
Research and application progress on key materials for sodium-ion batteries
Yongchang
Liu
ab,
Xiaobin
Liu
a,
Tianshi
Wang
a,
Li-Zhen
Fan
*a and
Lifang
Jiao
 *b
*b
aKey Laboratory of New Energy Materials and Technologies, Institute of Advanced Materials and Technology, University of Science and Technology Beijing, Beijing 100083, China. E-mail: fanlizhen@ustb.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China. E-mail: jiaolf@nankai.edu.cn
First published on 5th May 2017
Abstract
Sodium-ion batteries (SIBs) have been considered as a potential large-scale energy storage technology (especially for sustainable clean energy like wind, solar, and wave) owing to natural abundance, wide distribution, and low price of sodium resources. However, SIBs face challenges of low specific energy, unsatisfactory rate capability, and short cycling life caused by the heavy mass and large radius of Na+ ions. Therefore, developing promising host materials with the ability of fast, stable, and efficient sodium-ion insertion/extraction is key to promoting SIBs. Furthermore, the optimization of the electrolyte, the matching of cathode and anode materials, and the construction of sodium-ion full batteries with high-performance, high-safety, and low cost are urgently needed in order to make SIBs commercially available. In this review, we summarize the up-to-date research progress and insights on key materials (including cathode, anode, and electrolyte) for Na storage and some representative Na-ion full battery configurations will also be emphatically described. This should shed light on the fundamental research and practical applications of sodium-ion batteries.
 Yongchang Liu | Yongchang Liu is an Associate Professor at the University of Science and Technology Beijing (USTB), China. He received his BSc degree from Beijing Jiaotong University in 2011, and PhD degree in Materials Physics and Chemistry from Nankai University in 2016. His research interest focuses on rational design and efficient synthesis of micro/nano-materials for electrochemical energy storage/conversion (especially for sodium-ion batteries), and he has co-authored over 40 relevant peer-reviewed publications. |
 Li-Zhen Fan | Li-Zhen Fan received her PhD degree in Materials Science and Engineering from Tsinghua University in 2004, and then worked as a postdoctoral research fellow at Kyushu University in Japan from July 2004 to June 2005. After that, she worked as an Alexander von Humboldt fellow at the Max Planck Institute for Solid State Research in Germany from July 2005 to February 2007. She is a full professor at the University of Science and Technology Beijing (USTB), China now. Her research interest focuses on materials for energy storage and conversion, and she has co-authored nearly 120 relevant peer-reviewed publications. |
 Lifang Jiao | Lifang Jiao is an Associate Professor at Nankai University, China. She received her BSc (1999) and MSc (2002) degrees in chemistry from Hebei Normal University (China), and PhD degree from Nankai University (China) in 2005. She has authored more than 80 papers in refereed journals. Her current research is focused on energy conversion and storage (including lithium, sodium, and magnesium secondary batteries, and supercapacitors), hydrogen storage materials and electrocatalytic hydrogen evolution. |
1. Introduction
The early attention to sodium-ion batteries (SIBs) started in parallel with lithium-ion batteries (LIBs) in 1980s. LIBs have witnessed rapid developments and dominated the secondary battery (especially portable electronics) market since their first commercialization in 1991, while the progress on SIBs was relatively slow. In recent years, the increasing demand for emerging applications, such as electric vehicles and grid storage for intermittent sustainable clean energy like wind, solar, and wave, has raised concerns about the feasibility of continuing to increase LIB production. Due to limited and globally uneven distribution of lithium reserves (as shown in Table 1), it is estimated that the cost of LIBs will dramatically increase in the near future. Therefore, exploiting low-cost energy storage devices using naturally abundant raw materials is urgently required, especially for large-scale stationary applications. In this regard, SIBs have regained extensive popularity as a potential alternative to LIBs owing to the cost effectiveness and earth abundance of sodium resources (more than 1000 times higher than that of lithium).1–6| Category | Sodium | Lithium |
|---|---|---|
| Relative atomic mass (g mol−1) | 22.99 | 6.94 |
| Ionic radius (Å) | 1.02 | 0.76 |
| E o vs. SHE (V) | −2.71 | −3.04 |
| First ionization energy (kJ mol−1) | 495.8 | 520.2 |
| Melting point (°C) | 97.7 | 180.5 |
| Abundance in the earth crust (mg kg−1) | 23.6 × 103 | 20 |
| Distribution | Everywhere | 70% in South America |
| Cost of carbonate ($ per ton) | 250–300 | 5800 |
| Theoretical capacity (metal, mA h g−1) | 1166 | 3861 |
| Theoretical capacity (ACoO2, mA h g−1) | 235 | 274 |
Given the similarity between sodium and lithium chemistry, the working mechanism of SIBs is similar to that of LIBs. As shown in Fig. 1, the reversible charge/discharge of SIBs is realized through Na+ ions' shuttling between positive and negative electrode materials, which is also a so-called “rocking-chair” battery. However, Na+ ions (radius 1.02 Å, mass 22.99 g mol−1) are larger and heavier than Li+ ions (radius 0.76 Å, mass 6.94 g mol−1), resulting in sluggish diffusion kinetics and unsatisfactory rate capability. In addition, the Na-insertion process tends to bring large volume expansion to the electrode materials, sometimes even induce irreversible phase transformation, leading to capacity degradation. Furthermore, the less negative redox potential of Na+/Na as compared to Li+/Li (−2.71 and −3.04 V vs. standard hydrogen electrode) will reduce the operating voltage and lower the energy density. The above three drawbacks make it difficult to identify appropriate host materials with the ability of fast, stable, and efficient sodium-ion insertion/extraction. Thus, developing promising electrode materials with high energy/power density, good rate performance, and durable lifetime is key to promoting SIBs.7–10
 | ||
| Fig. 1 Working mechanism and potential applications of sodium-ion batteries. | ||
For a long time, research into Na-ion batteries has been sporadic. As the first turning point in the study for SIBs, Stevens and Dahn reported in 2000 that a reversible capacity of 300 mA h g−1, close to that for lithium insertion in graphite, has been achieved with hard carbon in a Na cell although its cyclability was insufficient at that time.11 Hard carbon is now extensively investigated as a potential anode material for SIBs. As the second important discovery, Okada and colleagues reported that NaFeO2 is electrochemically active in Na cells based on the Fe3+/Fe4+ redox couple.12 This discovery may correspond to the finding of LiCoO2 in 1980, which is still widely used as high-energy cathode materials for LIBs nowadays. On the basis of these important findings, in the past few years, there has been a resurgence of research interest in Na-ion battery chemistries because of its potential cost advantages. In fact, the number of publications on SIBs has drastically increased after 2010, and many different sodium insertion host materials have been discovered (and revisited) by many different research groups.13–15Fig. 2 summarizes the operating potentials and specific capacities of representative known Na-storage cathodes and anodes.16 Of which the cathode materials mainly include layered transition-metal oxides, polyanionic compounds (phosphate and fluorophosphate), and metal hexacyanometalates, whereas the anode materials mainly include hard carbon, metals/alloys, phosphorus, and metal sulfides/oxides. Currently, cathode materials show specific capacities of 120–200 mA h g−1 and operating potentials of 2.6–3.2 V (much lower than the oxidation potential of electrolytes, >4.0 V), while some important anode materials exhibit specific capacities of 300–600 mA h g−1 and operating potentials of 0.1–0.6 V (close to the potential of Na plating).
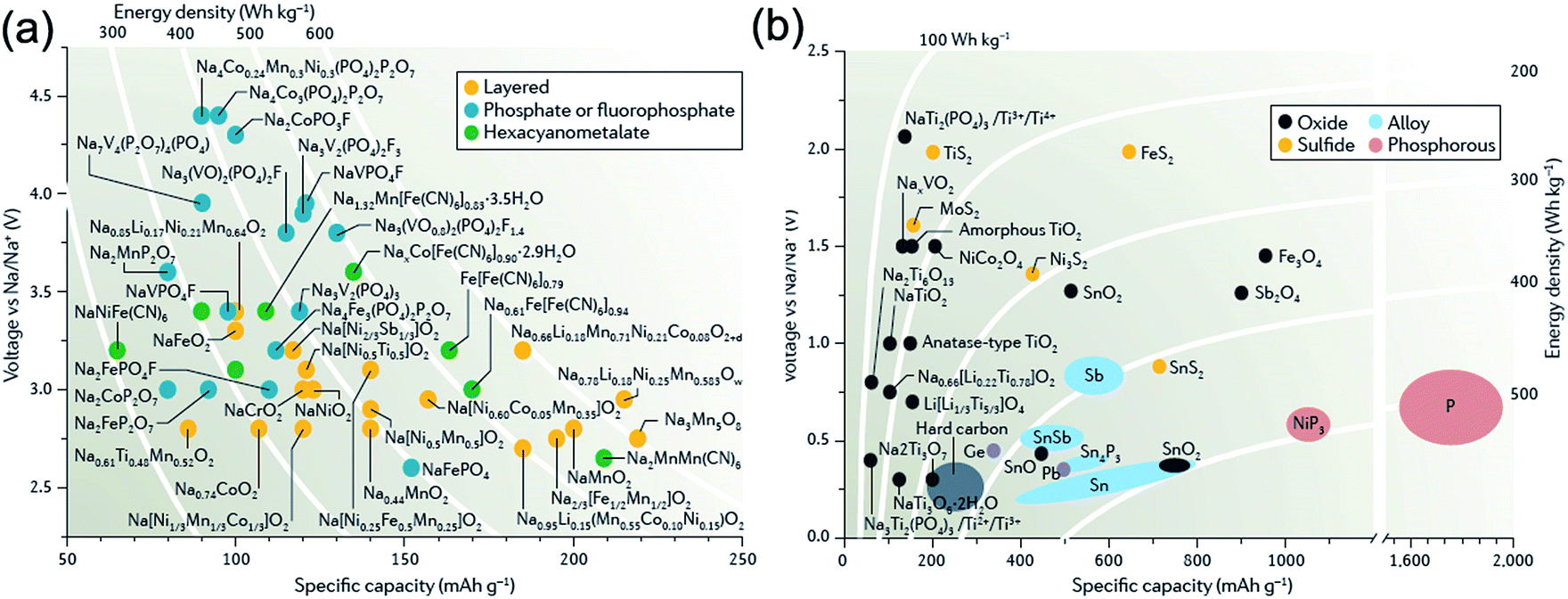 | ||
| Fig. 2 Relationships between specific capacity and voltage for present (a) cathode and (b) anode materials in Na-ion batteries. Reproduced with permission.16 Copyright Nature Publishing Group 2016. | ||
Aside from the Na insertion/extraction issues toward electrodes, the charge transfer in the electrode–electrolyte interface and Na+ ion transport through the solid electrolyte interphase (SEI) layer, are also fundamental scientific questions that need to be further elucidated. Therefore, along with the seeking for key electrode materials, exploring better electrolytes and designing a suitable interphase are also quite important. Moreover, matching the cathodes, anodes, and electrolytes to construct high-performance, high-safety, and low-cost Na-ion full batteries, as well as the deep understanding of Na-storage and failure mechanisms, thus making great breakthroughs in scale-up technology for application, is the main direction for further development of SIBs, and is also the only route to realize its commercialization.
Although several review papers have introduced the developments of SIBs, a review systematically summarizing the up-to-date research and application progress on key materials (including cathodes, anodes, and electrolytes) for Na-ion batteries is still needed. To make it a feasible task, in this paper, we will start by providing the most recent advances and prospects of cathode and anode materials. Since the electrochemical performance of electrode materials is tightly related with their structures, morphologies, and compositions, special focus on ingenious and reasonable strategies for enhancing electrochemical performance including structural and morphological engineering will be paid. Then, some achievements of electrolytes (including organic, aqueous, and solid systems) will also be described. Finally and most importantly, we will highlight some examples of Na-ion full batteries that hold potential for practical applications. It is believed that this review will guide readers to better understand the fundamental scientific problems and tackle the practical technological hurdles facing sodium-ion batteries.
2. Cathode materials
In Na-ion batteries, cathode materials offer active Na+ ions and high potential redox couples, which play a crucial role in determining the operating voltages and reversible capacities of batteries. Successful cathode materials should possess electrochemical features of high specific energy, excellent cycling life, and high rate capability. The specific energy is dominated by the specific capacity and potential of the cathode, the cycling life is governed by its structural stability, and the rate capability is affected by kinetic factors, mainly ionic diffusivity. The currently available positive electrode materials mainly include transition-metal oxides, polyanionic compounds, metal hexacyanometalates, and organic compounds.172.1 Transition-metal oxides
 | ||
| Fig. 3 Structural illustrations of (a) O3-type, (b) P2-type, and (c) tunnel-type transition-metal oxides. | ||
NaxCoO2 has been considered as a potential cathode for SIBs due to the success of its analogue LiCoO2 in LIBs.19 Delmas and coworkers selectively synthesized various phases of NaxCoO2 by altering the x value, and found that the structural transition of O3 ↔ O′3 ↔ P′3 phases is unfavorable to stable cycling.18 P2-phase NaxCoO2 exhibits relatively high cyclic stability. NaxMnO2 is more competitive than NaxCoO2 due to its high theoretical capacity of 243 mA h g−1, as well as its low cost.20 In addition to the O3 and P2 phases, the O2 and birnessite phases of NaxMnO2 also exhibit electrochemical activities for SIBs. Generally speaking, all of the layered NaxMnO2 can offer high practical capacities, but suffer from poor cycling performance due to their structural collapse and amorphization induced by continuous strains and distortions.21 NaFeO2 is another interesting positive Na-storage host material, owing to earth-abundant Fe resources and the highly active Fe3+/4+ couple.22 The electrochemical performance of NaFeO2 is closely associated with the cut-off voltage. When the cut-off voltage is limited to 3.4 V, NaFeO2 is able to deliver a stable capacity of 80–100 mA h g−1 with a potential plateau at 3.3 V. However, the reversibility is significantly deteriorated by charging it to above 3.5 V, due to irreversible structural change and Na conduction-path loss.23 Besides, some other layered oxides such as NaCrO2,24 NaNiO2,25 NaVxOy (ref. 26) have also been demonstrated to be electrochemically active in SIBs.
