
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mass-producible 2D-MoSe2 bulk modified screen-printed electrodes provide significant electrocatalytic performances towards the hydrogen evolution reaction†
Samuel J.
Rowley-Neale
a,
Christopher W.
Foster
 a,
Graham C.
Smith
b,
Dale A. C.
Brownson
a and
Craig E.
Banks
*a
a,
Graham C.
Smith
b,
Dale A. C.
Brownson
a and
Craig E.
Banks
*a
aFaculty of Science and Engineering, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, UK. E-mail: c.banks@mmu.ac.uk; Web: http://www.craigbanksresearch.com Fax: +44 (0) 1612476831; Tel: +44 (0) 1612471196
bFaculty of Science and Engineering, Department of Natural Sciences, University of Chester, Thornton Science Park, Pool Lane, Ince, Chester CH2 4NU, UK
First published on 25th January 2017
Abstract
We demonstrate a facile, low cost and reproducible methodology for the production of electrocatalytic 2D-MoSe2 incorporated/bulk modified screen-printed electrodes (MoSe2-SPEs). The MoSe2-SPEs outperform traditional carbon based electrodes, in terms of their electrochemical activity, towards the Hydrogen Evolution Reaction (HER). The electrocatalytic behaviour towards the HER of the 2D-MoSe2 within the fabricated electrodes is found to be mass dependent, with an optimal mass ratio of 10% 2D-MoSe2 to 90% carbon ink. MoSe2-SPEs with this optimised ratio exhibit a HER onset, Tafel value and a turn over frequency of ca. −460 mV (vs. SCE), 47 mV dec−1 and 1.48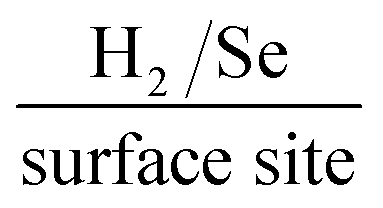 respectively. These values far exceed the HER performance of graphite (unmodified) SPEs, that exhibit a greater electronegative HER onset and Tafel value of ca. −880 mV and 120 mV dec−1 respectively. It is clear that impregnation of 2D-MoSe2 into the MoSe2-SPEs bulk ink/structure significantly increases the performance of SPEs with respect to their electrocatalytic activity towards the HER. When compared to SPEs that have been modified via a drop-casting technique, the fabricated MoSe2-SPEs exhibit excellent cycling stability. After 1000 repeat scans, a 10% modified MoSe2-SPE displayed no change in its HER onset potential of −450 mV (vs. SCE) and an increase of 31.6% in achievable current density. Conversely, a SPE modified via drop-casting with 400 mg cm−2 of 2D-MoSe2 maintained its HER onset potential of −480 mV (vs. SCE), however exhibited a 27.4% decrease in its achievable current density after 1000 scans. In addition to the clear performance benefits, the production of MoSe2-SPEs mitigates the need to post hoc modify an electrode via the drop-casting technique. We anticipate that this facile production method will serve as a powerful tool for future studies seeking to utilise 2D materials in order to mass-produce SPEs/surfaces with unique electrochemical properties whilst providing substantial stability improvements over the traditionally utilised technique of drop-casting.
respectively. These values far exceed the HER performance of graphite (unmodified) SPEs, that exhibit a greater electronegative HER onset and Tafel value of ca. −880 mV and 120 mV dec−1 respectively. It is clear that impregnation of 2D-MoSe2 into the MoSe2-SPEs bulk ink/structure significantly increases the performance of SPEs with respect to their electrocatalytic activity towards the HER. When compared to SPEs that have been modified via a drop-casting technique, the fabricated MoSe2-SPEs exhibit excellent cycling stability. After 1000 repeat scans, a 10% modified MoSe2-SPE displayed no change in its HER onset potential of −450 mV (vs. SCE) and an increase of 31.6% in achievable current density. Conversely, a SPE modified via drop-casting with 400 mg cm−2 of 2D-MoSe2 maintained its HER onset potential of −480 mV (vs. SCE), however exhibited a 27.4% decrease in its achievable current density after 1000 scans. In addition to the clear performance benefits, the production of MoSe2-SPEs mitigates the need to post hoc modify an electrode via the drop-casting technique. We anticipate that this facile production method will serve as a powerful tool for future studies seeking to utilise 2D materials in order to mass-produce SPEs/surfaces with unique electrochemical properties whilst providing substantial stability improvements over the traditionally utilised technique of drop-casting.
1. Introduction
The unpredictable and yet inevitable consequences of anthropogenic climate change cannot be mitigated by the refinement or increased efficiency and “greening” of the current fossil fuel (FF) based global energy economy. There must be a forced acceleration towards developing new and innovative non-polluting energy generation methods.1–7 In certain niches, such as for combined heat and power units,8,9 hydrogen fuel cells could offer non-polluting alternatives to current fossil based energy generating technologies.10,11 The major limiting factor to their implementation is the high cost of the energy per unit output, which is not economically competitive to that of comparable FF alternatives.12,13 Literature has tried to tackle this problem via increasing the ‘fuel stock input’ to ‘usable energy output’ efficiency as well as, obviously, lowering the cost of hydrogen production.1One of the most recently explored approaches for the production of hydrogen is the Hydrogen Evolution Reaction (HER) (2H+ + 2e− → H2), which is the focus of commercially available electrolysers. The efficiency of the HER is dependent on the choice of electrocatalyst.10 Platinum (Pt) is currently the most proficient catalyst for the HER with a near zero over-potential,14 however its high cost and sparse earth abundance makes large scale usage unattractive.10,13,16,17 Researchers have therefore focused on finding cheaper and more abundant carbon based electrocatalysts. For example, Liu et al.15 and Abbaspour et al.16 have observed relatively low HER overpotentials whilst utilising carbon based electrodes that were doped with nitrogen and Ni-ferrite nanoparticles respectively.
