
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triple bonds of niobium with silicon, germaniun and tin: the tetrylidyne complexes [(κ3-tmps)(CO)2Nb![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) E–R] (E = Si, Ge, Sn; tmps = MeSi(CH2PMe2)3; R = aryl)†
E–R] (E = Si, Ge, Sn; tmps = MeSi(CH2PMe2)3; R = aryl)†
Alexander C.
Filippou
 *,
David
Hoffmann
and
Gregor
Schnakenburg
*,
David
Hoffmann
and
Gregor
Schnakenburg
Institut für Anorganische Chemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Straße 1, 53121 Bonn, Germany. E-mail: filippou@uni-bonn.de
First published on 14th July 2017
Abstract
A systematic, efficient approach to first complexes containing a triple bond between niobium and the elements silicon, germanium or tin is reported. The approach involves a metathetical exchange of the niobium-centered nucleophile (NMe4)[Nb(CO)4(κ2-tmps)] (1) (tmps = MeSi(CH2PMe2)3) with a suitable organotetrel(II)halide. Compound 1 was obtained from (NMe4)[Nb(CO)6] and the triphosphane tmps by photodecarbonylation. Reaction of 1 with the disilene E-Tbb(Br)Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si(Br)Tbb in the presence of 4-dimethylaminopyridine afforded selectively the red-brown silylidyne complex [(κ3-tmps)(CO)2Nb
Si(Br)Tbb in the presence of 4-dimethylaminopyridine afforded selectively the red-brown silylidyne complex [(κ3-tmps)(CO)2Nb![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–Tbb] (2-Si, Tbb = 4-tert-butyl-2,6-bis(bis(trimethylsilyl)methyl)phenyl). Similarly, treatment of 1 with E(ArMes)Cl (E = Ge, Sn; ArMes = 2,6-mesitylphenyl) afforded after elimination of (NMe4)Cl and two CO ligands the deep magenta colored germylidyne complex [(κ3-tmps)(CO)2NbGe–ArMes] (3-Ge), and the deep violet, light-sensitive stannylidyne complex [(κ3-tmps)(CO)2NbSn–ArMes] (3-Sn), respectively. Formation of 3-Sn proceeds via the niobiastannylene [(κ3-tmps)(CO)3Nb–SnArMes] (4-Sn), which was detected by IR and NMR spectroscopy. The niobium tetrylidyne complexes 2-Si, 3-Ge and 3-Sn were fully characterized and their solid-state structures determined by single-crystal X-ray diffraction studies. All complexes feature an almost linear tetrel coordination and the shortest Nb–E bond lengths (d(Nb–Si) = 232.7(2) pm; d(Nb–Ge) = 235.79(4) pm; d(Nb–Sn) = 253.3(1) pm) reported to date. Reaction of 3-Ge with a large excess of H2O afforded upon cleavage of the Nb–Ge triple bond the hydridogermanediol Ge(ArMes)H(OH)2. Photodecarbonylation of [CpNb(CO)4] (Cp = η5-C5H5) in the presence of Ge(ArMes)Cl afforded the red-orange chlorogermylidene complex [Cp(CO)3NbGe(ArMes)Cl] (5-Ge). The molecular structure of 5-Ge features an upright conformation of the germylidene ligand, a trigonal–planar coordinated Ge atom, and a Nb–Ge double bond length of 251.78(6) pm, which lies in-between the Nb–Ge triple bond length of 3-Ge (235.79(4) pm) and a Nb–Ge single bond length (267.3 pm). Cyclic voltammetric studies of 2-Si, 3-Ge, and 3-Sn reveal several electron-transfer steps. One-electron oxidation and reduction of the germylidyne complex of 3-Ge in THF are electrochemically reversible suggesting that both the radical cation and radical anion of 3-Ge are accessible species in solution.
Si–Tbb] (2-Si, Tbb = 4-tert-butyl-2,6-bis(bis(trimethylsilyl)methyl)phenyl). Similarly, treatment of 1 with E(ArMes)Cl (E = Ge, Sn; ArMes = 2,6-mesitylphenyl) afforded after elimination of (NMe4)Cl and two CO ligands the deep magenta colored germylidyne complex [(κ3-tmps)(CO)2NbGe–ArMes] (3-Ge), and the deep violet, light-sensitive stannylidyne complex [(κ3-tmps)(CO)2NbSn–ArMes] (3-Sn), respectively. Formation of 3-Sn proceeds via the niobiastannylene [(κ3-tmps)(CO)3Nb–SnArMes] (4-Sn), which was detected by IR and NMR spectroscopy. The niobium tetrylidyne complexes 2-Si, 3-Ge and 3-Sn were fully characterized and their solid-state structures determined by single-crystal X-ray diffraction studies. All complexes feature an almost linear tetrel coordination and the shortest Nb–E bond lengths (d(Nb–Si) = 232.7(2) pm; d(Nb–Ge) = 235.79(4) pm; d(Nb–Sn) = 253.3(1) pm) reported to date. Reaction of 3-Ge with a large excess of H2O afforded upon cleavage of the Nb–Ge triple bond the hydridogermanediol Ge(ArMes)H(OH)2. Photodecarbonylation of [CpNb(CO)4] (Cp = η5-C5H5) in the presence of Ge(ArMes)Cl afforded the red-orange chlorogermylidene complex [Cp(CO)3NbGe(ArMes)Cl] (5-Ge). The molecular structure of 5-Ge features an upright conformation of the germylidene ligand, a trigonal–planar coordinated Ge atom, and a Nb–Ge double bond length of 251.78(6) pm, which lies in-between the Nb–Ge triple bond length of 3-Ge (235.79(4) pm) and a Nb–Ge single bond length (267.3 pm). Cyclic voltammetric studies of 2-Si, 3-Ge, and 3-Sn reveal several electron-transfer steps. One-electron oxidation and reduction of the germylidyne complex of 3-Ge in THF are electrochemically reversible suggesting that both the radical cation and radical anion of 3-Ge are accessible species in solution.
Introduction
Complexes of the general formula [LnME–R] (M = d-block metal; E = Si–Pb; R = singly bonded group (e.g. alkyl, aryl); Ln = ligand sphere) featuring a triple bond between a d-block metal and the tetrels Si/Ge/Sn/Pb are an intriguing class of compounds with an auspicious synthetic potential originating from the highly reactive, polar ME bond.1–4 Isolation of these compounds is very challenging and requires specific stereoelectronic properties of the metal fragment LnM as well as a steric protection of the electrophilic tetrel center by a tailor-made, bulky substituent R to circumvent a head-to-tail cyclodimerisation or unintentional intra- or intermolecular σ-bond activations destroying the ME–R functionality. Whereas earlier work concentrated exclusively on group 6 metals, recent studies have shown that also group 7,2l,3d,4d group 8 (ref. 1c and 5) and even group 10 metals6 can be incorporated into triple bonding with the tetrels Si–Pb. Extension of this chemistry to the group 5 elements V–Ta seemed attractive to investigate whether the lower electronegativity and larger metallic radii of these elements compared to Cr–W would have an effect on the ME functionality. Group 5 metal complexes featuring a triple bond to the heavier tetrels (E = Si–Pb) are presently not known, and even compounds with a ME double bond are very scarce and poorly characterized illustrating the challenge to make such compounds.7 We decided to address this issue, and present herein a systematic, efficient approach to the first complexes containing NbE (E = Si–Sn) triple bonds.