In general, NaxCoO2 possesses a relatively high ion-diffusion coefficient, NaxMnO2 shows high theoretical specific capacity, and NaFeO2 exhibits a high redox potential. However, they all face the problem of unsatisfactory cycling stability. To address this issue and combine the advantages of different layered oxides, a cation-substitution approach was adopted to fabricate multi-metal oxides towards improved Na-storage performance. O3–Na[Ni0.5Mn0.5]O2 consists of active Ni2+ and Mn4+ ions; it exhibits a capacity of 125 mA h g−1 in the voltage window of 2.2–3.8 V and a capacity retention of 75% after 50 cycles.27 Further substitution of appropriate Fe in the M layers can strengthen the structure, Na[Ni0.4Mn0.4Fe0.2]O2 (ref. 28) and Na[Ni0.25Mn0.25Fe0.5]O2 (ref. 29) can deliver a capacity retention of 95% over 30 cycles and 90.4% over 50 cycles, respectively. Additionally, Co substitution also has a positive effect in improving the electrochemical performance of Na[Ni0.5Mn0.5]O2 owing to synergetic effect of various metal ions. Representative Na[Ni1/3Mn1/3Co1/3]O2 displays a reversible capacity of 120 mA h g−1 in the potential range of 2.0–3.75 V with almost no capacity fading after 50 cycles.30 Another attractive multi-metal oxide P2–Na2/3[Fe1/2Mn1/2]O2 can deliver a reversible capacity as high as 190 mA h g−1 in the potential range of 1.5–4.3 V, with ∼82% capacity retained after 30 cycles.31
Recently, Li-containing multi-metal oxides that can effectively suppress the irreversible phase transformation upon charge/discharge have gained wide attention. For example, P2–Na0.85[Li0.17Ni0.21Mn0.64]O2 and O3–Na0.95[Li0.15Mn0.55Co0.10Ni0.15]O2 both showed good cycling performance with relatively high capacity.32,33 On this basis, Zhou and colleagues fabricated a layered oxide with P2 and O3 integration (Na0.66Li0.18Mn0.71Ni0.21Co0.08O2+d), which gave a high specific capacity (200 mA h g−1 at 0.1C), a good rate capability (134 mA h g−1 at 1C), and reasonable cycling life (a capacity retention of 84% over 50 cycles).34
2.2 Polyanionic compounds
Polyanionic compounds are identified to be very promising SIB cathodes owing to their structural diversity and stability, as well as the strong inductive effect of the anions. They are usually featured with high operating potentials and great cycling performance. Currently available polyanionic compounds mainly contain olivine NaFePO4, NASICON Na3V2(PO4)3, fluorophosphates (Na2MPO4F, Na3(VOx)2(PO4)2F3−2x), pyrophosphates (Na2MP2O7, Na4M3(PO4)2P2O7) (M = Fe, Co, Mn), and sulphates (Na2Fe2(SO4)3, Na2Fe(SO4)2·2H2O). The following puts an emphasis on the introduction of several most potential polyanionic cathode materials.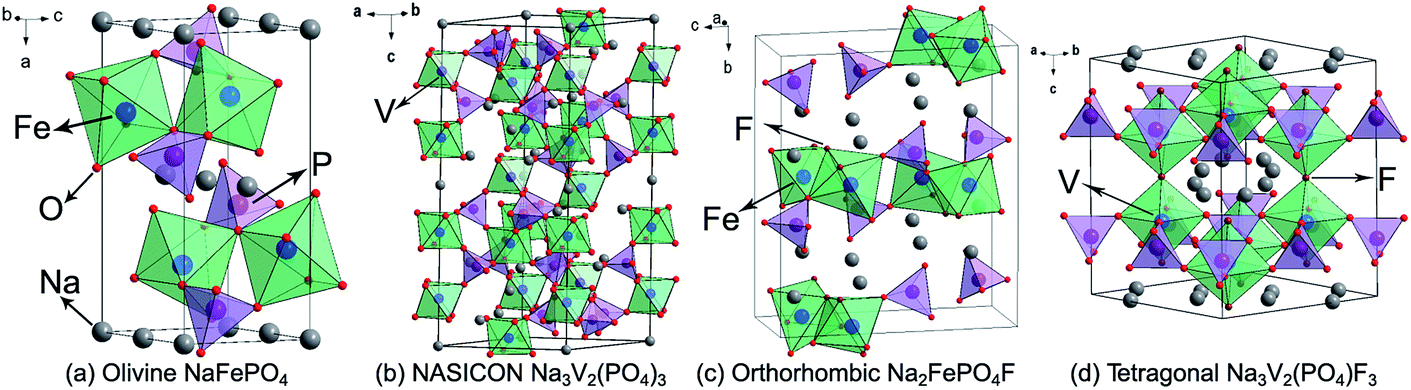 | ||
| Fig. 4 Structural illustrations of (a) olivine NaFePO4, (b) NASICON Na3V2(PO4)3, (c) orthorhombic Na2FePO4F, and (d) tetragonal Na3V2(PO4)F3. | ||
NASICON Na3V2(PO4)3 is widely regarded as a promising SIB cathode, because it provides a reversible and stable V3+/4+ redox reaction occurring at 3.4 V, as well as abundant ion-diffusion channels. As shown in Fig. 4b, in the Na3V2(PO4)3 structure, corner-sharing of PO4 tetrahedra and VO6 octahedra builds a three-dimensional (3D) open framework, which allows Na+ ions fast transportation. Two-thirds of the Na+ ions can be extracted from or inserted into the Na3V2(PO4)3 reversibly, corresponding to a theoretical capacity of 117 mA h g−1.44 Plashnitsa et al. first investigated Na3V2(PO4)3 as a Na-storage cathode, but the performance was not so good.45 Carbon-coating, nano-engineering, and creating a porous structure can significantly improve the electrochemical performance of Na3V2(PO4)3. Chen's group fabricated carbon-coated Na3V2(PO4)3 nanoparticles (∼40 nm) by a hydrothermal assisted sol–gel approach, displaying a capacity of 104.3 and 94.9 mA h g−1 at 0.5C and 5C, respectively, and a high capacity retention of 96.1% after 700 cycles.46 Saravanan et al. synthesized porous Na3V2(PO4)3/C using a novel solution-based method to further accelerate the Na-ion diffusion, the discharge capacity can reach 61.5 mA h g−1 even at 40C.47 Cationic substitution is also an effective strategy to enhance the electrochemical performance of Na3V2(PO4)3, for instance, Fe substituted Na3V2−xFex(PO4)3/C and Mg substituted Na3V2−xMgx(PO4)3/C both exhibit improved rate capability and cyclic stability.48,49 This originates from the optimization of ionic conductivity and structural stability. It is worth mentioning that Na3V2(PO4)3 can also be used as a Na-storage anode owing to its favorable interstitial sites and available V2+/3+ redox couple.
In 2003, Barker et al. first reported that the Na-ion battery constructed by NaVPO4F and hard carbon showed an operating voltage of 3.7 V, a charge capacity of 82 mA h g−1, and a capacity retention of 50% after 30 cycles.52 On this basis, Ruan et al. designed graphene modified NaVPO4F with significantly improved performance; the reversible capacity could reach 120.9 mA h g−1 with 97.7% retention after 50 cycles.53 Tetragonal Na3V2(PO4)2F3 is structurally made up of [V2O8F3] octahedral and [PO4] tetrahedral units, and they share oxygen ions to form open channels for Na+ ions diffusion (Fig. 4d). In the charge process, two plateaus occur at 3.7 V and 4.2 V, respectively, corresponding to a two-step electrochemical reaction of the V3+/4+ couple. Obviously, two-thirds of the Na+ ions can be extracted with the generation of a stable NaV2(PO4)2F3 phase, resulting in a theoretical capacity of 128 mA h g−1.54 Furthermore, the F element in Na3V2(PO4)2F3 can be substituted by O element to form Na3(VOx)2(PO4)2F3−2x (0 ≤ x ≤ 1) series materials. With the increasing of the oxygen content, the cell volume of Na3(VOx)2(PO4)2F3−2x gradually decreases due to lattice shrinkage caused by the replacement of larger V3+ ions by smaller V4+ ions. Meanwhile, the decrease of the F element will weaken the anionic inductive effect, leading to the drop of charge/discharge plateaus. Encouragingly, the existence of oxygen can stimulate the multi-electron redox reaction of vanadium. Kang's group discovered that the V3+/4+/V4+/5+ mixed redox couples in Na3(VO0.8)2(PO4)2F1.4 undergoes a 1.2-electron transfer, with a theoretical energy density as high as 600 W h kg−1.55
Aside from the aforementioned polyanionic compounds, pyrophosphate Na2MP2O7 and Na4M3(PO4)2P2O7 (M = Fe, Mn, Co) are also interesting polyanion-based cathode materials owing to their structural diversity and stability, as well as their good Na+ mobility.56–58 Besides, polyanionic sulfates are verified to be potential Na-storage cathodes. Goodenough's group found that the capacity of NaFe(SO4)2 can be maintained ∼80 mA h g−1 at 0.1C for 80 cycles, with a discharge plateau at ∼3 V vs. Na+/Na and a coulombic efficiency approaching 100%.59
2.3 Other cathode materials
Metal hexacyanometalates AxMM′(CN)6 (A = Na, K; M and M′ = Fe, Co, Mn, Ni, Zn) are a kind of new SIB cathode materials developed from Prussian blue (KFe[Fe(CN)6]) and its analogues, which possess merits of mild synthetic conditions and open frameworks for rapid Na-ion diffusion.60–63 This family mainly includes KM[Fe(CN)6], Na4[Fe(CN)6], Na2−xM[M(CN)6], NaM[Fe(CN)6], and M[Fe(CN)6]. For instance, Goodenough's team has identified Na1.72MnFe(CN)6 and Na1.92FeFe(CN)6 to be promising cathodes for SIBs because the Mn2+/3+ and Fe2+/3+ redox couples of the former were accessible to give a large capacity (134 mA h g−1) with a high reversible plateau (3.4 V),64 and the high Na concentration of the latter could overcome the sodium-deficiency problem.65 Metal fluorides are also determined to be potential Na-storage cathodes in recent years. Okada and coworkers first attested that NaMF3 (M = Fe, Mn, Ni) is electrochemically active in SIBs, of which the reversible capacity of NaFeF3 can reach 120 mA h g−1, while NaMnF3 and NaNiF3 showed inferior performance that needs to be further promoted.66 Afterwards, their group discovered that FeF3, TiF3, VF3, MnF3, and CoF3 could all display reversible Na-storage capacity to some degree.67In the past few years, organic cathode materials have received wide attention owing to their advantages of resource abundance, low cost, environmental friendliness, recyclability, and good designability.68,69 However, organic compounds are usually of low electrical conductivity and prone to dissolve into organic electrolyte solutions, leading to unsatisfactory electrochemical performance. In order to address these issues, adding conductive carbon, salt formation, polymerization, nanosizing, anchoring, and electrolyte optimizing are commonly adopted feasible strategies. For example, Chen's group designed organic tetrasodium salt (Na4C8H2O6) through a green one-pot method, based on the reversible uptake/removal of two Na+ ions in potential windows of 1.6–2.8 V (Na2C8H2O6/Na4C8H2O6) and 0.1–1.8 V (Na4C8H2O6/Na6C8H2O6), respectively; the as-prepared Na4C8H2O6 could be simultaneously used as positive and negative electrodes for SIBs, each delivering compatible and stable capacities of ca. 180 mA h g−1 with excellent cycling. More impressively, an all-organic Na-ion full battery based on Na4C8H2O6 displayed an average operation voltage of 1.8 V and a practical energy density of about 65 W h kg−1, showing some prospects for practical application.68
To give a comprehensive overview of the current families of SIB cathode materials, we comparatively summarize the practical discharge capacities, average operating potentials, specific energies, and cycling performance of some representative Na-storage cathodes in Table 2. Transition-metal oxides present relatively high reversible capacities (some can reach 200 mA h g−1), but often suffer from poor cyclic stability and low operating potentials (ca. 2.8 V). In comparison, polyanionic compounds exhibit long-term cyclability and high operating potentials (some reach 4.3 V), but their specific capacities are relatively low (80–120 mA h g−1). Metal hexacyanometalates and organic compounds provide a wide distribution of operating potential and reversible capacity because of their compositional and structural diversity.
