
Biomass-derived carbon electrode materials for supercapacitors
Hao
Lu
 and
X. S.
Zhao
*
and
X. S.
Zhao
*
School of Chemical Engineering, The University of Queensland, St Lucia, Brisbane, QLD 4072, Australia. E-mail: george.zhao@uq.edu.au
First published on 17th May 2017
Abstract
This review provides a summary of recent research progress towards biomass-derived carbon electrode materials, including specific cellulose-, lignin- and hemicellulose-derived carbon electrode materials, for supercapacitors. Various lab-scale methods for preparing biomass-derived carbons, including carbonisation and/or activation conditions are discussed. Control over the pore structure, electrical conductivity, and surface functional groups of biomass-derived carbons for enhancing electrocapacitive performance is analysed. Emphasis is made on discussing cellulose-, lignin- and hemicellulose-derived carbon electrode materials for supercapacitor applications. Future research trends in this field are projected.
1. Introduction
1.1 Supercapacitors
The development of sustainable and clean energy, such as solar and wind energy, relies significantly upon advanced energy storage systems, such as rechargeable batteries and supercapacitors (SCs).1–3 On the other hand, the rapidly growing market for portable electronic devices requires more and more energy storage systems with a high performance.4 SCs with distinctive advantages such as high power density, long cycle life (>100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles), and safety5 will find more and more applications in the future as an alternative energy storage resource for rechargeable batteries.6,7 A couple of excellent review articles have discussed the device configuration and energy storage mechanism of SCs.1,5,8,9
000 cycles), and safety5 will find more and more applications in the future as an alternative energy storage resource for rechargeable batteries.6,7 A couple of excellent review articles have discussed the device configuration and energy storage mechanism of SCs.1,5,8,9
SCs have already found wide applications as an energy storage system and/or a power source for hybrid and electric vehicles, smart grids, uninterruptible power supply (UPS) systems and many consumer electronics.10 Their excellent reliability under severe climate conditions, combined with the growing popularity of the automatic start–stop technology, enables SCs to become more and more popularly used in buses, trucks, trains, trams and metros.10 In these applications, SCs capture power from the regenerative braking system and release energy to assist in acceleration.11 Besides, SCs can offer rapid storage and efficient delivery of energy in heavy-duty applications even under harsh conditions. For instance, burst power and good low temperature operation performance make SCs widely used for hybrid forklift and cranes.6
Recent research interest has also been focused on integrating photovoltaics (PVs) with SCs, in which the SC serves as both an energy reservoir and a power buffer.12 As a clean and renewable energy supply, PV solar cells are expected to play a leading role in future global sustainable energy development. However, the energy converted from a solar cell is intermittent. SCs can alleviate the intermittence, making sustainable energy supply available at night and on cloudy days.13 In the past decade, the integration system packed as a parallel combination of PVs and SCs or a photo-supercapacitor has witnessed substantial progress at the laboratory scale.14,15 Another interesting application of SCs is for the intelligent wireless sensor (IWS). Currently, battery-powered or wired sensors that can provide different kinds of data from our surroundings are widely used. In the near future, wireless sensors needed to be installed in remote areas or incorporated in some special structures. The energy supply of the IWS can be vibration, radiofrequency, thermal and electromagnetic energy harvesting.16,17 To store the harvested energy for the IWS, the energy storage system needs to fulfill several properties, i.e., a sufficient number of charge and discharge cycles without deterioration during the lifetime of the device, high energy density and a low self-discharge.16
One of the key issues of SCs is their low energy density. Currently commercially available SCs can only provide an energy density of less than 10 W h kg−1. This is much inferior than that from lithium-ion batteries (LIBs), which can provide an energy density of more than 180 W h kg−1.18,19 Therefore, both the scientific community and companies are eager to enhance the energy density of SCs by developing new electrode materials, novel electrolytes with a wide operation voltage window, or an ingenious device design.18,19 In this review, we will mainly focus on the development of electrode materials from biomass.
1.2 Electrode materials for supercapacitors
SCs store energy via two mechanisms; one is the electric double-layer (EDL) mechanism and the other is the redox reaction mechanism. Thus there are two types of SCs, i.e., EDL capacitors (EDLCs) and pseudocapacitors. The electrode materials for the former are mainly activated carbons (ACs) while for the latter they can be metal oxides and conducting polymers. The electrode material plays a significant role in the electrocapacitive performance of SCs.The criteria for evaluating SC electrode materials include specific capacitance, stability against the electrolyte and cycling, and cost. In terms of specific capacitance, the specific surface area (SSA), pore size and geometry, and electronic and ionic conductivity are important parameters to consider.19,20
Generally, pseudocapacitive materials exhibit a higher specific capacitance than EDL electrodes; thus pseudocapacitors always have a higher energy density than EDLCs. However, carbon-based materials tend to have a better cycle stability and rate capability, and thus EDLCs can be generally operated at high charge and discharge rates with a lifetime of over a million cycles.20 Therefore, carbon materials have been the focus on searching for advanced electrode materials for SCs.
1.3 Biomass and biomass-derived carbon materials
Biomass is an energy source most often refers to plants or plant-derived, animal-derived and marine organism-derived materials.21,22 Because biomass is earth abundant, renewable, and low cost, biomass and biomass-derived materials have been exploited for various applications, such as CO2 capture,23,24 hydrogen storage,25–27 water treatment,28,29 catalysis and energy storage.30–35 This review mainly focuses on plants or plant-derived biomass as a source for making SC electrode materials.Plants or plant-derived biomass is often referred to as lignocellulose, which is mainly composed of carbohydrate polymers (cellulose and hemicellulose) and aromatic polymers (lignin and tannin).36 Carbon materials can be prepared directly from lignocellulose or its derivatives, such as cellulose, lignin and hemicellulose. Recently, Zhang et al.21 reviewed the applications of cellulose and alginate for both LIBs and SCs. Jabbour et al.37 summarised the progress of cellulose and cellulose derivative based LIBs. Ma et al.38 reviewed carbon-based materials derived from waste for water remediation and energy storage. Dutta et al.39 presented biomass-biopolymer derived hierarchically porous carbons (HPCs) for applications ranging from CO2 capture and carbon photonic crystal sensors to Li–S batteries and SCs. In this article, we provide an overview of biomass-derived carbon materials as electrodes for SCs.
2. Biomass-derived carbon for supercapacitors
2.1 Methods
To convert biomass into carbon, different carbonisation methods (e.g., pyrolysis and hydrothermal carbonisation) and activation methods (e.g., physical and chemical activations) can be used. As illustrated in Scheme 1, both physical and chemical means, or a combination of both, have been employed to transfer biomass into value-added carbon materials. By controlling parameters such as temperature, time, and reagents involved in the preparation, carbon materials with different porosities, surface properties, morphologies, and costs can be obtained. | ||
| Scheme 1 Common methods for converting biomass to carbon materials. | ||
Pyrolysis and hydrothermal carbonisation (HTC) are the two common methods used to carbonise biomass. Pyrolysis is carried out in an inert or limited oxygen atmosphere at elevated temperatures while HTC refers to a thermo-chemical process used to convert biomass to carbonaceous materials.40,41
The main pyrolysis products obtained from biomass depend on the temperature, temperature ramping rate, particle size and catalyst used.42 HTC is performed in a pressurized aqueous environment at a relatively low temperature, typically in the range of 120–250 °C,43 with or without the aid of a catalyst.44 It mimics the natural coalification of biomass, although the reaction rate is higher and the reaction time is shorter compared to the hundreds of years of slow nature coalification of biomass.40 In recent years, several review articles regrading hydrothermal conversion of biomass have been published.30,41,43,45 As a thermo-chemical conversion technique, the HTC can be influenced by several parameters, such as temperature, residence time, precursor concentration and catalyst. It uses subcritical water for the conversion of a biomass to carbonaceous products, resulting in efficient hydrolysis and dehydration of precursors and bestowing hydrochars with a high and tunable content of oxygen-containing functional groups (OFG).41 Other functionalities, e.g., nitrogen-containing groups, can also be introduced into hydrochars by using dopant-containing precursors or additives.45 The use of HTC for the conversion of biomass into carbon materials has received considerable attention for various applications such as catalysis,46,47 CO2 capture,48,49 and energy storage.43,50–55
Activation is a process of converting carbonaceous materials into AC. Both physical and chemical activation means can be used.5 Physical activation is usually conducted immediately after the pyrolysis step at high temperatures (up to 1200 °C) in an atmosphere of steam or CO2.25 Chemical activation is implemented with a chemical agent at a temperature typically ranging from 450 to 900 °C. The commonly used chemical activation agents include KOH, NaOH, ZnCl2, FeCl3, H3PO4, and K2CO3.
2.2 Carbonisation
Zhu et al.65 proposed and demonstrated a hydrothermally assisted pyrolysis procedure to produce fungi-derived electrode materials for SCs. By HTC at 120 °C for 6 h, the product was characterised by small particle sizes (50–200 nm), high oxygen content (13.4 wt%) and a low surface area (14 m2 g−1). To increase the porosity and improve the electronic conductivity, the hydrochar was further pyrolysed at 700 °C for 3 h. The ultimately obtained carbon material had a SSA of 80 m2 g−1 and an oxygen content of 5 wt%. In spite of the still low SSA after the pyrolysis, the material exhibited a specific capacitance of 196 F g−1 at a scan rate of 1 mV s−1, a value comparable to that of commercial ACs such as Maxsorb.
Actually, highly porous carbon materials based on hydrochar normally have a relatively high amount of heteroatoms.71–73 Thus, the hydrochar from the HTC process is an excellent precursor for the production of carbon with tuneable surface functionality and porosity.74 Torres et al.75 prepared AC materials doped with N and O heteroatoms by activation of hydrochars with KOH. The presence of nitrogen and oxygen groups was found to improve both the capacitance and charge transfer especially at high current densities. Wei et al.70 demonstrated that hydrochars with networks of uniformly distributed oxygen can be efficiently transformed into microporous carbons with a high SSA and large pore volume of interconnected pores. In their experiment, the transformation was accomplished via HTC at 230–250 °C for 2 h and subsequent chemical activation at 700–800 °C for 1 h. The carbon electrode produced from wood saw dust exhibited both a high capacitance of 236 F g−1 and a rapid charge/discharge capability in a symmetric two-electrode SC using an organic electrolyte.
2.3 Activation
Chemical and physical activation are the two mainly employed methods to obtain biomass-derived ACs. Compared with chemical activation, the latter one is simpler and more environmentally friendly but usually conducted at a higher temperature. Currently, chemical activation is more commonly used because of lower activation temperature, less activation time, higher yield, higher SSA and well-developed pores. Anyhow, the chemical activation method does have several disadvantages, such as the need for water for the necessary post-activation washing to thoroughly wash away any impurities generated during the activation process and the handling of the contaminated water.20,22,76,77000 cycles. The air activation step has also been shown to significantly influence the porous properties of biomass-derived carbon.79–81
Steam is a readily available activating agent used for biomass materials due to its low cost and the lack of post-activation process to remove by-products.8 The steam activation process is always combined with pyrolysis as a single step. It can generate rich surface oxygen-containing groups (defects), which lead to a poor electrical conductivity of the resulting carbon. Some researchers have systematically studied steam activation of biomass.82–87 Li's group studied the influence of the steam activation time and water flow rate on the texture and electrochemical performance of ACs derived from coconut shell86 and corncob residue.85 They found that the mesoporosity increased considerably with the increase of activation time and water flow rate, which enabled the sample to have high rate capability and cycle stability. Jin et al.87 investigated in detail the effect of steam activation time on the porosity and surface area of the activated carbon fibres (ACFs). Steam activation of liquefied wood with various activation times (20, 60, 100, 140, 180 and 220 min) was conducted. By controlling the activation time, the mesopore/micropore ratio could be effectively tuned and the micropore and mesopore surface area increased with the rising activation time before burn-off. The sample with the highest micropore surface area and a relatively high proportion of mesopores in the range of 3–4 nm presented a specific capacitance of 280 F g−1 at 0.5 A g−1 in 0.5 M H2SO4 and excellent rate performance as well as good cyclic stability.
