
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal exchange in lithiocuprates: implications for our understanding of structure and reactivity†
Andrew J.
Peel
,
Ryan
Ackroyd
and
Andrew E. H.
Wheatley
 *
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW UK. E-mail: aehw2@cam.ac.uk; Fax: +44(0) 1223 336362
First published on 4th May 2017
Abstract
New reagents have been sought for directed ortho cupration in which the use of cyanide reagents is eliminated. CuOCN reacts with excess TMPLi (TMP = 2,2,6,6-tetramethylpiperidide) in the presence of limited donor solvent to give crystals that are best represented as (TMP)2Cu0.1Li0.9(OCN)Li2(THF) 8, whereby both Lipshutz-type lithiocuprate (TMP)2Cu(OCN)Li2(THF) 8a and trinuclear (TMP)2(OCN)Li3(THF) 8b are expressed. Treatment of a hydrocarbon solution of TMP2CuLi 9a with LiOCN and THF gives pure 8a. Meanwhile, formation of 8b is systematized by reacting (TMPH2)OCN 10 with TMPH and nBuLi to give (TMP)2(OCN)Li3(THF)211. Important to the attribution of lower/higher order bonding in lithiocuprate chemistry is the observation that in crystalline 8, amide-bridging Cu and Li demonstrate clear preferences for di- and tricoordination, respectively. A large excess of Lewis base gives an 8-membered metallacycle that retains metal disorder and analyses as (TMP)2Cu1.35Li0.659 in the solid state. NMR spectroscopy identifies 9 as a mixture of (TMP)2CuLi 9a and other copper-rich species. Crystals from which the structure of 8 was obtained dissolve to yield evidence for 8b coexisting in solution with in situ-generated 9a, 11 and a kinetic variant on 9a (i-9a), that is best viewed as an agglomerate of TMPLi and TMPCu. Moving to the use of DALi (DA = diisopropylamide), (DA)2Cu0.09Li0.91(Br)Li2(TMEDA)212 (TMEDA = N,N,N′,N′-tetremethylethylenediamine) is isolated, wherein (DA)2Cu(Br)Li2(TMEDA)212a exhibits lower-order Cu coordination. The preparation of (DA)2Li(Br)Li2(TMEDA)212b was systematized using (DAH2)Br, DAH and nBuLi. Lastly, metal disorder is avoided in the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 lithium amide:Lipshutz-type monomer adduct (DA)4Cu(OCN)Li4(TMEDA)213.
:1 lithium amide:Lipshutz-type monomer adduct (DA)4Cu(OCN)Li4(TMEDA)213.
Introduction
Interest in methods for refining the regioselective functionalization of aromatics by using more selective bases has grown significantly since the inception1 of what have become known as ‘synergic bases’.2 These heterobimetallic systems – in their simplest form RmM(NR′2)nAM (R = organyl; m = 0, 2, 3; M = less polarizing metal; NR′2 = amide; n = 1, 2, 3; AM = more polarizing (alkali) metal) – have afforded new levels of reactivity,3 regioselectivity4 and functional group tolerance5 not hitherto available through traditional main group organometallic bases. Recently, a fruitful area of synergic base chemistry that has developed has involved directed cupration using lithium cuprate reagents.6 The structural variability manifest in lithium cuprate chemistry, which derives in part from the potential to enhance reactivity by introducing an alkali metal salt, highlights the need to better understand structural variability in synergic systems. The corresponding elucidation of synergic bases was initially dominated by crystallographic determination.7 However, their heterobimetallic composition has led to the need for solution analysis to establish the nature of competition between structure retention of the heterobimetallic moiety versus the cooperative action of individually monometallic reagents8 and the permutations for dynamic deaggregation/recombination.7c,9First developed 50 years ago,10 lithium cuprates have been subsequently modified in two main ways. First, lithium amidocuprates have been developed, offering often unique reactivities as well as the potential of the amido group as a non-transferable ligand and as a chiral auxiliary.9,11 Second, yield enhancements have been sought by combining so-called Gilman lithium cuprates (R2CuLi) with lithium salts. For the deployment of LiCN as the latter,12 the resulting cuprates (R2Cu(CN)Li2) were coined Lipshutz cuprates and spectroscopic studies suggested the possibility that transfer of cyanide to Cu would give a higher order (tricoordinate) metal.13 However, the alternative of retention of lower order (dicoordinate) copper was suggested on theoretical,14 spectroscopic15 and, more recently, X-ray diffraction16 grounds. The isolation and characterization of lithium cyanocuprates 1a–c, which were prepared using amidolithium reagents with CuCN, revealed the bonding characteristics of Cu in systems that could demonstrate either higher or lower order structural properties. In line with theoretical expectations,14 lower order (TMP)2Cu(CN)Li2(L) (L = OEt2, THF, THP = tetrahydropyran)17,18 structures were observed crystallographically, with agglomeration giving essentially planar dimers based on lithium–nitrogen cores (Scheme 1). The observation that, whilst in other respects 1a–c pertained to Lipshutz cuprate characteristics, they lack a Cu–CN interaction led to investigation of the generality of this motif. To this end (TMP)2Cu(X)Li2(L) (X = Cl 2, Br 3, I 4, L = OEt22a–4a, THF 2b–4b) were isolated (Scheme 1).19,20 As with the previously reported cyanide structures, these revealed lower order Cu in the solid state. The extension of monoatomic bridges to the field of triatomic inorganic anions was achieved by the development of thiocyanatocuprates. These utilize SCN− in place of CN− and formed (TMP)2Cu(SCN)Li2(L) (L = OEt25a, THF 5b, THP 5c), which demonstrated 8-membered metallacyclic (LiSCN)2 cores supported by 6-membered N3CuLi2 rings in the solid state (Scheme 2). In these structures, significant solvent effects were noted through the adoption of either planar, boat or chair conformers for the 8-membered core depending on whether the cuprate was additionally stabilized by OEt2, THF or THP, respectively.21
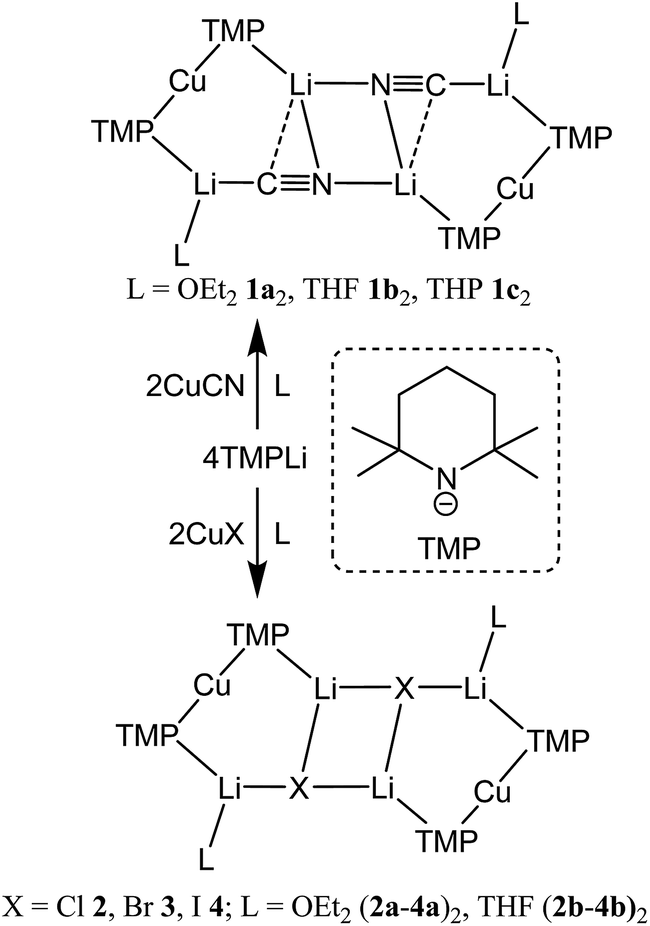 | ||
| Scheme 1 Formation of the dimers of cyano- and halogenocuprates 1–4. | ||
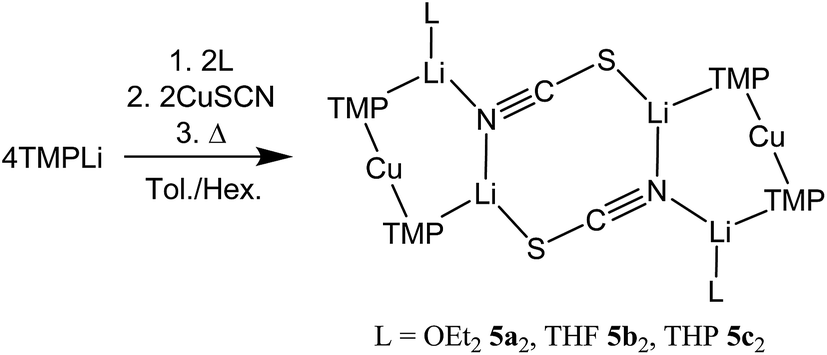 | ||
| Scheme 2 Formation of the dimers of thiocyanatocuprates 5. | ||
The recent observation that cyanide-incorporating Lipshutz cuprates exhibit solvent-controlled conversion to their Gilman counterparts7c,h by the abstraction of LiCN22 has been superceded by the discovery that this process is likely to be responsible for the in situ creation of active species in solution.23 This has reinvigorated interest in the properties of lithium amidocuprates and it has been noted that the ability of bimetallics of the type (R2N)2CuLi to dimerize, coupled with the acknowledged issue of metal exchange between Li and Cu, as elucidated by van Koten and co-workers,16a,b,24 means that the field of Gilman cuprate chemistry is closely interwoven with that of copper–nitrogen metallacycles. These have been observed to constitute tetrameric ketimides,25 hydrazides,25 phosphinimides26 mixed amides/guanidinates27 and amides9 with the last of these ligand-types also supporting 3D clusters.28 Copper amides, first prepared a century ago29 and more recently the focus of intense study,11a,30 have proven to be synthetically important, with solid state tetramers shown to deaggregate and participate in modified Ullmann amination31 with arenes in the presence of 1,10-phenanthroline.9 Meanwhile, Gilman lithium amidocuprates have been established to constitute the active species in directed aromatic cupration.17 However, synthetic studies having established that as-prepared Gilman cuprates are ineffectual, the need to convert Lipshutz(-type) cuprates to their Gilman counterparts in solution has been shown.23
The current work sees further efforts to study the use of polyatomic inorganic salts in lithium amidocuprate chemistry, through the deployment of the rarely used cyanate ligand. This accesses a range of new lithium cyanatocuprates. Metal disorder in some examples of these new materials sheds important new light on the predilection of Cu for lower/higher order structure formation. Advances are also reported in our understanding of the solution behaviour of lithium cuprates.
Results and discussion
Synthesis and solid state analysis
Cu(NO3)2(H2O)3 was reacted sequentially with LiOAc(H2O)2, and LiOCN (see ESI†). The addition of SO2 gave a green solution from which CuOCN 7 precipitated.32 The ability to prepare 7 offers the possibility of analyzing the cyanate analogue of recently reported (TMP)2Cu(SCN)Li2(THF).21 A hexane solution of TMPLi was therefore reacted with CuOCN in a 2:1 ratio in the presence of THF (1 eq. wrt Cu). This gave a modest yield of crystalline blocks. IR spectroscopy showed a strong peak at 2208 cm−1 and X-ray diffraction pointed to the formulation (TMP)2M(OCN)Li2(THF) but revealed disorder at the TMP-bridging metal site. The optimal crystallographic refinement suggests M = Cu0.1Li0.9, and therefore a 1:9 co-crystalline mixture of (TMP)2Cu(OCN)Li2(THF) 8a and (TMP)2(OCN)Li3(THF) 8b (Scheme 3 and Fig. 1).
