
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning brighter near-infrared fluorescent proteins: insights from structural and biochemical studies†
Mikhail
Baloban‡
a,
Daria M.
Shcherbakova‡
a,
Sergei
Pletnev‡
b,
Vladimir Z.
Pletnev
c,
J. Clark
Lagarias
 d and
Vladislav V.
Verkhusha
*ae
d and
Vladislav V.
Verkhusha
*ae
aDepartment of Anatomy and Structural Biology and Gruss-Lipper Biophotonics Center, Albert Einstein College of Medicine, Bronx, New York 10461, USA. E-mail: vladislav.verkhusha@einstein.yu.edu
bMacromolecular Crystallography Laboratory, National Cancer Institute, Leidos Biomedical Research Inc., Basic Research Program, Argonne, Illinois 60439, USA
cShemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow 117997, Russian Federation
dDepartment of Molecular and Cellular Biology, University of California in Davis, California 95616, USA
eDepartment of Biochemistry and Developmental Biology, Faculty of Medicine, University of Helsinki, Helsinki 00029, Finland
First published on 4th May 2017
Abstract
Brighter near-infrared (NIR) fluorescent proteins (FPs) are required for multicolor microscopy and deep-tissue imaging. Here, we present structural and biochemical analyses of three monomeric, spectrally distinct phytochrome-based NIR FPs, termed miRFPs. The miRFPs are closely related and differ by only a few amino acids, which define their molecular brightness, brightness in mammalian cells, and spectral properties. We have identified the residues responsible for the spectral red-shift, revealed a new chromophore bound simultaneously to two cysteine residues in the PAS and GAF domains in blue-shifted NIR FPs, and uncovered the importance of amino acid residues in the N-terminus of NIR FPs for their molecular and cellular brightness. The novel chromophore covalently links the N-terminus of NIR FPs with their C-terminal GAF domain, forming a topologically closed knot in the structure, and also contributes to the increased brightness. Based on our studies, we suggest a strategy to develop spectrally distinct NIR FPs with enhanced brightness.
Introduction
Near-infrared (NIR) fluorescent proteins (FPs) engineered from bacterial phytochrome photoreceptors (BphPs) have become invaluable probes for in vivo imaging, and also allow the visualization of several biological processes by multiplexing with traditional fluorescent probes developed from FPs of the green fluorescent protein (GFP) family.1 With both absorbance and fluorescence close to or within the so called “near-infrared imaging window” of tissue transparency (650–900 nm), NIR FPs are preferable for imaging deep into tissues, because of low absorbance and scattering of NIR light and low autofluorescence in this spectral region.2 NIR FPs are already widely used probes in many areas of biology and medicine, including cancer studies, parasitology, stem cells, and neuroscience.1 Still, development of brighter and more spectrally diverse probes is required to enhance imaging sensitivity and to design NIR biosensors.BphPs belong to the phytochrome family of red/far-red photoreceptors found in plants, algae, fungi, cyanobacteria and bacteria.3–5 Their primary function is a light-driven signal transduction. Phytochromes are multi-domain protein photoswitches consisting of conserved PAS (Per-ARNT-Sim), GAF (cGMP phosphodiesterase/adenylate cyclase/FhlA) and PHY (phytochrome-specific) tri-domain light sensing modules that regulate diverged effector output domains. Effector domains define the signaling output function of phytochromes and can confer light-regulated enzymatic activities or binding sites for targeted protein–protein interactions. All phytochromes autocatalytically incorporate linear tetrapyrroles (bilins) as chromophores.6,7 BphPs bind biliverdin IXα (BV), whereas plant and cyanobacterial phytochromes bind the more reduced phytobilins, phytochromobilin (PΦB) or phycocyanobilin (PCB).8,9 BV is the most red-shifted natural chromophore and binds to BphP apoprotein autocatalytically. Importantly, BV is abundant in mammalian cells and tissues as an intermediate product of heme degradation to bilirubin.10–12 For these reasons, BphPs have received the most attention as molecular templates for development of NIR FPs.
Absorption of light initiates a chain of structural transformations in photosensory modules (PAS, GAF, and PHY) and signaling domains that result in activation of all phytochromes. These structural changes are best understood for BphPs,13,14 where the PAS and GAF domains are minimally required for chromophore binding,3 whereas the PHY domains are critical for photoproduct spectral tuning and light-driven signal transduction for all phytochromes.15 Although the BV chromophore resides in the GAF domain of BphPs, it is covalently attached to a conserved Cys located in an N-terminal extension of the PAS domain.3,16–18 In contrast, a different Cys residue located in the GAF domain is responsible for phytobilin chromophore attachment in plant and cyanobacterial phytochromes.6 Recent studies revealed that the introduction of the conserved plant/cyanobacterial GAF-domain's Cys into BphPs by mutagenesis yields strongly fluorescent proteins with covalently bound BV.19
Most phytochromes photoswitch between Pr (red absorbing, 15/16Z (cis) double bond) and Pfr (far-red absorbing, 15/16E (trans) double bond) states.6 For this reason, wild-type phytochromes are only weakly fluorescent.20,21 Fluorescence emission of phytochromes was previously enhanced by chromophore substitution22 and by mutagenesis.23 For engineering of NIR FPs from BphPs, the intrinsic photoisomerization has been suppressed by removal of the PHY domain and by introduction of key mutations in the GAF-domain's chromophore binding pocket, such as residues 201 and 202 (amino acid positions are numbered according to Rhodopseudomonas palustris RpBphP1).20,24 Such mutations appear to minimize the non-radiative energy dissipation pathways, which compete with fluorescence emission.21 In addition to NIR absorbance at the major band, called the Q band, phytochromes also absorb at 400 nm (called the Soret band) which is a characteristic band for tetrapyrroles.25
The first BphP-based NIR FP, termed IFP1.4, was described in 2009.11 Soon after the development of IFP1.4, it became clear that NIR FPs require further optimization to be used in mammalian cells. IFP1.4 is very dim and requires BV supply to be fluorescent in cells. Key properties needing optimization include (i) spectra, (ii) molecular brightness (a product of an extinction coefficient and a quantum yield), and (iii) fluorescence intensity in mammalian cells (termed effective brightness). Effective brightness depends on the number of chromophore-containing fluorescent molecules in the cell and is influenced by protein folding and stability, protein expression level, intracellular BV concentration, and specificity of NIR FP to BV chromophore. The last parameter is important because of the competition with other intracellular heme-related compounds, such as protoporphyrins.26–28
To date, directed molecular evolution approaches and structural studies have resulted in the engineering of different NIR FPs, including IFP1.4/2.0,11,29 IFP1.4rev,30 and Wi-Phy20 from Deinococcus radiodurans DrBphP, mIFP from Bradyrhizobium BrBphP,31 iRFP series from Rhodopseudomonas palustris RpBphP2 and RpBphP6,32–34 PAiRFPs from Agrobacterium tumefaciens AtBphP2,35 BphP1-FP,19 and most recently, the miRFP series of monomeric NIR FPs from RpBphP1.19,36 The iRFP and miRFP series were engineered to specifically incorporate endogenous BV in mammalian cells, resulting in their high effective brightness.
The miRFP series, consisting of miRFP670, miRFP703, and miRFP709 NIR FPs, allowed the protein labeling and development of biosensors in the NIR spectral region. miRFPs are of special interest for structural and biochemical studies, since they evolved from the same RpBphP1 template and differ only by several key amino-acid residues that define their properties. Here, we report their crystal structures, which reveal the roles played by specific amino acids near the BV chromophore in the spectral properties, molecular brightness, and effective brightness in mammalian cells. Our studies also identify a new chromophore in bright blue-shifted NIR FPs and suggest a rational approach for the design of brighter and spectrally distinct NIR FPs.