Modification of carbon based electrodes with electrocatalytic 2D-materials such as transition metal dichalcogenides (TMD), the most thoroughly researched being MoS2,1 has become a common approach towards tackling the problem of hydrogen production outlined above.1,17–20 2D-MoSe2 has been shown to be an effective electrocatalyst for the HER, however, literature is comparatively sparse compared to 2D-MoS2, with Table 1 representing a thorough overview of all the studies currently available. 2D-MoSe2 is a typical TMD semiconducting material, having a structure which is analogous to 2D-MoS2, comprising a single layer of Mo sandwiched between two layers of Se atoms (with strong coordinate bonds).21 2D-MoSe2 monolayers are held together via weak van der waals forces.22 Theoretical studies suggest that it is the active edge planes of TMD nanosheets that exhibit the catalytic activity towards the HER, possessing exchange current densities close to that of the Pt-group metals, whilst the basal planes are relatively inert.23 Note that terminated edge planes and defect sites of the 2D-MoSe2 will comprise of both Mo and Se atoms, each having distinct electrochemical properties. In this case, it is the dangling bonds of the electronegatively charged Se atoms, located at the edge sites, which have an affinity for binding electropositive H+ atoms within the electrolyte (that arises from having a density functional theory calculated binding energy towards H+ of −0.05).24 Thus, these are likely the sites responsible for the 2D-electrochemical activity towards the HER.12
| Catalyst | Supporting electrode | Loading | Deposition technique | Cycling stability (CS) duration | Potential range of CS | CS performance | Electrolyte | HER onset (V) | Tafel (mV dec−1) | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| a —: value unknown, GN: graphene nanosheets, GC; glassy carbon, PECVD: plasma-enhanced chemical vapor deposition, a: the achievable current density is stable over the duration reported, RHE; reversible hydrogen electrode, b: degradation in the achievable current density over the duration reported, SCE; saturated calomel electrode, rGO: reduced graphene oxide, NW: nanowires, NF: nanofibers, EPD: electrophoresis deposition, *: mass of 2D-MoSe2 to mass of conductive carbon ink ratio, SPE; screen-printed electrode, c: increase in the achievable current density over the duration reported. | ||||||||||
| MoSe2/GN | GC | — | PECVD on a graphite disc | 6000 s | Continuous at −0.15 (V vs. RHE) | ca. 88% retention | 0.5 M H2SO4 | −0.05 (V vs. RHE) | 61 | 23 |
| MoSe2/rGO | GC | 0.16 mg cm−2 | Drop-casting | 7200 s | +0.10 to −0.35 (V vs. RHE) | b | 0.5 M H2SO4 | −0.05 (V vs. RHE) | 69 | 51 |
| MoSe2 nanofilm | Carbon NW and NF | — | RSP | 15000 cycles | +0.10 to −0.28 (V vs. RHE) | c | 0.5 M H2SO4 | −0.11 (V vs. RHE) | 60 | 45 |
| Vertically aligned MoSe2 | GC | 0.0135 μg cm−2 | Grown on GC | 1000 cycles | 0.00 to −0.45 (V vs. SCE) | b | 0.5 M H2SO4 | −0.20 (V vs. RHE) | 105–120 | 52 |
| MoS2(1−x)Se2x nanoflakes | GC | ca. 0.28 mg cm−2 | Drop-casting | 8000 cycles | 0.00 to −0.44 (V vs. SCE) | b | 0.5 M H2SO4 | −0.35 to −0.38 (V vs. SCE) | 45 | 53 |
| MoSe2 nanosheets | GC | — | Drop-casting | 2000 cycles | +0.10 to −0.30 (V vs. RHE) | b | 0.5 M H2SO4 | −0.08 (V vs. RHE) | 80 | 38 |
MoSe2/CoSe2 composite (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) :1 ratio) |
GC | 0.29 mg cm−2 | Drop-casting | 1000 cycles | +0.10 and −0.50 V (vs. RHE) | b | 0.5 M H2SO4 | −0.11 (V vs. RHE) | 73 | 54 |
| MoSe2–NiSe | GC | 0.29 mg cm−2 | Drop-casting | 1000 cycles | +0.20 and −0.30 V (vs. RHE) | b | 0.5 M H2SO4 | −0.15 (V vs. RHE) | 56 | 27 |
| MoSe2 nanosheets | Carbon cloth | — | EPD | 1000 cycles | +0.10 and −0.35 V (vs. RHE) | b | 0.5 M H2SO4 | −0.22 (V vs. RHE) | 76 | 55 |
| 2D-MoSe2 ink | SPE | 5%* | Screen-printed | 33600 s (1000 cycles) | 0.00 to −1.40 (V vs. SCE) | c | 0.5 M H2SO4 | −0.44 (V vs. SCE) | 49 | This work |
| 2D-MoSe2 ink | SPE | 10%* | Screen-printed | 33600 s (1000 cycles) | 0.00 to −1.40 (V vs. SCE) | c | 0.5 M H2SO4 | −0.45 (V vs. SCE) | 47 | This work |
| 2D-MoSe2 ink | SPE | 20%* | Screen-printed | 33600 s (1000 cycles) | 0.00 to −1.40 (V vs. SCE) | c | 0.5 M H2SO4 | −0.43 (V vs. SCE) | 63 | This work |
| 2D-MoSe2 ink | SPE | 40%* | Screen-printed | 33600 s (1000 cycles) | 0.00 to −1.40 (V vs. SCE) | c | 0.5 M H2SO4 | −0.43 (V vs. SCE) | 230 | This work |
Whilst the studies reported in Table 1 are diligent in their approach to exploring MoSe2 based catalysts towards the HER, there is a noticeable convention of solely utilising glassy carbon (GC) as a supporting electrode material with which to electrically “wire” the catalytic material, with limited attempts to utilise alternative supporting electrodes. Similar to Pt, GC is relatively expensive and thus limits its applicability within electrolysers and fuel cells. Conversely, screen-printed electrodes (SPEs) are significantly lower in cost and preferential in terms of tailorability and reducibility on mass. Resultantly, SPEs have recently been utilised (opposed to GC) as a preferential supporting electrode material within research focused on HER electrocatalysts. ESI Fig. 1(A)† shows the exact SPE design utilised herein whilst ESI Fig. 1(B)† shows an alternative SPE that could have been employed, with a much larger working area. The inclusion of the ESI Fig. 1† is to highlight the versatility capable in the design of SPEs, which is not capable by employing more traditional carbon based electrodes. Of note in Table 1 is the repeated use of the drop-casting technique as a method of modifying the supporting electrode with the desired 2D-nanomaterial based electrocatalyst. The drop-casting method, where a pipette is used to manually dispense a set volume of fluid containing the desired electrocatalyst onto an electrode's surface, is an easy/convenient technique to test the catalytic properties of a material. However, it has several disadvantages, including poor cycling stability, low levels of reproducibility and unevenly distributed coverage of the material deposited, which is especially problematic when using 2D nanomaterials that have anisotropic heterogeneous electrode transfer (HET) kinetics within their structure (i.e. varied properties can be observed at single nanosheet, intermediate and bulk forms).25 The poor cycling stability of the drop-casting technique is highlighted in Table 1, where all of the studies therein that deposit a MoSe2 based catalyst (via the drop-casting technique) are observed to have a degradation in the achievable current density over the duration of their cycling stability tests. A prevalent challenge within the literature has thus been to find an alternative method of modifying a supporting electrode such that one does not display the disadvantages associated with drop-casting.
In an attempt to produce a desirable alternative to GC and Pt electrodes currently utilised within electrolysers, this study aims to produce a facile and highly reproducible technique for the creation of highly electrocatalytic SPE variants by doping readily available carbon ink recipes with varying percentage (mass) contributions of 2D-MoSe2. We expect the fabricated electrodes to exhibit high reproducibility, excellent cycling stability and to demonstrate low overpotentials towards the HER. Several studies within the literature have previously used the term “ink”,26,27 when describing the slurry containing their particular electrocatalyst, however to the best of our knowledge we are the first to produce, characterise and implement a true screen-printable 2D-MoSe2 incorporated electrocatalytic ink.
2. Experimental section
All chemicals used were of analytical grade and were used as received from Sigma-Aldrich without any further purification. This includes the MoSe2 (product code: 1002015528) which has a reported purity of 99.9% on a trace metal basis.28 All solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm and were vigorously degassed prior to electrochemical measurements with high purity, oxygen free nitrogen. The above ensures the removal of any trace of oxygen from test solutions, which if present would convolute the observed results for HER with the competing oxygen evolution reaction; this is common practice in the literature.29,30 All measurements were performed in 0.5 M H2SO4 and the sulfuric acid solution utilised was of the highest possible grade available from Sigma-Aldrich (99.999%, double distilled for trace metal analysis).Electrochemical measurements were performed using an Ivium Compactstat™ (Netherlands) potentiostat. Measurements were carried out using a typical three electrode system with a Pt wire counter electrode and a saturated calomel electrode (SCE) reference. The working electrodes were screen-printed graphite electrodes (SPE), which have a 3 mm diameter working electrode. The SPEs were fabricated in-house with the appropriate stencils using a DEK 248 screen-printing machine (DEK, Weymouth, U.K.).31 These electrodes have been used extensively in previous studies.1,32–35 In their fabrication; first a carbon–graphite ink formulation (product code: C2000802P2; Gwent Electronic Materials Ltd., U.K.) was screen-printed onto a polyester (Autostat, 250 μm thickness) flexible film. This layer was then cured in a fan oven at 60 °C for 30 minutes. Next, a silver/silver chloride reference electrode was included by screen-printing Ag/AgCl paste (product code: C2030812P3; Gwent Electronic Materials Ltd., U.K.) onto the polyester substrates and a second curing step was undertaken where the electrodes were cured at 60 °C for 30 minutes. Finally, a dielectric paste (product code: D2070423D5; Gwent Electronic Materials Ltd., U.K.) was then printed onto the polyester substrate to cover the connections. After a final curing at 60 °C for 30 minutes the SPEs are ready to be used and were connected via an edge connector to ensure a secure electrical connection.36 The unmodified SPEs have been reported previously and shown to exhibit a heterogeneous electron transfer rate constant, ko, of ca. 10−3 cm s−1, as measured using the [Ru(NH3)6]3+/2+ outer-sphere redox probe.37 The 2D-MoSe2 ink was fabricated by incorporating 2D-MoSe2 into a graphite ink on the basis of the weight percent of MP to MI, where MP is the mass of particulate (in this case the 2D-MoSe2) and MI is the mass of the ink formulation used in the printing process, i.e. % = (MP/MI) × 100. The weight percent of MP to MI varied from 5, 10, 20 and 40%, which resulted four separate 2D-MoSe2 inks that could subsequently be individually screen-printed on top of the working SPE electrode (see above) and cured as described earlier (60 °C for 30 minutes). Note that the interaction between the graphite ink and the 2D-MoSe2 is physical blending, as XPS of the surface of a 10% MoSe2-SPE (see ESI Fig. 2†) demonstrates the same spectra as the 2D-MoSe2 powder described in the Results and discussion and that the 2D-MoSe2 is physical held by the binder in the ink. For the purpose of this work, electrochemical experiments were performed using the working electrode of the SPEs only and external reference and counter electrodes were utilised as detailed earlier to allow a direct comparison between all the electrodes utilised and with prior literature.
Transmission electron microscopy (TEM) images were obtained using a 200 kV primary beam under conventional bright-field conditions. The 2D-MoSe2 sample was dispersed onto a holey-carbon film supported on a 300 mesh Cu TEM grid. Raman spectroscopy was performed using a ‘Renishaw InVia’ spectrometer equipped with a confocal microscope (×50 objective) and an argon laser (514.3 nm excitation). Measurements were performed at a very low laser power level (0.8 mW) to avoid any heating effects. X-ray diffraction (XRD) was performed using an “X'pert powder PANalytical” model with a copper source of Kα radiation (of 1.54 Å) and Kβ radiation (of 1.39 Å), using a thin sheet of nickel with an absorption edge of 1.49 Å to absorb Kβ radiation. A reflection transmission spinner stage (15 rpm) was implemented to hold the commercially sourced MoSe2 nano-powder. The range was set between 10 and 100 2θ in correspondence with literature ranges.38 Additionally, to ensure well defined peaks, an exposure of 50 seconds per 2θ step was implemented with a size of 0.013°. The X-ray photoelectron spectroscopy (XPS) data was acquired using a bespoke ultra-high vacuum system fitted with a Specs GmbH Focus 500 monochromated Al Kα X-ray source, Specs GmbH Phoibos 150 mm mean radius hemispherical analyser with 9-channeltron detection, and a Specs GmbH FG20 charge neutralising electron gun.39 Survey spectra were acquired over the binding energy range 1100–0 eV using a pass energy of 50 eV and high resolution scans were made over the C 1s and O 1s lines using a pass energy of 20 eV. Under these conditions the full width at half maximum of the Ag 3d5/2 reference line is ca. 0.7 eV. In each case, the analysis was an area-average over a region approximately 1.4 mm in diameter on the sample surface, using the 7 mm diameter aperture and lens magnification of ×5. The energy scale of the instrument is calibrated according to ISO 15472, and the intensity scale is calibrated using an in-house method traceable to the UK National Physical Laboratory.40 Data were quantified using Scofield cross sections corrected for the energy dependencies of the electron attenuation lengths and the instrument transmission.41 Data interpretation was carried out using CasaXPS software v2.3.16.42
