
n/n junctioned g-C3N4 for enhanced photocatalytic H2 generation†
Minjie
Zhou
*ab,
Zhaohui
Hou
b,
Li
Zhang
b,
Yan
Liu
ac,
Qiongzhi
Gao
ad and
Xiaobo
Chen
*a
aDepartment of Chemistry, University of Missouri-Kansas City (UMKC), Kansas City, MO 64110, USA. E-mail: chenxiaobo@umkc.edu
bSchool of Chemistry and Chemical Engineering, Hunan Institute of Science and Technology, Yueyang 414000, China. E-mail: zmj0104@163.com
cCollege of Environment, Sichuan Agricultural University, Chengdu, Sichuan 611130, China
dInstitute of Biomaterial, College of Materials and Energy, South China Agricultural University, Guangzhou, 510642, China
First published on 9th January 2017
Abstract
Graphitic carbon nitride (g-C3N4) has been deemed as a promising metal-free catalyst for solar energy utilization toward water splitting. In the present work, homogeneous n/n junctioned g-C3N4/g-C3N4 is prepared by in situ annealing of two different precursors and its photocatalytic activity is studied for hydrogen generation. It exhibits a superior photocatalytic performance over its individual pure g-C3N4 moieties. The enhanced photocatalytic activity is explained by the synergistic effects between the two g-C3N4 in the homogeneous nanostructures, where a better charge separation is achieved in the homogeneous g-C3N4/g-C3N4 with many n/n junctions. Previously, synergistic effects were reported in the heterostructures of different materials or different phases. This study thus broadens the meaning of synergistic effects and demonstrates that synergistic effects can be obtained using the same materials of the same phase but made by different methods. Therefore, this finding is of general interest to the materials, chemistry, and renewable energy communities and will trigger a wide attention in the related research fields.
Introduction
Photocatalytic hydrogen production using solar energy has been regarded as one promising solution to the global energy and environmental crisis.1,2 Since the discovery of photoinduced decomposition of water on TiO2 electrodes in 1972,3 various semiconductor photocatalysts, such as TiO2,4,5 ZnO6 and CdS,7–9 have been developed for H2 production from water splitting. In the photocatalytic hydrogen generation process, photo-excited electrons and holes are first generated after the semiconductor absorbs light, and they separate and migrate to the surface of the semiconductor to photoreduce water into hydrogen.10,11 The overall photocatalytic efficiency is proportional to the efficiency of each step in the photocatalytic process. Specifically, the larger the amount of sunlight absorbed by the photocatalyst, the higher the photocatalytic efficiency expected, as more photoexcited electrons and holes will be produced. The more the electrons and holes reaching the surface of the photocatalyst, the higher the efficiency expected as well, as more charges can participate in the photooxidation or photoreduction reactions. The number of photoexcited electrons and holes on the surface is equal to the number produced by the light absorption minus the number consumed by various charge trapping or recombination processes before the charges reach the surface. Moreover, the photocatalytic activity depends on the surface area and surface facet of the photocatalyst. The larger the surface area, the higher the photocatalytic activity. And the more the active surface facet exposed, the higher the photocatalytic activity.Graphitic carbon nitride, g-C3N4, has been widely studied as a promising photocatalyst since its discovery by Wang, Domen and Antonietti.12,13 However, its photocatalytic efficiency is limited by its large interlayer resistance and fast charge recombination.14,15 In order to enhance its photocatalytic performance, various modification strategies have been explored, including morphology control,16–21 doping,22–24 and fabrication of composites25–27 and heterojunctions, to enhance the charge separation of semiconductors.25–40 To date, a variety of g-C3N4 based heterojunctions have been developed such as MoS2/g-C3N4,25 TiO2/g-C3N4,26,30 Ag3PO4/g-C3N4,27 graphene/g-C3N4,31,32 Bi spheres/g-C3N4,33 and g-C3N4/g-C3N4.34,35 For example, Dong et al. synthesized g-C3N4/g-C3N4via thermal treatment of urea and thiourea simultaneously and found that the heterojunction significantly improved the photocatalytic activity for NO removal.34 The g-C3N4/g-C3N4 was obtained by coupling two g-C3N4, similar to the anatase–rutile phase TiO2 heterojunction.39,40 A novel “phosphorylation” strategy has recently been demonstrated for boosting photocatalytic H2 production over g-C3N4 nanosheets under visible light with a rate of 947 μmol h−1 and an apparent quantum yield of 26.1% at 420 nm, benefiting from the synergy of enhanced proton reduction and improved hole oxidation.41
To the best of our knowledge, no study has been reported on the g-C3N4/g-C3N4 homojunction for photocatalytic hydrogen production as well as the synergistic effects in homogeneous structures. In this study, a homogeneous g-C3N4/g-C3N4 junction has been prepared by a facile thermal polymerization method using urea and thiourea as precursors.
The as-prepared nanojunction exhibits an enhanced photocatalytic activity for H2 evolution under stimulated sunlight irradiation. The enhanced photocatalytic performance is attributed to the better charge separation of photogenerated charge carriers due to the existence of the n/n junctions in the homogeneous g-C3N4. This is similar to that in the anatase/rutile composite, but between the g-C3N4 moieties of the same phase. Thus, this study may bring in a new concept in improving photocatalysts' performance by introducing synergistic effects in the same materials with the same phase or in homogeneous structures. Thus, this study may trigger more exciting discoveries in catalyst designs for photocatalytic hydrogen generation.
Results and discussion
Three types of g-C3N4 were synthesized for comparison, namely, U-CN, T-CN and M-CN, where U-CN stands for the pure g-C3N4 derived from urea, T-CN for the pure g-C3N4 derived from thiourea, and M-CN for the homogeneous g-C3N4/g-C3N4 derived from a mixture of urea and thiourea (molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Fig. 1A shows the X-ray diffraction (XRD) patterns of the U-CN, T-CN and M-CN. The XRD patterns confirmed the formation of graphitic stacking C3N4 layers.12 Two peaks with a well-developed g-C3N4 network were observed in the XRD patterns, and no additional peak was detected in all samples, suggesting that these samples produced by using different precursors had basically the same crystal structures. The small diffraction peak at around 13.2° was indexed to the (100) plane and assigned to the in-plane structural packing motif. The peak at around 27.5° was the typical (002) interlayer-stacking peak. In addition, there was a broad background overlapping the diffraction (002) peak for U-CN and M-CN. This broad peak hinted at the existence of a large distribution of the interlayer stacking and/or a large amount of structural disorder. The full width at the half maximum of the (002) peak decreased in the order of T-CN > M-CN > U-CN. This indicated that the average of the crystalline domains in these samples increased in the order of T-CN < M-CN < U-CN. The minor peak at around 56° was indexed to the (220) plane of the g-C3N4 (JCPDS no. 50-1512).
:1). Fig. 1A shows the X-ray diffraction (XRD) patterns of the U-CN, T-CN and M-CN. The XRD patterns confirmed the formation of graphitic stacking C3N4 layers.12 Two peaks with a well-developed g-C3N4 network were observed in the XRD patterns, and no additional peak was detected in all samples, suggesting that these samples produced by using different precursors had basically the same crystal structures. The small diffraction peak at around 13.2° was indexed to the (100) plane and assigned to the in-plane structural packing motif. The peak at around 27.5° was the typical (002) interlayer-stacking peak. In addition, there was a broad background overlapping the diffraction (002) peak for U-CN and M-CN. This broad peak hinted at the existence of a large distribution of the interlayer stacking and/or a large amount of structural disorder. The full width at the half maximum of the (002) peak decreased in the order of T-CN > M-CN > U-CN. This indicated that the average of the crystalline domains in these samples increased in the order of T-CN < M-CN < U-CN. The minor peak at around 56° was indexed to the (220) plane of the g-C3N4 (JCPDS no. 50-1512).
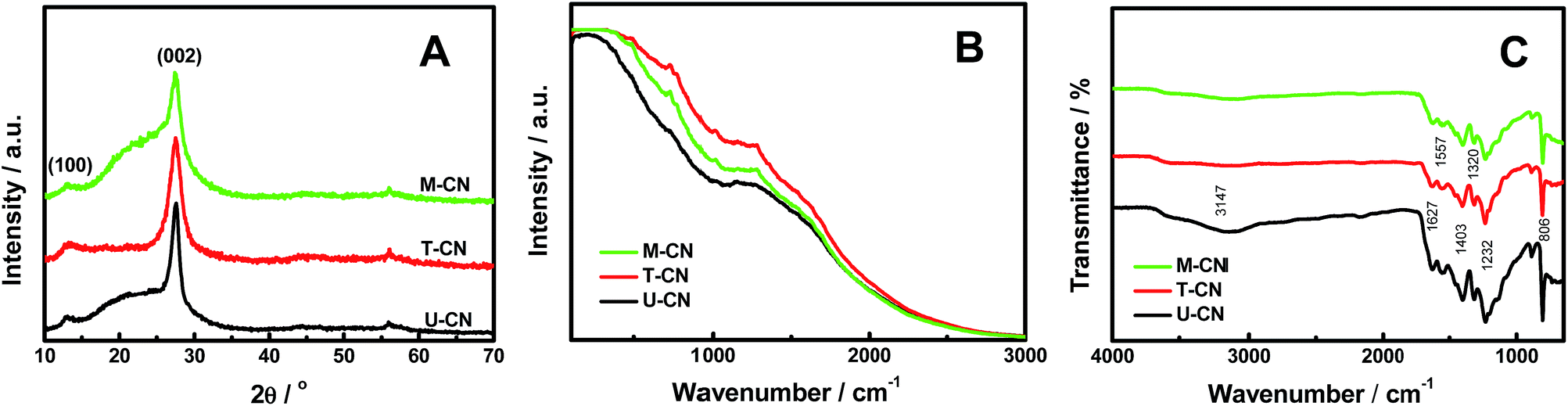 | ||
| Fig. 1 (A) XRD patterns, (B) Raman spectra and (C) FTIR spectra of U-CN, T-CN and M-CN. U-CN: pure g-C3N4 derived from urea, T-CN: pure g-C3N4 derived from thiourea, and M-CN: heterogeneous g-C3N4/g-C3N4 derived from a mixture of urea and thiourea. | ||
Raman spectroscopy was used to study the crystalline quality because of its sensitivity to slight variations in the lattice symmetry. The similar shape and intensity of the Raman spectra of U-CN, T-CN and M-CN shown in Fig. 1B suggested a similar amount of structural crystallinity and disorder in these samples, where the large and broad Raman spectra were likely the fluorescence background from defects. The amount of disorder/defects might slightly differ in each sample, as the intensity of the Raman spectrum increased in this order: U-CN < M-CN < T-CN. As the broad XRD background near the (002) peak increased in the order T-CN < U-CN < M-CN and there was no background for T-CN, the broad XRD background was likely due to the large distribution of the interlayer. Fig. 1C shows the Fourier transformed infrared (FT-IR) spectra of the U-CN, T-CN and M-CN. The peaks at 1232, 1320 and 1403 cm−1 were from the aromatic C–N stretching, and the peaks at 1557 and 1627 cm−1 were mainly due to the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds.12,34 The sharp peak at 806 cm−1 was attributed to the out-of-plane bending vibration characteristics of triazine rings.12,34 The broad peak at around 3147 cm−1 originated from stretching vibration modes of the –NH and hydroxyl of the adsorbed H2O.12,34 U-CN had an obvious –NH or OH absorption, and larger absorption in other regions as well.
N bonds.12,34 The sharp peak at 806 cm−1 was attributed to the out-of-plane bending vibration characteristics of triazine rings.12,34 The broad peak at around 3147 cm−1 originated from stretching vibration modes of the –NH and hydroxyl of the adsorbed H2O.12,34 U-CN had an obvious –NH or OH absorption, and larger absorption in other regions as well.
The TEM images of the U-CN, T-CN and M-CN are shown in Fig. 2. The U-CN consisted of many open and porous thinner nanosheets (Fig. 2A). The T-CN was composed of much denser and thicker layers. The morphological difference between U-CN and T-CN was likely from the difference in the formation and polycondensation processes of the g-C3N4 due to the different heteroatoms (oxygen in urea and sulfur in thiourea) in urea and thiourea.30Fig. 2C shows the morphology of the M-CN, where the dense and thick CN-T layers were closely packed on the surface of CN-U porous thin nanosheets, forming various g-C3N4/g-C3N4 homojunctions as clearly seen in the HRTEM image shown in Fig. 3D.
 | ||
| Fig. 2 TEM images of (A) U-CN, (B) T-CN, and (C) M-CN, and (D) HRTEM image of M-CN. | ||
 | ||
| Fig. 3 (A) Survey, (B) O 1s, (C) N 1s and (D) C 1s core-level XPS spectra of the U-CN, T-CN and M-CN. | ||
The X-ray photoelectron spectroscopy (XPS) survey spectra in Fig. 3A confirmed that all samples contained only C, N and O elements. No peak from sulfur species was observed for T-CN and M-CN, indicating the complete release of sulfur in thiourea during heating treatment. The O 1s peak at 532.7 eV (Fig. 3B) was from the adsorbed H2O on the catalyst surface.34 The dominant peak at 398.8 eV in the N 1s spectrum (Fig. 3C) was from the nitrogen in the aromatic N in triazine rings (CN–C), and two weak peaks at 401.3 eV and 404.5 eV were from the N–H groups and π-excitations, respectively.12,34 The large peak at 288.6 eV in the C 1s spectra (Fig. 3D) was from the tertiary carbon in the g-C3N4 lattice, and the small peak at 284.8 eV was from the carbon tape as the reference.
The UV-vis absorption of the U-CN, T-CN and M-CN shown in Fig. 4A displayed that all the samples had an intrinsic semiconductor-like absorption. T-CN had a larger absorption and an obvious red shift in the visible-light region compared to U-CN, consistent with the previous observation.30 The absorption of the M-CN was located between those of T-CN and U-CN. The optical bandgap (Eg) was estimated from the intercept of the tangents to the plots of (αhν)1/2vs. photon energy as shown in Fig. 4B. The bandgaps of U-CN, M-CN and T-CN were 2.85, 2.76 and 2.60 eV, respectively. The bandgap values were consistent with the values in the literature.12–34 The band structures of the U-CN, T-CN and M-CN were further examined by Mott–Schottky measurements. Based on the Mott–Schottky plots shown in Fig. 4C, the conduction band edge (CBE) position laid at −0.73, −0.86 and −0.92 V (relative to the normal hydrogen electrode – NHE) for U-CN, M-CN and T-CN, respectively. Thus, the valence band edge (VBE) position was at 2.12, 1.90 and 1.68 V for U-CN, T-CN and M-CN, respectively. The relative changes of the CBE and VBE positions of these three samples shown in Fig. 4D indicated that there was a linear change in the electronic structure from U-CN via M-CN to T-CN: the CBE and VBE of the homogeneous M-CN as well as its bandgap were in the middle of those of U-CN and T-CN. This linear change of the electronic band structures from U-CN to T-CN through M-CN was similar to that observed in many homogeneous alloyed semiconductor compounds, such as InPxAs1−x,42 ZnxCd1−xS,43 and MgxZn1−xTe,44 but different from that in heterogeneous composites, such as CdS/TiO2 (ref. 45) and TiO2/g-C3N4,26,30 where the overall optical bandgap of the composite was determined by the bandgap of the moiety with the smaller bandgap in the heterogeneous composite. Therefore, homogeneous, instead of heterogeneous, junctions were likely formed in the g-C3N4/g-C3N4 (M-CN) sample.
 | ||
| Fig. 4 (A) UV-vis absorption, (B) plots of (αhν)1/2vs. photon energy, (C) Mott–Schottky plots, and (D) VBE and CBE of the U-CN, T-CN and M-CN. | ||
Meanwhile, the positive slopes of the Mott–Schottky plots in Fig. 4C suggested that all the samples were n-type semiconductors. Thus, the M-CN sample featured many n/n homogeneous junctions inside. The donor density was calculated according to the equation:
| Nd = (2/e0ε0ε)[d(1/C2)/dV]−1 |
The photocatalytic hydrogen evolution activity of the U-CN, T-CN and M-CN was studied using 1.5 wt% Pt and 50% vol% methanol as the co-catalyst and hole sacrificial agent, respectively. As shown in Fig. 5A, all samples showed a good hydrogen evolution performance within the test time (6 h) under simulated sunlight irradiation. The amount of hydrogen increased linearly with irradiation time. The comparison of the specific activity is shown in Fig. 5B. The photocatalytic activity increased in the order of U-CN (337.2 μmol g−1 h−1) < T-CN (524.6 μmol g−1 h−1) < M-CN (780.5 μmol g−1 h−1). The hydrogen evolution activity of the M-CN is 2.3, which is 1.5 times higher than that of the U-CN and T-CN. In comparison, the photocatalytic activity of the g-C3N4 composites formed by physically mixing two different g-C3N4 particles was between those of U-CN and T-CN; thus no synergistic effects were observed. The surface areas calculated by the Brunauer–Emmett–Teller (BET) method were 119.53, 12.12, and 29.56 m2 g−1 for U-CN, T-CN and M-CN, respectively. The N2 isotherm adsorption and desorption and the Barrett–Joyner–Halenda (BJH) pore size distribution curves are shown in Fig. S1.† The adsorption–desorption isotherms showed that all the samples were of type IV, suggesting the presence of mesopores (2–50 nm). Compared with that of T-CN, the hysteresis loop of U-CN shifted to the region of lower pressure and the area of the hysteresis loop became large, indicating the formation of relatively large mesopores. The hysteresis loop of M-CN was changed accordingly with the introduction of urea in the precursor. The pore size distribution of the samples confirmed the formation of mesopores and a similar pore size distribution of all the samples. Apparently, the photocatalytic activity of the samples had no apparent correlation with their surface areas and pore size distribution. The higher activity of T-CN than U-CN was related to its large optical absorption, smaller crystalline domains (based on the XRD result in Fig. 1A), and lower donor density (based on the Mott–Schottky plots in Fig. 4C). The correlations between the photocatalytic activity with the donor concentration and optical bandgaps shown in Fig. 5C did not display apparent relationships. While the highest photocatalytic activity of the M-CN was not satisfactorily explained by its intermediate values of the optical absorption, structural disorder/defects, donor density, bandgap values and band edge positions among these three samples, it was successfully understood based on its homogeneous structure with many n/n junctions formed inside the material. The stability of the photocatalytic performance of the M-CN sample is shown in Fig. S2.† The result indicated that the M-CN sample was fairly stable during the 5 day test period, demonstrating its promising future for photocatalytic hydrogen generation.
 | ||
| Fig. 5 (A) Time profiles and (B) specific activity of the photocatalytic hydrogen generation using U-CN, M-CN and T-CN as the photocatalyst, and (C) comparison of the CBE and VBE of U-CN, M-CN and T-CN with the HER and OER potentials (at pH 0). (D) Illustration of the photocatalytic hydrogen generation using M-CN as the photocatalyst. | ||
Based on the electronic structure analysis, its enhanced photocatalytic activity was explained by the mechanism shown in Fig. 5D. In photocatalysis, as the M-CN was irradiated by the incident light, the electrons were photoexcited from the conduction band (CB) of both U-CN and T-CN. Due to the mismatch of the electronic band structure of U-CN and T-CN, there were build-in electrical fields and n/n junctions in the heterogeneous junctions.47–49 Thus, the photogenerated electrons were transferred from T-CN to U-CN, whereas the photogenerated holes moved from U-CN to T-CN. The potential difference drove a better charge separation. As a result, the excited electron/hole pairs were more effectively separated and transferred to the U-CN and T-CN, where the reduction and oxidation reactions took place, respectively. In addition, with effective separation of electron/hole pairs, the lifetime of photogenerated charge carriers was expected to be prolonged. The prolonged lifetime allowed fast charge transfer to the reactive substrates on the photocatalyst surface, enhancing the photocatalytic activity for hydrogen generation. Different from previous studies, where synergistic effects were found between different materials or phases, here, we reveal that synergistic effects can exist between the boundaries of the same materials of the same phase, or within the homogenous structures, but with different electronic structures. Thus, this study may trigger more exciting discoveries in catalyst designs for photocatalytic hydrogen generation.
Conclusions
In conclusion, homogeneous nanojunctioned g-C3N4 (M-CN) has been prepared through a thermal polymerization method using urea and thiourea as precursors. The differences in the electronic band structures of pure g-C3N4 (U-CN from urea and T-CN from thiourea) create mismatches in their conduction and valence band levels, forming many homogeneous n/n nanojunctions in the catalyst. A significant enhancement in the photocatalytic activity is observed for H2 evolution from water splitting under stimulated sunlight irradiation. This enhancement benefits from those n/n junctions for better charge separation, similar to the synergistic effects at the anatase/rutile interface, but across the interface between the g-C3N4 nanodomains of the same phase. Thus, a new concept on synergistic effects in photocatalysis is shown in this study that synergistic effects can be created not only in heterogeneous structures/composites, but also in homogeneous structures/composites. Thus, this study may trigger many new findings in the related materials, catalysis, and renewable energy fields.Acknowledgements
X. C. appreciates the financial support from the College of Arts and Sciences, University of Missouri – Kansas City and University of Missouri Research Board. M. Z. thanks the support from the Natural Science Foundation of China (Grant No. 51272075; 51372080). M. Z., T. L. and Y. L. thank the support from the China Scholarship Council for abroad research.Notes and references
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- M. Dahl, Y. Liu and Y. Yin, Chem. Rev., 2014, 114, 9853–9889 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- X. B. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- N. Liu, C. Schneider, D. Freitag, M. Hartmann, U. Venkatesan, J. Müller, E. Spiecker and P. Schmuki, Nano Lett., 2014, 14, 3309–3313 CrossRef CAS PubMed.
- X. Lu, G. Wang, S. Xie, J. Shi, W. Li, Y. Tong and Y. Li, Chem. Commun., 2012, 48, 7717–7719 RSC.
- J. Yang, H. Yan, X. Wang, F. Wen, Z. Wang, D. Fan, J. Shi and C. Li, J. Catal., 2012, 290, 151–157 CrossRef CAS.
- N. Bao, L. Shen, T. Takata and K. Domen, Chem. Mater., 2008, 20, 110–117 CrossRef CAS.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- X. H. Li, X. C. Wang and M. Antonietti, Chem. Sci., 2012, 3, 2170–2174 RSC.
- G. Han, B. Wang, Y. Zhao, C. Hu and L. Qu, Angew. Chem., 2015, 54, 11433–11437 CrossRef PubMed.
- F. He, G. Chen, Y. S. Zhou, Y. Yu, Y. Zheng and S. Hao, Chem. Commun., 2015, 51, 16244–16246 RSC.
- J. S. Zhang, Y. Chen and X. C. Wang, Energy Environ. Sci., 2015, 8, 3092–3108 CAS.
- Y. Zheng, L. H. Lin, B. Wang and X. C. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- J. S. Zhang and X. C. Wang, Angew. Chem., Int. Ed., 2015, 54, 7230–7232 CrossRef CAS PubMed.
- J. S. Xu, T. J. K. Brenner, Z. P. Chen, M. Antonietti and M. Shalom, ACS Appl. Mater. Interfaces, 2014, 6, 16481–16486 CAS.
- J. Liu, Y. Liu, N. Y. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 16850–16856 CAS.
- Y. Hou, Z. H. Wen, S. M. Cui, X. Guo and J. Chen, Adv. Mater., 2013, 25, 6291–6297 CrossRef CAS PubMed.
- Y. F. Chen, W. X. Huang, D. L. He, Y. Situ and H. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 14405–14414 CAS.
- Y. M. He, L. H. Zhang, B. T. Teng and M. Fan, Environ. Sci. Technol., 2015, 49, 649–656 CrossRef CAS PubMed.
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- S. J. A. Moniz, S. A. Shevlin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 CAS.
- W. K. Jo, T. Adinaveen, J. J. Vijaya and N. C. S. Selvam, RSC Adv., 2016, 6, 10487–10497 RSC.
- J. J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931–940 CrossRef CAS PubMed.
- L. Xu, W. Q. Huang, L. L. Wang, Z.-A. Tian, W. Hu, Y. Ma, X. Wang, A. Pan and G.-F. Huang, Chem. Mater., 2015, 27, 1612–1621 CrossRef CAS.
- F. Dong, Z. W. Zhao, Y. J. Sun, Y. X. Zhang, S. Yan and Z. B. Wu, Environ. Sci. Technol., 2015, 49, 12432–12440 CrossRef CAS PubMed.
- F. Dong, Z. W. Zhao, T. Xiong, Z. Ni, W. Zhang, Y. Sun and W. K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CAS.
- F. Dong, Z. L. Ni, P. D. Li and Z. B. Wu, New J. Chem., 2015, 39, 4737–4744 RSC.
- J. S. Zhang, M. W. Zhang, R. Q. Sun and X. C. Wang, Angew. Chem., Int. Ed., 2012, 51, 10145–10149 CrossRef CAS PubMed.
- H. L. Wang, L. S. Zhang, Z. G. Chen, J. Q. Hu, S. J. Li, Z. H. Wang, J. S. Liu and X. C. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- H. Q. Li, Y. X. Liu, X. Gao, C. Fu and X. C. Wang, ChemSusChem, 2015, 8, 1189–1196 CrossRef CAS PubMed.
- J. Zhang, Q. Xu, Z. C. Feng, M. Li and C. Li, Angew. Chem., Int. Ed., 2008, 47, 1766–1769 CrossRef CAS PubMed.
- T. Xia, N. Li, Y. Zhang, M. B. Kruger, J. Murowchick, A. Selloni and X. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 9883–9890 CAS.
- G. Liu, T. Wang, H. Zhang, X. Meng, D. Hao, K. Chang, P. Li, T. Kako and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 13561 CrossRef CAS PubMed.
- M. Balkanski and R. F. Wallis, Semiconductor physics and applications, Oxford University Press, New York, 2000 Search PubMed.
- S.-J. Chung, E.-K. Suh and Y.-K. Jun, J. Korean Phys. Soc., 2005, 46, 963–967 CAS.
- J. Shi, H. Yan, X. Wang, Z. Feng, Z. Lei and C. Li, Solid State Commun., 2008, 146, 249–252 CrossRef CAS.
- X. Li, T. Xia, C. Xu, J. Murowchick and X. Chen, Catal. Today, 2014, 225, 64–73 CrossRef CAS.
- R. Asahi, Y. Taga, W. Mannstadt and A. J. Freeman, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 7459 CrossRef CAS.
- T. Xia, C. Zhang, N. A. Oyler and X. Chen, Adv. Mater., 2013, 25, 6905–6910 CrossRef CAS PubMed.
- T. Xia, C. Zhang, N. A. Oyler and X. Chen, J. Mater. Res., 2014, 29, 2198–2210 CrossRef CAS.
- T. Xia, Y. Cao, N. A. Oyler, J. Murowchick, L. Liu and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 10407–10413 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6se00004e |
| This journal is © The Royal Society of Chemistry 2017 |