
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pH responsive complexation-based drug delivery system for oxaliplatin†
Bin
Li
ab,
Zhao
Meng
b,
Qianqian
Li
a,
Xiayang
Huang
a,
Ziyao
Kang
b,
Huajin
Dong
b,
Junyi
Chen
ab,
Ji
Sun
a,
Yansheng
Dong
b,
Jian
Li
a,
Xueshun
Jia
a,
Jonathan L.
Sessler
 *a,
Qingbin
Meng
*b and
Chunju
Li
*a
*a,
Qingbin
Meng
*b and
Chunju
Li
*a
aDepartment of Chemistry, Center for Supramolecular Chemistry and Catalysis, Shanghai University, Shanghai 200444, P. R. China. E-mail: sessler@mail.utexas.edu; cjli@shu.edu.cn
bState Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, Beijing 100850, P. R. China. E-mail: nankaimqb@sina.com
First published on 19th April 2017
Abstract
A responsive drug delivery system (DDS) for oxaliplatin (OX) has been designed with a view to overcoming several drawbacks associated with this anticancer agent, including fast degradation/deactivation in the blood stream, lack of tumor selectivity, and low bioavailability. The present approach is based on the direct host–guest encapsulation of OX by a pH-responsive receptor, carboxylatopillar[6]arene (CP6A). The binding affinities of CP6A for OX were found to be pH-sensitive at biologically relevant pH. For example, the association constant (Ka) at pH 7.4 [Ka = (1.02 ± 0.05) × 104 M−1] is 24 times larger than that at pH 5.4 [Ka = (4.21 ± 0.06) × 102 M−1]. Encapsulation of OX within the CP6A cavity did not affect its in vitro cytotoxicity as inferred from comparison studies carried out in several cancer cells (e.g., the HepG-2, MCF-7, and A549 cell lines). On the other hand, complexation by CP6A serves to increase the inherent stability of OX in plasma by 2.8-fold over a 24 h incubation period. The formation of a CP6A⊃OX host–guest complex served to enhance in a statistically significant way the ability of OX to inhibit the regrowth of sarcoma 180 (S180) tumors in Kunming (KM) mice xenografts. The improved anticancer activity observed in vivo for CP6A⊃OX is attributed to the combined effects of enhanced stability of the host–guest complex and the pH-responsive release of OX. Specifically, it is proposed that OX is protected as the result of complex formation and then released effectively in the acidic tumor environment.
Introduction
Platinum-based anticancer agents1 are a mainstay of oncology. Over 50% of all chemotherapeutic regimens administered to cancer patients include a platinum drug.2 Oxaliplatin (OX), the third platinum drug to obtain FDA approval, exhibits a number advantages compared to cisplatin and carboplatin, including a broad range of treatable tumours,1c,3 low toxicity (e.g., the absence of nephrotoxicity and reduced ototoxicity),4 and little or no cross resistance to cisplatin or carboplatin.5 Currently, OX in combination with other agents represents the standard of care for advanced colorectal cancer.6 Nevertheless, OX still has some major drawbacks, such as little inherent selectivity for neoplastic sites, cumulative sensory neurotoxicity,7 rapid degradation/deactivation following administration,8 and low bioavailability. In this work, we show that a pH responsive carboxylatopillar[6]arene (CP6A) may be used to form a pH-dependent complex with OX and that the resulting construct acts as an effective drug delivery system (DDS) that both improves the stability of OXin vitro and in vivo and enhances its in vivo anticancer activity in a mouse model.The high off-target toxicity of classic cancer chemotherapeutics, coupled with their general lack of tumor localizing capability and, in some instances, poor solubility or stability profiles has prompted considerable effort to develop so-called drug delivery systems (DDS)9 that might overcome some or all of these deficiencies. Promising approaches include the use of liposomes,10 vesicles,11 nanoparticles,12 hydrogels,13 carbon nanotubes,14 metal–organic frameworks,15 and supramolecular organic frameworks.16 Several of these systems are “smart” enough to realize the controllable release in response to environmental changes (pH, temperature and enzyme, etc.) or external noninvasive activation (e.g., heating or photo-irradiation), thus permitting a degree of cancer specificity.
Direct host–guest encapsulating of drugs by supramolecular macrocyclic molecules represents another promising approach to DDS development.17 In this context, cyclodextrins and cucurbiturils have received particular attention as receptors that can increase the aqueous solubility of drugs and improve bioavailability. In fact, several cyclodextrin derivatives, including α-cyclodextrin, β-cyclodextrin, hydroxypropyl β-cyclodextrin, and the trade named derivatives Captisol18 and Sugammadex19 have been clinically approved by the FDA. Very recently, Isaacs' group20 has used acyclic cucurbituril analogues to enhance the aqueous solubility of insoluble drugs. Using this latter drug ⊂ receptor complexation-based approach, the aqueous solubility of paclitaxel could be increased by 2750-fold. Although direct host–guest DDS strategies are simple and convenient, they often suffer from an inability to control drug release and a lack of cancer targeting selectivity. We felt that some of these issues could be addressed by using a receptor that responds to changes in pH.