| Cathode materialref. | Practical capacity (mA h g−1) | Operating potential (V vs. Na+/Na) | Specific energy (mW h g−1) | Capacity retention | |
|---|---|---|---|---|---|
| Transition-metal oxides | Na0.74CoO2 (ref. 70) | 107 | 2.8 | 300 | ∼96% (40 cycles) |
| Na0.67MnO2 (ref. 71) | 175 | 2.8 | 490 | ∼86% (25 cycles) | |
| NaFeO2 (ref. 23) | 100 | 3.3 | 330 | 50% (30 cycles) | |
| NaCrO2 (ref. 24) | 120 | 3.0 | 360 | 84% (50 cycles) | |
| Na[Ni1/2Mn1/2]O2 (ref. 72) | 140 | 2.9 | 406 | ∼85% (40 cycles) | |
| Na[Ni1/3Mn1/3Co1/3]O2 (ref. 30) | 120 | 2.8 | 336 | ∼100% (50 cycles) | |
| Na[Ni0.25Fe0.5Mn0.25]O2 (ref. 29) | 140 | 3.1 | 434 | 90.4% (50 cycles) | |
| Na2/3[Fe1/2Mn1/2]O2 (ref. 31) | 190 | 2.75 | 523 | ∼82% (30 cycles) | |
| Na[Ni1/2Ti1/2]O2 (ref. 73) | 121 | 3.1 | 375 | 52.8% (50 cycles) | |
| Na[Ni2/3Sb1/3]O2 (ref. 74) | 117 | 3.2 | 374 | 95% (50 cycles) | |
| Na0.85[Li0.17Ni0.21Mn0.64]O2 (ref. 32) | 100 | 3.4 | 340 | 98% (50 cycles) | |
| Na0.95[Li0.15Mn0.55Co0.10Ni0.15]O2 (ref. 33) | 200 | 2.8 | 560 | 85% (40 cycles) | |
| Na0.66Li0.18Mn0.71Ni0.21Co0.08O2+d (ref. 34) | 185 | 3.2 | 592 | 84% (50 cycles) | |
| Na0.44MnO2 (ref. 35) | 140 | 2.8 | 392 | 50% (50 cycles) | |
| Na0.44MnO2 (ref. 37) | 120 | 2.8 | 336 | 100% (100 cycles) | |
| Na0.44MnO2 (ref. 38) | 128 | 2.8 | 358 | 77% (1000 cycles) | |
| Na0.61Ti0.48Mn0.52O2 (ref. 39) | 86 | 2.9 | 249 | 81% (100 cycles) | |
| Polyanionic compounds | NaFePO4 (ref. 42) | 148 | 2.8 | 414 | 95% (200 cycles) |
| NaFePO4 (ref. 43) | 152 | 2.6 | 395 | 94.9% (300 cycles) | |
| Na3V2(PO4)3 (ref. 46) | 104 | 3.4 | 354 | 96.1% (700 cycles) | |
| Na3V2(PO4)3 (ref. 75) | 113 | 3.4 | 384 | 86.7% (1000 cycles) | |
| Na2FePO4F76 | 110 | 3.0 | 330 | 75% (20 cycles) | |
| Na2CoPO4F51 | 100 | 4.3 | 430 | ∼85% (5 cycles) | |
| NaVPO4F53 | 120.9 | 3.95 | 478 | 97.7% (50 cycles) | |
| Na3V2(PO4)2F3 (ref. 77) | 120 | 3.9 | 468 | 94% (40 cycles) | |
| Na3(VO0.8)2(PO4)2F1.4 (ref. 55) | 130 | 3.8 | 494 | 84% (500 cycles) | |
| Na2FeP2O7 (ref. 57) | 92 | 3.0 | 276 | ∼100% (80 cycles) | |
| NaFe(SO4)2 (ref. 59) | 99 | 3.0 | 297 | ∼98% (80 cycles) | |
| Hexacyanometalates and organics | Na4[Fe(CN)6]61 | 90 | 3.4 | 306 | 88% (500 cycles) |
| Na2MnMn(CN)6 (ref. 62) | 209 | 2.65 | 554 | 75% (100 cycles) | |
| KFe2(CN)6 (ref. 60) | 100 | 3.1 | 310 | ∼100% (30 cycles) | |
| Fe[Fe(CN)6]63 | 120 | 3.0 | 360 | 87% (500 cycles) | |
| Na1.72MnFe(CN)6 (ref. 64) | 134 | 3.4 | 456 | ∼90% (30 cycles) | |
| Na4C8H2O6 (ref. 68) | 183 | 2.3 | 421 | 84% (100 cycles) | |
| PTCDA69 | 130 | 2.2 | 286 | ∼77% (195 cycles) | |
3. Anode materials
The negative electrode is also a vital functional part of Na-ion batteries, which offers low potential redox couples.78–81 The currently available anode materials mainly include carbonaceous materials, alloy-type materials (Sn, Sb, P, Ge, SnSb, Sn4P3etc.), metal oxides/sulfides based on conversion reaction (Fe2O3, CuO, Co3O4, MoS2, SnS2etc.), titanium-based compounds (Na2Ti3O7, TiO2etc.), and organic compounds. Several most promising categories of anode materials will be emphatically described in the next section. Since the anode potential is generally low, around 0–1 V vs. Na+/Na, decomposition of electrolyte solution at the solid–liquid interface is a severe issue to realize long cycle life. Selection of binders, additives, and electrolyte, therefore, has a significant impact on the electrochemical behavior of negative electrodes, which will also be discussed.3.1 Carbonaceous materials
Carbonaceous materials are the most popular anodes for SIBs by virtue of their natural abundance, renewability, and environmental friendliness. Graphite is the commercial negative electrode of LIBs, but it is electrochemically less active in traditional ester-based electrolytes for Na storage. Encouragingly, in 2014, Adelhelm's group first reported that graphite showed electrochemical activity as a SIB anode in a diglyme-based electrolyte, and the discharge product was a ternary intercalation compound with an estimated stoichiometry of Na(diglyme)2C20.82 What should be pointed out is that the stability at higher voltages and the volumetric energy density affected by the expansion of graphite need to be further improved. Taking advantage of the co-intercalation effect of solvated Na ions (Fig. 5a), Chen's group investigated the Na-storage behavior of natural graphite in some linear ether-based electrolytes, achieving unprecedented cyclability (95% capacity retention after 6000 cycles), along with exceptional rate capability (more than 100 mA h g−1 at 10 A g−1).83 By conducting kinetic studies, they found that the pseudocapacitive behavior, namely, the coordinated solvent molecule decreased the reactivity of Na+ and thus the inserted charge might be electrochemically adsorbed in the graphene layer rather than atomically bonded to carbon, was responsible for the obtained superior electrode properties. More impressively, Wang's team fabricated expanded graphite with an enlarged interlayer distance of 4.3 Å as an anode for SIBs, showing a high capacity of 284 mA h g−1 in a conventional ester-based electrolyte.84 The mechanism of Na+ insertion into the expanded graphite is different from other carbonaceous materials. Most Na+ ions intercalate into the interlayer of graphite with reversible interlayer expansion/shrinkage during sodiation/desodiation, which is confirmed by cyclic voltammetry (CV) and in situ transmission electron microscopy (TEM, Fig. 5b).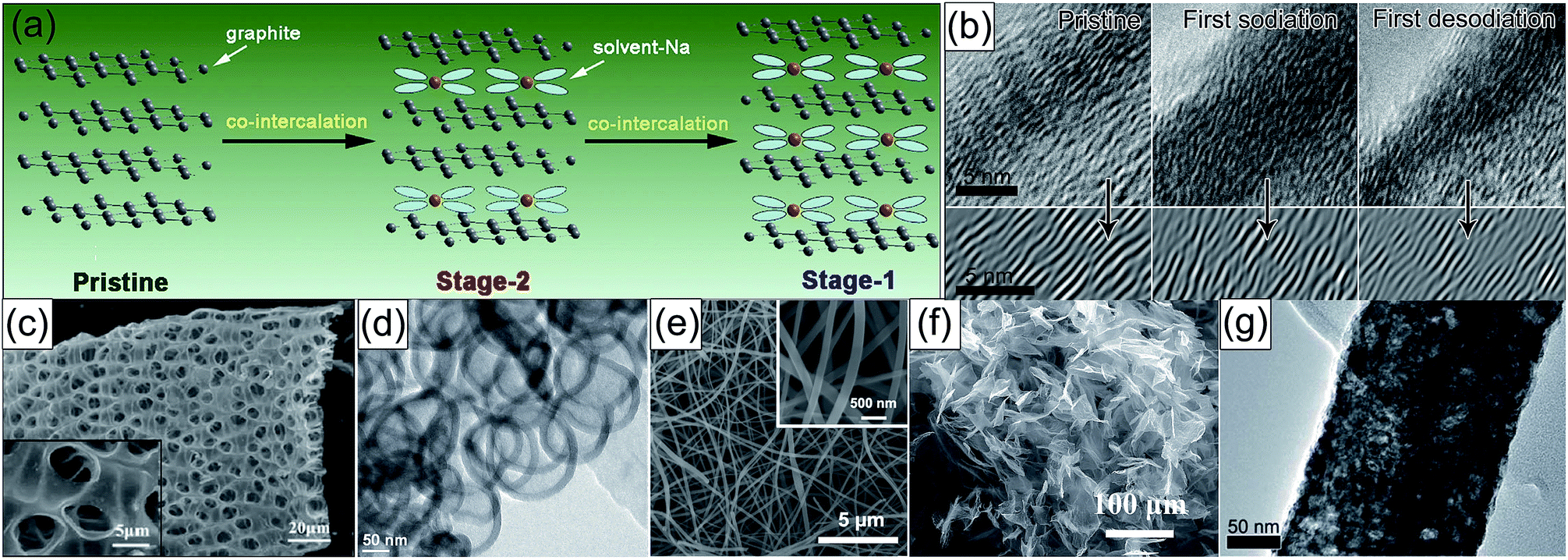 | ||
| Fig. 5 (a) Co-intercalation mechanism of solvent Na ions into graphite layers in linear ether-based electrolytes. Reproduced with permission.83 Copyright Elsevier 2015. (b) In situ TEM graphs of the expanded graphite at the pristine, first sodiation, and first desodiation states. Reproduced with permission.84 Copyright Nature Publishing Group 2014. TEM or scanning electron microscopy (SEM) images of (c) macroporous carbon nanosheets, reproduced with permission.95 Copyright American Chemical Society 2013. (d) Hollow carbon nanospheres, reproduced with permission.98 Copyright Wiley 2012. (e) N-Doped carbon nanofibers, reproduced with permission.99 Copyright Wiley 2016. (f) 3D N-doped graphene foams, reproduced with permission.102 Copyright Wiley 2015. (g) Exfoliated graphene/porous carbon nanofibers. Reproduced with permission.104 Copyright Royal Society of Chemistry 2017. | ||
As an important member of the carbon family, hard carbon (nongraphitic carbon) is favorable for Na-storage owing to its highly disordered structure (e.g. defects and vacancies) and large lattice distance. In 2000, Dahn and co-workers obtained hard carbon thorough pyrolysis of glucose, innovatively realizing a reversible capacity of ∼300 mA h g−1 in Na-ion batteries,11 which boosted the research of using hard carbon as SIB anodes to a new level. Afterwards, their team studied the underlying mechanism of Na+ insertion into hard carbon by in situ X-ray diffraction (XRD),85 which was subsequently confirmed by Alcántara et al. and Gotoh et al. using solid state 23Na nuclear magnetic resonance (NMR),86,87 revealing Na+ insertion inside turbostratic nanodomains (sloping region) and into nanopores (plateau region) of hard carbon. Besides, there are also some discussions on the Na-ion storage mechanism of amorphous carbon that deviate from the intercalation and pore-filling model.88–90 For example, Ji and coworkers proposed that the storage mechanism in the sloping part of potential curve could be better explained through storage at defect sites, and the storage mechanism in the low voltage plateau region was due to the intercalation between graphenic sheets and minor phenomenon of Na-ion adsorption on pore surfaces.89 Komaba et al. investigated the effect of different electrolytes on the Na-storage ability of hard carbon and found that a fluorinated ethylene carbonate (FEC) additive contributed to the generation of a stable passivated film on the hard carbon surface, which could strengthen its structural stability.91 Additionally, they observed that NaPF6-based electrolytes provided superior reversibility and cyclability of sodium insertion into hard carbon compared to NaClO4-based ones.92 Mitlin's group identified that the low coulombic efficiency of nongraphitic carbon during the initial several cycles was attributed to not only the SEI formation, but also the charge carriers trapped at the graphene defects and between the graphene planes.93 Then Ji's team demonstrated that the capacity fading of hard carbon upon cycling was mainly caused by kinetic problems at the electrolyte/electrode interface, which originated from SEI formation and electrolyte degradation.94 On the whole, although the hard carbons reported at early times could deliver a reversible capacity of 100–300 mA h g−1 at low current densities, their rate capability and cyclic stability were insufficient and needed to be further improved.
As effective strategies, rational morphological and structural engineering (e.g. designing micro/nano-structures, hollow structures, and porous structures) can significantly enhance the Na-storage performance of hard carbon. For instance, Mitlin's group prepared macroporous carbon nanosheets (Fig. 5c) through carbonization of peat moss,95 Cao et al. obtained hollow carbon nanowires by pyrolyzation of a hollow polyaniline nanowire precursor,96 Yu's group fabricated porous carbon nanofibers (CNFs) through electrospinning and pyrolysis processes,97 and Maier and colleagues synthesized hollow carbon nanospheres (Fig. 5d) based on hydrothermal carbonization of glucose in the presence of latex templates.98 These tailored nanostructures exhibited excellent electrochemical performance as anodes for Na-ion batteries, owing to their short ionic transport distance and strong stress tolerance. Heteroatom doping is also an efficient approach because the introduced heteroatom could modify the reactivity and electric conductivity of carbon. Very recently, David Lou's group fabricated a free-standing flexible nitrogen-doped carbon nanofiber film (Fig. 5e) by electrospinning, displaying admirable cycle life (99% capacity retention after 7000 cycles) and rate capability (154 mA h g−1 even at 15 A g−1).99 Zhou and co-workers prepared S-doped N-rich carbon nanosheets by treating N-rich carbon nanosheets with H2S/Ar mixed gas, which also demonstrated high capacity and excellent rate performance as the SIB anode.100 Graphene, an unique two-dimensional carbon material with large surface area, excellent electronic conductivity, good flexibility, and high chemical stability,101 has also shown fascinating Na-storage performance (especially high reversible capacity due to the abundant exposed active sites). For example, Dai and co-workers used three-dimensional (3D) N-doped graphene foams (Fig. 5f) as the SIB anode and achieved a high initial reversible capacity of 852.6 mA h g−1 at 500 mA g−1 with 69.7% capacity retention after 150 cycles.102 Guo and Wan developed a sandwich-like hierarchical carbon/graphene composite and obtained a high initial reversible capacity of 670 mA h g−1 at 50 mA g−1 with 400 mA h g−1 retained after 100 cycles.103 To incorporate the high capacity of graphene, as well as the high cyclic stability and ionic mobility of porous nanostructured carbon, our group creatively dispersed exfoliated graphene layers in porous carbon nanofibers (Fig. 5g) via electrospinning, rendering high reversible capacity (432.3 mA h g−1 at 100 mA g−1), unprecedented rate capability (261.1 mA h g−1 even at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mA g−1), and ultralong lifespan (91% capacity retention over 1000 cycles).104
000 mA g−1), and ultralong lifespan (91% capacity retention over 1000 cycles).104
In contrast to the non-graphitizable hard carbon, soft carbon represents a graphitizable nongraphitic carbon with higher electronic conductivity, whose graphitization degrees and interlayer distance can be tuned by a thermal treatment. Generally speaking, the graphenic layers in hard carbon are very much curved and not aligned, whereas the graphenic layers in soft carbon exhibit less curvature and are much better aligned. The pioneering study by Doeff et al. demonstrated that reversible sodiation of a petroleum coke soft carbon resulted in the formation of NaC24 by delivering a capacity of ∼90 mA h g−1.105 Then Ji and colleagues investigated the Na-ion storage properties of a soft carbon derived from organic perylene tetracarboxylic dianhydride (PTCDA, C24H8O6), observing a high reversible capacity of 200 mA h g−1 at 20 mA g−1 and 114 mA h g−1 at 1000 mA g−1.106 Furthermore, their team brought the following insights into the Na-storage mechanism of soft carbon, revealing that soft carbon displayed a generally sloping potential profile that lacked the low-potential plateau. As a result, the major trunk of its reversible capacity was from the potentials well above where Na plating may occur, thereby alleviating the concerns of dendrite formation, polarization, and electrolyte degradation.107
3.2 Alloy-type materials
Alloy-type materials are deemed to be very promising SIB anodes due to their high theoretical capacity and good electrical conductivity. They can alloy with sodium to form rich alloy phases, yielding a much higher capacity than carbonaceous materials. This family mainly contains Sn (Na15Sn4, 847 mA h g−1), Sb (Na3Sb, 660 mA h g−1), P (Na3P, 2596 mA h g−1), Ge (NaGe, 369 mA h g−1), and Bi (Na3Bi, 385 mA h g−1). What should be mentioned is that Si is a widely popular anode material (Li4.4Si, 4140 mA h g−1) in LIBs, but present reports indicate that Si is less active for Na storage; its potentiality needs to be further exploited. In most of the cases, alloying reactions are accompanied with large volume expansion, resulting in continuous pulverization of active materials and then their loss of electrical contact with current collectors, deteriorating the capacity and cyclability. In order to buffer the volume fluctuation and minimize the deformation stress, nanosizing, carbon coating, and alloying are feasible strategies.000 mA g−1) and ultra-long cycling life (90% capacity retention over 1300 cycles).115 Encouragingly, Tarascon's team recently discovered that microsized Sn particles (5–50 μm) could also display high capacity and stable cyclability in a glyme-based electrolyte, mainly due to the formation of protecting films that prevent the generation of insulating domains in active materials.116
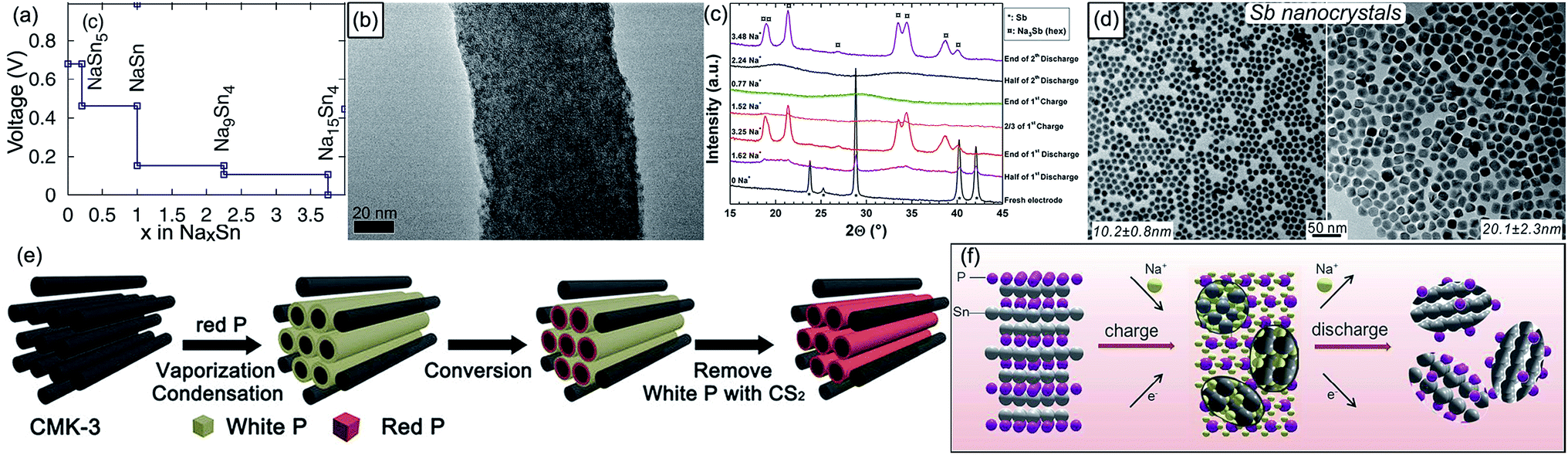 | ||
| Fig. 6 (a) Na–Sn voltage curve calculated using DFT. Reproduced with permission.108 Copyright ECS 2011. (b) TEM image of Sn nanodots@porous N-doped carbon nanofibers. Reproduced with permission.115 Copyright Wiley 2015. (c) Selected operando XRD patterns at various discharge and charge stages of a Sb/Na cell. Reproduced with permission.117 Copyright American Chemical Society 2012. (d) TEM images of monodisperse Sb nanocrystals with tunable size in the 10–20 nm range. Reproduced with permission.118 Copyright American Chemical Society 2014. (e) Schematic illustration of the preparation process for the red P@CMK-3 composite. Reproduced with permission.133 Copyright American Chemical Society 2016. (f) Schematic illustration of the Na-storage mechanism in the Sn4P3 electrode. Reproduced with permission.136 Copyright American Chemical Society 2014. | ||
Apart from Sn and Sb, some other metals like Ge, Bi, In, and Pb can also alloy with Na to be potential SIB anode materials. Noticeably, Mulder's team prepared amorphous Si nanoparticles (∼20 nm) from expanding thermal plasma chemical vapor deposition of silane, first realizing the reversible electrochemical Na ion uptake in Si, but the obtained capacity is relatively low (279 mA h g−1 at 10 mA g−1).121 Lately, Huang and co-workers reported the sodiation/desodiation mechanism of nanocrystalline silicon for the first time, verifying that an irreversible crystal structure conversion from crystalline Si to amorphous Si takes place during the first discharge process, leading to reversible Na+ insertion in the newly generated amorphous silicon. By designing the Si/C composite, a reversible capacity of more than 400 mA h g−1 at 50 mA g−1 and a durable lifespan of 2000 cycles were achieved.122 In addition, some micro/nano-structured alloys such as Ni3Sn2 microcages,123 Sn0.9Cu0.1 nanoparticles,124 SnSb/C nanofibers,125 Sn–Sb-Ge thin films, etc.,126 could also effectively accommodate the volume variations, thus rendering promising Na-storage performance.
3.3 Other anode materials
Metal oxides (MOx) are important SIB anode materials due to their low cost and high theoretical capacities based on multi-electron conversion reaction.141 The Na-storage mechanism of MOx can be classified into two types: if M is an electrochemically inactive element (e.g. Fe, Co, Ni, Cu, and Mn), metal oxides react with Na+ through a one-step conversion reaction to generate elementary metal and Na2O; if M is an electrochemically active element (e.g. Sn and Sb), metal oxides firstly undergo conversion reaction and then the generated active metals undergo alloying reaction. However, the intrinsic electrical conductivity of MOx is usually low, and the conversion reaction often brings a large volume change (sometimes even irreversible phase transformation) to the active materials, leading to unsatisfactory rate performance and rapid capacity decay. Designing carbon-containing composites and porous micro/nano-structures can significantly improve the electrochemical performance of metal oxides. For example, Chen's group prepared a 3D porous γ-Fe2O3@C nanocomposite with ultrasmall Fe2O3 nanograins of ∼5 nm (Fig. 7a and b) using an aerosol spray pyrolysis technology, when applied as an anode material for Na-ion batteries; this unique nanostructure displayed high rate capability (more than 800 mA h g−1 at 200 mA g−1 in comparison with 317 mA h g−1 at 8000 mA g−1) and long-term cycling stability (1400 cycles).142 Jian et al. reported the fabrication of monodispersed hierarchical Co3O4 spheres intertwined with CNTs (Fig. 7c and d); this novel composite showed good Na-storage performance with a capacity of up to 230 and 184 mA h g−1 at currents of 1600 and 3200 mA g−1, respectively.143 Zhang's team developed aligned porous CuO nanorod arrays grown on Cu foil as a binder-free anode for SIBs, achieving a high reversible capacity of around 600 mA h g−1.144 Wang's group synthesized a SnO2@graphene composite by an in situ hydrothermal method, obtaining a high reversible capacity of more than 700 mA h g−1 for 100 cycles at 20 mA g−1.145 In addition, Jiang and coworkers found that the synergistic effect of Sb2O3 involving conversion and alloying reactions promoted stabilizing the active material structure and accelerating the reaction kinetics, and therefore the Sb2O3 film exhibited a high capacity of 550 mA h g−1 at 50 mA g−1 and 265 mA h g−1 at 5000 mA g−1, as well as 414 mA h g−1 at 500 mA g−1 after 200 cycles.146 Recently, our group turned the research interest to spinel MnFe2O4, discovering a “self-buffering” effect in mixed transition-metal oxides that contributed to an ultra-long cycling life (4200 cycles).147 | ||
| Fig. 7 TEM, high resolution TEM (HRTEM), or SEM images of (a and b) the 3D porous γ-Fe2O3@C nanocomposite, reproduced with permission.142 Copyright Wiley 2015. (c and d) Co3O4@CNT sample, reproduced with permission.143 Copyright Royal Society of Chemistry 2014. (e–h) Ultrathin MoS2 embedded in the carbon nanofibers, reproduced with permission.151 Copyright Wiley 2014. and (i and j) WS2 nanowires. Reproduced with permission.152 Copyright Wiley 2015. | ||
Metal sulfides have also gained wide attention as potential SIB anodes; this family includes layered metal disulfides and non-layered metal sulfides. Generally, layered metal disulfides (MS2, M = Mo, W, Sn, Ti etc.) firstly undergo Na intercalation reaction at a high potential and then conversion reaction at a low potential; alloying reaction will also occur for SnS2 at lower potential.148–150 Apparently, the conversion reaction is accompanied with a large volume fluctuation that would destroy the layered structure, resulting in unstable cycling. Strategies like carbon coating and elevating the cut-off voltage to avoid conversion reaction are commonly adopted to address this issue. For instance, Maier and Yu embedded single-layered ultrasmall MoS2 nanoplates (ca. 4 nm) in carbon nanofibers (Fig. 7e–h) by electrospinning, the restriction to ultrasmall reaction domains allowed for an almost diffusion-less and nucleation-free “conversion”, thereby resulting in a high capacity (854 mA h g−1 at 100 mA g−1) and a remarkable cycling performance (100 cycles).151 Our group reported the synthesis and anode application for SIBs of WS2 nanowires with a very thin diameter of ∼25 nm and expanded interlayer spacing of 0.83 nm (Fig. 7i and j); after controlling the terminal voltage to 0.5 V, the Na–WS2 battery followed a reversible intercalation-type mechanism without conversion reaction, and a long-term lifespan of 1400 cycles was achieved.152 Qu et al. produced SnS2-reduced graphene oxide (rGO) nanocomposites by a facile hydrothermal route, delivering a high charge capacity of 630 mA h g−1 at 0.2 A g−1 and a stable cycling of 400 cycles in Na-ion batteries.153 In addition, some non-layered metal sulfides such as FeS/FeS2,154,155 Sb2S3,156 NiS/Ni3S2,157,158 CoS/Co9S8,159,160 and so on, have also been demonstrated to be electrochemically active for Na storage. Moreover, the electrolyte of NaSO3CF3 dissolved in an ether-based solvent was verified to show a magical effect in promoting the cycling performance of metal sulfides.155,161
At present, various titanium-based compounds (e.g. TiO2, Li4Ti5O12, Na2Ti3O7, Na2Ti6O13, Na0.66Li0.22Ti0.78O2, and NaTi2(PO4)3) have been explored as potential sodium insertion hosts due to their low cost, nontoxicity, appropriate operating potential, low strain, and excellent cyclability. However, Ti-based compounds usually provide low specific capacity (generally lower than 200 mA h g−1) because of their limited Na-storage sites. Hu's group systemically studied the Na-storage behavior of layered Na2Ti3O7, revealing that the microsized Na2Ti3O7 electrode could deliver a reversible capacity of 188 mA h g−1 in 1 M NaFSI/PC electrolyte with sodium alginate as the binder.162
Organic compounds are enjoying growing popularity as green and sustainable anode materials, owing to their advantages of abundant resources from biomass, tremendous molecular diversity, structural flexibility and possible multi-electron reactions. However, they also suffer from high solubility and sluggish kinetics. Hu's group first evaluated disodium terephthalate (Na2C8H4O4) as an anode material for SIBs, which exhibited a low Na-insertion voltage at 0.29 V vs. Na+/Na and a high reversible capacity of 250 mA h g−1 with two-electron transfer. It was found that the Na storage performance could be further improved by a thin layer of Al2O3 coating on the Na2C8H4O4 electrode surface.163
We comparatively summarized the electrochemical properties of some typical anodes for Na-ion batteries in Table 3. On the whole, carbonaceous materials with the lowest cost and alloy-type materials with the highest capacity are the most promising candidates in the future.
| Anode materialref. | Rate capability | Cyclic stability |
|---|---|---|
| Natural graphite83 | ∼110 mA h g−1 at 100 mA g−1 | 95% capacity retention at 200 mA g−1 (6000 cycles) |
| 102 mA h g−1 at 10000 mA g−1 |
||
| Expanded graphite84 | 284 mA h g−1 at 20 mA g−1 | 73.92% capacity retention at 100 mA g−1 (2000 cycles) |
| 184 mA h g−1 at 100 mA g−1 | ||
| 91 mA h g−1 at 200 mA g−1 | ||
| Hard carbon91 | ∼250 mA h g−1 at 25 mA g−1 | ∼240 mA h g−1 at 25 mA g−1 (100 cycles) |
| Macroporous carbon nanosheets95 | 306 mA h g−1 at 50 mA g−1 | 255 mA h g−1 at 100 mA g−1 (200 cycles) |
| 298 mA h g−1 at 100 mA g−1 | ||
| 150 mA h g−1 at 1000 mA g−1 | ||
| 66 mA h g−1 at 5000 mA g−1 | ||
| Hollow carbon nanowires96 | 252 mA h g−1 at 50 mA g−1 | 206.3 mA h g−1 at 50 mA g−1 (400 cycles) |
| 216 mA h g−1 at 250 mA g−1 | ||
| 149 mA h g−1 at 500 mA g−1 | ||
| Porous carbon nanofibers97 | 280 mA h g−1 at 50 mA g−1 | 266 mA h g−1 at 50 mA g−1 (100 cycles) |
| 200 mA h g−1 at 1000 mA g−1 | ||
| 60 mA h g−1 at 10000 mA g−1 |
140 mA h g−1 at 500 mA g−1 (1000 cycles) | |
| 40 mA h g−1 at 20000 mA g−1 |
||
| Hollow carbon nanospheres98 | 223 mA h g−1 at 50 mA g−1 | ∼160 mA h g−1 at 100 mA g−1 (100 cycles) |
| 120 mA h g−1 at 1000 mA g−1 | ||
| 75 mA h g−1 at 5000 mA g−1 | ||
| ∼50 mA h g−1 at 10000 mA g−1 |
||
| Nitrogen-doped carbon nanofiber film99 | 315 mA h g−1 at 500 mA g−1 | 377 mA h g−1 at 100 mA g−1 (100 cycles) |
| ∼275 mA h g−1 at 1000 mA g−1 | ||
| ∼170 mA h g−1 at 10000 mA g−1 |
210 mA h g−1 at 5000 mA g−1 (7000 cycles) | |
| 154 mA h g−1 at 15000 mA g−1 |
||
| S-Doped N-rich carbon nanosheets100 | 350 mA h g−1 at 50 mA g−1 | 211 mA h g−1 at 1000 mA g−1 (1000 cycles) |
| 250 mA h g−1 at 500 mA g−1 | ||
| 150 mA h g−1 at 5000 mA g−1 | ||
| 110 mA h g−1 at 10000 mA g−1 |
||
| 3D nitrogen-doped graphene foams102 | 1057.1 mA h g−1 at 100 mA g−1 | 594 mA h g−1 at 500 mA g−1 (150 cycles) |
| 815.2 mA h g−1 at 500 mA g−1 | ||
| 244.7 mA h g−1 at 2000 mA g−1 | ||
| 137.7 mA h g−1 at 5000 mA g−1 | ||
| Sandwich-like hierarchically carbon/graphene composite103 | 670 mA h g−1 at 50 mA g−1 | 400 mA h g−1 at 50 mA g−1 (100 cycles) |
| 250 mA h g−1 at 1000 mA g−1 (1000 cycles) | ||
| Graphene/Porous Carbon Nanofibers104 | 429.4 mA h g−1 at 100 mA g−1 | 382.8 mA h g−1 at 500 mA g−1 (300 cycles) |
| 371.5 mA h g−1 at 500 mA g−1 | ||
| 324.5 mA h g−1 at 2000 mA g−1 | 300.8 mA h g−1 at 2000 mA g−1 (1000 cycles) | |
| 291.4 mA h g−1 at 5000 mA g−1 | ||
| 261.1 mA h g−1 at 10000 mA g−1 |
||
| Sn nanofibers112 | 808.19 mA h g−1 at 84.7 mA g−1 | 776.26 mA h g−1 at 84.7 mA g−1 (100 cycles) |
| 784.53 mA h g−1 at 847 mA g−1 | ||
| ∼650 mA h g−1 at 1694 mA g−1 | ||
| ∼260 mA h g−1 at 4235 mA g−1 | ||
| Sn nanodots@porous N-doped carbon nanofibers115 | 633 mA h g−1 at 200 mA g−1 | 578 mA h g−1 at 500 mA g−1 (300 cycles) |
| 588 mA h g−1 at 500 mA g−1 | ||
| 543 mA h g−1 at 2000 mA g−1 | 483 mA h g−1 at 2000 mA g−1 (1300 cycles) | |
| 450 mA h g−1 at 10000 mA g−1 |
||
| Monodisperse Sb nanocrystals118 | ∼600 mA h g−1 at 660 mA g−1 | Almost no capacity fading at 660 mA g−1 (100 cycles) |
| ∼500 mA h g−1 at 13200 mA g−1 |
||
| Sb nanoparticles@porous carbon spheres120 | 435 mA h g−1 at 100 mA g−1 | 88% capacity retention at 100 mA g−1 (500 cycles) |
| 355 mA h g−1 at 1000 mA g−1 | ||
| 270 mA h g−1 at 4000 mA g−1 | ||
| Si/C composite122 | 438 mA h g−1 at 50 mA g−1 | 75% capacity retention at 5000 mA g−1 (2000 cycles) |
| ∼270 mA h g−1 at 5000 mA g−1 | ||
| Ni3Sn2 microcages123 | 540 mA h g−1 at 212 mA g−1 | ∼300 mA h g−1 at 847 mA g−1 (300 cycles) |
| 447 mA h g−1 at 1694 mA g−1 | ||
| 276 mA h g−1 at 8470 mA g−1 | ||
| SnSb-CNFs125 | 392 mA h g−1 at 50 mA g−1 | 99.4% capacity retention at 100 mA g−1 (2000 cycles) |
| 350 mA h g−1 at 100 mA g−1 | ||
| 198 mA h g−1 at 5000 mA g−1 | ||
| 113 mA h g−1 at 10000 mA g−1 |
||
| Amorphous red P/C127 | 1800 mA h g−1 at 250 mA g−1 | 1500 mA h g−1 at 250 mA g−1 (20 cycles) |
| 1380 mA h g−1 at 1000 mA g−1 | ||
| 930 mA h g−1 at 2000 mA g−1 | 1000 mA h g−1 at 250 mA g−1 (140 cycles) | |
| 640 mA h g−1 at 4000 mA g−1 | ||
| Red P/graphene nanosheets130 | 2164 mA h g−1 at 130 mA g−1 | 95% capacity retention at 260 mA g−1 (60 cycles) |
| 2077 mA h g−1 at 260 mA g−1 | ||
| ∼750 mA h g−1 at 2600 mA g−1 | ||
| ∼520 mA h g−1 at 5200 mA g−1 | ||
| Red P-single walled carbon nanotube132 | 711 mA h g−1 at 50 mA g−1 | Almost no capacity decay at 500 mA g−1 (200 cycles) |
| 580 mA h g−1 at 500 mA g−1 | ||
| 500 mA h g−1 at 1000 mA g−1 | 80% capacity retention at 2000 mA g−1 (2000 cycles) | |
| 300 mA h g−1 at 2000 mA g−1 | ||
| Red P@CMK-3 (ref. 133) | 2591 mA h g−1 at 519 mA g−1 | 2188 mA h g−1 at 519 mA g−1 (60 cycles) |
| 1984 mA h g−1 at 6228 mA g−1 | ||
| 1718 mA h g−1 at 12456 mA g−1 |
1020 mA h g−1 at 12975 mA g−1 (210 cycles) |
|
| 1042 mA h g−1 at 18943 mA g−1 |
||
| 650 mA h g−1 at 25431 mA g−1 |
||
| SnP3/C composite139 | 810 mA h g−1 at 150 mA g−1 | Almost no capacity decay at 150 mA g−1 (150 cycles) |
| ∼400 mA h g−1 at 2560 mA g−1 | ||
| Porous 3D γ-Fe2O3@C142 | ∼560 mA h g−1 at 120 mA g−1 | 444 mA h g−1 at 120 mA g−1 (200 cycles) |
| ∼275 mA h g−1 at 1200 mA g−1 | 214.8 mA h g−1 at 1200 mA g−1 (1400 cycles) | |
| 190 mA h g−1 at 4800 mA g−1 | ||
| Co3O4@CNTs143 | 425 mA h g−1 at 160 mA g−1 | 90% capacity retention at 160 mA g−1 (300 cycles) |
| 230 mA h g−1 at 1600 mA g−1 | ||
| 184 mA h g−1 at 3200 mA g−1 | ||
| CuO nanorod arrays144 | 640 mA h g−1 at 200 mA g−1 | 45.2% capacity retention at 200 mA g−1 (450 cycles) |
| ∼200 mA h g−1 at 1000 mA g−1 | ||
| SnO2@graphene nanocomposites145 | ∼700 mA h g−1 at 20 mA g−1 | 638 mA h g−1 at 40 mA g−1 (100 cycles) |
| 302 mA h g−1 at 160 mA g−1 | ||
| 143 mA h g−1 at 640 mA g−1 | ||
| MnFe2O4@CNFs147 | 504 mA h g−1 at 100 mA g−1 | 440 mA h g−1 at 500 mA g−1 (300 cycles) |
| 449 mA h g−1 at 500 mA g−1 | ||
| 391 mA h g−1 at 2000 mA g−1 | 360.8 mA h g−1 at 2000 mA g−1 (4200 cycles) | |
| 340 mA h g−1 at 5000 mA g−1 | ||
| 305 mA h g−1 at 10000 mA g−1 |
||
| Single-layered MoS2-carbon nanofiber151 | 854 mA h g−1 at 100 mA g−1 | 484 mA h g−1 at 1000 mA g−1 (100 cycles) |
| 623 mA h g−1 at 1000 mA g−1 | ||
| 436 mA h g−1 at 5000 mA g−1 | ||
| 331 mA h g−1 at 10000 mA g−1 |
||
| 224 mA h g−1 at 20000 mA g−1 |
||
| 75 mA h g−1 at 50000 mA g−1 |
||
| WS2 nanowires152 | ∼415 mA h g−1 at 200 mA g−1 | ∼330 mA h g−1 at 1000 mA g−1 (1400 cycles) |
| ∼370 mA h g−1 at 500 mA g−1 | ||
| ∼330 mA h g−1 at 1000 mA g−1 | ||
| SnS2–rGO hybrid153 | 630 mA h g−1 at 200 mA g−1 | 500 mA h g−1 at 1000 mA g−1 (400 cycles) |
| 544 mA h g−1 at 2000 mA g−1 | ||
| Na2Ti3O7 (ref. 162) | 188 mA h g−1 at 0.1C | 60 mA h g−1 at 0.1C (60 cycles) |
| 64 mA h g−1 at 2C | ||
| Na2C8H4O4/Ketjen Black163 | 258 mA h g−1 at 0.1C | 74.4% capacity retention at 0.1C (60 cycles) |
4. Electrolyte materials
The electrolyte is the medium that allows Na+ ion transportation between positive and negative electrodes; its physicochemical properties such as viscosity, ionic conductivity, thermostability, and potential window are tightly associated with the electrochemical performance and safety of Na-ion batteries. In organic electrolyte systems, NaClO4, NaPF6, and NaSO3CF3 are the common solutes, while the solvents mainly include ester-based propylene carbonate (PC), ethylene carbonate (EC), diethyl carbonate (DEC), ether-based diethylene glycol dimethyl ether (DEGDME), and tetraethylene glycol dimethyl ether (TEGDME). As for aqueous electrolytes, the solute is usually Na2SO4 and the solvent is H2O. Additionally, ionic liquid electrolytes (e.g. NaFSI and NaTFSI) were also proposed to address the safety problem as they are practically nonflammable.4,9Compared with organic electrolyte liquids, solid state electrolyte features much better mechanical strength and thermal stability; meanwhile, the progress of the solid state electrolyte is the key to developing all-solid-state sodium-ion batteries. Polymer electrolytes (PEO–NaClO4, PEO–NaCF3SO3, PMMA–SiO2, etc.) have displayed promising Na-ion transmittability in the past few years. Inorganic NASICON-type AnM2(XO4)3 (A = alkali metal, M = transition metal, X = Si4+, P5+, S6+, etc.) also holds great potential due to their 3D channels for fast Na+ ion diffusion,164 for example, the ionic conductivity of Na3Zr2−x/4Si2−xP1+xO12 (0 < x < 2) can reach 6 × 10−4 S cm−1 at room temperature. Recently, the Na3PS4-series solid state electrolytes have attracted a great interest owing to their superionic conductivity.165 On this basis, the further developed Na10SnP2S12, Na3PSe4, and Na3P0.62As0.38S4 electrolytes showed a superb ambient-temperature ionic conductivity of 0.4 mS cm−1, 1.16 mS cm−1, and 1.46 mS cm−1, respectively.166–168 Until now, research studies focusing on solid state electrolytes for SIBs are still relatively less; however, in view of its high safety, pursuing more suitable solid state electrolytes is an important direction for future development of Na-ion batteries.
5. Na-ion full batteries
With the advancing of fundamental research in key materials (cathodes, anodes, and electrolytes), developing Na-ion full batteries with practical value is imperative. In recent years, various Na-ion full cells have sprung up and showed application potentialities, which provided a basis for their future commercialization.5.1 Full batteries with organic electrolytes
Full batteries with organic electrolytes are the most studied SIB system, in which the anode is usually hard carbon, and the cathodes mainly concentrate on transition-metal oxides and polyanionic compounds. Komaba et al. constructed a NaNi0.5Mn0.5O2/hard carbon full cell with a reversible capacity of more than 200 mA h g−1 and an operating voltage of ∼3 V (Fig. 8a), its energy density could reach 60% of the LiCoO2/graphite LIBs.91 Afterwards, Johnson's team designed Na(Ni1/3Fe1/3Mn1/3)O2/hard carbon Na-ion batteries that could steadily run 150 cycles.169 Hu's group developed a novel layered O3–Na0.9[Cu0.22Fe0.30Mn0.48]O2 metal oxide, when matched with hard carbon to form full cells; the reversible capacity could reach 300 mA h g−1 (based on anodic mass) without obvious fading after 100 cycles, since the operation voltage was 3.2 V, the cell energy density was calculated to be 210 W h kg−1 based on the total mass of cathode and anode materials.170 Tang et al. evaluated the electrochemical performance of olivine NaFePO4/hard carbon full cells, obtaining a good capacity retention of 84% after 60 cycles.171 Chen's group used Na3V2(PO4)3@C as the cathode, natural graphite as the anode, and 1 M NaCF3SO3 in tetraglyme (TGM) as the electrolyte. The resultant full battery displayed a reversible capacity of 90 mA h g−1 (based on graphite mass) and an operation voltage of 2.2 V; more impressively, excellent rate and cycling performance (80% capacity retention after 400 cycles at 0.2 A g−1) was also achieved.83 Mai's team simultaneously applied the Na3V2(PO4)3/C nanocomposite as both the cathode and anode to design a symmetric Na-ion full battery. After comparing various adopted carbon sources, they found that acetylene black showed the best effect in promoting the electrochemical performance: a capacity retention of 80% after 200 cycles at 1C and tolerance of a 10C rate were demonstrated.172 Palacín's group matched the NaV2(PO4)2F3 cathode with the hard carbon anode, with the electrolyte optimized to be 1 M NaClO4 in EC0.45PC0.45DMC0.1, and they constructed a Na-ion full cell that exhibited a high operating voltage of 3.65 V, a stable reversible capacity of 97 mA h g−1 (based on NaV2(PO4)2F3) for 100 cycles, and a high rate capability of 70 mA h g−1 even at 5C (Fig. 8b).173 Inspiringly, Ma's team recently realized mass production of the layered NaNi1/3Fe1/3Mn1/3O2 cathode material, by using hard carbon as the anode counterpart, and they established promising soft package Na-ion batteries with a capacity of 1 A h, a capacity retention of 73% after 500 cycles, and an energy density of 100 W h kg−1 (Fig. 8c and d). After examining the electrochemistry and safety performance, SIB packs of 10 A h were further fabricated as a new portable energy storage device. This Na-storage device has a good prospect on household electrical energy storage, military power supply, smart grid, low-speed electric vehicle, etc.174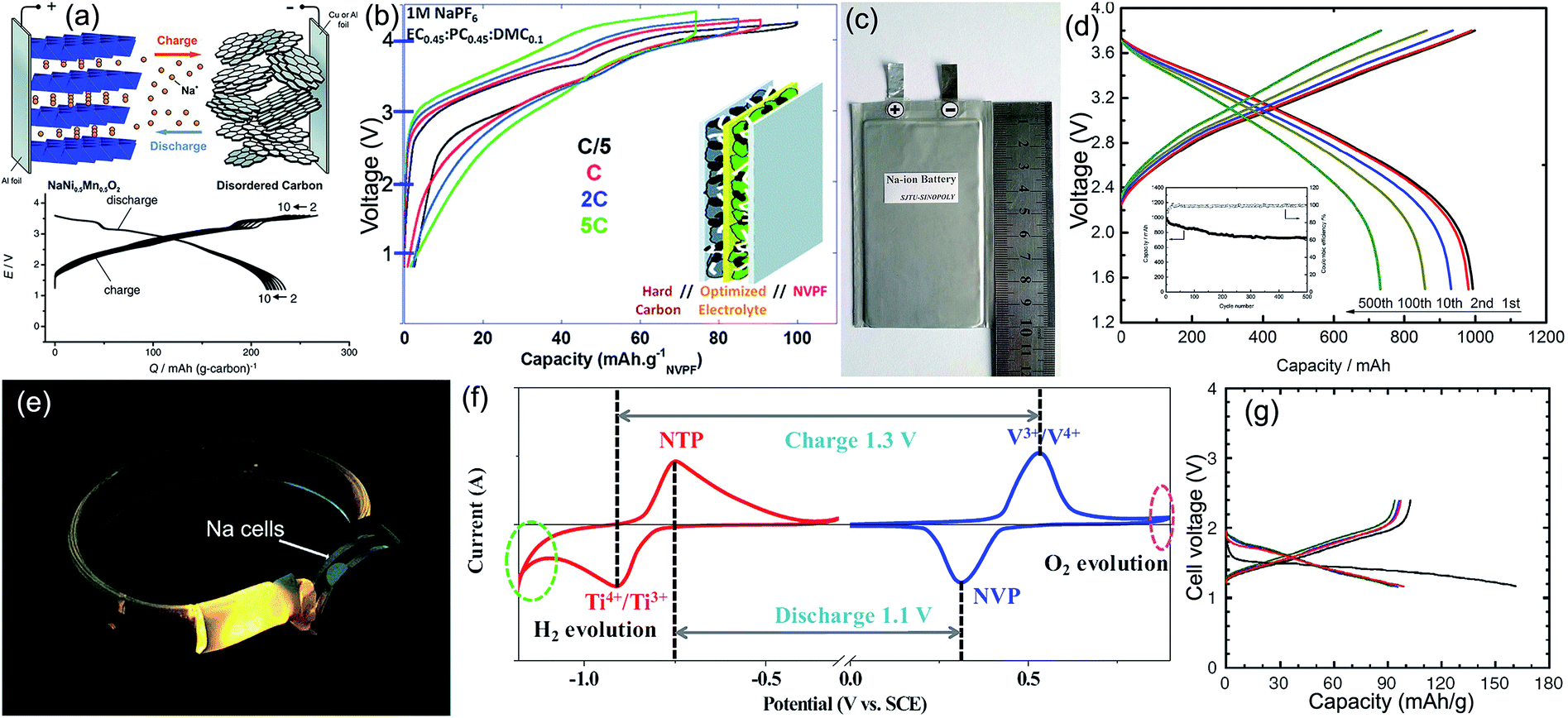 | ||
| Fig. 8 (a) Structural illustration and charge/discharge curves of the NaNi0.5Mn0.5O2/hard carbon full battery. Reproduced with permission.91 Copyright Wiley 2011. (b) Voltage versus capacity profiles of NaV2(PO4)2F3/hard carbon full Na-ion cells cycled in 1 M NaPF6 in EC0.45:PC0.45:DMC0.1 recorded at different rates. Reproduced with permission.173 Copyright Royal Society of Chemistry 2013. (c) Digital picture and (d) voltage profiles of the prepared 1 Ah soft-packed NaNi1/3Fe1/3Mn1/3O2/hard carbon sodium ion battery. Reproduced with permission.174 Copyright ECS 2016. (e) Na-ion batteries light up LED arrays in a wrist strap. Reproduced with permission.182 Copyright American Chemical Society 2016. (f) CV curves of the NaTi2(PO4)3 aqueous half cell at a scan rate of 1 mV s−1 and the Na3V2(PO4)3 aqueous half cell at a scan rate of 5 mV s−1. Reproduced with permission.184 Copyright Elsevier 2016. (g) Charge–discharge curves of the all-solid-state rechargeable sodium cell Na–Sn/Na3PS4 glass-ceramic/TiS2. The black, red, blue, and green lines, respectively, denote the first, second, third, and tenth cycles. Reproduced with permission.165 Copyright Nature Publishing Group 2012. | ||
Apart from the above examples, some other electrode materials have also manifested their application potential in SIBs. For instance, the Na-ion full cell configurations such as NaVPO4F–C/Sn@C,115 Prussian blue@C/hard carbon,175 Na3V2(PO4)3–rGO/Sb–rGO,176 Na0.66Ni0.26Zn0.07Mn0.67O2/P–C,131 Na3V2(PO4)2F3–C/MnFe2O4@C,147 Na0.80Li0.12Ni0.22Mn0.66O2/SnS2–graphene,153 Na3V2(PO4)3/NaTi2(PO4)3,177 Na1.0Li0.2Ni0.25Mn0.75Oδ/TiO2,178 Na3V2(PO4)2F3–C/Na2Ti6O13 (ref. 179) and so on, have exhibited favorable electrochemical performance. Another interesting finding is that Li and colleagues developed an iron-based NaxFeFe(CN)6/FeOx-CNTs soft package sodium-ion full battery, which was able to stably run 400 cycles.180 Very recently, an anode-free sodium full battery using a pre-sodiated pyrite cathode and a nanocarbon nucleation layer modified Al current collector was demonstrated, achieving an extremely high energy density of ∼400 W h kg−1 with long-term durability exceeding 1000 cycles.181 In addition, it is important to highlight that designing wearable Na-ion batteries based on flexible electrodes (Fig. 8e) is also a hotspot nowadays.182
5.2 Full batteries with aqueous and solid electrolytes
Na-ion batteries with aqueous electrolytes feature merits of high safety, environmental friendliness, low cost, long lifespan, and easy manipulation. Aqueous SIBs are anticipated to meet the requirements of large-scale energy storage, and its fundamental research and industrial development is getting more and more attention in recent years. However, aqueous electrolytes face side reactions of oxygen evolution on the cathode and hydrogen evolution on the anode. The theoretical decomposition potential of water is 1.23 V; therefore the voltage of aqueous Na-ion batteries is usually below 1.5 V to avoid the electrolysis of water. Undoubtedly, electrode materials must be water-insoluble and do not react with water; moreover, the reaction potential between Na and cathode should be lower than the oxygen evolution potential of water, and the reaction potential between Na and anode should be higher than the hydrogen evolution potential of water (as illustrated by the CV curves in Fig. 8f). NaTi2(PO4)3 is identified to be the most appropriate anode material for aqueous Na-ion batteries due to its typical NASICON structure that allows fast ion diffusion and, more importantly, its slightly higher Na-insertion potential than the hydrogen evolution potential of water. However, the intrinsic conductivity of NaTi2(PO4)3 is low; therefore it was usually composited with carbonaceous materials to improve the electrical conductivity and utilization rate. As for cathode materials, transition-metal oxides and polyanionic compounds are the most common choices. For instance, Li et al. established the Na0.44MnO2/NaTi2(PO4)3–C aqueous full cell, showing ultrafast rate capability (>100C) and superior cyclic stability (>1500 cycles), as well as a materials-only energy density of 127 W h L−1 and a cell-level energy density of ∼65 W h L−1.183 Li's group designed all NASICON structured Na3V2(PO4)3/NaTi2(PO4)3 aqueous full battery, demonstrating a flat discharge plateau at 1.2 V, an energy density of 29 W h kg−1 and a high power density of 5145 W kg−1.184 Currently, the Enpower Energy company (China) and Aquion Energy company (United States) have started the mass production of aqueous sodium-ion batteries with the major raw materials of Mn-based oxides, NaTi2(PO4)3, and Na2SO4; their competitive advantages are high safety, low cost, long life, and eco-friendliness.An all-solid-state battery is considered as the safest battery system without the hazards of liquid leakage, volatilization, and flammability. The NASICON-type Na3Zr2Si2PO12 solid state electrolyte and Na3V2(PO4)3 electrodes (serving as both the cathode and anode) were matched to construct an all-solid-state Na-ion battery, the ionic conductivity of the electrolyte and electrode was 1.5 × 10−3 S cm−1 and 1.9 × 10−4 S cm−1 at 200 °C, respectively, and the full cell exhibited a relatively high operation voltage (1.8 V) and energy density (1.87 × 10−3 W h cm−2).185 Hayashi et al. developed a superionic glass-ceramic electrolyte of Na3PS4; its ambient temperature conductivity could reach 2 × 10−4 S cm−1, when assembled with the TiS2 cathode and Na–Sn alloy anode, which is the first case that room-temperature operation of an all-solid-state rechargeable sodium battery was realized, displaying an operating voltage of ∼1.6 V and a reversible capacity of more than 90 mA h g−1 based on TiS2 (Fig. 8g).165Table 4 lists some representative examples of Na-ion full batteries, with their specific configurations and electrochemical performance summarized.
| Cathode | Anode | Electrolyte | Operating potential | Electrochemical performance | Ref. |
|---|---|---|---|---|---|
| NaNi0.5Mn0.5O2 | Hard carbon | 1 M NaN(SO2CF3)2 in PC | ∼3.0 V | Over 200 mA h g−1 at 25 mA g−1 (anode), 60% energy density of LiCoO2/graphite | 91 |
| Na(Ni1/3Fe1/3Mn1/3)O2 | Hard carbon | 1 M NaClO4 in PC | ∼2.75 V | 100 mA h g−1 for 150 cycles at 75 mA g−1 (cathode) | 169 |
| O3–Na0.9(Cu0.22Fe0.3Mn0.48)O2 | Hard carbon | 0.8 M NaPF6 in EC/DMC (1:1 vol) |
3.2 V | 300 mA h g−1 for 100 cycles at 50 mA g−1 (anode), 210 W h kg−1 (cathode and anode) | 170 |
| Olivine NaFePO4 | Hard carbon | 1 M NaClO4 in PC/EC (1:1 vol) + 5 vol% FEC |
∼1.8 V | 140 mA h g−1 at 0.1C, 84% capacity retention after 60 cycles at 1C (cathode) | 171 |
| Na3V2(PO4)3/C | Graphite | 1 M NaCF3SO3 in TGM | ∼2.2 V | 90 mA h g−1 at 0.1 A g−1, 80% capacity retention after 400 cycles at 2 A g−1 (anode) | 83 |
| Na3V2(PO4)3/acetylene carbon | Na3V2(PO4)3/acetylene carbon | 1 M NaClO4 in EC/DMC (1:2 w/w) |
1.75 V | 100.9 mA h g−1 at 0.5C, 42.2 mA h g−1 at 10C, 80% capacity retention after 200 cycles at 1C (cathode) | 172 |
| Na3V2(PO4)2F3 | Hard carbon | 1 M NaClO4 in EC/PC/DMC (9:9:2 vol) |
3.65 V | 97 mA h g−1 after 120 cycles at 0.2C, 70 mA h g−1 at 5C (cathode), 78 W h kg−1 (cathode and anode) | 173 |
| NaVPO4F/C | Sn nanodots@porous N-doped C | 1 M NaClO4 in PC + 5 vol% FEC | ∼2.0 V | 540 mA h g−1 at 0.5 A g−1, 460 mA h g−1 after 100 cycles at 0.5 A g−1 (anode) | 115 |
| Na0.66Ni0.26Zn0.07Mn0.67O2 | Black P/C | 1 M NaPF6 in PC + 2 vol% FEC | ∼2.5 V | 100 mA h g−1 after 100 cycles at 12 mA g−1, 80 mA h g−1 after 100 cycles at 60 mA g−1 (cathode) | 131 |
| Na3V2(PO4)2F3/C | MnFe2O4@C | 1 M NaClO4 in PC + 5 vol% FEC | ∼2.3 V | 406 mA h g−1 at 0.5 A g−1, 96.5% capacity retention after 100 cycles (anode), 77.8 W h kg−1 (whole battery) | 147 |
| Na0.80Li0.12Ni0.22Mn0.66O2 | SnS2–rGO | 1 M NaPF6 in EC/DEC, (1:1 vol) |
∼2.5 V | 423 mA h g−1 at 0.2 A g−1 and 74% capacity retention after 50 cycles (anode) | 153 |
| Prussian blue@C | Hard carbon | 1 M NaPF6 in EC/DEC, (1:1 vol) |
∼2.8 V | ∼120 mA h g−1 after 25 cycles at 100 mA g−1 (cathode) | 175 |
| Na3V2(PO4)3–rGO | Sb–rGO | 1 M NaClO4 in EC/DMC, (1:1, w/w) |
∼1.8 V | ∼400 mA h g−1 after 100 cycles at 100 mA g−1 (anode) | 176 |
| Na3V2(PO4)3 | NaTi2(PO4)3 | 1 M NaClO4 in EC/DMC (1:1 vol) + 5 vol% FEC |
∼1.2 V | 80 mA h g−1 at 50C, 96.9% capacity retention after 300 cycles at 5C (cathode) | 177 |
| Na1.0Li0.2Ni0.25Mn0.75Oδ | TiO2 nanotube | 1 M NaClO4 in PC | ∼1.8 V | 80 mA h g−1 at 11 mA g−1, 56 mA h g−1 at 0.56 A g−1 (anode) | 178 |
| Na3V2(PO4)2F3/C | Na2Ti6O13 | 1 M NaClO4 in EC/PC (1:1 vol) |
∼2.5 V | 42 mA h g−1 at 0.2C (anode) | 179 |
| NaxFeFe(CN)6 | CNT/FeOx | 1 M NaClO4 in EC/DEC (1:1 vol) + 8 vol% FEC |
∼2.0 V | 125 mA h g−1 at 0.25 A g−1, 70 mA h g−1 at 1 A g−1 (cathode), 136 W h kg−1 (cathode and anode) | 180 |
| Na0.44MnO2 | NaTi2(PO4)3 | 1 M Na2SO4 aqueous solution | ∼1.0 V | 50% capacity retention at 11.2 mA cm−2 after 1600 cycles, 65 W h L−1 (cathode and anode) | 183 |
| Na3V2(PO4)3 | NaTi2(PO4)3 | 1 M Na2SO4 aqueous solution | 1.2 V | 58 mA h g−1 at 10 A g−1, 50% capacity retention at 10 A g−1 for 50 cycles (anode), 29 W h kg−1 (cathode and anode) | 184 |
| Na3V2(PO4)3 | Na3V2(PO4)3 | Na3Zr2Si2PO12 (solid electrolyte) | 1.8 V | 1.87 × 10−3 W h cm−2 and 1.04 mA h cm−2 at 0.1C at 200 °C (cathode and anode) | 185 |
| TiS2 | Na–Sn alloy | Na3PS4 (solid electrolyte) | 1.6 V | 90 mA h g−1 for 10 cycles at 13 A cm−2 (cathode) | 165 |
6. Conclusions and outlook
Sodium-ion batteries have attracted extensive attention and witnessed great breakthroughs in the past few years owing to their analogous working mechanism to LIBs and plentiful resources. The potential markets of SIBs mainly lie in large-scale devices where durability and cost are more essential factors than energy density, such as stationary electric storage for sustainable and clean energies. This review summarizes the state-of-the-art research progress on Na-storage key materials including cathodes, anodes, and electrolytes; meanwhile, representative Na-ion full batteries with application value are also introduced. Among the positive electrode materials, layered transition-metal oxides have long been a shining point due to their high theoretical capacities and electrical conductivity. However, their cyclic stability is unsatisfactory induced by gliding of the transition-metal layers, cation-substitution is an effective strategy to enhance the structural stability of layered metal oxides and therefore their cycling performance. Polyanionic sodium salts featured with high operating potential and good stability (both thermal and structural) have also been regarded as a promising family of Na-storage cathode materials. The drawbacks are their relatively low specific capacities and electrical conductivity, carbon coating, nano-engineering, and creating porous structure are valid countermeasures.Among negative electrodes, carbon-based and alloy-based materials have shown the most application prospects. Although the specific capacities of carbonaceous anodes are relatively low, their abundant reserves, low price, and outstanding cyclability endow them with great commercial potential. Designing micro/nano, hollow, or porous structures is capable of further exploiting the Na-storage ability of carbon-based hosts. Generally, alloy-based materials feature high theoretical capacities and superb electrical conductivity, but the large volume expansion along with sodium insertion would cause serious capacity degradation. Modifications like reducing the dimension to nanoscale, carbon coating, and alloying are efficient strategies to buffer the volume fluctuation during repeated sodiation/desodiation. In addition, NaTi2(PO4)3 possesses open channels for rapid Na-ion diffusion and, more impressively, it is highly stable in water and would not induce the side hydrolysis reactions; therefore, NaTi2(PO4)3 is considered as the most appropriate anode material for aqueous SIBs.
Na-ion batteries are now enjoying fast development with the electrochemical performance being largely improved. Nevertheless, insight into the reaction and failure mechanisms, as well as in-depth understanding of the interactions at the electrode/electrolyte interface needs to be further gained. In addition, considerable studies are required not only for active materials but also for electrolytes, additives, and binders, which can significantly influence the electrode performance. To date, the majority of the reports regarding SIBs with great enhancement are mainly based on only experimental observations. The industrial-scale production and manipulation of functional materials with high electrochemical activity still face difficulties. In the future, optimization of the preparation technology to realize scale-up, matching of the electrode and electrolyte materials to construct high-performance and high-safety full cells, understanding of the correlation between structure/composition and property/performance, and establishment of mathematical modeling and theoretical simulation to direct purposeful design are highly anticipated towards more prosperous of sodium-ion batteries.
Acknowledgements
This work was supported by the Natural Scientific Foundation of China (51532002, 51372022, 51575030, 51622102, 51231003, and 51571124), the National Basic Research Program of China (2015CB932500), the Ministry of Education of China (IRT-13R30), MOST (2016YFB0901502), the National Postdoctoral Program for Innovative Talents (BX201600014), the Fundamental Research Funds for the Central Universities (FRF-TP-16-078A1), the China Postdoctoral Science Foundation (2016M600042), and the 111 Project (B12015).Notes and references
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- J. B. Goodenough, Acc. Chem. Res., 2013, 46, 1053–1061 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 CAS.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3431–3448 CrossRef CAS PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- S. P. Ong, V. L. Chevrier, G. Hautier, A. Jain, C. Moore, S. Kim, X. Ma and G. Ceder, Energy Environ. Sci., 2011, 4, 3680–3688 CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N.-S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067–2081 CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271–1273 CrossRef CAS.
- S. Okada, Y. Takahashi, T. Kiyabu, T. Doi, J.-I. Yamaki and T. Nishida, 210th ECS Meeting Abstracts, 2006, MA2006-02, p. 201 Search PubMed.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312–2337 CAS.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- Y. Li, Y. Lu, C. Zhao, Y.-S. Hu, M.-M. Titirici, H. Li, X. Huang and L. Chen, Energy Storage Mater., 2017, 7, 130–151 CrossRef.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- X. Xiang, K. Zhang and J. Chen, Adv. Mater., 2015, 27, 5343–5364 CrossRef CAS PubMed.
- C. Delmas, J.-J. Braconnier, C. Fouassier and P. Hagenmuller, Solid State Ionics, 1981, 3–4, 165–169 CrossRef CAS.
- Y. Lei, X. Li, L. Liu and G. Ceder, Chem. Mater., 2014, 26, 5288–5296 CrossRef CAS.
- J. Billaud, R. J. Clément, A. R. Armstrong, J. Canales-Vázquez, P. Rozier, C. P. Grey and P. G. Bruce, J. Am. Chem. Soc., 2014, 136, 17243–17248 CrossRef CAS PubMed.
- D. Su, C. Wang, H.-J. Ahn and G. Wang, Chem.–Eur. J., 2013, 19, 10884–10889 CrossRef CAS PubMed.
- J. Zhao, L. Zhao, N. Dimov, S. Okada and T. Nishida, J. Electrochem. Soc., 2013, 160, A3077–A3081 CrossRef CAS.
- N. Yabuuchi, H. Yoshida and S. Komaba, Electrochemistry, 2012, 80, 716–719 CrossRef CAS.
- S. Komaba, C. Takei, T. Nakayama, A. Ogata and N. Yabuuchi, Electrochem. Commun., 2010, 12, 355–358 CrossRef CAS.
- P. Vassilaras, X. Ma, X. Li and G. Ceder, J. Electrochem. Soc., 2013, 160, A207–A211 CrossRef CAS.
- Y. Dong, S. Li, K. Zhao, C. Han, W. Chen, B. Wang, L. Wang, B. Xu, Q. Wei, L. Zhang, X. Xu and L. Mai, Energy Environ. Sci., 2015, 8, 1267–1275 CAS.
- S. Komaba, N. Yabuuchi, T. Nakayama, A. Ogata, T. Ishikawa and I. Nakai, Inorg. Chem., 2012, 51, 6211–6220 CrossRef CAS PubMed.
- D. D. Yuan, Y. X. Wang, Y. L. Cao, X. P. Ai and H. X. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 8585–8591 CAS.
- S.-M. Oh, S.-T. Myung, C. S. Yoon, J. Lu, J. Hassoun, B. Scrosati, K. Amine and Y.-K. Sun, Nano Lett., 2014, 14, 1620–1626 CrossRef CAS PubMed.
- M. Sathiya, K. Hemalatha, K. Ramesha, J.-M. Tarascon and A. S. Prakash, Chem. Mater., 2012, 24, 1846–1853 CrossRef CAS.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef CAS PubMed.
- D. Kim, S.-H. Kang, M. Slater, S. Rood, J. T. Vaughey, N. Karan, M. Balasubramanian and C. S. Johnson, Adv. Energy Mater., 2011, 1, 333–336 CrossRef CAS.
- R. Kataoka, T. Mukai, A. Yoshizawa and T. Sakai, J. Electrochem. Soc., 2013, 160, A933–A939 CrossRef CAS.
- S. Guo, P. Liu, H. Yu, Y. Zhu, M. Chen, M. Ishida and H. Zhou, Angew. Chem., Int. Ed., 2015, 54, 5894–5899 CrossRef CAS PubMed.
- F. Sauvage, L. Laffont, J.-M. Tarascon and E. Baudrin, Inorg. Chem., 2007, 46, 3289–3294 CrossRef CAS PubMed.
- J.-P. Parant, R. Olazcuaga, M. Devalette, C. Fouassier and P. Hagenmuller, J. Solid State Chem., 1971, 3, 1–11 CrossRef CAS.
- M. Xu, Y. Niu, C. Chen, J. Song, S. Bao and C. M. Li, RSC Adv., 2014, 4, 38140–38143 RSC.
- Y. Cao, L. Xiao, W. Wang, D. Choi, Z. Nie, J. Yu, L. V. Saraf, Z. Yang and J. Liu, Adv. Mater., 2011, 23, 3155–3160 CrossRef CAS PubMed.
- S. Guo, H. Yu, D. Liu, W. Tian, X. Liu, N. Hanada, M. Ishida and H. Zhou, Chem. Commun., 2014, 50, 7998–8001 RSC.
- S.-M. Oh, S.-T. Myung, J. Hassoun, B. Scrosati and Y.-K. Sun, Electrochem. Commun., 2012, 22, 149–152 CrossRef CAS.
- Y. Zhu, Y. Xu, Y. Liu, C. Luo and C. Wang, Nanoscale, 2013, 5, 780–787 RSC.
- J. Kim, D.-H. Seo, H. Kim, I. Park, J.-K. Yoo, S.-K. Jung, Y.-U. Park, W. A. Goddard III and K. Kang, Energy Environ. Sci., 2015, 8, 540–545 CAS.
- C. Li, X. Miao, W. Chu, P. Wu and D. G. Tong, J. Mater. Chem. A, 2015, 3, 8265–8271 CAS.
- Z. Jian, C. Yuan, W. Han, X. Lu, L. Gu, X. Xi, Y.-S. Hu, H. Li, W. Chen, D. Chen, Y. Ikuhara and L. Chen, Adv. Funct. Mater., 2014, 24, 4265–4272 CrossRef CAS.
- L. S. Plashnitsa, E. Kobayashi, Y. Noguchi, S. Okada and J.-I. Yamaki, J. Electrochem. Soc., 2010, 157, A536–A543 CrossRef CAS.
- W. Duan, Z. Zhu, H. Li, Z. Hu, K. Zhang, F. Cheng and J. Chen, J. Mater. Chem. A, 2014, 2, 8668–8675 CAS.
- K. Saravanan, C. W. Mason, A. Rudola, K. H. Wong and P. Balaya, Adv. Energy Mater., 2013, 3, 444–450 CrossRef CAS.
- M. J. Aragón, P. Lavela, G. F. Ortiz and J. L. Tirado, J. Electrochem. Soc., 2015, 162, A3077–A3083 CrossRef.
- H. Li, X. Yu, Y. Bai, F. Wu, C. Wu, L.-Y. Liu and X.-Q. Yang, J. Mater. Chem. A, 2015, 3, 9578–9586 CAS.
- B. L. Ellis, W. R. M. Makahnouk, Y. Makimura, K. Toghill and L. F. Nazar, Nat. Mater., 2007, 6, 749–753 CrossRef CAS PubMed.
- K. Kubota, K. Yokoh, N. Yabuuchi and S. Komaba, Electrochemistry, 2014, 82, 909–911 CrossRef CAS.
- J. Barker, M. Y. Saidi and J. L. Swoyer, Electrochem. Solid-State Lett., 2003, 6, A1–A4 CrossRef CAS.
- Y.-L. Ruan, K. Wang, S.-D. Song, X. Han and B.-W. Cheng, Electrochim. Acta, 2015, 160, 330–336 CrossRef CAS.
- R. A. Shakoor, D.-H. Seo, H. Kim, Y.-U. Park, J. Kim, S.-W. Kim, H. Gwon, S. Lee and K. Kang, J. Mater. Chem., 2012, 22, 20535–20541 RSC.
- Y.-U. Park, D.-H. Seo, H.-S. Kwon, B. Kim, J. Kim, H. Kim, I. Kim, H.-Y. Yoo and K. Kang, J. Am. Chem. Soc., 2013, 135, 13870–13878 CrossRef CAS PubMed.
- P. Barpanda, S.-I. Nishimura and A. Yamada, Adv. Energy Mater., 2012, 2, 841–859 CrossRef CAS.
- H. Kim, R. A. Shakoor, C. Park, S. Y. Lim, J.-S. Kim, Y. N. Jo, W. Cho, K. Miyasaka, R. Kahraman, Y. Jung and J. W. Choi, Adv. Funct. Mater., 2013, 23, 1147–1155 CrossRef CAS.
- M. Nose, H. Nakayama, K. Nobuhara, H. Yamaguchi, S. Nakanishi and H. Iba, J. Power Sources, 2013, 234, 175–179 CrossRef CAS.
- P. Singh, K. Shiva, H. Celio and J. B. Goodenough, Energy Environ. Sci., 2015, 8, 3000–3005 CAS.
- Y. Lu, L. Wang, J. Cheng and J. B. Goodenough, Chem. Commun., 2012, 48, 6544–6546 RSC.
- J. Qian, M. Zhou, Y. Cao, X. Ai and H. Yang, Adv. Energy Mater., 2012, 2, 410–414 CrossRef CAS.
- H.-W. Lee, R. Y. Wang, M. Pasta, S. W. Lee, N. Liu and Y. Cui, Nat. Commun., 2014, 5, 5280 CrossRef CAS PubMed.
- X. Wu, W. Deng, J. Qian, Y. Cao, X. Ai and H. Yang, J. Mater. Chem. A, 2013, 1, 10130–10134 CAS.
- L. Wang, Y. Lu, J. Liu, M. Xu, J. Cheng, D. Zhang and J. B. Goodenough, Angew. Chem., Int. Ed., 2013, 52, 1964–1967 CrossRef CAS PubMed.
- L. Wang, J. Song, R. Qiao, L. A. Wray, M. A. Hossain, Y.-D. Chuang, W. Yang, Y. Lu, D. Evans, J.-J. Lee, S. Vail, X. Zhao, M. Nishijima, S. Kakimoto and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2548–2554 CrossRef CAS PubMed.
- I. D. Gocheva, M. Nishijima, T. Doi, S. Okada, J.-I. Yamaki and T. Nishida, J. Power Sources, 2009, 187, 247–252 CrossRef CAS.
- M. Nishijima, I. D. Gocheva, S. Okada, T. Doi, J.-I. Yamaki and T. Nishida, J. Power Sources, 2009, 190, 558–562 CrossRef CAS.
- S. Wang, L. Wang, Z. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 5892–5896 CrossRef CAS PubMed.
- W. Luo, M. Allen, V. Raju and X. Ji, Adv. Energy Mater., 2014, 4, 1400554 CrossRef.
- J. J. Ding, Y. N. Zhou, Q. Sun, X. Q. Yu, X. Q. Yang and Z. W. Fu, Electrochim. Acta, 2013, 87, 388–393 CrossRef CAS.
- J. Billaud, G. Singh, A. R. Armstrong, E. Gonzalo, V. Roddatis, M. Armand, T. Rojo and P. G. Bruce, Energy Environ. Sci., 2014, 7, 1387–1391 CAS.
- N. Yabuuchi, M. Yano, H. Yoshida, S. Kuze and S. Komaba, J. Electrochem. Soc., 2013, 160, A3131–A3137 CrossRef CAS.
- H. Yu, S. Guo, Y. Zhu, M. Ishida and H. Zhou, Chem. Commun., 2014, 50, 457–459 RSC.
- D. Yuan, X. Liang, L. Wu, Y. Cao, X. Ai, J. Feng and H. Yang, Adv. Mater., 2014, 26, 6301–6306 CrossRef CAS PubMed.
- Q. Wang, B. Zhao, S. Zhang, X. Gao and C. Deng, J. Mater. Chem. A, 2015, 3, 7732–7740 CAS.
- Y. Kawabe, N. Yabuuchi, M. Kajiyama, N. Fukuhara, T. Inamasu, R. Okuyama, I. Nakai and S. Komaba, Electrochem. Commun., 2011, 13, 1225–1228 CrossRef CAS.
- K. Chihara, A. Kitajou, I. D. Gocheva, S. Okada and J.-I. Yamaki, J. Power Sources, 2013, 227, 80–85 CrossRef CAS.
- Y. Kim, K.-H. Ha, S. M. Oh and K. T. Lee, Chem.–Eur. J., 2014, 20, 11980–11992 CrossRef CAS PubMed.
- H. Kang, Y. Liu, K. Cao, Y. Zhao, L. Jiao, Y. Wang and H. Yuan, J. Mater. Chem. A, 2015, 3, 17899–17913 CAS.
- M. Dahbi, N. Yabuuchi, K. Kubota, K. Tokiwa and S. Komaba, Phys. Chem. Chem. Phys., 2014, 16, 15007–15028 RSC.
- J. Cui, S. Yao and J.-K. Kim, Energy Storage Mater., 2017, 7, 64–114 CrossRef.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed.
- Z. Zhu, F. Cheng, Z. Hu, Z. Niu and J. Chen, J. Power Sources, 2015, 293, 626–634 CrossRef CAS.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033 CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803–A811 CrossRef CAS.
- R. Alcántara, P. Lavela, G. F. Ortiz and J. L. Tirado, Electrochem. Solid-State Lett., 2005, 8, A222–A225 CrossRef.
- K. Gotoh, T. Ishikawa, S. Shimadzu, N. Yabuuchi, S. Komaba, K. Takeda, A. Goto, K. Deguchi, S. Ohki, K. Hashi, T. Shimizu and H. Ishida, J. Power Sources, 2013, 225, 137–140 CrossRef CAS.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- C. Bommier, T. W. Surta, M. Dolgos and X. Ji, Nano Lett., 2015, 15, 5888–5892 CrossRef CAS PubMed.
- J. M. Stratford, P. K. Allan, O. Pecher, P. A. Chater and C. P. Grey, Chem. Commun., 2016, 52, 12430–12433 RSC.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakatama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- M. Dahbi, T. Nakano, N. Yabuuchi, S. Fujimura, K. Chihara, K. Kubota, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, ChemElectroChem, 2016, 3, 1856–1867 CrossRef CAS.
- E. M. Lotfabad, P. Kalisvaart, A. Kohandehghan, D. Karpuzov and D. Mitlin, J. Mater. Chem. A, 2014, 2, 19685–19695 Search PubMed.
- C. Bommier, D. Leonard, Z. Jian, W. F. Stickle, P. A. Greaney and X. Ji, Adv. Mater. Interfaces, 2016, 3, 1600449 CrossRef.
- J. Ding, H. L. Wang, Z. Li, A. Kohandehghan, K. Cui, Z. W. Xu, B. Zahiri, X. H. Tan, E. M. Lotfabad, B. C. Olsen and D. Mitlin, ACS Nano, 2013, 7, 11004–11015 CrossRef CAS PubMed.
- Y. L. Cao, L. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. Nie, L. V. Saraf, Z. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS PubMed.
- W. Li, L. Zeng, Z. Yang, L. Gu, J. Wang, X. Liu, J. Cheng and Y. Yu, Nanoscale, 2014, 6, 693–698 RSC.
- K. Tang, L. Fu, R. J. White, L. Yu, M.-M. Titirici, M. Antonietti and J. Maier, Adv. Energy Mater., 2012, 2, 873–877 CrossRef CAS.
- S. Wang, L. Xia, L. Yu, L. Zhang, H. H. Wang and X. W. (David) Lou, Adv. Energy Mater., 2016, 6, 1502217 CrossRef.
- J. Yang, X. Zhou, D. Wu, X. Zhao and Z. Zhou, Adv. Mater., 2017, 29, 1604108 CrossRef PubMed.
- L. M. Dai, Acc. Chem. Res., 2013, 46, 31–42 CrossRef CAS PubMed.
- J. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. X. Dou and L. M. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS PubMed.
- Y. Yan, Y.-X. Yin, Y.-G. Guo and L.-J. Wan, Adv. Energy Mater., 2014, 4, 1301584 CrossRef.
- Y. Liu, L.-Z. Fan and L. Jiao, J. Mater. Chem. A, 2017, 5, 1698–1705 CAS.
- M. M. Doeff, Y. Ma, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 1993, 140, L169–L170 CrossRef CAS.
- W. Luo, Z. Jian, Z. Xing, W. Wang, C. Bommier, M. M. Lerner and X. Ji, ACS Cent. Sci., 2015, 1, 516–522 CrossRef CAS PubMed.
- Z. Jian, C. Bommier, L. Luo, Z. Li, W. Wang, C. Wang, P. A. Greaney and X. Ji, Chem. Mater., 2017, 29, 2314–2320 CrossRef CAS.
- V. L. Chevrier and G. Ceder, J. Electrochem. Soc., 2011, 158, A1011–A1014 CrossRef CAS.
- L. D. Ellis, T. D. Hatchard and M. N. Obrovac, J. Electrochem. Soc., 2012, 159, A1801–A1805 CrossRef CAS.
- J. W. Wang, X. H. Liu, S. X. Mao and J. Y. Huang, Nano Lett., 2012, 12, 5897–5902 CrossRef CAS PubMed.
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657–1665 CrossRef CAS PubMed.
- D.-H. Nam, T.-H. Kim, K.-S. Hong and H.-S. Kwon, ACS Nano, 2014, 8, 11824–11835 CrossRef CAS PubMed.
- Y. Xu, Y. Zhu, Y. Liu and C. Wang, Adv. Energy Mater., 2013, 3, 128–133 CrossRef CAS.
- Y. Liu, Y. Xu, Y. Zhu, J. N. Culver, C. A. Lundgren, K. Xu and C. Wang, ACS Nano, 2013, 7, 3627–3634 CrossRef CAS PubMed.
- Y. Liu, N. Zhang, L. Jiao and J. Chen, Adv. Mater., 2015, 27, 6702–6707 CrossRef CAS PubMed.
- B. Zhang, G. Rousse, D. Foix, R. Dugas, D. A. D. Corte and J.-M. Tarascon, Adv. Mater., 2016, 28, 9824–9830 CrossRef CAS PubMed.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805–20811 CrossRef CAS PubMed.
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255–1262 CrossRef CAS PubMed.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, Energy Environ. Sci., 2014, 7, 323–328 CAS.
- N. Zhang, Y. Liu, Y. Lu, X. Han, F. Cheng and J. Chen, Nano Res., 2015, 8, 3384–3393 CrossRef CAS.
- Y. Xu, E. Swaans, S. Basak, H. W. Zandbergen, D. M. Borsa and F. M. Mulder, Adv. Energy Mater., 2016, 6, 1501436 CrossRef.
- L. Zhang, X. Hu, C. Chen, H. Guo, X. Liu, G. Xu, H. Zhong, S. Cheng, P. Wu, J. Meng, Y. Huang, S. Dou and H. Liu, Adv. Mater., 2017, 29, 1604708 CrossRef PubMed.
- J. Liu, Y. Wen, P. A. van Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 6387–6392 CrossRef CAS PubMed.
- Y.-M. Lin, P. R. Abel, A. Gupta, J. B. Goodenough, A. Heller and C. B. Mullins, ACS Appl. Mater. Interfaces, 2013, 5, 8273–8277 CAS.
- L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J.-G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed.
- B. Farbod, K. Cui, W. P. Kalisvaart, M. Kupsta, B. Zahiri, A. Kohandehghan, E. M. Lotfabad, Z. Li, E. J. Luber and D. Mitlin, ACS Nano, 2014, 8, 4415–4429 CrossRef CAS PubMed.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633–4636 CrossRef CAS PubMed.
- Y. Kim, Y. Park, A. Choi, N.-S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS PubMed.
- W.-J. Li, S.-L. Chou, J.-Z. Wang, H.-K. Liu and S.-X. Dou, Nano Lett., 2013, 13, 5480–5484 CrossRef CAS PubMed.
- J. Song, Z. Yu, M. L. Gordin, S. Hu, R. Yi, D. Tang, T. Walter, M. Regula, D. Choi, X. Li, A. Manivannan and D. Wang, Nano Lett., 2014, 14, 6329–6335 CrossRef CAS PubMed.
- G.-L. Xu, Z. Chen, G.-M. Zhong, Y. Liu, Y. Yang, T. Ma, Y. Ren, X. Zuo, X.-H. Wu, X. Zhang and K. Amine, Nano Lett., 2016, 16, 3955–3965 CrossRef CAS PubMed.
- Y. Zhu, Y. Wen, X. Fan, T. Gao, F. Han, C. Luo, S.-C. Liou and C. Wang, ACS Nano, 2015, 9, 3254–3264 CrossRef CAS PubMed.
- W. Li, Z. Yang, M. Li, Y. Jiang, X. Wei, X. Zhong, L. Gu and Y. Yu, Nano Lett., 2016, 16, 1546–1553 CrossRef CAS PubMed.
- L. Pei, Q. Zhao, C. Chen, J. Liang and J. Chen, ChemElectroChem, 2015, 2, 1652–1655 CrossRef CAS.
- J. Sun, H.-W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–985 CrossRef CAS PubMed.
- J. Qian, Y. Xiong, Y. Cao, X. Ai and H. Yang, Nano Lett., 2014, 14, 1865–1869 CrossRef CAS PubMed.
- W. Li, S.-L. Chou, J.-Z. Wang, J. H. Kim, H.-K. Liu and S.-X. Dou, Adv. Mater., 2014, 26, 4037–4042 CrossRef CAS PubMed.
- J. Liu, P. Kopold, C. Wu, P. A. van Aken, J. Maier and Y. Yu, Energy Environ. Sci., 2015, 8, 3531–3538 CAS.
- X. Fan, J. Mao, Y. Zhu, C. Luo, L. Suo, T. Gao, F. Han, S.-C. Liou and C. Wang, Adv. Energy Mater., 2015, 5, 1500174 CrossRef.
- S. Komaba, Y. Matsuura, T. Ishikawa, N. Yabuuchi, W. Murata and S. Kuze, Electrochem. Commun., 2012, 21, 65–68 CrossRef CAS.
- J. Jiang, M. Hu, D. Zhang, T. Yuan, W. Sun, B. Xu and M. Yan, Nano Energy, 2014, 5, 60–66 CrossRef.
- N. Zhang, X. Han, Y. Liu, X. Hu, Q. Zhao and J. Chen, Adv. Energy Mater., 2015, 5, 1401123 CrossRef.
- Z. Jian, P. Liu, F. Li, M. Chen and H. Zhou, J. Mater. Chem. A, 2014, 2, 13805–13809 CAS.
- S. Yuan, X.-L. Huang, D.-L. Ma, H.-G. Wang, F.-Z. Meng and X.-B. Zhang, Adv. Mater., 2014, 26, 2273–2279 CrossRef CAS PubMed.
- D. Su, H.-J. Ahn and G. Wang, Chem. Commun., 2013, 49, 3131–3133 RSC.
- M. Hu, Y. Jiang, W. Sun, H. Wang, C. Jin and M. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 19449–19455 CAS.
- Y. Liu, N. Zhang, C. Yu, L. Jiao and J. Chen, Nano Lett., 2016, 16, 3321–3328 CrossRef CAS PubMed.
- Y. Liu, Y. Li, H. Kang, T. Jin and L. Jiao, Mater. Horiz., 2016, 3, 402–421 RSC.
- Y. Liu, H. Kang, L. Jiao, C. Chen, K. Cao, Y. Wang and H. Yuan, Nanoscale, 2015, 7, 1325–1332 RSC.
- P. Zhou, X. Wang, W. Guan, D. Zhang, L. Fang and Y. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 6979–6987 CAS.
- C. Zhu, X. Mu, P. A. van Aken, Y. Yu and J. Maier, Angew. Chem., Int. Ed., 2014, 53, 2152–2156 CrossRef CAS PubMed.
- Y. Liu, N. Zhang, H. Kang, M. Shang, L. Jiao and J. Chen, Chem.–Eur. J., 2015, 21, 11878–11884 CrossRef CAS PubMed.
- B. Qu, C. Ma, G. Ji, C. Xu, J. Xu, Y. S. Meng, T. Wang and J. Y. Lee, Adv. Mater., 2014, 26, 3854–3859 CrossRef CAS PubMed.
- S. Y. Lee and Y. C. Kang, Chem.–Eur. J., 2016, 22, 2769–2774 CrossRef CAS PubMed.
- Z. Hu, Z. Zhu, F. Cheng, K. Zhang, J. Wang, C. Chen and J. Chen, Energy Environ. Sci., 2015, 8, 1309–1316 CAS.
- D. Y. W. Yu, P. V. Prikhodchenko, C. W. Mason, S. K. Batabyal, J. Gun, S. Sladkevich, A. G. Medvedev and O. Lev, Nat. Commun., 2013, 4, 2922 Search PubMed.
- D. Zhang, W. Sun, Y. Zhang, Y. Dou, Y. Jiang and S. X. Dou, Adv. Funct. Mater., 2016, 26, 7479–7485 CrossRef CAS.
- X. Song, X. Li, Z. Bai, B. Yan, D. Li and X. Sun, Nano Energy, 2016, 26, 533–540 CrossRef CAS.
- S. Peng, X. Han, L. Li, Z. Zhu, F. Cheng, M. Srinivansan, S. Adams and S. Ramakrishna, Small, 2016, 12, 1359–1368 CrossRef CAS PubMed.
- Y. N. Ko and Y. C. Kang, Carbon, 2015, 94, 85–90 CrossRef CAS.
- K. Zhang, Z. Hu, X. Liu, Z. Tao and J. Chen, Adv. Mater., 2015, 27, 3305–3309 CrossRef CAS PubMed.
- H. Pan, X. Lu, X. Yu, Y.-S. Hu, H. Li, X.-Q. Yang and L. Chen, Adv. Energy Mater., 2013, 3, 1186–1194 CrossRef CAS.
- L. Zhao, J. Zhao, Y.-S. Hu, H. Li, Z. Zhou, M. Armand and L. Chen, Adv. Energy Mater., 2012, 2, 962–965 CrossRef CAS.
- W. Zhou, Y. Li, S. Xin and J. B. Goodenough, ACS Cent. Sci., 2017, 3, 52–57 CrossRef CAS PubMed.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 856 CrossRef PubMed.
- W. D. Richards, T. Tsujimura, L. J. Miara, Y. Wang, J. C. Kim, S. P. Ong, I. Uechi, N. Suzuki and G. Ceder, Nat. Commun., 2016, 7, 11009 CrossRef CAS PubMed.
- L. Zhang, K. Yang, J. Mi, L. Lu, L. Zhao, L. Wang, Y. Li and H. Zeng, Adv. Energy Mater., 2015, 5, 1501294 CrossRef.
- Z. Yu, S.-L. Shang, J.-H. Seo, D. Wang, X. Luo, Q. Huang, S. Chen, J. Lu, X. Li, Z.-K. Liu and D. Wang, Adv. Mater., 2017, 29, 1605561 CrossRef PubMed.
- D. Kim, E. Lee, M. Slater, W. Lu, S. Rood and C. S. Johnson, Electrochem. Commun., 2012, 18, 66–69 CrossRef CAS.
- L. Mu, S. Xu, Y. Li, Y.-S. Hu, H. Li, L. Chen and X. Huang, Adv. Mater., 2015, 27, 6928–6933 CrossRef CAS PubMed.
- W. Tang, X. Song, Y. Du, C. Peng, M. Lin, S. Xi, B. Tian, J. Zheng, Y. Wu, F. Pan and K. P. Loh, J. Mater. Chem. A, 2016, 4, 4882–4892 CAS.
- S. Li, Y. Dong, L. Xu, X. Xu, L. He and L. Mai, Adv. Mater., 2014, 26, 3545–3553 CrossRef CAS PubMed.
- A. Ponrouch, R. Dedryvère, D. Monti, A. E. Demet, J. M. A. Mba, L. Croguennec, C. Masquelier, P. Johansson and M. R. Palacín, Energy Environ. Sci., 2013, 6, 2361–2369 CAS.
- H. Wang, X.-Z. Liao, Y. Yang, X. Yan, Y.-S. He and Z.-F. Ma, J. Electrochem. Soc., 2016, 163, A565–A570 CrossRef CAS.
- Y. Jiang, S. Yu, B. Wang, Y. Li, W. Sun, Y. Lu, M. Yan, B. Song and S. Dou, Adv. Funct. Mater., 2016, 26, 5315–5321 CrossRef CAS.
- W. Zhang, Y. Liu, C. Chen, Z. Li, Y. Huang and X. Hu, Small, 2015, 11, 3822–3829 CrossRef CAS PubMed.
- W. Ren, Z. Zheng, C. Xu, C. Niu, Q. Wei, Q. An, K. Zhao, M. Yan, M. Qin and L. Mai, Nano Energy, 2016, 25, 145–153 CrossRef.
- H. Xiong, M. D. Slater, M. Balasubramanian, C. S. Johnson and T. Rajh, J. Phys. Chem. Lett., 2011, 2, 2560–2565 CrossRef CAS.
- A. Rudola, K. Saravanan, S. Devaraj, H. Gong and P. Balaya, Chem. Commun., 2013, 49, 7451–7453 RSC.
- H. Ye, Y. Wang, F. Zhao, W. Huang, N. Han, J. Zhou, M. Zeng and Y. Li, J. Mater. Chem. A, 2016, 4, 1754–1761 CAS.
- A. P. Cohn, N. Muralidharan, R. Carter, K. Share and C. L. Pint, Nano Lett., 2017, 17, 1296–1301 CrossRef CAS PubMed.
- S. Fu, J. Ni, Y. Xu, Q. Zhang and L. Li, Nano Lett., 2016, 16, 4544–4551 CrossRef CAS PubMed.
- Z. Li, D. Young, K. Xiang, W. C. Carter and Y.-M. Chiang, Adv. Energy Mater., 2013, 3, 290–294 CrossRef CAS.
- Q. Zhang, C. Liao, T. Zhai and H. Li, Electrochim. Acta, 2016, 196, 470–478 CrossRef CAS.
- F. Lalere, J. B. Leriche, M. Courty, S. Boulineau, V. Viallet, C. Masquelier and V. Seznec, J. Power Sources, 2014, 247, 975–980 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |