
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein labeling for live cell fluorescence microscopy with a highly photostable renewable signal†
Nina G.
Bozhanova
 ab,
Mikhail S.
Baranov
a,
Natalia V.
Klementieva
b,
Karen S.
Sarkisyan
ac,
Alexey S.
Gavrikov
a,
Ilia V.
Yampolsky
ad,
Elena V.
Zagaynova
b,
Sergey A.
Lukyanov
abd,
Konstantin A.
Lukyanov
ab and
Alexander S.
Mishin
*ab
ab,
Mikhail S.
Baranov
a,
Natalia V.
Klementieva
b,
Karen S.
Sarkisyan
ac,
Alexey S.
Gavrikov
a,
Ilia V.
Yampolsky
ad,
Elena V.
Zagaynova
b,
Sergey A.
Lukyanov
abd,
Konstantin A.
Lukyanov
ab and
Alexander S.
Mishin
*ab
aShemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Moscow, Russia. E-mail: mishin@ibch.ru
bNizhny Novgorod State Medical Academy, Nizhny Novgorod, Russia
cCentre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Dr Aiguader 88, 08003 Barcelona, Spain
dPirogov Russian National Research Medical University, Moscow, Russia
First published on 3rd August 2017
Abstract
We present protein-PAINT – the implementation of the general principles of PAINT (Point Accumulation for Imaging in Nanoscale Topography) for live-cell protein labeling. Our method employs the specific binding of cell-permeable fluorogenic dyes to genetically encoded protein tags. We engineered three mutants of the bacterial lipocalin Blc that possess different affinities to a fluorogenic dye and exhibit a strong increase in fluorescence intensity upon binding. This allows for rapid labeling and washout of intracellular targets on a time scale from seconds to a few minutes. We demonstrate an order of magnitude higher photostability of the fluorescence signal in comparison with spectrally similar fluorescent proteins. Protein-PAINT ensures prolonged super-resolution fluorescence microscopy of living cells in both single molecule detection and stimulated emission depletion regimes.
Introduction
Fluorescence labeling of target proteins in live cells provides opportunities to visualize their intracellular distribution, interactions, and dynamics. Green Fluorescent Protein (GFP) and GFP-like proteins are most often used for this purpose as fully genetically encoded labels.1 Also, methods of protein labeling based on protein tags that covalently bind chemical fluorophores have been developed.2–7 In comparison to labeling with fluorescent proteins, these methods provide more flexibility in choosing the spectral characteristics of the dyes and the time of labeling, but require additional steps of prolonged incubation and washing out the excess of unbound dye.These steps could be avoided by using fluorogenic dyes – those that are non-fluorescent in solvents but become fluorescent in a complex with the molecule of interest.8–13 The first system for specific protein labeling using exogenous synthetic fluorogens consisted of single-chain antibodies (FAPs) against thiazole orange or malachite green.14 More recently, the Y-FAST15 protein was engineered on the basis of the blue-light photoreceptor from Halorhodospira halophila to reversibly bind fluorogenic rhodanine derivatives. However, both labeling systems do not provide an increase in photostability, which could be expected from the exchange of the protein-bound and free dye molecules from solution.
Constant exchange of the dye provides a method for efficient single molecule super-resolution microscopy, since the binding is detected as a flash of fluorescence signal. The original demonstration of this principle on membranes with the Points Accumulation for Imaging in Nanoscale Topography (PAINT) technique13 was further extended to the labeling of arbitrary targets with pairs of DNA strands16 (DNA-PAINT), one of which is fluorescent and the other one is conjugated to the target or binder (e.g. antibody). However, even the latest-generation17,18 multiplexed DNA-PAINT variants are still limited by design to fixed samples.
Here, we created novel fluorogen–protein pairs for live-cell protein labeling and validated them in widefield, confocal and super-resolution microscopy applications. With these pairs we demonstrate the protein-PAINT principle: the combination of the genetically encoded protein tag and the rapid exchange of the fluorogenic dye allows for on-demand fluorescent labeling of target proteins in living cells and is compatible with various super-resolution techniques.