Results
Crystal structures of miRFPs
Structures of the PAS–GAF domains of miRFPs are similar to those of BphPs and BphP-based NIR FPs3,20 (Fig. 1A and S1†). Asymmetric units for all three miRFPs contain one monomer. Crystallization as monomers was expected for miRFPs, since their monomeric state was determined by biochemical analysis and their good performance in protein fusions in live cells.36 Residues 1–34 of the PAS domain have an irregular secondary structure stretching away from the PAS domain core along the surface of the GAF domain. These residues are threaded through the loop insertion of the GAF domain (residues 221–252), forming a figure-eight knot characteristic of the chromophore-binding domain of DrBphP.3 A random-coil linker formed by residues 123–132 connects the PAS and GAF domains. The linear tetrapyrrole chromophore resides in the chromophore-binding pockets of the GAF domain formed by the central β-sheet and two α-helices. The chromophore is covalently bound to Cys20 in miRFP703 and miRFP709. In miRFP670, a mixture of two types of chromophores is detected. Both of them are bound to Cys253, and one is additionally bound to Cys20 (Fig. 1B).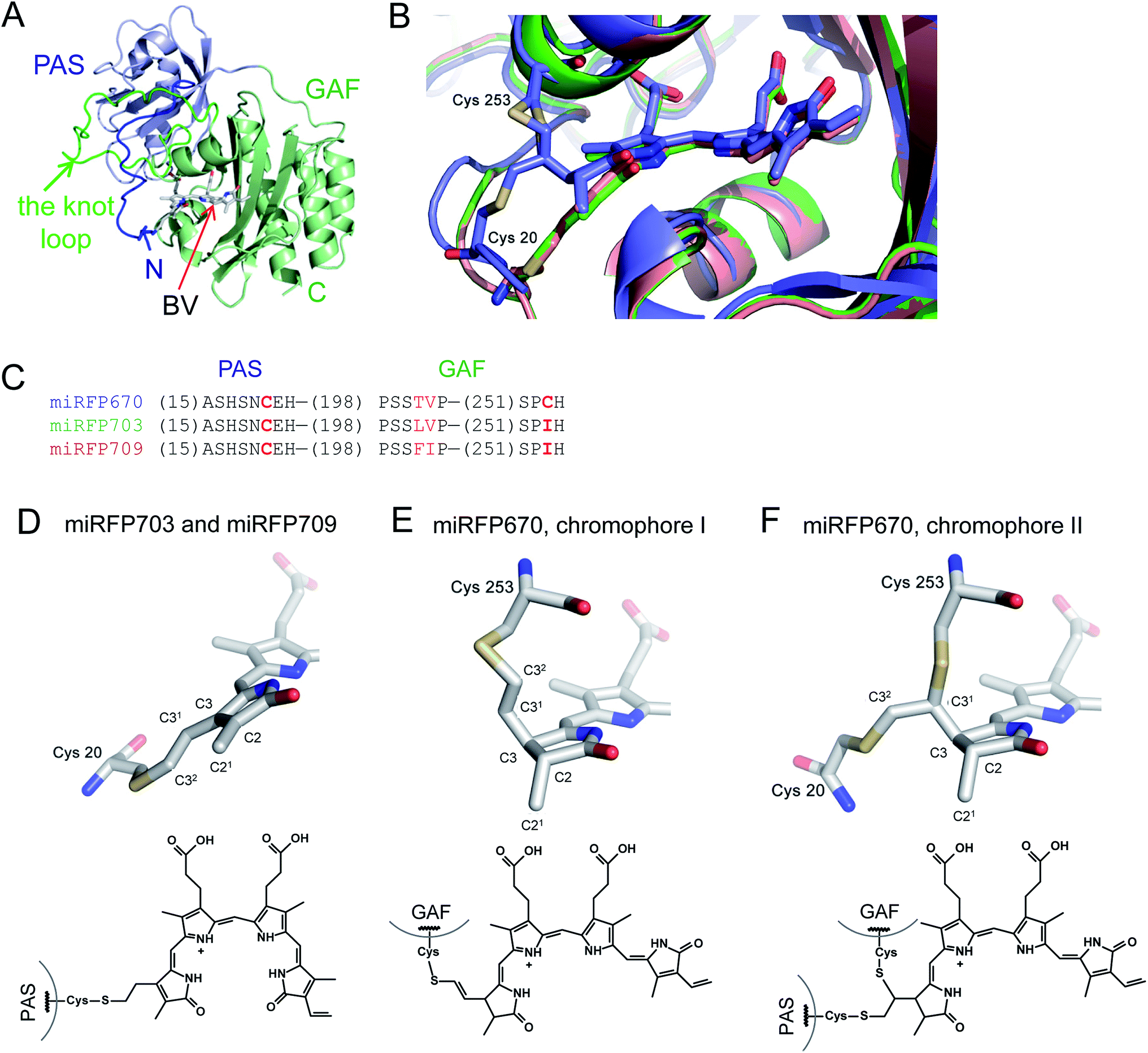 | ||
| Fig. 1 Overall structure and chromophores in miRFPs. (A) Structure of miRFP703 (PDB ID code 5VIK). The PAS domain is in blue, the GAF domain is in green. (B) Superimposed structures of miRFP670 (PDB ID code 5VIV, in blue), miRFP703 (PDB ID code 5VIK, in green), and miRFP709 (PDB ID code 5VIQ, in red) showing the chromophores in the binding pocket. (C) Partial sequence alignment of miRFPs showing critical mutations defining their properties. (D–F) Chromophores (rings A and B are only shown) in miRFPs and their respective chemical formulas. Carbon, nitrogen, oxygen, sulfur atoms are in grey, blue, red, yellow, respectively. Chemical formulas are shown below each structure. (D) Chromophore species in miRFP703 and miRFP709. (E and F) Two chromophore species observed in miRFP670. In the chromophore I, BV is bound via the C32 atom to the Cys253 residue in the GAF domain (E). In the chromophore II, BV is bound via the C31 atom to the Cys253 residue in the GAF domain and via C32 to the Cys20 residue in the PAS domain (F). | ||
Chromophores in miRFPs
miRFPs differ by three amino acid residue positions (201, 202, and 253) that define their spectral properties and brightness (Fig. 1C). Crystal structures of all three miRFPs enabled us to compare the chromophores and to identify how key residues in the immediate BV environment influence NIR FP properties.All miRFP chromophores adopt C5-Zsyn, C10-Zsyn, C15-Zanti conformations, typical of BphPs in the Pr state (Fig. 1C); whereas their B and C rings are coplanar, the rings A and D are tilted out of plane by (29° and 34°), (27° and 37°) and (19° and 37°) for miRFP703/miRFP709 and miRFP670, respectively. These values are in line with the angles reported earlier for natural BphPs in the Pr state.37–39 Similar to those of natural BphPs, BV chromophores of miRFP703 and miRFP709 are both covalently attached to the protein by a thioether bond between a conserved Cys20 in the PAS domain and the A-ring C32 of bilin4,17 (Fig. 1B, salmon and green colors; Fig. 1D). C2 and C3 atoms of miRFP703 and miRFP709 chromophores are in plane with the rest of the atoms of the ring A, indicating sp2 hybridization (Fig. 1D). Consistent with this interpretation, the carbon atom of the C21 methyl group of both FPs also lies in plane with atoms C2 and C3, as seen in the native parental RpBphP1 construct.39 These structures contrast with those of DrBphP and NIR FPs derived therefrom, which have a double bond between C3 and C31 of BV. Despite these structural differences, the presence of a double bond between C2 and C3 or between C3 and C31 confers similar spectral properties for both DrBphP-based and RpBphP1-based scaffolds.
Unlike single chromophore species in miRFP703 and miRFP709, the chromophore of miRFP670 exists as a mixture of two species (Fig. 1B, blue color). One species has a single covalent bond between the GAF domain Cys253 and C32 atom of the BV chromophore (Fig. 1E), and the other species has two covalent bonds between the PAS domain Cys20 and the GAF domain Cys253 and C32 and C31 atoms of BV, respectively (Fig. 1F). To confirm the presence of the unusual chromophore species covalently linked to two cysteine residues, we calculated the 2Fobs − Fcalc electron density map. The respective electron density map is well defined for the BV chromophore, Cys253, and the side chain of Cys20 (Fig. S2A†). To additionally prove the presence of a covalent bond between Cys20 and the chromophore, we calculated the composite OMIT difference electron density map in the absence of Cys20, Cys253, and BV species (Fig. S2B†). Similar to the 2Fobs − Fcalc map, the OMIT map shows a clearly defined density for the side chain of Cys20 and the Cys20–BV thioether bond.
Both C31 and C21 atoms are out of the plane of the ring A, suggesting the tetrahedral geometry (sp3-hybridization) of C2 and C3 atoms in a saturated pyrrole ring with the C2 carbon adopting an R stereochemistry in both species of miRFP670. Interestingly, the structure in Fig. 1E is similar to one of two singly-linked chromophore species found in the blue-shifted BphP1-FP/C20S protein.19 In this NIR FP, the chromophore species have a double bond between C31 and C32, which is out of conjugation with the rest of the π-electron system, resulting in a blue-shift of the spectra. The miRFP670 structure – albeit novel and unexpected – also is consistent with the blue-shifted spectrum of this NIR FP. Both chromophore systems in miRFP670 are one double bond shorter than that of miRFP703 and miRFP709, thus accounting well for the spectral blue shift.