Results and discussion
Two methods have been employed so far for the formation of transition metal-tetrel (Si–Pb) triple bonds. The first method, abbreviated as the “salt elimination method”, involves a substitution reaction of a suitable anionic 18 VE metal complex with an organotetrel(II) halide, as exemplified by the synthesis of Cp-substituted group 6 metal tetrylidyne complexes (Scheme 1).2a,2b,2i,2j,2m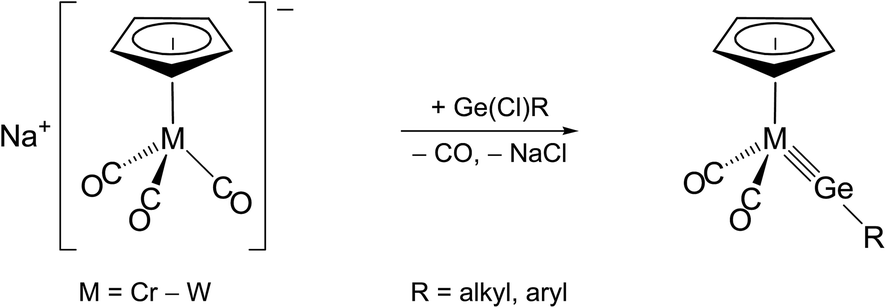 | ||
| Scheme 1 Preparation of half-sandwich group 6 metal germylidyne complexes by the salt elimination method. | ||
The second method, commonly termed “N2/PMe3 elimination method”, takes advantage of the exchange of labile ligands (mostly N2 or PMe3) in neutral 18 VE metal complexes by suitable organotetrel(II) halides. This approach may afford directly neutral ylidyne complexes, as demonstrated by the syntheses of phosphane-substituted group 6 and 7 metal tetrylidyne complexes (Scheme 2).2f,2g,2l,3a,4a,4b
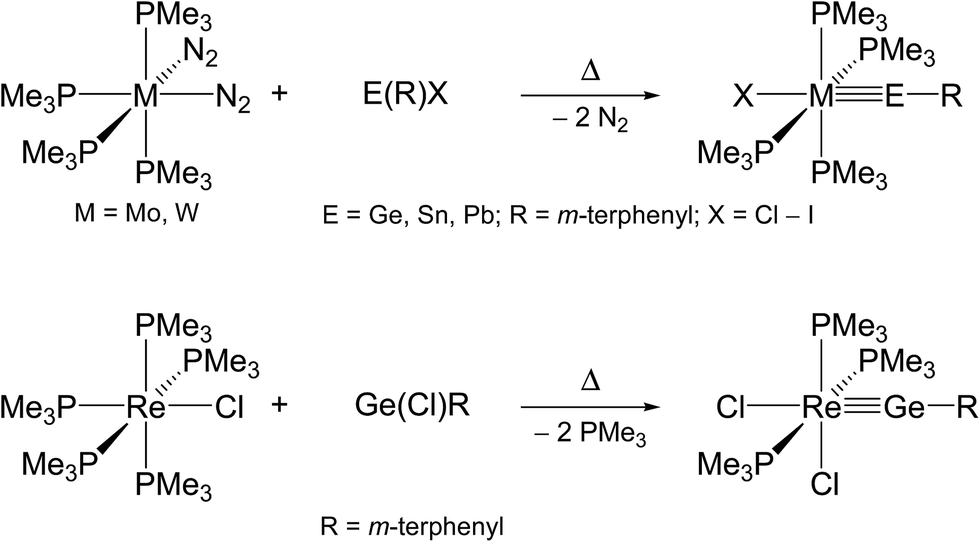 | ||
| Scheme 2 Preparation of neutral group 6 and 7 metal tetrylidyne complexes by the N2/PMe3 elimination method. | ||
Alternatively, haloylidene complexes are initially obtained by this method, which are subsequently converted to cationic ylidyne complexes by halide abstraction. Examples demonstrating this reaction path include the preparation of group 8 and 10 ylidyne complexes (Scheme 3).5,6
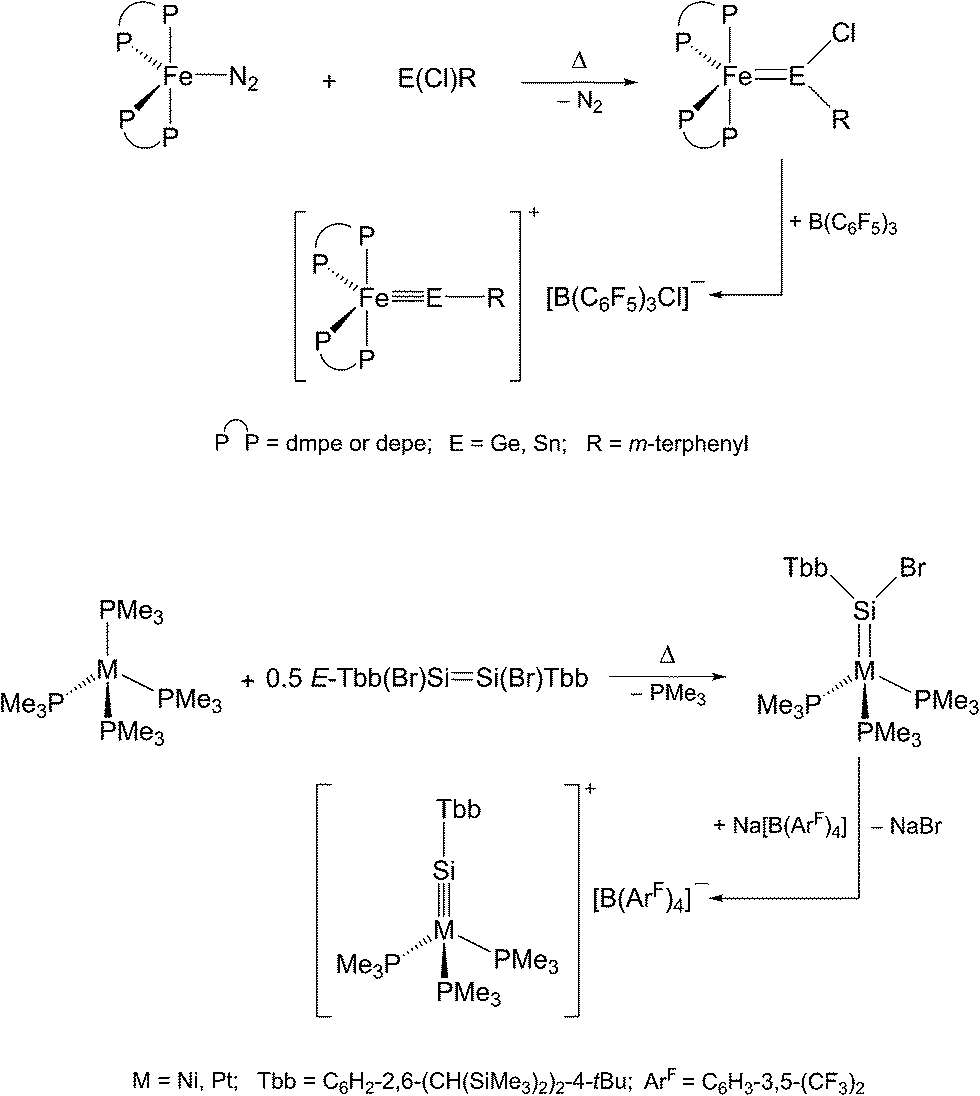 | ||
| Scheme 3 Preparation of group 8 and 10 metal tetrylidyne complexes via haloylidene complexes using the N2/PMe3 elimination method. | ||
We decided to apply the first method, given the availability of anionic niobium carbonyl complexes.8 At first, the homoleptic carbonyl niobate [Nb(CO)6]− was chosen. For this purpose the canary yellow salts (NR4)[Nb(CO)6] (R = Me, Et) were prepared, following the method developed by J. E. Ellis et al.9 However, these compounds proved to be unreactive towards the m-terphenyltetrel(II)halides E(ArMes)Cl (E = Ge, Sn; ArMes = 2,6-mesitylphenyl; mesityl (Mes) = 2,4,6-trimethylphenyl).10 For example, IR monitoring of the reaction of (NEt4)[Nb(CO)6] with Ge(ArMes)Cl in refluxing toluene did not provide any evidence for a conversion of the niobate even after prolonged heating, probably due to the poor nucleophilicity of [Nb(CO)6]−. Therefore, as next we turned our attention to niobates containing ligands with a higher σ-donor/π-acceptor ratio than CO, such as trialkyl- or triarylphosphanes. Various carbonyl(phosphane) niobates of the general formula [Nb(CO)4L2]− (L2 = bidentate di- or oligo-arylphosphane ligand) have been accessed from [Nb(CO)6]− upon photolytic CO substitution.11 In order to increase the electron density at the metal centre, we decided to use the highly basic, albeit, very oxygen-sensitive, tripodal alkylphosphane MeSi(CH2PMe2)3 (tmps).12
Photolysis of (NMe4)[Nb(CO)6] was carried out in the presence of one equivalent of tmps in THF at room temperature. A high-power blue light LED (λ = 465 nm) was used instead of a high-pressure mercury UV-lamp. The use of a nearly monochromatic source with an exciting wavelength close to the longest-wavelength absorption maximum of [Nb(CO)6]− (λmax = 440 nm in CH2Cl2)13 was conceived to be advantageous preventing the formation of insoluble brown decomposition products formed during the photolysis using a high-pressure mercury-lamp.11a
In fact, IR-monitoring of the reaction revealed a slow, but very selective conversion into the tetracarbonyl niobate [Nb(CO)4(κ2-tmps)]− proceeding via the pentacarbonyl intermediate [Nb(CO)5(κ1-tmps)]− (ν(CO) in THF: 1966 (m), 1821 (vs) cm−1). After work-up the salt (NMe4)[Nb(CO)4(κ2-tmps)] (1) was isolated in nearly quantitative yield (97%) as an orange, analytically pure, very air-sensitive powder, which decolorizes immediately upon exposure to air. The salt decomposes upon heating at 142 °C to a dark brown mass, and is well soluble in acetonitrile and tetrahydrofurane (THF), but only moderately soluble in benzene, toluene, and diethyl ether. Attempts to grow suitable single crystals of 1 for an X-ray diffraction study failed, however unambiguous proof for the composition and structure of 1 was provided by elemental analysis, IR spectroscopy and 1H, 13C{1H}, 31P{1H} and 29Si{1H} NMR spectroscopy. The IR spectrum of 1 in THF displays four ν(CO) absorption bands at 1900, 1787, 1764 and 1732 cm−1 (Fig. 1a), the band pattern being typical for octahedral cis-disubstituted metal tetracarbonyl complexes with a local C2v symmetry of the M(CO)4 fragment.14 All ν(CO) bands of 1 are shifted to lower frequencies than those of [Nb(CO)4(Ph2PCH2CH2PPh2)]− (ν(CO) in THF = 1908, 1806, 1782 and 1746 cm−1) or related disubstituted arylphosphane-carbonyl niobates.11b This shift to lower frequencies evinces the stronger +I effect of the P-bonded alkyl substituents in 1, which enhances the electron density at the metal center and leads to a stronger Nb(dπ) → CO(π*) backbonding and softening of the CO bonds in 1. The NMR spectra of 1 corroborate the presence of an overall Cs symmetric complex, in which one of the arms of the tripodal ligand tmps is pendant and the other two arms are bonded to the niobium center. For example, the 31P{1H} NMR spectrum of 1 displays a sharp singlet for the 31P nucleus of the pendant CH2PMe2 arm, which appears at almost the same position (δ(PA) = −55.8 ppm in benzene-d6) as that of the non-coordinated (“free”) tmps (δ(P) = −55.1 ppm in benzene-d6), and a very broad signal for the two symmetry-equivalent Nb-bonded 31P nuclei at considerably lower field (δ(PB) = −11.6 ppm in benzene-d6) (Fig. 1b). The broadness of the second signal (Δν1/2 (full width at half maximum) = 696 Hz) is caused by the quadrupole moment of the 93Nb nucleus (Q = −0.32 × 10−28 m2; I = 9/2, 100% natural abundance) and its effect on the relaxation time.15 Further structural information was provided by the 29Si{1H} NMR spectrum of 1, which shows a sharp signal for the bridgehead Si atom, that is split to a doublet of triplets (Fig. 1c) due to coupling with the two chemically different types of 31P nuclei in the integral ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (2J(Si,PA) = 14.7 Hz, 2J(Si,PB) = 8.2 Hz). A positional exchange of the pendant and the Nb-bonded arms of the tmps ligand in 1 was not observed in solution at 298 K.