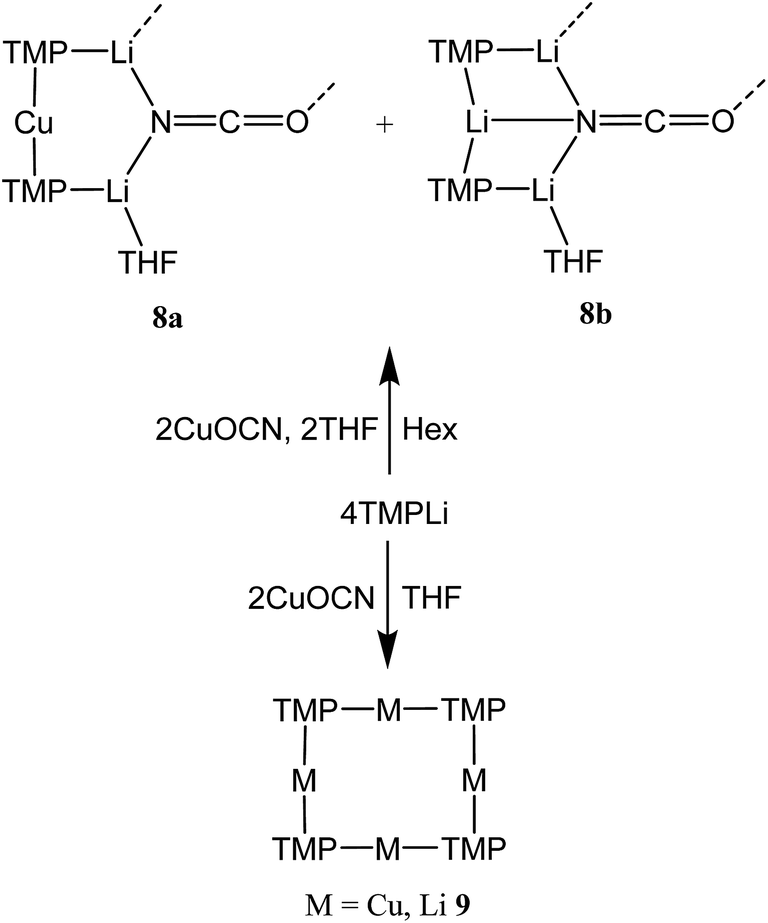 | ||
| Scheme 3 Solvent-dependent synthesis of 8a/b and 9. | ||
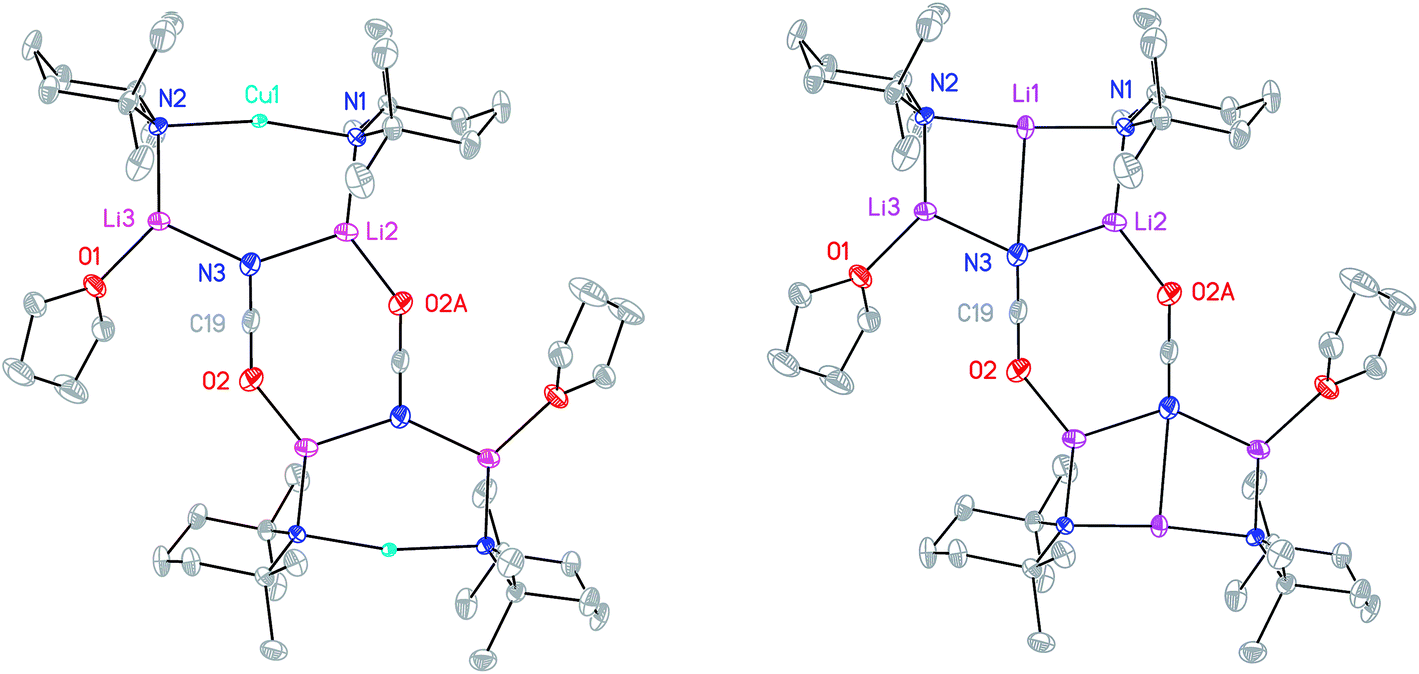 | ||
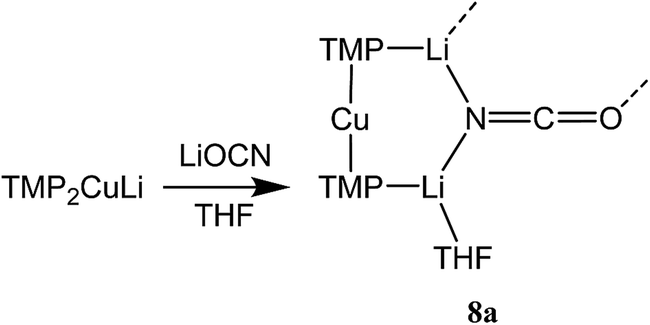
| Fig. 1 Molecular structures of 8a2 (left) and 8b2 (right, 30% probability and with H-atoms omitted). Selected bond lengths (Å) and angles (°): N1–Cu1 1.968(11), N2–Cu1 2.029(11), N3⋯Cu1 2.93(1), N1–Li1 1.970(16), N2–Li1 2.009(16), N3–Li1 2.597(11), N1–Li2 1.969(3), N2–Li3 1.935(3), N3–Li2 2.056(4), N3–Li3 2.040(4), O2–Li2A 1.904(3), O2–C19 1.211(3), N3–C19 1.168(3), N1–Cu1–N2 167.3(4), N1–Li1–N2 173.4(6), N1–Li1–N3 94.5(5), N2–Li1–N3 92.1(5). | ||
The X-ray diffraction structure of 8 reveals dimerization based on an 8-membered metallacyclic (LiOCN)2 core that incorporates a 3-coordinate Li centre (Li2). This core is supported by two 6-membered N3MLi2 rings, within which the coordination sphere of Li3 is completed by THF. However, in contrast to the previously noted THF-solvated thiocyanate 5b2,2182 reveals not a boat conformation but an essentially planar arrangement, with only TMP and THF deviating significantly from a plane defined by the three conjoined metallacycles and the O(THF)–Li bonds (ESI, Fig. S1†). Of most interest is the observation that whereas the cores of 8a2 and 8b2 are essentially identical, the geometries of the 6-membered N3MLi2 rings vary significantly. In 8a (M = Cu) the obtuse angle of 167.3(4)° at Cu1 results in a non-bonding N3⋯Cu1 distance of 2.93(1) Å. Insofar as this distance is consistent with a dicoordinate (lower) order Cu centre this bonding pattern is consistent with the recent characterization of Lipshutz cuprates 1a–c,17,18,21 halogenocuprates 2–4 (ref. 19 and 20) and thiocyanatocuprates 5a–c.21 In contrast, Li1 in the N3Li3 metallacycle of 8b is located significantly closer to N3 than is Cu1 in 8a, resulting in a reflex N1–Li1–N2 angle of 186.6(5)°. The observation of a N3–Li1 2.597(11) Å distance suggests a weak interaction between these two centres and yields a motif reminiscent of intercepted ladder-type structures reported in the past for amidolithium aggregates.33
Reasoning that the depletion of copper in 8 may have been caused by the rapid sequestration of in situ formed LiOCN by TMPLi, we attempted to prepare 8a by a route in which these two components were not present simultaneously. A solution of donor-free TMP2CuLi (9a) was therefore prepared using hydrocarbon solvent. This was combined with LiOCN and THF was introduced, later being replaced with hexane. The resulting solution gave radiating fans of crystals (Scheme 4). X-ray crystallography reveals the expected centrosymmetric dimer of 8a (ESI, Fig. S3†). Though pure 8a2 and its counterpart component of 82 are not strictly isostructural, the differences in molecular structure are small. In particular, marginally reduced planarity in pure 8a2 when compared to its partner component of 82, results in a larger value of mean deviation from the plane of 0.205 Å (excluding THF and TMP-carbons, cf. 0.085 Å for 8a2 in 82). These observations suggest that although weak, the N3⋯Li1 transannular interaction in component 8b2 of 82 exerts an influence, via crystal packing, over minor component 8a2 in the same structure.
 | ||
| Scheme 4 Synthesis of 8a by a route that eliminates the possibility of LiOCN abstraction by TMPLi. | ||
To attempt the synthesis of pure 8b, two different routes were pursued. The documented use of a secondary ammonium salt to generate a mixed lithium halide–lithium amide aggregate in situ34 led us to synthesize cyanate salt (TMPH2)OCN 10 (see ESI†). This was then reacted with TMPH and nBuLi in a 1:1:3 ratio. The deployment of bulk THF as reaction medium followed by its removal and replacement with hexane gave crystals. Alternatively, direct combination of LiOCN with TMPLi (1:2) in THF followed by recrystallization from hexane provided the same product. 1H NMR spectroscopy revealed these to comprise TMP and THF in a 1:1 ratio and IR spectroscopy gave a strong peak at 2207 cm−1. X-ray diffraction confirmed the formulation (TMP)2(OCN)Li3(THF)211 (Scheme 5), with dimerization occurring in the same way as with 8b (Fig. 1). The observation that this O–Li interaction is longer in 11 (O3A–Li2 1.968(3)°; Fig. 2) than in 8b is explained by additional THF rendering Li2 tetracoordinate. The result of this THF-inclusion on the wider structure is significant. With Li2 now pseudo-tetrahedral, 112 adopts a chair conformation in which only the core (LiOCN)2 ring occupies a single plane. In contrast to the structure of 82 (ESI, Fig. S1†) the lithium–nitrogen heterocycles and the THF molecules now deviate significantly above or below this central plane. Peripheral to the structure core, the geometry of the (NLi)3 arrangement in 11 imitates that in 8b, though the expanded N1–Li1–N2 reflex angle of 196.61(16)° allows a transannular interaction (N3–Li1 = 2.198(3) Å) shorter than that in 8b and similar to other N–Li bonds in the structure. A further point of contrast between 8b and 11 lies in the orientation of the amido groups. The ability of these to adopt different orientations for steric reasons has been noted previously in lithiocuprate systems.18 In the current work, the TMP ligands in 8b mimic the amido groups in lithiocuprate 5b whereby the 6-membered rings of the two TMP groups bonded to a Cu centre are each oriented away from one another and so lie flat or endo, endo with respect to the (LiOCN)2 structure core (Fig. 1). However, the inclusion of additional THF appears instrumental in causing one TMP ligand to adopt an upright orientation in 11,35 giving an exo, endo ligand pattern instead (Fig. 2).