It is well documented that the pH value in areas of tumor tissues is lower than that in most physiological environments (pH 7.4), such as the blood stream and in normal tissues.21 We have now succeeded in creating a responsive DDS through direct, pH-sensitive host–guest encapsulation. Toward this end, we have employed carboxylatopillar[6]arene (CP6A), which bears multiple carboxylate groups on its rim-like portals, as a pH responsive receptor. Oxaliplatin (OX) was chosen as a model drug in light of its clinical importance and the fact that it produces a strong complex with CP6A, as detailed below (Scheme 1).
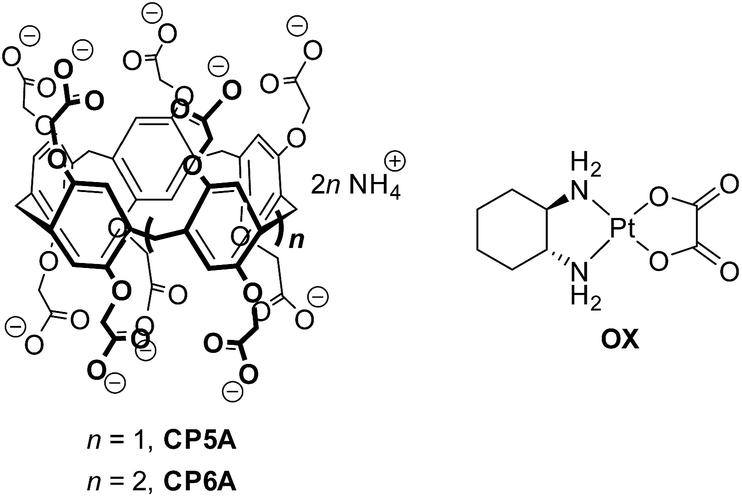 | ||
| Scheme 1 The structures of CP5A, CP6A, and OX. | ||
Pillar[n]arenes have become recognized as an important family of macrocyclic hosts since their discovery in 2008.22,23 Carboxylatopillar[n]arenes (CPnAs) are readily accessible water-soluble pillararene derivatives24 that serve as good hosts for suitably sized cationic guests. They typically display pH dependent recognition since the carboxylate substituents are weakly basic and, in their deprotonated forms, augment substrate binding through electrostatic anion–cationic guest interactions.22e,24e
Results and discussion
Host–guest complexation and pH-sensitivity
The design predicate underlying our proposed CP6A-based recognition strategy is that this carboxylate-functionalized pillararene would bind OX well at physiological pH and release it effectively at lower pH. We thus performed 1H NMR spectroscopy and isothermal microcalorimetric titrations (ITC) experiments to examine the host–guest inclusion and its pH response. Fig. 1 shows the spectra of OX in D2O recorded in the absence and presence of one equivalent (equiv.) of the putative CP6A host. The proton signals of OX (Ha, Hb, Hb′, Hc and Hc′) undergo a substantial upfield shift and experience considerable broadening when mixed with CP6A. Such spectral differences, which reflect shielding effects, are consistent with the cyclohexyl unit of OX being located within the CP6A host cavity. In addition, the peaks for the CP6A host shift to lower field (Δδ = 0.14 and 0.20 ppm for H1 and H2), presumably as a consequence of inclusion-induced deshielding effects. Variable-temperature 1H NMR spectral studies of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest mixture revealed a reduction in the spectral broadening ascribed to complexation when the spectra were recorded at 60 °C and 80 °C (Fig. S1†). A mole ratio plot for CP6A and OX was also constructed; it proved consistent with the proposed 1:1 host–guest binding stoichiometry (Fig. S2†).
:1 host–guest mixture revealed a reduction in the spectral broadening ascribed to complexation when the spectra were recorded at 60 °C and 80 °C (Fig. S1†). A mole ratio plot for CP6A and OX was also constructed; it proved consistent with the proposed 1:1 host–guest binding stoichiometry (Fig. S2†).