The covalent bond to Cys253 also alters the position of the ring A in miRFP670 by moving it 1.0–1.5 Å closer to helix α4 (Fig. S1† and 1B). The width of the chromophore-binding pocket is slightly decreased in miRFP670. In all other respects, the chromophore environment of miRFP670 remains almost the same as in miRFP703 and miRFP709. All three chromophoric systems maintain the similar hydrogen bond network and have the same pattern of hydrophobic contacts as the nearby residues (Fig. S3†). Similar to other BphP structures, the pyrrole nitrogen of the rings B and C forms hydrogen bonds with the main chain carbonyl of the amino acid in position 201 and with the conserved “pyrrole” water molecule.20,37
Spectral red-shifts and rational design of red-shifted NIR FPs
Residues Phe and Ile in positions 201 and 202 are responsible for the spectral red shift (10 nm in excitation and 6 nm in emission) of miRFP709 (excitation/emission maxima are at 683 nm/709 nm) relative to miRFP703 (excitation/emission maxima are at 673 nm/703 nm) (Table 1). These residues were found using random mutagenesis and screening for the red-shifted fluorescence. Interestingly, in the case of miRFPs, Phe201 and Ile202 coincide with residues introduced during the evolution of the most red-shifted BphP-based NIR FP, termed iRFP720.34 Phe202 and Ile203 residues in iRFP720, which is iRFP713/E180S/T202F/V203I/V254N, are mainly responsible for its 7 nm spectral shift, compared to its precursor iRFP713. Through overlay of the structures of miRFP703 and miRFP709 (Fig. 2A), we found that there is an overlap between the π-electron systems of F201 and the ring A, although they do not form well defined parallel stacking. Moreover, the L201F substitution in miRFP709 improves the planarity of the A ring with the rest of the chromophore. These two effects are likely responsible for the spectral red-shift of miRFP709 emission relative to that of miRFP703.| NIR FP | Parental natural BphP | Oligomeric state | Abs, nm | Ex, nm | Em, nm | Extinction coefficient, M−1 cm−1 | Quantum yield, % | Molecular brightness vs. miRFP670 | Effective brightness in HeLa cells vs. miRFP670 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| iRFP670 | RpBphP6 | Dimer | 644 | 643 | 670 | 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
11.7 | 110 | 165.3 | 40 |
| iRFP670/C10S | 641 | 640 | 669 | 85400 |
9.8 | 72 | 47.2 | |||
| iRFP682 | RpBphP2 | 663 | 660 | 682 | 90000 |
10.6 | 82 | 145.8 | ||
| iRFP682/C15S | 659 | 658 | 683 | 53800 |
4.8 | 22 | 37.5 | |||
| miRFP670 | RpBphP1 | Monomer | 644 | 642 | 670 | 87400 |
13.4 | 100 | 100 | 36 |
| miRFP670/C20S | 644 | 644 | 671 | 71300 |
11.1 | 68 | 41.7 | This work | ||
| miRFP670/C20A | 644 | 643 | 672 | 73800 |
9.8 | 62 | 64.2 | This work | ||
| miRFP670/AAANAEA | 644 | 643 | 672 | 81500 |
10.9 | 76 | 100.8 | This work | ||
| miRFP670/AAANCEA | 644 | 644 | 672 | 89300 |
13.7 | 104 | 123.1 | This work | ||
| miRFP703 | 676 | 674 | 703 | 90900 |
8.2 | 64 | 51.9 | 36 | ||
| miRFP709 | 684 | 683 | 709 | 78400 |
5.2 | 35 | 41.8 | |||
| miRFP709/Y210F | 687 | 687 | 713 | 71900 |
4.4 | 27 | 12.4 | This work |
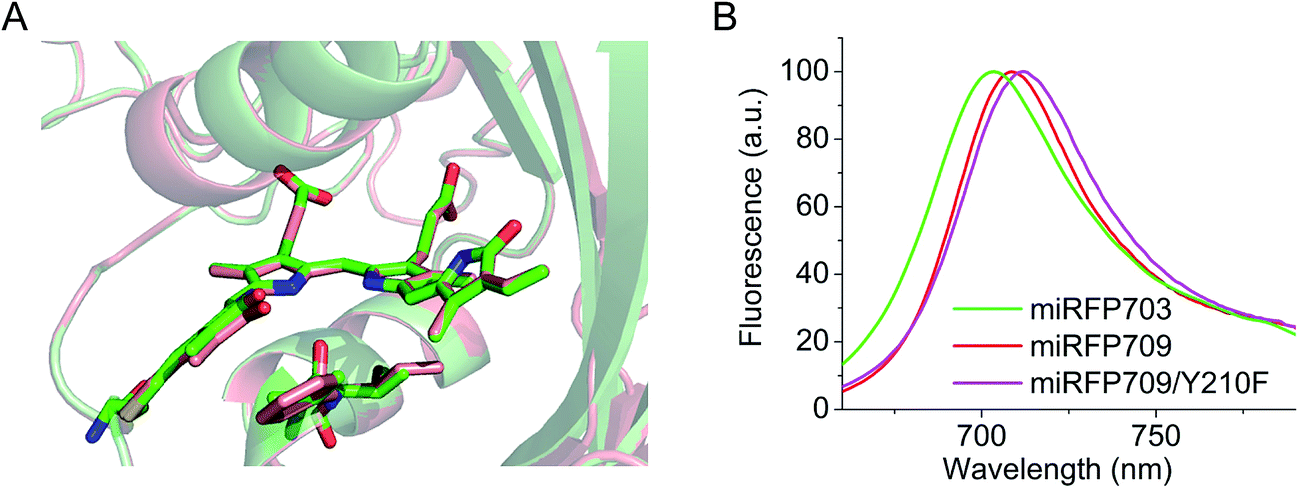 | ||
| Fig. 2 Spectral red-shift in miRFPs. (A) Superimposed structures of miRFP703 (in green) and miRFP709 (in red) showing chromophores in the binding pocket and residues L201, V202 in miRFP703, and F201, I202 in miRFP709. (B) Overlay of emission spectra of miRFP703 (in green), miRFP709 (in red), and miRFP709/Y210F with emission peak shifted to 713 nm (in magenta). | ||
In an attempt to further shift miRFP709 to the NIR part of the spectrum, we introduced aromatic residues in the immediate BV environment that could potentially form additional stacking interactions with the chromophore. While one of these mutants, such as miRFP709/H254Y, showed a red-shifted emission peak at 714 nm, the brightness decreased dramatically and the signal was barely visible (Fig. S4†). However, we were able to red-shift miRFP709's emission 4 nm further into the NIR spectral region by introducing neutral aromatic Phe instead of polar aromatic Tyr in the immediate chromophore environment. The resulting miRFP709/Y210F variant possesses an extinction coefficient of 71900 M−1 cm−1 and a quantum yield of 4.6% (Fig. 2B and Table 1). Further directed molecular evolution should improve the molecular and cellular brightness of this new monomeric NIR FP.
Biochemical assays detect miRFP670 molecules with BV bound to two Cys residues
To further characterize miRFP670, we compared its biochemical properties with mutants lacking the Cys residue in the PAS domain, i.e. miRFP670/C20S and miRFP670/C20A. The spectra of all three miRFP670 variants looked similar (Fig. 3A and B). However, the absorbance spectra differed by the ratios of the Q band to the Soret band, resulting in a decreased extinction coefficient of the C20S and C20A mutants (Fig. 3A). Excitation and emission spectra of all three miRFP670 variants were also quite similar, except that the emission spectra of the C20S and C20A mutants were slightly broader (Fig. 3B), which was consistent with observations on other NIR FPs.19,40,41 The molecular brightness of the C20S and C20A mutants were also lower than that of parental miRFP670, with the decrease in extinction coefficient and quantum yield (Table 1). The similar relationships between molecular brightness of NIR FPs with two Cys residues and their mutants with single Cys residue in the GAF domain only were previously observed for dimeric blue-shifted iRFP670 and iRFP682 developed from other BphPs.40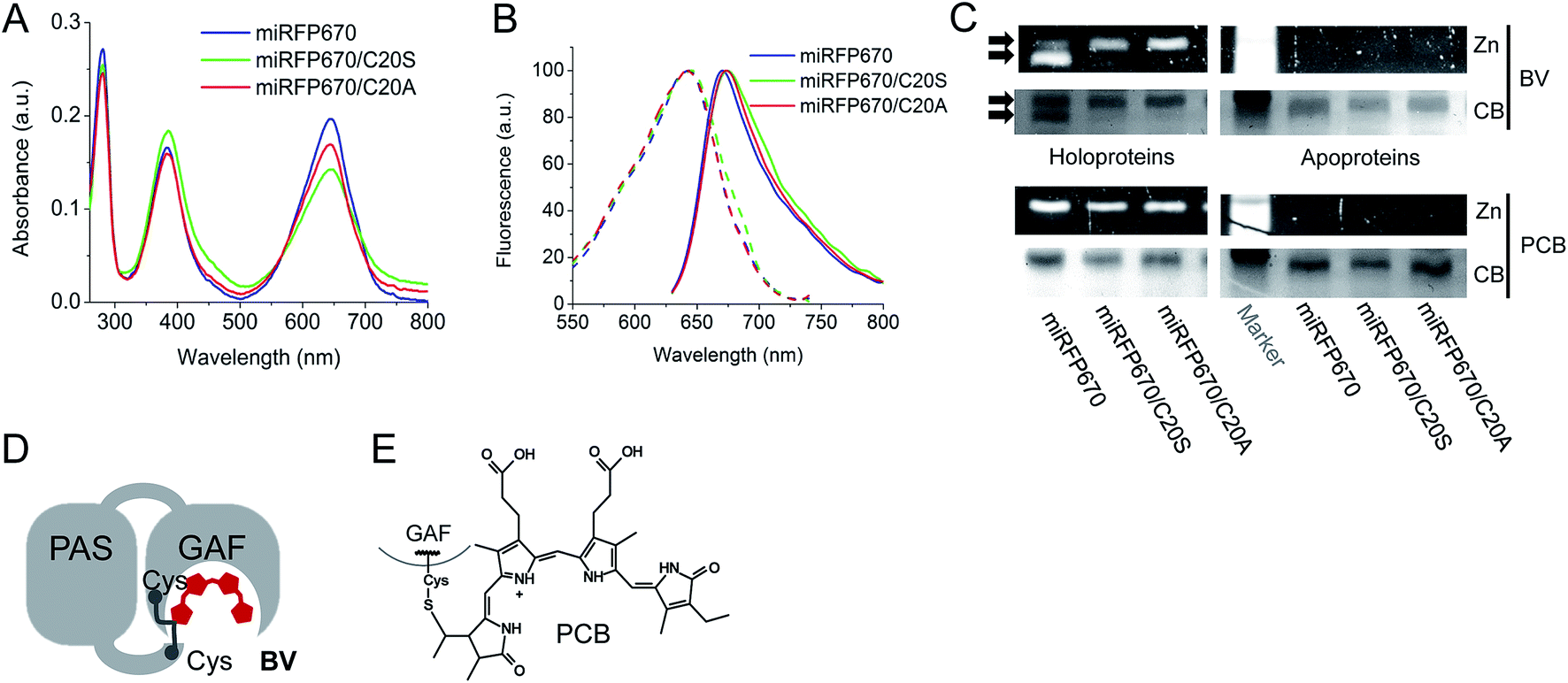 | ||
| Fig. 3 Biochemical analysis of miRFP670 and its mutants without Cys20 in the PAS domain. (A) Overlay of absorbance spectra of miRFP670 (in blue) and its C20S (in green) and C20A (in red) mutants. (B) Overlays of excitation spectra (dashed lines) and emission spectra (solid lines) of miRFP670 (in blue), and its C20S (in green) and C20A (in red) mutants. (C) SDS-PAGE and ZnCl2 staining assay of miRFP670 and its C20S and C20A mutants. Zn2+-induced fluorescence is observed if the bilin chromophore is covalently attached to the protein molecule. Holoproteins (left panels) assembled with BV (upper panel) or PCB (lower panel) and apoproteins (right panels) were analyzed. Two bands were observed only in the case of the miRFP670 holoprotein assembled with BV. (D) Schematics of the covalently closed loop in the miRFP670 molecule with BV bound to two Cys residues. (E) Chemical structure of PCB bound to the GAF domain. | ||
When miRFP670 and its mutants were analyzed by SDS-PAGE and Zn2+-dependent chromophore-binding assay, an additional band was detected in the miRFP670 sample (Fig. 3C). Interestingly, two bands were observed earlier for iRFP670 and iRFP682, both of which contained two Cys residues.40 The additional band, which migrated faster than the singly linked BV chromophore, is consistent with a more compact protein that is internally cross-linked and thereby experiences a lower frictional drag during electrophoresis. Moreover, this additional band was not present in miRFP670 apoprotein (Fig. 3C), demonstrating a crucial role for BV in the appearance of the second band. The formation of a topologically closed loop in the protein is thus consistent with the presence of two covalent bonds between the BV chromophore and the Cys residues in the PAS and the GAF (Fig. 3D and 1F). In this case, the protein cannot be fully linearized during sample preparation, since its N-terminus is covalently linked to the –PCH– motif of the GAF domain through BV, forming a closed loop.
To confirm that the additional band in the miRFP670 sample originates from the presence of a BV adduct bound to two Cys residues, we tested the same miRFP670 variants with another bilin chromophore PCB. PCB is reduced compared to BV and is able to covalently bind only to the Cys residue in the GAF domain of NIR FPs (Fig. 3E).19,42 Analysis of miRFP670 and its mutants assembled with PCB confirmed that PCB binds covalently, and also revealed that there is no additional band in the miRFP670 sample with PCB (Fig. 3C). Thus, only BV supports the formation of the additional faster running band that is dependent upon both Cys residues in NIR FPs.
Proteolytic assays support the presence of an intramolecular crosslink in miRFP670
To provide additional evidence that the faster running band observed in miRFP670 (Fig. 3C) corresponds to crosslinked protein arising from covalent BV binding to both the PAS and GAF domain's Cys residues, we inserted a TEV (tobacco etch virus) protease cleavage site in the loop between the PAS and GAF domains of miRFP670, miRFP670/C20S and miRFP709 (Fig. 4A). TEV protease cleavage, combined with SDS-PAGE, was then used to unambiguously address this hypothesis. We reasoned that TEV protease cleavage of the miRFP670 species with two thioether linkages would yield a single crosslinked polypeptide, whereas cleavage of the single-Cys-linked species, as well as those of miRFP670/C20S and miRFP709, would yield two smaller polypeptides each.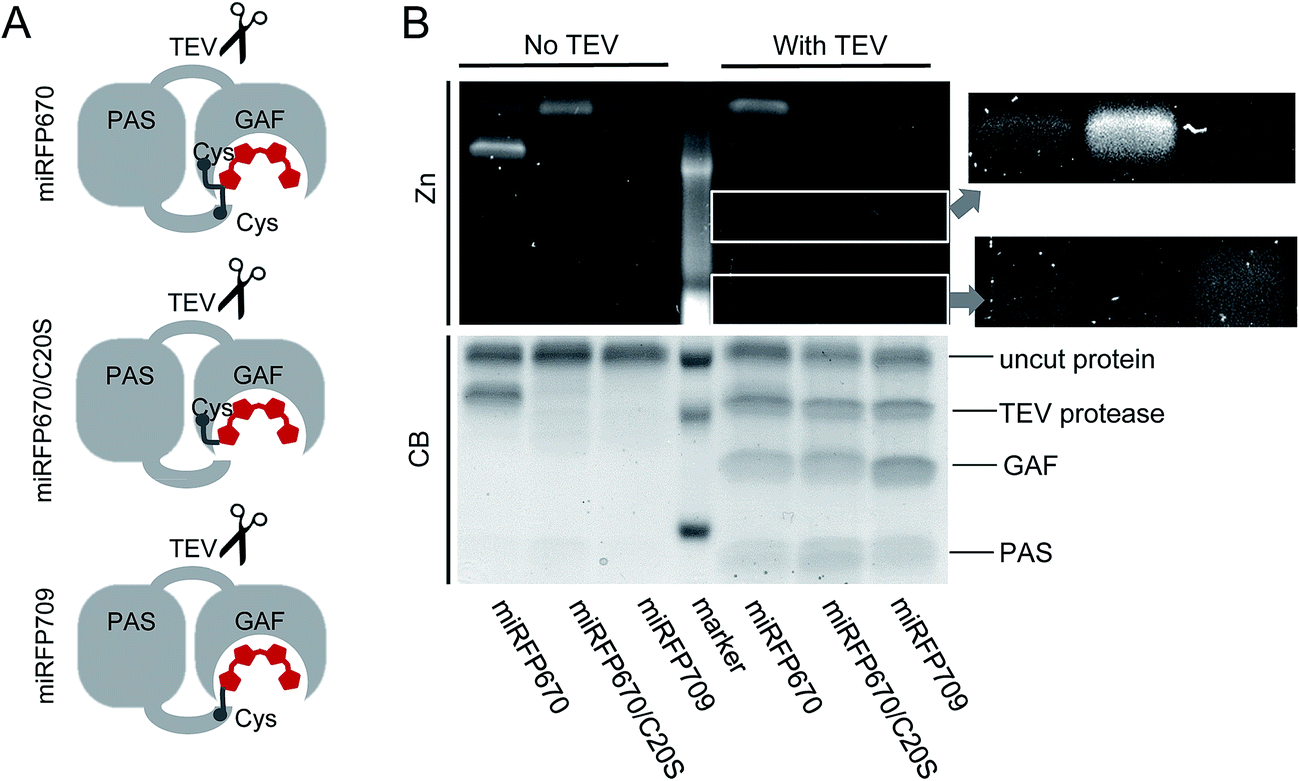 | ||
| Fig. 4 TEV protease assay of miRFP670 and its mutants with Cys either in the PAS domain or in the GAF domain. (A) Schematics of miRFP670 with BV bound to two Cys, miRFP670/C20S with BV bound to Cys in the GAF domain, and miRFP709 with BV bound to Cys in the PAS domain. The TEV protease site (GENLYFQG) was introduced between the PAS and the GAF domain. (B) SDS-PAGE and ZnCl2 staining assay of the proteins shown in (A) treated (with TEV) and untreated (no TEV) by TEV protease. Zn2+-induced fluorescence is observed if BV is covalently attached to the protein molecule. Areas of interest in the lower part of the gel are magnified and enhanced to better visualize low molecular weight BV-binding protein fragments. | ||
SDS-PAGE analysis confirmed that this was indeed the case, despite incomplete cleavage of the three proteins by TEV protease (Fig. 4B). Coomassie blue staining showed that all TEV-treated NIR FPs contained two smaller polypeptides, which can be assigned to the separated PAS and GAF domains, based on their molecular weights (Fig. 4B). For miRFP670 protein, these two bands represent the cleavage products derived from the singly-linked species (Fig. 1E) because the similar bands were also present in the cleavage mixtures of singly-linked miRFP670/C20S and miRFP709 proteins. As expected, Zn2+-dependent staining showed that BV was bound to the cleaved GAF domain in miRFP670/C20S, and to the cleaved PAS domain in miRFP709. In miRFP670, the minor fraction of the BV chromophore was bound to the GAF domain, traces were bound to the PAS domain, and the majority of the bound BV was re-distributed to the upper band. Interestingly, in untreated miRFP670 the relative Zn2+-dependent fluorescence intensity of the lower band, which corresponds to double linked species, was more intense than that of the upper band, which corresponds to singly-linked species in untreated miRFP670. The brighter Zn2+-dependent staining of BV bound to two Cys residues may result from its lower exposure to the SDS denaturing agent. Comparative Zn2+-dependent fluorescence intensities of two upper bands in the untreated and TEV-treated miRFP670 sample suggests that the cleavage of the miRFP670 species with BV bound to two Cys residues yields a linearized crosslinked product that co-migrates with the upper, uncleaved protein band. Taken together, the TEV protease cleavage analysis experimentally confirms the presence of two types of BV chromophores observed in the crystal structure of miRFP670: (i) the chromophore with a covalent bond between C32 and Cys253, and (ii) the chromophore with two covalent bonds between C31 and Cys253 in the GAF domain and C32 and Cys20 in the PAS domain.