 | ||
| Scheme 5 Lithiation of 10 and TMPH in bulk THF to give 11. | ||
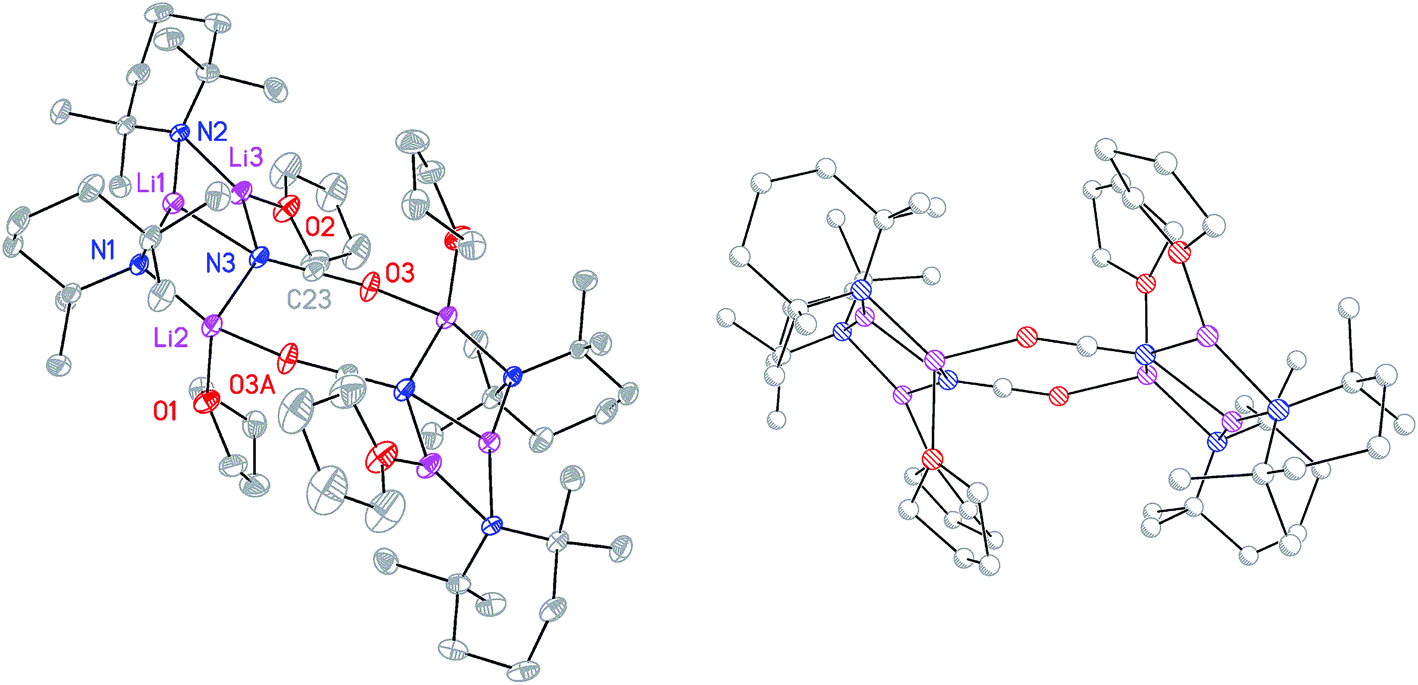 | ||
| Fig. 2 Molecular structure of 112 (30% probability and with H-atoms omitted). Selected bond lengths (Å) and angles (°): N1–Li1 2.029(3), N2–Li1 2.088(3), N3–Li1 2.198(3), N1–Li2 2.030(3), N2–Li3 1.930(3), N3–Li2 2.228(3), N3–Li3 2.036(3), O3A–Li2 1.968(3), O3–C27 1.208(2), N3–C27 1.172(2), N1–Li1–N2 163.39(16), N1–Li1–N3 100.56(12), N2–Li1–N3 95.39(12). | ||
The ability to isolate 8 proved solvent dependent; an equivalent synthesis in bulk THF (Scheme 3) yielded a crystalline material, 9, which analyses in the solid state as an 8-membered metallacyclic copper-rich material TMPm+nCumLin (in 9, m + n = 4; Fig. 3). The contribution of different species to the overall composition of 9 could only be elucidated by solution studies (see below). The structure shown in Fig. 3 can be rationalized in conjunction with the observation of that of 8; the excess THF solvating NCOLi and abstracting it from Lipshutz-type 8 to give 9. This view explains the deficiency of Cu in 8.
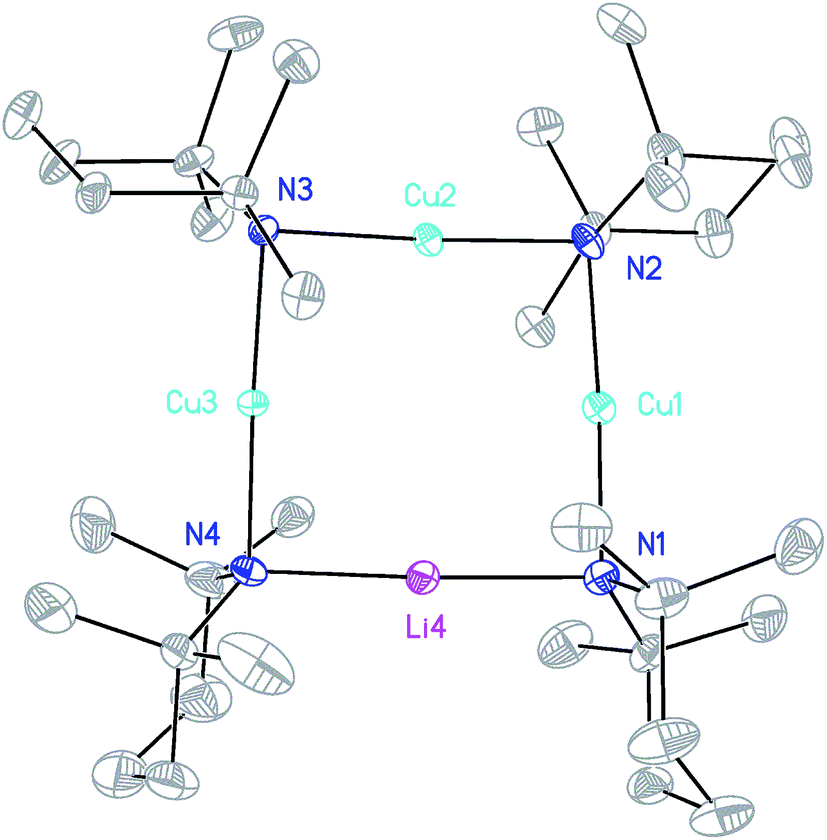 | ||
| Fig. 3 Molecular structure of 9 (30% probability and with H-atoms omitted, major metal occupancies shown). Selected bond lengths (Å): N1–M1 1.953(3), N2–M1 1.973(3), N2–M2 1.931(3), N3–M2 1.922(3), N3–M3 1.961(3), N4–M3 1.955(3), N4–M4 2.001(4), N1–M4 2.018(4). | ||
We next investigated the use of stronger Lewis bases as solvents for lithiocuprates. TMEDA (N,N,N′,N′-tetremethylethylenediamine) was introduced to 2:1 mixtures of amidolithium and Cu(I) reagents. Using DAH (= diisopropylamine), crystals were obtained incorporating different inorganic anions. As with prior work in the field of lithiocuprate chemistry, the use of CuBr as copper source enabled the isolation of a product containing both amide (in this case DA) and Lewis base (in this case TMEDA). This was achieved by treating DAH and TMEDA with nBuLi and CuBr in a 2:2:2:1 ratio in hydrocarbon media. 1H NMR spectroscopy suggested that the resulting isolable material incorporated DA and TMEDA in a 1:1 ratio, pointing to possible isolation of the Lipshutz-type cuprate (DA)2Cu(Br)Li2(TMEDA)2. X-ray diffraction revealed a more complex picture, displaying disorder at the amide-bridging metal site, with the best crystallographic refinement suggesting a Cu0.09Li0.91 population, i.e. a ca. 1:9 co-crystalline mixture of Lipshutz-type (DA)2Cu(Br)Li2(TMEDA)212a and (DA)2Li(Br)Li2(TMEDA)212b (collectively 12, Scheme 6).
 | ||
| Scheme 6 Anion-dependent synthesis of 12a/b and 13. | ||
X-ray diffraction for 12 reveals a monomer, with the bromide bridging two TMEDA-solvated Li+ centres to yield a 6-membered N2MLi2Br core. For M = Cu (Fig. 4, left) this is a motif known in bis(amido)lithiocuprate chemistry, though previously the use of monodentate Lewis bases yielded dimers,19–21 with the exception of (Ph2N)2Cu(NPh2)Li2(OEt2)2.11a In contrast to 8, but in common with previous reports of Lipshutz-type bis(amido)lithiocuprates incorporating 6-membered metallacycles, the Cu centre in 12a is essentially linear (N1–Cu1–N2 176.0(7)°) and the Cu1⋯Br1 distance (3.073(10) Å) is non-bonding. The dicoordinate (lower order) nature of Cu is highlighted by comparison with 12b, in which Li1 is located significantly closer to Br1 (2.624(14) Å) than Cu1 is in 12a. As with 8b, this displacement of the alkali metal results in a transannular interaction, the reflex N1–Li1–N2 angle being 208.4(8)°.
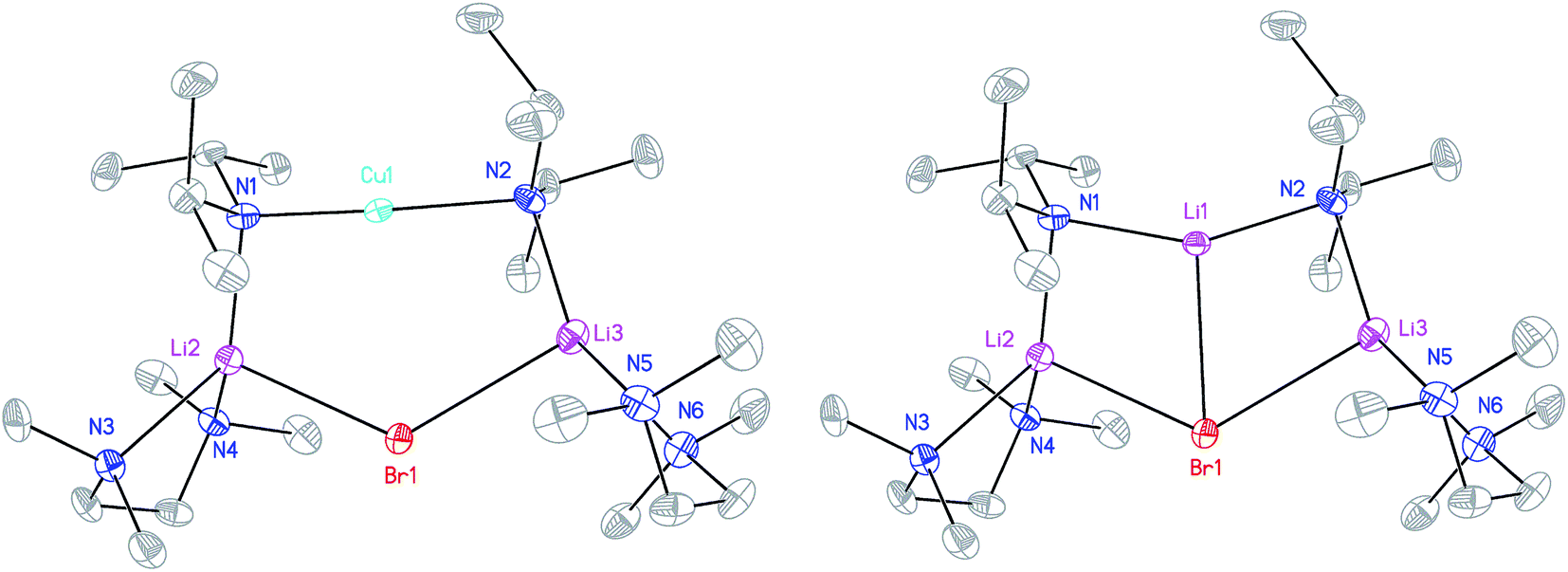 | ||
| Fig. 4 Molecular structure of the monomers of 12a and 12b (30% probability and with H-atoms omitted). Selected bond lengths (Å) and angles (°): N1–Cu1 1.823(13), N2–Cu1 2.064(13), Br1⋯Cu1 3.073(10), N1–Li1 2.009(16), N2–Li1 1.997(16), Br1–Li1 2.624(14), N1–Li2 2.047(9), N2–Li3 2.022(10), Br1–Li2 2.640(8), Br1–Li3 2.688(9), N1–Cu1–N2 176.0(7), N1–Li2–Br1 101.1(4), N2–Li3–Br1 102.5(4), Li2–Br1–Li3 125.5(3). | ||
The structure of 12b bears comparison with the highly unusual lithium amide–lithium chloride adduct (DA)2Li(Cl)Li2(TMEDA)2 (ref. 34) and, in the same way that the synthesis of amidolithium–cyanatolithium 8b could be systematized using the cyanate salt 10, it proved possible to achieve a synthesis of pure 12b by reacting (DAH2)Br with nBuLi in the presence of TMEDA (Scheme 7 and ESI, Fig. S4†). The removal of metal disorder to target pure cuprate 12a was also attempted; DAH was treated with nBuLi and TMEDA and then with CuOCN in hexane. The resulting crystalline material yielded NMR spectroscopic data that pointed to the presence of DA and TMEDA in a 2:1 ratio, while IR spectroscopy revealed a cyanate peak at 2208 cm−1. These data were inconsistent with the structure type exhibited by 12a and, in due course, X-ray diffraction revealed 13 to be a 2:1 adduct between DALi and the cuprate (DA)2Cu(OCN)Li2(TMEDA)2. Crystallography pointed to a complete lack of metal disorder at the DA-bridging position (Fig. 5a). It is immediately apparent that the behaviour of the cyanate ligand contrasts with that noted in 8a, where the bonding mode adopted by the ligand yielded both a 6-membered cuprate ring and facilitated dimerization. This variability in cyanate coordination is similar to that noted recently for the thiocyanate ligand21 and, in the current case, the cyanate ligand participates in the formation of an 8-membered N2CuLi2NCO ring. The capture of two molecules of DALi gives 4-membered N2Li2 and NOLi2 rings. Whereas 8a incorporates an essentially flat N2CuLi2N ring, the 8-membered metallacycle in 13 deviates significantly from planarity, allowing the DALi molecules to project above and below the plane incorporating the N–Cu–N unit and the cyanate C-centre (Fig. 5b).