3. Results and discussion
3.1 Characterisation of the MoSe2
Independent physicochemical characterisation was performed on the commercially sourced MoSe2 and is reported below.28Fig. 1 depicts typical TEM images of the MoSe2, where upon close inspection it is clear that each 2D-MoSe2 nanosheet has an average lateral size of ca. 100–500 nm, with an inter-nanosheet distance of ca. 3 nm, which strongly corresponds with literature.38 It is evident that there is some aggregation; however, this is the case for all nanosheet materials upon dispersion from their supporting solvents.1,44 | ||
| Fig. 1 TEM images of the commercially sourced 2D-MoSe2. (A) Scale bar: 100 nm, (B) scale bar: 500 nm. | ||
XRD analysis obtained for the 2D-MoSe2 utilised herein is presented in Fig. 2(A). The characteristic diffraction peaks at 2θ = 13.7°, 37.8°, 47.5°, 55.8° and 56.7° are assigned to the (002), (103), (105), (110) and (008) faces of the hexagonal face of MoSe2 (2H crystallinity) respectively.38 Raman analysis was also implemented (see Fig. 2(B)), with a Raman spectra showing the A1g and E12g vibrational bands at ca. 238 and 283 cm−1 respectively, which are two of the most prominent peaks associated with MoSe2, agreeing well with literature.23,43,51 The A1g peak corresponds to the out-of-plane Mo–Se phonon mode whilst the E12g vibrational band is the in-plane mode.23,43,51
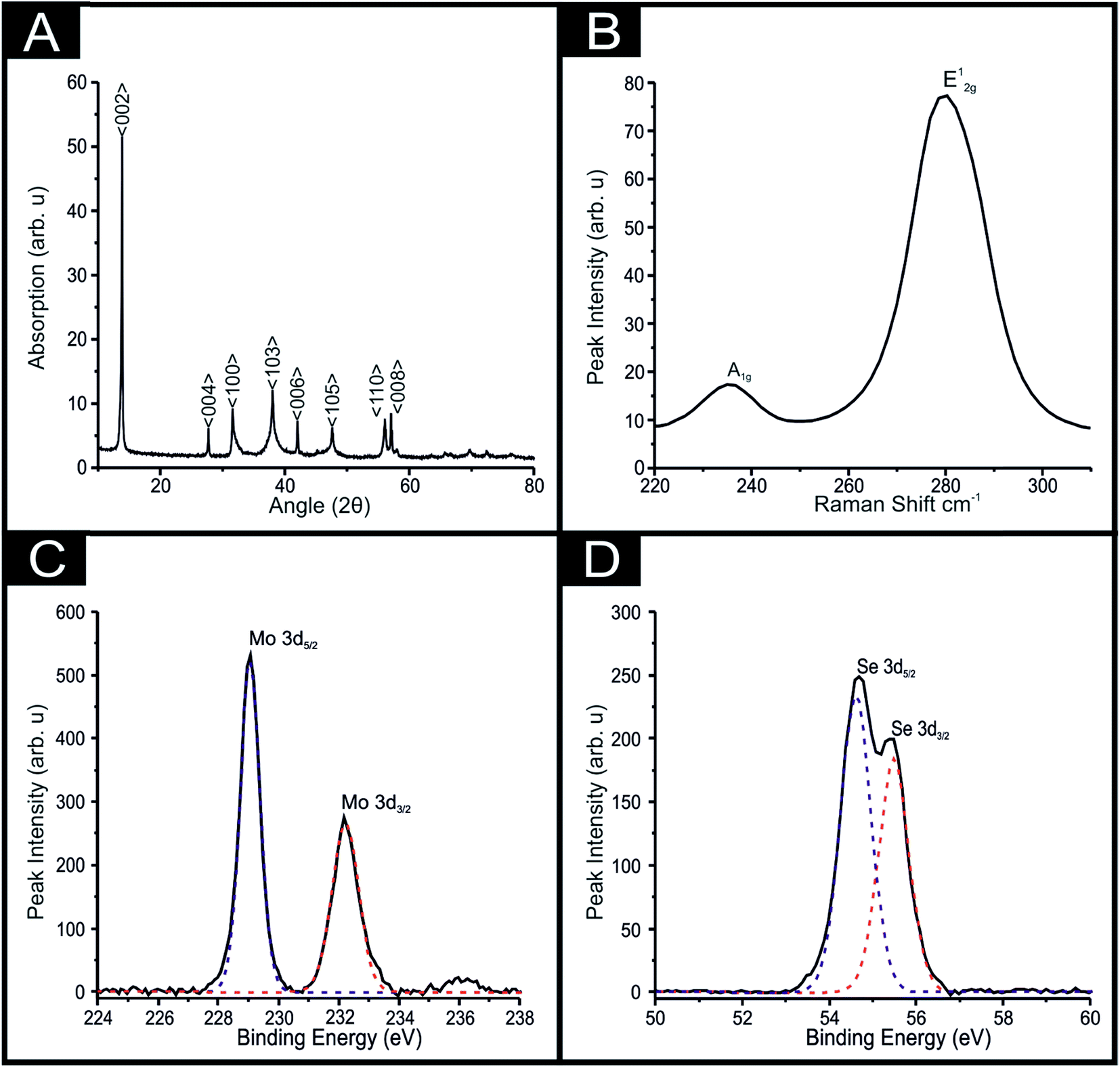 | ||
| Fig. 2 Characterisation of the commercially sourced 2D-MoSe2; (A) XRD spectra of the 2D-MoSe2, (B) Raman spectra 2D-MoSe2 deposited onto a silicon wafer. High-resolution XPS spectra of Mo 3d and Se 3d regions of MoSe2 (C and D respectively). | ||
The 2D-MoSe2 nanosheets utilised to fabricate the MoSe2-SPEs were next investigated by XPS. Upon inspection of Fig. 2(C) binding energies are evident for both Mo 3d5/2 and Mo 3d3/2 at 229.0 and 232.3 eV respectively, such analysis reveals a Mo4+ oxidation state. Also shown within Fig. 2(D) are binding energies for Se 3d5/2 and Se 3d3/2 positioned at 54.6 and 55.4 eV, indicating a Se2− oxidation state.45 De-convolution of the XPS spectra reveals a Mo/Se stoichiometric ratio of 1:1.8, which could be attributed to a slightly defected 2D-MoSe2 structure but agrees well with that expected of 1:2 respectively.
Overall, the 2D-MoSe2 utilised in this work to produce the novel electrocatalytic inks has been fully characterised and revealed to comprise of high quality 2D-MoSe2 nanosheets for implementation as an electrocatalyst towards the HER.
3.2 Application of the MoSe2-SPEs towards the HER
Previous work has shown that SPEs can be used as a preferential alternative to GC electrodes as a supporting material towards the HER.1 The typical method of modifying a supporting electrode with a desired electrocatalyst is the drop-casting technique (see Table 1), which has several disadvantages (as outlined in the introduction). In order to overcome these disadvantages we have created novel conductive inks via the incorporation of 5, 10, 20 and 40% 2D-MoSe2 to carbon ink on a % mass ratio (i.e. MP to MI × 100). Subsequently, we have utilised these novel inks to create 2D-MoSe2 containing screen-printed electrodes (MoSe2-SPE) via the screen-printing technique described in the Experimental section, which we now explore towards the HER and examine their electrocatalytic performance for future implementation.It was first essential to benchmark the electrochemical behaviour of a bare/unmodified SPE towards the HER in 0.5 M H2SO4, as is common within the literature.46Fig. 3(A) shows linear sweep voltammetry (LSV) for a bare/unmodified SPE as well as for 5, 10, 20 and 40% MoSe2-SPEs. The bare/unmodified SPE exhibited a HER onset of −880 mV and a current density of 0.095 mA cm−2 at a potential of −0.75 V. As expected, this is far more electronegative than the HER onset of Pt (ca. −0.25 V). The observed small electronegative HER onset potential for Pt is due to it being a pure metal that has a very small binding energy for H+.10 Note that the HER onset is analysed as the potential at which the observed current deviates from the background current by 25 μA cm−2, as is common within the literature.46
 | ||
| Fig. 3 (A) Linear sweep voltammetry (LSV) of a bare/unmodified SPE and 5%, 10%, 20% and 40% MoSe2-SPEs, showing the onset of the HER. Scan rate: 25 mV s−1 (vs. SCE). Solution composition: 0.5 M H2SO4. (B) Tafel analysis; potential vs. ln of current density for the Faradaic section of the LSV presented in (A). (C) Cyclic stability examination of a 10% MoSe2-SPE via LSV (scan rate: 100 mV s−1 (vs. SCE)) was performed between the potential range of 0 to −1.4 V, repeated for 1000 cycles, this figure shows the initial (yellow line), 10th (green line) scans, 100th (dark green) and 1000th scan (black line). | ||
It is clearly observable that all the MoSe2-SPEs have a HER onset potential which occurs at ca. −430 mV (see Fig. 3(A)). This value is significantly less electronegative than that of a bare/unmodified SPE and closer to the optimal of Pt. There is also a substantial increase in the recorded current density with the 5, 10, 20 and 40% MoSe2-SPEs exhibiting a current density at −0.75 V of 1.7, 1.8, 1.9 and 1.5 mA cm−2 respectively, indicating a far greater production of the desired H2 (gas) compared to the bare/unmodified SPE. The decrease in the electronegativity of the HER over-potential and the increase in observed current density indicates that the 2D-MoSe2 incorporated ink is an effective electrocatalyst towards the HER. Whilst the 40% MoSe2-SPE has a comparable HER onset to the other MoSe2-SPEs, it appears to produce H2 (gas) at a slower rate, represented by the smaller current density at −0.75 V.
As the MoSe2-SPEs demonstrate a greater proficiency at catalysing the HER, it can be inferred that the electrochemical reaction mechanism occurring must be altering to account for this. A common method within the literature of assessing the HER mechanism is via Tafel analysis.1 Literature has suggested three possible steps in the reaction, each of which are capable of being the rate-determining step of the HER. The initial H+ discharge step being the Volmer reaction, leading to the following equation:47–49
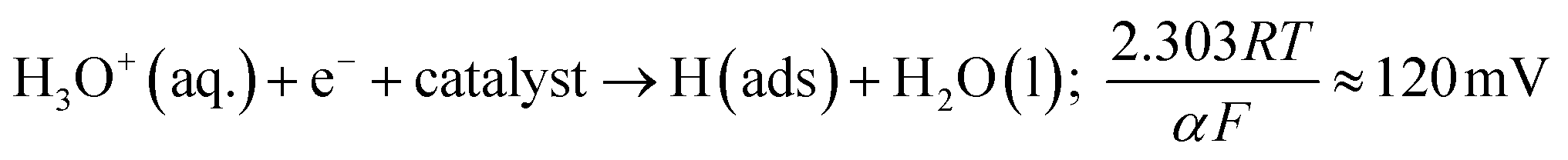
The Volmer step can then be followed by one of two possible steps; either the Heyrovsky step:
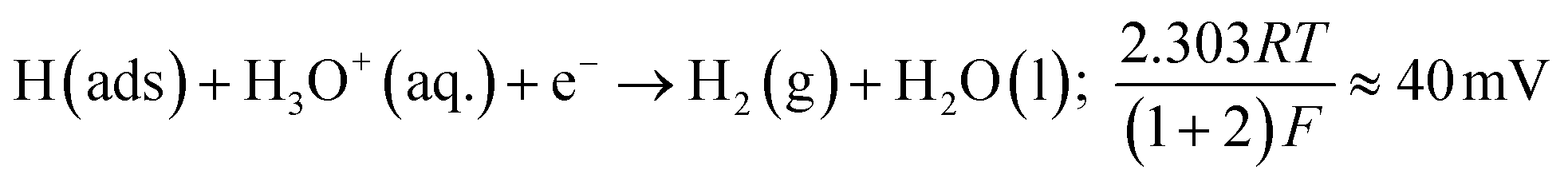

Tafel analysis was performed on the Faradaic sections of the LSVs shown in Fig. 3(A), with the resultant Tafel slopes being exhibited in Fig. 3(B). The Tafel slope values obtained for the bare/unmodified SPE, 5, 10, 20 and 40% MoSe2-SPEs correspond to 120, 49, 47, 63 and 230 mV dec−1. Interpretation of these values suggests that the rate limiting step of the HER reaction mechanism on the SPE is the “adsorption Volmer” step, whereas it is likely the “discharge Heyrosky step” for the 5, 10 and 20% MoSe2-SPEs. This change in the HER mechanism is indicative of a reduction in the free energy barrier of the discharge step, resulting in an improvement of the observed HER activity.45 The 40% MoSe2-SPE Tafel slope value is too large to be accurately described by the Tafel analysis, however the size of the value implies very poor HER activity with the initial step of H+ adsorption being the rate limiting step, with a small surface coverage of adsorbed hydrogen.
It is clear from the above analysis that there is an optimal ratio of 2D-MoSe2 to carbon ink. In this case the optimal ratio is between 10% and 20%. At this optimal ratio it is likely that the structural model of the 2D-MoSe2-ink has proportionally the greatest number of exposed electrocatalytic edge planes to relatively inert basal planes of 2D-MoSe2. Thus, resulting in the least electronegative HER onsets and the highest achievable current densities. Once the percentage mass of 2D-MoSe2 within the ink has exceeded the optimal ratio there is a severe reduction in the observed current density, as observed for the 40% MoSe2-SPE. This reduction in the achievable current density is possibly a result of the structure of 2D-MoSe2 at percentages greater than 20% recombining into bulk forms within the SPE ink and results in an increase in the charge transfer resistance and a smaller number of exposed edge planes for H+ binding.
In order to assess the intrinsic catalytic activity being displayed by the 2D-MoSe2 upon the surface of the MoSe2-SPE on a “per active site basis”, the turn over frequency (ToF) was deduced (the methodology by which the ToF values were deduced is presented within the ESI Fig. 3 and 4†). The resultant ToF values for the 5, 10, 20 and 40% MoSe2-SPEs were found to correspond to 2.70, 1.48, 0.92 and  respectively. It is evident that with an increased percentage of 2D-MoSe2 there is a decrease in the ToF value obtained. This could be a result of larger ratios of 2D-MoSe2 to conductive carbon ink leading to competition for available H+ between the active edge plane sites present on the surface. It could also be as a result of the larger masses of 2D-MoSe2 forming a structure where there is increased shielding of the electronegative Se atoms located at the active edge planes by the relatively inactive basal planes.21
respectively. It is evident that with an increased percentage of 2D-MoSe2 there is a decrease in the ToF value obtained. This could be a result of larger ratios of 2D-MoSe2 to conductive carbon ink leading to competition for available H+ between the active edge plane sites present on the surface. It could also be as a result of the larger masses of 2D-MoSe2 forming a structure where there is increased shielding of the electronegative Se atoms located at the active edge planes by the relatively inactive basal planes.21
It was essential to ascertain whether the printing technique implemented herein resulted in a complete and uniform coverage of 2D-MoSe2 onto a SPE's surface. We therefore employ a Raman mapping technique encompassing the entire surface of a SPE and separately a MoSe2-SPE. At each of the points within the Raman grid a comparison was made between the observed intensity of the peak at 240 cm−1 (characteristic of 2D-MoSe2) and the underlying graphite peak at 1580 cm−1, thus allowing one to observe the coverage effect of the 2D-MoSe2. It is evident upon inspection of Fig. 4 that increasing the percentage mass of 2D-MoSe2 within the graphitic ink, used to produce the MoSe2-SPEs surface, results in an increased intensity of the 240 cm−1 Raman peak. Signifying that there is a positive correlation between the percentage of 2D-MoSe2 within the separate inks and the 2D-MoSe2 on the surface of the electrode. The visible red (the Raman map of a bare/unmodified SPE) coverage in Fig. 4(A) signifies that the incorporation of 5% 2D-MoSe2 into the graphitic ink is not a sufficient mass to result in a complete coverage of the underlying SPE's surface. At 10% 2D-MoSe2 incorporation within the ink, the number of points in which the underlying red is visible has greatly diminished. We therefore suggest that the 10% MoSe2-SPE has complete and uniform coverage upon the electrodes surface. Note that trying to visually assess 2D-MoSe2 on the surface of an SPE via SEM analysis was found to be inconclusive given that the 2D-MoSe2 proved to be indistinguishable from the underlying SPE surface, as evident in ESI Fig. 5.†
 | ||
| Fig. 4 Raman maps showing the surface of (A) 5% MoSe2-SPE, (B) 10% MoSe2-SPE, (C) 20% MoSe2-SPE and (D) 40% MoSe2-SPE. Each point shows the intensity ratio between the sum of the characteristic MoSe2 peak areas (240 cm−1) against the area of the underlying graphite peak (1580 cm−1). The green maps are the MoSe2-SPE and the underlying red in each map represents an unmodified electrode surface. The X and Y axis values correspond to μm. | ||
The intra-repeatability of the MoSe2-SPEs were next evaluated (N = 3). The percentage relative standard deviation (% RSD) for the observed HER onset potential and current density values at −0.75 V were found to correspond to 0.74, 1.41, 1.82 and 1.95% and 5.30, 3.64, 4.68 and 3.91% for the 5, 10, 20 and 40% MoSe2-SPEs respectively. With respect to the observed HER onset, there is clearly a trend of increasing % RSD corresponding to an increase in the percentage of 2D-MoSe2 within the MoSe2-SPEs. We postulate that this is due to a greater percentage of 2D-MoSe2 present leading to a larger number of variations within the orientation of the modified 2D-MoSe2 structure, whereby there will be a different ratio of active edge planes to comparatively inert basal planes.23 The small % RSD values observed for the HER onset and current density give rise to the high/favourable reproducibility of the screen-printing technique utilised herein to produce the MoSe2-SPEs. In order to emphasize the advantages in terms of reproducibility displayed by the MoSe2-SPEs over drop-casting the MoSe2, we examined the RSD value corresponding to the current density values at −0.75 V for bare SPEs with 400 mg cm−2 of MoSe2. This was observed to be 11.43%, which is significantly larger than any of the RSD values (see above) displayed by a MoSe2-SPE.
Electrochemical impedance spectroscopy (EIS) was used to determine the impedance of the electrode system as the percentage of 2D-MoSe2 increased within the SPE ink. ESI Fig. 6† indicates that the charge transfer resistance (Ω) for all of the MoSe2-SPEs decreased in comparison to the bare/unmodified SPE, which had a Ω value of 6.7 × 103 Ω. The Ω value decreased from 4.1 × 103 to 2.3 × 103 Ω with a change in the percentage 2D-MoSe2 ink composition from 5% to 10% respectively. A subsequent further increase was observed corresponding with the 20% and 40% to 3.0 and 4.3 Ω respectively. The 40% MoSe2-SPE exhibited the highest Ω value of all the MoSe2-SPEs examined herein, which again is attributed to the MoSe2 forming bulk structures and resulting in resistance changes. Error values for the aforementioned results were recorded as 6.9 × 10−1, 4.4 × 10−1, 5.9 × 10−1, 3.4 × 10−1 and 7.7 × 10−1 Ω for the SPE, 5, 10, 20 and 40% MoSe2-SPEs respectively. The EIS data presented above supports the prior inference that MoSe2-SPEs are effective electrocatalysts for the HER and that the optimal mass of ink modification is ca. 10%.
Next it was essential to assess the electrocatalytic stability of MoSe2-SPEs towards the HER. This is a vital consideration in the utilisation of this technology for industrial applications where concerns over stability and longevity are at the forefront. Using a 10% MoSe2-SPE as a representative example for all of the MoSe2-SPEs, it is evident upon inspection of Fig. 3(C) that the 10% MoSe2-SPE retains its HER onset of ca. −430 mV. This electrode exhibits a 31.6% increase in the current density from 103.1 to 148.6 μA at the 1st to the 1000th repeat scan (based upon the current at −0.75 V and a cyclic voltammetry (CV) potential range of 0 to −1.4 V). The observed increase in current density could be a result of several contributing factors, such as the prolonged exposure of the MoSe2-SPE to the acidic electrolyte, partially corroding polymers found within the carbon ink and thus leading to the exposure of a greater number of 2D-MoSe2 active edge planes. Another possible explanation is the chemical alteration of the electrodes surface leading to more favourable HER activity. ESI Table 1† shows the elemental compositions, deduced via XPS, of a 10% MoSe2-SPE pre and post the 1000 repeat scans described above. There is a trace amount of Pt (0.04%) on the post 1000 scan MoSe2-SPE that is not present on the pre-scan MoSe2-SPE, which may have contributed to the increased current. ESI Fig. 7† shows that the increase in achievable current cannot solely be prescribed to Pt contamination as there is also an increase of 34.7% in achievable current when a carbon counter is utilised during the cycling stability test. The Pt contamination likely arose from in situ deposition of the Pt counter onto the working electrode via the mechanism described by Gottlieb et al.50 This is supported by ESI Table 1† which shows the lack of any Pt in the elemental analysis of a MoSe2-SPE surface which underwent a 1000 scan cycling stability study whilst utilising a carbon counter, rather than a Pt counter. Further work to ascertain the exact reason of the observed increase in current density would be of interest and is planned for future studies. A small reduction peak at −0.3 V is observed on the 1000th scan, which is not present in the 1st, 10th and 100th scan. This observation can be attributed to the oxygen reduction reaction occurring, as oxygen is likely to have permeated the 0.5 M H2SO4 electrolyte over the course of the cycling stability experiment.46 In order to further explore the stability of the 10% MoSe2-SPE, we performed chronoamperometry at −0.75 V for 36000 seconds as reported within ESI Fig. 8.† The achieved maximum current was 174 μA at 1214 seconds after which there was a gradual decrease in the current to 160 μA at 36000 seconds. It was essential to assess whether the observed stability of the 10% 2D-MoSe2 was due to the incorporation of the 2D-MoSe2 into the bulk SPE ink and that drop-casting the 2D-MoSe2 would not lead to a similar stability being observed. ESI Fig. 9† shows the observed CVs of a SPE that had 400 mg cm−2 of 2D-MoSe2 drop-cast onto its surface and then subsequently underwent a cycling stability test under identical conditions as implemented above. It is clear upon inspection of this figure that the HER onset potential remained stable at ca. −480 mV (vs. SCE) across the duration of the 1000 scans, however there is a 27.4% reduction evident in the observed current (from 41.9 μA at the 1st scan to 30.3 μA at the 1000th scan, based upon the current at −0.75 V and a CV potential range of 0 to −1.4 V). This reduction of the achievable current is likely due to the delamination of the 2D-MoSe2 from the electrode's surface over the course of 1000 scans. Given the results of the cycling stability studies, it can be interfered that incorporating the 2D-MoSe2 into the bulk SPE ink (rather then drop-casting the 2D-MoSe2) results in a greater stability of the achievable current. Of note within the literature is the inconsistent approach to testing the electrochemical stability of a potential electrocatalyst towards the HER. Each separate study alters the potential range, duration (time) and number of cycles implemented. This point is particularly evident upon inspection of Table 1. Future studies should endeavour to produce and adhere to a universal literature standard methodology that not only would vigorously test the cycling stability of an electrocatalyst, but also allow for easy comparisons to be established between distinct studies.
SPEs have several advantages over traditional carbon based electrodes, such as their reproducibility, tailorability and vast economy of scales. They have however historically lacked in comparable and competitive HER activity. The results presented herein demonstrate that this problem can addressed via the utilisation of 2D-MoSe2 within printable inks. Fabricated MoSe2-SPEs exhibit low HER overpotentials and large current densities, with the distinct advantage of having the desired electrocatalyst incorporated into their ink/structure resulting in excellent cycling stability. Whilst the MoSe2-SPEs still have a more electronegative HER onset potential compared to Pt based electrodes, their low production cost and short manufacturing time make them desirable alternatives for situations where the efficiency of Pt based electrodes is not an adequate trade-off for their high cost. It would be of interest in future studies to alter the size, morphology and surface composition of the MoSe2 utilised to produce the MoSe2-inks. Then to observe whether making these variations to the MoSe2 has an effect upon their ability to catalyse the HER. The electrodes presented in this work mitigate the need to post hoc modify an electrode via the drop-casting technique (that has previously been shown to demonstrate poor cycling stability).1
4. Conclusions
We have reported, for the first time, the production of a 2D-MoSe2 electrocatalytic ink which can be utilised to produce screen-printed electrodes/surfaces that exhibit low HER onset, high reproducibility and excellent cycling stability. The use of screen-printing allows a mass scalable approach to produce 2D-MoSe2 electrocatalytic surfaces. Incorporating the known electrocatalyst, 2D-MoSe2, into the screen-printed ink at an optimal ratio of 10% 2D-MoSe2 mass (90% carbon ink) results in a HER onset, Tafel value and a turn over frequency of ca. −460 mV (vs. SCE), 47 mV dec−1 and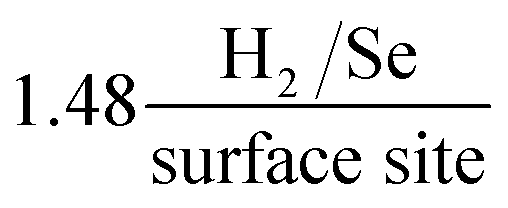 respectively. These values show that the 10% MoSe2-SPE is significantly more electrocatalytic towards the HER in comparison to a bare/unmodified SPE, which has a HER onset and Tafel value of −880 mV (vs. SCE) and 120 mV dec−1 respectively. The fabricated MoSe2-SPEs exceed, in terms of the HER activity, any previous studies that have utilised SPEs towards the HER.
respectively. These values show that the 10% MoSe2-SPE is significantly more electrocatalytic towards the HER in comparison to a bare/unmodified SPE, which has a HER onset and Tafel value of −880 mV (vs. SCE) and 120 mV dec−1 respectively. The fabricated MoSe2-SPEs exceed, in terms of the HER activity, any previous studies that have utilised SPEs towards the HER.
This work demonstrates via the use of Raman mapping and cycling stability that the herein produced MoSe2-SPEs have a uniform and stable coverage of 2D-MoSe2. Exhibiting no degradation in observed HER onset over the course of 1000 repeat scans, with, in fact, a 31.6% increase in the achievable current density over this period. This remarkable stability, which arises from the incorporation/anchoring of the 2D-MoSe2 into the SPE ink, exceeds the stability observed when 400 mg cm−2 of 2D-MoSe2 was drop-casted onto a SPE (which, displayed a 27.4% decrease over the same experimental duration), as well as the studies highlighted within this paper that utilise the drop-casting technique to modify their electrodes (see Table 1).
We have provided insights into the electrochemistry occurring and the HER mechanism prevalent at the novel MoSe2-SPEs, showing that they have clear potential to be utilised as beneficial alternatives to GC and Pt in future research and industrial applications. Few studies have such a direct transferability to their desired field whilst also opening a vast number of avenues for future research that seeks to utilise 2D-nanomaterials in order to produce cheap, stable and tailorable electrocatalytic electrodes. This work can be readily extended to allow the incorporation of other 2D electrocatalytic materials; this fabrication approach provides substantial stability improvements over the traditionally employed drop-casting technique. This approach can be readily extended to allow the incorporation of other 2D electrocatalytic materials.
Acknowledgements
The authors acknowledge a British Council Institutional Link grant (No. 172726574) for the support of this research as well as the Ramsay Memorial Fellowships Trust and the Engineering and Physical Sciences Research Council (Ref. EP/N001877/1).References
- S. J. Rowley-Neale, D. A. C. Brownson, G. C. Smith, D. A. G. Sawtell, P. J. Kelly and C. E. Banks, Nanoscale, 2015, 7, 18152–18168 RSC.
- A. J. López-Menéndez, R. Pérez and B. Moreno, J. Environ. Manage., 2014, 145, 368–373 CrossRef PubMed.
- Y. Zhao, Y. Zhang, Z. Yang, Y. Yan and K. Sun, Sci. Technol. Adv. Mater., 2013, 14, 043501 CrossRef PubMed.
- T. Wang, L. Liu, Z. Zhu, P. Papakonstantinou, J. Hu, H. Liu and M. Li, Energy Environ. Sci., 2013, 6, 625–633 CAS.
- M. Gratzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef PubMed.
- J. P. Barton and D. G. Infield, IEEE Transactions on Energy Conversion, 2004, 19, 441–448 CrossRef.
- F. Blaabjerg, Z. Chen and S. B. Kjaer, IEEE Trans Power Electron, 2004, 19, 1184–1194 CrossRef.
- J. Wu, X. Z. Yuan, J. J. Martin, H. Wang, J. Zhang, J. Shen, S. Wu and W. Merida, J. Power Sources, 2008, 184, 104–119 CrossRef CAS.
- M. Jouin, R. Gouriveau, D. Hissel, M. C. Péra and N. Zerhouni, IFAC-PapersOnLine, 2015, 48, 26–31 CrossRef.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- M. G. Schultz, T. Diehl, G. P. Brasseur and W. Zittel, Science, 2003, 302, 624–627 CrossRef CAS PubMed.
- C. Xu, S. Peng, C. Tan, H. Ang, H. Tan, H. Zhang and Q. Yan, J. Mater. Chem. A, 2014, 2, 5597–5601 CAS.
- M. Gara and R. C. Compton, New J. Chem., 2011, 35, 2647–2652 RSC.
- W. Sheng, M. Myint, C. G. Chen and Y. Yan, Energy Environ. Sci., 2013, 6, 1509–1512 CAS.
- Y. Liu, H. Yu, X. Quan, S. Chen, H. Zhao and Y. Zhang, Sci. Rep., 2014, 4, 6843 CrossRef CAS PubMed.
- A. Abbaspour and E. Mirahmadi, Fuel, 2013, 104, 575–582 CrossRef CAS.
- D. Y. Chung, S.-K. Park, Y.-H. Chung, S.-H. Yu, D.-H. Lim, N. Jung, H. C. Ham, H.-Y. Park, Y. Piao, S. J. Yoo and Y.-E. Sung, Nanoscale, 2014, 6, 2131–2136 RSC.
- S. Ji, Z. Yang, C. Zhang, Z. Liu, W. W. Tjiu, I. Y. Phang, Z. Zhang, J. Pan and T. Liu, Electrochim. Acta, 2013, 109, 269–275 CrossRef CAS.
- C. Ataca, M. Topsakal, E. Aktürk and S. Ciraci, J. Phys. Chem. C, 2011, 115, 16354–16361 CAS.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS PubMed.
- H. Wang, X. Wang, L. Wang, J. Wang, D. Jiang, D. Li, Y. Zhang, H. Zhong and Y. Jiang, J. Phys. Chem. C, 2015, 119, 10197–10205 CAS.
- J. Morales, J. Santos and J. L. Tirado, Solid State Ionics, 1996, 83, 57–64 CrossRef CAS.
- S. Mao, Z. Wen, S. Ci, X. Guo, K. Ostrikov and J. Chen, Small, 2015, 11, 414–419 CrossRef CAS PubMed.
- C. Tsai, K. Chan, F. Abild-Pedersen and J. K. Norskov, Phys. Chem. Chem. Phys., 2014, 16, 13156–13164 RSC.
- C. W. Foster, R. O. Kadara and C. E. Banks, Screen-Printing Electrochemical Architectures, Springer, Berlin, 2016 Search PubMed.
- J. Zhou, H. Xiao, B. Zhou, F. Huang, S. Zhou, W. Xiao and D. Wang, Appl. Surf. Sci., 2015, 358, 152–158 CrossRef CAS.
- X. Zhou, Y. Liu, H. Ju, B. Pan, J. Zhu, T. Ding, C. Wang and Q. Yang, Chem. Mater., 2016, 28, 1838–1846 CrossRef CAS.
- Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/aldrich/778087?lang=en%26region=GB, accessed 07/09/2016.
- A. B. Laursen, A. S. Varela, F. Dionigi, H. Fanchiu, C. Miller, O. L. Trinhammer, J. Rossmeisl and S. Dahl, J. Chem. Educ., 2012, 89, 1595–1599 CrossRef CAS.
- D. Marin, F. Medicuti and C. Teijeiro, J. Chem. Educ., 1994, 71, A277 CrossRef CAS.
- N. A. Choudry, D. K. Kampouris, R. O. Kadara and C. E. Banks, Electrochem. Commun., 2010, 12, 6–9 CrossRef CAS.
- L. R. Cumba, J. P. Smith, D. A. C. Brownson, J. Iniesta, J. P. Metters, D. R. D. Carmo and C. E. Banks, Analyst, 2015, 140, 1543–1550 RSC.
- C. W. Foster, J. Pillay, J. P. Metters and C. E. Banks, Sensors, 2014, 14, 21905–21922 CrossRef CAS PubMed.
- C. W. Foster, J. P. Metters and C. E. Banks, Electroanalysis, 2013, 25, 2275–2282 CAS.
- J. P. Metters, M. Gomez-Mingot, J. Iniesta, R. O. Kadara and C. E. Banks, Sens. Actuators, B, 2013, 177, 1043–1052 CrossRef CAS.
- F. E. Galdino, C. W. Foster, J. A. Bonacin and C. E. Banks, Anal. Methods, 2015, 7, 1208–1214 RSC.
- S. J. Rowley-Neale, D. A. C. Brownson and C. E. Banks, Nanoscale, 2016, 8, 15241–15251 RSC.
- Z. Lei, S. Xu and P. Wu, Phys. Chem. Chem. Phys., 2016, 18, 70–74 RSC.
- Innovation in Surface Spectroscopy and Microscopy Systems, http://www.specs.de/cms/front_content.php?idcat=209, accessed 20/03/2016.
- M. P. Seah and S. J. Spencer, J. Electron Spectrosc. Relat. Phenom., 2006, 151, 178–181 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- CasaXPS, http://www.casaxps.com/, accessed 2/02/2016.
- D. Nam, J.-U. Lee and H. Cheong, Sci. Rep., 2015, 5, 17113 CrossRef CAS PubMed.
- A. F. Khan, D. A. C. Brownson, E. P. Randviir, G. C. Smith and C. E. Banks, Anal. Chem., 2016, 88, 9729–9737 CrossRef CAS PubMed.
- H. Wang, D. Kong, P. Johanes, J. J. Cha, G. Zheng, K. Yan, N. Liu and Y. Cui, Nano Lett., 2013, 13, 3426–3433 CrossRef CAS PubMed.
- S. J. Rowley-Neale, D. A. C. Brownson, J. M. Fearn, G. C. Smith, X. Ji and C. E. Banks, Nanoscale, 2016, 8, 14767–14777 RSC.
- N. M. Marković, B. N. Grgur and P. N. Ross, J. Phys. Chem. B, 1997, 101, 5405–5413 CrossRef.
- N. M. Marković and P. N. Ross Jr, Surf. Sci. Rep., 2002, 45, 117–229 CrossRef.
- A. B. Laursen, S. Kegnaes, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577–5591 CAS.
- E. Gottlieb, M. Kopeć, M. Banerjee, J. Mohin, D. Yaron, K. Matyjaszewski and T. Kowalewski, ACS Appl. Mater. Interfaces, 2016, 8, 21531–21538 CAS.
- H. Tang, K. Dou, C.-C. Kaun, Q. Kuanga and S. Yang, J. Mater. Chem. A, 2014, 2, 360–364 CAS.
- D. Kong, H. Wang, J. J. Cha, M. Pasta, K. J. Koski, J. Yao and Y. Cui, Nano Lett., 2013, 13, 1341–1347 CrossRef CAS PubMed.
- Q. Gong, L. Cheng, C. Liu, M. Zhang, Q. Feng, H. Ye, M. Zeng, L. Xie, Z. Liu and Y. Li, ACS Catal., 2015, 5, 2213–2219 CrossRef CAS.
- C. H. Mu, H. X. Qi, Y. Q. Song, Z. P. Liu, L. X. Ji, J. G. Deng, Y. B. Liao and F. Scarpa, RSC Adv., 2016, 6, 23–30 RSC.
- Y. D. Liu, L. Ren, Z. Zhang, X. Qi, H. X. Li and J. X. Zhong, Sci. Rep., 2016, 6, 9 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6se00115g |
| This journal is © The Royal Society of Chemistry 2017 |