Molecular and cellular brightness of blue-shifted NIR FPs and the design of brighter NIR FPs
Besides the peak wavelength of fluorescence emission, the effective brightness of NIR FPs in mammalian cells is one of the most important properties to optimize. Among miRFPs, miRFP670 is the brightest FP so far (Table 1). Despite similar absorption spectra, miRFP670 is more than twice as bright as the singly linked C20S mutant. The higher cellular brightness of other blue-shifted NIR FPs with two chromophore-binding Cys residues has also been reported for iRFP670 and iRFP682 engineered from other BphPs (Table 1). For comparative purposes, we determined the brightness of miRFP670, miRFP670/C20S and a newly constructed miRFP670/C20A mutant (Fig. 5). Although lower than miRFP670, the effective brightness of C20A in cells was higher than that of C20S (64% and 42% of that of miRFP670, respectively; Fig. 5B and C and Table 1). In contrast, the purified C20A and C20S variants exhibited comparable molecular brightness (62% and 68% of miRFP670, respectively; Fig. 5D).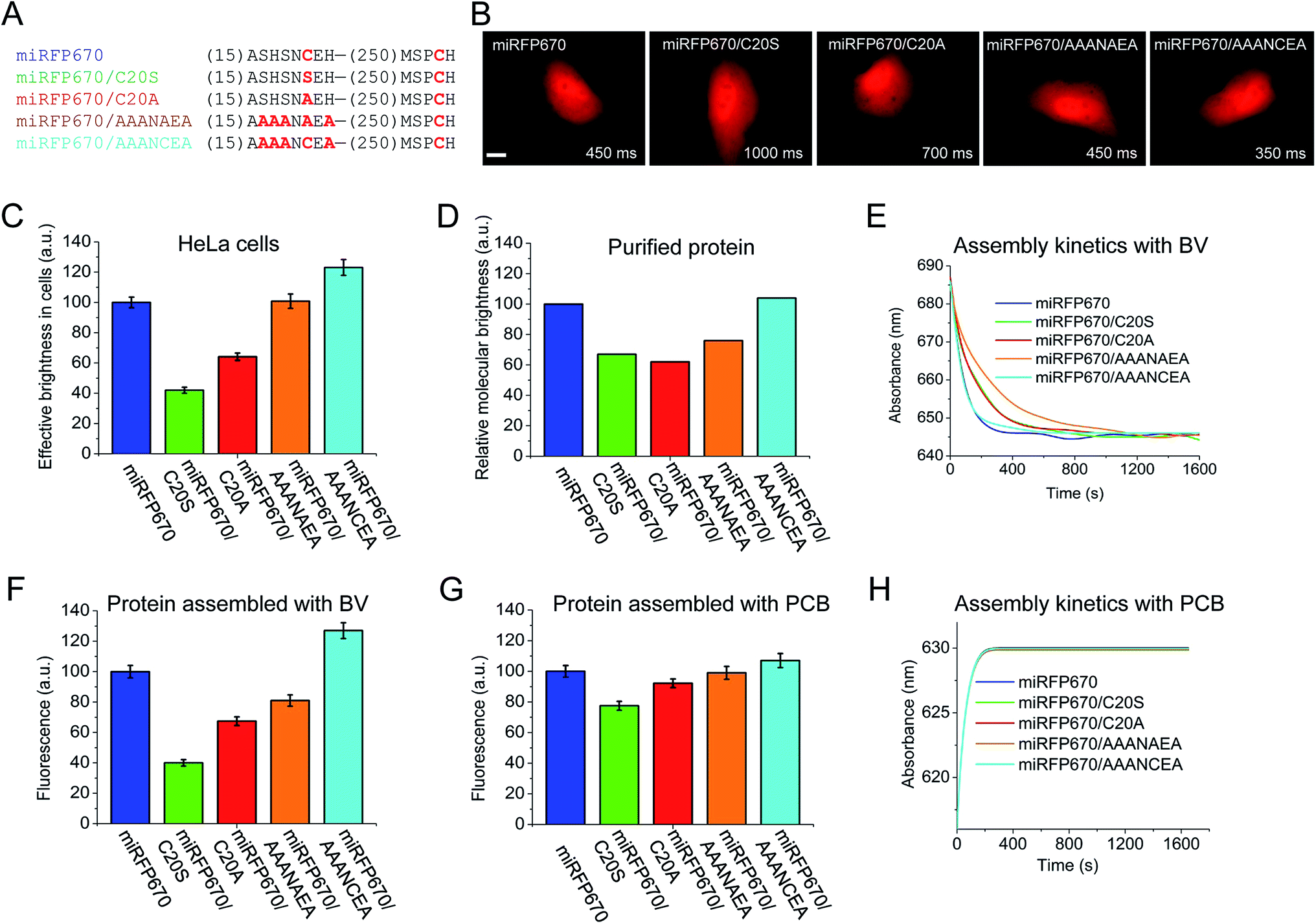 | ||
| Fig. 5 Analysis of miRFP670 and its mutants in vitro and in live mammalian cells. (A) Partial sequence alignment of miRFP670 and its mutants showing introduced mutations. (B) Representative fluorescence images of miRFP670 and its mutants expressed in HeLa cells. Acquisition time for each image is indicated. Scale bar, 10 μm. (C) Effective brightness of miRFP670 and its mutants in HeLa cells analyzed by flow cytometry. The NIR fluorescence was normalized to transfection efficiency by using green fluorescence of co-transfected EGFP. (D) Molecular brightness of miRFP670 and its mutants normalized to that of miRFP670. (E) Kinetics of assembly of apoproteins corresponding to miRFP670 and its mutants with BV (ratio 1.5 to 1) in vitro. The change in the absorbance maximum is shown over time (full spectra are presented in Fig. S3†). (F) Fluorescence signals of equal amounts of apoproteins corresponding to miRFP670 and its mutants fully assembled with BV, normalized to the signal of miRFP670 sample. (G) Same as in (F), but for the proteins assembled with PCB. (H) Same as in (E), but for the proteins assembled with PCB (full spectra are shown in Fig. S4†). | ||
To understand the molecular basis of these differences, we set up an in vitro assembly experiment. Upon mixing of equal amounts of miRFP670 apoprotein or the two mutant apoproteins with BV in the ratio of 1.5 to 1 (apoprotein to BV), we monitored the kinetics of the BV–apoprotein assembly by measuring time dependent changes in absorbance (Fig. 5E and S5†). For all the studied proteins, the BV binding reached steady state after 30 min (Fig. 5E). We also detected the magnitude of the fluorescence signal of the fully assembled NIR FPs after 2 h (Fig. S5† and 5F). These measurements showed that miRFP670, which contains two chromophore-binding Cys residues, assembled faster than both its C20A and C20S mutants (Fig. 5E). However, there was no difference between the C20A and C20S variants. Interestingly, the fluorescence intensity of the three fully assembled miRFP670 variants mirrored their relative brightness levels observed in mammalian cells (Fig. 5F). This suggested that the apoprotein molecules of miRFP670/C20S incorporated BV less efficiently than miRFP670/C20A in vitro and in live cells.
The data indicate that the substitutions of Cys20 with Ser (and with Ala to a lesser extent) are inhibitory to BV binding. To test the influence of the residue at position 20, we performed a comparative assembly experiment using PCB instead of BV. Since PCB binding requires only Cys253 in the GAF domain, these experiments were designed to understand the influence of the amino acid at position 20 on the thioether linkage formation to Cys253 (Fig. S6†). Unlike the previous BV assembly experiments, there were only slight differences in the fluorescence intensities of the PCB-reconstituted miRFP670 or the two Cys20 mutant holoproteins (Fig. 5G). The kinetics of the PCB binding was indistinguishable among the three miRFP670 variants (Fig. 5H).
To further test this effect, we replaced two upstream Ser residues and two flanking His residues with Ala in both miRFP670/C20A and the original miRFP670 to create miRFP670/AAANAEA and miRFP670/AAANCEA mutants, respectively (Fig. 5A). Both in vivo and in vitro analyses revealed that these substitutions improved the fluorescence brightness of the resulting holoproteins (Fig. 5C, D, F and G). The brightness of in vitro assembled miRFP670/AAANAEA with BV was still lower than miRFP670, whereas its brightness assembled with PCB was nearly equal to that of miRFP670 (Fig. 5F and G).
Importantly, the miRFP670/AAANCEA mutant combines two favorable features, i.e. chromophore binding Cys residue in the PAS and the optimized N-terminus. This NIR FP showed 120% effective brightness in mammalian cells, compared to that of the original miRFP670 (Fig. 5B and C and Table 1), thus becoming the brightest monomeric NIR FP available.
Discussion
The structural and biochemical analyses of three miRFP proteins engineered from Rhodopseudomonas palustris RpBphP1 revealed the importance of the key residues in the chromophore environment on spectral properties and brightness of NIR FPs: the chromophore binding Cys residues in the PAS and the GAF, the residues 201 and 202, and the residues at the N-terminal flanking the chromophore binding Cys in the PAS.Amino acid residues in positions 201 and 202 influence the spectral properties and brightness of NIR FPs. Both red-shifted NIR FPs, miRFP703 and miRFP709 contain non-polar amino acids in position 201. Although only the side chain of residue 201 is directly involved in hydrogen bond interaction with BV, the nature of this residue is critical for NIR FP fluorescence. It was suggested that the non-polar amino acid in position 201 prevents the formation of a polar hydrogen-bonding network.43 This network is believed to participate in the formation of excited-state intermediates in light-induced chromophore photoswitching.24,41 Phe and Ile residues in positions 201 and 202 shift the spectra further into the NIR region and allow the most red-shifted NIR FPs to be obtained from different BphP templates.34,36 Here, we found that Phe201 causes the red-shift through its interaction with the ring A of the chromophore. Introduction of aromatic amino acids, which could form stacking interactions with the chromophore, shifted the spectra 5 nm further into the NIR, but also resulted in significantly decreased brightness (Fig. S4†). However, by changing polar Tyr210 with neutral Phe in the immediate chromophore environment, we obtained a relatively bright miRFP709/Y210F mutant shifted 4 nm further into the NIR (Fig. 2 and Table 1).
The structure of miRFP670 confirms the key role of the Cys residue introduced into the GAF domain in the spectral blue-shift in NIR FPs.19 This Cys covalently binds BV to yield a mixed chromophore species with one less double bond in the π-conjugated system than the NIR FP mutants with the chromophore-binding Cys residue in the PAS domain only. The resulting sp3 hybridization (tetrahedral geometry) of the C3 atom of the BV adduct apparently allows efficient binding to this GAF-domain's Cys residue, which is located above the chromophore plane. The discovery that the introduction of the GAF-domain's Cys leads to blue-shifted NIR FPs was already applied for the development of NIR FPs from various natural BphP templates, including RpBphP2 and RpBphP6,34 RpBphP1,36 and BrBphP.44
In the present study, structural and biochemical analyses of miRFP670 unexpectedly revealed the presence of a new chromophore species in blue-shifted NIR FPs. In these species, the PAS-domain's Cys also participates in the BV binding, together with the Cys in the GAF domain, to produce a topologically crosslinked knot structure in which BV is dually linked to both Cys20 and Cys253 (Fig. 6A). This crosslink is well detected by SDS-PAGE (Fig. 3 and 4), and we estimate that it is present in approximately half of the miRFP670 molecules. Similar crosslinks have also been detected in other NIR FPs, including dimeric iRFP670, iRFP682 and iRFP713/V256C (ESI Fig. 2† in ref. 40), suggesting the presence of a blue-shifted dual-linked chromophore species in these NIR FPs too. Interestingly, the topologically trapped knot is very uncommon in proteins.18,45
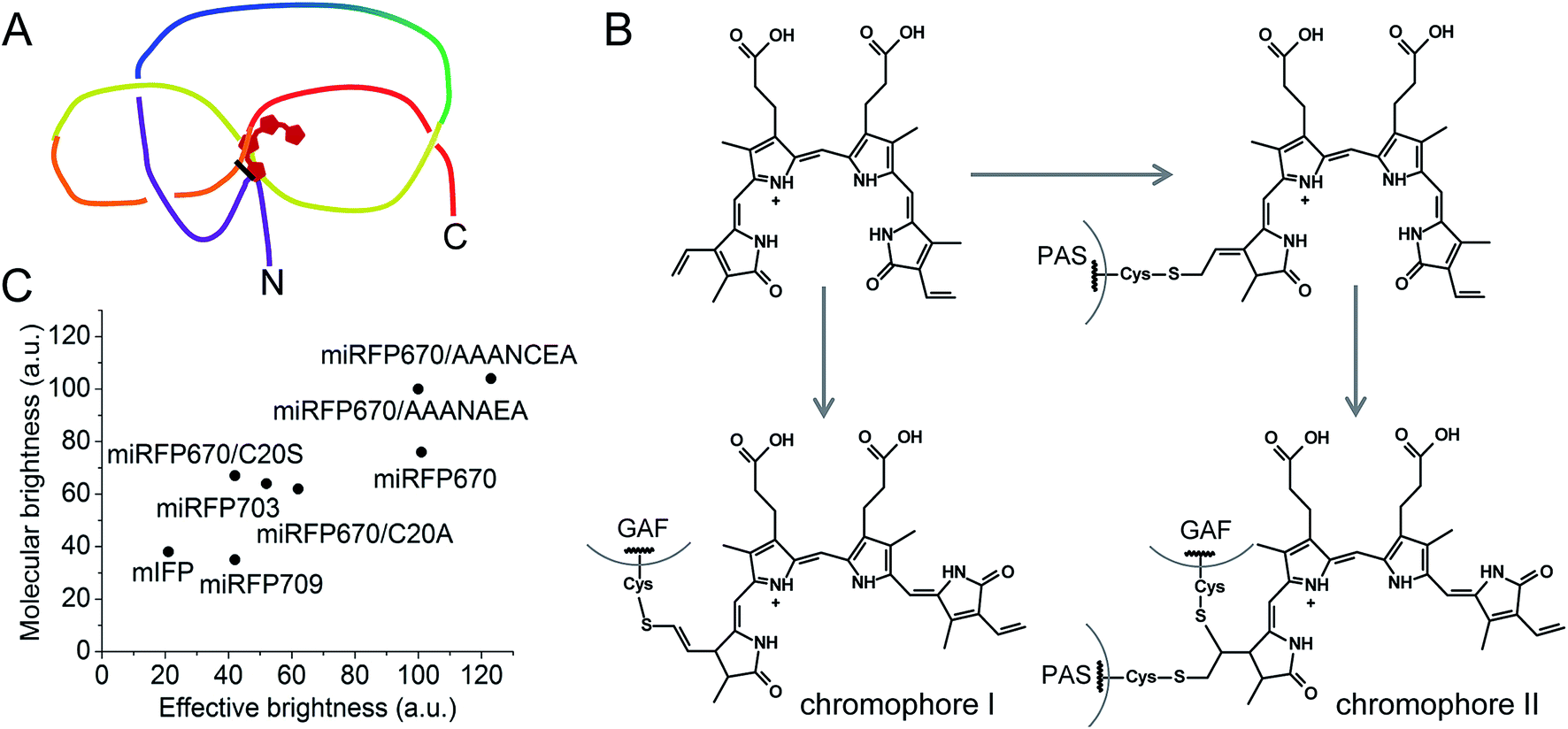 | ||
| Fig. 6 Features of brightest NIR FPs. (A) Schematics of a figure-eight knot with a closed loop in blue-shifted NIR FP molecules with BV covalently bound to two Cys residues in the PAS domain and the GAF domain. (B) Proposed schematics of the chromophore formation in miRFP670 and other blue-shifted NIR FPs with two chromophore-binding Cys residues. Chromophore I and chromophore II species are the same as observed in miRFP670 (Fig. 1E and F). (C) Comparison of effective brightness in mammalian cells and molecular brightness of monomeric NIR FPs and their mutants. | ||
In addition to the dual-linked BV chromophore species (Fig. 1F), a single-linked BV chromophore bound via C32 to the GAF-domain's Cys (Fig. 1E) is present in approximately half of the miRFP670 molecules. We propose that the formation of the dual-linked BV adduct occurs by addition of the PAS-domain's Cys to the least substituted vinyl carbon at C32, followed by addition of the GAF domain Cys at C31 (Fig. 6B). In contrast, formation of the singly-linked species in which the GAF-domain's Cys is linked to the C32 and similar to that found in BphP1-FP/C20S19 requires a different mechanism in which the GAF-domain's Cys initially attaches to the C32 position of BV (Fig. 6B).
Dual Cys linkages have been previously observed in cyanobacteriochrome (CBCR) photoreceptors46–49 and in phycobiliproteins,50 but not in BphPs. However, the structures of these adducts differ significantly from those of miRFP670, and they are likely to have different mechanisms for their formation. Indeed, CBCRs and phycobiliproteins incorporate phytobilins, such as PCB, instead of BV as chromophore precursors. A family of dual-linked CBCRs includes PixJ from Thermosynechococcus elongatus, which contains two Cys residues, one in a conserved –DXCF– motif, in addition to the chromophore-binding Cys in the –XCH– motif, which is also conserved in phytochromes. These photoreceptors exhibit blue-to-green photoswitching. This striking blue-shift arises from binding of the Cys residue in the –DXCF– motif to the C10 atom located between the B and C rings of the PCB chromophore,49 which is quite distinct from the dual thioether linkages found in NIR FPs in this paper.
Importantly, blue-shifted NIR FPs with two chromophore-binding Cys residues in the PAS and the GAF domains have higher molecular brightness and effective brightness, compared to their mutants that lack the PAS-domain's Cys. Thus, the new chromophore species bound to two Cys residues appear to be mainly responsible for the increase in the quantum yield. Most probably, two covalent bonds between the ring A and two Cys residues in the PAS and GAF domains fix the position of the ring A in the binding pocket. Hence, more rigid dual-linked chromophore species appear to be responsible for the higher quantum yield observed in blue-shifted NIR FPs with two Cys. Higher extinction coefficient also may be a result of improved BV incorporation in NIR FPs possessing both Cys residues.
Analysis of the cellular brightness of miRFP670 and its mutants clearly demonstrates that molecular brightness of NIR FPs purified from bacteria producing excess of BV does not always correlate with effective brightness in mammalian cells. Surprisingly, the assembly assay of an apoprotein with BV better predicts the effective brightness of closely related NIR FP variants (Fig. 5). This assay allowed us to uncover the negative influence of Ser20 residue and neighbor residues on NIR FP brightness. The actual role of these residues remains unclear. We do see that the rate of chromophore binding is slower for miRFP670/AAANAEA, however, this rate is the same for miRFP670/C20A and miRFP670/C20S. Thus, Ser20 might not be directly involved in chromophore binding. The data in Fig. 5 show that Ser20 and neighboring Ser residues may interfere with efficient chromophore incorporation in the pocket and/or binding to Cys in the GAF domain. Substitution of these residues with Ala considerably increased the molecular and cellular brightness of miRFP670 mutants (Fig. 5). This region likely plays a regulatory role in the knot formation between the PAS and GAF domains; hence, mutations therein are expected to alter the efficiency of chromophorylation. It is also possible that inefficient chromophore binding results in the incorporation of small amounts of non-covalently bound BV, which contribute to a decrease in molecular brightness. This hypothesis is consistent with the appearance of broader absorption spectra of mutants with Cys in the PAS replaced with Ser or Ala residues (Fig. 3B and ref. 40 and 41).
Conclusions
Based on the above results, we suggest two approaches to boost the molecular and cellular brightness of the blue-shifted NIR FPs: (i) to keep both Cys residues in the PAS and GAF domains during molecular evolution of a NIR FP, and (ii) to replace Ser residues in the vicinity of the Cys in the PAS domain with Ala residues. The first approach should result in an increase in quantum yield and likely, an improvement of extinction coefficient. The second approach should mostly influence the extinction coefficient by making BV incorporation more efficient. A combination of these two approaches has resulted in the brightest monomeric NIR FP in vitro and in live mammalian cells (Fig. 6C).Overall, this work sets a foundation for the rational design of NIR FPs. Introduction of specific amino acids into the BV binding pocket of the GAF domain and in the N-terminus of the PAS domain will direct the molecular evolution toward the development of brighter multispectral NIR FPs from a variety of the natural BphP templates.