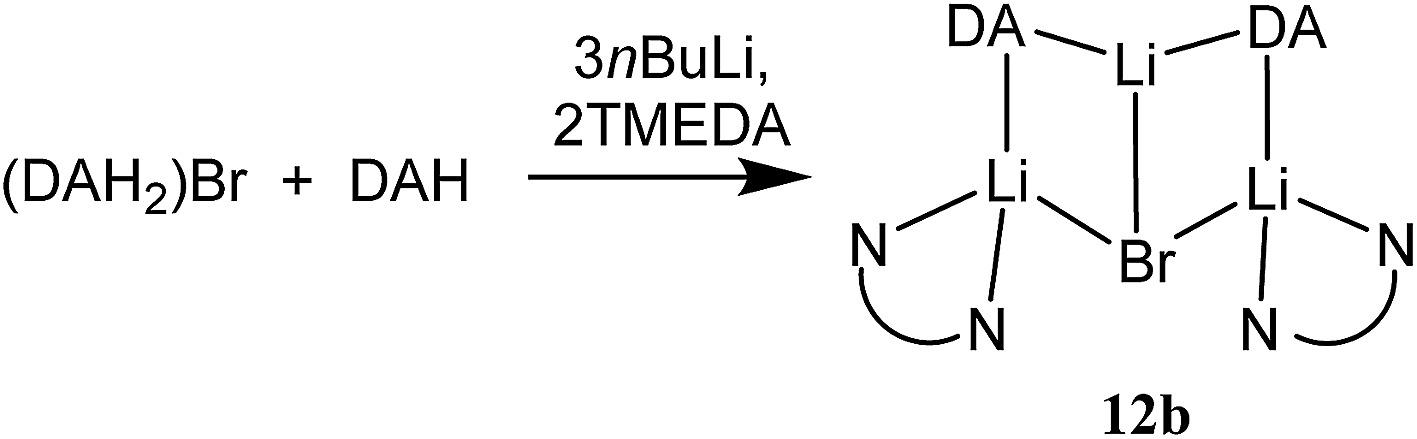 | ||
| Scheme 7 Lithiation of (DAH2)Br and DAH to give pure 12b. | ||
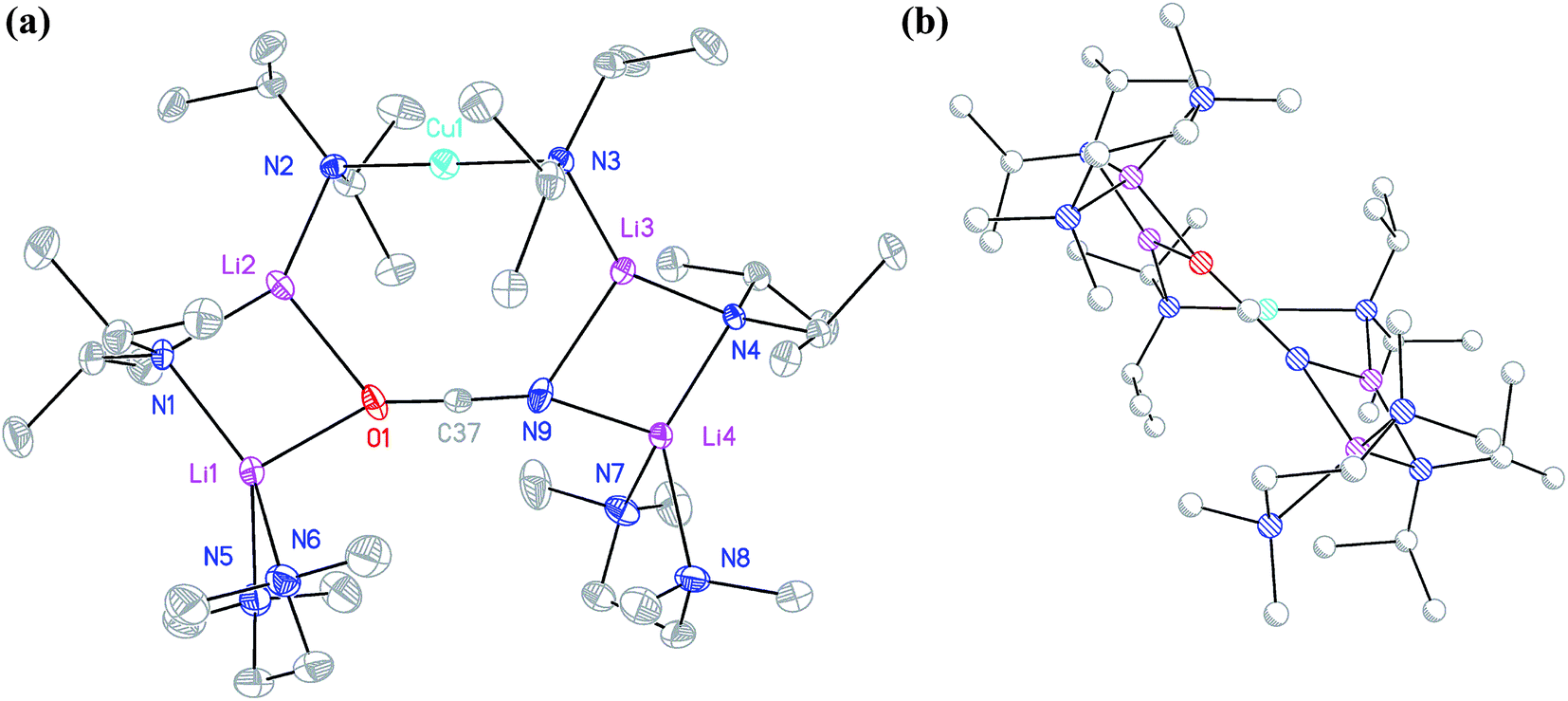 | ||
| Fig. 5 (a) Molecular structure of 13 (30% probability and with minor disorder and H-atoms omitted). Selected bond lengths (Å) and angles (°): N2–Cu1 1.910(2), N3–Cu1 1.909(2), N2–Li2 2.058(5), N1–Li2 1.991(5), N1–Li1 2.044(5), N3–Li3 2.053(5), N4–Li3 1.985(5), N4–Li4 2.048(5), N9–Li3 2.159(6), N9–Li4 2.026(5), O1–Li2 2.137(6), O1–Li1 2.009(5), N9–C37 1.202(4), O1–C37 1.205(4), N2–Cu1–N3 179.09(11), N2–Li2–O1 117.7(2), N3–Li3–N9 117.4(2), Li2–O1–C37 126.3(2), Li2–O1–Li1 81.6(2), Li2–N1–Li1 84.4(2), N1–Li1–O1 98.0(2), Li3–N9–C37 121.9(2), Li3–N9–Li4 81.3(2), Li3–N4–Li4 85.1(2), N4–Li4–N9 97.9(2); (b) 13 viewed along the C37⋯Cu1 vector and with the N3–Cu1–N2 unit oriented horizontally. | ||
NMR spectroscopy
Interpretation of the convoluted solution behaviour for the systems described above has been attempted. It was anticipated that compound 8 would yield two species in solution – 8a and 8b. In the event, NMR spectroscopy of the bulk material could not be reconciled with this simple model of metal disorder. 7Li NMR spectroscopy, for example, revealed multiple solution species (Fig. 6). With NMR spectra of 9a reported previously,19 its presence at as a minor component of this mixture at δ 0.90 ppm was easily established. To aid the assignment of other components, the spectroscopy of (TMP)2(OCN)Li3(THF)211 was examined. The reproducible in situ reformation of a limited amount of TMPH in all TMP-based systems is attributed to the presence of trace moisture in the deuterated solvent in spite of its storage over molecular sieves (3 Å). This notwithstanding, 13C NMR spectroscopy revealed the existence of 11 as the only species observable in solution (Fig. 7).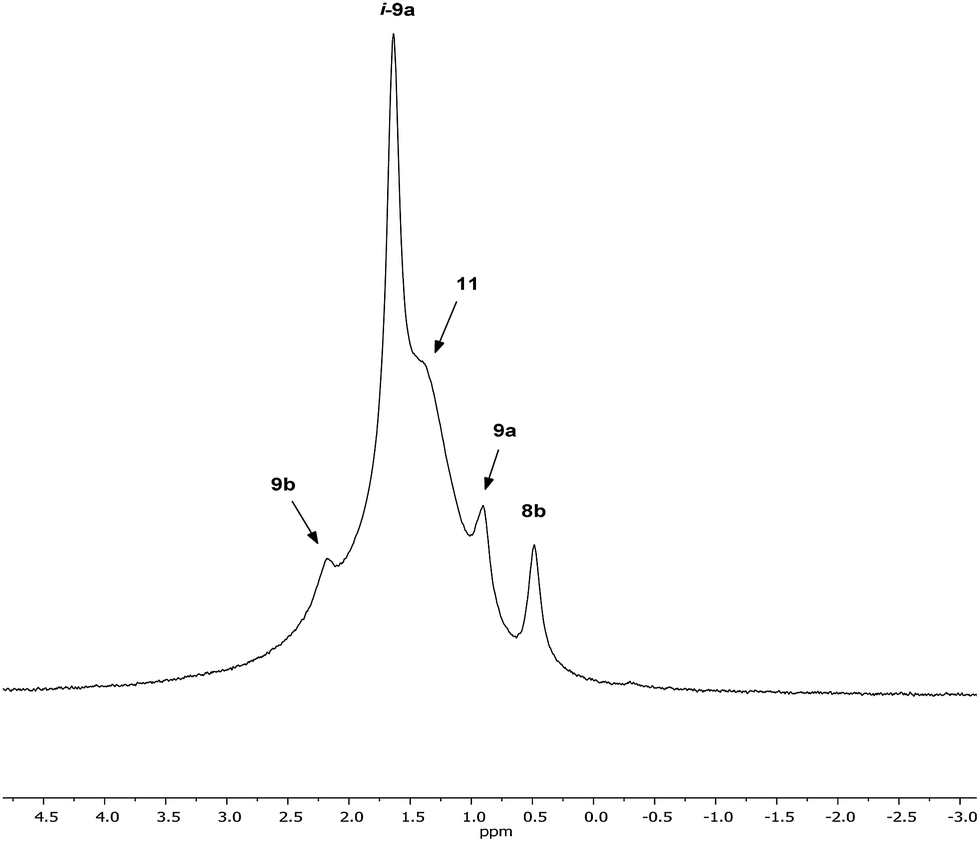 | ||
| Fig. 6 7Li NMR spectrum of bulk 8 in C6D6. | ||
 | ||
| Fig. 7 13C NMR spectrum of 11 in C6D6 (main); 7Li NMR spectrum (inset). *TMPH. | ||
Returning to the 7Li NMR spectrum of bulk 8, the broad features at δ 2.18 and 1.41 ppm can now be accounted for by 11 and, by comparison with an authentically prepared sample (see ESI†), 9b, respectively, leaving two signals to be identified at δ 1.64 ppm and at δ 0.48 ppm. To assign the first of these, 1H NMR spectroscopy was used. Firstly, THF notwithstanding, the 1H NMR spectrum of 11 was dominated by three singlets, at δ 1.76, 1.57 and 1.39 ppm in a 1:2:1 integral ratio. HSQC spectroscopy identified these as belonging to TMP-Me groups, and comparison with the TMP-Me resonances in authentic samples of 9a–c (see ESI†) revealed that each of the aforementioned singlets falls within the expected chemical shift range for these compounds. Overall, these data suggest an isomeric variant of the previously reported dimer of Gilman cuprate 9a (Fig. 8a).19 We propose inverse-9a (i-9a); a tetranuclear metallacycle incorporating adjacent pairs of Li and Cu centres (Fig. 8b) best viewed as resulting from the agglomeration of dimers of TMPLi 9b and TMPCu 9c. The arrangement of i-9a yields three distinct TMP environments (Fig. 8). We hypothesise i-9a to be a kinetic product since it results formally from the catenation of dinuclear units of 9b and 9cwithout requiring the formation of Gilman cuprate 9a. This thesis suggests that under suitable conditions, i-9a will rearrange to 9a2. To test this, a typical sample of bulk 8 was heated to reflux and allowed to stand at room temperature. Fine needles crystallized, which were shown to be (thermodynamically preferred) Gilman cuprate by NMR spectroscopy.
