
A phenyl disulfide@CNT composite cathode for rechargeable lithium batteries†
Amruth
Bhargav
 a,
Shravan V.
Patil
a and
Yongzhu
Fu
*ab
a,
Shravan V.
Patil
a and
Yongzhu
Fu
*ab
aDepartment of Mechanical Engineering, Indiana University-Purdue University Indianapolis, Indianapolis, IN 46202, USA. E-mail: yongfu@iupui.edu
bRichard G. Lugar Center for Renewable Energy (LCRE), Indiana University-Purdue University Indianapolis, Indianapolis, IN 46202, USA
First published on 11th May 2017
Abstract
Rechargeable lithium batteries are playing an important role in portable electronics, electric vehicles, and renewable energy storage. Cathode materials in lithium batteries are mainly inorganic; however, some organic materials are also promising for these applications due to their diversified properties. Organosulfides are a unique class of materials which are low cost, abundant, and could provide high capacities. In this work, a phase extraction technique is used to prepare a core–sheath structured composite consisting of carbon nanotubes (CNTs) coated with a layer of phenyl disulfide (PDS, C6H5SSC6H5), which is designated as PDS@CNT. It forms a free-standing and binder-free paper which can be made in a scalable way with high mass loading. In lithium batteries, the S–S bonds in the composite can reversibly break and form in the CNT matrix, enabling good cycling performance. The half-cell can maintain a stable open circuit voltage over a week and deliver a specific capacity of 218 mA h g−1 at 1C while retaining 70% of the initial capacity after 150 cycles.
The world energy consumption is projected to grow by 40% by the year 2020.1,2 To meet this staggering increase in energy demand, mankind has to incorporate renewable energy sources in every sector. This concept seems to be promising due to the increased proliferation of renewable sources owing to the scientific advancements in the recent past. In part, energy storage devices coupled with intermittent energy sources such as wind and solar have strengthened their commercial viability for widespread use. Rechargeable batteries, such as lithium-ion (Li-ion) and redox-flow battery technologies, have gathered popular scientific interest in this arena.3
Promising candidates for cathode materials are organosulfur compounds which are environmentally benign, inexpensive and can be obtained from natural sources.4,5 Additionally, they offer specific energies up to 1600 W h kg−1 and power densities of nearly 400 W kg−1, making them suitable cathodes for rechargeable lithium batteries for large-scale energy storage.6–8 Furthermore, the ability to tailor the electrochemistry adds to the appeal of organic compounds.9 To this end, organodisulfides (RSSR, where R is an alkyl or aryl group) were extensively explored by Visco and co-workers in the early 1990s including phenyl disulfide (PDS, C6H5SSC6H5).10–13PDS was also explored by Inamasu and co-workers14 and Maddanimath and co-workers.15 Although these studies establish the viability of PDS as a cathode material, they report that the performance of PDS in a battery is poor which is attributed to the sluggish kinetics of PDS, i.e., S–S bond breaking and formation. This conclusion faded interest in organosulfur cathodes leading to no recent exploration of organodisulfides as cathode materials to the best of our knowledge. Recent advances in lithium–sulfur (Li–S) batteries using ether-based electrolyte systems inspire new interest in re-examining PDS as a cathode material in rechargeable lithium batteries. In this work, we utilize a phase extraction technique to prepare a core–sheath structured PDS@carbon nanotube (CNT) composite material, which shows promising performance in rechargeable lithium batteries using ether-based electrolytes.