:2 (2J(Si,PA) = 14.7 Hz, 2J(Si,PB) = 8.2 Hz). A positional exchange of the pendant and the Nb-bonded arms of the tmps ligand in 1 was not observed in solution at 298 K.
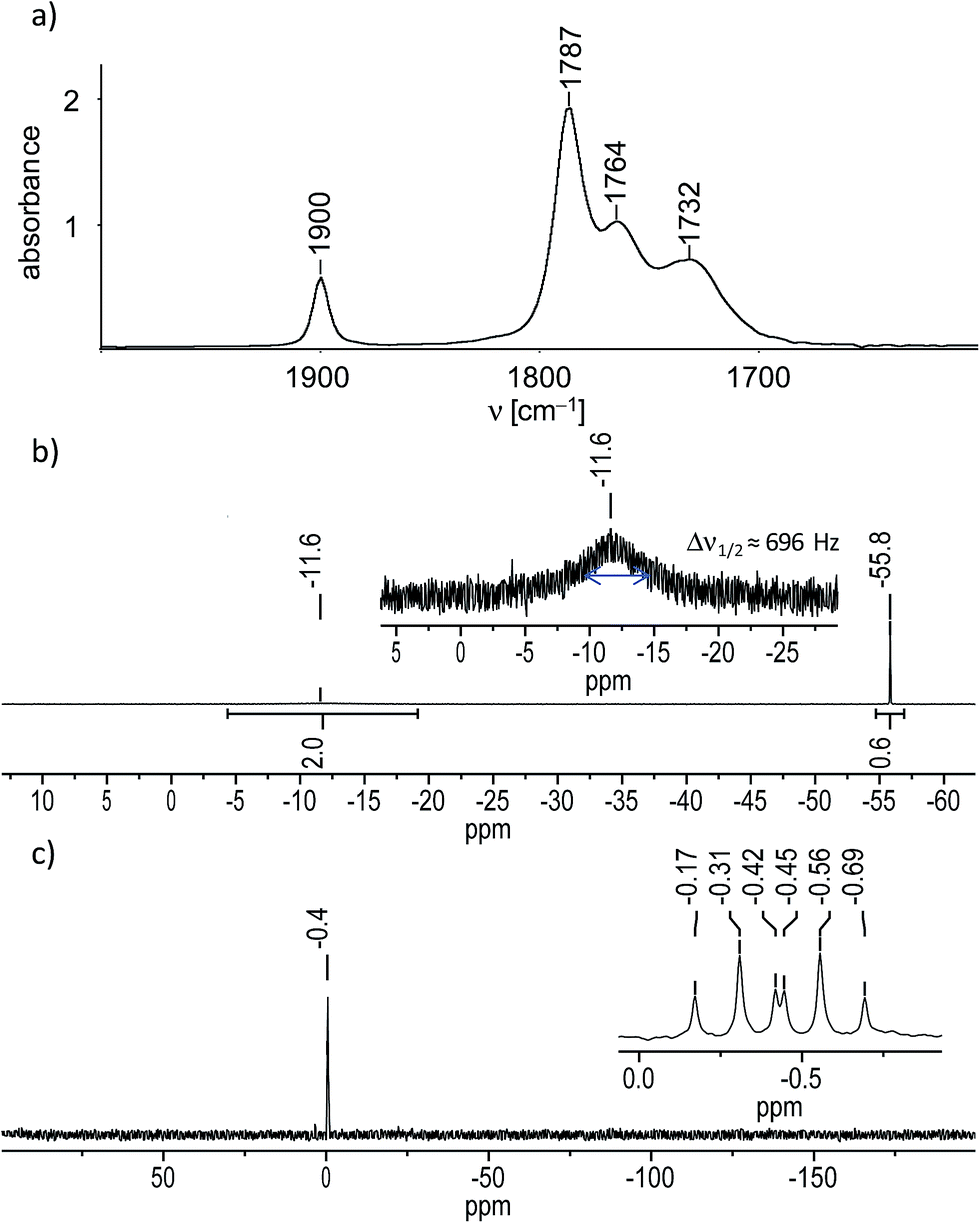 | ||
| Fig. 1 (a) FT-IR spectrum of 1 in THF in the range of 2000–1600 cm−1. (b) 31P{1H} NMR spectrum of 1 in benzene-d6; an enlarged excerpt with the broad signal at δ = −11.6 ppm is shown in the inset. (c) 29Si{1H} NMR spectrum of 1 in THF-d8; an enlarged excerpt with the signal at δ = −0.44 ppm is depicted in the inset. | ||
Complex 1 was found to be a very suitable nucleophile for the formation of NbE triple bonds (E = Si–Sn). Thus addition of a freshly prepared, orange-colored solution of a mixture of the 1,2-dibromodisilene E-Tbb(Br)SiSi(Br)Tbb16 and 4-dimethylamino pyridine (4-DMAP) (molar ratio 1:4), to a solution of one equiv. of 1 in toluene at ambient temperature was accompanied by an immediate color change to red-brown, and precipitation of a white solid ((NMe4)Br). IR monitoring revealed a complete and selective conversion to the silylidyne complex [(κ3-tmps)(CO)2NbSi–Tbb] (2-Si, Scheme 4). After work-up, complex 2-Si was isolated in 59% yield as a red-brown, extremely air-sensitive, microcrystalline solid, which decolorizes immediately upon exposure to air. Compound 2-Si is remarkably thermostable, and decomposes to a dark brown mass at 258 °C. It is moderately soluble in n-pentane, but readily soluble in benzene, toluene and THF.
 | ||
| Scheme 4 Synthesis of the niobium silylidyne complex 2-Si and the germylidyne complex 3-Ge. | ||
Similarly, treatment of complex 1 with the m-terphenylgermanium(II) chloride Ge(ArMes)Cl in toluene at −40 °C followed by warming to room temperature afforded rapidly and selectively the germylidyne complex [(κ3-tmps)(CO)2NbGe–ArMes] (3-Ge) (Scheme 4). Compound 3-Ge was isolated as a deep-magenta, very air-sensitive, thermally stable powder (dec. at 284 °C), that is moderately soluble in benzene and toluene, and well soluble in THF. No evidence for the formation of the putative metallogermylene intermediate [(κ3-tmps)(CO)3Nb–GeArMes] could be obtained during IR monitoring of the reaction of 1 with Ge(ArMes)Cl in toluene, the reaction starting at −35 °C and proceeding rapidly with CO evolution below 0 °C.
In comparison, reaction of the analogous m-terphenyltin(II)chloride Sn(ArMes)Cl with 1 in toluene afforded after stirring at ambient temperature the brick-red metallostannylene [(κ3-tmps)(CO)3Nb–SnArMes] (4-Sn) with a small amount of the stannylidyne complex [(κ3-tmps)(CO)2NbSnArMes] (3-Sn) (Scheme 5). Prolonged heating at 80 °C and periodic evacuation of the reaction tube was necessary to remove the released CO and to convert 4-Sn almost quantitatively into the stannylidyne complex 3-Sn, which after work-up was isolated as a dark violet, very air-sensitive powder in 70% yield. Complex 3-Sn is as 3-Ge thermally stable and decomposes upon heating at 266 °C. However, unlike 3-Ge, complex 3-Sn was found to be extremely light sensitive. Thus exposure of the deep-violet solutions of 3-Sn to fluorescent, ambient light or sun light lead to deposition of a tin mirror and formation of tmps and 1,3-dimesitylbenzene as evidenced by 1H NMR spectroscopy. Therefore, all operations during the synthesis, isolation and characterization of 3-Sn had to be carried out under exclusion of light.
 | ||
| Scheme 5 Synthesis of the niobium stannylidyne complex 3-Snvia the niobiastannylene 4-Sn. | ||
Decarbonylation of 4-Sn to afford 3-Sn is a remarkable, new type of reaction in the chemistry of metallostannylenes. In fact previous attempts to transform the metallostannylenes [Cp(CO)3M–SnR] (M = Cr, Mo, W; R = ArMes, ArTrip; ArTrip = C6H3–2,6-Trip2, Trip = C6H2–2,4,6-iPr3),17 [Cp(CO)2Fe–SnR] (R = ArDipp, ArTrip; ArDipp = C6H3–2,6-Dipp2, Dipp = C6H3–2,6-iPr2)18 or [Cp*(CO)3W–Sn(IDipp)]+ (Idipp = C[N(Dipp)CH]2, Dipp = C6H3–2,6-iPr2)2n into terminal stannylidyne complexes failed. We assume, that the increased steric pressure imposed by the tripodal ligand at the metal center weakens the Nb–CO bonds in the seven-coordinate complex 4-Sn and decreases thereby the barrier for a CO dissociation. In addition, formation of a strong NbSn triple bond resulting from the higher energy and larger radial extension of the d orbitals, which are engaged in the Nb(dπ) → SnR(π*) back bonding, may be also a driving force for the reaction.