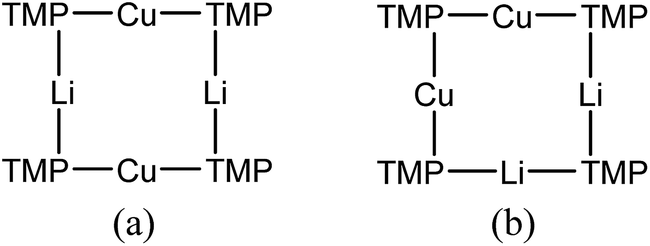 | ||
| Fig. 8 (a) The dimer of 9a reveals one distinct TMP environment, whereas (b) dimers of 9b and 9c can aggregate to give i-9a, which reveals three. | ||
Lastly, the minor signal at δ 0.48 ppm in Fig. 6 most logically belonged to either 8a or 8b. The spectroscopic analysis of pure 8a was therefore carried out. This revealed a system whose behaviour paralleled that of previously reported 5a,21 with the deposition of a fine white powder occurring upon sample dissolution in C6D6 (presumed LiOCN in this case). Except for the presence of THF, the signals matched those of 9a (7Li NMR δ 0.90 ppm). This suggests that the signal at δ 0.48 ppm in Fig. 6 is due to 8b not 8a.
Both the metal disorder in single crystal 8 and the presence of multiple products in the bulk reaction mixture are unusual features in amidocuprate systems. However, they can be explained by reactions (1)–(3) below. In (1), CuOCN reacts with 9b to expel LiOCN. This is rapidly sequestered in (2) at a rate competitive with (3). Remaining 9b reacts with in situ formed 9c(3) to then generate any compound in the series TMPm+nCumLin (e.g.i-9a). The failure to isolate i-9a in previous lithium amidocuprate work is best explained by a combination of the kinetic properties of i-9a and the ability of 9a (but not i-9a) to also be generated by the dissociation of Lipshutz-type cuprates.
Formation of TMPCu 9c
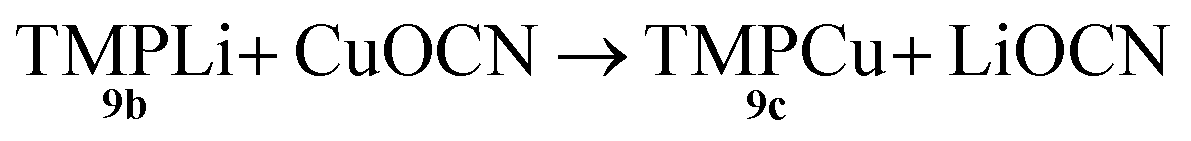 | (1) |
Formation of TMP2Li3OCN(THF)211
 | (2) |
Formation of TMP2CuLi 9a and i-9a
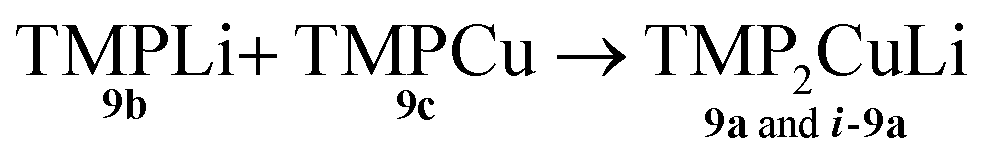 | (3) |
Moving to the behaviour of lithium amidocuprates in more polar reaction media, the 2:1 reaction of TMPLi and CuOCN in THF followed by recrystallization from hexane gave 9, which crystallographic refinement indicated to be rich in Cu (Fig. 3), though the individual contributions of TMPm+nCumLin could not be established by solid state analysis. In contrast, 7Li NMR spectroscopy in C6D6 (see ESI†) identified three Li-containing species in solution: a minor signal at δ 1.65 ppm which matched i-9a, a dominant resonance at δ 0.91 ppm corresponding to 9a and a shoulder at δ 0.94 ppm. Since 9a and i-9a contain equal amounts of Cu and Li, it was anticipated that the unidentified species should be rich in Cu to ensure that the sample was Cu-rich overall. Strong contenders include TMP3Cu2Li and TMP4Cu3Li – since literature precedents exist for trimeric and tetrameric variants of 9b36–38 and 9c.39,40
Lastly, the solution behaviour of co-crystalline (DA)2Cu(Br)Li2(TMEDA)212a/(DA)2Li(Br)Li2(TMEDA)212b was probed in conjunction with that of pure 12b. The data for pure 12b proves relatively straightforward. It suggests one species dominant in solution (alongside minimal reformation of DAH,41Fig. 9). Most clearly, 7Li NMR spectroscopy reveals signals in the expected 1:2 ratio at δ 2.28 and 1.26 ppm (ESI, Fig. S11c†). Moving to co-crystalline 12, peaks corresponding to 12b are added to by signals for 12a (ESI, Fig. S12a–f†). 1H NMR spectroscopic data for multiple samples of 12 clearly show the levels of 12a and 12b to be variable (Fig. 9), pointing to the possibility of incorporating higher levels of Cu than evidenced by X-ray crystallography.
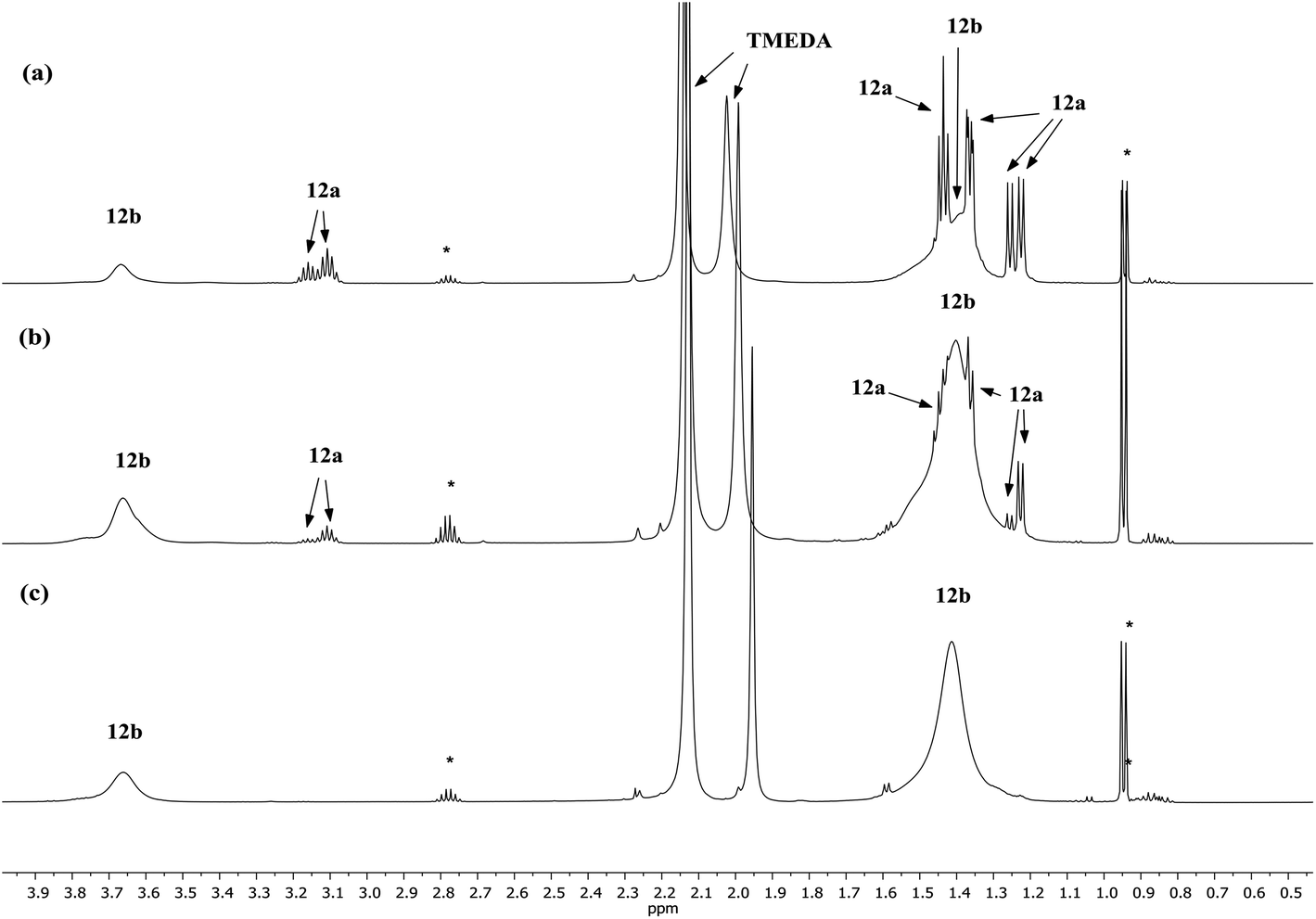 | ||
| Fig. 9 1H NMR spectra in C6D6 of (a) 12 (b) 12 (c) 12b. *DAH. | ||
Conclusions
The recent development of thiocyanatocuprate reagents has been extended to yield cyanatocuprates. Copper–lithium exchange in this new field has been evidenced by the observation of co-crystalline products of the type (TMP)2M(OCN)Li2 where M = Cu, Li. An analysis of the geometry at M reinforces the view, recently expressed for X = CN, halide, and SCN,21,23 that in (amido)2M(X)Li2 systems (X = inorganic anion) Cu prefers a linear, dicoordiante geometry (viz.8). This disinclination for higher order structure formation is emphasised when, for M = Li, a tendency for transannulation emerges and M becomes tricoordinate within the same structure-type (viz.11). These data are reinforced by attempts to fabricate monomeric Lipshutz-type cuprates, with Cu linear in (DA)2Cu(Br)Li2(TMEDA)212a and amidolithium-lithiocuprate adduct 13 and Li trigonal in (DA)2(Br)Li3(TMEDA)212b.Solution studies have helped elucidate the complex chemistry on offer. In particular, insights into the behaviour of bulk 8 and also of 11 establish the in situ formation of Gilman cuprate 9a. We show that this can form directly or through the rearrangement of a newly observed species (i-9a) that we propose to form kinetically from the combination of amidolithium 9b and amidocopper 9c. In the case of 12, results from crystallographic refinement could be combined with spectroscopic studies of 12b to distinguish copper-containing Lisphutz-type monomers from their lithium-only congeners. Work is being initiated to deconvolute the complex solution chemistry of 13 and to establish the synthetic portfolio of cyantocuprates. This is focusing on enhancing atom efficiency in directed ortho cupration19 and halopyridine21 derivatization by combining Gilman dimer 9a2 with substoichiometric LiOCN. The current work demonstrates, for the first time, the ability of an inorganic salt to combine with a dimeric Gilman cuprate to yield a Lipshutz-type dimer. Taking this together with previous work establishing Lipshutz-type dimers as a source of reactive Gilman monomers19 alongside reformed lithium salt, we are now testing the non-stoichiometric deployment of LiOCN with a view to furthering recent interest in transferring main group polar organometallic chemistry to the catalytic regime.7d,42
Experimental section
General synthetic and analytical details
Reactions were carried out under dry nitrogen, using double manifold and glove-box methods. Solvents were distilled off sodium (toluene) or sodium–potassium amalgam (THF, hexane) immediately before use. 2,2,6,6-Tetramethylpiperidine (TMPH) was purchased from Alfa Aesar and stored over molecular sieves (4 Å). Other chemicals were used as received. nBuLi (1.6 M in hexanes) was purchased from Acros and used as received. For details of the syntheses of Ba(OCN)26,32 CuOCN 7 (ref. 32) and (TMPH2)OCN 10 see the ESI.† The syntheses of reference materials TMPLi 9b and TMPCu 9c were based on the literature36,43 and details are provided in the ESI.† IR spectra were collected on a Perkin Elmer Spectrum One FT IR spectrometer. The abbreviations used are: m = medium, s = strong. NMR data were collected on a Bruker Avance III HD 500 MHz Smart Probe FT NMR spectrometer (500.200 MHz for 1H, 125.775 MHz for 13C, 194.397 for 7Li). Spectra were obtained at 25 °C using deuterated solvent stored over molecular sieves (3 Å). For 1H and 13C, chemical shifts are internally referenced to deuterated solvent and calculated relative to TMS. For 7Li, an external reference was used (1 M LiCl in D2O). Chemical shifts are expressed in δ ppm. The following abbreviations are used: br = broad, m = multiplet, s = singlet, sh = shoulder.Crystallographic details