Results and discussion
GFP chromophore analogs have been previously reported to be fluorogenic with RNA aptamers10 and protein hosts.19–21 Many of them combine the ability to penetrate cell membranes with low brightness in the unbound state, and can dramatically increase the quantum yield when conformationally locked in a pocket of a host macromolecule.Application of a chromophore-containing solution to cells expressing host proteins or RNA allows for the fast formation of fluorescent complexes, but also – if the dye is not bound to the host too tightly – for efficient exchange of dye molecules with the pool of free molecules. Therefore, photobleached dye molecules can be quickly exchanged for the fluorescent ones. Taking this as a starting point, we designed and synthesized the library of GFP chromophore analogs, GFP-like aminated chromophores, and conformationally locked compounds22 (see ESI Methods†). For the protein counterpart we have chosen the lipocalin family of proteins as a scaffold capable of reversibly binding various small-molecule ligands.23–26 To avoid undesirable specific protein–protein interactions in eukaryotic systems we focused on bacterial proteins and selected the monomeric lipocalin Blc from E. coli without intramolecular disulphide bonds.27
We performed in silico mutagenesis of amino acids within the ligand-binding pocket of Blc (Fig. 1a) and then computationally screened the generated library using molecular docking of GFP chromophores. Mutants with an incompatible pocket size were filtered out as previously described.21 As a result, we selected nineteen top-scoring mutants (Fig. S1†) for cloning, expression and screening in vitro against the chromophore library.
 | ||
| Fig. 1 Characterization of the Blc mutants in a complex with chromophore M739in vitro and in cellulo. (a) Locations of the amino acid sites of Blc analyzed in this work, highlighted in the wild-type crystal structure (PDB ID: 1QWD). Pocket-facing amino acid positions are colored. Sites with mutations tested in vitro are shown in blue. (b) Changes in fluorescence upon complex formation. Shown are emission spectra of the same concentration of M739 (free or in the presence of saturating amounts of the corresponding proteins) normalized for the DiB1 maximal value. (c) Sequential staining and washout of H2B with DiBs in live HeLa cells. Top-to-bottom: DiB3, DiB2 and DiB1. The green filled and black hollow rectangles above the curves designate addition of the M739 solution (0.5 μM) or washout with HBSS buffer, respectively. Multiple intensity profiles correspond to different cells; on-to-off signal ratios are approximately 30, 15, and 30 for DiB3, DiB2, and DiB1, respectively. (d–f) Confocal fluorescence microscopy of DiBs in a HeLa cell line; (d) α-actinin-DIB1 in the presence of 0.25 μM M739 (excitation: 488 nm, emission: 520–560 nm); (e) α-actinin-DIB2 in the presence of 1 μM M739 (excitation: 488 nm, emission: 520–560 nm); (f) α-actinin-DIB3 in the presence of 5 μM M739 (excitation: 543 nm, emission: 560–600 nm). Scale bars – 20 μm. | ||
We observed an increase in fluorescence intensity in a number of protein–chromophore pairs (Fig. S2†). We excluded chromophores that showed too broad cross-reactivity with mutant proteins (e.g.1167 in Fig. S2†) from further characterisation. We also filtered out proteins with intense visible coloring or autofluorescence in vitro or in cell culture (Fig. S2,† right column).
For further characterisation we selected the three best-performing complexes of proteins with chromophore M739 that possessed different dissociation constants and called them DiB1 (“Dye in Blc 1”, Kd = 0.1 μM), DiB2 (Kd = 4 μM) and DiB3 (Kd = 9 μM) (Tables 1 and S1†). Within the protein complexes, M739 showed blue- (for DiB1 and DiB2) or red-shifted (for DiB3) absorption spectra with similar extinction coefficients in the range of 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–51000 M−1 cm−1 (Fig. S3,†Table 1). Compared to free M739, these complexes possessed an ∼5–10-fold increase in the fluorescence quantum yield.