The tetrylidyne complexes 2-Si, 3-Ge and 3-Sn were characterized by elemental analyses, IR spectroscopy and 1H, 13C{1H}, 31P{1H}, 29Si{1H} and 119Sn{1H} NMR spectroscopy. In addition their molecular structures were determined by single-crystal X-ray crystallography (Fig. 2 and 3). All complexes are distorted octahedral and feature a tridentate (κ3-bonded) tmps ligand, which spans three facial coordination sites with the P–Nb–P bite angles varying in a small range (85.3–87.9°). A view along the Si⋯Nb vector reveals that the CH2 groups connecting the bridgehead Si atom with the P donors are twisted out creating a local C3 symmetric, right or left-handed conformation, which reduces the bite of the chelating triphosphane ligand and optimizes the bonding with the niobium center (Fig. 3b). In solution, however, a rapid interchange of the two conformational enantiomers occurs according to NMR spectroscopy leading to averaged Cs symmetric structures.
 | ||
| Fig. 2 DIAMOND plot of the molecular structure of the silylidyne complex 2-Si in the solid state. Thermal ellipsoids were set at 30% electronic probability at 100 K. Hydrogen atoms and the methyl groups of the C2,6–CH(SiMe3)2 substituents were omitted for clarity. Selected bond lengths [pm] and angles [°]: Nb–Si1 232.7(2), Nb–P1 259.9(2), Nb–P2 258.4(2), Nb–P3 259.3(2), Nb–C35 206.8(9), Nb–C36 206.3(7), Si1–C1 189.0(7), C35–O1 117.6(8), C36–O2 117.9(7); Nb–Si1–C1 159.2(2), P1–Nb–P2 85.61(7), P1–Nb–P3 85.31(6), P2–Nb–P3 87.92(6), C35–Nb–C36 93.3(3). | ||
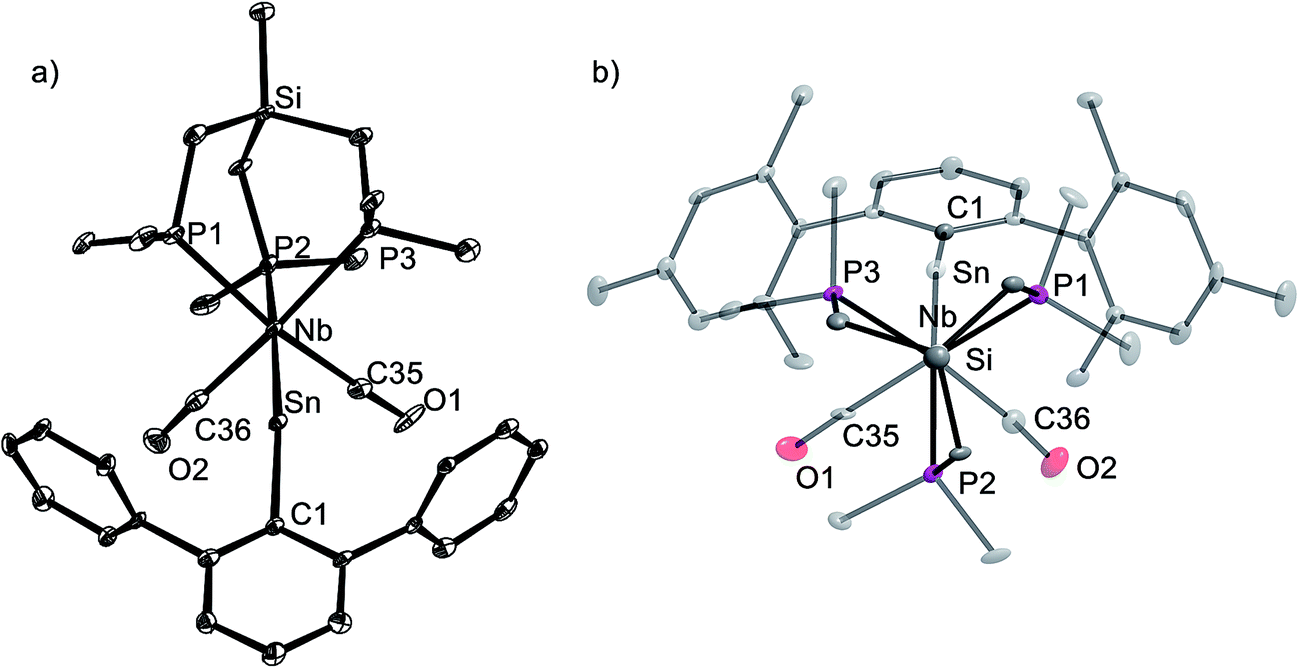 | ||
| Fig. 3 (a) DIAMOND plot of the molecular structure of the stannylidyne complex 3-Sn (toluene) in the solid state. Thermal ellipsoids were set at 30% electronic probability at 100 K. Hydrogen atoms and methyl groups of the ArMes substituent were omitted for clarity. Selected bond lengths [pm] and angles [°] of 3-Sn (toluene) (bond lengths and angles for 3-Ge (THF) are given in brackets): Nb–Sn 253.3(1) [235.79(4)], Nb–P1 260.6(4) [259.5(1)], Nb–P2 255.1(3) [258.0(1)], Nb–P3 258.6(4) [261.2(1)], Nb–C35 205.7(14) [206.0(5)], Nb–C36 207.1(16) [206.5(5)], Sn–C1 214.2(1) [196.3(4)], C35–O1 116.8(18) [116.8(6)], C36–O2 115.4(18) [115.5(6)]; Nb–Sn–C1 160.9(3) [164.0(1)], P1–Nb–P2 87.7(1) [86.30(4)], P1–Nb–P3 85.9(1) [86.08(4)], P2–Nb–P3 87.4(1) [87.82(4)], C35–Nb–C36 90.9(5) [92.5(2)]. (b) Top view of 3-Sn along the Si⋯Nb vector illustrating the C3-symmetric twist of the tmps ligand. | ||
The tetrylidyne complexes 2-Si, 3-Ge and 3-Sn feature the shortest Nb–Si, Nb–Ge and Nb–Sn bonds reported to date. In practice, the Nb–Si bond of 2-Si (232.7(2) pm) is ca. 28 pm shorter than the Nb–Si single bonds of silyl complexes (d(Nb–Si)mean of 28 structurally characterized complexes = 261.3 pm),19 and the Nb–Ge bond of 3-Ge (235.79(4) pm) ca. 31 pm shorter than a Nb–Ge single bond (d(Nb–Ge)mean = 267.3 pm).20 Similarly, the Nb–Sn bond of 3-Sn (253.3(1) pm) is ca. 30 pm shorter than a Nb–Sn single bond (d(Nb–Sn)mean 282.9 pm).21 Notably, a comparison of the Nb–E triple bond lengths of 2-Si, 3-Ge and 3-Sn with those of related molybdenum tetrylidyne complexes (e.g. d(MoSi) in [Cp(CO)2MoSi–ArTrip] = 222.41(7) pm;1ad(MoGe) in [Cp(CO)2MoGe–R] (R = C(SiMe3)3, ArMes, ArTrip) = 227–228 pm;2a,2b,2i,2md(MoSn) in trans-[X(PMe4)MoSn–ArMes] (X = Cl, Br, I) = 248–249 pm)22) reveals that the differences in the ME triple bond lengths (E = Si: 10 pm; E = Ge: 8–9 pm; E = Sn: 5–6 pm) compare reasonably well with the difference (7 pm) of the metallic radii of the two elements (rNb = 147 pm, rMo = 140 pm; radii for a coordination number of 12).23 A series of additive triple bond radii for most elements of the periodic table have been predicted by P. Pyykkö et al.24 The experimental Nb–E triple bond lengths 2-Si, 3-Ge and 3-Sn, are however, longer than the sum of the theoretically predicted triple bond radii (d(NbE)calc = Si: 218 pm, Ge: 230 pm, Sn: 248 pm).