For details of data collections see Table 1. Crystals were transferred from the mother liquor to a drop of perfluoropolyether oil mounted upon a microscope slide under cold nitrogen gas.44 Suitable crystals were attached to the goniometer head via a MicroLoop™, which was then centred on the diffractometer. Data were collected on a Bruker D8 Quest (Cu-Kα, λ = 1.54184 Å), each equipped with an Oxford Cryosystems low-temperature device (T = 180(2) K). Structures were solved using SHELXT,45 with refinement, based on F2, by full-matrix least squares.46 Non-hydrogen atoms were refined anisotropically (for disorder, standard restraints and constraints were employed as appropriate) and a riding model with idealized geometry was employed for the refinement of H-atoms. Crystals of 8a grew as two-component non-merohedral twins (see ESI†). Data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications CCDC 1540281 (8), 1540280 (8a), 1540286 (9), 1540282 (11), 1540284 (12), 1540285 (12b) and 1540283 (13).| 8 2 | 8a 2 | 9 | 11 2 | 12 | 12b | 13 | |
|---|---|---|---|---|---|---|---|
| a Based on agreement between observed single and composite intensities and those calculated from refined unique intensities and twin fractions. | |||||||
| Formula | C46H88Cu0.21Li5.79N6O4 | C46H88Cu2Li4N6O4 | C36H72Cu2.7Li1.3N4 | C54H104Li6N6O6 | C24H60BrCu0.09Li2.91N6 | C24H60BrLi3N6 | C37H88CuLi4N9O |
| M | 842.74 | 944.06 | 741.41 | 975.07 | 538.60 | 533.51 | 766.46 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c |
P |
P21/c | P21/c |
P |
| a | 7.9156(3) | 8.4118(4) | 11.6661(3) | 11.8638(3) | 17.7591(8) | 17.7718(4) | 11.9447(8) |
| b | 13.4775(5) | 11.5807(5) | 22.7624(5) | 12.0430(3) | 12.2389(6) | 12.1985(3) | 13.7635(9) |
| c | 13.8111(6) | 13.5645(6) | 15.2664(4) | 12.2033(3) | 17.2889(8) | 17.2945(5) | 16.4214(11) |
| α | 110.986(2) | 98.376(2) | 90 | 82.4990(10) | 90 | 90 | 80.989(3) |
| β | 94.546(2) | 95.058(2) | 108.6770(10) | 66.9060(10) | 118.227(2) | 118.2370(10) | 78.164(3) |
| γ | 104.331(2) | 94.955(2) | 90 | 72.4890(10) | 90 | 90 | 67.824(3) |
| V | 1309.70(9) | 1295.58(10) | 3840.48(17) | 1529.35(7) | 3310.9(3) | 3303.09(15) | 2437.5(3) |
| Z | 1 | 1 | 4 | 1 | 4 | 4 | 2 |
| ρ calcd | 1.068 | 1.210 | 1.282 | 1.059 | 1.081 | 1.073 | 1.044 |
| μ | 0.583 | 1.344 | 1.952 | 0.511 | 1.878 | 1.823 | 0.881 |
| Data | 17570 |
4430 | 19840 |
16485 |
25251 |
26193 |
35010 |
| Unique data | 4631 | 4430 | 5479 | 5374 | 5837 | 5811 | 8630 |
| R int | 0.0395 | 0.0361a | 0.0469 | 0.0303 | 0.0843 | 0.0562 | 0.0420 |
| θ (°) | 3.488–66.840 | 3.311–66.793 | 3.621–59.106 | 3.849–66.691 | 2.824–66.842 | 2.822–66.687 | 2.760–66.920 |
| wR2 | 0.1388 | 0.1026 | 0.1167 | 0.1400 | 0.1524 | 0.0901 | 0.1881 |
| R | 0.0515 | 0.0351 | 0.0501 | 0.0502 | 0.0825 | 0.0414 | 0.0572 |
| GoF | 1.026 | 1.082 | 1.055 | 1.035 | 1.117 | 1.023 | 1.048 |
| Parameters | 298 | 289 | 417 | 343 | 317 | 307 | 513 |
| Peak/hole (eÅ−3) | 0.431/−0.450 | 0.261/−0.452 | 0.787/−0.382 | 0.535/−0.313 | 0.510/−0.632 | 0.277/−0.345 | 0.547/−0.874 |
Synthesis and characterization of (TMP)2Cu0.1Li0.9(OCN)Li2(THF) 8
To a stirred solution of TMPH (0.34 mL, 2 mmol) and THF (0.08 mL, 1 mmol) in hexane (4 mL) was added nBuLi (1.25 mL, 1.6 M in hexanes, 2 mmol) at −78 °C. The mixture was returned to room temperature to give a yellow solution. This was added to a suspension of CuOCN (0.11 g, 1 mmol) in hexane (1 mL) at −78 °C. The mixture was returned to room temperature to give a pale yellow suspension. Filtration gave a yellow solution and storage at −27 °C gave crystalline material from which a well-faceted, block-like crystal of (TMP)2Cu0.1Li0.9(OCN)Li2(THF) was selected for X-ray diffraction. The following characterization refers to the bulk crystalline product. Yield 54 mg, melting point dec. from 125 °C. Selected IR spectroscopy (nujol)![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) 2208 (s, CN), 1340 (m, CO), 1229 (m, CO) cm−1. 1H NMR spectroscopy (500 MHz, C6D6) δ 3.56 (m, 8H, THF), 2.17–1.77 (br, 8H, TMP-4), 1.75 (s, 6H, TMP-Me), 1.74–1.60 (br, m, 12H, TMP-3,5, TMP-Me), 1.57 (s, 12H, TMP-Me), 1.56–1.41 (br, m, 12H, TMP-3,5, TMP-4, TMP-Me), 1.39 (s, 6H, TMP-Me), 1.36 (m, 8H, THF), 1.34–1.13 (br, 12H, TMP-3,5), 1.12–1.07 (m, 4H, TMP-3,5), 1.06 (s, 2H, TMPH-Me). 13C NMR (125 MHz, C6D6) δ 64.8 (THF), 56.9 (TMP-2,6 i-9a), 54.2 (TMP-2,6 i-9a/TMP-2,6 9a), 53.4 (TMP-2,6 8), 52.0 (TMP-2,6 i-9a), 51.6 (TMP-2,6 11), 49.2 (TMPH-2,6), 42.6 (TMP-3,5 i-9a), 42.5 (TMP-3,5 i-9a), 42.4 (TMP-3,5 8), 42.1 (TMP-3,5 i-9a/TMP-3,5 9a), 41.9 (TMP-3,5 11), 40.5 (TMP-Me 8), 40.2 (TMP-Me 9a), 39.7 (TMP-Me i-9a), 38.1 (TMPH-3,5), 37.5 (br, TMP-Me 11), 37.0 (TMP-Me i-9a), 36.8 (TMP-Me i-9a), 35.7 (TMP-Me 8), 34.5 (TMP-Me 9a), 34.2 (TMP-Me i-9a), 33.5 (br, TMP-Me 11), 31.6 (TMPH-Me), 25.1 (THF), 20.1 (TMP-4 11), 19.6 (TMP-4 i-9a), 19.2 (TMP-4 i-9a/TMP-4 9a), 19.1 (TMP-4 8), 19.1 (TMP-4 i-9a), 18.4 (TMPH-4). 7Li NMR (194 MHz, C6D6) δ 2.17 (br, s, 1Li, 9b), 1.64 (s, 1.5Li, i-9a), 1.39 (br, s, 2Li, 11), 0.90 (s, 0.5Li, 9a), 0.48 (s, 0.5Li, 8).
2208 (s, CN), 1340 (m, CO), 1229 (m, CO) cm−1. 1H NMR spectroscopy (500 MHz, C6D6) δ 3.56 (m, 8H, THF), 2.17–1.77 (br, 8H, TMP-4), 1.75 (s, 6H, TMP-Me), 1.74–1.60 (br, m, 12H, TMP-3,5, TMP-Me), 1.57 (s, 12H, TMP-Me), 1.56–1.41 (br, m, 12H, TMP-3,5, TMP-4, TMP-Me), 1.39 (s, 6H, TMP-Me), 1.36 (m, 8H, THF), 1.34–1.13 (br, 12H, TMP-3,5), 1.12–1.07 (m, 4H, TMP-3,5), 1.06 (s, 2H, TMPH-Me). 13C NMR (125 MHz, C6D6) δ 64.8 (THF), 56.9 (TMP-2,6 i-9a), 54.2 (TMP-2,6 i-9a/TMP-2,6 9a), 53.4 (TMP-2,6 8), 52.0 (TMP-2,6 i-9a), 51.6 (TMP-2,6 11), 49.2 (TMPH-2,6), 42.6 (TMP-3,5 i-9a), 42.5 (TMP-3,5 i-9a), 42.4 (TMP-3,5 8), 42.1 (TMP-3,5 i-9a/TMP-3,5 9a), 41.9 (TMP-3,5 11), 40.5 (TMP-Me 8), 40.2 (TMP-Me 9a), 39.7 (TMP-Me i-9a), 38.1 (TMPH-3,5), 37.5 (br, TMP-Me 11), 37.0 (TMP-Me i-9a), 36.8 (TMP-Me i-9a), 35.7 (TMP-Me 8), 34.5 (TMP-Me 9a), 34.2 (TMP-Me i-9a), 33.5 (br, TMP-Me 11), 31.6 (TMPH-Me), 25.1 (THF), 20.1 (TMP-4 11), 19.6 (TMP-4 i-9a), 19.2 (TMP-4 i-9a/TMP-4 9a), 19.1 (TMP-4 8), 19.1 (TMP-4 i-9a), 18.4 (TMPH-4). 7Li NMR (194 MHz, C6D6) δ 2.17 (br, s, 1Li, 9b), 1.64 (s, 1.5Li, i-9a), 1.39 (br, s, 2Li, 11), 0.90 (s, 0.5Li, 9a), 0.48 (s, 0.5Li, 8).
Synthesis and characterization of (TMP)2Cu(OCN)Li2(THF) 8a
To a stirred solution of TMPH (0.68 mL, 4 mmol) in hexane (2 mL) and toluene (2 mL) was added nBuLi (2.5 mL, 1.6 M in hexanes, 4 mmol) at −78 °C. The solution was warmed to room temperature and transferred to a suspension of CuSCN (0.243 g, 2 mmol) in hexane (2 mL) and toluene (2 mL). The suspension was warmed to room temperature then heated to reflux whereupon a grey discolouration was observed. To remove LiSCN, this mixture was filtered whilst hot onto LiOCN (0.10 g, 2 mmol) and the solvent was removed in vacuo. THF (6 mL) was added and the suspension was stirred at room temperature for 30 minutes, during which time partial dissolution of the LiOCN occurred. The THF was removed in vacuo to give a sticky solid, which dissolved when hexane (6 mL) was added. The solution was filtered and the filtrate stored at −27 °C for 1 week during which time 8a deposited as radiating fans of crystals. Yield 310 mg (33% wrt. CuSCN), melting point 192–194 °C. Elemental analysis C24H48CuLi2N3O2, requires (%) C, 58.52; H, 9.40; N, 8.90. Found (%) C, 58.06; H, 9.38; N, 9.01. Selected IR spectroscopy (nujol) 2241 (s, CN), 2208 (s, CN), 1340 (m, CO), 1228 (m, CO) cm−1. 1H NMR spectroscopy (500 MHz, C6D6) δ 3.57 (br, m, 4H, THF), 1.89–1.76 (br, m, 2H, TMP-4), 1.67–1.61 (m, 4H, TMP-3,5), 1.60 (s, 12H, TMP-Me), 1.59–157 (m, 2H, TMP-4), 1.56 (s, 12H, TMP-Me), 1.41 (br, m, 4H, THF), 1.09 (m, 4H, TMP-3,5), 1.06 (s, 1.3H, TMPH-Me). 13C NMR spectroscopy (125 MHz, C6D6) δ 67.4 (br, THF), 54.2 (TMP-2,6), 49.2 (TMPH-2,6), 42.1 (TMP-3,5), 40.1 (TMP-Me), 38.2 (TMPH-3,5), 34.5 (TMP-Me), 31.6 (TMPH-Me), 25.3 (br, THF), 19.2 (TMP-4), 18.4 (TMPH-4). 7Li NMR spectroscopy (194 MHz, C6D6) δ 0.90.