CO2 activation, which is based on the controlled gasification of a char with CO2 gas at a high temperature, is the most commonly used physical activation process. Studies have shown that smaller sized microporous carbons exhibit a larger capacitance.7 However, CO2 activation-derived carbon tends to have a high fraction of mesorpores and an increased average pore size, as the large dimension of CO2 molecules tends to restrict the accessibility of CO2 into micropores.8,74,84,88,89 Besides, researchers found that, compared with KOH activation, as seen in Fig. 1, CO2 activation could result in a little higher degree of graphitisation with apparently oriented multilayer domains and graphene sheets stacked in parallel in the structure of the porous carbons.90
 | ||
| Fig. 1 XRD patterns of the activated carbon materials with CO2 and KOH activation. The inset is a sketch map for the calculation of the R values.90 | ||
Systematic research has been conducted focusing on CO2 activation of polymer-derived carbons84 and hard/soft-template carbons89,91 as well as their applications for SCs92,93 and CO2 adsorption.94,95 However, research focusing on CO2 activation of biomass-derived carbons for SCs is still relatively less.96,97
Compared with steam and CO2 activation, air activation needs a lower temperature. But the former two are more commonly used than the latter as the ACs produced with steam or CO2 tend to have a wider pore size distribution.98,99 Research on the former has further shown that steam activation produces a larger development of mesopores and macropores than CO2 activation.98,100 However, research for physical activation towards biomass is still relatively insufficient as chemical activation is more commonly used. For instance, Osswald88 and Yan91et al. have systematically investigated the porosity, SSA and stability of carbide-derived carbon materials physically activated using CO2, air or steam. By contrast, such detailed research on physical activation for biomass is little, especially combined with their further application for SC electrodes.
2.3.2.1 KOH activation. KOH is a most often used chemical for activating biomass-derived carbons. A rough carbon surface could be induced by the activation of KOH, which brings about a high SSA and a porous structure that are advantageous in charge storage.101 The detailed mechanism of KOH activation has not been fully understood because of the complex variables including the experimental parameters as well as the reactivity of different carbon precursors.90,102–105 In a general view, the development of a large SSA and high porosity in KOH-activated carbons is the result of the synergistic, comprehensive actions, including chemical activation and carbon lattice expansion by metallic K intercalation.76
For a given carbon precursor, experimental variables of KOH activation include the mass ratio of KOH/biomass, heating rate, activation temperature and time. The normally adopted variables are the following: (1) the KOH/biomass mass ratio ranges from 2 to 5; (2) a heating rate of 3–10 °C min−1; (3) the activation temperature and time are 550–900 °C and 1–4 h, respectively. Karthikeyan et al.106 studied the chemical activation of pine cone petal powders with KOH/biomass mass ratios of 1, 3 and 5, respectively. The mixture was then pyrolysed at 750 °C under an Ar flow at a heating rate of 5 °C min−1 for 1.5 h. The highest SSA was obtained from the sample prepared with a KOH/biomass ratio of 5. A symmetric SC fabricated with this carbon showed an energy density of ∼61 W h kg−1 at a power density of ∼0.39 kW kg−1 with an excellent capacitance retention of ∼90% after 20000 cycles in an organic electrolyte.
KOH-activated, in a single or two-step process, biomass-derived carbons of good performance have been reported. Li et al.107 described a one-step process for the preparation of nitrogen-doped activated carbon with corncob, KOH, and NH3 as the carbon source, activating agent and nitrogen source, respectively. The corncob powders were mixed with KOH in a 1:3 mass ratio. Then the mixture was heated to the desired temperature under a N2 or NH3 flow for a certain time. The obtained sample had a narrow micro- to meso-pore distribution ratio and showed a high SSA of 2900 m2 g−1 with a moderate N content of 4 wt%, and delivered a specific capacitance of up to 185 F g−1 in an organic electrolyte at a current density of 0.4 A g−1.
In comparison, the two-step KOH activation method is more often used to prepare biomass-derived carbon materials for SCs. Biomass is always pre-treated by HTC,52–55,67,70,73,75,108 or pre-carbonisation68,109–112 or pyrolysis78,113–120 before KOH activation. Qian et al.109 prepared heteroatom doped porous carbon flakes from human hair fibres using the two-step KOH method, combining pre-carbonisation with KOH activation. In their experiment, hair fibres were firstly pre-carbonised at 300 °C for 1.5 h and then mixed with KOH (WKOH/Wcarbon = 2:1) and further activated at 700, 800 or 900 °C, respectively. In the pre-carbonisation step, some surface functional groups or unstable component of human hairs, which act as active sites in the chemical activation with KOH, are likely to decompose, with a moderate amount of N, O and S retained.68,112 The material activated at 800 °C has a SSA of 1306 m2 g−1 with a doping of N, O and S, 4.38, 5.39, 1.51%, respectively. The temperature of pre-carbonisation noticeably affects the chemical composition, surface area and porosity development.111,112 Further research is needed to study the influence of the pre-carbonisation temperature on the performance of biomass-derived carbons as SC electrodes.
Hou et al.116 prepared carbons with a micro/mesopore interconnected structure through pyrolysis and KOH activation in a two-step process. Rice brans were carbonised under a N2 atmosphere at a rate of 3 °C min−1 from room temperature to 700 °C for 1 h and was then KOH-activated at 850 °C for 1 h. Porous carbons with a SSA of 2475 m2 g−1 and a pore volume of 1.21 cm3 g−1 (40% for mesopores) were obtained. It exhibited high specific capacitance especially at large current densities in 6 M KOH electrolyte, i.e., 265 and 182 F g−1 at 10 and 100 A g−1 respectively. Moreover, an energy density of 70 W h kg−1 and a power density of 1223 W kg−1 were obtained in an ionic liquid (IL). Wang et al.114 used waste celtuce leaves to prepare porous carbons. Pyrolysed at 600 °C, followed by KOH activation at 800 °C for 1 h, the as-prepared carbon has a high SSA of 3404 m2 g−1 and a large pore volume of 1.88 cm3 g−1. As an electrode, it exhibited specific capacitances of 421 and 273 F g−1 at a current density of 0.5 A g−1 in three and two-electrode systems, respectively.
Conventionally, precursors and solid KOH, with different mass ratios, are thoroughly blended or ground in an agate mortar and then carbonised to a certain temperature at a certain rate and maintained for several hours.52–54,68,70,73,75,78,106–109,112,113,116–118,120,121 Some researchers have tried to modify the conventional method by impregnation of the precursor with KOH aqueous solution and some good results were obtained.67,110,111,114,115,119,122–125 Precursors were first pyrolysed or pre-carbonised and then dispersed and stirred in aqueous KOH with different mass ratios of KOH/C, followed by an evaporation step until a stable slurry or colloidal solution was obtained. Subsequently, the mixture was annealed under the same conditions of the conventional method. Ruoff's125 and Huang's122 groups both used such a modified KOH activation method in their experiments, using graphene oxide and polypyrrole micro-sheets as the precursor respectively. It is believed that the phase separation between hydrophobic carbon and water during the activation process leads to both mesopores and macropores. Therefore 3D HPCs of high SSA with excellent electrocapacitive performance were obtained. However, detailed research on modified KOH activation using biomass as the precursor is still less.
2.3.2.2 ZnCl2 activation. ZnCl2 is another commonly used chemical activation agent for converting biomass-derived carbons into porous ACs.126–136 It acts as a dehydrating agent during the activation process and it also has a deoxygenation effect at high temperatures by removing oxygen in the form of water as well as by carbothermal reduction.137,138 ZnCl2-activated biomass-derived carbons of high performance, through a simple one-step process, have been reported.
Rufford et al.133 employed the ZnCl2 activation method to prepare porous carbon with a SSA as high as 1000 m2 g−1. The carbon prepared at 750 °C with a ZnCl2 to sugar cane bagasse weight ratio of 1 delivered the highest specific capacitance at low current densities. At current densities greater than 1 A g−1, however, the carbon with mesopores that was prepared at 900 °C with a ZnCl2 to bagasse ratio of 3.5 showed the most stable electrochemical performance. These results demonstrate the benefit of mesopores to energy storage at fast charge–discharge rates, i.e., acting as reservoirs for electrolyte ions and facilitating ion transport through the carbon pore network.139
Hou and co-workers129 prepared hierarchically porous nitrogen-doped carbon via simultaneous ZnCl2 activation and graphitisation, in a one-step process. Natural silk was mixed with ZnCl2 and FeCl3 solution in a certain ratio, followed by annealing at 900 °C for 1 h. The as-obtained carbon consisted of 2D nanosheet architecture with a hierarchical porosity, high SSA (2494 m2 g−1), rich N-doping (4.7%), and defects. The synergistic effect of these characteristics enables the as-obtained carbon to display high energy storage performance. Tested in an IL electrolyte two-electrode system, it exhibited a capacitance of 242 F g−1, an energy density of 90 W h kg−1 at a power density of 875 W kg−1 and high cycling life stability (9% loss after 10000 cycles).
2.3.2.3 Other activation reagents. Besides KOH and ZnCl2, several other reagents are also used for biomass chemical activation for SCs, such as H3PO4 (ref. 140–142) and KHCO3.143 Some researchers have also employed microwave-induced activation144–148 or physicochemical activation149–157 consisting of a chemical activation step followed by physical activation to further control the porosity development and tune the PSD of activated carbons.22,25 However, there is still relatively less research on their application for biomass-derived SC electrodes.
KOH, ZnCl2 and H3PO4 are the three mainly used chemical activation agents. In comparison, H3PO4-activated carbons usually have a relatively low SSA of below 1000 m2 g−1 while both KOH and ZnCl2 activation can easily produce a higher SSA. To enhance the SSA, KOH is a kind of oxidant, while ZnCl2 is a dehydrating and deoxygenation agent. Currently, KOH-activation is preferred to be used for biomass-derived ACs for SC electrodes as it can easily produce hierarchical porous carbons with an even high SSA of beyond 3000 m2 g−1 (Table 1).
| Materials | Pre-activation treatment | Activation agent | Activation temperature and time (°C)/(h) | SSA (m2 g−1) | C (F g−1) (symmetric SCs) | Measurements at | Electrolyte | Ref. |
|---|---|---|---|---|---|---|---|---|
| Seaweed | Pyrolysis | NA | NA | 746 | 264 | 2 mV s−1 | 1 M H2SO4 | 58 |
| Eggplant | Freeze drying/pyrolysis | NA | NA | 950 | 121 | 5 mV s−1 | 6 M KOH | 60 |
| Dead Neem leaves | Pyrolysis | NA | NA | 1230 | 400 | 0.5 A g−1 | 1 M H2SO4 | 59 |
| Eggshell membranes | Pyrolysis | Air | 300/2 | 221 | 205 | 2 A g−1 | 1 M H2SO4 | 78 |
| Corncob | NA | Steam | 850/0.75 | 1210 | 120 | 1 A g−1 | 6 M KOH | 85 |
| Wood | NA | Steam | 850/3 | 3223 | 247 | 0.5 A g−1 | 1 M H2SO4 | 87 |
| Coconut shell | NA | Steam | 800/1 | 1532 | 192 | 1 A g−1 | 6 M KOH | 86 |
| Coffee endocarp | Pyrolysis | CO2 | 800/2 | 709 | 176 | — | 1 M H2SO4 | 97 |
| Fungi | HTC/pyrolysis | NA | NA | 80 | 196 | 1 mV s−1 | 6 M KOH | 65 |
| Wood saw dust | HTC | KOH | 800/1 | 2967 | 236 | 1 mV s−1 | TEABF4/AN | 70 |
| Pollen | HTC | KOH | 900/1 | 3037 | 185 | 1 A g−1 | TEABF4/AN | 54 |
| Tobacco rods | HTC | KOH | 800/1 | −2000 | 263 | 0.5 A g−1 | 6 M KOH | 52 |
| Microalgae | HTC | KOH | 700/— | 2130 | 200 | 0.1 A g−1 | 6 M LiCl | 73 |
| Human hair | Pre-carbonisation | KOH | 800/2 | 1306 | 340 | 1 A g−1 | 6 M KOH | 109 |
| Cornstalk core | Pre-carbonisation | KOH | 800/3 | 2139 | 317 | 1 mV s−1 | 6 M KOH | 110 |
| Bean dregs | Pre-carbonisation | KOH | 700/1 | 2876 | 280 | 0.1 A g−1 | 1 M H2SO4 | 68 |
| Rice bran | Pyrolysis/600 | KOH | 850/1 | 2475 | 323 | 0.1 A g−1 | 6 M KOH | 116 |
| Ginkgo shells | Pyrolysis/600 | KOH | 700/1 | 1775 | 237 | 2 mV s−1 | 6 M KOH | 113 |
| Celtuce | Pyrolysis/600 | KOH | 88/1 | 3404 | 273 | 0.5 A g−1 | 6 M KOH | 114 |
| Broad beans | Pyrolysis/800 | KOH | 650/1 | 655 | 202 | 0.5 A g−1 | 6 M KOH | 115 |
| Pine cone petal | NA | KOH | 750/1.5 | 3850 | 198 | 0.25 A g−1 | 1 M LiPF6 | 106 |
| Corncob | NA | KOH/NH3 | 400/— | 2900 | 185 | 0.4 A g−1 | 1.2 M LiPF6 | 107 |
| Silk fibroin | NA | KOH | 800/3 | 2557 | 264 | 0.1 A g−1 | 1 M H2SO4 | 101 |
| Coffee beans | NA | ZnCl2 | 900/1 | 1021 | 134 | 0.05 A g−1 | 1 M TEABF4/AN | 131 |
| Coffee beans | NA | ZnCl2 | 900/1 | 1019 | 368 | 0.05 A g−1 | 1 M H2SO4 | 132 |
| Sugar cane bagasse | NA | ZnCl2 | 900/1 | 1788 | 300 | 0.25 A g−1 | 1 M H2SO4 | 133 |
| Banana fiber | NA | ZnCl2 | 800/1 | 1097 | 296 | 0.5 A g−1 | 1 M Na2SO4 | 126 |
| Chestnut shell | NA | ZnCl2 | 800/1.5 | 1987 | 92 | 10 A g−1 | 6 M KOH | 127 |
| Silk | NA | ZnCl2 | 900/1 | 2494 | 242 | 0.1 A g−1 | EMIMBF4 | 129 |
| Coconut shell | NA | ZnCl2 | 900/1 | 1874 | 276 | 1 A g−1 | 6 M KOH | 130 |
| Coffee bean | NA | H3PO4 | 800/0.5 | 742 | 160 | 1 A g−1 | 1 M H2SO4 | 140 |
| Cotton stalk | NA | H3PO4 | 800/2 | 1481 | 114 | 0.5 A g−1 | 1 M TEABF4/AN | 141 |
| Bamboo | NA | KHCO3 | 800/1 | 1425 | 187 | 0.5 A g−1 | 6 M KOH | 143 |
| Rice husk | NA | Microwave-assisted ZnCl2 | 600 W/1/3 | 1552 | 94 | 0.05 A g−1 | 1 M Et4NBF4/PC | 146 |
| Bagasse pith | NA | Microwave-assisted ZnCl2 | 700 W/0.25 | — | 138 | 0.2 A g−1 | 1 M EMIMBF4 | 158 |
| Cassava peel | NA | KOH/CO2 | 800/3 | 1186 | 264 | — | 0.5 M H2SO4 | 153 |
| 800/1 | ||||||||
| Oil palm | Pre-carbonisation | KOH/CO2 | 800/— | 1704 | 150 | — | 1 M H2SO4 | 157 |
| 800/3 |
2.4 Structure control
High SSA biomass-derived activated carbons contain predominantly micropores with a high pore tortuosity, which poses a large resistance to ion transport, leading to poor power density and rate capability of SC devices.25,159 In addition, pore geometry and dimension as well as electrical conductivity are also important parameters determining SC performance.143,160,161 Therefore, pore structure control and graphitisation degree are important for the electrocapacitive performance of biomass-derived activated carbons.162The pore structure of carbon materials can be made to have hierarchical pores in two dimensions (2D) or three dimensions (3D) to facilitate ion transport and provide a robust interface for charge storage.162 The transport behaviour of electrolyte ions in pores is significantly determined by pore length, pore size and tortuosity. The ion transport time (τ) is given by equation τ = L2/d,163 where L refers to the ion transport distance and d is the ion transport coefficient. A porous carbon with macropores, mesopores and micropores well-distributed in 2D or 3D with low-resistant ion-transport paths is ideal for EDLCs. This enables active ions in micropores to have nanometre transport distances from adjacent mesopores and macropores, thus shortening the transport time.139,162,164 Through careful control over the carbonisation and activation conditions, biomass-derived carbon electrodes with 2D53,129,164–167 or 3D78,116,118,143,168–170 hierarchical pore structures of high SSA have been reported, as shown in Table 2, which exhibited both high specific capacitance and excellent rate capability.