Phenyl disulfide, the simplest aryl disulfide, has a theoretical specific capacity of 245.5 mA h g−1 by taking two electrons per molecule and a theoretical specific energy of about 552 W h kg−1. As PDS is soluble in ether electrolytes, it faces challenges similar to Li–S batteries such as inadequate confinement which could lead to a redox shuttle effect causing low coulombic efficiency and poor cyclability.16 To circumvent this issue, the cathode we have developed utilizes CNTs as the porous and conductive carbon matrix that has been shown to efficiently trap soluble polysulfides in Li–S batteries.17 They also promote active material utilization through adequate electrolyte infiltration while providing efficient pathways for charge transfer and accommodating volume expansion.17,18
A “phase-extraction” technique, which utilizes the difference in solubility of PDS in methanol and water, as a cathode fabrication process is schematically illustrated in Fig.1a. Firstly, PDS and CNTs in the ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight were vigorously stirred to dissolve PDS in methanol. This solution was then ultrasonicated to cause the CNTs to disperse and interweave. Following ultrasonication, water was added dropwise into the solution. PDS being insoluble in the water phase was extracted from the methanol phase and conformally deposited on the interwoven CNT bundles, giving the “phase-extracted” PDS@CNT composite. The dropwise addition of water avoids agglomeration of extracted PDS particles and enables their conformal coating on the CNTs. Vacuum filtration and subsequent drying of this composite yield a free-standing and flexible cathode film, as can be seen in the optical image in Fig. 1a, which can be used directly in lithium batteries without additional current collectors or binders. This reduces the mass of inactive cathode components and makes it attractive for use in flexible batteries.19 This cathode preparation technique is easily scalable and tunable as cathodes with high active material loading (>10 mg cm−2) can be fabricated simply by proportionally increasing the quantity of starting materials. This enables facile sizing-up of synthesis to practical levels.
:1 by weight were vigorously stirred to dissolve PDS in methanol. This solution was then ultrasonicated to cause the CNTs to disperse and interweave. Following ultrasonication, water was added dropwise into the solution. PDS being insoluble in the water phase was extracted from the methanol phase and conformally deposited on the interwoven CNT bundles, giving the “phase-extracted” PDS@CNT composite. The dropwise addition of water avoids agglomeration of extracted PDS particles and enables their conformal coating on the CNTs. Vacuum filtration and subsequent drying of this composite yield a free-standing and flexible cathode film, as can be seen in the optical image in Fig. 1a, which can be used directly in lithium batteries without additional current collectors or binders. This reduces the mass of inactive cathode components and makes it attractive for use in flexible batteries.19 This cathode preparation technique is easily scalable and tunable as cathodes with high active material loading (>10 mg cm−2) can be fabricated simply by proportionally increasing the quantity of starting materials. This enables facile sizing-up of synthesis to practical levels.
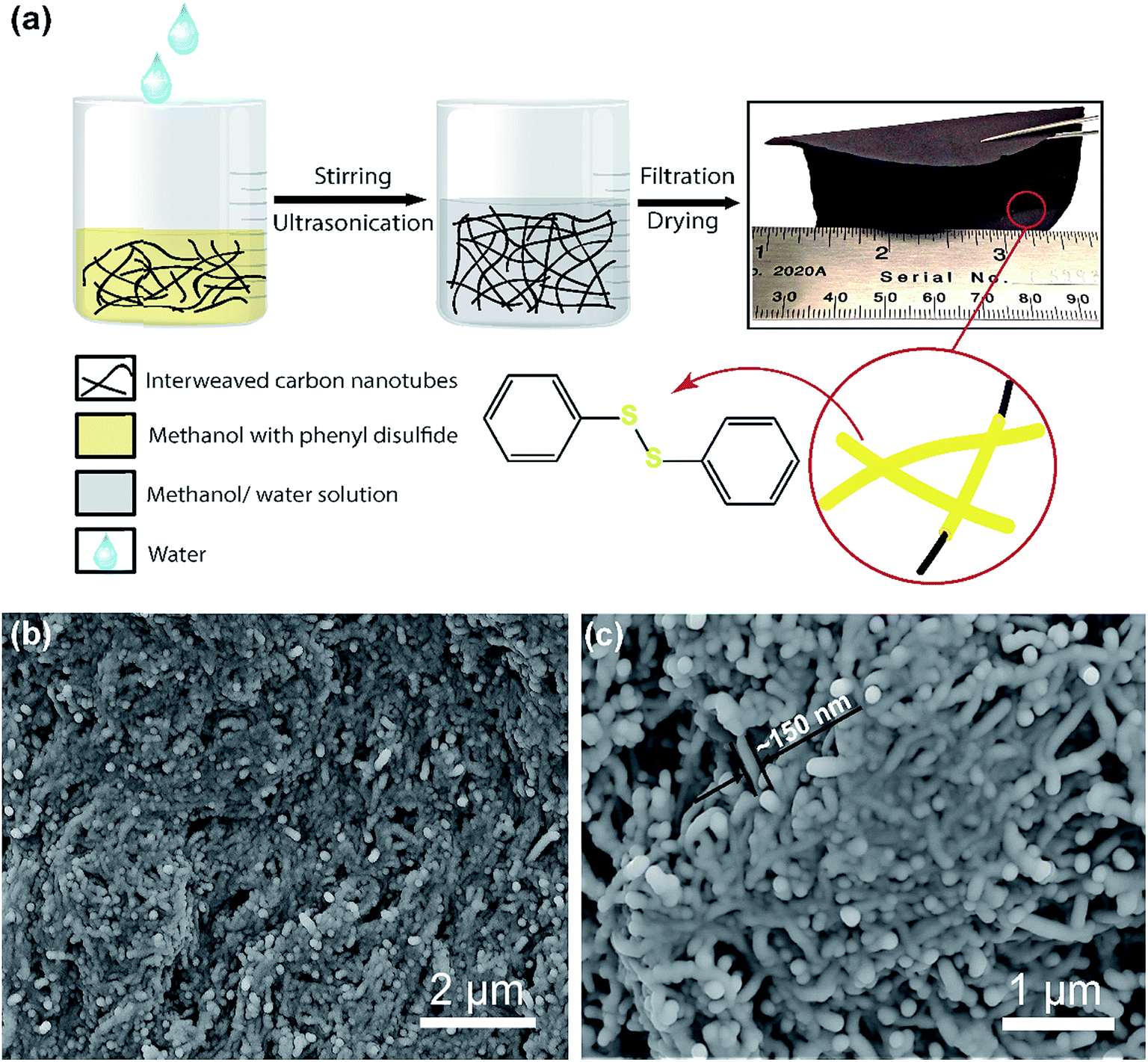 | ||
| Fig. 1 (a) Schematic illustration of the “phase-extraction” method of the PDS@CNT cathode preparation. (b) Low magnification SEM image and (c) high magnification SEM image of the cathode showing uniform deposition of PDS on CNTs leading to its increased thickness. | ||
Once prepared, scanning electron microscopy (SEM) was carried out to determine the morphology of the cathode. When the process was carried out in the absence of PDS, a CNT paper with morphology as shown in Fig. S1(ESI)† is obtained. These pristine CNT fibrils have an average diameter of about 50 nm and are several micrometers in length. In contrast, the PDS@CNT cathode (Fig. 1b and c) shows bundles of conformally coated CNTs with an average diameter of nearly 150 nm. The SEM images reveal a core–sheath type structure in the cathode composite with CNTs at the core with PDS forming the sheath as depicted in the inset illustration in Fig. 1a. Thermogravimetric analysis of the cathode was performed to determine its composition. The TGA profile of the cathode in Fig. S2 (ESI)† shows that 75% of the cathode is composed of PDS. This also confirms that all of the PDS was successfully extracted from the methanol solution by water, demonstrating the versatility of this technique.
The synthesized cathodes were then subjected to electrochemical measurements using standard CR-2032 coin-cells, as described in the ESI. A solution of 1.0 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL) (1:1 v/v) with 0.2 M lithium nitrate (LiNO3) as an additive to passivate the Li metal anode was used as the electrolyte. The established stability of this electrolyte with sulfur-based systems affords appropriate evaluation of PDS as a cathode material as compared to previous reports.20 Cyclic voltammetry (CV) was performed to understand the redox behavior of the compound (Fig. 2a). The CV curve shows a reduction peak at about 2 V in the cathodic scan corresponding to the homolytic cleavage of the disulfide bond on assimilation of two electrons leading to the formation of two thiophenolate radicals (C6H5S˙).8,21 These radicals would take two Li+ ions and electrons forming two lithium thiophenolates (C6H5SLi).21 These processes would occur simultaneously as evidenced by a single reduction peak. During oxidation in the anodic scan, on releasing one electron and one Li+ ion each, the two thiophenolate radicals would combine at 2.4 V to yield PDS. The CV curves overlap consecutively over 10 cycles showing that this conversion reaction is highly reversible.
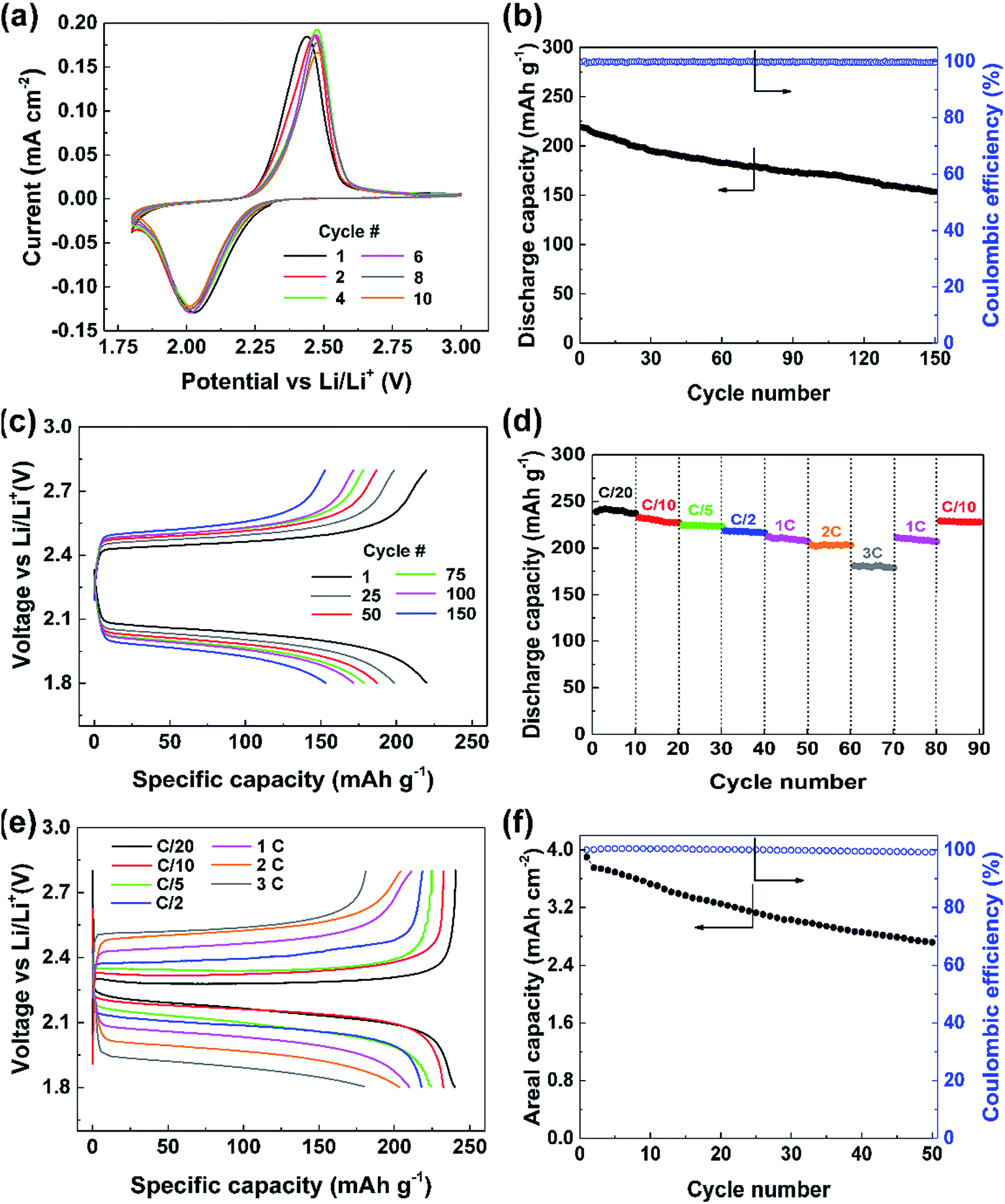 | ||
| Fig. 2 (a) Cyclic voltammogram of the PDS@CNT cathode carried out at a scan rate of 0.2 mV s−1 showing single step conversion reaction. (b) Cycle life of the cathode with ∼5 mg cm−2 loading cycled at 1C and (c) voltage profile of the corresponding cell. (d) Performance of the cathode at different rates demonstrating its fast lithium storage behaviour with (e) corresponding voltage profiles. (f) Performance of the cathode with high areal mass loading (∼19 mg cm−2) cycled at C/10. 1C corresponds to 245 mA g−1 of PDS. | ||
The galvanostatic cycling performance of the cathode at 1C reflected in Fig. 2b shows a high initial discharge capacity of over 218 mA h g−1 corresponding to 89% of the theoretical capacity. The performance is quite stable while retaining 70% of its initial capacity, i.e., 153 mA h g−1, after 150 cycles demonstrating the stability of the cathode in retaining the intermediate species. The cycling occurs at excellent coulombic efficiencies of over 99% through all 150 cycles. The voltage profile of this cell (Fig. 2c) clearly shows the discharge occurring at 2.1 V due to the disulfide bond cleavage, which requires 54.5 kcal mol−1.22 The following charge occurs at 2.4 V on disulfide bond reformation as depicted by the CV curve. We can see that the charge and discharge overpotentials increase slowly with cycles probably due to the increased dead spots in the CNT matrix upon deposition of discharge products.
The robustness of this cathode design was verified by comparing against a conventional slurry cast cathode. The slurry cast cathode contained 70% PDS and an areal loading similar to that of the PDS@CNT composite (see ESI† for information on cathode preparation). Electrochemical impedance spectroscopy (EIS) was performed to compare the charge transfer efficacy of the PDS@CNT cathode. As seen in Fig. S3,† the charge transfer resistance of the slurry cast cathode is nearly 50 Ω higher than that of the PDS@CNT cathode which more closely resembles that of pure CNT paper at ∼20 Ω. This shows that the intimate CNT matrix provides significantly better electron conduction to PDS and thus improves its kinetics. On cycling, the slurry cast cathode yields only 209 mA h g−1 despite operating at a lower rate of C/5 (as shown in Fig. S4, ESI†) and loses 56% of its initial capacity in 150 cycles revealing that the PDS@CNT cathode design offers improved kinetics and material utilization and can better retain the active material at the cathode. To demonstrate the capability of the CNTs to sequester the soluble species, the open circuit voltage (OCV) of the cell was measured over time. The cell exhibits a stabilized OCV for one week as shown in Fig. S5 (ESI†). Discharging this cell yields 215 mA h g−1 (Fig. S6, ESI†) thus demonstrating that the porous CNT framework can well contain the soluble species within the cathode. In addition, the low electrolyte to active material ratio (discussed later) also helps to reduce the solubility of PDS in the electrolyte which contributes to the stability of OCV.
The PDS@CNT cathode clearly demonstrates its viability for long cycle life. However, its rate performance is also imperative. Therefore, the cathode was cycled at different C-rates to analyze its rate performance. Fig. 2d clearly shows that the cathode can yield 240 mA h g−1, which is nearly its full theoretical capacity, when cycled at C/20. It delivers 230 mA h g−1, 223 mA h g−1, and 217 mA h g−1 at C/10, C/5, and C/2, respectively, showing excellent material utilization and storage capability at low rates. At high rates, such as 1C, 2C, and 3C, the cathode yields high capacities of 209 mA h g−1, 202 mA h g−1, and 180 mA h g−1, respectively indicating excellent power capabilities. On inspecting the corresponding voltage profile in Fig. 2e, we can see that a voltage efficiency of 90% is maintained at low rates of C/20 and C/10 while still exhibiting a voltage efficiency of 75% at rates as high as 3C thus adding to its high-power capability.
An important attribute of a cathode fabrication technique is its ability to be scaled up efficiently. An areal capacity between 3 and 5 mA h cm−2 is considered essential for practical applications.23 The “phase-extraction” technique is ideal for this scaling-up as high areal loading cathodes can be prepared by simply starting with proportionally more material in the cathode fabrication process. Cathodes with ultra-high areal loadings of PDS amounting to 19.3 mg cm−2 were easily fabricated. The performance of such a cathode is shown in Fig. 2f wherein an areal capacity of 3.9 mA h cm−2 is obtained. Despite this high loading and the usage of an electrolyte to active material ratio of only 4 μL mg−1, the cell was able to sustain cycling with relative ease. A low amount of electrolyte also promotes lesser dissolution of the active material, thus facilitating good rechargeability. In this cell, 2.72 mA h cm−2 of areal capacity was still retained after 50 cycles. This excellent “practical-level” performance of the cathode clearly denotes the ability of this preparation technique and the cathode to be applied to practical use upon further optimization.
Noting that this cathode is capable of excellent performance, it is essential to understand the fundamental chemical, structural, and morphological changes occurring in the cathode during cycling. X-ray diffraction (XRD) of the cathode at different stages was performed to trace the structural changes occurring in the cathode. The XRD pattern (Fig. 3a) of the PDS@CNT cathode carries forward the defining peaks of PDS as seen in its commercial sample signifying that PDS remains unaltered in the “phase-extraction” process.21,24 The 26.4° peak of the (002) plane of CNTs is clearly diminished owing to the coating of CNTs with PDS. Upon discharging the cathode, we can observe that new peaks appear at 22.7°, 27.5°, and 30.8° and between 37° to 40° with low intensities signaling the conversion of PDS to low-crystalline lithium thiophenolate. At the end of the following charge step, the cathode reverts back to its PDS state displaying its characteristic peaks except for some changes in the peak intensities owing to the slightly changed crystallographic orientation of the organic molecule. The 26.4° peak of CNTs is prominent at this stage due to better exposure of CNT fibrils. To confirm the complete conversion of disulfide to thiophenolate, Fourier transform infrared spectroscopy (FT-IR) was performed on the cathode upon discharge and recharge. Fig. 3b shows the FT-IR spectra which focuses on the 400–600 cm−1 region where the disulfide bond vibration is observed. The spectrum of the synthesized cathode has a peak at 463 cm−1 corresponding to the disulfide bond.24 Upon discharge, this peak completely disappears, thus confirming the complete conversion of the disulfides. The peak reappears on recharging corresponding to the reformation of PDS. These confirm that the reaction Ph2S2 + 2Li ↔ 2PhSLi occurs during battery cycling.
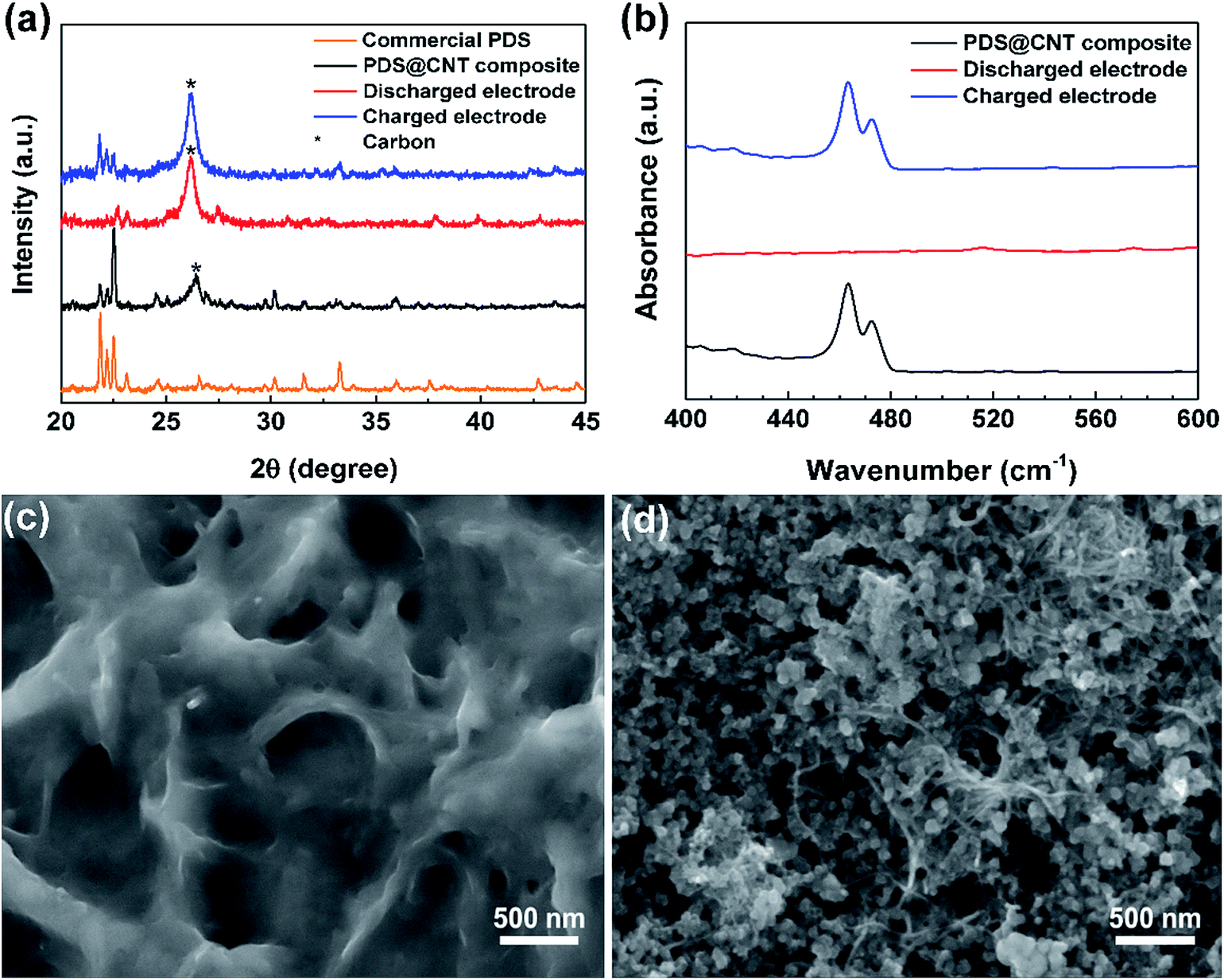 | ||
| Fig. 3 (a) X-ray diffractograms of a commercial PDS sample along with the PDS@CNT cathode at different stages of cycling. (b) FT-IR spectra of the cathode at different stages of cycling focusing on the region of disulfide bonds between 400 and 600 cm−1. SEM images showing the morphology of the cathode after (c) discharge and after (d) recharge. | ||
On confirming the successful chemical conversion occurring on the cathode, we focus on its morphological changes. A SEM image of the cathode taken upon discharge (Fig. 3c) reveals that the core–sheath-like PDS on conversion to lithium thiophenolate is coated on the entire cathode surface. The entire available surface area is utilized upon expansion in the cathode during discharge. A SEM micrograph of the recharged cathode reveals that the cathode does not return to its original core–sheath-like structure. Instead the reformed PDS forms platelet-like particles confined in the mesh of the CNT framework. This morphology is uncannily similar to Li2O2 formation in Li–O2 systems wherein it occurs due to the solution phase formation and growth of the product crystals.25,26 This provides a convincing explanation for the observed morphological change as the recharge product, i.e., PDS, is soluble in the electrolyte leading to toroid and platelet-like crystal growth. Following this, to further analyze the morphology change over extended cycling, the cathode was extracted after 150 cycles and partially washed to observe the changes in the CNTs. The SEM micrograph in Fig. S7 (ESI)† reveals that the formation of platelet-like PDS persists on repeated cycling. It also shows that the CNT network is able to well-confine the active material and provides a flexible yet highly conductive framework that can accommodate the repeated volume changes in the cathode. The self-weaving behavior of CNTs allows them to rearrange themselves during cycling. This might lead to loss of active material due to dissolution in the electrolyte over cycling causing the “dead-spots”. This problem seems to be aggravated at higher loadings. Although the CNT cathode presented here is robust, we note that it is not yet optimal with scope for improvements through modifications such as polymer coating,27 doping,28 and introduction of novel materials29,30 could improve the performance of this cathode especially at high areal loading. Furthermore, the redox potential can also be altered by tuning the energy of disulfide bonds by utilizing different substituents in the phenyl ring.22
In summary, we report a simple, scalable approach to fabricate an organodisulfide cathode based on PDS. The solution based “phase-extraction” technique proves to be versatile in synthesizing free-standing, flexible cathode films of the PDS@CNT composite. Electrochemical performance shows that this cathode can deliver a high specific capacity of over 200 mA h g−1 at 1C and can sustain extended cycling at excellent coulombic efficiencies. Performance of ultra-high loading cells demonstrates the ease of cathode operation under conditions where other cathodes may fail.26 Materials characterization through XRD and FT-IR sufficiently confirms the complete conversion of PDS to lithium thiophenolates on discharge and back to PDS on charging. Investigation on morphology change through SEM helps shed light on the performance behavior of the cathode. Thus, PDS is able to offer performance comparable to if not better than Li-ion technology in terms of capacity and specific energy. We hope that this work leads to further studies on the development and optimization of such cathodes which would make organodisulfides a promising candidate for inexpensive, environmentally benign, and intrinsically safe cathode materials for applications in rechargeable lithium batteries.
Acknowledgements
This work was supported by the startup grant from Purdue School of Engineering and Technology and Department of Mechanical Engineering at Indiana University-Purdue University Indianapolis (IUPUI). The authors would like to acknowledge the Integrated Nanosystems Development Institute (INDI) for use of their Bruker D8 Discover X-Ray Diffraction Instrument, which was awarded through the NSF grant MRI-1429241 and for use of their JEOL7800F Field Emission Scanning Electron Microscope, which was awarded through NSF grant MRI-1229514.References
- International Energy Outlook 2016, U.S. Energy Information Administration, Washington, DC, 2016.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- R. Steudel, Chem. Rev., 2002, 102, 3905–3946 CrossRef CAS PubMed.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- P. Poizot and F. Dolhem, Energy Environ. Sci., 2011, 4, 2003–2019 CAS.
- M. Wu, Y. Cui, A. Bhargav, Y. Losovyj, A. Siegel, M. Agarwal, Y. Ma and Y. Fu, Angew. Chem., Int. Ed., 2016, 55, 10027–10031 CrossRef CAS PubMed.
- D. Wu, Z. Xie, Z. Zhou, P. Shen and Z. Chen, J. Mater. Chem. A, 2015, 3, 19137–19143 CAS.
- M. Liu, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 1989, 136, 2570–2575 CrossRef CAS.
- M. Liu, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 1990, 137, 750–759 CrossRef CAS.
- S. J. Visco, M. Liu and L. C. De Jonghe, J. Electrochem. Soc., 1990, 137, 1191–1192 CrossRef CAS.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef.
- T. Inamasu, D. Yoshitoku, Y. Sumi-otorii, H. Tani and N. Ono, J. Electrochem. Soc., 2003, 150, A128–A132 CrossRef CAS.
- T. Maddanimath, Y. B. Khollam, M. Aslam, I. S. Mulla and K. Vijayamohanan, J. Power Sources, 2003, 124, 133–142 CrossRef CAS.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- Y. Fu, Y.-S. Su and A. Manthiram, Angew. Chem., Int. Ed., 2013, 52, 6930–6935 CrossRef CAS PubMed.
- Y.-S. Su, Y. Fu and A. Manthiram, Phys. Chem. Chem. Phys., 2012, 14, 14495–14499 RSC.
- G. Zhou, F. Li and H.-M. Cheng, Energy Environ. Sci., 2014, 7, 1307–1338 CAS.
- S. Zhang, K. Ueno, K. Dokko and M. Watanabe, Adv. Energy Mater., 2015, 5, 1500117 CrossRef.
- M. Wu, A. Bhargav, Y. Cui, A. Siegel, M. Agarwal, Y. Ma and Y. Fu, ACS Energy Lett., 2016, 1, 1221–1226 CrossRef CAS.
- Y.-M. Yang, H.-Z. Yu, X.-H. Sun and Z.-M. Dang, J. Phys. Org. Chem., 2016, 29, 6–13 CrossRef.
- K. G. Gallagher, S. E. Trask, C. Bauer, T. Woehrle, S. F. Lux, M. Tschech, P. Lamp, B. J. Polzin, S. Ha, B. Long, Q. Wu, W. Lu, D. W. Dees and A. N. Jansen, J. Electrochem. Soc., 2016, 163, A138–A149 CrossRef CAS.
- P. Das, S. Ray, A. Bhaumik, B. Banerjee and C. Mukhopadhyay, RSC Adv., 2015, 5, 6323–6331 RSC.
- A. Bhargav, W. Guo and Y. Fu, Chem. Commun., 2016, 52, 5678–5681 RSC.
- N. B. Aetukuri, B. D. McCloskey, J. M. García, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2015, 7, 50–56 CrossRef CAS PubMed.
- Y. Cui and Y. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 20369–20376 CAS.
- J. Song, M. L. Gordin, T. Xu, S. Chen, Z. Yu, H. Sohn, J. Lu, Y. Ren, Y. Duan and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 4325–4329 CrossRef CAS PubMed.
- Q. Pang, D. Kundu and L. F. Nazar, Mater. Horiz., 2016, 3, 130–136 RSC.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section along with additional data. See DOI: 10.1039/c7se00135e |
| This journal is © The Royal Society of Chemistry 2017 |