Synthesis and characterization of (TMP)2Cu1.35Li0.659
To a stirred solution of TMPH (0.34 mL, 2 mmol) and THF (2 mL) was added nBuLi (1.25 mL, 1.6 M in hexanes, 2 mmol) at −78 °C. The mixture was left to reach room temperature. The resulting yellow solution was transferred to a suspension of CuOCN (0.11 g, 1 mmol) in THF (1 mL) at −78 °C. The mixture was returned to room temperature to give a pale yellow suspension. The solvent was removed and the resulting yellow solid dissolved in hexane (4 mL). Filtration gave a yellow solution that was stored at −27 °C to give a crystalline aggregate which analysed as (TMP)2Cu1.35Li0.65 by X-ray diffraction. Yield 65 mg. 1H NMR spectroscopy (500 MHz, 298 K, C6D6) δ 1.89–1.80 (m, 4H, TMP-4), 1.79 (s, 6H, TMP-Me), 1.76 (s, 8H, TMP-Me), 1.73 (s, br, 6H, TMP-Me), 1.72–1.69 (br, m, 4H, TMP-4), 1.68–1.61 (m, 4H, TMP-3,5), 1.59 (s, 12H, TMP-Me; TMP-Me 9a), 1.57 (s, 10H, TMP-Me; TMP-3,5; TMP-4), 1.56 (s, 12H, TMP-Me 9a), 1.10 (m, 6H, TMP-3,5), 1.06 (s, 1H, TMPH-Me).13C NMR spectroscopy (125 MHz, 298 K, C6D6) δ 56.9 (TMP-2,6), 54.2 (TMP-2,6), 54.2 (TMP-2,6 9a), 49.2 (TMPH-2,6), 42.6 (TMP-3,5), 42.5 (TMP-3,5), 42.1 (TMP-3,5 9a), 42.1 (TMP-3,5), 40.1 (TMP-Me 9a), 39.7 (TMP-Me), 38.2 (TMPH-3,5), 37.6 (br, TMP-Me), 37.2 (br, TMP-Me), 36.6 (br, TMP-Me), 34.8 (TMP-Me), 34.5 (TMP-Me 9a), 31.6 (TMPH-Me), 19.3 (TMP-4), 19.2 (TMP-4 9a), 19.2 (TMP-4), 18.4 (TMPH-4).7Li NMR spectroscopy (194 MHz, 298 K, C6D6) δ 1.65 (s, 0.16Li), 0.94 (sh, 0.8Li), 0.91 (s, 1.0Li, 9a).Synthesis and characterization of (TMP)2(OCN)Li3(THF)211
2207 (s, CN), 1352 (s, CO), 1226 (s, CO) cm−1. 1H NMR spectroscopy (500 MHz, C6D6) δ 3.56 (m, 8H, THF), 2.27–1.37 (br, m, 30H, TMP-3,4,5,Me), 1.35 (s, 6H, TMP-Me), 1.34 (m, 8H, THF), 1.06 (s, 3.2H, TMPH-Me), 0.31 (br, s, 0.31H, TMPH-NH). 13C NMR (126 MHz, C6D6) δ 67.8 (THF), 52.0 (TMP-2,6 9b), 51.7 (TMP-2,6 11), 49.2 (TMPH-2,6), 42.4 (TMP-3,5 9b), 41.9 (TMP-3,5 11), 38.2 (TMPH-3,5), 37.5 (br, TMP-Me 11), 36.5 (TMP-Me 9b), 33.5 (br, TMP-Me 11), 31.6 (TMPH-Me), 25.0 (THF), 20.2 (TMP-4 11), 18.4 (TMPH-4). 7Li NMR (194 MHz, C6D6) δ 2.21 (s, br, 1Li, 9b), 1.48 (s, br, 2Li, 11), −1.54 (s, 0.2Li, unidentified).
Synthesis and characterization of (DA)2Cu0.09Li0.91(Br)Li2(TMEDA)212
nBuLi (2.5 mL, 1.6 M in hexanes, 4 mmol) was added to a stirred solution of DAH (0.56 mL, 4 mmol) and TMEDA (0.6 mL, 4 mmol) in hexane (4 mL) at −78 °C. The resulting solution was returned to room temperature to give a yellow solution that was transferred to a −78 °C suspension of CuBr (0.28 g, 2 mmol) in hexane (2 mL). The mixture was returned to room temperature to give a brown suspension. Filtration gave a pale yellow solution. Storage of this at −27 °C for 24 hours gave colourless blocks of cocrystalline Lipshutz-type (DA)2Cu(Br)Li2(TMEDA)212b and (DA)2(Br)Li3(TMEDA)212a. Yield 350 mg.Synthesis and characterization of (DA)2(Br)Li3(TMEDA)212b
To a suspension of DAH·HBr (0.18 g, 1 mmol) in hexane (6 mL) was added DAH (0.14 mL, 1 mmol) and TMEDA (0.30 mL, 2 mmol). The mixture was cooled to −78 °C, treated with nBuLi (1.9 mL, 1.6 M in hexanes, 3 mmol) and returned to room temperature to give a pale yellow solution. The solution was filtered, concentrated (to ca. 4 mL) and stored at −27 °C for 1 day after which colourless block-like crystals of 12b were deposited. Yield 320 mg (60% wrt. Br), melting point 76–78 °C. Elemental analysis, C24H60BrLi3N6 requires (%) C 54.03, H 11.34, N 15.75; found (%) C 54.09, H 11.66, N 15.58. 1H NMR spectroscopy (500 MHz, C6D6) δ 3.66 (s, br, 2H, DA–CH), 2.79 (octet, 3JHH = 6 Hz, 0.06H, DAH–CH), 2.13 (s, 24H, TMEDA–Me), 1.96 (s, 8H, TMEDA–CH2), 1.41 (s, br, 24H, DA–Me), 0.95 (d, 3JHH = 6 Hz, 1.35H, DAH–Me). 13C NMR (126 MHz, C6D6) δ 56.9 (TMEDA–CH2), 48.8 (DA–CH), 46.5 (TMEDA–CH3), 44.9 (DAH–CH), 26.2 (DA–Me), 23.2 (DAH–Me). 7Li NMR (194 MHz, C6D6) δ 2.28 (s, br, 1Li, (DA)2Li), 1.26 (s, 2Li, Li(TMEDA)).Synthesis and characterization of (DA)4Cu(OCN)Li4(TMEDA)213
nBuLi (1.25 mL, 1.6 M in hexanes, 2 mmol) was added to a stirred solution of DAH (0.28 mL, 2 mmol) and TMEDA (0.3 mL, 2 mmol) in hexane (4 mL) at −78 °C. The resulting solution was returned to room temperature to give a yellow solution that was transferred to a suspension of CuOCN (0.11 g, 1 mmol) in hexane (1 mL) at −78 °C. The mixture was returned to room temperature to give a grey suspension, which was filtered to give a pale yellow solution. Storage at +5 °C for 24 hours gave white needle-like crystals of 13. Yield 0.11 g (14% wrt. CuOCN), melting point dec. ca. 95 °C. Elemental analysis, C37H88CuLi4N9O requires (%) C, 57.98; H, 11.57; N, 16.45. Found: C, 57.77; H, 11.68; N, 16.76. Selected IR spectroscopy (nujol) 2207 (s, CN), 1353 (m, CO), 1293 (m, CO) cm−1. 1H NMR spectroscopy (500 MHz, C6D6) δ 3.88–3.40 (br, m, 6.2H, DA–CH), 3.28 (br, m, 0.53H, DA–CH), 3.14 (m, 0.15H, DA–CH), 3.11 (septet, 3JHH = 6 Hz, 1.12H, DA–CH), 2.78 (octet, 3JHH = 6 Hz, 0.14H, DAH–CH), 2.01 (s, 24H, TMEDA–Me), 1.94 (s, 8H, TMEDA–CH2), 1.71–1.38 (br, m, 22H, DA–Me), 1.37 (d, 3JHH = 6 Hz, 3H, DA–Me), 1.35–1.25 (br, m, 16H, DA–Me), 1.23 (d, 3JHH = 6 Hz, 3H, DA–Me), 1.22–1.14 (br, m, 4H, DA–Me), 0.94 (d, 3JHH = 6 Hz, 0.8H, DAH–Me). 13C NMR (125 MHz, C6D6) δ 57.3 (TMEDA–CH2), 50.1 (DA–CH), 49.8 (DA–CH), 49.6 (DA–CH), 49.2 (DA–CH), 48.6 (DA–CH), 48.3 (DA–CH), 45.9 (TMEDA–Me), 44.9 (DAH–CH), 44.8 (DAH–CH), 28.2 (DA–Me), 27.8 (DA–Me), 27.7 (DA–Me), 27.0 (DA–Me), 26.0 (DA–Me), 25.8 (DA–Me), 25.2 (DA–Me), 25.1 (DA–Me), 23.2 (DAH–Me), 23.1 (DAH–Me). 7Li NMR (194 MHz, C6D6) δ 2.34 (br, s, 0.78Li), 2.14 (sh, 0.22Li), 1.58 (s, 0.94Li), 0.84 (s, 0.40Li), 0.37 (s, 1.66Li).
Acknowledgements
This work was supported by the U.K. EPSRC through grants EP/J500380/1 and EP/K039520/1. Thanks go also to Dr Andrew Bond (University of Cambridge) for help with crystallographic analysis. Detailed supporting data for this paper are available at the University of Cambridge data repository (see https://doi.org/10.17863/CAM.9469).References
- Y. Kondo, M. Shilai, M. Uchiyama and T. Sakamoto, J. Am. Chem. Soc., 1999, 121, 3539–3540 CrossRef CAS.
- (a) R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743–755 CrossRef CAS PubMed; (b) R. E. Mulvey, Dalton Trans., 2013, 42, 6676–6693 RSC.
- (a) R. E. Mulvey, V. L. Blair, W. Clegg, A. R. Kennedy, J. Klett and L. Russo, Nat. Chem., 2010, 2, 588–591 CrossRef CAS PubMed; (b) A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834–837 CrossRef PubMed.
- (a) M. Uchiyama, H. Naka, Y. Matsumoto and T. Ohwada, J. Am. Chem. Soc., 2004, 126, 10526–10527 CrossRef CAS PubMed; (b) F. Mongin and M. Uchiyama, Curr. Org. Chem., 2011, 15, 2340–2361 CrossRef CAS; (c) A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470–7562 CrossRef CAS PubMed; (d) F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7727 CrossRef PubMed.
- (a) M. Uchiyama, T. Miyoshi, Y. Kajihara, T. Sakamoto, Y. Otani, T. Ohwada and Y. Kondo, J. Am. Chem. Soc., 2002, 124, 8514–8515 CrossRef CAS PubMed; (b) M. Uchiyama, Y. Matsumoto, D. Nobuto, T. Furuyama, K. Yamaguchi and K. Morokuma, J. Am. Chem. Soc., 2006, 128, 8748–8750 CrossRef CAS PubMed; (c) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802–3824 CrossRef CAS PubMed; (d) F. García, M. McPartlin, J. V. Morey, D. Nobuto, Y. Kondo, H. Naka, M. Uchiyama and A. E. H. Wheatley, Eur. J. Org. Chem., 2008, 644–647 CrossRef; (e) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794–9824 CrossRef CAS PubMed.
