
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbnormal room temperature phosphorescence of purely organic boron-containing compounds: the relationship between the emissive behaviorand the molecular packing, and the potential related applications†
Zhaofei
Chai
a,
Can
Wang
a,
Jinfeng
Wang
a,
Fan
Liu
a,
Yujun
Xie
a,
Yong-Zheng
Zhang
b,
Jian-Rong
Li
 b,
Qianqian
Li
a and
Zhen
Li
*a
b,
Qianqian
Li
a and
Zhen
Li
*a
aDepartment of Chemistry, Hubei Key Lab on Organic and Polymeric Opto-Electronic Materials, Wuhan University, Wuhan 430072, China. E-mail: lizhen@whu.edu.cn
bDepartment of Chemistry and Chemical Engineering, Beijing University of Technology, Beijing 100124, China
First published on 9th October 2017
Abstract
Purely organic materials with the characteristic of room-temperature phosphorescence (RTP) under ambient conditions demonstrate potential benefits in advanced optoelectronic applications. Exploration of versatile and efficient RTP compounds with low prices is full of challenges due to the slow intersystem crossing process and ultrafast deactivation of the active excited states of organic compounds. Here, a series of boron-containing phosphors were found to present RTP with long-lived lifetimes. Among these commercially available and cheap compounds, (4-methoxyphenyl)boronic acid (PBA-MeO) exhibits long-lived RTP, with a lifetime of 2.24 s, which is among the longest lifetimes of single-component small molecules. Our extensive experiments illustrate that both a rigid conformation and expanded conjugation induced by molecular alignment contribute to the persistent RTP. Because of strong intermolecular interactions via hydrogen bonds, these arylboronic acids easily form crystals and are quite appropriate for anti-forgery materials. Subsequently, we develop a precise, speedy and convenient inkjet printing technology for the fabrication of optoelectronic displays. Furthermore, PBA-MeO is used as an additive to feed Bombyx mori silkworms and shows low toxicity over inorganic materials. Our findings may pave a new way for the development of RTP phosphors and promote their use in practical applications.
1. Introduction
Recently, organic room-temperature phosphorescence (RTP) materials have attracted great attention for their potential applications in optoelectronic devices as well as chemical and biological detection.1–3 So far, only a few reports describe single-component small molecules with RTP properties (>100 ms) under ambient conditions. Mainly, there are three key prerequisites to achieve long-lived RTP: (a) functional groups capable of enhancing the singlet-to-triplet transitions; (b) a rigid conformation in the solid state to decrease the rapid rate of nonradiative decay; (c) relatively large conjugation to stabilize the triplet states.4 As shown in Fig. 1A and S1,† the functional groups of most organic phosphors are confined to carbonyls, sulfones and the heavy atoms of halogens, to promote spin-orbital coupling for the efficient generation of triplet excitons.5–7 On the one hand, tremendous efforts have been made to minimize nonradiative deactivation processes of triplet excitons and avoid collisions with quenching species of oxygen and moisture, including the use of host–guest doping, crystallization and metal–organic frameworks.4 However the systematic study and practical applications of versatile, abundant and efficient RTP compounds are still very rare.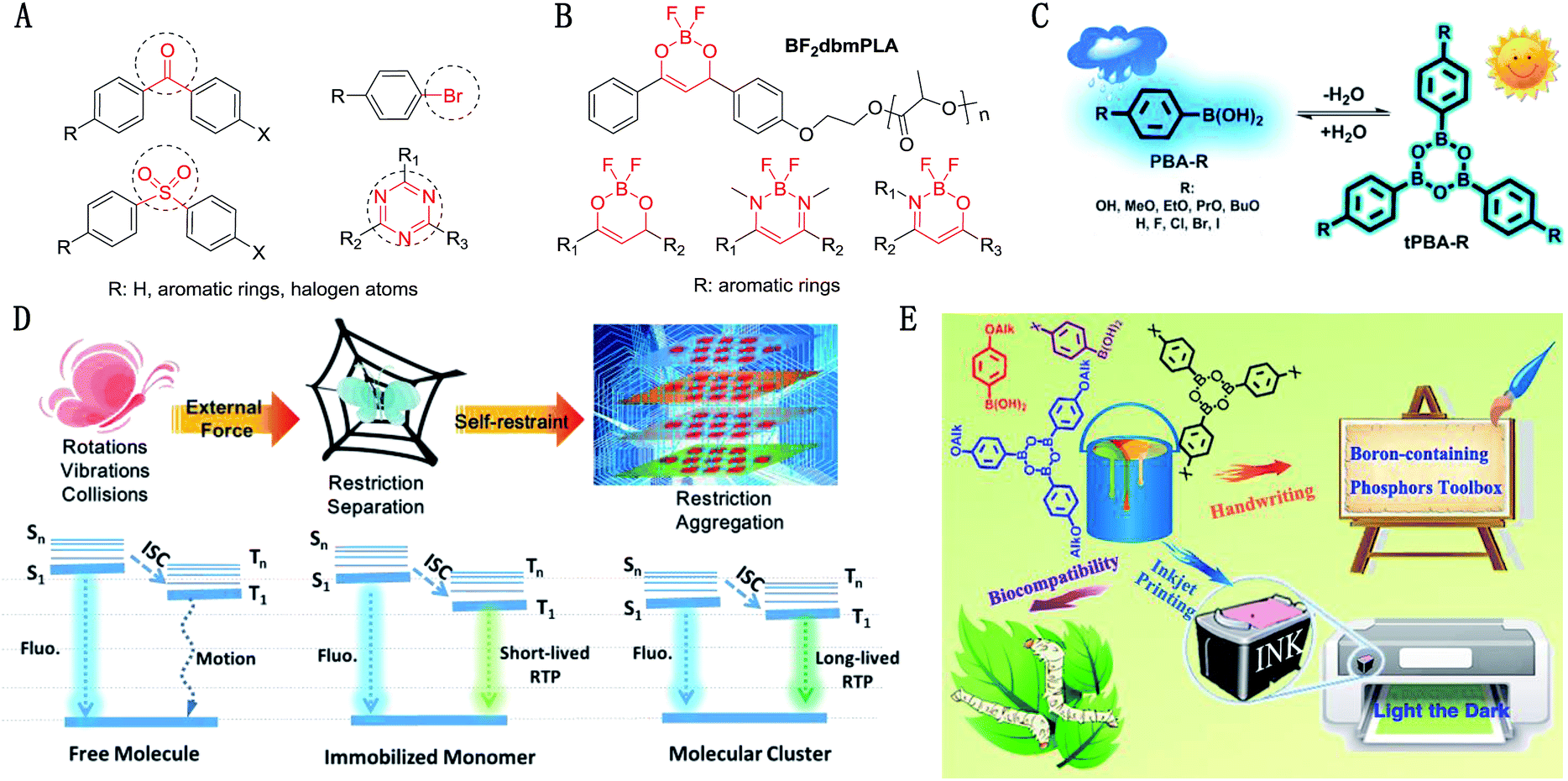 | ||