In all complexes the tetrylidyne ligand is slightly bent at the tetrel center as evidenced by the bonding angle Nb–E–C1 (2-Si: 159.2(2)°, 3-Ge: 164.0(1)°, 3-Sn: 160.9(3)°). Bending occurs in all cases towards the CO ligands. It is presently unclear, whether this phenomenon, which is also observed in a series of group 6 metal dicarbonyl ylidyne complexes, is of steric or electronic origin or both. No clear evidence for steric congestion is at least provided by the molecular structures of 2-Si, 3-Ge and 3-Sn. For example, the closest van der Waals contacts were found in 2-Si between the methyl groups of the tmps ligand and the SiMe3 methyl groups of the Tbb substituent (d(H⋯H) = 244 pm). These contacts are longer than twice the van der Waals radius of hydrogen (rvdW(H) = 110 pm).25 It should be also taken into consideration, that deviation of the ME–R atom sequence from linearity does not require a lot of energy, indicating that subtle electronic effects may cause such a bending.26
Further structural information was obtained from the IR and NMR spectra of the tetrylidyne complexes. The IR spectra of 2-Si, 3-Ge and 3-Sn display two ν(CO) bands of almost equal intensity, which are typical for cis-dicarbonyl complexes and can be assigned to the in-phase (A′ symmetric) and out-of-phase (A′′ symmetric) CO stretching modes assuming local Cs symmetry of the M(CO)2 fragment (Fig. 4a). The ν(CO) bands of 3-Sn appear at lower frequencies (1851 and 1791 cm−1 in toluene) than those of 3-Ge (1868 and 1805 cm−1 in toluene), which suggests that the stannylidyne ligand SnArMes has a higher σ-donor/π-acceptor ratio than the germylidyne ligand GeArMes. Notably, the ν(CO) bands of 2-Si appear also at lower wavenumbers (1855 and 1790 cm−1 in toluene) than those of 3-Ge. This shift can be rationalized with the stronger +I effect of the Tbb substituent, leading to a higher σ-donor/π-acceptor ratio of the silylidyne ligand SiTbb than that of the germylidyne ligand GeArMes. The low-frequency position of the ν(CO) bands of 2-Si, 3-Ge and 3-Sn suggests the presence of an electron-rich Nb center that is engaged in strong Nb(dπ) → CO(π*) backbonding. Additional evidence for a strong Nb(dπ) → CO(π*) backbonding is provided by the 13C{1H} NMR spectra, which all display a broad CO signal at even lower field (δCO = 238.7 ppm (2-Si), 239.2 ppm (3-Ge), 238.9 ppm (3-Sn)) than that of 1 (δCO = 226.5 ppm).27 The number and relative intensity of the NMR signals indicate an averaged Cs symmetry of the tetrylidyne complexes in solution and a rapid rotation of the tetrel-bonded aryl group about the E–Caryl bond. The signals of all nuclei directly attached to the quadrupolar 93Nb nucleus are significantly broadened due to fast relaxation (vide supra). For example, the 29Si{1H} NMR spectrum of 2-Si displays at 298 K a very broad signal (Δν1/2 = 130 Hz) for the NbSi nucleus at δ = 267.8 ppm, for which the 2J(29Si,31P) coupling could not be resolved. In comparison, the remote positioned bridgehead Si atom of the tmps ligand and the SiMe3 groups of the Tbb substituent give rise to sharp signals at δ = −0.7 ppm and +1.5 ppm, respectively, with the first of these signals being split into a quartet due to coupling to the three 31P nuclei (2J(29Si,31P) = 9.7 Hz) (Fig. S16 and S17 (ESI†)). Similarly, the 31P{1H} NMR spectra of 2-Si and 3-Ge show only one broad signal at δ = −13.0 ppm (Δν1/2 ≈ 182 Hz at 298 K) and −10.7 ppm (Δν1/2 ≈ 187 Hz at 283 K), respectively, instead of two 31P NMR signals expected for an AX2 spin system (Fig. 4b). Broadness of the signals can be influenced by the temperature given the well known relationship between the quadrupole-coupled nuclear relaxation time and the temperature dependent molecular correlation time.28 In fact, lowering of the temperature lead to a “decoupling” of the Nb nucleus and allowed to resolve the two 31P NMR signals and their 2J(P,P) coupling of 20.9 Hz as illustrated by the 31P NMR spectrum of 3-Ge at 193 K (Fig. 4c). Taking advantage of the same effect, also the 119Sn resonance of 3-Sn, that was not observable in THF-d8 at room temperature, could be detected at 243 K as a very broad signal (Δν1/2 ≈ 1297 Hz) at δ = 829.7 ppm (Fig. S36 (ESI†)).
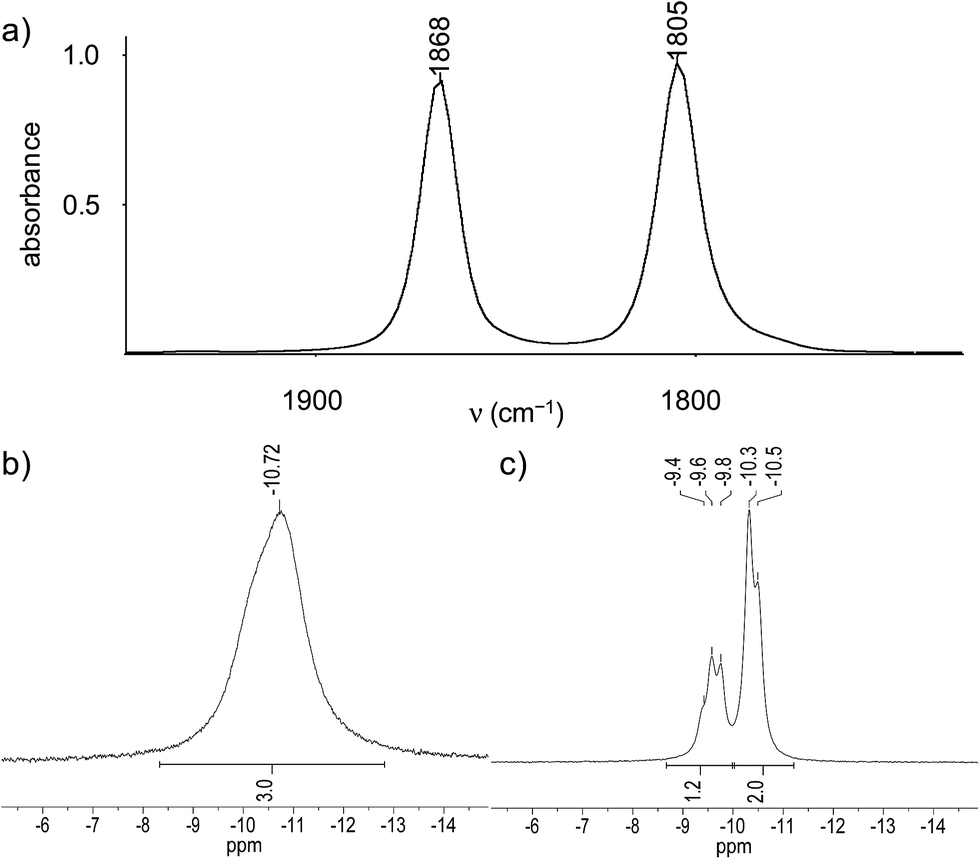 | ||
| Fig. 4 (a) IR ν(CO) absorption bands of the germylidyne complex 3-Ge in toluene. (b) 31P{1H} NMR signal of the germylidyne complex 3-Ge in THF-d8 at 283 K. (c) 31P{1H} NMR signals of the germylidyne complex 3-Ge in THF-d8 at 193 K. | ||
First studies reveal a marked difference in the reactivity of the niobium germylidyne complex 3-Ge and the related molybdenum germylidyne complexes [Cp(CO)2MoGe–R] (R = C(SiMe3)3, ArMes, ArTrip). Thus treatment of [Cp(CO)2MoGe–R] with H2O or MeOH (one equiv.) in diethyl ether at 0 °C followed by warming to ambient temperature afforded within one hour selectively the brown hydroxy/methoxygermylidene complexes [Cp(CO)2(H)MoGe(OR′)R] (R′ = H, Me), which were fully characterized.2m In contrast, no reaction of 3-Ge with H2O (one equiv.) was observed in THF even at 60 °C. The inertness of 3-Ge can be rationalized with the stronger metal-germylidyne Nb(dπ) → GeR(π*) back bonding, which reduces the electrophilicity of the Ge center in 3-Ge, and increases in combination with the steric protection of the metal center by the tridentate tmps ligand the activation barrier for the H2O addition at the NbGe bond. In fact, a large excess of water (925 equiv.) and prolonged heating (3 h) was necessary to effectuate a full conversion of 3-Ge accompanied by a color change of the reaction solution from magenta to orange. IR monitoring of the reaction did not provide any evidence for the formation of the anticipated H2O addition products. Instead, a continuous decrease in intensity of the two ν(CO) bands of 3-Ge was observed suggesting the formation of mainly CO-free products. Benzene extraction of the orange-brown solid obtained after solvent evaporation afforded a benzene soluble, pale-orange part containing mainly the germanediol Ge(ArMes)H(OH)2, as well as a benzene-insoluble brownish part. The unprecedented hydridogermanediol29 was isolated as a pale yellow solid and characterized by IR and 1H NMR spectroscopy. Its IR spectrum displays two ν(OH) bands at 3600 and 3398 cm−1 and a characteristic ν(Ge–H) band at 2104 cm−1, the latter one appearing at a close position to that of GeBr2HMes (ν(Ge–H) = 2105 cm−1).30 In the 1H NMR spectrum a distinctive doublet signal is observed for the Ge(OH)2 protons at δ = 0.91 ppm and a triplet signal for the Ge–H functionality at δ = 5.61 ppm (2J(H,H) = 3.5 Hz) in the integral ratio of 2:1. Notably, the Ge–OH protons of the germanetriol Ge(ArTrip)(OH)3 have a similar chemical shift (δ = 0.77 ppm in CDCl3).29k
Attempts were also undertaken to access cationic tetrylidyne complexes. For this purpose, [CpNb(CO)4]31 was prepared using a slightly modified procedure32 and irradiated in THF with a high-power blue light LED (λ = 465 nm) in the presence of one equivalent of Ge(ArMes)Cl. IR monitoring of the reaction revealed a quite selective decarbonylation leading to the chlorogermylidene complex 5-Ge, which after work-up was isolated as red-orange, air-sensitive crystals in 25% yield (Scheme 6). Remarkably attempts to abstract the chloride from 5-Ge and to form the putative germylidyne complex cation [Cp(CO)3NbGeArMes]+ were not successful so far. For example, no reaction of 5-Ge with Na[B(ArF)4] (ArF = C6H3–3,5-(CF3)2) was observed in C6H5F at room temperature.
 | ||
| Scheme 6 Synthesis of the niobium chlorogermylidene complex 5-Ge. | ||
Complex 5-Ge is the first niobium germylidene complex to be reported. Its solid-state molecular structure was determined by single-crystal X-ray crystallography (Fig. 5). The four-legged piano stool complex is Cs symmetric and features a trigonal–planar coordinated Ge-atom (sum of angles at the Ge atom = 360.0°). The symmetry plane passes through the atoms Nb, Ge, C1 and Cl, and bisects the CpNb(CO)3 fragment and the central ring of the m-terphenyl substituent.