- R. P. Davies, Coord. Chem. Rev., 2011, 255, 1226–1251 CrossRef CAS.
- e.g. (a) J. Garcia-Álvarez, A. R. Kennedy, J. Klett and R. E. Mulvey, Angew. Chem., Int. Ed., 2007, 46, 1105–1109 CrossRef PubMed; (b) J. García-Álvarez, E. Hevia, A. R. Kennedy, J. Klett and R. E. Mulvey, Chem. Commun., 2007, 2402–2404 RSC; (c) R. P. Davies, S. Hornauer and P. B. Hitchcock, Angew. Chem., Int. Ed., 2007, 46, 5191–5194 CrossRef CAS PubMed; (d) Y. Kondo, J. V. Morey, J. M. Morgan, P. R. Raithby, D. Nobuto, M. Uchiyama and A. E. H. Wheatley, J. Am. Chem. Soc., 2007, 129, 12734–12738 CrossRef CAS PubMed; (e) H. Naka, J. V. Morey, J. Haywood, D. J. Eisler, M. McPartlin, F. García, H. Kudo, Y. Kondo, M. Uchiyama and A. E. H. Wheatley, J. Am. Chem. Soc., 2008, 130, 16193–16200 CrossRef CAS PubMed; (f) W. Clegg, J. Garcia-Álvarez, P. Garcia-Álvarez, D. V. Graham, R. W. Harrington, E. Hevia, A. R. Kennedy, R. E. Mulvey and L. Russo, Organometallics, 2008, 27, 2654–2663 CrossRef CAS; (g) A. R. Kennedy, J. Klett, R. E. Mulvey and D. S. Wright, Science, 2009, 326, 706–708 CrossRef CAS PubMed; (h) R. Bomparola, R. P. Davies, T. Gray and A. J. P. White, Organometallics, 2009, 28, 4632–4635 CrossRef CAS.
- D. R. Armstrong, E. Crosbie, E. Hevia, R. E. Mulvey, D. L. Ramsay and S. D. Robertson, Chem. Sci., 2014, 5, 3031–3045 RSC.
- S. Sung, D. C. Braddock, A. Armstrong, C. Brennan, D. Sale, A. J. P. White and R. P. Davies, Chem.–Eur. J., 2015, 21, 7179–7192 CrossRef CAS PubMed.
- (a) H. Gilman, R. Jones and L. Woods, J. Org. Chem., 1952, 17, 1630 CrossRef CAS; (b) H. House, W. Respess and G. Whitesides, J. Org. Chem., 1966, 31, 3128–3141 CrossRef CAS.
- (a) P. Reiss and D. Fenske, Z. Anorg. Allg. Chem., 2000, 626, 1317–1331 CrossRef CAS; (b) R. M. Gschwind, Chem. Rev., 2008, 108, 3029–3053 CrossRef CAS PubMed.
- G. Whitesides, W. Fisher, J. San Filipo, R. Bashe and H. House, J. Am. Chem. Soc., 1969, 91, 4871–4882 CrossRef CAS.
- (a) B. H. Lipshutz, R. S. Wilhelm and D. M. Floyd, J. Am. Chem. Soc., 1981, 103, 7672–7674 CrossRef CAS; (b) B. H. Lipshutz, S. Sharma and E. L. Ellsworth, J. Am. Chem. Soc., 1990, 112, 4032–4034 CrossRef CAS; (c) B. Lipshutz and B. James, J. Org. Chem., 1994, 59, 7585–7587 CrossRef CAS.
- (a) S. Bertz, J. Am. Chem. Soc., 1990, 112, 4031–4132 CrossRef CAS; (b) J. Snyder, D. Spangler, J. Behling and B. Rossiter, J. Org. Chem., 1994, 59, 2665–2667 CrossRef CAS; (c) J. Snyder and S. Bertz, J. Org. Chem., 1995, 60, 4312–4315 CrossRef CAS; (d) T. L. Stemmler, T. M. Barnhart, J. E. Penner-Hahn, C. E. Tucker, P. Knochel, M. Bohme and G. Frenking, J. Am. Chem. Soc., 1995, 117, 12489–12497 CrossRef CAS.
- G. van Koten and J. G. Noltes, J. Am. Chem. Soc., 1979, 101, 6593–6605 CrossRef CAS.
- (a) G. van Koten and J. G. Noltes, J. Chem. Soc., Chem. Commun., 1972, 940–941 RSC; (b) G. van Koten, J. T. B. H. Jastrzebski, F. Muller and C. H. Stam, J. Am. Chem. Soc., 1985, 107, 697–698 CrossRef CAS; (c) C. Kronenberg, J. Jastrzebski, A. Spek and G. van Koten, J. Am. Chem. Soc., 1998, 120, 9688–9689 CrossRef; (d) G. Boche, F. Bosold, M. Marsch and K. Harms, Angew. Chem., Int. Ed., 1998, 37, 1684–1686 CrossRef CAS.
- S. Usui, Y. Hashimoto, J. V. Morey, A. E. H. Wheatley and M. Uchiyama, J. Am. Chem. Soc., 2007, 129, 15102–15103 CrossRef CAS PubMed.
- P. J. Harford, A. J. Peel, J. P. Taylor, S. Komagawa, P. R. Raithby, T. P. Robinson, M. Uchiyama and A. E. H. Wheatley, Chem.–Eur. J., 2014, 20, 3908–3912 CrossRef CAS PubMed.
- S. Komagawa, S. Usui, J. Haywood, P. J. Harford, A. E. H. Wheatley, Y. Matsumoto, K. Hirano, R. Takita and M. Uchiyama, Angew. Chem., Int. Ed., 2012, 51, 12081–12085 CrossRef CAS PubMed.
- (a) N. Marquise, P. J. Harford, F. Chevallier, T. Roisnel, A. E. H. Wheatley, P. C. Gros and F. Mongin, Tetrahedron Lett., 2013, 54, 3154–3157 CrossRef CAS; (b) N. Marquise, P. J. Harford, F. Chevallier, T. Roisnel, V. Dorcet, A.-L. Gagez, S. Sablé, L. Picot, V. Thiéry, A. E. H. Wheatley, P. C. Gros and F. Mongin, Tetrahedron, 2013, 69, 10123–10133 CrossRef CAS.
- A. J. Peel, M. Hedidi, G. Bentabed-Ababsa, T. Roisnel, F. Mongin and A. E. H. Wheatley, Dalton Trans., 2016, 45, 6094–6104 RSC.
- J. Haywood, J. V. Morey, A. E. H. Wheatley, C.-Y. Liu, S. Yasuike, J. Kurita, M. Uchiyama and P. R. Raithby, Organometallics, 2009, 28, 38–41 CrossRef CAS.
- P. J. Harford, A. J. Peel, F. Chevallier, R. Takita, F. Mongin, M. Uchiyama and A. E. H. Wheatley, Dalton Trans., 2014, 43, 14181–14203 RSC.
- (a) G. van Koten and J. G. Noltes, J. Am. Chem. Soc., 1979, 101, 6593–6605 CrossRef CAS; (b) C. M. P. Kronenburg, J. T. B. H. Jastrzebski, J. Boersma, M. Lutz, A. L. Spek and G. van Koten, J. Am. Chem. Soc., 2002, 124, 11675–11683 CrossRef CAS PubMed.
- R. A. D. Soriaga, S. Javed and D. M. Hoffman, J. Cluster Sci., 2010, 21, 567–575 CrossRef CAS.
- T. P. Robinson, R. D. Price, M. G. Davidson, M. A. Fox and A. L. Johnson, Dalton Trans., 2015, 44, 5611–5619 RSC.
- S. D. Bunge and J. L. Steele, Inorg. Chem., 2009, 48, 2701–2706 CrossRef CAS PubMed.
- C. Wagner and K. Merzweiler, Z. Anorg. Allg. Chem., 2014, 640, 2198–2202 CrossRef CAS.
- E. C. Franklin, Proc. Natl. Acad. Sci. U. S. A., 1915, 1, 68–70 CrossRef CAS.
- (a) S. Gambarotta, M. Bracci, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Chem. Soc., Dalton Trans., 1987, 1883–1888 RSC; (b) H. Chen, M. M. Olmstead, S. C. Shoner and P. P. Power, J. Chem. Soc., Dalton Trans., 1992, 451–457 RSC; (c) A. M. James, R. K. Laxman, F. R. Fronczek and A. W. Maverick, Inorg. Chem., 1998, 37, 3785–3791 CrossRef CAS PubMed; (d) B. S. Lim, A. Rahtu, J.-S. Park and R. G. Gordon, Inorg. Chem., 2003, 42, 7951–7958 CrossRef CAS PubMed.
- (a) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed; (b) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed; (c) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed; (d) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- E. Söderbäck, Acta Chem. Scand., 1957, 11, 1622–1634 CrossRef.
- (a) K. Gregory, P. v. R. Schleyer and R. Snaith, Adv. Inorg. Chem., 1991, 37, 47–142 CrossRef CAS; (b) R. E. Mulvey, Chem. Soc. Rev., 1991, 20, 167–209 RSC.
- F. S. Mair, W. Clegg and P. A. O'Neil, J. Am. Chem. Soc., 1993, 115, 3388–3389 CrossRef CAS.
- P. L. Hall, J. H. Gilchrist, A. T. Harrison, D. J. Fuller and D. B. Collum, J. Am. Chem. Soc., 1991, 113, 9575–9585 CrossRef CAS.
- For 9b3 see: (a) E. Hevia, A. R. Kennedy, R. E. Mulvey, D. L. Ramsay and S. D. Robertson, Chem.–Eur. J., 2013, 19, 14069–14075 CrossRef CAS PubMed. For general trimeric lithium amides see: (b) D. R. Armstrong, R. E. Mulvey, G. T. Walker, D. Barr, R. Snaith, W. Clegg and D. Reed, J. Chem. Soc., Dalton Trans., 1988, 617–628 RSC; (c) D. R. Armstrong, K. W. Henderson, A. R. Kennedy, W. J. Kerr, F. S. Mair, J. H. Moir, P. H. Moran and R. Snaith, J. Chem. Soc., Dalton Trans., 1999, 4063–4068 RSC; (d) S. R. Dubberley, P. Mountford and N. Adams, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, 58, m342 CAS; (e) R. M. Porter and A. A. Danopoulos, Dalton Trans., 2004, 2556–2562 RSC; (f) V. H. Gessner and C. Strohmann, Organometallics, 2010, 29, 1858–1861 CrossRef CAS.
- For 9b4 see: (a) M. F. Lappert, M. J. Slade, A. Singh, J. L. Atwood, R. D. Rogers and R. Shakir, J. Am. Chem. Soc., 1983, 105, 302–304 CrossRef CAS; (b) ref. 36 .
- For general tetrameric lithium amides see: J. Jubb, P. Berno, S. Hao and S. Gambarotta, Inorg. Chem., 1995, 34, 3563–3566 CrossRef CAS.
- For trimeric copper amides see: B. S. Lim, A. Rahtu, J.-S. Park and R. G. Gordon, Inorg. Chem., 2003, 42, 7951–7958 CrossRef CAS PubMed.
- For 9c4 see: ref. 9. For general tetrameric copper amides see: (a) H. Hope and P. P. Power, Inorg. Chem., 1984, 23, 936–937 CrossRef CAS; (b) S. Gambarotta, M. Bracci, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Chem. Soc., Dalton Trans., 1987, 1883–1888 RSC.
- J. Garcìa-Àlvarez, D. V. Graham, E. Hevia, A. R. Kennedy and R. E. Mulvey, Dalton Trans., 2008, 1481–1486 RSC.
- (a) W. Clegg, B. Conway, E. Hevia, M. D. McCall, L. Russo and R. E. Mulvey, J. Am. Chem. Soc., 2009, 131, 2375–2384 CrossRef CAS PubMed; (b) S. Tamba, K. Ide, K. Shono, A. Sugie and A. Mori, Synlett, 2013, 24, 1133–1136 CrossRef CAS.
- D. R. Armstrong, R. David, P. Garcia-Alvarez, A. R. Kennedy, R. E. Mulvey, E. Robert and S. D. Robertson, Chem.–Eur. J., 2011, 17, 6725–6730 CrossRef CAS PubMed.
- T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1540280–1540286. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01423f |
| This journal is © The Royal Society of Chemistry 2017 |