
Photoelectrochemical H2 evolution using TiO2-coated CaFe2O4 without an external applied bias under visible light irradiation at 470 nm based on device modeling†
Shintaro
Ida
 *ac,
Kara
Kearney
bc,
Takamitsu
Futagami
a,
Hidehisa
Hagiwara
ac,
Takaaki
Sakai
a,
Motonori
Watanabe
c,
Angus
Rockett
cd and
Tatsumi
Ishihara
ac
*ac,
Kara
Kearney
bc,
Takamitsu
Futagami
a,
Hidehisa
Hagiwara
ac,
Takaaki
Sakai
a,
Motonori
Watanabe
c,
Angus
Rockett
cd and
Tatsumi
Ishihara
ac
aDepartment of Applied Chemistry, Faculty of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: s_ida@chem.kumamoto-u.ac.jp
bDepartment of Materials Science and Engineering, University of Illinois, 1304 West Green Street, Urbana, IL 61801, USA
cInternational Institute for Carbon Neutral Energy Research (I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
dDepartment of Metallurgy and Materials Engineering, Colorado School of Mines, 201 Hill Hall, 1500 Illinois Street, Golden, CO 80401, USA
First published on 9th March 2017
Abstract
CaFe2O4 (CFO) can be used as a photocathode to evolve H2 from water in a photoelectrochemical cell. However, CFO degrades during operation and an external voltage is necessary for PEC H2 evolution because the onset potential is less than the potential required for water oxidation considering the overpotential at the counter electrode. In order to develop a reliable CFO electrode with a greater onset potential, improvement of chemical stability and suppression of surface recombination is necessary. In this study, a chemically stable electrode structure with a greater onset potential was achieved by coating the [00l]-oriented CFO with a thin layer of titanium dioxide (TiO2). A CFO|TiO2 electrode was designed using a device simulator. The simulation results predict that coating CFO with TiO2 produces a positive or negative shift in the onset potential under visible and ultraviolet light irradiation, respectively. The experimental onset potentials matched the simulation prediction. The observed onset potential for TiO2-coated CFO was around (1.6 V vs. RHE) under visible light (470 nm) and 0.9 V under ultraviolet light (300 nm), compared to 1.2–1.3 V vs. RHE for a bare CFO electrode. The onset potential (1.6 V) under visible light irradiation is the most positive onset potential among the oxide photocathodes ever reported for PEC water splitting. Using the TiO2-coated CFO as the photocathode and RuO2-loaded Pt as the anode, stable photocurrent was observed under 470 nm excitation without an external voltage and evolution of H2 from the system was confirmed.
Introduction
Hydrogen (H2) is an ideal energy carrier for CO2-free combustion. However, most H2 is produced from fossil fuels by steam reforming, which results in substantial CO2 emission. Photoelectrochemical (PEC) water-splitting is a promising solar-driven, carbon-free H2 production method.1–3 The first report of PEC H2 production utilized a wide gap n-type TiO2–Pt system limited to UV absorption.4 Subsequent studies have replaced TiO2 with materials that can absorb longer wavelengths of light such as TaON,5,6 BiVO4,7–10 Ta3N5,6,11,12 SiC,13,14 CuOx,15 Rh-doped SrTiO3,16 Fe2O3,17–21 CaFe2O4,22–30 LaFeO331 and ZnFe2O4.32 However, PEC systems have more practical problems to be addressed such as efficiency, stability, and cost.CaFe2O4 (CFO) is an attractive material composed of Earth-abundant elements with low handling and purification costs.33 CFO is a p-type semiconductor (bandgap: 1.9 eV) that can function as a photocathode in a PEC cell.34 The onset potential for CFO photocathodic current is reported to be ∼1.2 V vs. a reference hydrogen electrode (RHE), which is higher than other p-type semiconductors such as CuOx, SiC, and chalcogenide-based materials.35–38 However, an external applied voltage is necessary to decompose water using a two-electrode system such as CFO and Pt. To produce H2 and O2 without an external applied bias, the onset potential for the CFO electrode must be increased to ∼1.5–1.6 V vs. RHE considering the overpotential required for water oxidation at the counter electrode.
Coating a photoelectrode with a metal oxide such as TiO239,40 or ZnO15 is often used to shift the onset potential for p-type semiconducting materials in addition to effectively protecting the surface. However, depending on the deposition conditions of the metal oxide,41 the onset potential may be reduced rather than increased. The magnitude and direction of the induced shift depends on the generation, recombination, and charge transport across the metal oxide, which are sensitive to electrical properties such as mobility and charge carrier density.
In this study, a PEC system consisting of a TiO2-coated CFO photocathode (CFO|TiO2) and Pt counter electrode was designed and demonstrated to evolve H2 without an external applied bias. The design was developed using wxAMPS – a free device physics simulation package previously used to model the performance of dye-sensitized solar cells and functionalized Si(111) photoelectrodes, among other applications.42–47 The simulation results predicted that excitation with wavelengths exclusively >420 nm (i.e. avoiding excitation of the TiO2) would increase the onset potential of the CFO|TiO2 photocathode past ∼1.5–1.6 V vs. RHE and produce H2 without an external applied bias. The prediction was validated experimentally for a CFO|TiO2–Pt system under 470 nm excitation.
Experimental
Materials synthesis
Results and discussion
Fig. 1a shows the current–potential curves of a bare CFO electrode in 0.1 M NaOH solution under chopped AM1.5 solar illumination. The onset potential for photocathodic current was around 1.30 V vs. RHE. Although the onset potential is slightly larger than the water oxidation potential (1.23 V) it is still not large enough to realize PEC H2 evolution without an external applied voltage in a two-electrode system such as CFO–Pt. The necessary onset potential would be ∼1.5–1.6 V vs. RHE due to the overpotential of the water oxidation at the Pt electrode. However, the valence band position determined from the photoelectron spectroscopy in air spectrum was 1.58 V vs. NHE as shown in Fig. 1b. This indicates that increasing the onset potential to >1.5 V is indeed possible in theory, implying that PEC H2 evolution without an external applied voltage can be realized.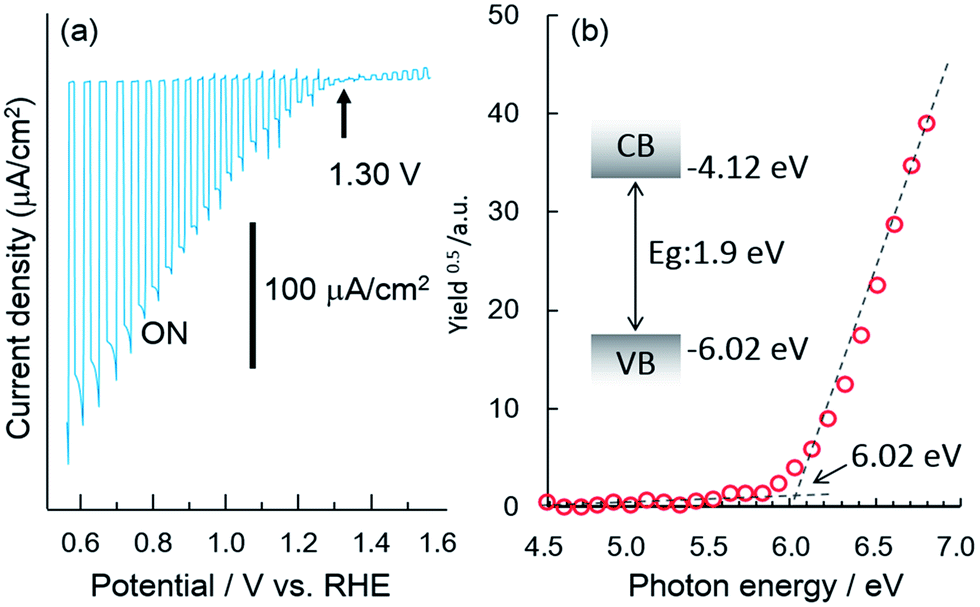 | ||
| Fig. 1 (a) Current–potential curve of a bare CFO electrode in 0.1 M NaOH solution under chopped illumination (solar simulator, AM1.5) and (b) photoelectron spectroscopy in air spectrum and band structure model of CaFe2O4. | ||
To develop a method of achieving a larger (more positive) onset potential, a drift-diffusion solid state device simulator, wx-AMPS was used to predict the performance. In this simulation, information of band structure (bandgap: 1.9 eV, CBM: −0.32 V vs. NHE, VBM: 1.58 V vs. NHE), carrier concentration, mobility, defects, and surface recombination rate is necessary. The input values for the carrier concentration (NA = 1016 cm3) and hole mobility (μp = 0.05 cm2 V−1 s−1) were estimated from the Mott–Schottky plot as shown in Fig. S1.† To model trap states in the TiO2, donor defects were added at midgap with a concentration of 1016 cm−3 and capture cross sections of 5 × 1012 cm2 for both electrons and holes. In general, it was difficult to measure the actual surface recombination rate. Therefore, in this study, the surface recombination rate of CFO was estimated by comparing the actual onset potential for photocathodic current of CFO with the simulated open circuit voltage as a function of surface recombination rate. Fig. 2a shows the simulated relationship between open circuit voltage and surface recombination rate obtained using wxAMPS. The open circuit voltage decreased with increasing surface recombination rate, which indicates that less surface recombination will improve the open circuit voltage. For a surface recombination rate between 104 and 108 cm2, the open circuit voltage remained constant at about 1.3 V vs. NHE, which corresponded to the actual onset potential (1.30 V vs. NHE) for photocathodic current of CFO in Fig. 1a. In the following simulations, the surface recombination rate was set as 107 cm s−1, which is approximately equal to the thermal velocity of an electron. Fig. 2b shows current–potential curves with or without light illumination of CaFe2O4 simulated using the above-mentioned input parameters with the photocurrent normalized at 0.0 V as a reference. Additional input parameters are summarized in Table S1.†
 | ||
| Fig. 2 (a) Simulated open circuit voltage (onset potential) as a function of surface recombination rate of CaFe2O4 and (b) simulated current–potential curve of CaFe2O4 (surface recombination rate: 107 cm s−1). | ||
In this study, a TiO2 passivation layer was used to improve the onset potential for photocathodic current. First, current–potential curves of TiO2-coated CFO at different wavelengths of excitation were simulated using wxAMPS. The simulated photocurrent versus applied potential under 300, 470, and 532 nm excitations are shown in Fig. 3. The calculated band diagrams of CFO and TiO2-coated CFO photoelectrodes are shown in ESI Fig. S2 and S3.† The photocurrents were normalized using the photocurrent at 0.0 V as a reference. The open circuit voltage (Voc) for bare CFO was ∼1.35 V vs. NHE for all three wavelengths of excitation, which agrees well with the experimentally observed onset potential of 1.30 V. For the TiO2-coated CFO photoelectrode, the Voc was strongly influenced by the wavelength of excitation. The Voc for 300 nm, 470 nm, and 532 nm excitation was 1.06 V, 1.92, and 1.87 V vs. NHE, respectively. Therefore, the TiO2 induces a decrease in Voc for 300 nm excitation (UV light) and an increase in the Voc for 470/532 nm excitation (visible light). The simulation results indicate that the Voc of TiO2-coated CFO under longer wavelength irradiation is sufficient to realize PEC H2 evolution without an external applied voltage.
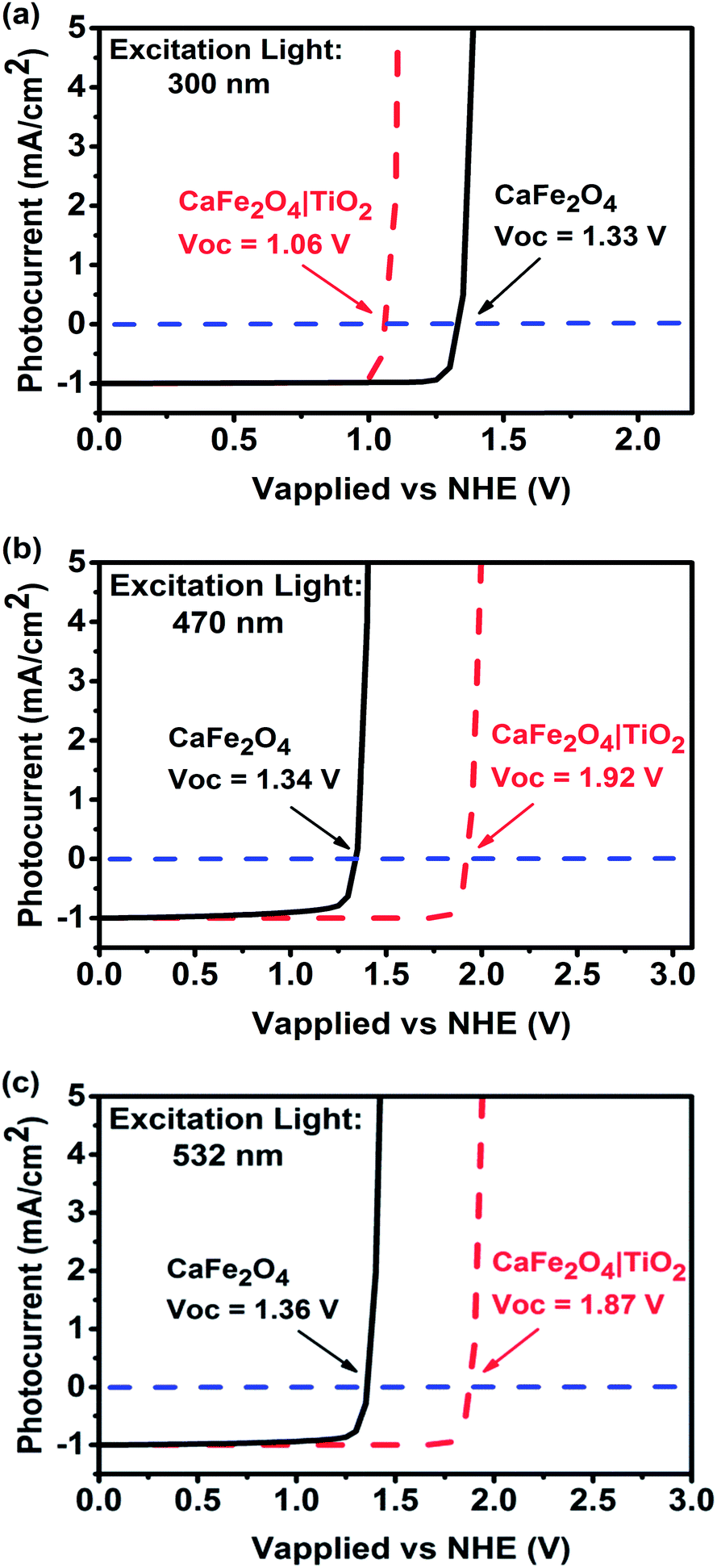 | ||
| Fig. 3 Simulated photocurrent versus applied voltage for CFO and TiO2-coated CFO electrodes under (a) 300, (b) 470, and (c) 500 nm excitation. | ||
We propose the following explanation for the change in sign of the photo-induced shift in Voc by the TiO2 coating under UV and visible light irradiation. Under UV illumination, the photon energy is sufficient to generate carriers in the TiO2. These accumulate in surface traps resulting in an electric field in the TiO2 that reduces the Voc. Under visible light illumination, the photon energy is insufficient to excite carriers in the TiO2 and all the carrier generation occurs in the CFO. The field in the CFO efficiently separates the electron–hole pairs, and the conduction and valence band offsets between the CFO and TiO2 create an electron-selective contact, resulting in a large Voc. The proposed mechanism for the TiO2-coated CFO photoelectrode under visible light excitation is shown in Fig. 4. Additionally, the simulations shown in Fig. 2a reveal that a lower Voc may be caused by a high surface recombination rate. In solar cell technology, it is well known that introducing a passivation layer is effective at reducing the surface recombination rate. Therefore, the presence of TiO2 may contribute to a higher Voc by lowering the surface recombination rate of the electrode.
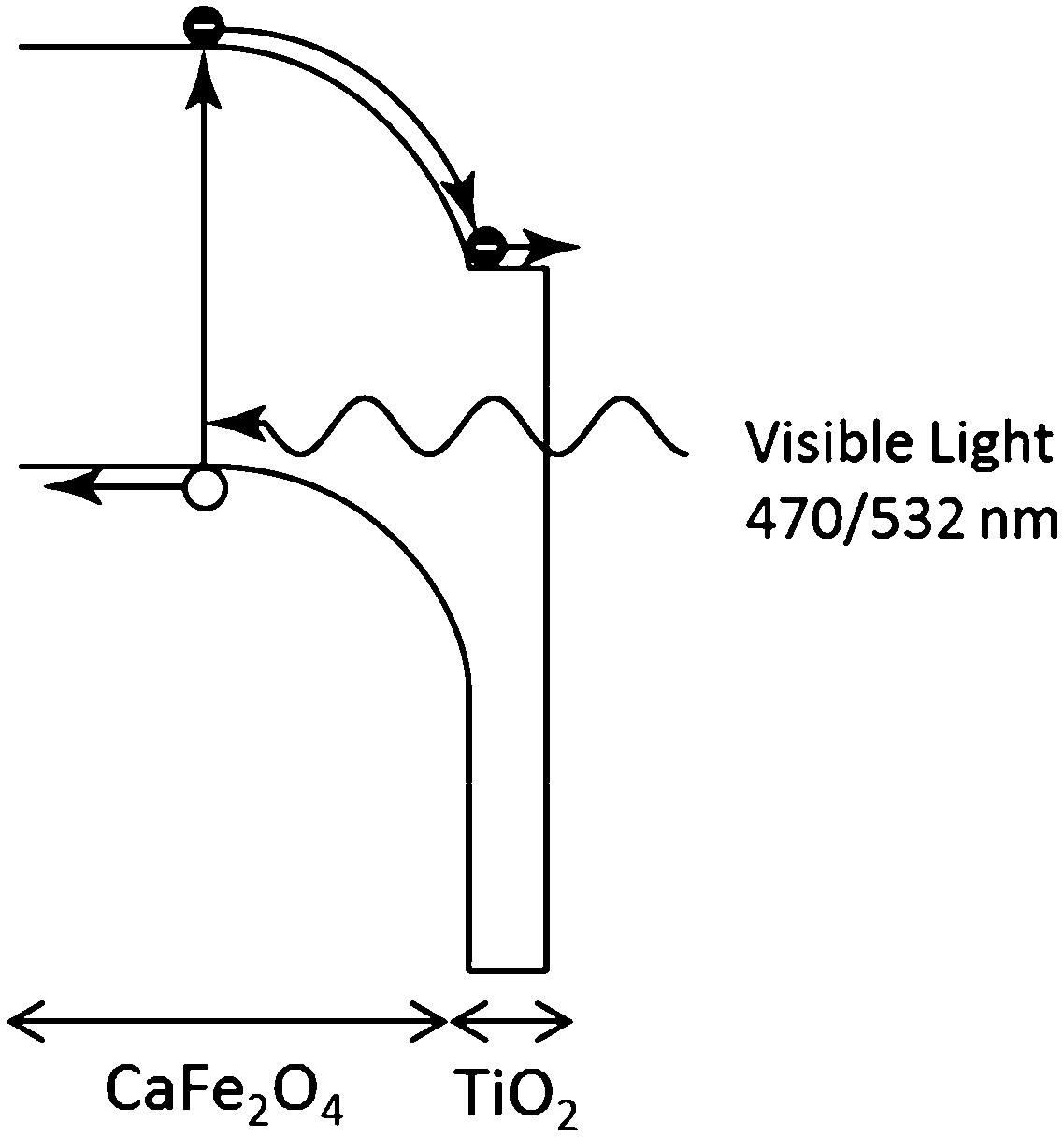 | ||
| Fig. 4 Proposed charge transfer mechanism for TiO2-coated CaFe2O4 photoelectrode under visible light illumination. | ||
To validate the simulation prediction, the PEC performance of a TiO2 (20 nm)-coated CFO electrode was experimentally observed. Fig. 5 shows a structural model and typical SEM images of the CFO|TiO2 electrode. A dense 50 μm film of CFO was prepared on a Pt substrate and 20 nm of TiO2 was deposited on top. The resistivity of TiO2 films deposited by PLD at room temperature was 0.13 Ωm, which is much lower than that of bulk TiO2 (>109 Ωm). These results indicate that a TiO2 layer with a high concentration of donor-like defects was prepared. Fig. 6a shows out-of-plane XRD pattern of TiO2 (20 nm)-coated CFO electrode. One strong diffraction peak assigned to (004) and/or (302) faces of CFO was observed (ICSD-16695, CaFe2O4). Fig. 6b shows pole figure for diffraction peak at 33.4°. Many diffraction spots were observed at around 60 degrees. Since the (004) face is inclined at 60° with respect to the (302) face, the diffraction spots at around 60° were assigned to (004) and/or (302) faces. A spot-like pattern in the pole figure means that the CFO film is composed of crystal grains with a relatively large lateral size. To confirm whether the CFO film is oriented to the [004] or [302] direction, wide-angle reciprocal space mapping of the CFO film was measured. Fig. 6c shows the reciprocal space mapping of the CFO film. Characteristic diffraction spots assigned to (112), (113), (114), (115) and (116) faces were observed in the mapping, which is a characteristic reciprocal space mapping assigned to (004)-oriented CFO. This indicates that the diffraction pattern at around 33.4° in the XRD pattern (Fig. 6a) is assigned to the (004) face. Thus, it was found that the CFO film is mainly oriented to [001] direction as shown in the structural image of the CFO film in the inset of Fig. 6a.
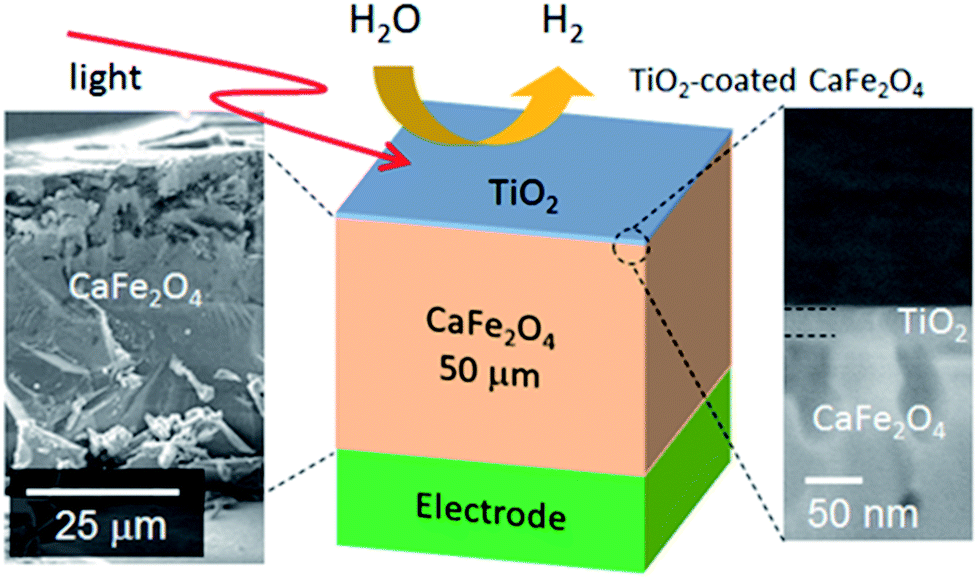 | ||
| Fig. 5 Structural model and SEM image of TiO2-coated CFO electrode. | ||
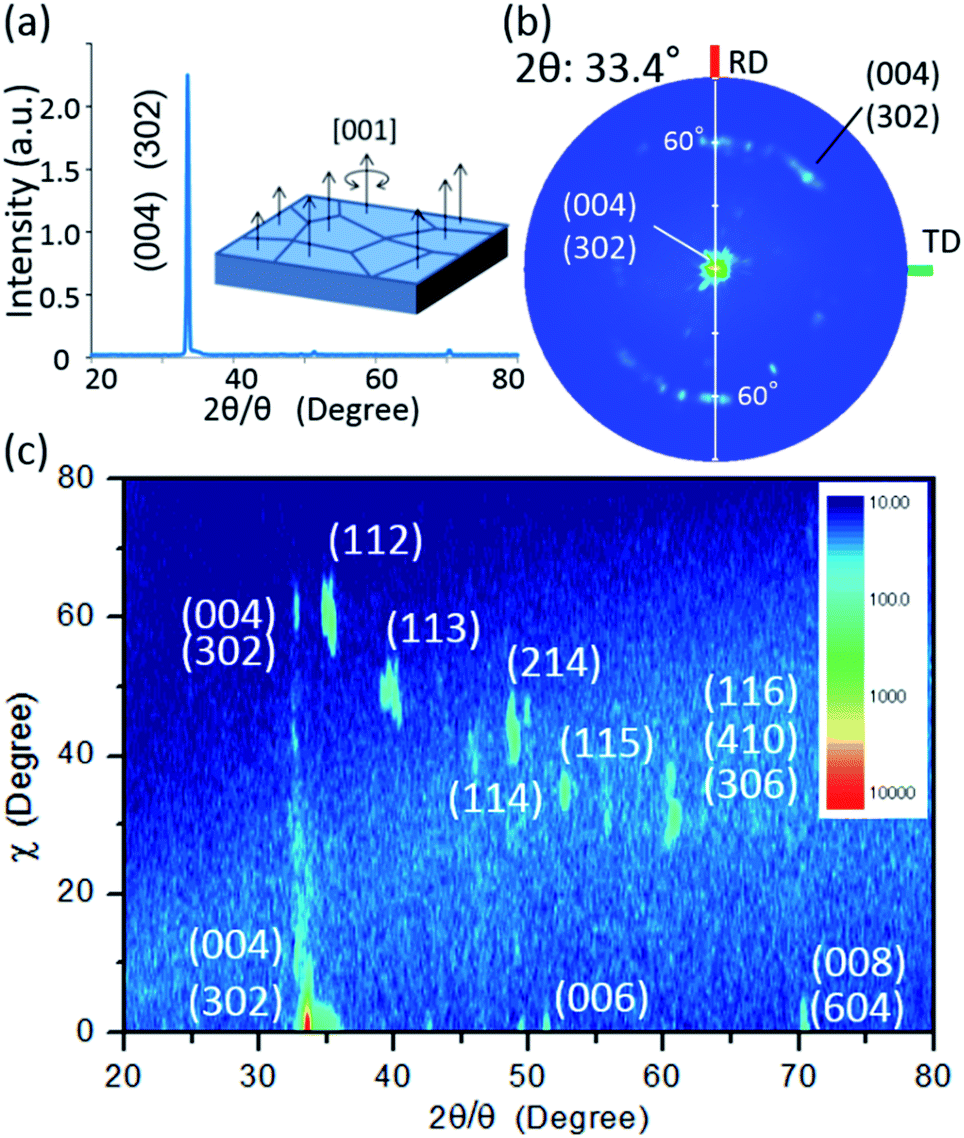 | ||
| Fig. 6 (a) Out-of-plane XRD pattern with an inset of the structural image of CFO, (b) pole figure for 33.4°, (c) wide-angle reciprocal mapping. | ||
With regard to the crystal structure of the TiO2 film prepared by PLD, the diffraction pattern assigned to TiO2 was not observed in the out-of-plane XRD. Therefore, the crystal structure of the prepared TiO2 was analyzed by in-plane XRD. Fig. 7 shows the in-plane XRD pattern of the TiO2-coated CFO electrode. Although the diffraction patterns assigned to (210), (410) and (610) faces for CFO were observed, there is no clear XRD pattern for the TiO2, indicating that it has an amorphous structure.
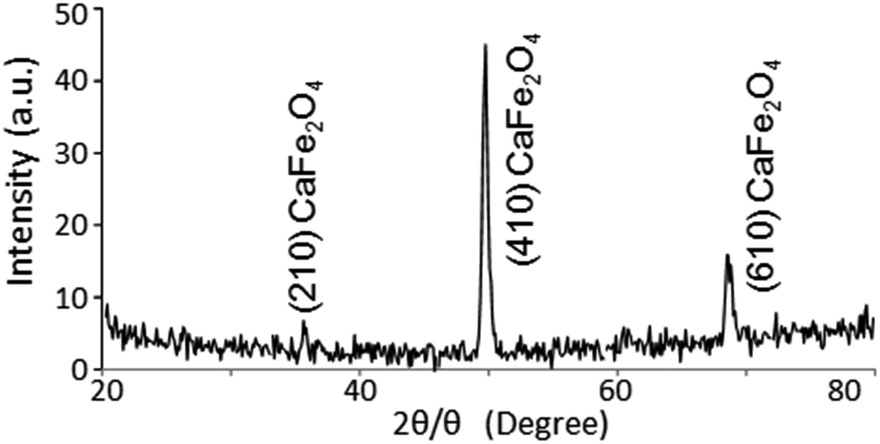 | ||
| Fig. 7 In-plane XRD pattern of TiO2 (20 nm)-coated CaFe2O4. | ||
The surface coverage of the TiO2 film was analyzed using XPS. Fig. 8 shows survey and narrow Ti 2p XPS spectra of CFO and TiO2-coated CFO. The bare CFO electrode showed signals assigned to Fe, Ca, and O (Fig. 8a). The TiO2-coated CFO electrode showed signals assigned only to Ti and O, a very weak Ca signal, and no signal for Fe (Fig. 8b). This indicates that most of the surface of the CFO was coated with TiO2. The binding energy of the Ti 2p3/2 peak was 458.4 eV, which is attributed to Ti4+ species in the TiO2 matrix.
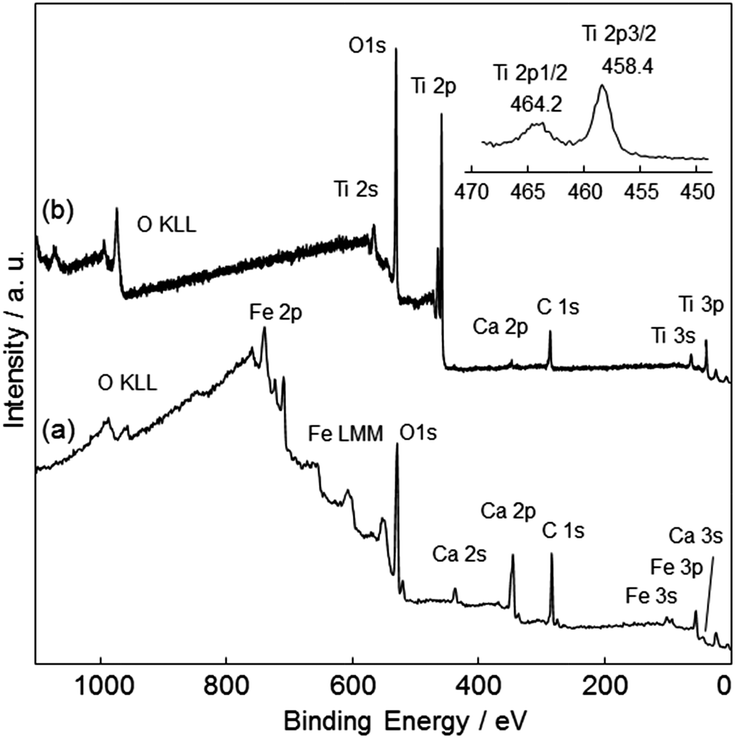 | ||
| Fig. 8 Survey and narrow Ti 2p XPS spectra of (a) CFO and (b) TiO2-coated CFO. | ||
Fig. 9 shows experimental current–potential curves of CFO and TiO2-coated CFO electrodes in 0.1 M NaOH solution under chopped illumination (300, 470, 532 nm). Under 300 nm light, the onset potentials of bare CFO and TiO2-coated CFO were 1.27 and 0.92 V vs. RHE, respectively, as shown in Fig. 9a and b. Thus, under 300 nm excitation, TiO2 induces a negative shift in the onset potential which agrees very well with the simulated current–potential curve as shown in Fig. 3a. When the electrode is illuminated with 300 nm light, which corresponds to the bandgap excitation of TiO2, photo-excited carriers are generated in the TiO2 layer. Thus, an electric field is formed in the TiO2 by the photo-excited carriers that accumulate in trap states, which decreases the onset potential.
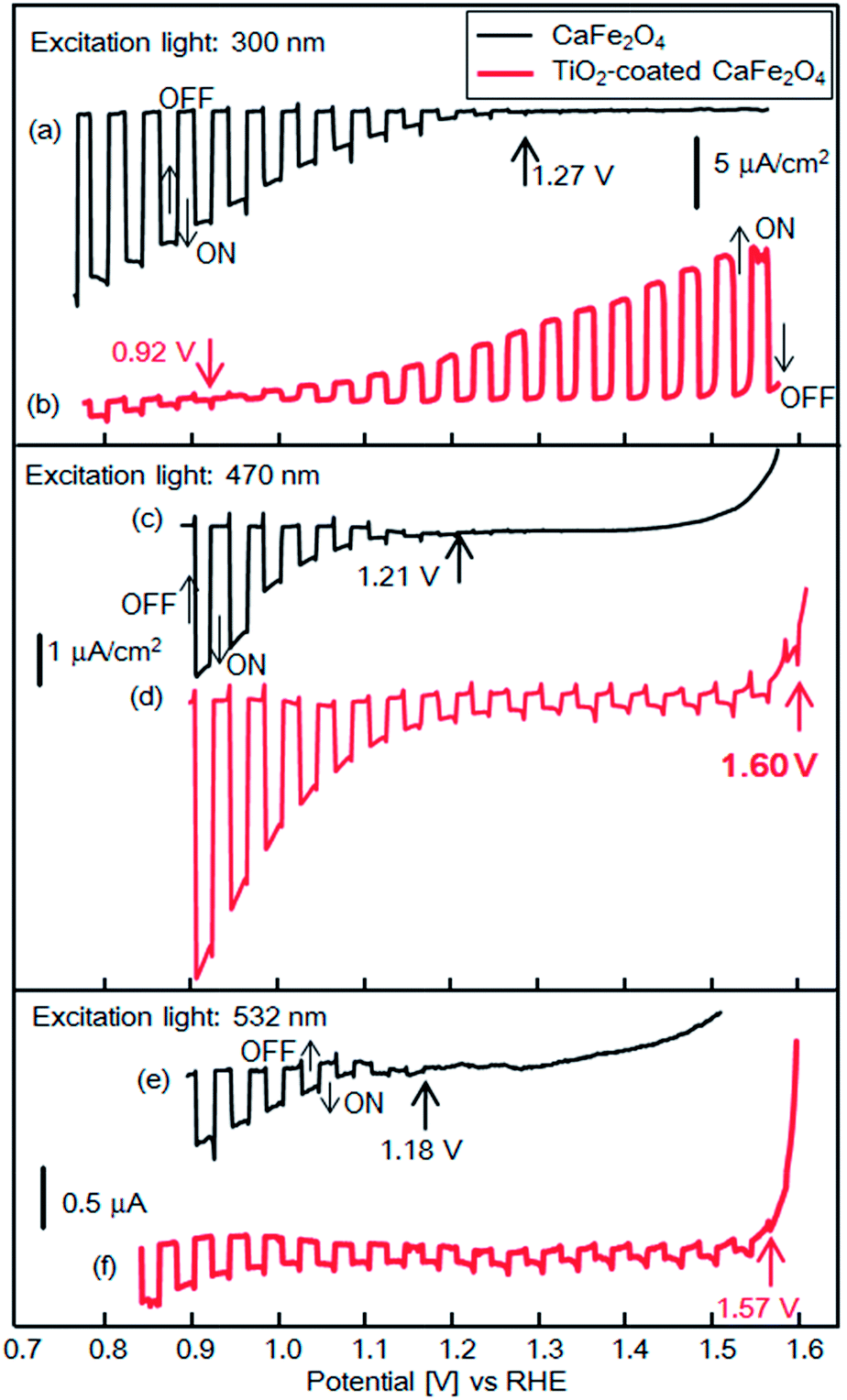 | ||
| Fig. 9 Current–potential curves of CFO and TiO2-coated CFO electrodes in 0.1 M NaOH solution under chopped light illumination (300, 470, 532 nm); black line: CFO, red line: TiO2-coated CFO. | ||
With 470 nm excitation the onset potentials of bare CFO and CFO|TiO2 were 1.21 and 1.60 V vs. RHE, respectively, as shown in Fig. 9c and d. Although the incident photon to current conversion efficiency (IPCE) at 1.23 V for TiO2-coated CFO was 0.002%, the IPCEs from 1.2 to 1.6 V was apparently improved compared with bare CFO (Fig. S4†). The onset potentials (vs. RHE) in 0.1 M NaOH was the almost same as those in 0.1 M Na2SO4 or 0.1 H2SO4 aqueous solution as shown in Fig. S5.† For 532 nm excitation the onset potentials of bare CFO and CFO|TiO2 were 1.18 V and 1.57 V vs. RHE (Fig. 9e and f). However, the actual onset potentials are at minimum 1.60 and 1.57 V vs. RHE for 470 and 532 nm, respectively. For example, in the case of 470 nm excitation, photocathodic current is observed at 1.70 V vs. RHE as shown in Fig. S6,† but background current due to water electrolysis interferes with the exact determination of the onset potential. Thus, under visible irradiation, TiO2 induces an increase in the onset potential, as predicted by the simulation results as shown in Fig. 3b and c. As mentioned above, in this system, the TiO2 layer acts as a passivation layer for the CFO surface and as an electron-selective contact between CFO and the electrolyte, which results in a decrease in the surface recombination rate of CFO and an increase in the onset potential.
In the case of powdered photocatalysis, TiO2 usually requires a co-catalyst for photocatalytic H2 evolution. However, in the present case, adding a co-catalyst is not necessary to produce H2 on TiO2. When TiO2-coated CFO is illuminated by visible light, carriers are only generated in the CFO and not in the TiO2. The degree of band bending in the CaFe2O4 combined with proper band-edge alignment between TiO2 and CaFe2O4 produces a driving force sufficient enough to evolve H2 without requiring a catalyst.
To the best of our knowledge, the TiO2-coated CFO has the most positive onset potential among the oxide photocathodes ever reported for PEC water splitting. Another important result is that the onset potential is strongly affected on the excitation wavelength. In the case of TiO2-coated CFO, the experimentally observed potential difference between 300 nm illumination and 470 nm illumination was 680 mV. This result indicates that the excitation wavelength for PEC H2 evolution should be considered according to the optical property of the passivation layer.
Next, PEC H2 evolution without an external applied voltage was attempted for a TiO2-coated CFO and RuO2–Pt two-electrode system. RuO2-loaded Pt electrode was used as the anode electrode for O2 evolution because the overpotential on RuO2-loaded Pt was slightly lower than that of pure Pt. Fig. 10a shows the I–V curve of a photocell with TiO2-coated CaFe2O4 and RuO2–Pt electrodes in 0.1 M NaOH aqueous solution under 470 nm light illumination. The short-circuit current (V = 0) was 18 μA cm−2.
 | ||
| Fig. 10 (a) Current–potential curve of a photocell with TiO2-coated CaFe2O4 and RuO2–Pt electrode electrodes in 0.1 M NaOH aqueous solution under 470 nm-light illumination and (b) CFO–Pt electrodes system under visible light irradiation (470 nm) in 0.1 M NaOH aqueous solution. | ||
The photocurrent was still observed at 1.6 V, although it is difficult to see due to large background currents for water decomposition. The onset potential of TiO2-coated CaFe2O4 under 470 nm light irradiation in Fig. 9 was around 1.6 V, which matches the voltage observed photocurrent in the TiO2-coated CFO and RuO2–Pt electrode system shown in Fig. 10a. Fig. 10b shows the current–time curve under 470 nm light irradiation without an external applied bias. Although a rapid decrease in photocurrent was observed initially, the system showed a stable photocurrent (around 500 nA cm−2) for over 15 h. On the other hand, in the case of bare CFO, the photocurrent decreased immediately and became negligible (Fig. 10b).
Fig. 11 shows the time evolution of H2 production from a TiO2-coated CFO and RuO2–Pt electrode system without an external applied bias under 470 nm irradiation. H2 gas evolution was confirmed by gas chromatography, indicating that PEC H2 evolution reaction occurred at the TiO2-coated CFO electrode. The amount of evolved H2 corresponds to the theoretical amount estimated from Faraday's law. Evolution of H2 was observed for over 70 h indicating that the TiO2-coated CFO electrode is stable during the PEC reaction. With regard to oxygen, reliable results for oxygen evolution (nanomole amount of oxygen) were not obtained due to contamination of very small amount of air into the reaction chamber. In the case of bare CFO electrode, H2 evolution was not observed without an external bias. Thus, a positive shift in the onset potential of CFO from 1.3 V to 1.6 V by coating the electrode with TiO2 results in H2 evolution without an external applied voltage under visible light illumination.
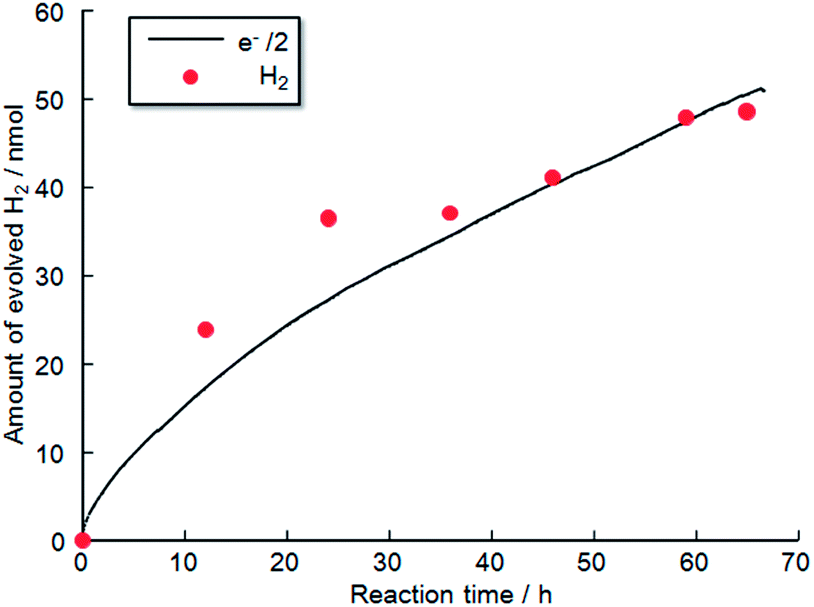 | ||
| Fig. 11 Time evolution of H2 production from photoelectrochemical cell using TiO2 coated CFO electrode without an external applied bias in 0.1 M NaOH aqueous solution under visible light irradiation (470 nm); anode: RuO2-loaded Pt electrode. | ||
To analyze the chemical stability of the TiO2-coated CFO electrode, PEC H2 production was conducted under irradiation from a solar simulator (400 mW cm−2) applying an external bias (1.2 V) to accelerate the PEC reaction on the electrode surface. For the bare CFO (cathode)–Pt (anode) system, the photocurrent decreased with increasing reaction time as shown in Fig. 12a due to the decomposition of the CFO surface. Before the reaction, CFO has a flat surface; yet after the PEC reaction, many cracks were observed on the surface of the CFO as shown in Fig. 13a. Also, the color of the electrode surface changed from black to brown due to the loss of Ca2+ ions into the aqueous solution. For the TiO2 coated-CFO (cathode)–Pt (anode) system, although the initial photocurrents were smaller than that for the bare CFO, the decrease in the photocurrent was suppressed (Fig. 12a). The photocurrent increased during the first 4 hours of PEC H2 production but then remained constant throughout the reaction. The initial increase in photocurrent may be due to a change in the surface condition of the TiO2 or the interfacial structure between the amorphous-like TiO2 layer and CFO during the PEC reaction. With regard to chemical stability, no cracks were evident on the electrode surface of TiO2-coated CFO after 14 h of H2 production as shown in Fig. 13b.
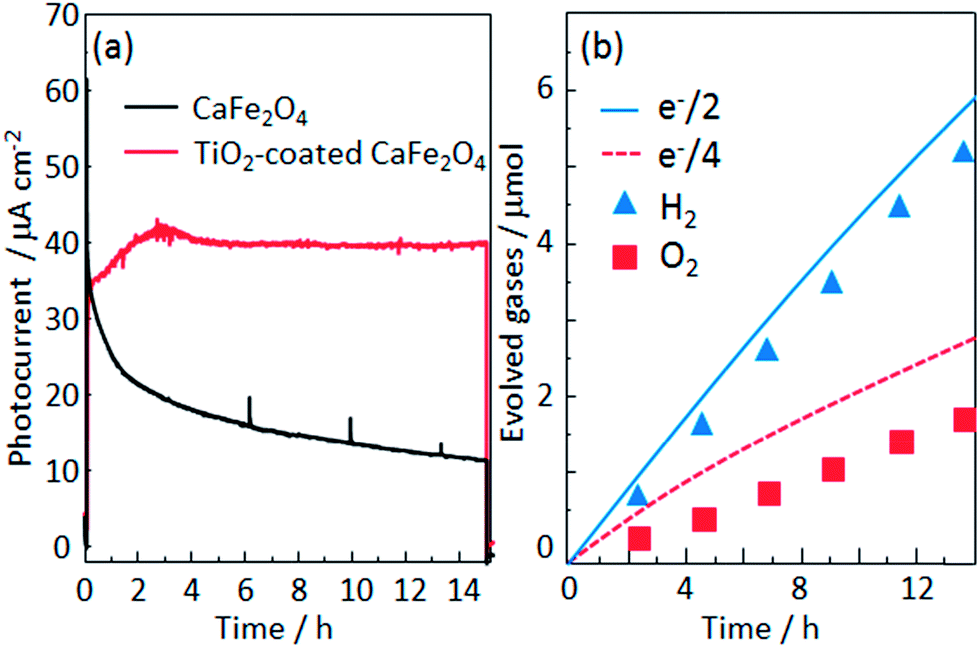 | ||
| Fig. 12 (a) Current–time evolution for the CaFe2O4 (cathode)–Pt (anode) electrodes in a 0.1 M NaOH aqueous solution under irradiation from a solar simulator (400 mW cm−2) applying an external bias (1.2 V) and (b) time evolution of H2 and O2 production from TiO2-coated CFO and Pt electrode system. | ||
 | ||
| Fig. 13 FE-SEM images electrode surface after PEC reaction for 14 h of (a) CaFe2O4 and (b) TiO2-coated CaFe2O4 electrodes. | ||
The time evolution of H2 and O2 production for the TiO2-coated-CFO (cathode)–Pt (anode) system is shown in Fig. 12b. Faraday efficiencies for the evolved H2 and O2 were 92% and 70%, respectively. Backward reaction may be a main reason why the amounts of H2 and O2 evolved did not correspond to the quantity of electricity that flowed. The total amount of evolved O2 was not sufficiently much and then the solubility in water of oxygen is larger than that of hydrogen, which might be related to the low faradaic efficiency of oxygen compared with hydrogen. Thus, it was confirmed that coating with TiO2 is effective to improve the chemical stability of CFO and onset potential under visible light irradiation.
Conclusions
A TiO2-coated CFO photocathode was designed to achieve H2 evolution without an external voltage using a device simulator. The simulation results predicted that coating CFO with TiO2 gives an increase or decrease in the onset potential under visible and ultraviolet irradiation, respectively. Under 300 nm light the observed onset potential shifted from 1.27 to 0.92 V vs. RHE while under visible irradiation (470 & 532 nm) the onset potential shifted from 1.2 to 1.6 V vs. RHE. Thus, the simulation results predicted the actual performance of the TiO2-coated CFO electrode. Using TiO2-coated CFO as the photocathode and RuO2-loaded Pt as the anode, stable photocurrent was observed under 470 nm excitation without an external voltage. Evolution of H2 from the system was confirmed. Chemical stability of CFO was also improved by coating the surface with TiO2. To our knowledge, TiO2-coated CFO is the first example of a photocathode that exhibits a greater onset potential than that required for photocathodic water reduction.Acknowledgements
This work was supported by the JST PRESTO program, JSPS KAKENHI Grant Numbers 15H00879. We thank Prof. T. Nakanotani for discussion about STEM data.Notes and references
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- R. Abe, M. Higashi and K. Domen, J. Am. Chem. Soc., 2010, 132, 11828–11829 CrossRef CAS PubMed.
- M. Higashi, K. Domen and R. Abe, Energy Environ. Sci., 2011, 4, 4138–4147 CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459–11467 CrossRef CAS.
- K. Sayama, A. Nomura, T. Arai, T. Sugita, R. Abe, M. Yanagida, T. Oi, Y. Iwasaki, Y. Abe and H. Sugihara, J. Phys. Chem. B, 2006, 110, 11352–11360 CrossRef CAS PubMed.
- Y. H. Ng, A. Iwase, A. Kudo and R. Amal, J. Phys. Chem. Lett., 2010, 1, 2607–2612 CrossRef CAS.
- W. Luo, Z. Yang, Z. Li, J. Zhang, J. Liu, Z. Zhao, Z. Wang, S. Yan, T. Yu and Z. Zou, Energy Environ. Sci., 2011, 4, 4046–4051 CAS.
- J. Hou, Z. Wang, C. Yang, H. Cheng, S. Jiao and H. Zhu, Energy Environ. Sci., 2013, 6, 3322–3330 CAS.
- Y. Li, T. Takata, D. Cha, K. Takanabe, T. Minegishi, J. Kubota and K. Domen, Adv. Mater., 2013, 25, 125–131 CrossRef CAS PubMed.
- T. Inoue and T. Yamase, Chem. Lett., 1985, 7, 869 CrossRef.
- M. Kato, T. Yasuda, K. Miyake, M. Ichimura and T. Hatayama, Int. J. Hydrogen Energy, 2014, 39, 4845–4849 CrossRef CAS.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- K. Iwashina and A. Kudo, J. Am. Chem. Soc., 2011, 133, 13272–13275 CrossRef CAS PubMed.
- C. Leygraf, M. Hendewerk and G. A. Somorjai, J. Phys. Chem., 1982, 86, 4484–4485 CrossRef CAS.
- J. E. Turner, M. Hendewerk and G. A. Somorjai, Chem. Phys. Lett., 1984, 105, 581–585 CrossRef CAS.
- A. Duret and M. Grätzel, J. Phys. Chem. B, 2005, 109, 17184–17191 CrossRef CAS PubMed.
- D. K. Zhong, J. Sun, H. Inumaru and D. R. Gamelin, J. Am. Chem. Soc., 2009, 131, 6086–6087 CrossRef CAS PubMed.
- W. B. Ingler Jr, J. P. Baltrus and S. U. M. Khan, J. Am. Chem. Soc., 2004, 126, 10238–10239 CrossRef PubMed.
- S. Ida, K. Yamada, T. Matsunaga, H. Hagiwara, Y. Matsumoto and T. Ishihara, J. Am. Chem. Soc., 2010, 132, 17343–17345 CrossRef CAS PubMed.
- Y. Matsumoto, M. Omae, K. Sugiyama and E. I. Sato, J. Phys. Chem., 1987, 91, 577–581 CrossRef CAS.
- Z. Liu, Z. G. Zhao and M. Miyauchi, J. Phys. Chem. C, 2009, 113, 17132–17137 CAS.
- J. Cao, T. Kako, P. Li, S. Ouyang and J. Ye, Electrochem. Commun., 2011, 13, 275–278 CrossRef CAS.
- S. Ida, K. Yamada, T. Matsunaga, H. Hagiwara, T. Ishihara, T. Taniguchi, M. Koinuma and Y. Matsumoto, Electrochemistry, 2011, 79, 797–800 CrossRef CAS.
- S. Ida, K. Yamada, M. Matsuka, H. Hagiwara and T. Ishihara, Electrochim. Acta, 2012, 82, 397–401 CrossRef CAS.
- J. Cao, J. Xing, Y. Zhang, H. Tong, Y. Bi, T. Kako, M. Takeguchi and J. Ye, Langmuir, 2013, 29, 3116–3124 CrossRef CAS PubMed.
- E. S. Kim, N. Nishimura, G. Magesh, J. Y. Kim, J. W. Jang, H. Jun, J. Kubota, K. Domen and J. S. Lee, J. Am. Chem. Soc., 2013, 135, 5375–5383 CrossRef CAS PubMed.
- K. Sekizawa, T. Nonaka, T. Arai and T. Morikawa, ACS Appl. Mater. Interfaces, 2014, 6, 10969–10973 CAS.
- Q. Yu, X. Meng, T. Wang, P. Li, L. Liu, K. Chang, G. Liu and J. Ye, Chem. Commun., 2015, 51, 3630–3633 RSC.
- K. J. McDonald and K. S. Choi, Chem. Mater., 2011, 23, 4863–4869 CrossRef CAS.
- M. G. Ahmed, T. A. Kandiel, A. Y. Ahmed, I. Kretschmer, F. Rashwan and D. Bahnemann, J. Phys. Chem. C, 2015, 119, 5864–5871 CAS.
- Y. Matsumoto, J. Solid State Chem., 1996, 126, 227–234 CrossRef CAS.
- K. Ramanathan, M. A. Contreras, C. L. Perkins, S. Asher, F. S. Hasoon, J. Keane, D. Young, M. Romero, W. Metzger, R. Noufi, J. Ward and A. Duda, Prog. Photovoltaics Res. Appl., 2003, 11, 225–230 CrossRef CAS.
- I. Repins, M. A. Contreras, B. Egaas, C. DeHart, J. Scharf, C. L. Perkins, B. To and R. Noufi, Prog. Photovoltaics Res. Appl., 2008, 16, 235–239 CrossRef CAS.
- P. Jackson, D. Hariskos, E. Lotter, S. Paetel, R. Wuerz, R. Menner, W. Wischmann and M. Powalla, Prog. Photovoltaics Res. Appl., 2011, 19, 894–897 CrossRef CAS.
- L. Zhang, T. Minegishi, J. Kubota and K. Domen, Phys. Chem. Chem. Phys., 2014, 16, 6167–6174 RSC.
- B. Seger, T. Pedersen, A. B. Laursen, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Am. Chem. Soc., 2013, 135, 1057–1064 CrossRef CAS PubMed.
- B. Seger, S. D. Tilley, T. Pedersen, P. C. K. Vesborg, O. Hansen, M. Grätzel and I. Chorkendorff, J. Mater. Chem. A, 2013, 1, 15089–15094 CAS.
- Y. Lin, R. Kapadia, J. Yang, M. Zheng, K. Chen, M. Hettick, X. Yin, C. Battaglia, I. D. Sharp, J. W. Ager and A. Javey, J. Phys. Chem. C, 2015, 119, 2308 CAS.
- Y. Liu, Y. Sun and A. Rockett, Sol. Energy Mater. Sol. Cells, 2012, 98, 124 CrossRef CAS.
- Solar Cells Simulation, https://wiki.cites.illinois.edu/wiki/display/solarcellsim, accessed March 2, 2016.
- M. Chitambar, Z. Wang, Y. Liu, A. Rockett and A. Maldonado, J. Am. Chem. Soc., 2012, 134(25), 10670 CrossRef CAS PubMed.
- S. J. Fonash, J. Arch, J. Cuiffi, J. Hou, W. Howland, P. J. McElheny, A. Moquin, M. Rogosky, T. Tran, H. Zhu and F. Rubinelli, A Manual for AMPS-1D for Windows 95/NT, The Pennsylvania State University, Happy Valley, PA, 1997 Search PubMed.
- K. L. Kearney and A. Rockett, J. Electrochem. Soc., 2016, 163(7), H598 CrossRef CAS.
- H. J. Kim, K. L. Kearney, L. H. Le, L. Z. J. Haber, A. A. Rockett and M. J. Rose, J. Phys. Chem. C, 2016, 120(45), 25697–25708 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00084g |
| This journal is © The Royal Society of Chemistry 2017 |