| Fig. 1 (A) Typical functional groups of organic phosphors; (B) examples and fundamental structures of boron-containing materials; (C) molecular structures and their interconversions in the main research. Detailed structures of (A–C) are shown in Fig S1–S3.† (D) An illustration of the achievement of RTP from free molecules in solution, immobilized molecules embedded in a polymer host and a molecular cluster, and their corresponding energy levels; (E) a summary of the potential practical applications of our work, OAlk: OH, EtO, PrO, BuO; X: H, F, Cl, Br, I. | ||
Among solid state luminescent materials, boron-containing materials have attracted great attention for their intense fluorescence and unusual RTP.3,8–12 The RTP of these tetracoordinate compounds (Fig. 1B) is sensitive to the ambient environment due to intramolecular motions and collisions with quenching species. As a result, the external force provided by covalently bonded polymers or extra polymer matrices is needed to provide restricted and separated conditions, for the realization of RTP. According to the literature and our recent work,13–15 the intermolecular interactions in self-restricted aggregates are key for the realization of RTP properties of single-component small molecules. Besides, the rigidification of molecules and intramolecular interactions of aggregates may also decrease the energy gap (ΔEST) and enhance the intersystem crossing (ISC) process.7 Thus, it is anticipated that denser molecular packing, induced by specific functional groups, might be sufficient for boron-containing materials to emit RTP, instead of requiring the assistance of polymers, accompanied by the much reduced complexity of the RTP system. From the basic textbook Organic Chemistry, hydrogen bonds can result in strong intermolecular interactions, sometimes even causing the formation of association complexes. Accordingly, it is possible that the introduction of hydrogen bonds into boron-containing compounds may offer additional denser molecular packing through strong intermolecular interactions,16–18 this would provide the required conditions for the realization of RTP, partially by suppressing the vibration in crystals (crystallization-induced phosphorescence),15 and prolonging the triplet lifetime.
In this regard, we tested the RTP properties of commercially available phenylboronic acid and its derivatives (PBA-R) to validate our idea (Fig. 1C). Amazingly, most of them showed long-lived RTP in the solid state, with a lifetime longer than 100 ms without any pre-treatment, which constitutes a new class of single-component small molecules bearing RTP properties. Interestingly, two related works were reported during the preparation of this manuscript. However, their interpretations focused more heavily on single molecule phosphorescence, while their experiments were mainly conducted with crystals or powders in the aggregate state (Fig. S3†). Possibly due to the lack of X-ray single crystal diffraction data, the considerably different RTP behaviors of two similar molecules could not be explained well. Additionally it is a pity that the descriptions of triphenyl boroxine (tPBA) were not right. In this contribution, we carefully investigated the luminescence behavior in different media, mainly based on the representative 4-methoxyphenylboronic acid (PBA-MeO). We established the relationship between the phosphorescence behavior and packing modes in the solid state, based on a series of phenylboronic acids with different substituents, and demonstrated some potential applications of this kind of RTP material, including investigating its use in inkjet printing technology and its biotoxicity in silkworms (Fig. 1E).
2. Results and discussion
2.1 Emission properties of alkoxy-substituted phenylboronic acids
Out of five PBA-OAlks compounds, PBA-MeO exhibited the longest RTP lifetime of 2.24 s in the crystalline-state, which was also the longest lifetime ever reported for single-component small molecules. So, PBA-MeO was taken as a representative compound for discussion. It displayed very bright luminescence in the solid state upon ultraviolet irradiation at 254 nm (Fig. 2A). The emission peak located at a short wavelength (λmax = 302 nm, Fig. 2B) was attributed to the localized excited state of phenylboronic acid, which was further verified by the measurement of its weak fluorescence in solution (λmax = 299 nm, Fig. S4†). However, the emission band from 310 to 430 nm was more likely derived from the enlarged π–π stacking. Transient PL decay was subsequently carried out at room temperature (300 K) to investigate the nature of the excited-state. The lifetimes of these emission bands were in the magnitude of nanoseconds (<10 ns), confirming that the emission bands corresponded to fluorescence (Fig. S5†). After removal of the ultraviolet source, the emission changed from deep sky blue to cyan and slowly faded. From the phosphorescence spectrum, two resolved emission peaks located at 457 and 488 nm were observed, these showed single exponential decays with remarkably long lifetimes of 2.24 s and 2.19 s, respectively. In addition, the intensity and peak profiles displayed little change in argon or ambient atmosphere conditions (Fig. 2D), illustrating the fact that the triplet states were insensitive to oxygen. From the X-ray single-crystal diffraction data discussed later, the molecules were well immobilized through the H-bonds formed by the boric acid and methoxyl group. So, the rigid crystal matrix may partly inhibit the movement of molecules and provide rigorous protection from the quenching of triplet oxygen. Compared to the examples of the above tetracoordinate compounds, the achievement of RTP under ambient conditions could surely promote the wide practical applications of PBA-MeO. In solution, due to the increased vibrational freedom, PBA-MeO did not demonstrate phosphorescence in the presence or absence of O2 at room temperature. However, at 77 K, frozen dilute MeOH solutions of PBA-MeO showed phosphorescence, accompanied by distinct blue shifts and longer lifetimes upon the increase of concentration (Fig. 2F, S6 and S7†). It was inferred that once molecules got closer to some extent, the orbitals of adjacent molecules overlapped and redistributed, possibly resulting in the activation of triplet states in higher energy levels and the stabilization of excitons. Specifically, the phosphorescence spectrum of PBA-MeO in the solid state at 77 K was similar to that measured in frozen dilute MeOH solution (10−5 mol L−1), probably indicating that the micro-environments of the two states for the domination of the strong phosphorescence at low temperatures should be almost the same. Also, it is possible that the phosphorescence properties converged towards single-molecule behavior, as reconfirmed by their almost identical lifetimes (1.50 s in solution and 1.51 s in crystals).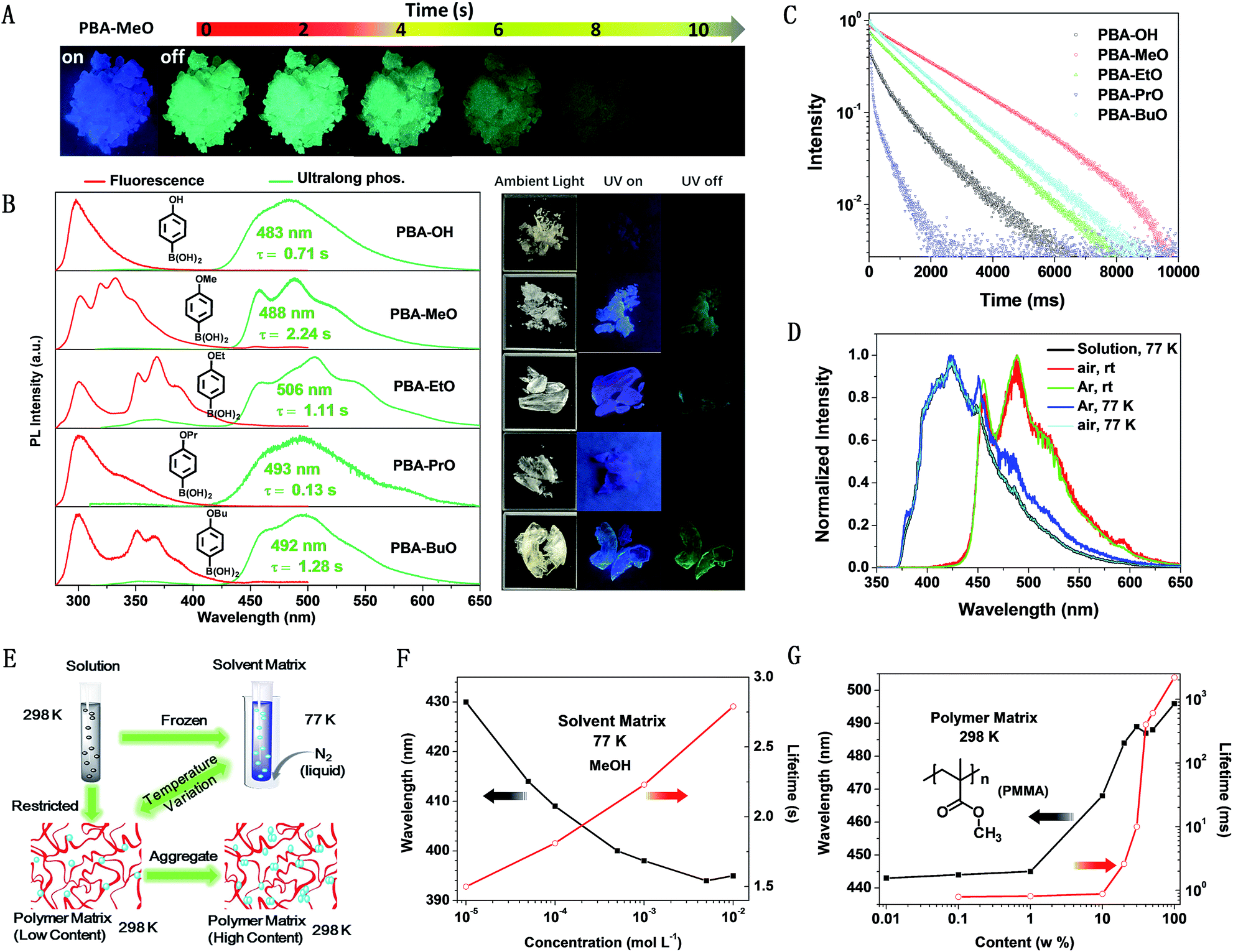 | ||
| Fig. 2 (A) Photographs of PBA-MeO (crystals) taken before and after irradiation (254 nm) under ambient conditions; (B) fluorescence and phosphorescence spectra of the PBA-OAlk crystals. The room temperature fluorescence spectra were recorded at 254 nm, while the phosphorescence spectra were recorded at their optimum excitation wavelengths (290–295 nm). The right insets show the corresponding photographs taken under ambient light (left), 254 nm UV light (middle) and just after stopping UV light irradiation (right); (C) room temperature phosphorescence decay profiles of PBA-OAlks excited at their optimum excitation wavelengths and detected at their maximum emission wavelengths; (D) phosphorescence spectra of model molecule (PBA-MeO) under different conditions. The concentration of methanol solution is 1 × 10−5 mol L−1 and crystalline-state samples were used for the other tests; (E) design idea for the investigation of the RTP properties from single molecule to aggregated states. (F) Maximum phosphorescence wavelengths and lifetimes of PBA-MeO in MeOH under different concentrations (λex = 280 nm, 77 K, in air). (G) Maximum phosphorescence wavelengths and lifetimes of PBA-MeO embedded in PMMA in different amounts (λex = 280 nm, room temperature, in air). When the concentration is higher than 20%, the content of PBA-MeO is too high to form uniform films, resulting in irregular changes. The complete data set and details of (F) and (G) are available in the ESI.† | ||
This phenomenon inspired us to explore the phosphorescence of single molecules at room temperature by embedding PBA-MeO into polymer films, with PMMA as the host.19,20 Under this circumstance, the molecules were also well restricted as in solution at low temperatures or in crystals (Fig. 2E).21 To our surprise, the phosphorescence was detected at a low content (0.01 wt%), which is very rare for organic compounds as guests without carbonyl groups or heavy halogen atoms.22 With the increase of PBA-MeO content in PMMA, new fluorescence bands at longer wavelengths were observed, revealing that the molecules aggregated with enlarged orbitals through effective intermolecular interactions. The red-shift of the maximum phosphorescence wavelengths and the longer lifetimes of the aggregates were close to that of the crystalline state, suggesting that the RTP spectra of the crystals were determined by the aggregation conformations, instead of the single molecules. The lifetime changing tendency in both polymer and frozen solvent matrices demonstrated that the triplet state greatly benefited from the aggregated states with longer lifetimes.
From the above discussion of PBA-MeO, the glassy solvent and polymer apparently influenced the interaction and alignment of molecules. However the details were hard to analyze using experimental methods. The packing modes may also be affected by the hydrogen or alkyl chains of phenylboronic acid, resulting in different energy levels and radiative/nonradiative pathways. To have a deep understanding of this phenomenon, five alkoxy-substituted phenylboronic acids (PBA-OAlks), including PBA-MeO, were carefully investigated. Upon excitation at 254 nm, the five compounds exhibited two different luminescence behaviors in the solid state: the crystalline samples of PBA-MeO, PBA-EtO and PBA-BuO displayed very bright luminescence, while luminescence from the other two samples (PBA-OH and PBA-PrO) was very weak. From Fig. 2B, all of them showed a typical emission peak (ca. 300 nm) of phenylboronic acid (PBA). Except PBA-OH, the other four compounds also exhibited new peaks at wavelengths longer than 320 nm, in the sequence of PBA-PrO < PBA-MeO < PBA-EtO ≈ PBA-BuO. Similar cases of RTP were observed after the UV lamp was turned off. PBA-MeO, PBA-EtO and PBA-BuO showed persistent RTP with remarkably long luminescence lifetimes (2.24 s, 1.11 s and 1.28 s, respectively). The RTP of PBA-OH was dimly lit and quickly disappeared (τ = 0.713 s), while that of PBA-PrO was hardly seen by the naked eye (τ = 0.129 s).
2.2 The influence of molecular arrangement on emission properties
Considering the similar electronic structures of the above five compounds, their different luminescence behaviors could be ascribed to their different packing modes. X-ray single-crystal diffraction analysis was performed to determine their spatial configurations. In view of the overall stacking mode, the unit cell of PBA-PrO displayed a much more complicated conformation compared to the other compounds (Fig. S10†), which will be discussed in later parts. The detailed interactions and packing modes viewed from different orientations were extracted from crystals (Fig. 3). Numerous hydrogen bonds were found in the crystals with different types and bond-lengths (Fig. S13†). The boric acid (weak electron acceptor, A) showed a preferred conformation of the syn–anti type (BO–H⋯OB, 1.9–2.2 Å) as anticipated, forming different polygons, similar to those of –COOH and –CONH2.14,23,24 Obviously, the shape of the polygon was also affected by the phenolic hydroxyl and alkoxyl groups (weak electron donor, D). The D parts of PBA-OH and PBA-MeO demonstrated suitable spatial positions and formed hydrogen bonds through the interactions of CO–H⋯OC (1.993, 2.009 Å), CO⋯H–C (2.793, 2.804 Å) and OC–H⋯OC (2.718 Å), leading to nearly two-dimensional planes. Once the alkyl chains were long enough for PBA-EtO and PBA-BuO, the planes were clipped into nanoribbons by the alkyl chains, this was further confirmed by the analysis of PBA-HexO (Fig. S11†). However, if the alkyl chains next to the chromophore were not fixed, it also led to the quenching of the triplet states and thus short-lived lifetimes. These layers (Plane 1) were connected to form vertical surfaces (Plane 2) through C–H⋯OB and C–H⋯OC interactions, in which the hydrogen bond lengths were longer than those in Plane 1. Thus, the intramolecular motions were restricted to a large extent, which minimized the nonradiative loss of excitons and boosted the phosphorescence emission. The hydrogen bonds in Plane 1 contributed little to the π–π stacking and energy of the excited states for the ineffective overlap of molecular orbitals, as shown in the latter parts, which could not fully explain the difference in their spectra and lifetimes. So, the typical arrangement of molecules in Plane 2 (Fig. S12†), perpendicular to Plane 1, was analyzed. The stacking mode for PBA-MeO, PBA-EtO and PBA-BuO was constructed from two parallel neighboring molecules with opposite D–A directions, bringing about the formation of dimers with twist angles of 70.6°, 70.34° and 0° between adjacent molecules. The molecules in the dimer of PBA-OH maintained a shorter distance of 4.76 Å but an angle of 68.58°, which surely influenced the intermolecular π–π stacking. Together with the distance and angle of the dimer, the strength of the π–π stacking interactions of these compounds were in the order of PBA-OH < PBA-MeO < PBA-EtO ≈ PBA-BuO, which was consistent with the fluorescence spectra. For single crystals of PBA-PrO viewed along the c axis (sketch of H-bonds), the herringbone-type structure was arranged in the shape of a rhombus, and no flat layers could be found. When the colored part was enlarged and viewed along the b axis, the two parallel molecules (Plane 1) on one line were isolated by the surrounding molecules with large twist angles (>70°). When one of the rhombus layers was extracted, there were two parallel dimers, which consisted of two molecules with the same D–A direction and small dihedral angle (0.4°). While the dimers in two neighboring rhombuses showed a large twist angle of about 75°, bad π–π stacking resulted, which led to a very weak luminescence. Combined with the crystal densities of the five compounds, this arrangement was also harmful to dense packing and resulted in an increase in the molecular vibrational degrees of freedom and the quenching of triplet states. Thus, both the well restricted conformation and long-range π–π stacking interactions contributed to the RTP lifetime and intensity. | ||
| Fig. 3 Packing modes of alkoxy-substituted phenylboronics. Blue dotted lines represent H-bonds and dn is the density of the crystals. | ||
2.3 Theoretical calculations
On the basis of the preceding measurements and analysis, both single molecules and molecular clusters (including dimers and larger long-range π–π stacking interaction systems) of the five compounds could emit RTP, once the molecular conformations were locked and rigidified. To understand the possible mechanism, theoretical calculations were conducted using TD-DFT to investigate the single molecule and aggregated structures (simplified to dimers) in both the singlet and triplet excited states. Taking PBA-MeO for example, monomers and dimers with minimum (P1) and maximum (V1) π–π stacking interactions were chosen as typical conformations to illustrate the possible ISC processes and the importance of effective π–π stacking, as shown in Fig. 4 and Table 1 (other conformations of dimers and the calculated results are presented in Fig. S15 and Table S3†). According to the Franck–Condon principle, the small energy gap (ΔEST) and spin–orbit coupling between the singlet (Sn) and triplet (Tn) states were the two main factors that determine the rate of the ISC process.14 There were four Tn states for M and P1, and two for V1, which all existed below the S1 state to thermodynamically permit the ISC process and had the same transition configurations, enhancing the probability of spin–orbit coupling. However, only the V1 conformation contained two energy transition channels with a ΔEST smaller than or close to 0.3 eV (ref. 34) (S1 → T4, ΔEST = 0.223 eV and S1 → T2, ΔEST = 0.355 eV), more than one energy transition channel for M (S1 → T4, ΔEST = 0.144 eV) and P1 (S1 → T4, ΔEST = 0.147 eV). These results suggested that effective π–π stacking could decrease the singlet excited state energy and ΔEST, thereby enhancing the probability of spin–orbit coupling and thus the ISC process.25 The molecular orbitals also agreed well with the above discussion that the orbitals of two molecules could overlap only in the dimers (V1 and V2) with effective π–π stacking (Fig. S14†). The effective overlap of orbitals would increase the electronic delocalization and stabilize the triplet excitons, leading to a longer lifetime of RTP. Thus, when the content of PBA-MeO in PMMA increased, the molecules were gradually coming together to form expanded π–π stacking, resulting in longer lifetimes of RTP. These theoretical calculations further explain the importance of the π–π stacking interactions and different luminescence behaviors of PBA-OAlks. The stronger π–π stacking interactions brought about red-shifted fluorescence and longer RTP lifetimes of PBA-MeO, PBA-EtO and PBA-BuO, compared to those of PBA-OH and PBA-PrO (Table 2).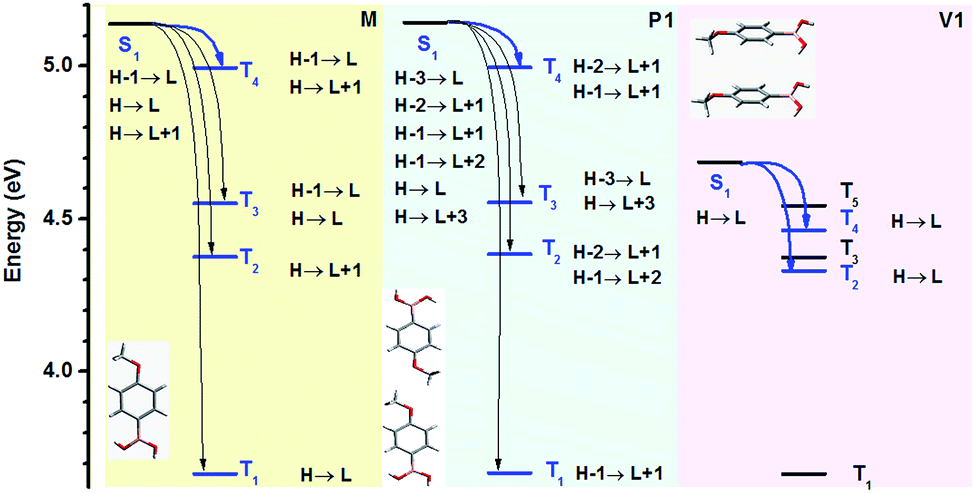 | ||
| Fig. 4 Schematic diagrams showing the TD-DFT-calculated energy levels of the singlet (S1) and triplet (Tn) states (PBA-MeO). Note that H and L refer to the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), respectively, and only the Tn states lower than S1 are shown. The conformations were derived from single-crystal diffraction data. The blue arrows represent the probable ISC processes occurring from the S1 state to the Tn state, while the black arrows refer to transitions to the other triplet states available, but with a larger ΔEST (>0.4 eV). | ||
| Compound | λ ex (nm) | λ maxem (nm) | 〈τ〉 (s) |
|---|---|---|---|
| a Measurements were conducted with crystalline-state samples at room temperature. nd: very weak and not determined. | |||
| PBA-OH | 291 | 491 | 0.71 |
| PBA-MeO | 295 | 483 | 2.24 |
| PBA-EtO | 290 | 488 | 1.11 |
| PBA-PrO | 290 | 506 | 0.13 |
| PBA-BuO | 291 | 492 | 1.28 |
| PBA | 287 | 493 | 0.89 |
| PBA-F | 280 | 492 | 1.34 |
| PBA-Cl | 285 | 593 | 0.37 |
| PBA-Br | 300 | 546 | 0.073 |
| PBA-I | 327 | 540 | nd |
| tPBA-OH | 280 | 486 | 0.58 |
| tPBA-MeO | 301 | 503 | 0.71 |
| tPBA-EtO | 282 | 500 | nd |
| tPBA-PrO | 302 | 492 | nd |
| tPBA-BuO | 306 | 444 | nd |
| tPBA | 280 | 494 | 0.16 |
| tPBA-F | 280 | 500 | 1.96 |
| tPBA-Cl | 282 | 512 | 0.25 |
| tPBA-Br | 302 | 480 | 0.17 |
| tPBA-I | 324 | 520 | nd |
| PyBA | 370 | 502 | 0.28 |
2.4 Empirical verifications and compound survey
Subsequently, the basic structure of phenyl boric acid (PBA) was investigated. As expected, the freshly prepared sample displayed RTP with a lifetime of 0.89 ms and a similar luminescence spectrum to that of PBA-OH (Fig. S16†). However, phenyl boric acid could easily be converted into triphenyl boroxine (tPBA) via the heating process reported in the previous literature.26 To clarify the RTP properties of the two kinds compounds, a traditional method of oil–water separation was conducted to get pure tPBA.27 RTP could not be seen by the naked eye in either the powder or the crystals, due to its weak intensity (the lifetime of the crystalline sample was 157 ms, much shorter-lived than that of a previous report with a lifetime of 460 ms)28 (Fig. S17†). This may be a good illustration for understanding the effect of H-bonds on the RTP properties. Fortunately, the single crystals of both PBA and tPBA were obtained, which provided detailed information about the molecular arrangement in the solid state (Fig. S28†). As shown in Fig. S19,†the molecules of PBA were well restricted by H-bonds and appeared in the form of dimers. However, the vertical structure of every two parallel dimers led to finite π–π stacking, similar to that of PBA-OH and PBA-PrO, resulting in dim RTP compared to PBA-MeO. As for tPBA, the reduced H-bonds were only present in the molecules (Fig. S22†), which increased the molecular vibrational degrees of freedom and caused the quenching of triplet states. Meanwhile, the longer distance (parallel to molecular plane, 3.39 Å) and larger plane angle (perpendicular to molecular plane, 79.1°) between the neighboring layers were undesirable for the effective overlap of molecular orbitals to stabilize the triplet excitons, bringing about the weaker intensities and shorter lifetime of RTP (Fig. S23†). If this was the key point, the corresponding boroxine derivatives of PBA-OH and PBA-MeO should have good opportunities to emit RTP, because of the H-bonds formed by their D parts (–OH and –OMe). In reality, both tPBA-OH and tPBA-MeO displayed RTP with lifetimes of 583 and 710 ms, respectively. Additionally, there were H-bonds and short interlayer spaces present in the single crystal of tPBA-MeO as expected (Fig. S24†). As for the other three boroxine derivatives of PBA-OAlks, only weak RTP was detected (Fig. S18†).Adhering to the above basic principles, halogenated phenyl boric acids (PBA-X, X: F, Cl, Br, I) and the corresponding boroxine derivatives (tPBA-X) were further investigated (Fig. S16 and S17†). Due to the presence of H-bonds through the C–H⋯F and C–H⋯Cl interactions, PBA-F/Cl and tPBA-F/Cl exhibited RTP with different lifetimes. Unexpectedly, PBA-Br and PBA-I only showed weak and short-lived RTP. Generally, the heavy-halogens can promote the spin–orbit coupling between the massive nucleus and excitons to enhance the singlet-triplet conversion, as reported in the previous literature.29 In comparison with PBA, the average number of H-bonds per molecule decreased, and Br and I were fixed by weaker halogen bonds,30 which would increase the nonradiative relaxation of triplet excitons (Fig. S20 and S21†). Meanwhile, the large distance of the dimers and the absence of long-range effective π–π stacking could not stabilize the excited states for bright luminescence. After dehydration, tPBA-Br miraculously brightened, with a very bright yellow phosphorescence. The lifetime also extended from 73 to 170 ms. As shown in Fig. S26,† the phenyl group of tPBA-Br was restricted by intramolecular H-bonds, as other boroxines were. Also, stronger C–H⋯π and C–Br⋯π networks of tPBA-Br were found due to the reduced distance between the two layers (2.351 Å) compared to PBA-Br. This stacking mode also facilitated the π–π stacking interactions, which, along with the rigid conformation, increased the lifetime and intensity of phosphorescence. It is reasonable to ascribe the weak luminescence and short-lived RTP lifetime of tPBA-I to its loose stacking mode, possibly due to the larger size of I and the acceleration of both the singlet-to-triplet and triplet-to-singlet ISC processes induced by the heavy atom.22
The above discussion strengthened the concept of “rigid conformation and effective π–π stacking”, which will be useful to search for new phosphors with the prospect of building a “Boron-containing Phosphors Toolbox”. Besides the interaction of O⋯H, F⋯H and Cl⋯H, N⋯H is another type of H-bond, which has been widely used in molecular recognition and supramolecular synthesis.31,32 As expected, a commercially available product of pyridin-4-ylboronic acid (PyBA) showed long-lived RTP with a lifetime of 280 ms. Indeed, many aryl boronic acids or esters exhibited RTP once the molecules were immobilized and possessed effective π–π stacking (Fig. S29–S40†).
2.5 Practical applications
During our experiment, we accidentally discovered that the RTP intensities of the crystals and freshly prepared samples made via rotary evaporation were almost the same, which indicated that the latter probably also formed little crystals due to strong intermolecular interactions mainly derived from H-bonds. To illustrate this phenomenon, we tried to observe the process of fast crystallization using an inverted optical microscope (Olympus IX71). As shown in Fig. 5A and a video (ESI†), after a small drop of PBA-MeO in solution (ca. 1.5 mol L−1) was dripped on the glass slide, massive grains came into being and formed hexagonal crystals like those grown from slow evaporation (Fig. 5B). The newly-formed crystal thin films with different thicknesses, probably as thin as the wavelength of visible light, resulted in interference of light, which created the iridescence of the crystals. With volatilization of solvent, the size and number of crystals increased in a very short time and the crystals emitted persistent RTP. The further XRD experiment confirmed that the sample was in the crystalline state, as it showed sharp peaks in the spectrum (not shown). The above results revealed the easy crystallization and strong interaction of boric acids. Actually, the luminescence of PBA-MeO was resistant to crushing, and showed little difference before and after grinding. In the XRD spectra (Fig. 5D), a peak located at 23.63° was observed in both the powder crystals and ground sample, indicating that some special layers were strongly linked via noncovalent bonds and could not be destroyed even by hard mechanical grinding. Combined with the previous X-ray single-crystal diffraction data, the sharp peak was more likely ascribed to the lattice planes in the layers built by H-bonds, which fulfilled the Bragg equation. | ||
| Fig. 5 (A) The fast crystallization process of the model molecule (PBA-MeO) under ambient conditions over time. To shorten the crystallization time, concentrated solutions in highly volatile acetone were used (a larger image and fluorescence photo are shown in Fig. S39 and S40,† the video is in the ESI.† The photos and video were taken in the bright field by a 10× lens, Olympus IX71); (B) photographs of crystals grown by the slow evaporation of the acetone solution (PBA-MeO) under ambient light (top) and UV light (bottom); (C) fluorescence and phosphorescence spectra of the PBA-MeO powder crystals and ground sample; (D) XRD patterns of the two states, insets showing photographs of PBA-MeO crystals (top) and ground sample (bottom) taken under 254 nm UV light (left) and just after stopping UV light (right); (E) the stacking mode corresponding to the sharp diffraction peak located at 23.63° as assumed, the dashed blue lines representing H-bonds. (F) Demonstration of boron-containing phosphor-encoding for security documents with (t)PBA-MeO, (t)PBA-Cl and (t)PBA-Br before and after heating; (G) rooster pattern printed with PBA-MeO for 3 cycles; (H) panda pattern printed with PBA-MeO for 8 cycles. | ||
This characteristic was quite useful for practical application and no extra procedures or treatments were needed to carefully capture the RTP-active crystals.5,33–35 Actually, many boric acids were obtained by recrystallization from water and emitted highly efficient RTP, confirming that this kind of compound was more likely to form dense aggregates without the disturbance of residual moisture (note that PBA-F could be dissolved in water). Thus, no special carriers or conditions were required for the preparation of some smart materials. For example, the various and long-lived phosphors would be quite appropriate in anti-forgery documents. At first, the patterns were written by PBA-AlkOs and PBA-Xs in acetone solutions (ca. 1.0 mol L−1) on printing paper to check their luminescence behavior. As shown in Fig. S43,† the luminescence of these compounds displayed little difference under 254 nm light at lower loading amounts, but the same RTP characteristics were observed, corresponding to their powder crystals. Combined with the aforementioned boroxines, the photoluminescent behavior further changed and exhibited different trends during the process of dehydration. Thus, different messages could be conveyed by changing the samples and treating methods. As shown in Fig. 5F, the letters in the word “CHEMISTRY” did not show distinct differences of luminescence upon excitation at 254 nm; but bright RTP of “CHEM” and faint “IS” were observed after turning off the UV lamb; however, the “CHEM” disappeared, “IS” became brighter and “TRY” emerged after heating the paper at 80 °C for 2 h.
The boron-containing materials, previously reported in the literature, required careful adjustment of the molecular weight or the ratio of polymers, and sometimes inert gas, to find the best system for the realization of RTP, restricting their practical applications to some degree. However, boric acids could be easily prepared, and many of them have been obtained at a low price (e.g., PBA-MeO, 100 g/600¥). Furthermore, boron-containing materials represent a class of compounds with potential RTP properties, which may confer advantages in optoelectronic applications and biological imaging which demand long-lived phosphorescence. Thus, there is an urgent need to establish an easier way to prepare materials using a fast, automated and widely used technique. As an easy approach, with low cost and technical convenience, inkjet printing technology was considered. This printing method adopts the piezo-electric ‘drop-on-demand’ deposition of nominal 10 picoliter sized droplets, which not only allows the production of uniform patterns, but also makes it possible to adjust the loading amount for detection. After 3 repeated printing cycles, the patterns could be easily recognized after the UV-lamp was turned off (Fig. 5G). The brightness could be easily enhanced by increasing the printing times as shown in Fig. 5H, demonstrating precise control and good reproducibility of the printing process. Subsequent folding or extrusion of the paper did not influence the RTP properties, showing its potential for practical use.
To test the biotoxicity of PBA-MeO, we investigated its effect on silkworm growth. The silkworms were fed on mulberry leaves spread with PBA-MeO solution or an inorganic suspension (SrAl2O4:Eu2+, Dy3+). After about one and a half months, all the silkworms secreted silk to form cocoons, which showed no remarkable difference in color or luminescence under UV light. But the cocoon weights of silkworms fed with PBA-MeO were closer to the normal level, while those fed with the inorganic suspension were not (the details are presented in ESI†), indicating its low toxicity and potential biological applications.
3. Conclusions
In summary, we presented a “Boron-containing Phosphors Toolbox”, which exhibited long-lived room-temperature phosphorescence in the crystalline state. PBA-MeO, with a simple structure, emitted very bright RTP with a lifetime of 2.24 s, the longest lifetime of RTP for a single-component organic phosphor reported so far. Careful investigations on the relationship between the packing mode and RTP properties demonstrated that both the rigid conformation which decreases the rapid rate of nonradiative decay, and the effective π–π stacking which stabilizes the triplet states, are of great importance to achieve bright and persistent RTP for small molecular systems. The commercial availability, suitability for use in inkjet printing technology, low biological toxicity and convenient handling give these materials huge potential for applications in different fields of organic optoelectronics, bio-imaging and anti-forgery materials. Deep understanding of their abnormal RTP would shed new light, further developing this area.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21325416 and 51573140) and the Open Fund of the State Key Laboratory of Luminescent Materials and Device in South China University of Technology (no. 2017-skllmd-04).References
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- Y. Shao and Y. Yang, Adv. Mater., 2005, 17, 2841–2844 CrossRef CAS.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- M. Baroncini, G. Bergamini and P. Ceroni, Chem. Commun., 2017, 53, 2081–2093 RSC.
- C.-R. Wang, Y.-Y. Gong, W.-Z. Yuan and Y.-M. Zhang, Chin. Chem. Lett., 2016, 27, 1184–1192 CrossRef CAS.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS PubMed.
- P. Lehner, C. Staudinger, S. M. Borisov and I. Klimant, Nat. Commun., 2014, 5, 4460 CAS.
- C. A. DeRosa, S. A. Seaman, A. S. Mathew, C. M. Gorick, Z. Fan, J. N. Demas, S. M. Peirce and C. L. Fraser, ACS Sens., 2016, 1, 1366–1373 CrossRef CAS PubMed.
- M. Koch, K. Perumal, O. Blacque, J. A. Garg, R. Saiganesh, S. Kabilan, K. K. Balasubramanian and K. Venkatesan, Angew. Chem., Int. Ed., 2014, 53, 6378–6382 CrossRef CAS PubMed.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS PubMed.
- Y. Xie, Y. Ge, Q. Peng, C. Li, Q. Li and Z. Li, Adv. Mater., 2017, 29, 1606829 CrossRef PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CAS.
- L. Wang, J. Zhang, B. Kim, J. Peng, S. N. Berry, Y. Ni, D. Su, J. Lee, L. Yuan and Y. T. Chang, J. Am. Chem. Soc., 2016, 138, 10394–10397 CrossRef CAS PubMed.
- Y. Gong, L. Zhao, Q. Peng, D. Fan, W. Z. Yuan, Y. Zhang and B. Z. Tang, Chem. Sci., 2015, 6, 4438–4444 RSC.
- S. Kuno, H. Akeno, H. Ohtani and H. Yuasa, Phys. Chem. Chem. Phys., 2015, 17, 15989–15995 RSC.
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Nat. Chem., 2015, 6, 8947 Search PubMed.
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS PubMed.
- J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589–15593 CrossRef CAS PubMed.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- V. R. Pedireddi and N. SeethaLekshmi, Tetrahedron Lett., 2004, 45, 1903–1906 CrossRef CAS.
- G. Zhang, F. Rominger and M. Mastalerz, Cryst. Growth Des., 2016, 16, 5542–5548 CAS.
- D. Beljonne, Z. Shuai, G. Pourtois and J. L. Bredas, J. Phys. Chem. A, 2001, 105, 3899–3907 CrossRef CAS.
- M. K. Smith and B. H. Northrop, Chem. Mater., 2014, 26, 3781–3795 CrossRef CAS.
- Q. Xiao, L. Tian, R. Tan, Y. Xia, D. Qiu, Y. Zhang and J. Wang, Org. Lett., 2012, 14, 4230–4233 CrossRef CAS PubMed.
- Y. Shoji, Y. Ikabata, Q. Wang, D. Nemoto, A. Sakamoto, N. Tanaka, J. Seino, H. Nakai and T. Fukushima, J. Am. Chem. Soc., 2017, 139, 2728–2733 CrossRef CAS PubMed.
- J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2016, 55, 15589–15593 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- J. H. Fournier, T. Maris, J. D. Wuest, W. Guo and E. Galoppini, J. Am. Chem. Soc., 2003, 125, 1002–1006 CrossRef CAS PubMed.
- I. Georgiou, S. Kervyn, A. Rossignon, F. De Leo, J. Wouters, G. Bruylants and D. Bonifazi, J. Am. Chem. Soc., 2017, 139, 2710–2727 CrossRef CAS PubMed.
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195–6201 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y. C. Wu, P. Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed.
- B. J. Xu, H. Z. Wu, J. R. Chen, Z. Yang, Z. Y. Yang, Y. C. Wu, Y. Zhang, C. J. Jin, P. Y. Lu, Z. G. Chi, S. W. Liu, J. R. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909–1914 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1547113, 1547361, 1547292, 1547293–1547295, 1547338, 1547298, 1519392 and 1547297. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04098a |
| This journal is © The Royal Society of Chemistry 2017 |