 | ||
| Fig. 1 1H NMR spectra (400 MHz, 293 K D2O) of (A) OX (4.9 mM), (B) OX (4.8 mM) + CP6A (5.0 mM), and (C) CP6A (5.0 mM). | ||
For comparison purposes, the interaction of OX with smaller CP5A was also tested. Upon addition of CP5A, no obvious NMR spectral changes were observed for either the guest or host signals (Fig. S3†). We thus suggest that little or no inclusion of OX occurs in the case of this smaller pillararene. Such a finding is consistent with our previous work that revealed that linear alkanes, but not cycloalkanes such as the cyclohexyl moiety present in OX, are good substrates for pillar[5]arenes.25
In order to study quantitatively the effect of pH on the inferred binding of OX by CP6A, ITC studies were carried out in aqueous phosphate buffer solution (PBS) at different pH (7.4, 6.5, 6.0, and 5.4). Using a 1:1 binding model as supported by the mole ratio plot, the association constants (Ka) and corresponding thermodynamic parameters (enthalpy and entropy changes, ΔH° and ΔS°) could be derived (Fig. S4–S7†). From an analysis of the data, it was found that at pH 7.4 Ka = (1.02 ± 0.05) × 104 M−1. This Ka value proved 5.2, 11 and 24 times larger than the corresponding values determined at pH = 6.5, 6.0, and 5.4, respectively (Table 1). Qualitative support for these findings came from 1H NMR spectroscopic experiments carried out at different pDs. In particular, greater complexation-induced NMR shifts for both the host and the guest signals at pD 7.4 than at 5.4 (Fig. S8†).
| pH | K a (M−1) | −ΔH° | TΔS° |
|---|---|---|---|
| 7.4 | (1.02 ± 0.05) × 104 | 17.7 | 5.19 |
| 6.5 | (1.98 ± 0.03) × 103 | 16.3 | 2.47 |
| 6.0 | (9.08 ± 0.06) × 102 | 13.1 | 3.82 |
| 5.4 | (4.21 ± 0.06) × 102 | 12.0 | 2.96 |
Based on the thermodynamic parameters obtained from the ITC studies, the host–guest complexation appears synergistically enabled by both enthalpy and entropy effects. The observed negative enthalpy contributions (−ΔH° = 17.7–12.0 kJ mol−1) are ascribed to electrostatic interactions between the anionic host and the cationic PtII, while the favorable entropy changes (TΔS° = 5.19–2.47 kJ mol−1) are considered to reflect complexation induced desolvation. The enthalpic contributions decrease with pH 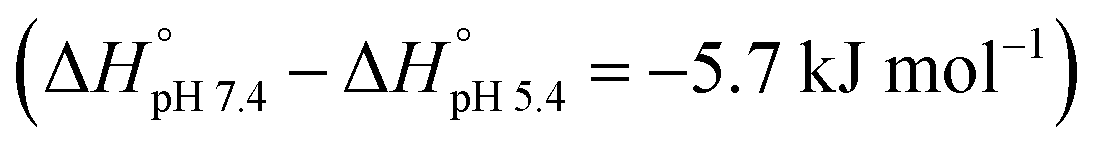 , as expected. The strong variation in the binding affinities over a relatively small pH range and at values corresponding to those expected to pertain in normal biological environments and cancerous tissues, respectively, was considered to augur well for the use of CP6A to create a viable DDS for OX. Efforts were thus made to explore this possibility in greater detail.
, as expected. The strong variation in the binding affinities over a relatively small pH range and at values corresponding to those expected to pertain in normal biological environments and cancerous tissues, respectively, was considered to augur well for the use of CP6A to create a viable DDS for OX. Efforts were thus made to explore this possibility in greater detail.
Biocompatibility of CP6A
Prior to testing its efficacy as a DDS, the cytotoxicity of CP6A was assessed in 293T (a normal cell line), as well as the HepG-2 (hepatocellular carcinoma), MCF-7 (breast carcinoma) and A549 (non-small cell lung cancer) cell lines. This was done using a commercially available Cell Counting Kit-8 (CCK-8) using different pillarene concentrations (Fig. S9†). CP6A displays minimal cytotoxicity against these four cell lines even at relatively high concentrations. For example, in the case of the 293T cells, viability at the 93.0 (±6.8) and 89.8 (±2.6)% level was maintained after incubating for 48 h with 160 and 320 μM of CP6A, respectively.Acute toxicity experiments were then carried out with Kunming (KM) mice to evaluate in preliminary fashion whether CP6A could be safely administered in vivo. The mice were randomly divided into seven groups with 10 mice (5 male and 5 female) in each group. The mice were subject to a single intravenous injection of CP6A in saline at doses of 80, 160, 320, 360, 420, 640, 1280 mg kg−1, respectively, followed by observing for 14 days. On the basis of these studies, the LD50 was calculated to be ≥378 mg kg−1 using the SPSS (Statistical Product and Service Solutions) software.
The above in vitro cytotoxicity and in vivo acute toxicity results led us to consider that CP6A could be applied safely as a drug carrier. We thus set out to test whether it would provide a benefit in the specific case of OX for which evidence of pH-dependent 1:1 complexation had been obtained (vide supra).
Plasma and in vivo stability
As noted in the introduction, one of the limitations of OX is its poor stability in the blood. As a consequence, higher-than-optimal dose levels are required and poor patient compliance is observed. OX molecules also interact with plasma proteins and red blood cells,26 for example thiol containing proteins through Pt–S bonds; this reduces the amount of drug that arrives at the tumor sites after intravenously administration. Given the ca. 104 M−1Ka value for the binding of OX by CP6A at pH of 7.4, we considered it reasonable that appreciable host–guest interactions would occur under conditions of administration and that the resulting complex would be less prone to react with plasma proteins and red blood cells. The stability of OX in plasma and in vivo (rats) in the absence and presence of 1.0 equiv. of CP6A was evaluated using reversed-phase high-performance liquid chromatography (RP-HPLC) and inductively coupled plasma mass spectrometry (ICP-MS), respectively. The results of these studies are detailed below.In the serum stability studies, the initial concentration of OX is 940 μM. For a Ka value of (1.02 ± 0.05) × 104 M−1, the OX present in the form of the corresponding CP6A complex was calculated to be 73%. As shown in Fig. 2A, in the presence of CP6A, OX exhibited much greater stability than on its own. For example, after being incubated with diluted plasma (rat plasma diluted with water (1:3, v/v)) for 24 h, the residual percentage of drug was only 13% in the case of OX alone; this value increased to 35% in the presence of the host. This >2× enhancement in stability was taken as support for the hypothesis underlying this work, namely that the host-guest inclusion makes OX less prone to react with plasma proteins.
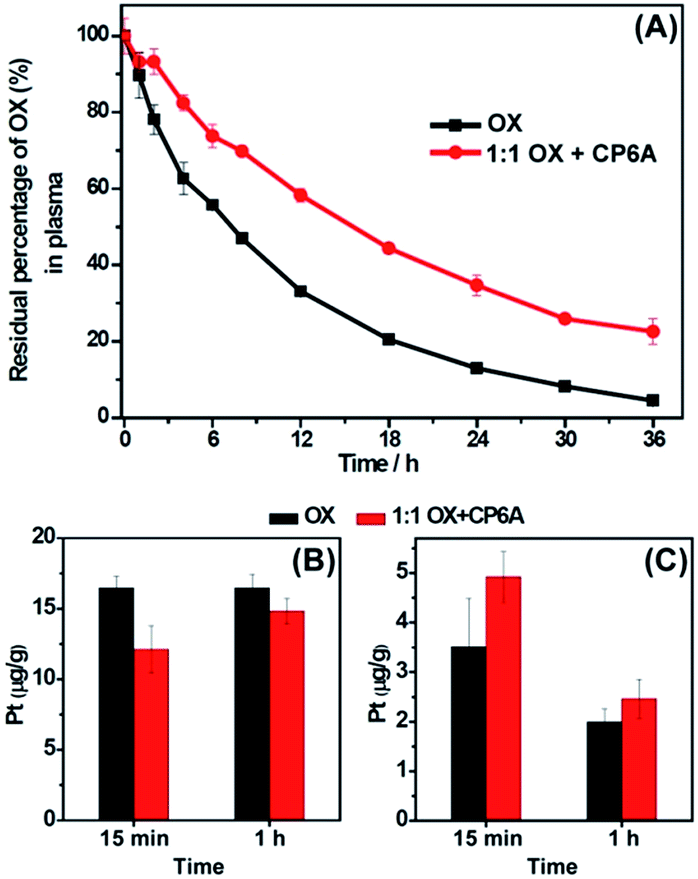 | ||
| Fig. 2
In vitro and in vivo stability of OX in the absence and presence of 1.0 equiv. of CP6A. (A) In vitro stability. Residual percentage of OX in plasma in the absence and presence of 1.0 equiv. of CP6A as determined by RP-HPLC. The initial concentration of OX is 940 μM. (B) and (C) In vivo stability experiments. Pt content in red blood cells (B) and in plasma (C) at time intervals of 15 min and 1 h after injection of OX or a 1:1 mixture of CP6A and OX into rats as determined by ICP-MS. The dose of OX was 15 mg kg−1 and the initial concentration of the solutions used for injection was 4.72 mM. | ||
The stability of OXin vivo was then tested using ICP-MS. Ten rats were randomly divided into two groups with five rats in each group. The rats were injected with OX and a 1:1 mixture of OX and CP6Avia tail vein, respectively, with an equivalent OX dose of 15.0 mg kg−1 ([OX] = 4.72 mM). The initial fraction of OX that exists in the form of its CP6A complex was calculated to be 87% in the injection solution. It is recognized that after injection, the concentrations of OX and CP6A would decrease due to dilution in the blood stream making the actual percentage bound in vivo difficult to determine. Nevertheless, the operative assumption was that significant amounts of complex would pertain in vivo and this would translate into enhanced plasma localization and improved stability.
To test the above hypothesis, blood samples were collected at time intervals of 15 min and 1 h after injection, and then centrifuged to separate plasma and red blood cells. As expected, in the animals co-dosed with CP6A, the platinum content in red blood cell samples was lower than in its absence (Fig. 2B), while that in the plasma increased (Fig. 2C). For example, at a time interval of 15 min, the platinum concentration in red blood cells of animals dosed with OX alone (16.5 ± 0.85 μg g−1) proved to be 36% larger than that in the presence of the host (12.1 ± 1.7 μg g−1), leading us to conclude that encapsulation of OX by CP6A militate against OX being taken up into, or reacting within, red blood cells. These findings are also consistent with the presence of significant OX ⊂ CP6Ain vivo after injection of a 1:1 mixture of OX and CP6A. More importantly, the enhancement in stability for OX seen in the presence of CP6A both in vitro and in vivo was expected to prolong the retention time of OX in the blood stream, potentially improving its bioavailability while reducing off-target interactions and deleterious side effects.
In vitro cytotoxicity
The cytotoxicity of OX + CP6A in the MCF-7, HepG-2 and A549 cell lines was assessed by CCK-8, with free OX being used as a positive agent. The results are provided in Fig. 3. Concentration-dependent cell death was observed for both the 1:1 OX + CP6A mixture and the free drug (OX). The cytotoxic activity of the OX + CP6A mixture proved to be nearly equal to that of OX alone over a range of concentrations (Fig. 3), although the actual IC50 values were found to be slightly better in the case of OX in the MCF-7 and HepG-2 cell lines (Table 2).
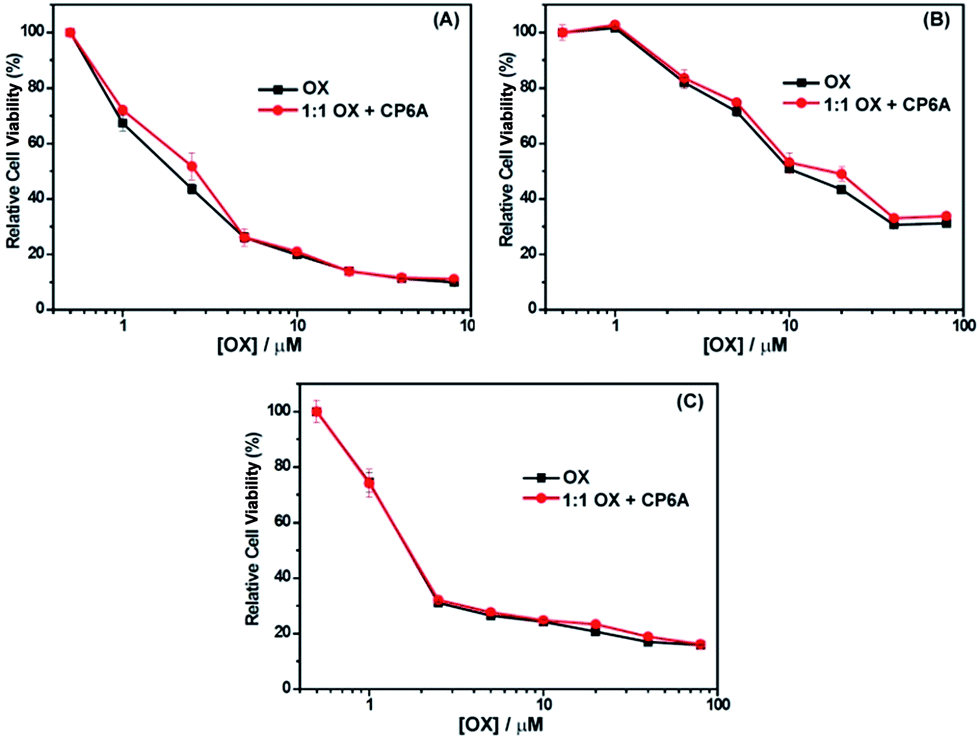 | ||
| Fig. 3 Antiproliferative activity of free OX and a 1:1 mixture of OX and CP6A as seen in (A) MCF-7, (B) HepG-2, and (C) A549 cells as determined using CCK-8. Cells were treated with either OX or OX and CP6A for 72 h followed by incubation for 24 h in fresh medium. | ||
:1 mixture of OX and CP6A against the A549, HepG-2, and MCF-7 cell lines
In vivo antitumor efficacy
Based on our design expectations, the in vivo activity of OX was expected to be enhanced as the result of the pH responsive host–guest complexation provided by CP6A.27 To test this hypothesis, the in vivo inhibitory effect on tumor growth was evaluated in xenograft KM mice bearing S180 tumors. The models were established by injection of S180 cells into the abdominal cavity of the KM mice. After 24 h, the mice were randomly divided into four groups with 12 mice in each group. Mice were injected with a 1:1 mixture of OX and CP6A through the tail vein four times (on day 1, 3, 5, and 7). Two dose groups for this mixture were tested; these involved total OX dosages of 15 mg kg−1 and 35 mg kg−1, respectively. A single dose group with OX alone at a total dosage of 35 mg kg−1 was examined for comparison. Saline was used as the negative control. The final tumor weights were measured after treatment and as a function of time.
Analysis of the results (cf.Fig. 4B and C) revealed that the normalized tumor weights were much lower in the case of both the OX alone and the OX + CP6A groups at the 35 mg kg−1 drug level than observed in the case of the negative control. It was notable that at this dose level, administration of a 1:1 mixture of OX + CP6A proved much more efficacious in terms of tumor inhibition than OX alone. The normalized tumor weight of the OX group (0.85 ± 0.24 g) was 77% higher than that of the corresponding OX + CP6A group (0.48 ± 0.24 g) at the 35 mg kg−1 drug level (p < 0.001). In the case of the 1:1 OX + CP6A mixture, a dose dependence was seen. Specifically, when the OX + CP6A mixture dose level increased from 15 to 35 mg kg−1, the anticancer effect was improved by about 70% as inferred from the normalized tumor weights.
 | ||
| Fig. 4
In vivo antitumor experiments. Change in body weight observed for S180 xenograft mice (A), normalized tumor weights after treatment (B), and pictures of the tumors (C) excised from S180 xenograft mice treated with saline (control), free OX, and a 1:1 mixture of OX and CP6A at an OX dose of 15 or 35 mg kg−1 as indicated. ***p < 0.001. | ||
The enhancement seen for OX + CP6A relative to OX alone at the 35 mg kg−1 dose level is ascribed to two factors. First, encapsulation of OX with CP6A to form OX ⊂ CP6A, although not expected to be carried fully to equilibrium under the conditions of the in vivo studies,27 serves to improve drug stability and decrease off-target binding after injection (Fig. 2). Second, as the result of the pH effect on binding (vide supra and Table 1), free OX should be released from OX ⊂ CP6A upon reaching the mildly acidic environment of the tumor site. The benefit of these putative effects is shown schematically in Fig. 5.
 | ||
| Fig. 5 Schematic illustration of the DDS effect expected to operative in the case of a 1:1 mixture of OX and CP6A. | ||
In addition to looking at tumor regrowth, the body weights of the xenograft mice were measured every 2 days (Fig. 4A). There was no statistically difference in the body weight between the OX + CP6A and free OX groups at the 35 mg kg−1 dose level.
Taken in concert, the in vivo results provide support for the conclusion that, as proposed, inclusion of OX within CP6A improves the anti-tumor activity of OXin vivo without increasing its toxicity.
Acute toxicity study
Acute toxicity tests of a 1:1 mixture of OX and CP6A were also performed. For this study, mice were randomly divided into seven groups with 10 mice (5 male and 5 female) in each group. Mixtures of OX and CP6A, as well as OX alone, were then administrated by intravenous injection at the 1.75, 5.50, 17.5, 30.9, 41.3, 55.0, 158 mg kg−1 dose level (on a per OX basis) followed by observing for 14 days. Studies of free OX were performed under the same conditions to provide a positive control. The LD50 value of the 1:1 OX + CP6A mixture was determined to be 21.4 mg kg−1 (on a per OX basis) by SPSS. This value proved almost equal to that for free OX (22.5 mg kg−1). These results are thus taken as further support for the conclusion reached above (cf.Fig. 2A), namely that a 1:1 mixture of OX and CP6A is not appreciably more toxic than OX alone and that CP6A alone exhibits good biocompatibility, both in vitro and in vivo (cf. Fig. S9†).
Conclusions
We have put forward a simple DDS strategy that involves the direct encapsulation of a specific cationic drug, OX, by an appropriately sized pH-responsive receptor, namely CP6A. This water soluble pillararene interacts more strongly with OX at pH 7.4 (Ka = (1.02 ± 0.05) × 104 M−1) than when the pH is lowered slightly. The partial formation of a presumed 1:1 inclusion complex with CP6A did not affect significantly the cytotoxicity of OXin vitro as determined using the MCF-7, HepG-2, and A549 cancer cell lines. Nor did it change appreciably the acute toxicity in vivo as compared to OX. On the other hand, a statistically significant improvement in in vivo tumor regrowth activity was seen. The enhancement of antitumor efficiency in vivo is consistent with the two key predicative suggestions underlying this study, namely that complexation leads to an increase in drug stability in plasma and in vivo (rat models) and that drug release is enhanced in the lower pH environments that characterize most tumors. The fact that CPnAs are known in a variety of sizes (n = 5–10) and are capable of encapsulating efficiently a wide range of guests, leads us to suggest that the present pillararene-based DDS strategy could be generalized to include a wide range of chemotherapeutic guests. Studies directed to exploring this possibility are currently ongoing.
Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (21472122, 21272155, 81573354, and 81202465), the National Key Technologies R&D Program for New Drugs of China (2012ZX09301003), the Shanghai Pujiang Program (16PJD024), and center funds provided by Shanghai University.Notes and references
- (a) B. Rosenberg, L. VanCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed; (b) T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed; (c) E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- (a) E. Wexselblatt, E. Yavin and D. Gibson, Inorg. Chim. Acta, 2012, 393, 75–83 CrossRef CAS; (b) G. Thiabaud, J. F. Arambula, Z. H. Siddik and J. L. Sessler, Chem.–Eur. J., 2014, 20, 8942–8947 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- J. Cassidy and J.-L. Misset, Semin. Oncol., 2002, 29, 11–20 CrossRef CAS PubMed.
- B. Stordal, N. Pavlakis and R. Davey, Cancer Treat. Rev., 2007, 33, 347–357 CrossRef CAS PubMed.
- (a) A. Ibrahim, S. Hirschfeld, M. H. Cohen, D. J. Griebel, G. A. Williams and R. Pazdur, The Oncologist, 2004, 9, 8–12 CrossRef PubMed; (b) L. R. Wiseman, J. C. Adkins, G. L. Plosker and K. L. Goa, Drugs Aging, 1999, 14, 459–475 CrossRef CAS PubMed.
- L. M. Pasetto, M. R. D'Andrea, E. Rossi and S. Monfardini, Critical Reviews in Oncology/Hematology, 2006, 59, 159–168 CrossRef PubMed.
- During blood circulation, OX could be fast degraded or deactivated by nucleophilic plasma and thiol or thiolate-containing proteins. (a) H. Ehrsson, I. Wallin and J. Yachnin, Med. Oncol., 2002, 19, 261–265 CrossRef CAS PubMed; (b) J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS PubMed; (c) L. Pendyala and P. J. Creaven, Cancer Res., 1993, 53, 5970–5976 CAS.
- (a) J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2016, 17, 20–37 CrossRef PubMed; (b) C. J. Kearney and D. J. Mooney, Nat. Mater., 2013, 12, 1004–1017 CrossRef CAS PubMed; (c) N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed; (d) X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed; (e) G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed.
- (a) V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed; (b) D. Landesman-Milo and D. Peer, Bioconjugate Chem., 2016, 27, 855–862 CrossRef CAS PubMed.
- (a) J. F. Lovell, C. S. Jin, E. Huynh, H. Jin, C. Kim, J. L. Rubinstein, W. C. W. Chan, W. Cao, L. V. Wang and G. Zheng, Nat. Mater., 2011, 10, 324–332 CrossRef CAS PubMed; (b) Y. Cao, X.-Y. Hu, Y. Li, X. Zou, S. Xiong, C. Lin, Y.-Z. Shen and L. Wang, J. Am. Chem. Soc., 2014, 136, 10762–10769 CrossRef CAS PubMed; (c) X. Chi, X. Ji, D. Xia and F. Huang, J. Am. Chem. Soc., 2015, 137, 1440–1443 CrossRef CAS PubMed.
- (a) K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64, 37–48 CrossRef; (b) S. Angelos, N. M. Khashab, Y.-W. Yang, A. Trabolsi, H. A. Khatib, F. J. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 12912–12914 CrossRef CAS PubMed; (c) S. D. Brown, P. Nativo, J.-A. Smith, D. Stirling, P. R. Edwards, B. Venugopal, D. J. Flint, J. A. Plumb, D. Graham and N. J. Wheate, J. Am. Chem. Soc., 2010, 132, 4678–4684 CrossRef CAS PubMed; (d) Y. Li, T.-Y. Lin, Y. Luo, Q. Liu, W. Xiao, W. Guo, D. Lac, H. Zhang, C. Feng, S. Wachsmann-Hogiu, J. H. Walton, S. R. Cherry, D. J. Rowland, D. Kukis, C. Pan and K. S. Lam, Nat. Commun., 2014, 5, 4712 CrossRef CAS PubMed.
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed.
- P. Liu, Ind. Eng. Chem. Res., 2013, 52, 13517–13527 CrossRef CAS.
- C. He, D. Liu and W. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef CAS PubMed.
- J. Tian, T.-Y. Zhou, S.-C. Zhang, S. Aloni, M. V. Altoe, S.-H. Xie, H. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Nat. Commun., 2014, 5, 5574 CrossRef CAS PubMed.
- (a) K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045–2076 CrossRef CAS PubMed; (b) I. Ghosh and W. M. Nau, Adv. Drug Delivery Rev., 2012, 64, 764–783 CrossRef CAS PubMed; (c) Y. Liu, G.-S. Chen, L. Li, H.-Y. Zhang, D.-X. Cao and Y.-J. Yuan, J. Med. Chem., 2003, 46, 4634–4637 CrossRef CAS PubMed; (d) Y. J. Jeon, S.-Y. Kim, Y. H. Ko, S. Sakamoto, K. Yamaguchi and K. Kim, Org. Biomol. Chem., 2005, 3, 2122–2125 RSC; (e) L. Cao, G. Hettiarachchi, V. Briken and L. Isaacs, Angew. Chem., Int. Ed., 2013, 52, 12033–12037 CrossRef CAS PubMed; (f) Y. R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Chem. Sci., 2015, 6, 1189–1193 RSC.
- For FDA approval, see: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/207155Orig1s000_Orig2s000TOC.cfm.
- For FDA approval, see: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm477512.htm.
- D. Ma, G. Hettiarachchi, D. Nguyen, B. Zhang, J. B. Wittenberg, P. Y. Zavalij, V. Briken and L. Isaacs, Nat. Chem., 2012, 4, 503–510 CrossRef CAS PubMed.
- S. Zorbas-Seifried, C. G. Hartinger, K. Meelich, M. Galanski, B. K. Keppler and H. Zorbas, Biochemistry, 2006, 45, 14817–14825 CrossRef CAS PubMed.
- (a) T. Ogoshi, S. Kanai, S. Fujinami, T.-a. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed; (b) D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9721–9723 CrossRef CAS PubMed; (c) M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed; (d) T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed; (e) C. Li, Chem. Commun., 2014, 50, 12420–12433 RSC; (f) Y. Ma, Z. Zhang, X. Ji, C. Han, J. He, Z. Abliz, W. Chen and F. Huang, Eur. J. Org. Chem., 2011, 5331–5335 CrossRef CAS.
- (a) W. Si, Z.-T. Li and J.-L. Hou, Angew. Chem., Int. Ed., 2014, 53, 4578–4581 CrossRef CAS PubMed; (b) S.-H. Li, H.-Y. Zhang, X. Xu and Y. Liu, Nat. Commun., 2015, 6, 7590 CrossRef PubMed; (c) N. L. Strutt, D. Fairen-Jimenez, J. Iehl, M. B. Lalonde, R. Q. Snurr, O. K. Farha, J. T. Hupp and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 17436–17439 CrossRef CAS PubMed; (d) W. Wang, L.-J. Chen, X.-Q. Wang, B. Sun, X. Li, Y. Zhang, J. Shi, Y. Yu, L. Zhang, M. Liu and H.-B. Yang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5597–5601 CrossRef CAS PubMed.
- (a) T. Ogoshi, M. Hashizume, T. Yamagishi and Y. Nakamoto, Chem. Commun., 2010, 46, 3708–3710 RSC; (b) Z. Li, G. Yu, J. Yang, G. Yu, J. He, Z. Abliz and F. Huang, Chem. Commun., 2014, 50, 2841–2843 RSC; (c) G. Yu, X. Zhou, Z. Zhang, C. Han, Z. Mao, C. Gao and F. Huang, J. Am. Chem. Soc., 2012, 134, 19489–19497 CrossRef CAS PubMed; (d) G. Yu, J. Zhou and X. Chi, Macromol. Rapid Commun., 2015, 36, 23–30 CrossRef CAS PubMed; (e) C. Li, J. Ma, L. Zhao, Y. Zhang, Y. Yu, X. Shu, J. Li and X. Jia, Chem. Commun., 2013, 49, 1924–1926 RSC; (f) G. Yu, W. Yu, Z. Mao, C. Gao and F. Huang, Small, 2015, 11, 919–925 CrossRef CAS PubMed.
- (a) Y. Wang, G. Ping and C. Li, Chem. Commun., 2016, 52, 9858–9872 RSC; (b) C. Li, S. Chen, J. Li, K. Han, M. Xu, B. Hu, Y. Yu and X. Jia, Chem. Commun., 2011, 47, 11294–11296 RSC; (c) X. Shu, S. Chen, J. Li, Z. Chen, L. Weng, X. Jia and C. Li, Chem. Commun., 2012, 48, 2967–2969 RSC.
- (a) F. G. A. Jansman, D. T. Sleijfer, J. L. L. M. Coenen, J. C. De Graaf and J. R. B. J. Brouwers, Drug Saf., 2000, 23, 255–278 CrossRef CAS PubMed; (b) E. Gamelin, A. L. Bouil, M. Boisdron-Celle, A. Turcant, R. Delva, A. Cailleux, A. Krikorian, S. Brienza, E. Cvitkovic, J. Robert, F. Larra and P. Allain, Clin. Cancer Res., 1997, 3, 891–899 CAS; (c) Prescribing information of ELOXATIN® (oxaliplatin), see: http://products.sanofi.us/eloxatin/eloxatin.html.
- The initial concentrations of the solutions used for injection in the efficacy studies were 0.786 mM (2.50 mg kg−1), 3.15 mM (10.0 mg kg−1), 4.72 mM (15.0 mg kg−1), and 2.36 mM (7.5 mg kg−1) on days 1, 3, 5, and 7. Using these values the initial fraction of OX complexed by CP6A was calculated to be 70%, 84%, 87%, and 82% for these time points, respectively. It is recognized that after injection, the concentrations of OX and CP6A would decrease due to a dilution in the blood stream.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details, variable-temperature 1H NMR spectra, mole ratio plot, ITC titration curves, and in vitro cytotoxicity assay. See DOI: 10.1039/c7sc01438d |
| This journal is © The Royal Society of Chemistry 2017 |