Experimental section
Protein expression, purification and characterization
miRFP DNA was PCR amplified and cloned as BglII/EcoRI fragments into a pBAD/His-B vector (Life Technologies/Invitrogen). Site-specific mutagenesis was performed using a QuikChange mutagenesis kit (Stratagene). Plasmids encoding miRFPs were electroporated into TOP10 host (Invitrogen) containing the pWA23h plasmid34,51 encoding heme oxygenase from Bradyrhizobium ORS278 (hmuO) under the rhamnose promoter for expression of holoproteins. The pWA23h plasmid was removed from cell lines for expression of apoproteins. Bacterial cells were grown in RM medium supplemented with ampicillin and kanamycin (where required). Heme oxygenase was first induced with addition of 0.02% rhamnose for 5 h at 37 °C, followed by addition of 0.002% arabinose to induce FP protein expression. Bacterial cultures were then incubated for an additional 12 h at 37 °C, followed by 24 h at 18 °C. FPs were purified by immobilized metal affinity chromatography using Ni-NTA agarose (Qiagen) and 100 mM EDTA instead of imidazole for elution, followed by size exclusion chromatography using a Superdex 200 (16/60) column (Amersham Biosciences). For chromophore binding assays, purified FPs were separated by SDS/PAGE in the presence of 1 mM ZnCl2, imaged for zinc-dependent fluorescence using UV light and then visualized by staining with Coomassie blue.Fluorescence spectra were recorded with a FluoroMax-3 spectrofluorometer (Jobin Yvon). Absorbance spectra were measured with a Hitachi U-2000 spectrophotometer. Extinction coefficients were calculated by comparison of the long wavelength peak absorbance with that of the Soret band, using the assumption that Soret bands of all FPs possess the same extinction coefficient of 39900 M−1 cm−1 as BV.11,32 To determine the fluorescence quantum yield, the fluorescence intensity of each FP was compared with that of an equally absorbing acidic ethanol solution of Nile blue dye (quantum yield of 0.27 in acidic ethanol52). The obtained values were corrected for the difference in refractive indexes between water and ethanol.
For kinetic analysis of BV assembly with NIR FP apoproteins, purified apoprotein (15 μM) was mixed with 10 μM BV in PBS containing 1 mM DTT. Absorbance spectra were monitored immediately after mixing (in about 1 min) and at the indicated time points until the absorbance reached a steady state. For TEV protease digestion, the TEV recognition site flanked by Gly resides (–GENLYFQG–) was introduced into an unstructured protein region between the PAS and GAF domains. Purified NIR FPs were boiled for 5 min, cooled and then SelecTEV Protease (Lucigen) and DTT containing buffer were added. The reaction mixture was incubated overnight at 30 °C. The control samples contained no TEV protease.
Expression in mammalian cells
Mammalian expression plasmids pmiRFP670, pmiRFP703, pmiRFP709 and their mutants contained PCR-amplified AgeI–NotI gene fragments that were exchanged for the gene encoding EGFP in the pEGFP-N1 plasmid (Clontech). HeLa cells were grown in DMEM medium supplemented with 10% FBS, 0.5% penicillin–streptomycin and 2 mM glutamine (Life Technologies/Invitrogen). For fluorescence microscopy, cells were cultured in 35 mm glass-bottom Petri dishes with no. 1 coverglass (MatTek). Plasmid transfections were performed using an Effectene reagent (Qiagen).Fluorescence microscopy
Epifluorescence microscopy of live HeLa cells was performed 48 h after the transfection. HeLa cells were imaged using an Olympus IX81 inverted epifluorescence microscope equipped with a xenon-arc lamp Lambda LS (Sutter Instrument), 100× 1.4 NA oil immersion objective lens (UPlanSApo, Olympus), and Cy5.5 filter set (665/45 nm exciter and 725/50 nm emitter) (Chroma). SlideBook v.6 software (Intelligent Imaging Innovations) was used to operate the microscope.Flow cytometry
Fluorescence flow cytometry analysis was performed using a BD LSRII flow cytometer. HeLa cells co-transfected with EGFP and NIR FPs were analyzed using 488 nm and 640 nm laser excitation with 530/40 nm, 670/30 nm and 710/20 nm emission filters for detection. The cells were co-transfected with a plasmid coding for specific NIR FP and a plasmid coding for EGFP. To quantify fluorescence signals from cells, the mean fluorescence intensity in the NIR channel was divided by the mean fluorescence intensity of the same population of cells in the green channel, thus normalizing the NIR signal to the transfection efficiency. For each sample, 50000 events were analyzed. The NIR fluorescence intensity was also normalized to the excitation efficiency of each NIR FP with a 640 nm laser (% excitation relative to the excitation peak), and to the fluorescence signal of each NIR FP in the emission filter (the area of the emission spectra cut by the emission filter). The NIR effective brightness of miRFP670 was assumed to be 100%.
Protein crystallization
For crystallization, purified FPs were transferred to a buffer containing 20 mM Tris–HCl, 200 mM NaCl at pH 8.0 and concentrated to 16.3, 16.5, and 18.7 mg mL−1 for miRFP670, miRFP703 and miRFP709, respectively using Amicon Ultra-15 centrifugal filter units with 10 kDa molecular weight cutoff cellulose membrane (Millipore). A search for crystallization conditions was carried out using a Mosquito robotic crystallization system (TTP LabTech). Potentially promising crystallization conditions were further optimized using Hampton Research additive screens. Successful conditions were further optimized manually. Large-scale crystallization trials were performed using the hanging drop vapor diffusion method. Typically, 2 μL of the protein solution was mixed with 2 μL of the reservoir solution and incubated over 500 μL of the same reservoir solution at 20 °C for 2 weeks. The best crystals of miRFP670 were obtained from 3.4 M NaCl and 0.08 M Tris–HCl at pH 7.5. The best crystals of miRFP703 and miRFP709 were grown from 0.08 M Na citrate at pH 5.0, 24% v/v Jeffamine ED-2001 at pH 7.0, 0.4% n-octyl-β-D-glucopyranoside and 0.16 M MgCl2, 0.08 M Tris–HCl at pH 8.5, 20% w/v PEG 3350, and 3.2% 2,2,2-trifluoroethanol.Diffraction data collection and processing
X-ray diffraction data were collected on SER-CAT 22-BM and 22-ID beamlines (Advanced Photon Source, Argonne National Laboratory, Argonne, IL). Diffraction image intensities were registered on MAR 225 and Rayonix MX300HS CCD detectors. Prior to data collection the crystals were briefly soaked (5–10 s) in a cryoprotecting solution consisting of 20% glycerol and 80% of well solution and were flash-frozen in a 100 K nitrogen stream; throughout the diffraction experiment the cryogenic temperature was maintained by a CryoJetXL cooling device (Oxford Cryosystems). To minimize radiation damage of the crystals a helical data collection technique was used for all X-ray data acquisitions. Diffraction images were indexed, integrated and scaled with the HKL2000 software.53 The statistics and data processing are given in Tables S1 and S2.†Structure solution and refinement
Initial phases for miRFP670, miRFP703 and miRFP709 were obtained by the molecular replacement method with MOLREP54 using the structure of BphP1-FP/C20S (PDB ID: 4XTQ19) excluding its chromophore as a search model. To remove model bias, the chains were rebuilt with the ARP/wARP crystallographic molecular model building suite.55 Real space model correction and the search for the ordered solvent molecules was performed with COOT.56 Maximum likelihood structure refinement was performed with REFMAC.57 Structure validation was carried out with COOT and PROCHECK.58 The crystal structures of miRFP670, miRFP703 and miRFP709 were respectively refined to 1.33 Å, 1.35 Å and 1.34 Å. The crystals of miRFP670, miRFP703 and miRFP709 belonged to space groups P212121, P21, and P21, respectively (Tables S1 and S2†). The electron density of each p\FP was generally well defined for the entire molecule, except for the N-terminal His-tags and residues 9–18 (miRFP670), 1–18, and 315 (miRFP703, miRFP709) that were disordered. Superposition of the Cα atoms in miRFPs resulted in a root-mean-square deviation of only 1.0 Å, indicating a close similarity of all of the structures.Author contribution
M. B. and D. M. S. developed the proteins and their mutants and performed their biochemical characterization. S. P. crystallized the proteins and together with V. Z. P. performed their structural analysis. J. C. L. provided useful ideas and discussed the manuscript. V. V. V. planned and directed the project and together with M. B., S. P. and D. M. S. designed the experiments and analyzed the data. D. M. S. and V. V. V. wrote the manuscript.Acknowledgements
We thank Eric Giraud (Institute for Research and Development, Montpellier, France) for the RpBphP1 gene and Jinghang Zhang for assistance with flow cytometry. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. Research reported in this publication was supported in part with the US Federal funds from the National Cancer Institute of the National Institutes of Health (NIH) under contract HHSN261200800001E, the intramural research program of the NIH, the NIH grants GM068552 (J. C. L.), GM122567 and NS099573 (V. V. V.), the ERC-2013-ADG-340233 grant from EU FP7 (V. V. V.) and the 17-74-20100 grant from Russian Science Foundation (V. Z. P.). The content is solely the responsibility of the authors and does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.References
- D. M. Shcherbakova, M. Baloban and V. V. Verkhusha, Curr. Opin. Chem. Biol., 2015, 27, 52–63 CrossRef CAS PubMed.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed.
- J. R. Wagner, J. S. Brunzelle, K. T. Forest and R. D. Vierstra, Nature, 2005, 438, 325–331 CrossRef CAS PubMed.
- B. Karniol, J. R. Wagner, J. M. Walker and R. D. Vierstra, Biochem. J., 2005, 392, 103–116 CrossRef CAS PubMed.
- E. Giraud and A. Vermeglio, Photosynth. Res., 2008, 97, 141–153 CrossRef CAS PubMed.