| Precursor | SSA (m2 g−1) | Pore structure | S meso+macro/St | V meso+macro/Vt | C 1 (F g−1) (symmetric SCs) | C 2 (F g−1) (symmetric SCs) | Rate capability | Electrolyte | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Silk proteins | 2557 | Microporous | 0.34 | — | 264 (0.1 A g−1) | 162 (6.2 A g−1) | 61% | 1 M H2SO4 | 101 |
| Broad beans | 655 | Rich in micropores | — | — | 202 (0.25 A g−1) | 129 (10 A g−1) | 63% | 6 M KOH | 115 |
| Sucrose | 2094 | Microporous | — | — | 224 (0.2 A g−1) | 91 (20 A g−1) | 41% | 6 M KOH | 212 |
| Nutshell | 1069 | 2D microporous | — | 0.17 | 261 (0.2 A g−1) | 97 (8 A g−1) | 37% | 6 M KOH | 165 |
| Acacia gum | 1832 | Microporous | 0.11 | 0.19 | 272 (1 A g−1) | 160 (10 A g−1) | 59% | 6 M KOH | 213 |
| Glucose | 2600 | Oriented and interlinked 2D hierarchical porous | 0.31 | 0.69 | 257 (0.5 A g−1) | 184 (100 A g−1) | 72% | 6 M KOH | 166 |
| Corn gluten meal waste | 3353 | Interconnected meso/microporous | — | — | 298 (0.5 A g−1) | 215 (10 A g−1) | 72% | 6 M KOH | 214 |
| Bagasse | 2296 | Hierarchical porous | 0.26 | 0.33 | 180 (0.2 A g−1) | 128 (15 A g−1) | 71% | 6 M KOH | 215 |
| Bamboo-based industrial by-products | 1472 | Beehive-like hierarchical nanoporous | — | 0.21 | 301 (0.1 A g−1) (three electrode) | 192 (100 A g−1) (three electrode) | 64% | 6 M KOH | 216 |
| Corn husk | 867 | 3D hierarchical porous | 0.14 | 0.27 | 260 (1 A g−1) | 228 (10 A g−1) | 88% | 6 M KOH | 176 |
| Artemia Cyst shells | 1758 | 3D hierarchical porous | 0.21 | 0.30 | 369 (0.5 A g−1) (three electrode) | 334 (10 A g−1) (three electrode) | 91% | 1 M H2SO4 | 217 |
| Rice bran | 2475 | 3D porous | 0.15 | 0.39 | 323 (0.1 A g−1) | 265 (10 A g−1) | 82% | 6 M KOH | 116 |
| Waste wood shavings | 3223 | Porous carbon fibre | 0.29 | 0.49 | 247 (0.5 A g−1) | 227 (10 A g−1) | 92% | 1 M H2SO4 | 87 |
| Gelatin | 3012 | Hierarchical porous | — | 0.33 | 385 (0.05 A g−1) | 281 (5 A g−1) | 73% | 6 M KOH | 218 |
| Lignin-derived by-products | 2218 | Interconnected hierarchical porous | — | — | 312 (1 A g−1) | 254 (80 A g−1) | 81% | 6 M KOH | 207 |
| Lignin | 907 | 3D hierarchical porous | 0.15 | 0.21 | 165 (0.05 A g−1) | 124 (10 A g−1) | 75% | 1 M H2SO4 | 168 |
Using glucose as a precursor, Zheng et al.166 prepared 2D porous carbon nanosheets with an excellent capacitive performance. During the activation process, potassium species acted as not only an activator but also a melt template that led to the oriented nanosheet structure. The obtained 2D porous carbon exhibited a specific capacitance of 257 F g−1 at a current density of 0.5 A g−1 and it was maintained at 184 F g−1 at 100 A g−1. The large amount of micropores and small mesopores led to a high SSA of around 2600 m2 g−1 which gave rise to the high specific capacitance. The interlinked 2D hierarchical porous structure facilitated ion transport, thus enabling the ultrahigh rate capability. Hao et al.169 prepared 3D hierarchical porous carbon electrode materials using bagasse as the raw material. They assembled solid state symmetric SCs and due to the advantages of the aforementioned hierarchically porous structure, a comparable high specific capacitance of 142 F g−1 at a current density of 0.5 A g−1 was obtained. The solid state SCs displayed a good rate capability and an excellent capacitance retention of 93.9% over 5000 cycles.
Improving the graphitisation of AC simultaneously enhances the electric conductivity of the electrode and its surface wettability towards aqueous electrolytes, which can facilitate ion diffusion and electron transfer, thus improving the electrochemical performance.130 High-temperature treatment can enhance graphitisation but it is of high energy consumption. Besides, it will also decrease the SSA and pore volume of the AC.
Catalytic graphitisation by means of a transition metal is an effective way to obtain ACs with a certain graphitisation degree.113 Coupling chemical activation with catalytic graphitisation enables the preparation of porous carbons with a high SSA and an excellent electrocapacitive performance.53,113,128–130,167 Sun et al.130 synthesised porous graphene-like nanosheets (PGNSs) with a large SSA via a simultaneous activation-graphitisation route using coconut shell as the precursor. FeCl3 and ZnCl2, functioning as a graphitic catalyst and activating agent respectively, were simultaneously introduced into the skeleton of the coconut shell through coordination of the metal precursors with the functional groups in the coconut shell, thus simultaneously carrying out activation and graphitisation. The obtained PGNSs possessed good electrical conductivity due to a high graphitisation degree, a SSA of 1874 m2 g−1 and a pore volume of 1.21 cm3 g−1. With no addition of conductive additives, it exhibited a specific capacitance of 268 F g−1 at 1 A g−1 in KOH electrolyte. Besides, it also displayed a capacitance of 196 F g−1 at 1 A g−1 in an organic electrolyte. An energy density of 54.7 W h kg−1 was obtained at a power density of 10 kW kg−1 (Fig. 2).
 | ||
| Fig. 2 HR-TEM (a), Raman spectra (b), rate performance (c) and Ragone plot (d) of PGNS.130 | ||
2.5 Heteroatom-doped biomass-derived carbon
It has been a widely accepted strategy to enhance the specific capacitance of a carbon electrode without sacrificing the high rate capability and long cycle life through doping with heteroatoms, such as N, O, P, S, and B. Some researchers obtained heteroatom-doped biomass-derived carbon by mixing a biomass precursor with other heteroatom-containing precursors171–173 or through post-treatment.174However, as a naturally available resource, the most interesting and convenient spot for biomass is that it can be transformed into heteroatom-contained carbon by a simple one-step calcination without the addition of any other precursors.52,67,114,115,129,175–177
Hou et al.129 prepared nitrogen-doped hierarchically porous carbon nanosheets via simultaneous activation and graphitisation, using natural silk. The carbon material exhibited good performance for both SC and lithium-ion batteries. Xu et al.115 prepared sulfur and nitrogen dual-doped porous carbon materials using broad bean shells which are abundant in amino acids and vitamins as the precursor. Broad beans were thermally treated at 800 °C for 2 h and then activated with a KOH ethanol solution at 650 °C for 1 h under nitrogen with a heating rate of 3 °C min−1. The doped sulfur increased the space utilisation by a specific electrosorption of electrolyte ions. The incorporation of nitrogen increased the electrical conductivity as well as the wettability of the electrode. In addition, both sulphur- and nitrogen-containing groups contributed to pseudocapacitance. Therefore, the prepared sample exhibited a specific capacitance of 202 F g−1 at a current density of 0.5 A g−1 in 6 M KOH aqueous electrolyte and maintained 129 F g−1 at 10 A g−1 in spite of the moderate SSA (655 m2 g−1) of the carbon.
3. Lignocellulose-derived carbon
During the past decade, biomass especially lignocellulose has attracted researchers' huge interest and research worldwide.21,58 However, the dependence of electrochemical performance upon the composition of lignocellulose is still unclear. Therefore, it is necessary and a trend to separately study cellulose, lignin and hemicellulose, three main components of lignocellulose, for the transformation and application of biomass into SC electrodes.178 Some researchers have conducted such research to some extent and in the following part, we will give them a brief review separately.3.1 Cellulose
Sevilla and coworkers62,70,71,75 produced activated carbon materials with cellulose as the carbon precursor. By HTC at 230–250 °C for 2 h and subsequent KOH activation at 700–800 °C for 1 h, the carbon electrode produced displayed a capacitance of 140 F g−1 at 10 mV s−1 and an excellent rate capability. Tam et al.183 conducted a one-step pyrolysis on melamine-formaldehyde cellulose nanocrystals. The sample pyrolysed at 900 °C displayed a capacitance value of 352 F g−1 at a current density of 5 A g−1 in a three-electrode system with 1 M H2SO4 electrolyte. Ji's group also presented a simple one-step fabrication methodology for nitrogen-doped nanoporous carbon membranes via annealing cellulose filter paper under NH3.188 They discovered that the doped nitrogen played an important role in the activation of carbon under NH3, leading to a large SSA. Nitrogen doping (up to 10.3 at%) occurred during cellulose pyrolysis under NH3 at as low as 550 °C. At 700 °C or above, N-doped carbon further reacted with NH3, resulting in a large SSA. Compared with conventional AC (1533 m2 g−1), the N-doped nanoporous carbon (1326 m2 g−1) exhibited more than double the unit area capacitance (90 vs. 41 mF m−2).
An interesting route recently has been attracting researchers' increasing interest, i.e., dissolving cellulose in other solutions, especially in NaOH/urea solution.184,185,189 Zhao et al.184 prepared meso–microporous activated carbons via the template method in combination with pyrolysis and ZnCl2 chemical activation using cellulose and biowaste lignosulphonate as the precursors. Cellulose was first regenerated to couple with the silica template. Lignosulphonate was then cast into the composites to fill voids and produce mesoporous carbon. A post-ZnCl2 activation was employed to further optimize the pore structure. The as obtained sample exhibited specific capacitances of 286 F g−1 and 141 F g−1 at current densities of 0.25 A g−1 and 10 A g−1, respectively in 6 M KOH electrolyte. However, the whole procedure of this route is kind of complex when considering the practical application for cellulose.
Some researchers tried to isolate cellulose from lignocellulose using different isolation processes and then transform them into ACs as SC electrodes, as shown in Fig. 3.169,176,186,187 In the approach reported by Wan's group176 as seen in Fig. 3a, corn husks were first added into KOH solution and subsequently refluxed at 80 °C for 4 h. The obtained colloidal liquid was filtered by using a stainless steel mesh. The obtained solid residue, i.e., cellulose, was carbonised at 800 °C for 1 h. The final obtained AC possessed a 3D architecture, a SSA of 928 m2 g−1, uniform pore size and rich O-doping (17.1%). It exhibited a specific capacitance of 260 F g−1 at 1 A g−1 and maintained up to 228 F g−1 at 10 A g−1. Besides, it also displayed an energy density of 21 W h kg−1 at a power density of 875 W kg−1 in Na2SO4 aqueous electrolyte with a cell voltage of 1.8 V.
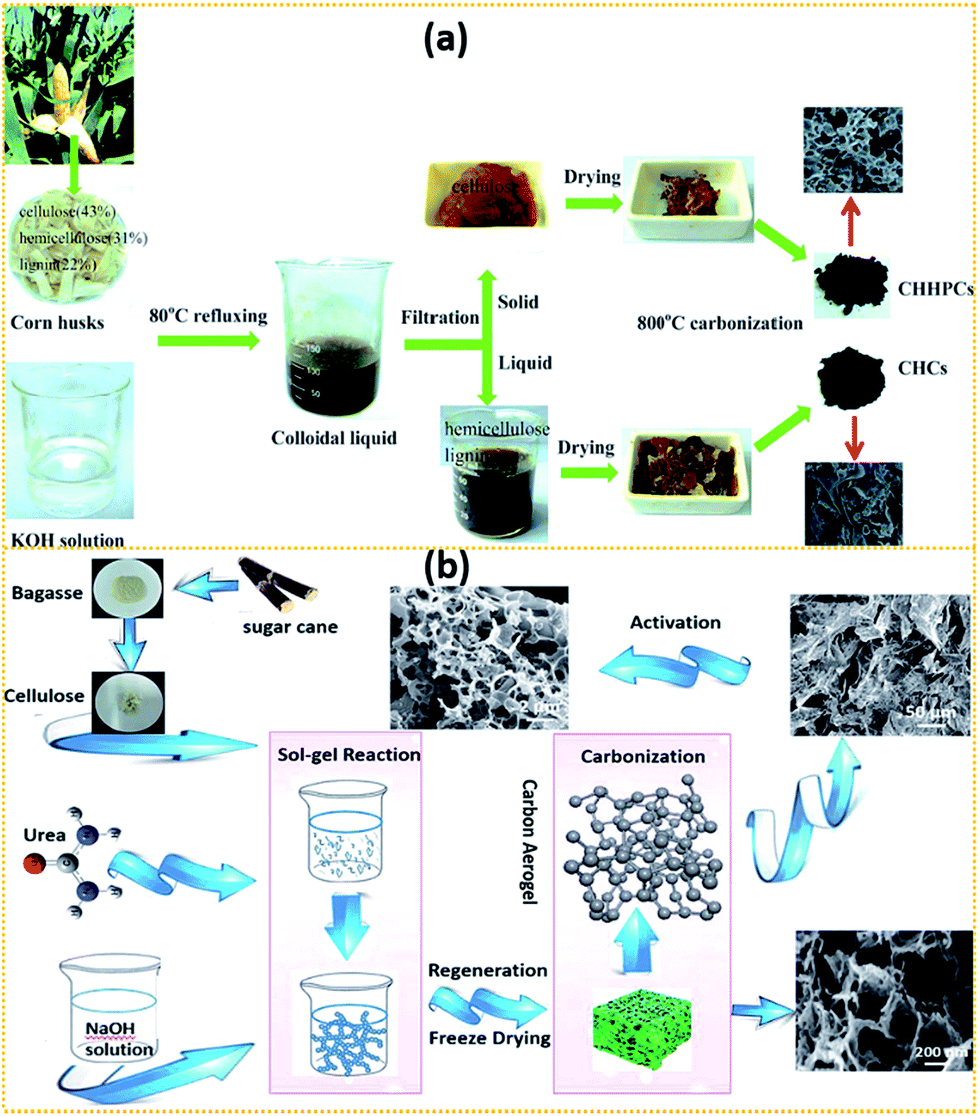 | ||
| Fig. 3 A scheme showing isolating cellulose from (a) corn husk176 and (b) bagasse169 using two different isolation processes. | ||
As shown in Fig. 3b, Hao et al.169 purified cellulose from bagasse by alkaline hydrolysis and then bleaching using a sodium chlorite/glacial acetic acid mixture. 7 wt% of the obtained cellulose was dispersed into the solvent mixture of NaOH/urea/H2O (7.5:11.5:81) precooled to −12 °C under vigorous stirring for 4 h at −6 °C. The cellulose sol was then freeze dried at −80 °C for 12 h. The obtained aerogel was pyrolysed and KOH-activated at different temperatures. At last, 3D hierarchical porous ACs were obtained and they exhibited good performance in solid state symmetric SCs.
3.2 Lignin
Generally, lignin can be classified into two main categories: sulfur lignin and sulfur-free lignin. Sulfur lignin includes Kraft lignin (alkali lignin) and lignosulfonates, while the latter includes soda lignin and organosolv lignin.
Lignin has a carbon to oxygen ratio of above 2:1, and thus is more energy dense than cellulose and hemicellulose. Compared with cellulose, there is a less detailed elucidation of lignin's structure by the experimental method. However, the currently commercially available lignin, which is the by-product of the cellulose industry, is more easily dissolved in aqueous solvent than cellulose.195 Therefore, it seems more easily for lignin acting as the carbon precursor for SC electrodes.
One of the interesting routes is the synthesis of lignin-based carbon fibre through electrospinning.197–200 Hu et al.197 prepared AC fibres through both NaOH (Na-ACFs) and KOH (K-ACFs) activation using low sulfonated alkali lignin as the precursor. The hydrophilic and high SSA ACFs exhibited large-size nanographites and good electrical conductivity to demonstrate a good electrochemical performance. K-ACFs showed a specific capacitance of 344 F g−1 at 10 mV s−1, much higher than that of Na-ACF. The superior electrochemical properties of SC constructed with K-ACF over Na-ACF were attributed mainly to the higher microporosity and more narrowly distributed pore size.
Another interesting route is to prepare lignin-derived porous carbons through a template201–205 or template-free168,206–209 method. As shown in Fig. 4a, Saha et al.201 synthesised mesoporous carbon using the surfactant Pluronic F127, a triblock copolymer, as the template. Subsequent CO2 physical activation and KOH chemical activation enhanced the SSA of the pristine mesoporous carbon to 624 and 1148 m2 g−1 respectively, with the present of a certain percent of micropores. The CO2-activated and KOH-activated mesoporous carbon exhibited a capacitance of 102.3 and 91.7 F g−1 respectively. Zhang et al.168 prepared lignin-derived HPCs (LHPCs) through a template-free method, as seen in Fig. 4b. Lignin was first dissolved in KOH solution and then solidified. In the composite, KOH crystallised into small particles with different sizes. Then the composites were pyrolysed at 700 °C for 2 h when the crystallised KOH particles act as both the template and activation agent. The finally obtained LHPCs consist of a 3D hierarchical porous network as well as a large amount of oxygen-containing groups, which contributes to the pseudocapacitance. It exhibited a capacitance of 165 F g−1 at 0.05 A g−1 and maintained 123 F g−1 at 10 A g−1. Besides, the hierarchical porous structure enables it to maintain over 97% of the initial value at 1 A g−1 after 5000 cycles.
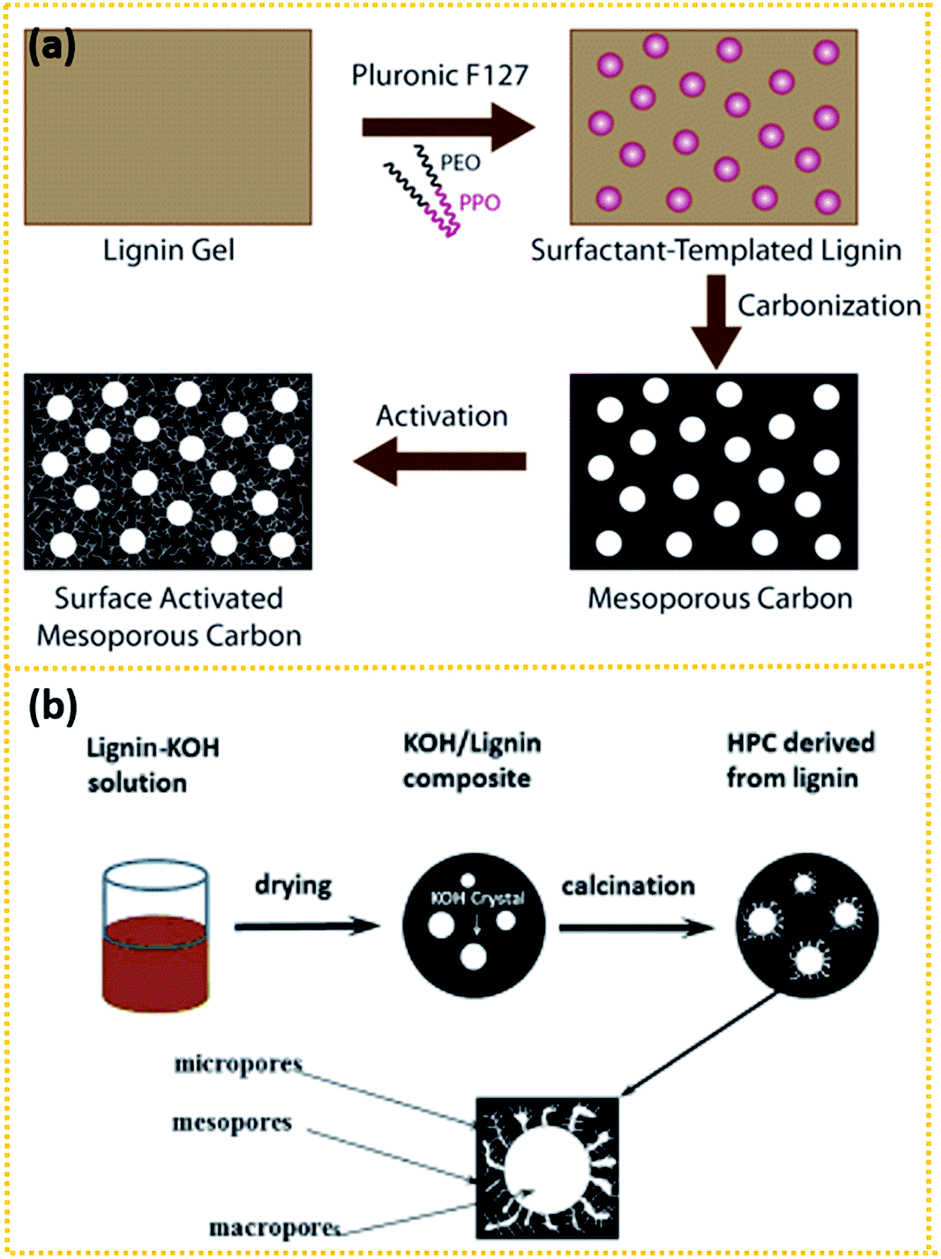
3.3 Hemicellulose
Hemicellulose, constituting approximately 20–35% of lignocellulose, is the second most common polysaccharide in nature. While cellulose is crystalline, strong, and resistant to hydrolysis, hemicellulose has a random, amorphous structure with little strength. Therefore, it can be easily hydrolysed in dilute acid or base.210 However, research on hemicellulose is less as most researchers focus on the other two main components of lignocellulose, i.e., cellulose and lignin. Therefore review on the application of hemicellulose for SC electrodes here will be relatively brief.Wang et al.211 and Falco et al.66 extracted hemicellulose from biomass through base and acid hydrolysis, respectively. In Wang's experiment, a certain amount of hemp stems powder was soaked in 6 wt% NaOH aqueous solution with stirring for 12 h at 40 °C. The hemicellulose was then isolated by precipitation of filtrate with two volumes of ethanol. The extracted hemicellulose was added into 5 wt% H2SO4 solution and was then HTC treated at 160 °C for 12 h. By contrast, Falco and coworkers impregnated corncob and spruce in diluted acid solution and the hydrolysed hemicellulose products was HTC treated at 200 °C for 24 h. Lastly, both samples were KOH-activated to increase the SSA and porosity. The finally obtained samples exhibited a specific capacitance of 240 F g−1 at 0.1 A g−1 in 6 M KOH and 315 F g−1 at 0.25 A g−1 in 0.5 M H2SO4, respectively. Anyhow, more research focusing on hemicellulose-derived carbon electrode materials for SCs is needed (Table 3).
| Main materials | Activation | Modification | SSA (m2 g−1) | C (F g−1) (symmetric SCs) | Measurements at | Electrolyte | Ref. |
|---|---|---|---|---|---|---|---|
| Lignosulphonate cellulose | ZnCl2 | NA | 856 | 286 | 0.25 A g−1 | 6 M KOH | 184 |
| Cellulose filter paper | NH3 | N-Doped | 1326 | 120 | 1 A g−1 | 2 M KOH | 188 |
| Bagasse-derived cellulose | KOH | NA | 1892 | 142 | 0.5 A g−1 | KOH/PVA gel | 169 |
| Corn husk-derived cellulose | NA | NA | 867 | 260 | 1 A g−1 | 6 M KOH | 176 |
| Cellulose acetate | Steam | MWNT | 1120 | 145 | 10 A g−1 | 6 M KOH | 178 |
| Paper cellulose | NA | SWNT | — | 200 | — | 1 M H2SO4 | 219 |
| Textiles | NA | SWNT | — | 140 | 20 μA cm−2 | Organic electrolyte | 220 |
| Bacterial nanocellulose paper | NA | CNT | — | 50.5 | 1 A g−1 | Ion gel | 221 |
| Cellulose nanofiber | NA | RGO | — | 207 | 5 mV s−1 | H2SO4/PVA | 222 |
| Cellulose nanocrystals | NA | PPy | — | 336 | — | 0.1 M KCl | 187 |
| Cellulose sponge | NA | MnO2/CNT | — | 1000 | 1 mV s−1 | 1 M Na2SO4 | 223 |
| Cellulose fibres | NA | CNT/MnO2 | 327 | 10 mV s−1 | 1 M Na2SO4 | 224 | |
| Cellulose nanofibers | NA | Ni(OH)2 | — | 172 (asymmetric) | 1 mV s−1 | 6 M KOH | 225 |
| Low sulfonated alkali lignin | KOH | NA | 1400 | 344 | 10 mV s−1 | 6 M KOH | 197 |
| Alcell lignin | NA | NA | 930 | 116 | 1 A g−1 | 1 M H2SO4 | 204 |
| Hardwood kraft lignin | CO2 | NA | 624 | 102 | 1 mV s−1 | 6 M KOH | 201 |
| Lignin | NA | NA | 803 | 208 | 0.1 A g−1 | 6 M KOH | 203 |
| Lignin-derived by-products | KOH | NA | 2218 | 141 | 1 A g−1 | EMI–BF4 | 207 |
| Alkali lignin | KOH | NA | 3775 | 286 | 0.2 A g−1 | 6 M KOH | 208 |
| Kraft lignin | KOH | NA | 1406 | 87 | 0.1 A g−1 | 1.5 M NEt4BF4/ACN | 206 |
| Solvent lignin | KOH | N-Doped | 3130 | 306 | 0.1 A g−1 | KOH/PVA | 196 |
| Softwood lignin | KOH | S/O-doped | 1800 | 231 | 1 A g−1 | EMI–BF4 | 226 |
| Lignin | NA | BC | 199 | 124 | 0.5 A g−1 | 6 M KOH | 227 |
| Solvent lignin | KOH | Aniline | 2265 | 336 | 1 A g−1 | 6 M KOH | 228 |
| Natural lignin | NA | rGO/PEDOT | — | 144 | 0.1 A g−1 | 0.1 M HClO4 | 229 |
| Sodium lignosulphonate | NA | NiO | 802 | 880 (three-electrode) | 1 A g−1 | 6 M KOH | 230 |
| Hemp-derived hemicellulose | KOH | NA | 3062 | 240 | 0.1 A g−1 | 6 M KOH | 211 |
| Corncob-derived hemicellulose | KOH | NA | 2300 | 315 | 0.25 A g−1 | 0.5 M H2SO4 | 66 |
3.4 Modification of lignocellulose-derived carbon
Researchers could conduct similar physical or chemical processes on lignocellulose to get a high SSA or hierarchically porous structure as depicted in Section 2.4. In this section, we will preferably focus on other modifications of lignocellulose-derived carbons.Cui's group made conductive and stretchable SWNT-paper219,231 and SWNT-textile220 electrodes for SCs by using simple solution processes, as shown in Fig. 5. In their study, paper or ordinary textiles were explored as a platform for SCs by integration with SWNTs via rod coating or dipping and drying methods. The coated cellulose films, functioning as both electrodes and current collectors, show high conductivity, porosity, and robust chemical and mechanical stability, which lead to high-performance SCs. Deng178 and Kang et al.221 also researched modification on cellulose-derived carbons with CNTs.
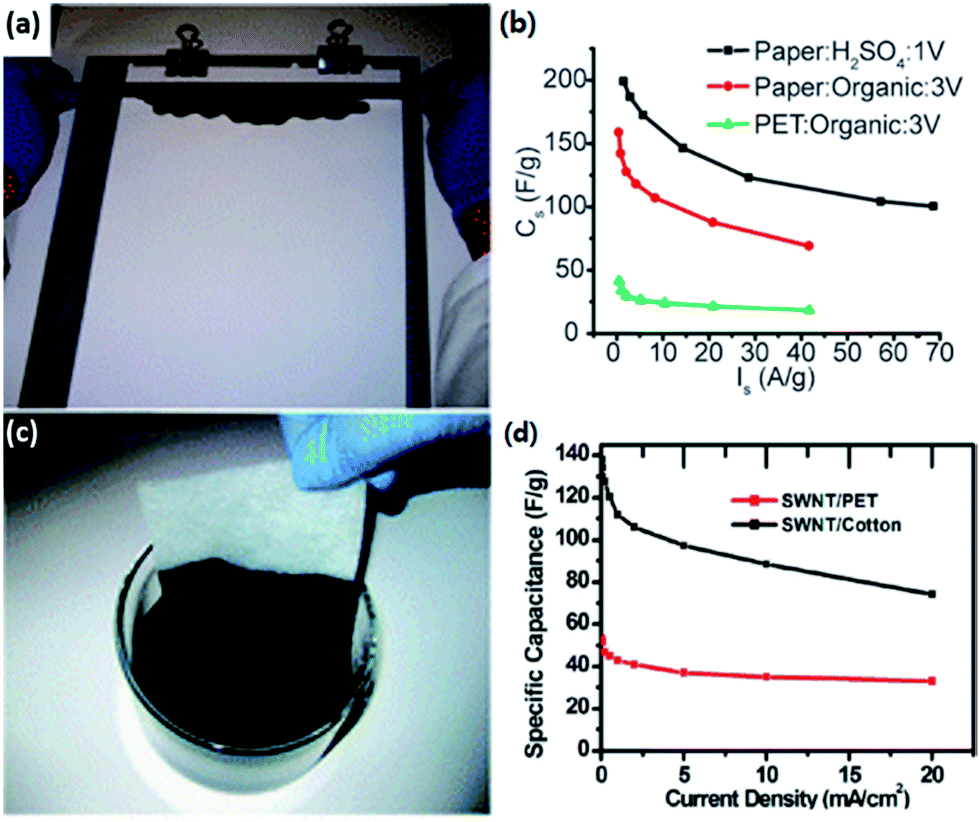 | ||
| Fig. 5 Cellulose/SWNT composite electrode through a simple rod coating219 (a) or dipping and drying220 (c) method, and their electrochemical performance respectively (b) and (d). | ||
Gao et al.222 reported a cellulose nanofiber-reduced graphene oxide (CNF–RGO) hybrid aerogel as the electrode material for all-solid-state flexible SCs. In their experiment, CNF–RGO hybrid hydrogels were prepared by acidizing homogeneous solution of CNFs and GO nanosheets with hydrochloric acid vapor. Then the hybrid aerogel was prepared by supercritical CO2 drying. The finally obtained flexible SCs exhibited a capacitance of 207 F g−1 at 5 mV s−1 in H2SO4/PVA gel electrolyte. It showed a good electrochemical stability under bent state. The capacitance remained at 207 F g−1 in bent state (180°) and did not change obviously after 100 bending cycles.
Biomass-based carbon aerogels represent an important novel research direction in aerogel development. However, lignin-derived aerogel is brittle and fragile. Xu et al.227 toughened lignin-resorcinol-formaldehyde (LRF) aerogel using bacterial cellulose (BC) through a catalyst-free process. The toughened and graphitised lignin-derived aerogel exhibited a core–shell nanostructure and it can undergo at least 20% reversible compressive deformation. The large mesopore ratio and core–shell nanostructure of the sample, with BC-derived carbon nanofiber as the backbone and LRF-converted carbon as the coating respectively, enable it to show a high areal capacitance of 62.2 μF cm−2 at 0.5 A g−1 with a relatively low SSA, 119.4 m2 g−1.
Both conducting polymer and metal oxides can provide higher capacitance than carbon materials. However, their electrochemical stability and conductivity are relatively poor compared with porous carbon. Therefore, researchers coated a conductive polymer187,228,229,232–235 or metal oxides199,223–225,230 on cellulose-derived or lignin-derived porous carbons, which function as the backbone.
Alshareef's group223 fabricated a “sponge supercapacitor” using a MnO2–CNT–sponge hybrid electrode. The macroporous nature of the cellulose-sponge as well as the porous nature of the electrodeposited MnO2 nanoparticles provided a double porous electrode structure, giving rise to good conductivity and full accessibility of the electrolyte to MnO2. The capacitance was dramatically increased to 1230 F g−1 (based on the mass of MnO2) at a scan rate of 1 mV s−1 in a three-electrode system. Besides, it also showed excellent rate capability and cycle stability.
Chen et al.230 incorporated NiO nanoparticles into lignin-derived mesoporous carbon (MPC) using a LC phase-templating preparation method. The obtained core–shell structured NiO@MPC composite not only increased the utilization of NiO, but also improved its electrical conductivity and mechanical strength. It exhibited a specific capacitance of 880.2 F g−1 at a current density of 1 A g−1, with enhanced rate capability and cycle stability—90.9% and 93.7% of the capacitance value was maintained at a current density of 10 A g−1 and after 1000 GCD cycles, respectively.
Another direction to increase the capacitance of lignocellulose-derived carbon materials is by heteroatom doping. Wide research has been focused on heteroatom doping, including N, O, S, and P, on polymer-derived or biomass-derived carbon electrodes. However, there is still little research on doping heteroatoms in specific cellulose,188 lignin196,205,226 or hemicellulose-derived carbons and more research is required.
4. Conclusions and prospects
Biomass is a good resource for making functional carbon materials for electrocapacitive energy storage applications such as electrodes, separators, or binders. Recent research has shown that affordable biomass-derived carbon materials with electrocapacitive properties comparable to commercial activated carbon can be prepared using simple carbonisation and/or activation methods. Advanced hierarchical porous biomass-derived carbons of excellent electrocapacitive performance can be obtained by using the chemical activation methods using, for example, potassium hydroxide. To enable biomass-derived carbon materials to find practical applications in energy storage, challenges and issues must be addressed.First, considering the diversity of biomass resources, fundamental research focusing on the effect of biomass composition on the electrocapacitive properties of resulting carbon is needed. Perhaps, it could be a good practice to do this kind of research with cellulose-, lignin- and hemicellulose-derived carbon materials.
Second, the majority of the reported biomass-derived electrode materials of excellent electrocapacitive performance were prepared at the lab scale. There is a need to do scale-up research to develop a process protocol for further development of the technology.
Third, while hierarchical porous biomass-derived carbons of high specific surface area can be prepared using the KOH-activation method, it has little control over the pore geometry, pore size and pore connection. Besides, the KOH-activation method is unfavourable for creating graphitic carbons, which determines the electrical conductivity and surface wettability towards an electrolyte. KOH activation in combination with thermal treatment may enable one to prepare biomass-derived carbons of a high specific surface area, appropriate pore structure, good electrical conductivity and good wettability towards electrolytes.
Forth, there has been an increasing demand for flexible supercapacitor devices with advantages of portability and flexibility for portable electronics applications. Biomass offers opportunities for making flexible electrodes as fibres or foams. This will open up a new research area.
Fifth, the energy density of supercapacitors can be enhanced by increasing electrode capacitance or widening the operation voltage. Adding pseudocapacitive materials to biomass-derived carbon such as heteroatoms, metal oxides, or conductive polymers, is a future research direction to increase the capacitance. Widening of the voltage window by using ionic liquids as electrolytes or configuring asymmetric cells or fabricating hybrid capacitors will be another effective way to improve both energy density and power density.
Acknowledgements
This work was supported by the Australian Research Council Discovery Project (DP 130101870) and the University of Queensland Research and Teaching Fellowship Program (2015000144). HL wishes to acknowledge the China Scholarship Council (CSC) and the University of Queensland for providing scholarship.References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. Zhang and X. Zhao, ChemSusChem, 2012, 5, 818–841 CrossRef CAS PubMed.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917–918 CrossRef CAS PubMed.
- C. Xu, B. Xu, Y. Gu, Z. Xiong, J. Sun and X. Zhao, Energy Environ. Sci., 2013, 6, 1388–1414 CAS.
- L. L. Zhang and X. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P.-L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- Y. P. Zhai, Y. Q. Dou, D. Y. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- W. Gu and G. Yushin, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 424–473 CrossRef CAS.
- P. Thounthong, V. Chunkag, P. Sethakul, S. Sikkabut, S. Pierfederici and B. Davat, J. Power Sources, 2011, 196, 313–324 CrossRef CAS.
- M. Yu, W. D. McCulloch, Z. Huang, B. B. Trang, J. Lu, K. Amine and Y. Wu, J. Mater. Chem. A, 2016, 4, 2766–2782 CAS.
- H. Chen, T. N. Cong, W. Yang, C. Tan, Y. Li and Y. Ding, Prog. Nat. Sci., 2009, 19, 291–312 CrossRef CAS.
- K. Q. Peng, X. Wang, L. Li, Y. Hu and S. T. Lee, Nano Today, 2013, 8, 75–97 CrossRef CAS.
- A. P. Cohn, W. R. Erwin, K. Share, L. Oakes, A. S. Westover, R. E. Carter, R. Bardhan and C. L. Pint, Nano Lett., 2015, 15, 2727–2731 CrossRef CAS PubMed.
- L. Staaf, P. Lundgren and P. Enoksson, Nano Energy, 2014, 9, 128–141 CrossRef CAS.
- L. E. Bell, Science, 2008, 321, 1457–1461 CrossRef CAS PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- Z. Yu, L. Tetard, L. Zhai and J. Thomas, Energy Environ. Sci., 2015, 8, 702–730 CAS.
- L. L. Zhang, Y. Gu and X. Zhao, J. Mater. Chem. A, 2013, 1, 9395–9408 CAS.
- L. Zhang, Z. Liu, G. Cui and L. Chen, Prog. Polym. Sci., 2015, 43, 136–164 CrossRef CAS.
- A. M. Abioye and F. N. Ani, Renewable Sustainable Energy Rev., 2015, 52, 1282–1293 CrossRef CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- N. Balahmar, A. C. Mitchell and R. Mokaya, Adv. Energy Mater., 2015, 5, 1500867 CrossRef.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 CAS.
- A. F. Dalebrook, W. Gan, M. Grasemann, S. Moret and G. Laurenczy, Chem. Commun., 2013, 49, 8735–8751 RSC.
- C. Zhang, Z. Geng, M. Cai, J. Zhang, X. Liu, H. Xin and J. Ma, Int. J. Hydrogen Energy, 2013, 38, 9243–9250 CrossRef CAS.
- Y. Matsumura, M. Sasaki, K. Okuda, S. Takami, S. Ohara, M. Umetsu and T. Adschiri, Combust. Sci. Technol., 2006, 178, 509–536 CrossRef CAS.
- X. Liao, C. Chen, Z. Wang, R. Wan, C.-H. Chang, X. Zhang and S. Xie, Process Biochem., 2013, 48, 312–316 CrossRef CAS.
- M. M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- N. Liu, K. Huo, M. T. McDowell, J. Zhao and Y. Cui, Sci. Rep., 2013, 3, 1919 CrossRef PubMed.
- A. Jain, V. Aravindan, S. Jayaraman, P. S. Kumar, R. Balasubramanian, S. Ramakrishna, S. Madhavi and M. Srinivasan, Sci. Rep., 2013, 3, 3002 CrossRef PubMed.
- D. Larcher and J. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- R. R. Gaddam, D. Yang, R. Narayan, K. Raju, N. A. Kumar and X. Zhao, Nano Energy, 2016, 26, 346–352 CrossRef CAS.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- L. Jabbour, R. Bongiovanni, D. Chaussy, C. Gerbaldi and D. Beneventi, Cellulose, 2013, 20, 1523–1545 CrossRef CAS.
- Q. Ma, Y. Yu, M. Sindoro, A. G. Fane, R. Wang and H. Zhang, Adv. Mater., 2017, 29, 1605361 CrossRef PubMed.
- S. Dutta, A. Bhaumik and K. C.-W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 CAS.
- K. Qian, A. Kumar, H. Zhang, D. Bellmer and R. Huhnke, Renewable Sustainable Energy Rev., 2015, 42, 1055–1064 CrossRef CAS.
- A. Jain, R. Balasubramanian and M. Srinivasan, Chem. Eng. J., 2016, 283, 789–805 CrossRef CAS.
- R. Saidur, E. Abdelaziz, A. Demirbas, M. Hossain and S. Mekhilef, Renewable Sustainable Energy Rev., 2011, 15, 2262–2289 CrossRef CAS.
- M. M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 Search PubMed.
- H. Wang, Z. Li and D. Mitlin, ChemElectroChem, 2014, 1, 332–337 CrossRef.
- M. M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- M. M. Titirici, M. Antonietti and A. Thomas, Chem. Mater., 2006, 18, 3808–3812 CrossRef CAS.
- R. Demir-Cakan, P. Makowski, M. Antonietti, F. Goettmann and M. M. Titirici, Catal. Today, 2010, 150, 115–118 CrossRef CAS.
- M. Sevilla, J. A. Maciá-Agulló and A. B. Fuertes, Biomass Bioenergy, 2011, 35, 3152–3159 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 CAS.
- L. Zhao, N. Baccile, S. Gross, Y. Zhang, W. Wei, Y. Sun, M. Antonietti and M.-M. Titirici, Carbon, 2010, 48, 3778–3787 CrossRef CAS.
- Y. Ren, Q. Xu, J. Zhang, H. Yang, B. Wang, D. Yang, J. Hu and Z. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 9689–9697 CAS.
- Y. Q. Zhao, M. Lu, P. Y. Tao, Y. J. Zhang, X. T. Gong, Z. Yang, G. Q. Zhang and H. L. Li, J. Power Sources, 2016, 307, 391–400 CrossRef CAS.
- H. Wang, Z. Xu, A. Kohandehghan, Z. Li, K. Cui, X. Tan, T. J. Stephenson, C. K. King'ondu, C. M. Holt and B. C. Olsen, ACS Nano, 2013, 7, 5131–5141 CrossRef CAS PubMed.
- L. Zhang, F. Zhang, X. Yang, K. Leng, Y. Huang and Y. Chen, Small, 2013, 9, 1342–1347 CrossRef CAS PubMed.
- J. Jiang, J. Zhu, W. Ai, Z. Fan, X. Shen, C. Zou, J. Liu, H. Zhang and T. Yu, Energy Environ. Sci., 2014, 7, 2670–2679 CAS.
- K. László, A. Bóta and L. G. Nagy, Carbon, 2000, 38, 1965–1976 CrossRef.
- E. Raymundo-Piñero, F. Leroux and F. Béguin, Adv. Mater., 2006, 18, 1877–1882 CrossRef.
- E. Raymundo-Piñero, M. Cadek and F. Béguin, Adv. Funct. Mater., 2009, 19, 1032–1039 CrossRef.
- M. Biswal, A. Banerjee, M. Deo and S. Ogale, Energy Environ. Sci., 2013, 6, 1249–1259 CAS.
- Z. Li, W. Lv, C. Zhang, B. Li, F. Kang and Q.-H. Yang, Carbon, 2015, 92, 11–14 CrossRef CAS.
- D. Puthusseri, V. Aravindan, S. Madhavi and S. Ogale, Energy Environ. Sci., 2014, 7, 728–735 CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281–2289 CrossRef CAS.
- M. M. Titirici, A. Thomas, S.-H. Yu, J.-O. Müller and M. Antonietti, Chem. Mater., 2007, 19, 4205–4212 CrossRef CAS.
- S. H. Yu, X. Cui, L. Li, K. Li, B. Yu, M. Antonietti and H. Cölfen, Adv. Mater., 2004, 16, 1636–1640 CrossRef CAS.
- H. Zhu, X. Wang, F. Yang and X. Yang, Adv. Mater., 2011, 23, 2745–2748 CrossRef CAS PubMed.
- C. Falco, J. M. Sieben, N. Brun, M. Sevilla, T. Van der Mauelen, E. Morallón, D. Cazorla-Amorós and M. M. Titirici, ChemSusChem, 2013, 6, 374–382 CrossRef CAS PubMed.
- T. Wei, X. Wei, Y. Gao and H. Li, Electrochim. Acta, 2015, 169, 186–194 CrossRef CAS.
- C. Ruan, K. Ai and L. Lu, RSC Adv., 2014, 4, 30887–30895 RSC.
- L. Zhao, L. Z. Fan, M. Q. Zhou, H. Guan, S. Qiao, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 5202–5206 CrossRef CAS PubMed.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Energy Mater., 2011, 1, 356–361 CrossRef CAS.
- M. Sevilla, A. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 4, 1400–1410 CAS.
- C. Falco, J. P. Marco-Lozar, D. Salinas-Torres, E. Morallón, D. Cazorla-Amorós, M.-M. Titirici and D. Lozano-Castelló, Carbon, 2013, 62, 346–355 CrossRef CAS.
- M. Sevilla, W. Gu, C. Falco, M. Titirici, A. Fuertes and G. Yushin, J. Power Sources, 2014, 267, 26–32 CrossRef CAS.
- S. Roman, J. V. Nabais, B. Ledesma, J. González, C. Laginhas and M. Titirici, Microporous Mesoporous Mater., 2013, 165, 127–133 CrossRef CAS.
- D. Salinas-Torres, D. Lozano-Castelló, M. M. Titirici, L. Zhao, L. Yu, E. Morallón and D. Cazorla-Amoros, J. Mater. Chem. A, 2015, 3, 15558–15567 CAS.
- J. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710–23725 RSC.
- M. Choi and R. Ryoo, J. Mater. Chem., 2007, 17, 4204–4209 RSC.
- Z. Li, L. Zhang, B. S. Amirkhiz, X. Tan, Z. Xu, H. Wang, B. C. Olsen, C. Holt and D. Mitlin, Adv. Energy Mater., 2012, 2, 431–437 CrossRef CAS.
- Y. Gong, H. Wang, Z. Wei, L. Xie and Y. Wang, ACS Sustainable Chem. Eng., 2014, 2, 2435–2441 CrossRef CAS.
- D. Jiménez-Cordero, F. Heras, M. A. Gilarranz and E. Raymundo-Piñero, Carbon, 2014, 71, 127–138 CrossRef.
- Y. Gong, Z. Wei, J. Wang, P. Zhang, H. Li and Y. Wang, Sci. Rep., 2014, 4, 6349 CrossRef CAS PubMed.
- X. H. Duan, C. Srinivasakannan, K. B. Yang, J. H. Peng and L. B. Zhang, Waste Biomass Valorization, 2012, 3, 131–139 CrossRef CAS.
- B. del-Campo, M. Morris, D. Laird, M. Kieffer and R. Brown, Technology, 2015, 3, 104–113 CrossRef.
- J. Górka and M. Jaroniec, Carbon, 2011, 49, 154–160 CrossRef.
- W. H. Qu, Y. Y. Xu, A. H. Lu, X. Q. Zhang and W. C. Li, Bioresour. Technol., 2015, 189, 285–291 CrossRef CAS PubMed.
- J. Mi, X. R. Wang, R. J. Fan, W. H. Qu and W. C. Li, Energy Fuels, 2012, 26, 5321–5329 CrossRef CAS.
- Z. Jin, X. Yan, Y. Yu and G. Zhao, J. Mater. Chem. A, 2014, 2, 11706–11715 CAS.
- S. Osswald, C. Portet, Y. Gogotsi, G. Laudisio, J. Singer, J. Fischer, V. Sokolov, J. Kukushkina and A. Kravchik, J. Solid State Chem., 2009, 182, 1733–1741 CrossRef CAS.
- D. Wu, Y. Liang, X. Yang, C. Zou, Z. Li, G. Lv, X. Zeng and R. Fu, Langmuir, 2008, 24, 2967–2969 CrossRef CAS PubMed.
- H. Wang, Q. Gao and J. Hu, J. Am. Chem. Soc., 2009, 131, 7016–7022 CrossRef CAS PubMed.
- Y. Yan, J. Wei, F. Zhang, Y. Meng, B. Tu and D. Zhao, Microporous Mesoporous Mater., 2008, 113, 305–314 CrossRef CAS.
- N. P. Wickramaratne, J. Xu, M. Wang, L. Zhu, L. Dai and M. Jaroniec, Chem. Mater., 2014, 26, 2820–2828 CrossRef CAS.
- M. Noked, S. Okashy, T. Zimrin and D. Aurbach, Angew. Chem., Int. Ed., 2012, 51, 1568–1571 CrossRef CAS PubMed.
- W. Hao, E. Björkman, M. Lilliestråle and N. Hedin, Appl. Energy, 2013, 112, 526–532 CrossRef CAS.
- E. David and J. Kopac, J. Anal. Appl. Pyrolysis, 2014, 110, 322–332 CrossRef CAS.
- E. Taer, M. Deraman, I. A. Talib, A. A. Umar, M. Oyama and R. M. Yunus, Current Applied Physics, 2010, 10, 1071–1075 CrossRef.
- J. V. Nabais, J. G. Teixeira and I. Almeida, Bioresour. Technol., 2011, 102, 2781–2787 CrossRef PubMed.
- M. Ruiz-Fernández, M. Alexandre-Franco, C. Fernández-González and V. Gómez-Serrano, Adsorption, 2011, 17, 621–629 CrossRef.
- M. Song, B. Jin, R. Xiao, L. Yang, Y. Wu, Z. Zhong and Y. Huang, Biomass Bioenergy, 2013, 48, 250–256 CrossRef CAS.
- C. A. Toles, W. E. Marshall, L. H. Wartelle and A. McAloon, Bioresour. Technol., 2000, 75, 197–203 CrossRef CAS.
- Y. S. Yun, S. Y. Cho, J. Shim, B. H. Kim, S. J. Chang, S. J. Baek, Y. S. Huh, Y. Tak, Y. W. Park and S. Park, Adv. Mater., 2013, 25, 1993–1998 CrossRef CAS PubMed.
- T. Otowa, R. Tanibata and M. Itoh, Gas Sep. Purif., 1993, 7, 241–245 CrossRef CAS.
- E. Raymundo-Pinero, P. Azais, T. Cacciaguerra, D. Cazorla-Amorós, A. Linares-Solano and F. Béguin, Carbon, 2005, 43, 786–795 CrossRef CAS.
- W. Qiao, S. H. Yoon and I. Mochida, Energy Fuels, 2006, 20, 1680–1684 CrossRef CAS.
- D. Lozano-Castello, J. Calo, D. Cazorla-Amoros and A. Linares-Solano, Carbon, 2007, 45, 2529–2536 CrossRef CAS.
- K. Karthikeyan, S. Amaresh, S. N. Lee, X. Sun, V. Aravindan, Y. G. Lee and Y. S. Lee, ChemSusChem, 2014, 7, 1435–1442 CrossRef CAS PubMed.
- B. Li, F. Dai, Q. Xiao, L. Yang, J. Shen, C. Zhang and M. Cai, Energy Environ. Sci., 2016, 9, 102–106 CAS.
- M. Sevilla, L. Yu, C. O. Ania and M. M. Titirici, ChemElectroChem, 2014, 1, 2138–2145 CrossRef CAS.
- W. Qian, F. Sun, Y. Xu, L. Qiu, C. Liu, S. Wang and F. Yan, Energy Environ. Sci., 2014, 7, 379–386 CAS.
- C. Liu, G. Han, Y. Chang, Y. Xiao, M. Li, W. Zhou, D. Fu and W. Hou, ChemElectroChem, 2016, 3, 323–331 CrossRef CAS.
- J. Song, W. Shen, J. Wang and W. Fan, Carbon, 2014, 69, 255–263 CrossRef CAS.
- C. Lu, S. Xu, Y. Gan, S. Liu and C. Liu, Carbon, 2005, 43, 2295–2301 CrossRef CAS.
- L. Jiang, J. Yan, L. Hao, R. Xue, G. Sun and B. Yi, Carbon, 2013, 56, 146–154 CrossRef CAS.
- R. Wang, P. Wang, X. Yan, J. Lang, C. Peng and Q. Xue, ACS Appl. Mater. Interfaces, 2012, 4, 5800–5806 CAS.
- G. Xu, J. Han, B. Ding, P. Nie, J. Pan, H. Dou, H. Li and X. Zhang, Green Chem., 2015, 17, 1668–1674 RSC.
- J. Hou, C. Cao, X. Ma, F. Idrees, B. Xu, X. Hao and W. Lin, Sci. Rep., 2014, 4, 7260 CrossRef CAS PubMed.
- Z. Li, Z. Xu, H. Wang, J. Ding, B. Zahiri, C. M. Holt, X. Tan and D. Mitlin, Energy Environ. Sci., 2014, 7, 1708–1718 CAS.
- S. H. Du, L. Q. Wang, X. T. Fu, M. M. Chen and C. Y. Wang, Bioresour. Technol., 2013, 139, 406–409 CrossRef CAS PubMed.
- C. Peng, X. B. Yan, R. T. Wang, J. W. Lang, Y. J. Ou and Q. J. Xue, Electrochim. Acta, 2013, 87, 401–408 CrossRef CAS.
- M. Galinski, K. Babeł and K. Jurewicz, J. Power Sources, 2013, 228, 83–88 CrossRef CAS.
- K. Kierzek, E. Frackowiak, G. Lota, G. Gryglewicz and J. Machnikowski, Electrochim. Acta, 2004, 49, 515–523 CrossRef CAS.
- L. Qie, W. Chen, H. Xu, X. Xiong, Y. Jiang, F. Zou, X. Hu, Y. Xin, Z. Zhang and Y. Huang, Energy Environ. Sci., 2013, 6, 2497–2504 Search PubMed.
- F. C. Wu, R. L. Tseng, C. C. Hu and C. C. Wang, J. Power Sources, 2005, 144, 302–309 CrossRef CAS.
- A. Elmouwahidi, Z. Zapata-Benabithe, F. Carrasco-Marín and C. Moreno-Castilla, Bioresour. Technol., 2012, 111, 185–190 CrossRef CAS PubMed.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806–1812 CrossRef CAS PubMed.
- V. Subramanian, C. Luo, A. Stephan, K. Nahm, S. Thomas and B. Wei, J. Phys. Chem. C, 2007, 111, 7527–7531 CAS.
- L. Cheng, P. Guo, R. Wang, L. Ming, F. Leng, H. Li and X. Zhao, Colloids Surf., A, 2014, 446, 127–133 CrossRef CAS.
- D. Zhai, H. Du, B. Li, Y. Zhu and F. Kang, Carbon, 2011, 49, 725–729 CrossRef CAS.
- J. Hou, C. Cao, F. Idrees and X. Ma, ACS Nano, 2015, 9, 2556–2564 CrossRef CAS PubMed.
- L. Sun, C. Tian, M. Li, X. Meng, L. Wang, R. Wang, J. Yin and H. Fu, J. Mater. Chem. A, 2013, 1, 6462–6470 CAS.
- T. E. Rufford, D. Hulicova-Jurcakova, E. Fiset, Z. Zhu and G. Q. Lu, Electrochem. Commun., 2009, 11, 974–977 CrossRef CAS.
- T. E. Rufford, D. Hulicova-Jurcakova, Z. Zhu and G. Q. Lu, Electrochem. Commun., 2008, 10, 1594–1597 CrossRef CAS.
- T. E. Rufford, D. Hulicova-Jurcakova, K. Khosla, Z. Zhu and G. Q. Lu, J. Power Sources, 2010, 195, 912–918 CrossRef CAS.
- Z. Li, K. Zhai, G. Wang, Q. Li and P. Guo, Materials, 2016, 9, 912 CrossRef.
- X. Wei, X. Jiang, J. Wei and S. Gao, Chem. Mater., 2016, 28, 445–458 CrossRef CAS.
- G. Ma, J. Li, K. Sun, H. Peng, E. Feng and Z. Lei, J. Solid State Electrochem., 2016, 1–11 Search PubMed.
- Z. L. Yu, G. C. Li, N. Fechler, N. Yang, Z. Y. Ma, X. Wang, M. Antonietti and S. H. Yu, Angew. Chem., 2016, 128, 14843–14847 CrossRef.
- Y. Huang, Z. Liu and G. Zhao, RSC Adv., 2016, 6, 78909–78917 RSC.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., 2008, 120, 379–382 CrossRef.
- C. Huang, T. Sun and D. Hulicova-Jurcakova, ChemSusChem, 2013, 6, 2330–2339 CrossRef CAS PubMed.
- M. Chen, X. Kang, T. Wumaier, J. Dou, B. Gao, Y. Han, G. Xu, Z. Liu and L. Zhang, J. Solid State Electrochem., 2013, 17, 1005–1012 CrossRef CAS.
- L. Wang, Y. Guo, B. Zou, C. Rong, X. Ma, Y. Qu, Y. Li and Z. Wang, Bioresour. Technol., 2011, 102, 1947–1950 CrossRef CAS PubMed.
- J. Deng, T. Xiong, F. Xu, M. Li, C. Han, Y. Gong, H. Wang and Y. Wang, Green Chem., 2015, 17, 4053–4060 RSC.
- T. Kim, G. Jung, S. Yoo, K. S. Suh and R. S. Ruoff, ACS Nano, 2013, 7, 6899–6905 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz and M. Thommes, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- X. He, P. Ling, J. Qiu, M. Yu, X. Zhang, C. Yu and M. Zheng, J. Power Sources, 2013, 240, 109–113 CrossRef CAS.
- K. Yang, J. Peng, C. Srinivasakannan, L. Zhang, H. Xia and X. Duan, Bioresour. Technol., 2010, 101, 6163–6169 CrossRef CAS PubMed.
- X. He, Y. Geng, J. Qiu, M. Zheng, S. Long and X. Zhang, Carbon, 2010, 48, 1662–1669 CrossRef CAS.
- F. Caturla, M. Molina-Sabio and F. Rodriguez-Reinoso, Carbon, 1991, 29, 999–1007 CrossRef CAS.
- M. Molina-Sabio, M. Gonzalez, F. Rodriguez-Reinoso and A. Sepúlveda-Escribano, Carbon, 1996, 34, 505–509 CrossRef CAS.
- F.-C. Wu and R.-L. Tseng, J. Colloid Interface Sci., 2006, 294, 21–30 CrossRef CAS PubMed.
- A. Arami-Niya, W. M. A. W. Daud and F. S. Mjalli, Chem. Eng. Res. Des., 2011, 89, 657–664 CrossRef CAS.
- A. E. Ismanto, S. Wang, F. E. Soetaredjo and S. Ismadji, Bioresour. Technol., 2010, 101, 3534–3540 CrossRef CAS PubMed.
- X. Li, W. Xing, S. Zhuo, J. Zhou, F. Li, S.-Z. Qiao and G.-Q. Lu, Bioresour. Technol., 2011, 102, 1118–1123 CrossRef CAS PubMed.
- M. J. Prauchner and F. Rodriguez-Reinoso, Microporous Mesoporous Mater., 2008, 109, 581–584 CrossRef CAS.
- Z. Hu and M. Srinivasan, Microporous Mesoporous Mater., 2001, 43, 267–275 CrossRef CAS.
- R. Farma, M. Deraman, A. Awitdrus, I. A. Talib, E. Taer, N. Basri, J. Manjunatha, M. Ishak, B. Dollah and S. Hashmi, Bioresour. Technol., 2013, 132, 254–261 CrossRef CAS PubMed.
- W. J. Si, X. Z. Wu, W. Xing, J. Zhou and S. P. Zhuo, J. Inorg. Mater., 2011, 26, 107–112 CrossRef CAS.
- Z. Li, D. Wu, Y. Liang, R. Fu and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 4805–4808 CrossRef CAS PubMed.
- K. Xia, Q. Gao, J. Jiang and J. Hu, Carbon, 2008, 46, 1718–1726 CrossRef CAS.
- Z. Lei, J. Zhang, L. L. Zhang, N. A. Kumar and X. Zhao, Energy Environ. Sci., 2016, 9, 1891–1930 CAS.
- X. Zheng, J. Luo, W. Lv, D. W. Wang and Q. H. Yang, Adv. Mater., 2015, 27, 5388–5395 CrossRef CAS PubMed.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, J. Phys. Chem. C, 2008, 112, 9950–9955 CAS.
- X. Fan, C. Yu, J. Yang, Z. Ling, C. Hu, M. Zhang and J. Qiu, Adv. Energy Mater., 2015, 5, 1401761 CrossRef.
- J. Xu, Q. Gao, Y. Zhang, Y. Tan, W. Tian, L. Zhu and L. Jiang, Sci. Rep., 2014, 4, 5545 CrossRef CAS PubMed.
- X. Zheng, W. Lv, Y. Tao, J. Shao, C. Zhang, D. Liu, J. Luo, D.-W. Wang and Q.-H. Yang, Chem. Mater., 2014, 26, 6896–6903 CrossRef CAS.
- L. Wang, G. Mu, C. Tian, L. Sun, W. Zhou, P. Yu, J. Yin and H. Fu, ChemSusChem, 2013, 6, 880–889 CrossRef CAS PubMed.
- W. Zhang, H. Lin, Z. Lin, J. Yin, H. Lu, D. Liu and M. Zhao, ChemSusChem, 2015, 8, 2114–2122 CrossRef CAS PubMed.
- P. Hao, Z. Zhao, J. Tian, H. Li, Y. Sang, G. Yu, H. Cai, H. Liu, C. Wong and A. Umar, Nanoscale, 2014, 6, 12120–12129 RSC.
- L. F. Chen, Z. H. Huang, H. W. Liang, H. L. Gao and S. H. Yu, Adv. Funct. Mater., 2014, 24, 5104–5111 CrossRef CAS.
- M. Seredych, D. Hulicova-Jurcakova, G. Q. Lu and T. J. Bandosz, Carbon, 2008, 46, 1475–1488 CrossRef CAS.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS.
- Z. Ling, Z. Wang, M. Zhang, C. Yu, G. Wang, Y. Dong, S. Liu, Y. Wang and J. Qiu, Adv. Funct. Mater., 2016, 26, 111–119 CrossRef CAS.
- D. Hulicova-Jurcakova, A. M. Puziy, O. I. Poddubnaya, F. Suárez-García, J. M. Tascón and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 5026–5027 CrossRef CAS PubMed.
- X. Hong, K. Hui, Z. Zeng, K. Hui, L. Zhang, M. Mo and M. Li, Electrochim. Acta, 2014, 130, 464–469 CrossRef CAS.
- S. Song, F. Ma, G. Wu, D. Ma, W. Geng and J. Wan, J. Mater. Chem. A, 2015, 3, 18154–18162 CAS.
- K. Wang, N. Zhao, S. Lei, R. Yan, X. Tian, J. Wang, Y. Song, D. Xu, Q. Guo and L. Liu, Electrochim. Acta, 2015, 166, 1–11 CrossRef CAS.
- L. Deng, R. J. Young, I. A. Kinloch, A. M. Abdelkader, S. M. Holmes, D. A. De Haro-Del Rio and S. J. Eichhorn, ACS Appl. Mater. Interfaces, 2013, 5, 9983–9990 CAS.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- M. Hamedi, E. Karabulut, A. Marais, A. Herland, G. Nyström and L. Wagberg, Angew. Chem., Int. Ed., 2013, 52, 12038–12042 CrossRef CAS PubMed.
- C. Salas, T. Nypelö, C. Rodriguez-Abreu, C. Carrillo and O. J. Rojas, Curr. Opin. Colloid Interface Sci., 2014, 19, 383–396 CrossRef CAS.
- L. Jiang, G. W. Nelson, H. Kim, I. Sim, S. O. Han and J. S. Foord, ChemistryOpen, 2015, 4, 586–589 CrossRef CAS PubMed.
- X. Wu, Z. Shi, R. Tjandra, A. J. Cousins, S. Sy, A. Yu, R. M. Berry and K. C. Tam, J. Mater. Chem. A, 2015, 3, 23768–23777 CAS.
- Z. Zhao, S. Hao, P. Hao, Y. Sang, A. Manivannan, N. Wu and H. Liu, J. Mater. Chem. A, 2015, 3, 15049–15056 CAS.
- Y. S. Yun, J. Shim, Y. Tak and H. J. Jin, RSC Adv., 2012, 2, 4353–4358 RSC.
- D. Bhattacharya, L. T. Germinario and W. T. Winter, Carbohydr. Polym., 2008, 73, 371–377 CrossRef CAS.
- S. Y. Liew, W. Thielemans and D. A. Walsh, J. Phys. Chem. C, 2010, 114, 17926–17933 CAS.
- W. Luo, B. Wang, C. G. Heron, M. J. Allen, J. Morre, C. S. Maier, W. F. Stickle and X. Ji, Nano Lett., 2014, 14, 2225–2229 CrossRef CAS PubMed.
- G. Zu, J. Shen, L. Zou, F. Wang, X. Wang, Y. Zhang and X. Yao, Carbon, 2016, 99, 203–211 CrossRef CAS.
- D. D. Laskar, B. Yang, H. Wang and J. Lee, Biofuels, Bioprod. Biorefin., 2013, 7, 602–626 CrossRef CAS.
- B. Yu, Z. Chang and C. Wang, Mater. Chem. Phys., 2016, 181, 187–193 CrossRef CAS.
- Z. Z. Chang, B. J. Yu and C. Y. Wang, J. Solid State Electrochem., 2016, 20, 1405–1412 CrossRef CAS.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- W. J. Liu, H. Jiang and H. Q. Yu, Green Chem., 2015, 17, 4888–4907 RSC.
- M. Graglia, J. Pampel, T. Hantke, T. P. Fellinger and D. Esposito, ACS Nano, 2016, 10, 4364–4371 CrossRef CAS PubMed.
- K. Wang, M. Xu, Y. Gu, Z. Gu and Q. H. Fan, J. Power Sources, 2016, 332, 180–186 CrossRef CAS.
- S. Hu, S. Zhang, N. Pan and Y. L. Hsieh, J. Power Sources, 2014, 270, 106–112 CrossRef CAS.
- C. Lai, Z. Zhou, L. Zhang, X. Wang, Q. Zhou, Y. Zhao, Y. Wang, X. F. Wu, Z. Zhu and H. Fong, J. Power Sources, 2014, 247, 134–141 CrossRef CAS.
- X. Ma, P. Kolla, Y. Zhao, A. L. Smirnova and H. Fong, J. Power Sources, 2016, 325, 541–548 CrossRef CAS.
- M. Ago, M. Borghei, J. S. Haataja and O. J. Rojas, RSC Adv., 2016, 6, 85802–85810 RSC.
- D. Saha, Y. Li, Z. Bi, J. Chen, J. K. Keum, D. K. Hensley, H. A. Grappe, H. M. Meyer III, S. Dai and M. P. Paranthaman, Langmuir, 2014, 30, 900–910 CrossRef CAS PubMed.
- R. Ruiz-Rosas, M. J. Valero-Romero, D. Salinas-Torres, J. Rodríguez-Mirasol, T. Cordero, E. Morallón and D. Cazorla-Amorós, ChemSusChem, 2014, 7, 1458–1467 CrossRef CAS PubMed.
- H. Li, D. Yuan, C. Tang, S. Wang, J. Sun, Z. Li, T. Tang, F. Wang, H. Gong and C. He, Carbon, 2016, 100, 151–157 CrossRef CAS.
- D. Salinas-Torres, R. Ruiz-Rosas, M. J. Valero-Romero, J. Rodríguez-Mirasol, T. Cordero, E. Morallón and D. Cazorla-Amorós, J. Power Sources, 2016, 326, 641–651 CrossRef CAS.
- J. Tian, Z. Liu, Z. Li, W. Wang and H. Zhang, RSC Adv., 2017, 7, 12089–12097 RSC.
- A. M. Navarro-Suárez, J. Carretero-González, V. Roddatis, E. Goikolea, J. Ségalini, E. Redondo, T. Rojo and R. Mysyk, RSC Adv., 2014, 4, 48336–48343 RSC.
- L. Zhang, T. You, T. Zhou, X. Zhou and F. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 13918–13925 CAS.
- W. Zhang, M. Zhao, R. Liu, X. Wang and H. Lin, Colloids Surf., A, 2015, 484, 518–527 CrossRef CAS.
- J. W. Jeon, L. Zhang, J. L. Lutkenhaus, D. D. Laskar, J. P. Lemmon, D. Choi, M. I. Nandasiri, A. Hashmi, J. Xu and R. K. Motkuri, ChemSusChem, 2015, 8, 428–432 CrossRef CAS PubMed.
- A. S. Mamman, J. M. Lee, Y. C. Kim, I. T. Hwang, N. J. Park, Y. K. Hwang, J. S. Chang and J. S. Hwang, Biofuels, Bioprod. Biorefin., 2008, 2, 438–454 CrossRef CAS.
- Y. Wang, R. Yang, M. Li and Z. Zhao, Ind. Crops Prod., 2015, 65, 216–226 CrossRef CAS.
- L. Mao, Y. Zhang, Y. Hu, K. H. Ho, Q. Ke, H. Liu, Z. Hu, D. Zhao and J. Wang, RSC Adv., 2015, 5, 9307–9313 RSC.
- Y. Fan, P. Liu, B. Zhu, S. Chen, K. Yao and R. Han, Int. J. Hydrogen Energy, 2015, 40, 6188–6196 CrossRef CAS.
- B.-H. Cheng, K. Tian, R. J. Zeng and H. Jiang, Sustainable Energy & Fuels, 2017, 1, 891–898 Search PubMed.
- H. Feng, H. Hu, H. Dong, Y. Xiao, Y. Cai, B. Lei, Y. Liu and M. Zheng, J. Power Sources, 2016, 302, 164–173 CrossRef CAS.
- W. Tian, Q. Gao, Y. Tan, K. Yang, L. Zhu, C. Yang and H. Zhang, J. Mater. Chem. A, 2015, 3, 5656–5664 CAS.
- Y. Zhao, W. Ran, J. He, Y. Song, C. Zhang, D.-B. Xiong, F. Gao, J. Wu and Y. Xia, ACS Appl. Mater. Interfaces, 2015, 7, 1132–1139 CAS.
- B. Xu, S. Hou, G. Cao, F. Wu and Y. Yang, J. Mater. Chem., 2012, 22, 19088–19093 RSC.
- L. Hu, J. W. Choi, Y. Yang, S. Jeong, F. La Mantia, L.-F. Cui and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21490–21494 CrossRef CAS PubMed.
- L. Hu, M. Pasta, F. L. Mantia, L. Cui, S. Jeong, H. D. Deshazer, J. W. Choi, S. M. Han and Y. Cui, Nano Lett., 2010, 10, 708–714 CrossRef CAS PubMed.
- Y. J. Kang, S. J. Chun, S. S. Lee, B. Y. Kim, J. H. Kim, H. Chung, S. Y. Lee and W. Kim, ACS Nano, 2012, 6, 6400–6406 CrossRef CAS PubMed.
- K. Gao, Z. Shao, J. Li, X. Wang, X. Peng, W. Wang and F. Wang, J. Mater. Chem. A, 2013, 1, 63–67 CAS.
- W. Chen, R. Rakhi, L. Hu, X. Xie, Y. Cui and H. N. Alshareef, Nano Lett., 2011, 11, 5165–5172 CrossRef CAS PubMed.
- Z. Gui, H. Zhu, E. Gillette, X. Han, G. W. Rubloff, L. Hu and S. B. Lee, ACS Nano, 2013, 7, 6037–6046 CrossRef CAS PubMed.
- J. Cai, H. Niu, Z. Li, Y. Du, P. Cizek, Z. Xie, H. Xiong and T. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 14946–14953 CAS.
- M. Klose, R. Reinhold, F. Logsch, F. Wolke, J. Linnemann, U. Stoeck, S. Oswald, M. Uhlemann, J. Balach and J. Markowski, ACS Sustainable Chem. Eng., 2017, 5, 4094–4102 CrossRef CAS.
- X. Xu, J. Zhou, D. H. Nagaraju, L. Jiang, V. R. Marinov, G. Lubineau, H. N. Alshareef and M. Oh, Adv. Funct. Mater., 2015, 25, 3193–3202 CrossRef CAS.
- K. Wang, Y. Cao, X. Wang, M. A. Castro, B. Luo, Z. Gu, J. Liu, J. D. Hoefelmeyer and Q. Fan, J. Power Sources, 2016, 307, 462–467 CrossRef CAS.
- A. M. Navarro-Suárez, N. Casado, J. C. González, D. Mecerreyes and T. Rojo, J. Mater. Chem. A, 2017, 5, 7137–7143 Search PubMed.
- F. Chen, W. Zhou, H. Yao, P. Fan, J. Yang, Z. Fei and M. Zhong, Green Chem., 2013, 15, 3057–3063 RSC.
- L. Hu, H. Wu and Y. Cui, Appl. Phys. Lett., 2010, 96, 183502 CrossRef.
- G. Nyström, A. Mihranyan, A. Razaq, T. Lindström, L. Nyholm and M. Strømme, J. Phys. Chem. B, 2010, 114, 4178–4182 CrossRef PubMed.
- S. Liu, T. Yu, Y. Wu, W. Li and B. Li, RSC Adv., 2014, 4, 34134–34143 RSC.
- G. Milczarek and O. Inganäs, Science, 2012, 335, 1468–1471 CrossRef CAS PubMed.
- F. Ajjan, N. Casado, T. Rębiś, A. Elfwing, N. Solin, D. Mecerreyes and O. Inganäs, J. Mater. Chem. A, 2016, 4, 1838–1847 CAS.
| This journal is © The Royal Society of Chemistry 2017 |