 | ||
| Fig. 5 DIAMOND plot of the molecular structure of 5-Ge in the solid state. Thermal ellipsoids were set at 30% electronic probability at 100 K, and hydrogen atoms were omitted for clarity. Selected bond lengths [pm] and angles [°]: Nb–Ge 251.78(6), Ge–C1 196.2(4), Ge–Cl 219.1(1), Nb–C17 207.4(3), Nb–C18 206.1(4), C17–O1 115.2(4), C18–O2 114.5(5); Nb–Ge–C1 141.4(1), Nb–Ge–Cl 118.75(4), Cl–Ge–C1 99.8(1). | ||
The germylidene ligand adopts an upright conformation, with the ArMes substituent pointing towards the cyclopentadienyl ring. The Nb–Ge distance (251.78(6) pm) of 5-Ge lies in-between that found for the Nb–Ge triple bond of 3-Ge (235.79(4) pm) (vide supra) and that of a Nb–Ge single bond (d(Nb–Ge) = 267.3 pm)20 indicating the presence of a Nb–Ge double bond in 5-Ge. The angles at the Ge atom differ markedly with the Nb–Ge–Caryl angle (141.4(1)°) being much larger than the Caryl–Ge–Cl angle (99.8(1)°). This distortion can be attributed to the large steric demand of the ArMes substituent and the low tendency of germanium for isovalent hybridization.1a,1b,2l,3d The Ge–Cl bond of 5-Ge (219.1(1) pm) compares well with that of Ge(ArTrip)Cl (220.3(2) pm),33 but is considerably shorter than those of chlorogermylidene complexes containing electron-rich metal centers, such as [(dmpe)2FeGe(ArMes)Cl] (d(Ge–Cl) = 232.2(1) pm),5 [(PMe3)3NiGe(ArMes)Cl] (d(Ge–Cl) = 230.03(8) pm)6 or [(PMe3)3PdGe(ArMes)Cl] (d(Ge–Cl) = 227.3(1) pm),6 in which a strong M(dπ) → Ge(pπ) back bonding is presumed to cause a strong polarization of Ge–Cl bond leading to a facile chloride abstraction by Lewis acids. The reduced polarization of the Ge–Cl bond of 5-Ge provides a rationale for its inertness towards mild chloride abstraction reagents.
The solution IR and NMR spectra of 5-Ge are fully consistent with its solid-state molecular structure. Thus, the IR spectrum of 5-Ge in THF displays three intense ν(CO) absorption bands at 1980, 1910 and 1899 cm−1, as expected for a Nb(CO)3 fragment with local Cs symmetry, which are assigned to the A′ (all three CO modes in phase), A′ (two COlat modes in phase; COdiag mode out-of-phase) and A′′ symmetric (two COlat modes out-of-phase) CO stretching modes, respectively. The ν(CO) absorption bands of 5-Ge are high-frequency shifted compared to those of [CpNb(CO)3THF] (ν(CO) in THF = 1961, 1840 cm−1)31 or [CpNb(CO)3PEt3] (ν(CO) in THF = 1953, 1850 cm−1),28b but appear at roughly the same position as those of [CpNb(CO)3N2] (ν(CO) in n-heptane = 1991, 1905 cm−1)34 suggesting a similar σ-donor/π-acceptor ratio of the germylidene GeArMesCl and the N2 ligand. The 1H and 13C{1H} NMR spectra also confirm the Cs symmetry of 5-Ge in solution. Rotation of the m-terphenyl substituent about the Ge–Caryl bond occurs fast on the NMR time-scale at ambient temperature leading to an exchange of the two diastereotopic ortho (C2,6) and meta (C3,5) positions of the enantiotopic mesityl substituents. Therefore, only one singlet signal is observed in the 1H NMR spectrum of 5-Ge for the C2,6-bonded methyl groups and C3,5-bonded protons of the mesityl substituents, respectively.
Electrochemical studies
Electrochemical studies of the tetrylidyne complexes 2-Si, 3-Ge and 3-Sn were carried out using cyclic voltammetry to elucidate the redox properties of these compounds. All complexes display a rich electrochemistry involving several electron-transfer steps (see ESI, chapter 3†). Remarkably, both the one-electron reduction and oxidation of the germylidyne complex 3-Ge are electrochemically reversible occurring at a half wave potential (E1/2) of −2.612 mV and −405 mV vs. the dmfc1+/0 redox couple (dmfc = decamethylferrocene), respectively (Fig. 6).35 | ||
| Fig. 6 Single-scan cyclic voltammograms of the reversible one-electron oxidation of 3-Ge at different scan rates in THF at −11 °C (supporting electrolyte: [NBu4][PF6] (0.1 M); reference electrode: 0.004 M [Fe(C5Me5)2]+1/0/0.1 M [NBu4][PF6]/THF). | ||
In comparison, the corresponding redox steps of 2-Si and 3-Sn are irreversible (ESI, chapter 3†), but one-electron oxidation 2-Si and 3-Sn occurs at similar potentials as that of 3-Ge (2: Epa + Epc/2 = −468 mV, 3-Sn: Epa + Epc/2 = −435 mV (scan rate = 100 mV s−1)). Evidence that the redox process at E1/2 = −405 mV involves a one electron oxidation of 3-Ge was provided by chemical means. Thus, no reaction of 3-Ge with the one-electron reducing agent cobaltocene (E1/2 of CoCp2 in DME = −740 mV) was observed in fluorobenzene even at 70 °C, whereas an instantaneous oxidation of 3-Ge occurred upon treatment with one equivalent of [Fe(η5-C5Me5)2][B(ArF)4] in fluorobenzene solution at −30 °C. Unfortunately, attempts to isolate the putative germylidyne complex radical cation [(κ3-tmps)(CO)2Nb(GeArMes)]+ failed so far.36 Notably, the redox potential for the one-electron oxidation of 3-Ge is slightly lower than that of the molybdenum tetrylidyne complexes trans-[ClMo(PMe3)4E–ArMes] (E = Ge: E1/2 in C6H5F = −340 mV; E = Sn: E1/2 in THF = −350 mV; E = Pb: E1/2 in THF = −358 mV) verifying the presence of an electron-rich Nb center in 3-Ge.
Conclusion
The synthesis of the tailor-made carbonyl-niobate (NMe4)[Nb(CO)4(κ2-tmps)] allowed to explore its reactivity towards a series of organotetrel(II) halides, which lead to the isolation of the first niobium complexes featuring triple bonds with the elements Si, Ge and Sn. Photochemical CO substitution in [CpNb(CO)4] (Cp = η5-C5H5) by Ge(ArMes)Cl afforded also the novel chlorogermylidene complex [Cp(CO)3NbGe(ArMes)Cl]. The structural, spectroscopic and electrochemical data of the tetrylidyne complexes [(κ3-tmps)(CO)2NbSi–Tbb] (2-Si), [(κ3-tmps)(CO)2NbGe–ArMes] (3-Ge) and [(κ3-tmps)(CO)2NbSn–ArMes] (3-Sn) suggest the presence of an electron-rich metal center that is engaged into strong metal (dπ) → ER(π*) and metal (dπ) → CO(π*) back bonding. Remarkably, one-electron oxidation and reduction of the germylidyne complex 3-Ge are electrochemically reversible.
Acknowledgements
We thank the Jürgen Manchot Stiftung and the University of Bonn for the financial support of this work. We also thank Mrs Charlotte Rödde, Dr Senada Nozinovic, Dipl.-Ing. Karin Prochnicki, Dr Burhanshah Lewall, Mrs Kerstin Kühnel-Lysek and Mrs Anna Martens for their contributions to the single-crystal X-ray diffraction studies, the NMR measurements, the cyclic voltammetric studies and the elemental analyses.References
- For experimental studies on compounds with MSi bonds (silylidyne complexes) see:
(a) A. C. Filippou, O. Chernov, K. W. Stumpf and G. Schnakenburg, Angew. Chem. Int. Ed., 2010, 49, 3296 (
Angew. Chem.
, 2010
, 122
, 3368
) CrossRef CAS PubMed;
(b) A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem. Int. Ed., 2011, 50, 1122 (
Angew. Chem.
, 2011
, 123
, 1154
) CrossRef CAS PubMed;
(c) P. G. Hayes, Z. Xu, C. Beddie, J. M. Keith, M. B. Hall and T. D. Tilley, J. Am. Chem. Soc., 2013, 135, 11780 CrossRef CAS PubMed;
(d) A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem. Int. Ed., 2014, 53, 565 (
Angew. Chem.
, 2014
, 126
, 576
) CrossRef CAS PubMed;
(e) T. Fukuda, T. Yoshimoto, H. Hashimoto and H. Tobita, Organometallics, 2016, 35, 921 CrossRef CAS;
(f) T. Yoshimoto, H. Hashimoto, N. Hayakawa, T. Matsuo and H. Tobita, Organometallics, 2016, 35, 3444 CrossRef CAS.
- For experimental studies on germylidyne complexes see: (a) R. S. Simons and P. P. Power, J. Am. Chem. Soc., 1996, 118, 11966 CrossRef CAS; (b) L. Pu, B. Twamley, S. T. Haubrich, M. M. Olmstead, B. V. Mork, R. S. Simons and P. P. Power, J. Am. Chem. Soc., 2000, 122, 650 CrossRef CAS; (c) A. C. Filippou, A. I. Philippopoulos, P. Portius and D. U. Neumann, Angew. Chem. Int. Ed., 2000, 39, 2778 ( Angew. Chem. , 2000 , 112 , 2881 ) CrossRef CAS; (d) A. C. Filippou, A. I. Philippopoulos, P. Portius and G. Schnakenburg, Organometallics, 2004, 23, 4503 CrossRef CAS; (e) A. C. Filippou, G. Schnakenburg, A. I. Philippopoulos and N. Weidemann, Angew. Chem. Int. Ed., 2005, 44, 5979 ( Angew. Chem. , 2005 , 117 , 6133 ) CrossRef CAS PubMed; (f) A. C. Filippou, N. Weidemann, A. I. Philippopoulos and G. Schnakenburg, Angew. Chem. Int. Ed., 2006, 45, 5987 ( Angew. Chem. , 2006 , 118 , 6133 ) CrossRef CAS PubMed; (g) A. C. Filippou, A. Barandov, G. Schnakenburg, B. Lewall, M. van Gastel and A. Marchanka, Angew. Chem., 2012, 124, 813 ( Angew. Chem. Int. Ed. , 2012 , 51 , 789 ) CrossRef; (h) H. Hashimoto, T. Fukuda, H. Tobita, M. Ray and S. Sakaki, Angew. Chem., Int. Ed., 2012, 51, 2930 ( Angew. Chem. , 2012 , 124 , 2984 ) CrossRef CAS PubMed; (i) A. C. Filippou, K. W. Stumpf, O. Chernov and G. Schnakenburg, Organometallics, 2012, 31, 748 CrossRef CAS; (j) J. Hicks, T. J. Hadlington, C. Schenk, J. Li and C. Jones, Organometallics, 2013, 32, 323 CrossRef CAS; (k) T. Fukuda, H. Hashimoto and H. Tobita, Chem. Commun., 2013, 49, 4232 RSC; (l) A. C. Filippou, U. Chakraborty and G. Schnakenburg, Chem.–Eur. J., 2013, 19, 5676 CrossRef CAS PubMed; (m) K. W. Stumpf, Germylidene and Germylidyne Complexes of Molybdenum, Dissertation, University of Bonn, Verlag Dr Hut, 2014, ISBN 978-3-8439-1849-7; (n) Y. Lebedev, U. Das, G. Schnakenburg and A. C. Filippou, Organometallics, 2017, 36, 1530 CrossRef CAS.
- For experimental studies on stannylidyne complexes see: (a) A. C. Filippou, P. Portius, A. I. Philippopoulos and H. Rohde, Angew. Chem. Int. Ed., 2003, 42, 445 ( Angew. Chem. , 2003 , 115 , 461 ) CrossRef CAS PubMed; (b) A. C. Filippou, A. I. Philippopoulos and G. Schnakenburg, Organometallics, 2003, 22, 3339 CrossRef CAS; (c) H. Rohde, M. Menzel, F. Renz and A. C. Filippou, Hyperfine Interact., 2008, 185, 129 CrossRef CAS; (d) A. C. Filippou, P. Ghana, U. Chakraborty and G. Schnakenburg, J. Am. Chem. Soc., 2013, 135, 11525 CrossRef CAS PubMed; (e) C. Lindlahr, Novel Tetrylidyne Complexes of Group 6 Metals with Trialkylphosphane Ligands, Dissertation, University of Bonn, Verlag Dr Hut, 2015, ISBN 978-3-8439-2126-8.
- For experimental studies on plumbylidyne complexes see: (a) A. C. Filippou, H. Rohde and G. Schnakenburg, Angew. Chem. Int. Ed., 2004, 43, 2243 ( Angew. Chem. , 2004 , 116 , 2293 ) CrossRef CAS PubMed; (b) A. C. Filippou, N. Weidemann, G. Schnakenburg, H. Rohde and A. I. Philippopoulos, Angew. Chem. Int. Ed., 2004, 43, 6512 ( Angew. Chem. , 2004 , 116 , 6674 ) CrossRef CAS PubMed; (c) A. C. Filippou, N. Weidemann and G. Schnakenburg, Angew. Chem. Int. Ed., 2008, 47, 5799 ( Angew. Chem. , 2008 , 120 , 5883 ) CrossRef CAS PubMed; (d) U. Chakraborty, Multiple Bonds between Group 7 Transition Metals and Heavier Tetrel Elements (Ge–Pb), Dissertation, University of Bonn, Dr Hut Verlag, 2013, ISBN 978-3-8439-1248-8.
- B. Blom, Reactivity of Ylenes at Late Transition Metal Centers, Dissertation, University of Bonn, Cuviller Verlag, 2011, ISBN 978-3-86955-907-0.
- I. Papazoglou, Unprecedented Tetrylidyne Complexes of Group 6 and Group 10 Metals, Dissertation, University of Bonn, Dr Hut Verlag, 2017, ISBN 978-3-8439-3155-7.
- (a) A. V. Lalov, M. P. Egorov, O. M. Nefedov, V. K. Cherkasov, N. L. Ermolaev and A. V. Piskunov, Russ. Chem. Bull., 2005, 54, 807 CrossRef CAS; (b) A. Shinohara, J. McBee, R. Waterman and T. D. Tilley, Organometallics, 2008, 27, 5717 CrossRef CAS; (c) H. Arp, J. Baumgartner, C. Marschner, P. Zark and T. Müller, J. Am. Chem. Soc., 2012, 134, 10864 CrossRef CAS PubMed.
- (a) A. Davison and J. E. Ellis, J. Organomet. Chem., 1972, 36, 113 CrossRef CAS; (b) F. Calderazzo, G. Pampaloni and G. Pelizzi, J. Organomet. Chem., 1982, 233, C41 CrossRef CAS; (c) K. M. Pfahl and J. E. Ellis, Organometallics, 1984, 3, 230 CrossRef CAS; (d) F. Calderazzo, M. Castellani, G. Pampaloni and P. F. Zanazzi, J. Chem. Soc., Dalton Trans., 1985, 1989 RSC; (e) E. M. Carnahan and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 4166 CrossRef CAS; (f) M. V. Barybin, J. E. Ellis, M. K. Pomije, M. L. Tinkham and G. F. Warnock, Inorg. Chem., 1998, 37, 6518 CrossRef CAS PubMed; (g) P. J. Daff, P. Legzdins and S. J. Rettig, J. Am. Chem. Soc., 1998, 120, 2688 CrossRef CAS.
- (a) C. G. Dewey, J. E. Ellis, K. L. Fjare, K. M. Pfahl and G. F. P. Warnock, Organometallics, 1983, 2, 388 CrossRef CAS; (b) F. Calderazzo, U. Englert, G. Pampaloni, G. Pelizzi and R. Zamboni, Inorg. Chem., 1983, 22, 1865 CrossRef CAS; (c) F. Calderazzo, G. Pampaloni and J. E. Ellis, Inorganic Syntheses, John Wiley & Sons, Hoboken, NJ, USA, 1990, p. 192 Search PubMed.
- R. S. Simons, L. Pu, M. M. Olmstead and P. P. Power, Organometallics, 1997, 16, 1920 CrossRef CAS.
- (a) A. Davison and J. E. Ellis, J. Organomet. Chem., 1971, 31, 239 CrossRef CAS; (b) H.-C. Bechthold and D. Rehder, J. Organomet. Chem., 1982, 233, 215 CrossRef CAS; (c) K. Bachmann and D. Rehder, J. Organomet. Chem., 1984, 276, 177 CrossRef CAS; (d) F. Calderazzo, G. Pampaloni, U. Englert and J. Strähle, J. Organomet. Chem., 1990, 383, 45 CrossRef CAS.
- H. H. Karsch and A. Appelt, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1983, 38, 1399 Search PubMed.
- M. S. Wrighton, D. I. Handeli and D. L. Morse, Inorg. Chem., 1976, 15, 434 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, Hoboken, 2009 Search PubMed.
- (a) P. Raghavan, At. Data Nucl. Data Tables, 1989, 42, 189 CrossRef CAS; (b) J. H. Nelson, Nuclear Magnetic Resonance Spectroscopy, Pearson Education, 2003 Search PubMed.
- T. Agou, N. Hayakawa, T. Sasamori, T. Matsuo, D. Hashizume and N. Tokitoh, Chem.–Eur. J., 2014, 20, 9246 CrossRef CAS PubMed.
- B. E. Eichler, A. D. Phillips, S. T. Haubrich, B. V. Mork and P. P. Power, Organometallics, 2002, 21, 5622 CrossRef CAS.
- H. Lei, J.-D. Guo, J. C. Fettinger, S. Nagase and P. P. Power, Organometallics, 2011, 30, 6316 CrossRef CAS.