- N. C. Rockwell, Y. S. Su and J. C. Lagarias, Annu. Rev. Plant Biol., 2006, 57, 837–858 CrossRef CAS PubMed.
- M. E. Auldridge and K. T. Forest, Crit. Rev. Biochem. Mol. Biol., 2011, 46, 67–88 CrossRef CAS PubMed.
- S. H. Bhoo, S. J. Davis, J. Walker, B. Karniol and R. D. Vierstra, Nature, 2001, 414, 776–779 CrossRef CAS PubMed.
- N. C. Rockwell and J. C. Lagarias, ChemPhysChem, 2010, 11, 1172–1180 CrossRef CAS PubMed.
- J. Kapitulnik and M. D. Maines, Front. Pharmacol., 2012, 3, 136 Search PubMed.
- X. Shu, A. Royant, M. Z. Lin, T. A. Aguilera, V. Lev-Ram, P. A. Steinbach and R. Y. Tsien, Science, 2009, 324, 804–807 CrossRef PubMed.
- M. T. Tran, J. Tanaka, M. Hamada, Y. Sugiyama, S. Sakaguchi, M. Nakamura, S. Takahashi and Y. Miwa, Exp. Anim., 2014, 63, 311–319 CrossRef CAS PubMed.
- X. Yang, J. Kuk and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14715–14720 CrossRef CAS PubMed.
- H. Takala, A. Bjorling, O. Berntsson, H. Lehtivuori, S. Niebling, M. Hoernke, I. Kosheleva, R. Henning, A. Menzel, J. A. Ihalainen and S. Westenhoff, Nature, 2014, 509, 245–248 CrossRef CAS PubMed.
- S. H. Wu and J. C. Lagarias, Biochemistry, 2000, 39, 13487–13495 CrossRef CAS PubMed.
- T. Lamparter, N. Michael, O. Caspani, T. Miyata, K. Shirai and K. Inomata, J. Biol. Chem., 2003, 278, 33786–33792 CrossRef CAS PubMed.
- T. Lamparter, M. Carrascal, N. Michael, E. Martinez, G. Rottwinkel and J. Abian, Biochemistry, 2004, 43, 3659–3669 CrossRef CAS PubMed.
- O. V. Stepanenko, G. S. Bublikov, D. M. Shcherbakova, V. V. Verkhusha, K. K. Turoverov and I. M. Kuznetsova, FEBS J., 2014, 281, 2284–2298 CrossRef CAS PubMed.
- D. M. Shcherbakova, M. Baloban, S. Pletnev, V. N. Malashkevich, H. Xiao, Z. Dauter and V. V. Verkhusha, Chem. Biol., 2015, 22, 1540–1551 CrossRef CAS PubMed.
- M. E. Auldridge, K. A. Satyshur, D. M. Anstrom and K. T. Forest, J. Biol. Chem., 2012, 287, 7000–7009 CrossRef CAS PubMed.
- K. C. Toh, E. A. Stojkovic, I. H. van Stokkum, K. Moffat and J. T. Kennis, Phys. Chem. Chem. Phys., 2011, 13, 11985–11997 RSC.
- J. T. Murphy and J. C. Lagarias, Curr. Biol., 1997, 7, 870–876 CrossRef CAS PubMed.
- A. J. Fischer and J. C. Lagarias, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17334–17339 CrossRef CAS PubMed.
- J. Zhu, D. M. Shcherbakova, Y. Hontani, V. V. Verkhusha and J. T. Kennis, Sci. Rep., 2015, 5, 12840 CrossRef CAS PubMed.
- S. Franzen and S. G. Boxer, J. Biol. Chem., 1997, 272, 9655–9660 CrossRef CAS PubMed.
- J. R. Wagner, J. Zhang, D. von Stetten, M. Gunther, D. H. Murgida, M. A. Mroginski, J. M. Walker, K. T. Forest, P. Hildebrandt and R. D. Vierstra, J. Biol. Chem., 2008, 283, 12212–12226 CrossRef CAS PubMed.
- H. Lehtivuori, I. Rissanen, H. Takala, J. Bamford, N. V. Tkachenko and J. A. Ihalainen, J. Phys. Chem. B, 2013, 117, 11049–11057 CrossRef CAS PubMed.
- A. J. Fischer, N. C. Rockwell, A. Y. Jang, L. A. Ernst, A. S. Waggoner, Y. Duan, H. Lei and J. C. Lagarias, Biochemistry, 2005, 44, 15203–15215 CrossRef CAS PubMed.
- D. Yu, W. C. Gustafson, C. Han, C. Lafaye, M. Noirclerc-Savoye, W. P. Ge, D. A. Thayer, H. Huang, T. B. Kornberg, A. Royant, L. Y. Jan, Y. N. Jan, W. A. Weiss and X. Shu, Nat. Commun., 2014, 5, 3626 CAS.
- S. Bhattacharya, M. E. Auldridge, H. Lehtivuori, J. A. Ihalainen and K. T. Forest, J. Biol. Chem., 2014, 289, 32144–32152 CrossRef CAS PubMed.
- D. Yu, M. A. Baird, J. R. Allen, E. S. Howe, M. P. Klassen, A. Reade, K. Makhijani, Y. Song, S. Liu, Z. Murthy, S. Q. Zhang, O. D. Weiner, T. B. Kornberg, Y. N. Jan, M. W. Davidson and X. Shu, Nat. Methods, 2015, 12, 763–765 CrossRef CAS PubMed.
- G. S. Filonov, K. D. Piatkevich, L. M. Ting, J. Zhang, K. Kim and V. V. Verkhusha, Nat. Biotechnol., 2011, 29, 757–761 CrossRef CAS PubMed.
- G. S. Filonov and V. V. Verkhusha, Chem. Biol., 2013, 20, 1078–1086 CrossRef CAS PubMed.
- D. M. Shcherbakova and V. V. Verkhusha, Nat. Methods, 2013, 10, 751–754 CrossRef CAS PubMed.
- K. D. Piatkevich, F. V. Subach and V. V. Verkhusha, Nat. Commun., 2013, 4, 2153 Search PubMed.
- D. M. Shcherbakova, M. Baloban, A. V. Emelyanov, M. Brenowitz, P. Guo and V. V. Verkhusha, Nat. Commun., 2016, 7, 12405 CrossRef CAS PubMed.
- J. R. Wagner, J. Zhang, J. S. Brunzelle, R. D. Vierstra and K. T. Forest, J. Biol. Chem., 2007, 282, 12298–12309 CrossRef CAS PubMed.
- X. Yang, E. A. Stojkovic, J. Kuk and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12571–12576 CrossRef CAS PubMed.
- D. Bellini and M. Z. Papiz, Structure, 2012, 20, 1436–1446 CrossRef CAS PubMed.
- O. V. Stepanenko, M. Baloban, G. S. Bublikov, D. M. Shcherbakova, O. V. Stepanenko, K. K. Turoverov, I. M. Kuznetsova and V. V. Verkhusha, Sci. Rep., 2016, 6, 18750 CrossRef CAS PubMed.
- Y. Hontani, D. M. Shcherbakova, M. Baloban, J. Zhu, V. V. Verkhusha and J. T. Kennis, Sci. Rep., 2016, 6, 37362 CrossRef CAS PubMed.
- K. A. Rumyantsev, D. M. Shcherbakova, N. I. Zakharova, A. V. Emelyanov, K. K. Turoverov and V. V. Verkhusha, Sci. Rep., 2015, 5, 18348 CrossRef CAS PubMed.
- H. Lehtivuori, S. Bhattacharya, N. M. Angenent-Mari, K. A. Satyshur and K. T. Forest, Front. Mol. Biosci., 2015, 2, 65 Search PubMed.
- D. Yu, Z. Dong, W. C. Gustafson, R. Ruiz-Gonzalez, L. Signor, F. Marzocca, F. Borel, M. P. Klassen, K. Makhijani, A. Royant, Y. N. Jan, W. A. Weiss, S. Guo and X. Shu, Protein Sci., 2016, 25, 308–315 CrossRef CAS PubMed.
- N. C. Lim and S. E. Jackson, J. Phys.: Condens. Matter, 2015, 27, 354101 CrossRef PubMed.
- N. C. Rockwell, S. L. Njuguna, L. Roberts, E. Castillo, V. L. Parson, S. Dwojak, J. C. Lagarias and S. C. Spiller, Biochemistry, 2008, 47, 7304–7316 CrossRef CAS PubMed.
- E. S. Burgie, J. M. Walker, G. N. Phillips Jr and R. D. Vierstra, Structure, 2013, 21, 88–97 CrossRef CAS PubMed.
- R. Narikawa, T. Ishizuka, N. Muraki, T. Shiba, G. Kurisu and M. Ikeuchi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 918–923 CrossRef CAS PubMed.
- N. C. Rockwell, S. S. Martin, K. Feoktistova and J. C. Lagarias, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11854–11859 CrossRef CAS PubMed.
- H. Scheer and K. H. Zhao, Mol. Microbiol., 2008, 68, 263–276 CrossRef CAS PubMed.
- K. D. Piatkevich, F. V. Subach and V. V. Verkhusha, Chem. Soc. Rev., 2013, 42, 3441–3452 RSC.
- R. Sens and K. H. Drexhage, J. Lumin., 1981, 24, 709–712 CrossRef.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- A. Vagin and A. Teplyakov, J. Appl. Crystallogr., 1997, 30, 1022–1025 CrossRef CAS.
- A. Perrakis, M. Harkiolaki, K. S. Wilson and V. S. Lamzin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 1445–1450 CrossRef CAS.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubak, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: All experimental details, crystallographic data collection and refinement statistics, full spectra, additional 6 figures and 2 tables. See DOI: 10.1039/c7sc00855d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |