and the phenyl ring cis and exo to the norbornyl group. The arene sits trans to the naphthyl backbone with short contacts between gold and two carbon atoms (Cipso and one Cortho). The Au–Cipso distance (2.416(2) Å) is shorter than the Au–Cortho distance (2.593(3) Å), indicating dissymmetric η2-coordination of the arene to gold(III). The interaction of the (p-OMe)-phenyl ring with the gold(III) center is also apparent from NMR (Fig. 1a). As for 4, the 13C signals for Cipso (δ 90.3 ppm) and one of the Cortho atoms (δ 115.7 ppm) of 4′ are shifted to high field compared to those of 2-(p-OMe)-phenylbicycloheptane26 (Δδ13C = 49 ppm for Cipso and 13 ppm for Cortho).30 Interestingly, the NMR signals for the (p-OMe)-phenyl ring are desymmetrized even at room temperature, suggesting tighter arene coordination in 4′vs.4.
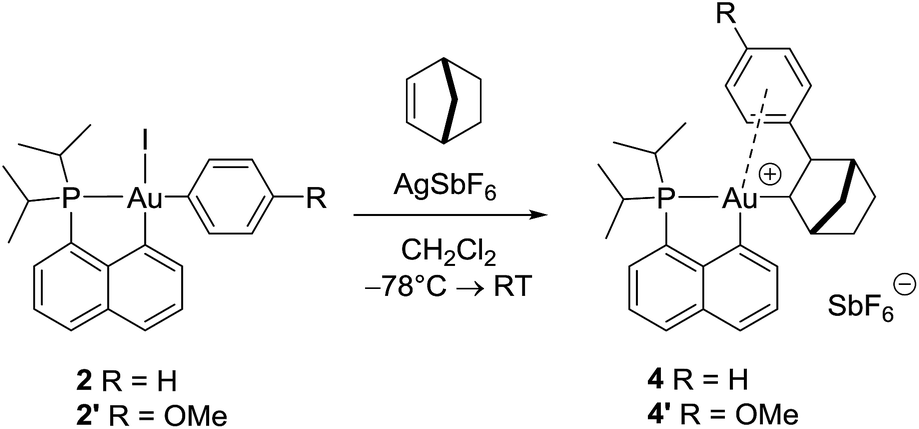 | ||
| Scheme 2 Formation of the gold(III) norbornyl complexes 4 and 4′ by insertion of norbornene into the Au(III)–Ar bond of 2 and 2′. | ||
DFT study of complex 4: structure and mechanism of formation
DFT calculations were performed at the B3PW91/SDD+f(Au),6-31G**(other atoms) level of theory. Two isomeric forms of complex 4 were located on the potential energy surface, corresponding to cis and trans arrangements of the two carbon atoms, Cnorbornyl and Cnaphthyl, at the gold (cis-4 and trans-4, respectively). In line with experimental observations, complex cis-4 was calculated to be thermodynamically more stable than trans-4 by 13.2 kcal mol−1.24 To confirm the experimental assignments, the 13C NMR data for complex cis-4 were computed using the GIAO method and the IGLO-II basis set (P, C, and H atoms).24 The coupling constant between the phosphorus and Cnorbornyl atoms (2JPC = 75.9 Hz) nicely matches that observed experimentally (72.5 Hz). Moreover, the Cipso and one of the Cortho atoms are significantly shielded compared to the other signals (Table S1†), in line with η2-coordination to gold.24 The optimized geometry of cis-4 (Fig. S26†) very much resembles that determined crystallographically for 4′. The gold center is 3-coordinated and adopts T-shaped geometry (PAuCNB and PAuCnaphthyl bond angles of 171.7° and 82.9°, respectively). The arene occupies the fourth coordination site of the gold(III) center and is η2-coordinated. Short contacts are observed with the Cipso and one of the Cortho atoms (2.62 and 2.63 Å, respectively). The coordination of the aryl ring to gold was further confirmed by Natural Bond Orbital (NBO) analysis. At the second-order perturbation level, donation from the π(CipsoCortho) orbital to the σ*(AuCnaphthyl) orbital was found with a delocalization energy ΔE(2) of 30.7 kcal mol−1 (Fig. 1c). This indicates the presence of a rather strong donor–acceptor interaction compared with those encountered in related [(P,C)Au(III)R]+/norbornene adducts [ΔE(2) of 9–18 kcal mol−1].8,24 Similar results were obtained for the p-OMe substituted complex 4′. Its optimized structure is consistent with that determined crystallographically, with a slightly more dissymmetric η2-coordination of the arene (CipsoAu and CorthoAu distances of 2.44 and 2.80 Å, respectively). In NBO, the delocalization energy ΔE(2) for the π(CipsoCortho) → σ*(AuCnaphthyl) interaction (36.6 kcal mol−1) is slightly higher than for 4, in line with a strengthening of the π interaction.24The reaction profile accounting for the formation of complex 4 from 3 was also computed. This involves the coordination and migratory insertion of norbornene into the Au–Ph bond, followed by cis/trans isomerization (Fig. 2). The insertion of NB proceeds via an in-plane 4-center transition state and gives the gold(III)–norbornyl complex trans-4. The associated activation barrier is low (ΔG≠ = 10.7 kcal mol−1), much lower than that computed for the reaction of NB with the corresponding Au(III)–Me complex (ΔG≠ = 18.7 kcal mol−1).31 Complex trans-4 is not observed experimentally. It spontaneously isomerizes (with a low barrier ΔG≠ of 14.3 kcal mol−1) into the more thermodynamically stable complex cis-4. The Gibbs free energy ΔG for the overall transformation cis-3 → cis-4 is −19.4 kcal mol−1.
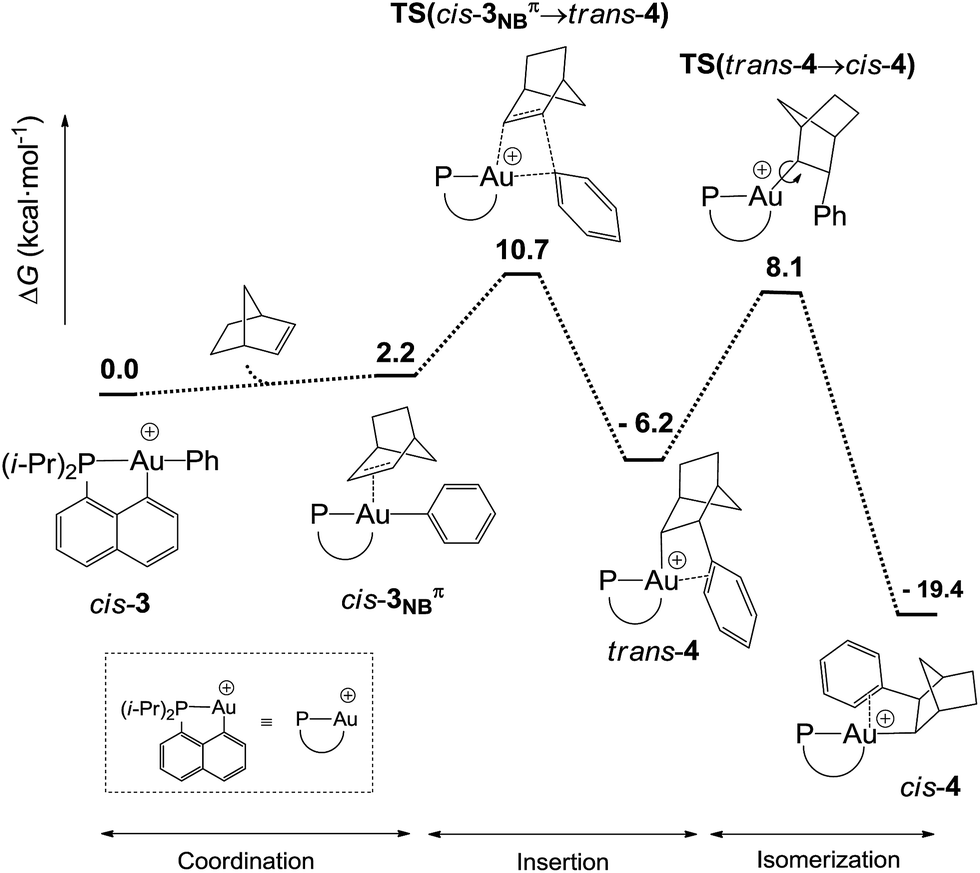 | ||
| Fig. 2 Reaction profile (ΔG in kcal mol−1) computed at the B3PW91/SDD+f(Au)/6-31G**(other atoms) level of theory for the formation of the gold(III)–arene complex 4 upon reaction of cis-3 with NB. | ||
Formation of the neutral cyclometalated complex 5: base-promoted C–H activation of 4
Given the propensity of electrophilic Au(III) complexes to activate aromatic C–H bonds, we were intrigued by the possibility of promoting the auration of the π-coordinated arene ring of 4. Heating a DCM solution at 45 °C led only to decomposition products, but in the presence of a base such as pyridine or a proton sponge, C–H activation proceeded rapidly (Scheme 3). The resulting cyclometalated complex 5 was isolated in 95% yield and fully characterized.24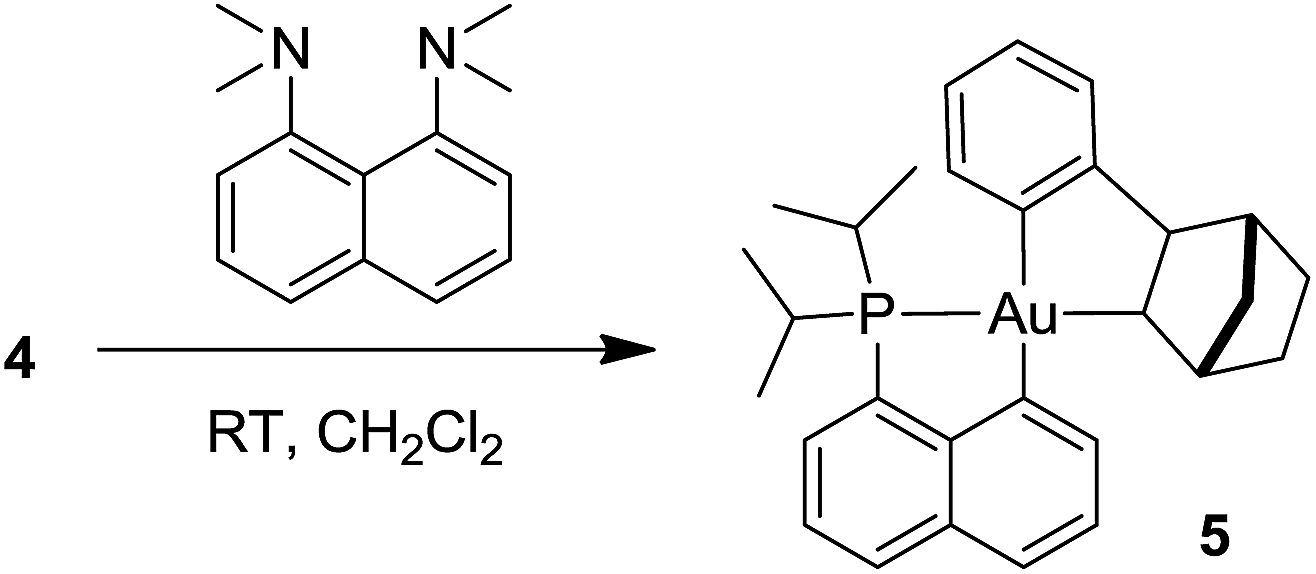 | ||
| Scheme 3 Cyclometallation of the Au(III)–arene complex 4 leading to 5. | ||
The insertion of NB into the Au–Ph bond of 3 is a very facile process. It affords a straightforward entry to the cationic gold(III) norbornyl complex 4 and to the neutral cyclometalated complex 5 following C–H activation. The η2-coordination of the phenyl ring to gold has been unambiguously authenticated in 4. This intramolecular gold(III)–arene interaction provides significant thermal stability to complex 4. Indeed, cationic gold(III) alkyl complexes tend to rapidly undergo reductive elimination or β-hydride elimination even at low temperature and only a few such species have been characterized so far.21,22 These observations prompted us to explore the reactivity of the cationic phenyl complex 3 towards ethylene and to thereby probe the impact of π–arene interactions on the reactivity of gold(III) alkyl species.
Reaction of the cationic (P,C)gold(III)–phenyl complex 3 with ethylene
Remarkably, complex 3 reacts swiftly with ethylene (1 bar) at low temperature whereas insertion of ethylene required 4 hours at 50 °C and 2 bars for the related gold(III) methyl complex.21 Moreover, the reaction of the [(P,C)AuMe]+ species with ethylene directly afforded higher olefins (propylene and butenes) and a cationic 2-coordinated bis(phosphine) gold(I) complex (δ31P 60 ppm), while complex 3 is instantaneously converted into a single gold(III) species 6, as indicated by 31P NMR spectroscopy (δ31P 95.9 ppm). The new compound 6 was unequivocally characterized by multinuclear NMR spectroscopy. In particular, 2D 1H–13C NMR data clearly indicated the presence of the sec-alkyl group –CH(CH3)Ph at the gold (Scheme 4).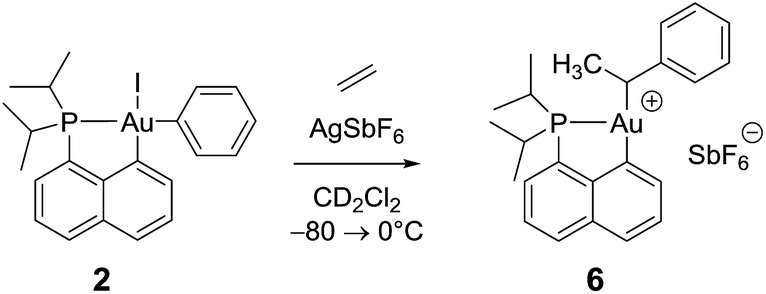 | ||
| Scheme 4 Formation of the gold(III) alkyl complex 6 upon reaction of the cationic (P,C)gold(III) phenyl complex with ethylene. | ||
The methine group at the gold resonates in 13C NMR at δ 68.6 ppm with a small 2JPC coupling constant of 7.7 Hz, indicating that the sec-alkyl moiety sits in this case in a trans position to the Cnaphthyl atom. The 13C NMR spectrum also revealed a significant shift to high field of the Cipso and one of the Cortho carbon atoms of the phenyl ring compared to those of ethylbenzene (Δδ13C = 20 ppm for Cipso and 16 ppm for Cortho, Fig. 3a).32 All the carbon atoms of the phenyl ring except for one of the Cmeta couple with phosphorus (4.4 < JPC < 10.3 Hz), and one of the Hortho resonance signal also display coupling to phosphorus (JPH = 3.4 Hz). All these data support η2-coordination of the phenyl ring to gold(III), as in the case of the gold(III) norbornyl complex 4.
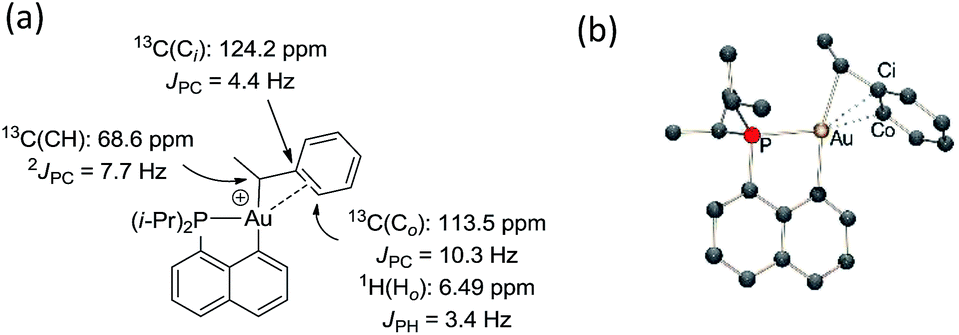 | ||
| Fig. 3 Complex 6: (a) diagnostic NMR data determined experimentally at 0 °C and (b) DFT-optimized structure of trans-6 calculated at the B3PW91/SDD+f(Au)/6-31G**(other atoms) level of theory: Au–Cipso 2.28 Å and Au–Cortho 2.55 Å. | ||
Complex 6 shows unusual thermal stability (no degradation is observed after 24 h at 0 °C) given the presence of hydrogen atoms in a β position and the proclivity of cationic 3-coordinated gold(III) alkyl complexes to undergo β-H elimination.21 Indeed, the corresponding n-propyl and n-butyl complexes were previously found to rapidly release propene and butenes at 0 °C. A similar process with the formation of styrene is observed with 6 but only after 1 h at room temperature. The higher stability of complex 6 is most likely due to the π-interaction of the phenyl ring with the gold(III) center.
The structure of complex 6 was further assessed by DFT and NBO calculations. Accordingly, complex 6 adopts a slightly distorted T-shaped geometry (Fig. 3b and Table S3†) with PAuC(H) and CnaphthylAuC(H) bond angles of 109.4 and 166.3°, respectively. The pendant phenyl ring is significantly bent towards the cationic [(P,C)Au]+ fragment [AuC(H)Cipso bond angle = 74.2°] and interacts with the gold center via the Cipso and Cortho carbons in an asymmetrical manner (Au–Cipso: 2.28 Å, Au–Cortho: 2.55 Å). The η2-coordination of the phenyl ring to gold(III) is corroborated by the 13C NMR data computed for the corresponding Cipso and Cortho carbon atoms (Table S3†). The respective resonance signals are shifted to high field (δ 124.6 and 120.6 ppm for Cipso and Cortho, respectively) and display JPC coupling constants of 5 and 10.7 Hz, respectively. NBO analysis (Fig. S31†) identified a donation from the π(CipsoCortho) orbital to the Au σ*(AuCnaphthyl) orbital with a delocalization energy ΔE(2) of 38.6 kcal mol−1, a value very close to that found in the gold(III) norbornyl complex 4. It is interesting to note that complex 6 is characterized experimentally in its trans form (the Cnaphthyl and Calkyl atoms are in a trans arrangement) and does not isomerize into the more thermodynamically stable cis-isomer (ΔG = −5.2 kcal mol−1 and ΔG≠ = 25 kcal mol−1, Fig. S30†).
To gain a deeper insight into the mechanism of formation of complex 6, the reaction of the cationic complex 3 with ethylene was thoroughly investigated by DFT calculations (Fig. 4 and S32†). Ethylene first coordinates to gold to give complex cis-3π (ΔG = 1.5 kcal mol−1) and then inserts into the Au–Ph bond to afford the Au(CH2CH2Ph) complex trans-A. The activation barrier for the migratory insertion is low (ΔG≠ = 9.5 kcal mol−1 from the initial reactants). In line with experimental observations, insertion into the Au–Ph bond is kinetically more facile than into the Au–Me bond (the corresponding activation barrier reaches 16.5 kcal mol−1). Complex trans-A displays some π–arene interaction [according to NBO, ΔE(2) = 10.5 kcal mol−1] (Fig. S33†) but coordination of the pendant phenyl ring to gold(III) is less stabilizing with the flexible CH2CH2 spacer than that encountered in complex 6. In line with the fact that trans-A is not observed experimentally, it easily undergoes β-hydride elimination (ΔG≠ = 4.8 kcal mol−1).33 The reaction is slightly exergonic and leads to intermediate cis-H. Finally, the Au(III)–H styrene complex readily undergoes (2,1) re-insertion of styrene into the Au–H bond to give complex trans-6 with an activation barrier of ΔG≠ = 8.0 kcal mol−1 from cis-H to trans-6 (styrene dissociation is comparatively disfavored with ΔG≠ = 14.5 kcal mol−1).34,35 As mentioned above, η2-coordination of the phenyl ring substantially stabilizes complex trans-6 (ΔG = −22.9 kcal mol−1), and the latter species is actually the thermodynamic product of ethylene insertion into the Au–Ph bond, enabling its experimental characterization. Note that in accordance with experimental observations, the arene coordination disfavors β-hydride elimination from complex trans-6 (Fig. S34†).24 The associated energy barrier ΔG≠ amounts to 16.5 kcal mol−1vs. 7.1 kcal mol−1 for the corresponding [(P,C)Au(III)(n-Bu)]+ complex.21 The arene coordination also disfavors isomerization of trans-6 into its more thermodynamically stable cis isomer (Fig. S30†). The latter process requires a high activation energy (ΔG≠ = 25 kcal mol−1) and is even less favored than β-hydride elimination.
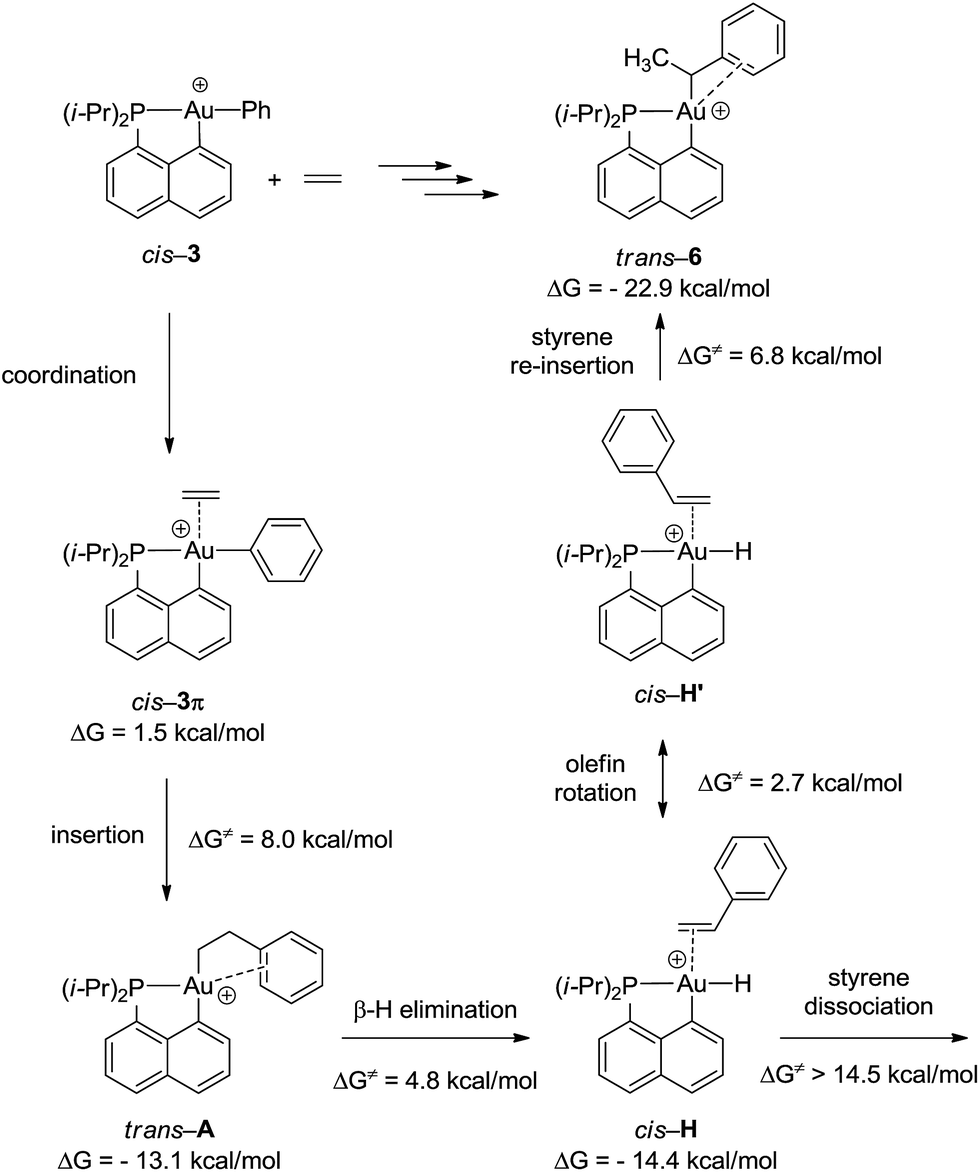 | ||
| Fig. 4 Reaction sequence computed at the B3PW91/SDD+f(Au)/6-31G**(other atoms) level of theory for the formation of the gold(III)–arene complex 6 (ΔG values set with the initial reactants cis-3 + ethylene as origin, while ΔG≠ values correspond to the activation barriers of each step). | ||
Conclusions
A suitable synthetic strategy based on olefin insertion into a Au(III)–Ph bond allowed us to experimentally characterize gold(III)–arene complexes for the first time. The η2-coordination of the remote phenyl ring to gold has been substantiated spectroscopically, crystallographically, and computationally. The isolation and characterization of such intermediates provides interesting information on the interaction between arenes and gold(III) species in the context of gold-catalyzed functionalization of aromatic C–H bonds.12,14This study also shows that migratory insertion of olefins such as norbornene and ethylene into the Au(III)–Ph bond is a very facile process. This transformation and the ensuing intermediates, which are stabilized intramolecularly by π–arene coordination, are relevant to a number of key catalytic processes (such as Mizoroki–Heck coupling, the Catellani reaction, and the hydroarylation of olefins).
Future work from our group will seek to take advantage of the increasing scope of gold(III) species and reactivities to develop valuable catalytic transformations.
Acknowledgements
Financial support from the Centre National de la Recherche Scientifique, the Université de Toulouse, the Agence Nationale de la Recherche (ANR-JCJC-2012-POGO) and IFP Energies nouvelles is gratefully acknowledged. Umicore AG & Co. is acknowledged for a generous gift of gold precursors. Université de Pau et des Pays de l'Adour, MCIA (Mésocentre de Calcul Intensif Aquitain), and IDRIS under Allocation 2016 (i2016080045) made by Grand Equipement National de Calcul Intensif (GENCI) are also acknowledged for computational facilities. L. Estevez thanks Xunta de Galicia for financial support through the I2C program.Notes and references
- (a) E. P. Kündig, Top. Organomet. Chem., 2004, 7 Search PubMed; (b) G. A. Solan, Organomet. Chem., 2005, 32, 314 CAS.
- (a) W. C. Zeise, Poggendorff's Ann. Phys., 1827, 9, 632 Search PubMed; (b) W. C. Zeise, Poggendorff's Ann. Phys., 1831, 21, 497 CrossRef.
- R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714 CrossRef CAS PubMed.
- D. A. Rosca, J. A. Wright and M. Bochmann, Dalton Trans., 2015, 44, 20785 RSC.
- N. Savjani, D. Rosca, M. Schormann and M. Bochmann, Angew. Chem., Int. Ed., 2013, 52, 874 CrossRef CAS PubMed.
- E. Langseth, M. L. Scheuermann, D. Balcells, W. Kaminsky, K. I. Goldberg, O. Eisenstein, R. H. Heyn and M. Tilset, Angew. Chem., Int. Ed., 2013, 52, 1660 CrossRef CAS PubMed.
- D. Balcells, O. Eisenstein, M. Tilset and A. Nova, Dalton Trans., 2016, 45, 5504 RSC.
- F. Rekhroukh, L. Estevez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Organometallics, 2016, 35, 995 CrossRef CAS.
- H. Schmidbaur and A. Schier, Arabian J. Sci. Eng., 2012, 37, 1187 CrossRef CAS.
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022 CrossRef CAS PubMed.
- R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994 CrossRef CAS PubMed.
- For recent reviews, see: (a) T. C. Boorman and I. Larrosa, Chem. Soc. Rev., 2011, 40, 1910 RSC; (b) T. de Haro and C. Nevado, Synthesis, 2011, 2530 CAS; (c) M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem.–Eur. J., 2011, 17, 8248 CrossRef CAS PubMed.
- For selected examples of stoichiometric gold-mediated C–H activation of arenes, see: (a) M. S. Kharasch and H. S. Isbell, J. Am. Chem. Soc., 1931, 53, 3053 CrossRef CAS; (b) D. A. Rosca, D. A. Smith and M. Bochmann, Chem. Commun., 2012, 48, 7247 RSC; (c) X. C. Cambeiro, T. C. Boorman, P. Lu and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 1781 CrossRef CAS PubMed; (d) M. Hofer and C. Nevado, Tetrahedron, 2013, 69, 5751 CrossRef CAS.
- For selected examples of gold-catalyzed C–H activation of arenes, see: (a) T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512 CrossRef CAS PubMed; (b) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed; (c) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, J. Am. Chem. Soc., 2014, 136, 254 CrossRef CAS PubMed; (d) X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636 CrossRef CAS PubMed; (e) A. J. Cresswell and G. C. Lloyd-Jones, Chem.–Eur. J., 2016, 22, 12641 CrossRef CAS PubMed; (f) T. J. A. Corrie, L. T. Ball, C. A. Russell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 245 CrossRef CAS PubMed.
- M. K. Ghosh, M. Tilset, A. Venugopal, R. H. Heyn and O. Swang, J. Phys. Chem. A, 2010, 114, 8135 CrossRef CAS PubMed.
- (a) F.-B. Xu, Q.-S. Li, L. Z. Wu, X.-B. Leng, Z.-C. Li, X.-S. Zeng, Y. L. Chow and Z.-Z. Zhang, Organometallics, 2003, 22, 633 CrossRef CAS; (b) Q.-S. Li, C.-Q. Wan, R.-Y. Zou, F.-B. Xu, H.-B. Song, X.-J. Wan and Z.-Z. Zhang, Inorg. Chem., 2006, 45, 1888 CrossRef CAS PubMed; (c) Q. L. Ni, X. F. Jiang, T. H. Huang, X. J. Wang, L. C. Gui and K. G. Yang, Organometallics, 2012, 31, 2343 CrossRef CAS.
- (a) E. Herrero-Gomez, C. Nieto-Oberhuber, S. Lopez, J. Benet-Buchholz and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 5455 CrossRef CAS PubMed; (b) P. Pérez-Galán, N. Delpont, E. Herrero-Gomez, F. Maseras and A. M. Echavarren, Chem.–Eur. J., 2010, 16, 5324 CrossRef PubMed; (c) D. V. Partyka, T. J. Robilotto, M. Zeller, A. D. Hunter and T. B. Gray, Organometallics, 2008, 27, 28 CrossRef CAS.
- V. Lavallo, G. D. Frey, S. Kousar, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13569 CrossRef CAS PubMed.
- Weak intermolecular Au⋯π interactions (3.5 < d < 4 Å) have also been reported to support supramolecular aggregation in the crystalline state: (a) I. Caracelli, J. Zukerman-Schpector and E. R. T. Tiekink, Gold Bull., 2013, 46, 81 CrossRef CAS; (b) E. R. T. Tiekink and J. Zukerman-Schpector, CrystEngComm, 2009, 11, 1176 RSC.
- F. Rekhroukh, R. Brousses, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 1266 CrossRef CAS PubMed.
- F. Rekhroukh, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 11920 CrossRef CAS PubMed.
- F. Rekhroukh, L. Estévez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2016, 55, 3414 CrossRef CAS PubMed.
- J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778 CrossRef CAS PubMed.
- See ESI for details.†.
- It was not possible at this stage to identify the precise structures of the two forms of complex 3. Coordination of the solvent or counteranion to gold may be involved. Aggregation involving intermolecular π–arene coordination is also conceivable.
- Y. Yamamoto and K. Itonaga, Chem.–Eur. J., 2008, 14, 10705 CrossRef CAS PubMed.
- (a) C. S. Li, C. H. Cheng, F. L. Liao and S. L. Wang, J. Chem. Soc., Chem. Commun., 1991, 710 RSC; (b) C.-S. Li, D.-C. Jou and C.-H. Cheng, Organometallics, 1993, 12, 3945 CrossRef CAS; (c) C.-H. Liu, C.-S. Li and C.-H. Cheng, Organometallics, 1994, 13, 18 CrossRef CAS.
- M. Catellani, C. Mealli, E. Motti, P. Paoli, E. Perez-Carreño and P. S. Pregosin, J. Am. Chem. Soc., 2002, 124, 4336 CrossRef CAS PubMed.
- D. I. Chai, P. Thansandote and M. Lautens, Chem.–Eur. J., 2011, 17, 8175 CrossRef CAS PubMed.
- Upon treatment with N(n-Bu)4Cl, the arene coordination is displaced and the neutral tetracoordinate complex 4′-Cl was isolated. The 13C NMR signals for Cipso and Co/Co′ atoms are no longer high-field shifted and appear at 141.8 and 129.8 ppm, respectively.24.
- The trend is very similar to that reported for ethylene insertion into Pd(II)–Ph vs. Pd(II)–Me bonds: H. von Schenck, S. Strömberg, K. Zetterberg, M. Ludwig, B. Akermark and M. Svensson, Organometallics, 2001, 20, 2813 CrossRef CAS.
- All the carbon atoms of the phenyl ring also seem to be affected by the presence of the gold center, albeit to a much lower extent. See ESI for details.†.
- Isomerization of trans-A into the most stable cis-A isomer involves a much higher activation barrier (ΔG≠ = 15.8 kcal mol−1). See ESI for details.†.
- An insertion/β-H elimination/re-insertion sequence has already been proposed to account for the formation of internal butenes from the [(P,C)Au(III)(n-Bu)]+ complex.21.
- For recent discussions on olefin rotation within gold(III) complexes, see ref. 7 and 21.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details (syntheses, analytical data and copies of 1H, 13C and 31P NMR spectra), crystallographic data of 2 and computational details. CCDC 1526613 and 1538015. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc00145b |
| ‡ Current address: Departamento de Química Física, Universidade de Vigo, Facultade de Química Lagoas-Marcosende s/n, 36310 Vigo, Galicia, Spain. |
| This journal is © The Royal Society of Chemistry 2017 |