- According to a a CSD survey (13.06.17) the Nb–Si bond lengths of silyl complexes range from 254.1 to 267.9 pm leading to a mean value of 261.3 pm and a median of 261.2 pm for the Nb–Si single bond length: (a) J. Arnold, T. D. Tilley, A. L. Rheingold and S. J. Geib, Organometallics, 1987, 6, 473 CrossRef CAS; (b) A. Antiñolo, F. Carrillo, M. Fajardo, A. Otero, M. Lanfranchi and M. A. Pellinghelli, Organometallics, 1995, 14, 1518 CrossRef; (c) G. I. Nikonov, L. G. Kuzmina, D. A. Lemenovskii and V. V. Kotov, J. Am. Chem. Soc., 1995, 117, 10133 CrossRef CAS; (d) G. I. Nikonov, L. G. Kuzmina, S. F. Vyboishchikov, D. A. Lemenovskii and J. A. K. Howard, Chem.–Eur. J., 1999, 5, 2947 CrossRef CAS; (e) V. I. Bakhmutov, J. A. K. Howard, D. A. Keen, L. G. Kuzmina, M. A. Leech, G. I. Nikonov, E. V. Vorontsov and C. C. Wilson, J. Chem. Soc., Dalton Trans., 2000, 1631 RSC; (f) G. I. Nikonov, P. Mountford, J. C. Green, P. A. Cooke, M. A. Leech, A. J. Blake, J. A. K. Howard and D. A. Lemenovskii, Eur. J. Inorg. Chem., 2000, 1917 CrossRef CAS; (g) G. I. Nikonov, L. G. Kuzmina and J. A. K. Howard, J. Chem. Soc., Dalton Trans., 2002, 3037 RSC; (h) G. I. Nikonov, P. Mountford and S. R. Dubberley, Inorg. Chem., 2003, 42, 258 CrossRef CAS PubMed; (i) K. Y. Dorogov, E. Dumont, N.-N. Ho, A. V. Churakov, L. G. Kuzmina, J.-M. Poblet, A. J. Schultz, J. A. K. Howard, R. Bau, A. Lledos and G. I. Nikonov, Organometallics, 2004, 23, 2845 CrossRef CAS; (j) K. Y. Dorogov, A. V. Churakov, L. G. Kuzmina, J. A. K. Howard and G. I. Nikonov, Eur. J. Inorg. Chem., 2004, 771 CrossRef CAS; (k) K. Y. Dorogov, M. Yousufuddin, N.-N. Ho, A. V. Churakov, L. G. Kuzmina, A. J. Schultz, S. A. Mason, J. A. K. Howard, D. A. Lemenovskii, R. Bau and G. I. Nikonov, Inorg. Chem., 2007, 46, 147 CrossRef CAS PubMed; (l) S. K. Ignatov, N. H. Rees, A. A. Merkoulov, S. R. Dubberley, A. G. Razuvaev, P. Mountford and G. I. Nikonov, Chem.–Eur. J., 2008, 14, 296 CrossRef CAS PubMed.
- A CSD survey (13.06.17) revealed that only two niobium germyl complexes have been structurally characterized. The Nb–Ge distances range from 260.8 to 271.0 pm, leading to a mean value of 267.3 pm and a median of 270.2 pm for the Nb–Ge single bond length: (a) A. Antiñolo, F. Carrillo-Hermosilla, A. Castel, M. Fajardo, J. Fernández-Baeza, M. Lanfranchi, A. Otero, M. A. Pellinghelli, G. Rima, J. Satgé and E. Villaseñor, Organometallics, 1998, 17, 1523 CrossRef; (b) G. I. Nikonov, A. V. Churakov and M. Y. Antipin, Organometallics, 2003, 22, 2178 CrossRef CAS.
- A CSD survey (13.06.17) revealed that 9 niobium stannyl complexes have been structurally characterized. The Nb–Sn bond lengths range from 276.3 to 286.2 pm, giving a mean value of 282.9 pm and a median of 283.0 pm for the Nb–Sn single bond length. The respective complexes can be found in [ref. 20a] and in the following references: (a) F. Näumann, J. Kopf and D. Rehder, J. Organomet. Chem., 1984, 267, 249 CrossRef; (b) Y. V. Skripkin, O. G. Volkov, A. A. Pasynskii, A. S. Antsyshkina, L. M. Dikareva, V. N. Ostrikova, M. A. Porai-Koshits, S. L. Davydova and S. G. Sakharov, J. Organomet. Chem., 1984, 263, 345 CrossRef CAS; (c) Y. V. Skripkin, O. G. Volkov, A. A. Pasynskii, M. A. Porai-Koshits, A. S. Antsyshkina, L. M. Dikareva and V. N. Ostrikova, Koord. Khim., 1985, 11, 1136 CAS; (d) M. L. H. Green, A. K. Hughes and P. Mountford, J. Chem. Soc., Dalton Trans., 1991, 1407 RSC; (e) M. L. H. Green, A. K. Hughes and P. Mountford, J. Chem. Soc., Dalton Trans., 1991, 1699 RSC; (f) G. I. Nikonov, L. G. Kuzmina, J. Lorberth and J. A. K. Howard, Eur. J. Inorg. Chem., 1999, 825 CrossRef CAS.
- H. Rohde, Synthesis and Reactivity Studies of Stannylidyne Complexes, Dissertation, University of Bonn, 2007.
- A. F. Holleman, E. Wiberg and N. Wiberg, Inorganic Chemistry, Academic Press, San Diego, 2001 Search PubMed.
- (a) P. Pyykkö, S. Riedel and M. Patzschke, Chem.–Eur. J., 2005, 11, 3511 CrossRef PubMed; (b) P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326 CrossRef PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806 CrossRef CAS PubMed.
- For example quantum chemical studies on the model compounds trans-[Cl(PH3)4WEPh] (E = Si–Pb) at the B3LYP/TZVPP(W,E)/6-311G*(C,H,P,Cl) level of theory reveal that the energy required for bending the tetrylidyne ligand up to 20° is rather small (max. 20 kJ mol−1) and decreases with increasing atomic number of E.
- L. J. Todd and J. R. Wilkinson, J. Organomet. Chem., 1974, 77, 1 CrossRef CAS.
- (a) P. S. Ireland, C. A. Deckert and T. L. Brown, J. Magn. Reson., 1976, 23, 485 CAS; (b) D. Rehder and H.-C. Bechthold, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1984, 39, 323 Search PubMed and references therein.
- Only few four-coordinated germanediols and two germanetriols (Ge(ArTrip/Dip)(OH)3) were reported so far: (a) H. Puff, S. Franken, W. Schuh and W. Schwab, J. Organomet. Chem., 1983, 254, 33 CrossRef CAS; (b) L. Fajarí, J. Carilla, L. Juliá, J. Riera, A. Párraga, M. Coll and X. Solans, J. Organomet. Chem., 1994, 474, 89 CrossRef; (c) K. Kishikawa, N. Tokitoh and R. Okazaki, Chem. Lett., 1996, 25, 695 CrossRef; (d) L. Pu, N. J. Hardman and P. P. Power, Organometallics, 2001, 20, 5105 CrossRef CAS; (e) T. Matsumoto, Y. Nakaya and K. Tatsumi, Organometallics, 2006, 25, 4835 CrossRef CAS; (f) Y. Sugiyama, T. Sasamori, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1023 CrossRef CAS PubMed; (g) E. Bonnefille, S. Mazières, C. Bibal, N. Saffon, H. Gornitzka and C. Couret, Eur. J. Inorg. Chem., 2008, 4242 CrossRef CAS; (h) E. Bonnefille, S. Mazières, N. Saffon and C. Couret, J. Organomet. Chem., 2009, 694, 2246 CrossRef CAS; (i) M. Saito, M. Nakamura and T. Tajima, Heterocycles, 2009, 78, 657 CrossRef CAS; (j) L. Li, T. Fukawa, T. Matsuo, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, Nat. Chem., 2012, 4, 361 CrossRef CAS PubMed; (k) L.-C. Pop, N. Kurokawa, H. Ebata, K. Tomizawa, T. Tajima, M. Ikeda, M. Yoshioka, M. Biesemans, R. Willem, M. Minoura and M. Saito, Can. J. Chem., 2014, 92, 542 CrossRef CAS.
- P. Rivière, M. Rivière-Baudet, A. Castel, J. Satgé and E. A. Lavabre, Synth. React. Inorg. Met.-Org. Chem., 1987, 17, 539 CrossRef.
- W. A. Herrmann and H. Biersack, J. Organomet. Chem., 1980, 191, 397 CrossRef CAS and refs. therein.
- T. E. Bitterwolf, S. Gallagher, J. T. Bays, B. Scallorn, A. L. Rheingold, I. A. Guzei, L. Liable-Sands and J. C. Linehan, J. Organomet. Chem., 1998, 557, 77 CrossRef CAS.
- L. Pu, M. M. Olmstead, P. P. Power and B. Schiemenz, Organometallics, 1998, 17, 5602 CrossRef CAS.
- G. I. Childs, D. C. Grills, S. Gallagher, T. E. Bitterwolf and M. W. George, J. Chem. Soc., Dalton Trans., 2001, 1711 RSC.
- All potentials in the text are given vs. the [Fe(η5-C5Me5)2]1+/0 (dmfc1+/0) redox couple, which was chosen as the reference standard for the CV experiments due to its favorable properties vs. the [Fe(η5-C5H5)2]1+/0 redox couple: (a) I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, J. Phys. Chem. B, 1999, 103, 6713 CrossRef CAS; (b) J. R. Aranzaes, M.-C. Daniel and D. Astruc, Can. J. Chem., 2006, 84, 288 CrossRef . For comparison reasons the half-wave potential of the [Fe(η5-C5H5)2]1+/0 redox couple was also determined under the same conditions (THF, −11 °C) and found to be +440 mV vs. the [Fe(η5-C5Me5)2]1+/0 redox couple.
- Rapid oxidation of 3-Ge was evidenced by the immediate disappearance of the green color of the fluorobenzene solution of [Fe(η5-C5Me5)2][B(ArF)4] at the dropping site and the change of the color of the reaction solution to brown-red. After evaporation of the solvent and washing of the residue with n-pentane to remove [Fe(η5-C5Me5)2], a brown oil was obtained, which displayed in the IR spectrum in THF mainly two intense, broad ν(CO) bands at 1954 and 1889 cm−1. The brown oil contained according to 31P NMR spectroscopy diamagnetic impurities and could not be crystallized.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses and analytical data of 1, 2-Si, 3-Ge, 3-Sn and 5-Ge, illustrations of the IR and heteronuclear magnetic resonance spectra of 1, 2-Si, 3-Ge 3-Sn and 5-Ge, details of the cyclic voltammetric studies of 2-Si, 3-Ge and 3-Sn, and crystal structure determination of 2-Si, 3-Ge (THF), 3-Sn (toluene) and 5-Ge. CCDC 1553387–1553389 and 1555671. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc02708g |
| This journal is © The Royal Society of Chemistry 2017 |