000–51000 M−1 cm−1 (Fig. S3,†Table 1). Compared to free M739, these complexes possessed an ∼5–10-fold increase in the fluorescence quantum yield.
| Pair name | Mutationsa | λ ex, nm | λ em, nm | FQYb, % | EC, M−1 cm−1 | λ-Dependent FI, foldc | K d, μM | Photostability (relative to)d |
|---|---|---|---|---|---|---|---|---|
| a For numbering and sequence information refer to ESI Fig. 1.† b Fluorescence quantum yield. c Fluorescence increase compared to free M739 under conditions of confocal or TIRF microscopy (excitation at 488 nm and detection at 500–530 nm for DiB1 and DiB2; excitation at 561 nm and detection at 580–620 nm for DiB3). d See Fig. 2. | ||||||||
| DiB1 | A36C/L141N | 513 | 542 | 32 | 45800 |
52 | 0.1 | 2× (EGFP) |
| DiB2 | A36C | 510 | 539 | 32 | 51000 |
64 | 4 | 6× (EGFP) |
| DiB3 | V74F/L141Q | 546 | 565 | 15 | 43000 |
11 | 9 | 10× (mKate) |
| Free M739 | N/A | 520 | 563 | 3.5 | 53500 |
N/A | ||
But since the binding also led to a blue-shift of the emission spectra of DiB1 and DiB2, the effective wavelength-dependent fluorescence increase of the complex was more than 50-fold (Fig. 1b and Table 1).
To test the DiBs' performance in mammalian cells we expressed them as fusions with various proteins of interest (histone H2B, cytokeratin, α-actinin and vimentin). We found that the two yellow-colored pairs (DiB1 and DiB2) performed well in both widefield and confocal fluorescence microscopy. The target intracellular structures of the live cells demonstrated bright fluorescence immediately upon addition of the fluorogen M739 at concentrations of 0.1–1 μM (Fig. 1d and e and S4†). Thanks to the cell-permeable M739 fluorogen, multiple staining and washout cycles could be repeated in rapid succession with the cell-perfusion system (Fig. 1c), which is not possible with labeling systems with ester-modified fluorogens.28 Likely due to the lower affinity of the complex, labeling with DiB3 required a higher fluorogen concentration, which resulted in a higher level of background fluorescence (Fig. S5†). Therefore, DiB3 was found to have a limited utility for widefield microscopy, while DiB1 and DiB2 performed almost as well as a fluorescent protein (Fig. S5†).
We compared the photostability of the fluorescence signal of DiB-labeled (1 μM M739) histone H2B in living cells with the photostability of spectrally similar fluorescent proteins. We found that the protein–fluorogen pairs with higher Kd values (DiB2 and DiB3) exhibited an order of magnitude higher photostability than the corresponding fluorescent proteins (Fig. 2a), in agreement with the original idea of a dye exchange-based photostability increase.28 Also, the washout time for DiB1 observed in labeling-washout cycles in live cells was notably higher than for DiB2 (Fig. 1c). By imaging the washout of fixed detergent-treated cells with a higher temporal resolution we estimated the apparent koff values of the DiBs to be 0.01 s−1 for DiB1, 0.1 s−1 for DiB2, and 0.3 s−1 for DiB3. We also observed the positive effect of the delay between the frames on the overall photostability (Fig. 2b–d), even at fluorogen concentrations far above the Kd (Fig. 2b). Prolonged time-series with no sample illumination in between the frames is a typical live-cell microscopy setup, for which labeling with DiBs may be particularly useful.
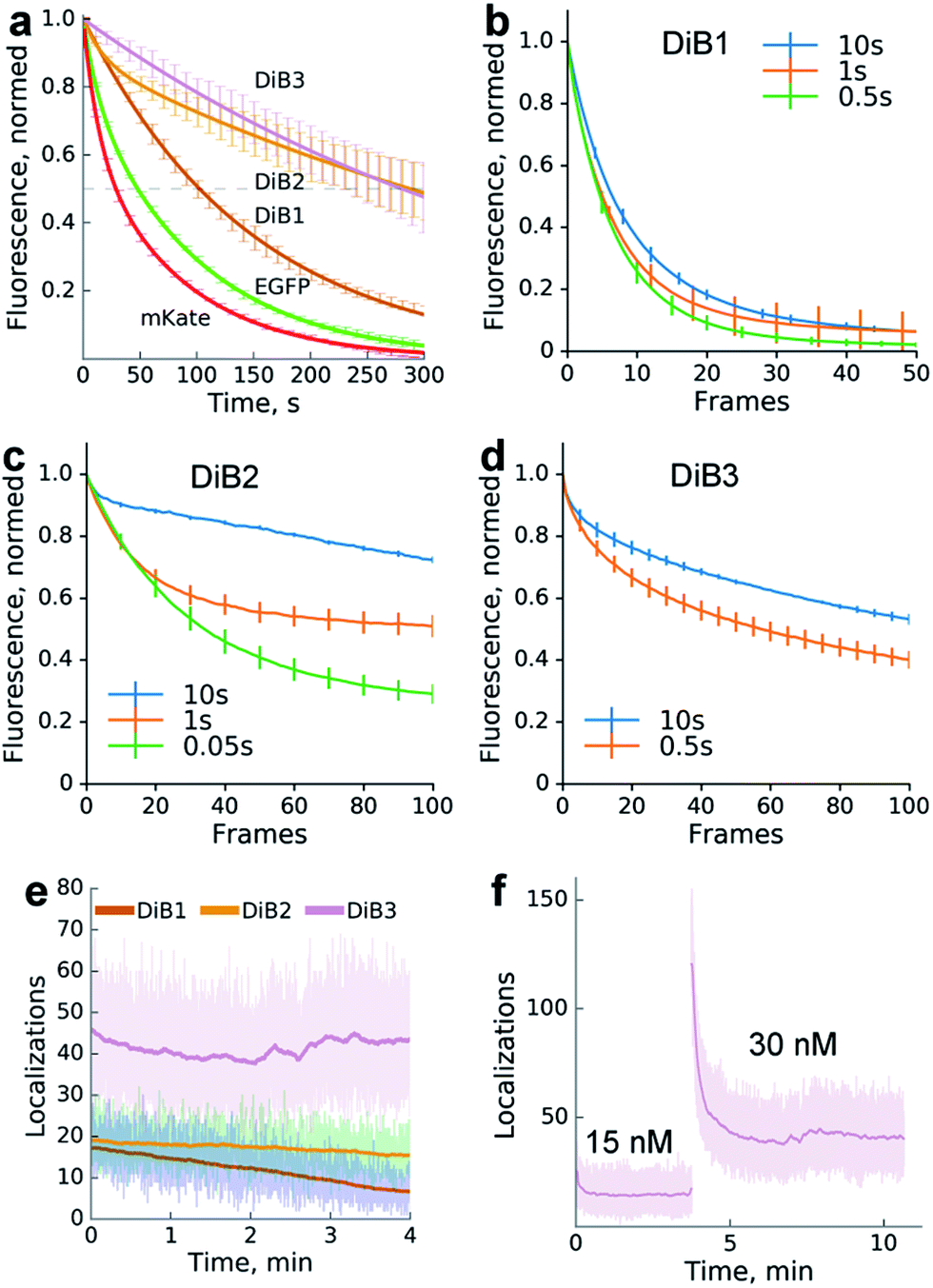 | ||
| Fig. 2 Photostability of the protein-PAINT labeling system. Live-cell performance of the protein-PAINT labeling system. (a) Photobleaching curves of HEK293T expressing H2B fused with Blc mutants and stained with chromophore M739 in a confocal setup. Curves for H2B-EGFP and H2B-mKate under the same imaging conditions are provided for comparison. mKate and DiB3: a 55.4 μm2 region was scanned with a 20 μW 543 nm laser. EGFP, DiB2 and DiB1: a 3542 μm2 region was scanned with a 100 μW 488 nm laser. Lines – two-term exponential fitting; error bars – s.d. (b–d) The impact of the imaging regime on DiBs photostability. Plots show the photobleaching curves obtained by collecting widefield images with different time gaps between ‘bleaching’ frames ((b and d) 1 s and (c) 0.1 s bursts of ∼60 W cm−2 light) for live HEK293T cells expressing H2B fused with DiB1, DiB2, and DiB3 in the presence of 0.5, 5, and 10 μM M739, respectively. (e and f) Photostability in the single-molecule imaging (TIRF) setup. The graph shows the number of localizations per frame (an ∼50 μm2 region of the frame is illuminated with 4.5 W cm−2 488 nm or 120 W cm−2 561 nm laser light in TIRF mode). The laser illumination occurred without intermittence, and the frames were taken with 16 ms exposure. (f) Prolonged single molecule imaging with DiB3 in the presence of 15 and 30 nM M739. Note that the addition of the chromophore solution to the cell medium immediately results in an increased number of localization events. | ||
To further test the utility of the DIBs we analysed their performance in single-molecule localization microscopy. We expected that application of the dye at a low concentration far below the Kd value would result in the stochastic formation of dye–protein complexes which would be detectable as sparse single-molecule fluorescence bursts, similarly to the FAP,28 PAINT13 or DNA-PAINT16 methods.
We therefore performed TIRF imaging of DiB-labeled cytoskeletal proteins (cytokeratin, α-actinin and vimentin) in live HeLa Kyoto cells with an EM-CCD camera, allowing for the detection of single molecule emitters (Fig. 2f).
Addition of the compound M739 at nanomolar concentrations led to the immediate appearance of sparsely distributed individual fluorescence spots (ESI Movies and Dataset†). The number of detected fluorescence bursts per frame increased with the increase in dye concentration (Fig. 2f). Remarkably, a dynamic equilibrium between dye entry into the cell, DiB complex formation, dissociation and bleaching resulted in a constant number of events per frame for DiB2 and DiB3 at the tested concentrations of the dye. In contrast, the number of DiB1 emitters declined rapidly in similar conditions (Fig. 2e), in accordance with the photostability data (Fig. 2a). The observed temporal stability of DiB2 and DiB3 labeling at the single-molecule level is in contrast with the previously reported time-traces of fluorogen activation by FAP,28 in which ever-increasing concentrations of fresh fluorogen were required for the same effect. Taken together, the DiBs' photostability data (at the single-molecule detection level, in a widefield and confocal mode) indicate the possible link between the residence time (determined by koff) of the fluorogen within the complex and the irreversible photobleaching. Indeed, photobleaching of GFP-like proteins occurs through multistep photochemical reactions of the chromophore, amino acids of the protein host and external compounds.30 With a number of possible long-lived and optically active fluorogen intermediates, the release of at least some of them into the solution before permanent damage of the protein host would improve photostability. The link between the photostability and the koff was also reported for some FAP variants.31 While all of the DiBs could be used for single molecule localization microscopy (Fig. 3 and S6†), only the low-affinity pairs (DiB2 and DiB3) exhibited the full advantages of the dynamic dye interchange. Thus, the protein-PAINT labeling method with DiB2 and DiB3 allows for the prolonged super-resolution imaging of live cells with the density of single-molecule fluorescence bursts controlled by the concentration of the dye in the imaging solution.
 | ||
| Fig. 3 Super-resolution imaging with the protein-PAINT labeling system under a moderate illumination power. Live HeLa Kyoto cells were transiently transfected with cytokeratin-DiB1 (a–c), vimentin-DiB2 (d–f) or α-actinin-DiB3 (g–i). Widefield images (a, d and g) and super-resolution reconstructions (b, e and h) from 5000 frames are shown; scale bars are 1 μm. (c, f and i) Normalized intensity profiles between the arrowheads shown on the widefield images (a, d and g); black curves – widefield and red curves – super-resolution. The fluorescence was excited with 488 nm (4.5 W cm−2, (a–f)) or 561 nm (120 W cm−2, (g–i)) laser lines. | ||
Importantly, a significant increase in image resolution (Fig. 3, right column) could be achieved with illumination intensities far below damaging levels,29 typical for most of the single-molecule localization microscopy techniques. However, higher photon numbers could be obtained, if needed, with an increase of the laser power (Fig. S7†). The high photostability of the DiBs is also beneficial for other light-intensive super-resolution modalities, such as stimulated emission depletion (STED). We observed a dramatic increase in the photostability (in comparison with a fluorescent protein) of DiB1 and DiB2 in STED mode on the same scale (Fig. S8†) as in the confocal mode (Fig. 2a).
Conclusions
To conclude, we engineered novel fluorogen–protein pairs exhibiting high photostabilities which were well-suited for single-molecule localization super-resolution microscopy. The fluorogenic GFP-like chromophores tested in this work showed rapid cell penetration and reversible labeling, allowing for simple on-demand tuning of the labeling density in living cells. Fluorogenic dyes greatly simplify cell staining, allowing for more sophisticated experimental design: in principle, various fluorogen-based labeling systems could be combined in the same experiment, with temporal separation in a single fluorescence channel, similarly to multiplexed DNA-PAINT,17,18 but in live cells. The Blc lipocalin, naturally evolved to bind various small molecules, proved to be a promising scaffold for the development of fluorogen-based labeling systems.Conflicts of interest
M. S. B., I. V. Y. and K. A. L. are the inventors the patent application covering the fluorogenic dyes used in this paper (WO2014031021 A1 and US20150177254 A1).Acknowledgements
We thank Timo Zimmermann and Raquel García Olivas from the CRG Advanced Light Microscopy Unit for assistance. This work was supported by the Russian Science Foundation grant 16-14-10364. KSS acknowledges support of the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2013-2017’ and the CERCA Programme/Generalitat de Catalunya. IVY thanks the MCB Program of the Russian Academy of Sciences for support.References
- D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Physiol. Rev., 2010, 90, 1103–1163 CrossRef CAS PubMed.
- C. Uttamapinant, K. A. White, H. Baruah, S. Thompson, M. Fernández-Suárez, S. Puthenveetil and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10914–10919 CrossRef CAS PubMed.
- D. S. Liu, L. G. Nivón, F. Richter, P. J. Goldman, T. J. Deerinck, J. Z. Yao, D. Richardson, W. S. Phipps, A. Z. Ye, M. H. Ellisman, C. L. Drennan, D. Baker and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4551–E4559 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa Jr, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- A. Keppler, H. Pick, C. Arrivoli, H. Vogel and K. Johnsson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9955–9959 CrossRef CAS PubMed.
- T. Gronemeyer, C. Chidley, A. Juillerat, C. Heinis and K. Johnsson, Protein Eng., Des. Sel., 2006, 19, 309–316 CrossRef CAS PubMed.
- Y. Hori, T. Norinobu, M. Sato, K. Arita, M. Shirakawa and K. Kikuchi, J. Am. Chem. Soc., 2013, 135, 12360–12365 CrossRef CAS PubMed.
- G. Lukinavičius, L. Reymond, E. D'Este, A. Masharina, F. Göttfert, H. Ta, A. Güther, M. Fournier, S. Rizzo, H. Waldmann, C. Blaukopf, C. Sommer, D. W. Gerlich, H.-D. Arndt, S. W. Hell and K. Johnsson, Nat. Methods, 2014, 11, 731–733 CrossRef PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299–16308 CrossRef CAS PubMed.
- I. Schoen, J. Ries, E. Klotzsch, H. Ewers and V. Vogel, Nano Lett., 2011, 11, 4008–4011 CrossRef CAS PubMed.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS PubMed.
- C. Szent-Gyorgyi, B. F. Schmidt, B. A. Schmidt, Y. Creeger, G. W. Fisher, K. L. Zakel, S. Adler, J. A. J. Fitzpatrick, C. A. Woolford, Q. Yan, K. V. Vasilev, P. B. Berget, M. P. Bruchez, J. W. Jarvik and A. Waggoner, Nat. Biotechnol., 2008, 26, 235–240 CrossRef CAS PubMed.
- M.-A. Plamont, E. Billon-Denis, S. Maurin, C. Gauron, F. M. Pimenta, C. G. Specht, J. Shi, J. Quérard, B. Pan, J. Rossignol, K. Moncoq, N. Morellet, M. Volovitch, E. Lescop, Y. Chen, A. Triller, S. Vriz, T. Le Saux, L. Jullien and A. Gautier, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 497–502 CrossRef CAS PubMed.
- R. Jungmann, C. Steinhauer, M. Scheible, A. Kuzyk, P. Tinnefeld and F. C. Simmel, Nano Lett., 2010, 10, 4756–4761 CrossRef CAS PubMed.
- F. Schueder, M. T. Strauss, D. Hoerl, J. Schnitzbauer, T. Schlichthaerle, S. Strauss, P. Yin, H. Harz, H. Leonhardt and R. Jungmann, Angew. Chem., Int. Ed., 2017, 56, 4052–4055 CrossRef CAS PubMed.
- S. S. Agasti, Y. Wang, F. Schueder, A. Sukumar, R. Jungmann and P. Yin, Chem. Sci., 2017, 8, 3080–3091 RSC.
- A. Baldridge, S. Feng, Y.-T. Chang and L. M. Tolbert, ACS Comb. Sci., 2011, 13, 214–217 CrossRef CAS PubMed.
- J.-S. Lee, A. Baldridge, S. Feng, Y. SiQiang, Y. K. Kim, L. M. Tolbert and Y.-T. Chang, ACS Comb. Sci., 2011, 13, 32–38 CrossRef CAS PubMed.
- N. V. Povarova, N. G. Bozhanova, K. S. Sarkisyan, R. Gritcenko, M. S. Baranov, I. V. Yampolsky, K. A. Lukyanov and A. S. Mishin, J. Mater. Chem. C, 2016, 4, 3036–3040 RSC.
- M. S. Baranov, K. M. Solntsev, N. S. Baleeva, A. S. Mishin, S. A. Lukyanov, K. A. Lukyanov and I. V. Yampolsky, Chem.–Eur. J., 2014, 20, 13234–13241 CrossRef CAS PubMed.
- R. E. Bishop, Biochim. Biophys. Acta, 2000, 1482, 73–83 CrossRef CAS.
- G. Beste, F. S. Schmidt, T. Stibora and A. Skerra, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1898–1903 CrossRef CAS.
- A. Skerra, FEBS J., 2008, 275, 2677–2683 CrossRef CAS PubMed.
- S. Schlehuber, G. Beste and A. Skerra, J. Mol. Biol., 2000, 297, 1105–1120 CrossRef CAS PubMed.
- A. Schiefner, L. Chatwell, D. A. Breustedt and A. Skerra, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 1308–1315 CrossRef CAS PubMed.
- Q. Yan, S. L. Schwartz, S. Maji, F. Huang, C. Szent-Gyorgyi, D. S. Lidke, K. A. Lidke and M. P. Bruchez, ChemPhysChem, 2014, 15, 687–695 CrossRef CAS PubMed.
- S. Wäldchen, J. Lehmann, T. Klein, S. van de Linde and M. Sauer, Sci. Rep., 2015, 5, 15348 CrossRef PubMed.
- A. Acharya, A. M. Bogdanov, B. L. Grigorenko, K. B. Bravaya, A. V. Nemukhin, K. A. Lukyanov and A. I. Krylov, Chem. Rev., 2017, 117, 758–795 CrossRef CAS PubMed.
- S. Saurabh, M. Zhang, V. R. Mann, A. M. Costello and M. P. Bruchez, ChemPhysChem, 2015, 16, 2974–2980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary methods, figures, movies, and data. See DOI: 10.1039/c7sc01628j |
| This journal is © The Royal Society of Chemistry 2017 |