
Long stable cycling of fluorine-doped nickel-rich layered cathodes for lithium batteries
Chandrasekar M.
Subramaniyam
 ab,
Hugo
Celio
b,
Konda
Shiva
b,
Hongcai
Gao
b,
John B.
Goodneough
*b,
Hua Kun
Liu
*a and
Shi Xue
Dou
a
ab,
Hugo
Celio
b,
Konda
Shiva
b,
Hongcai
Gao
b,
John B.
Goodneough
*b,
Hua Kun
Liu
*a and
Shi Xue
Dou
a
aInstitute for Superconducting and Electronic Materials, Australian Institute for Innovative Materials, Innovation Campus, University of Wollongong, North Wollongong, Australia. E-mail: hua@uow.edu.au
bTexas Materials Institute, Department of Mechanical Engineering, University of Texas at Austin, Texas, USA. E-mail: jgoodenough@mail.utexas.edu
First published on 5th June 2017
Abstract
Theoretically, layered Ni-rich metal oxides are capable of delivering 200 mA h g−1. However, their performances deteriorate due to an irreversible surface reaction with the electrolyte, which could be overcome by the partial substitution of fluorine for oxygen. Herein, a fluorine-doped, Ni-rich metal oxide with the composition LiNi0.7Co0.15Mn0.15O1.95F0.05 exhibited a capacity of 170 mA h g−1 after 100 cycles when tested against lithium.
Lithium-ion rechargeable batteries (LIBs) are considered the best near-term storage medium for electric power in electric vehicles that can compete with internal combustion engines powered by fossil fuels.1–3 This application requires improvements in safety, cycle life, and stored energy density over the LIBs that power electronic devices. The energy density is 〈V(q)〉. Q(I), where 〈V(q)〉 is the average voltage over the state of charge, q, of the battery, and Q(I) is the capacity at a delivered current, I, of the electric power stored per unit weight and/or volume of the battery stack of cells. Safety concerns and energy density requirements dictate the development of a dendrite-free lithium anode and a high-voltage cathode host into which lithium can be inserted reversibly over a large solid-state range. The safety issues, dendrite-free character, lithium anode and long cycle life are being addressed with the development of an insertion cathode host that provides a large cathode energy density.
Transition-metal oxides offer the highest voltage, and Li+ ions can be extracted reversibly at acceptable rates from oxides with close-packed oxygen arrays. Layered LiMO2, spinel LiM2O4, and olivine LiMPO4 with M containing a transition-metal cation with a stable redox energy have been of particular interest because they are stable at high voltages in the electrolyte they contact.4–15
Acceptable ordering of the layered LiMO2 oxides requires a sufficiently small average radius of the M cations relative to the radius of the Li+ ion. The Ni(III)/Ni(II) redox energy is at the top of the O2−:2p6 volume bands in an oxide; it is possible to have access not only to the Ni(III)/Ni(II) completely but also to host, if not all, of the Ni(IV)/Ni(III) couple pinned at the top of the O2−:2p6 valence bands with little, or no, energy gap between the two Ni couples. Therefore, the Ni-rich layered oxides Li(Ni(II)1−2xCo(III)xMn(IV)x)O2 have been investigated as LIB cathodes11,16–21 and easily give a cathode discharge capacity of 200 mA h g−1.17–21 However, the surface reactions with the liquid electrolyte of a conventional LIB are not totally suppressed by cation substitutions, including the Li2MnO3 interlayer 2D phase.22–29 This situation has led to the exploration of anion substitutions, particularly F− for O2−.30–41 Herein, we report an investigation of F− doping on the high-voltage Ni-rich layered cathodes Li(Ni0.7Co0.15Mn0.15)O2−xFx (x = 0.025, 0.05, 0.075) synthesized solvothermally followed by annealing. The sample with x = 0.05 delivered a capacity of 170 mA h g−1 at 200 mA g−1, even after 100 cycles.
All chemical precursors utilized were 99.99% pure and purchased from Sigma-Aldrich. The Ni0.7Co0.15Mn0.15(OH)2 (NCM) precursor was prepared by a solvothermal method. Ni(CH3COO)2·4H2O (7 mmol), Co(CH3COO)2·4H2O (1.5 mmol), and Mn(CH3COO)2·4H2O (1.5 mmol) were dissolved in 50 ml of absolute ethanol and water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The obtained homogeneous solution was then transferred to a 90 ml Teflon-lined stainless-steel autoclave and heated in a muffle furnace at 200 °C for 12 h. After cooling to room temperature, the obtained slurry was centrifuged and washed several times with ethanol and vacuum-dried at 60 °C overnight. The Ni0.7Co0.15Mn0.15(OH)2 (NCM) precursors were thoroughly blended with 5% Li excess of a stoichiometric amount of Li2CO3 and LiF as lithium and fluorine sources, respectively. The mixture was sintered at 480 °C and 800 °C in O2 flow for 5 and 12 h, respectively. For comparison, a pristine sample was prepared using the same sintering conditions but without adding LiF.
:1, v/v). The obtained homogeneous solution was then transferred to a 90 ml Teflon-lined stainless-steel autoclave and heated in a muffle furnace at 200 °C for 12 h. After cooling to room temperature, the obtained slurry was centrifuged and washed several times with ethanol and vacuum-dried at 60 °C overnight. The Ni0.7Co0.15Mn0.15(OH)2 (NCM) precursors were thoroughly blended with 5% Li excess of a stoichiometric amount of Li2CO3 and LiF as lithium and fluorine sources, respectively. The mixture was sintered at 480 °C and 800 °C in O2 flow for 5 and 12 h, respectively. For comparison, a pristine sample was prepared using the same sintering conditions but without adding LiF.
Both the pristine and F-doped Ni-rich layered cathodes were subjected to phase identification along with surface and electrochemical characterizations. X-ray diffraction (XRD, Rigaku Miniflex 600) equipped with Cu-Kα radiation was employed for phase identification with a scan rate of 2° min−1 and step size of 0.02°. Surface characterization was carried out using a commercial X-ray photoelectron spectrometer (XPS, Kratos Axis Ultra DLD, Manchester, U.K.) with a monochromated Al-Kα X-ray source (hν = 1486.5 eV) and X-ray power of 120 Watt. The spectrometer had hybrid optics (a magnetic and electrostatic lens used simultaneously) and a multi-channel plate detector coupled to a hemispherical photoelectron kinetic analyzer. The base pressure in the analysis chamber was typically 3 × 10−9 Torr. Spectra were collected with a pass energy of 20 eV and scanned at 0.1 eV per step. All peaks were calibrated with respect to adventitious carbon, C 1s, at 285 eV. Casa XPS analysis software was used for peak fitting analysis, and the stoichiometric ratios were determined from corrected peak areas by employing the Kratos sensitivity factors for each element of interest. A field-emission gun scanning electron microscope (Hitachi S5500 SEM/STEM) coupled with energy dispersive X ray spectroscopy (EDS) operated at 5 kV and 10 μA was used to visualize and study the cathode morphologies and compositions, respectively.
The electrochemical performances of the pristine and fluorine-doped Ni-rich layered cathodes were studied with CR2032 half-cell coin cells assembled in an argon-filled glove box (MBraun, Germany). All samples were blended individually with carbon Super P as a conducting agent and polyvinylidene fluoride (Sigma-Aldrich) as a binder in a weight ratio of 8:1:1 with N-methyl-2-pyrrolidone as a solvent. After mixing, the obtained slurry was tape-casted over a double-sided, carbon-coated aluminum current collector and vacuum-dried at 120 °C overnight. The dried electrodes were cut into circular discs and half cell coin cells were assembled with these cathodes and Li metal foil as the counter/reference electrode separated by a Celgard polypropylene film containing a few drops of commercially available 1 M LiPF6 in 1:1 (v/v) ethylene carbonate (EC):diethyl carbonate (DEC) as electrolyte. All the assembled cells were tested in a battery-testing analyzer (Landt, China CT2001A) at a constant specific current density (mA g−1) between 2.8 and 4.4 V. A Solartron electrochemical workstation was employed to perform potentiostatic electrochemical impedance spectroscopy (PEIS) in the frequency range of 1 MHz to 10 mHz against Li+/Li0.
The XRD pattern in Fig. 1 pertains to Ni0.7Co0.15Mn0.15(OH)2 (NCM) (JCPDF: #14-0117), confirming that the NCM precursor is a typical M(OH)2 oxide (where M = Ni, Co or Mn). The precursors contained nanometer-sized particle agglomerates, forming granular particles with sizes of ∼1 μm. The EDS results (data not shown) reveal that Ni, Co, Mn were disturbed uniformly with an atomic ratio of 0.73:0.14:0.13, which is close to the desired composition of 0.70:0:15:0:15. Fig. 2 shows the XRD patterns of the pristine and fluorine-doped LiNi0.7Co0.15Mn0.15O2−xFx, where x = 0, 0.025, 0.05 and 0.075. All the materials possessed the layered hexagonal rock-salt structure of α-NaFeO2 in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group. The intensity ratio I(003)/I(104) has been reported as a key determinant of the degree of ordering of the Li+ and Ni2+ ions into alternate (101) planes.9,12 After careful analysis of the XRD pattern (Fig. 2), the values of I(003)/I(104) were determined to be 1.83 (pristine), 1.19 (x = 0.025), 1.02 (x = 0.05) and 0.9 (x = 0.075). Therefore, it is understood that the intensity ratio I(003)/I(104) decreased with increasing x, indicating the increased disorder of Li+ and Ni2+ with increasing fluorine doping content. The (006)/(102) and (108)/(110) planes were clearly separated in the pristine sample but became indistinguishable as the fluorine doping content increased. From the XRD analysis, it is anticipated that a lower fluorine content in LiNi0.7Co0.15Mn0.15O2 would result in a better electrochemical Li+ insertion performance. The annealing of Ni0.7Co0.15Mn0.15(OH)2 with a stoichiometric amount of Li2CO3 and LiF at 480 °C and 800 °C for 5 and 12 h, respectively, led to highly crystalline particles with an insignificant change in their lattice parameters with fluorine doping. Further, the changes in the lattice parameters a and c of the annealed pristine and F-doped samples were determined using XRDA 3.1 software and are tabulated in Table 1.
m space group. The intensity ratio I(003)/I(104) has been reported as a key determinant of the degree of ordering of the Li+ and Ni2+ ions into alternate (101) planes.9,12 After careful analysis of the XRD pattern (Fig. 2), the values of I(003)/I(104) were determined to be 1.83 (pristine), 1.19 (x = 0.025), 1.02 (x = 0.05) and 0.9 (x = 0.075). Therefore, it is understood that the intensity ratio I(003)/I(104) decreased with increasing x, indicating the increased disorder of Li+ and Ni2+ with increasing fluorine doping content. The (006)/(102) and (108)/(110) planes were clearly separated in the pristine sample but became indistinguishable as the fluorine doping content increased. From the XRD analysis, it is anticipated that a lower fluorine content in LiNi0.7Co0.15Mn0.15O2 would result in a better electrochemical Li+ insertion performance. The annealing of Ni0.7Co0.15Mn0.15(OH)2 with a stoichiometric amount of Li2CO3 and LiF at 480 °C and 800 °C for 5 and 12 h, respectively, led to highly crystalline particles with an insignificant change in their lattice parameters with fluorine doping. Further, the changes in the lattice parameters a and c of the annealed pristine and F-doped samples were determined using XRDA 3.1 software and are tabulated in Table 1.
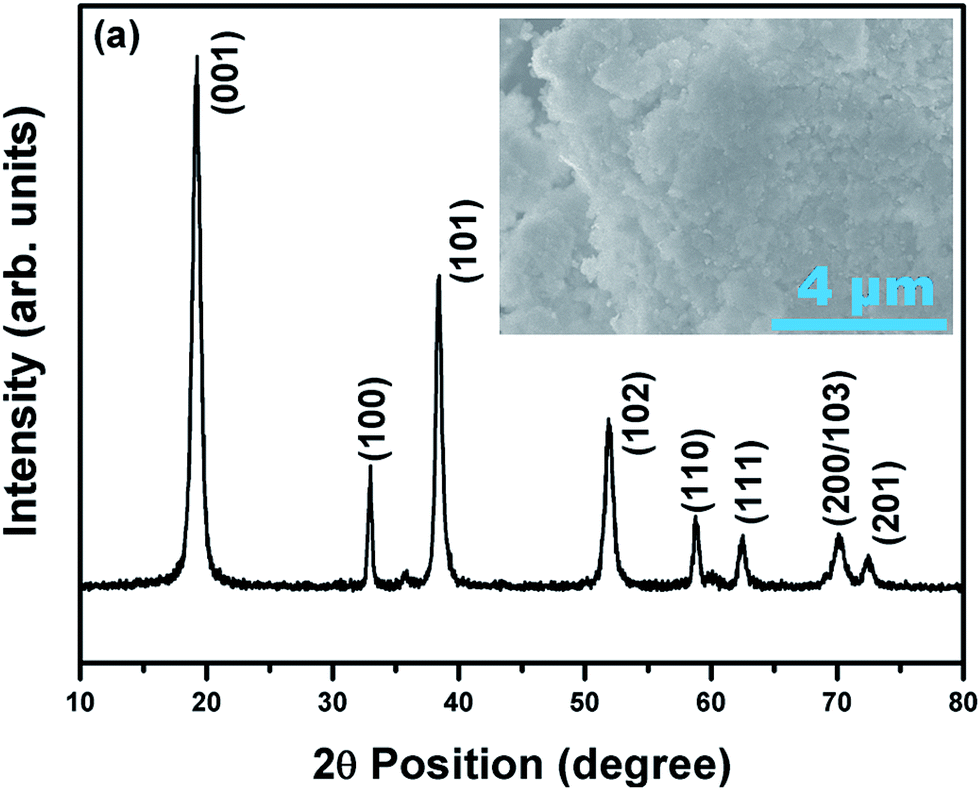 | ||
| Fig. 1 XRD pattern and FESEM image (insert) showing the phase and morphology, respectively, of the as-prepared NCM precursor. | ||
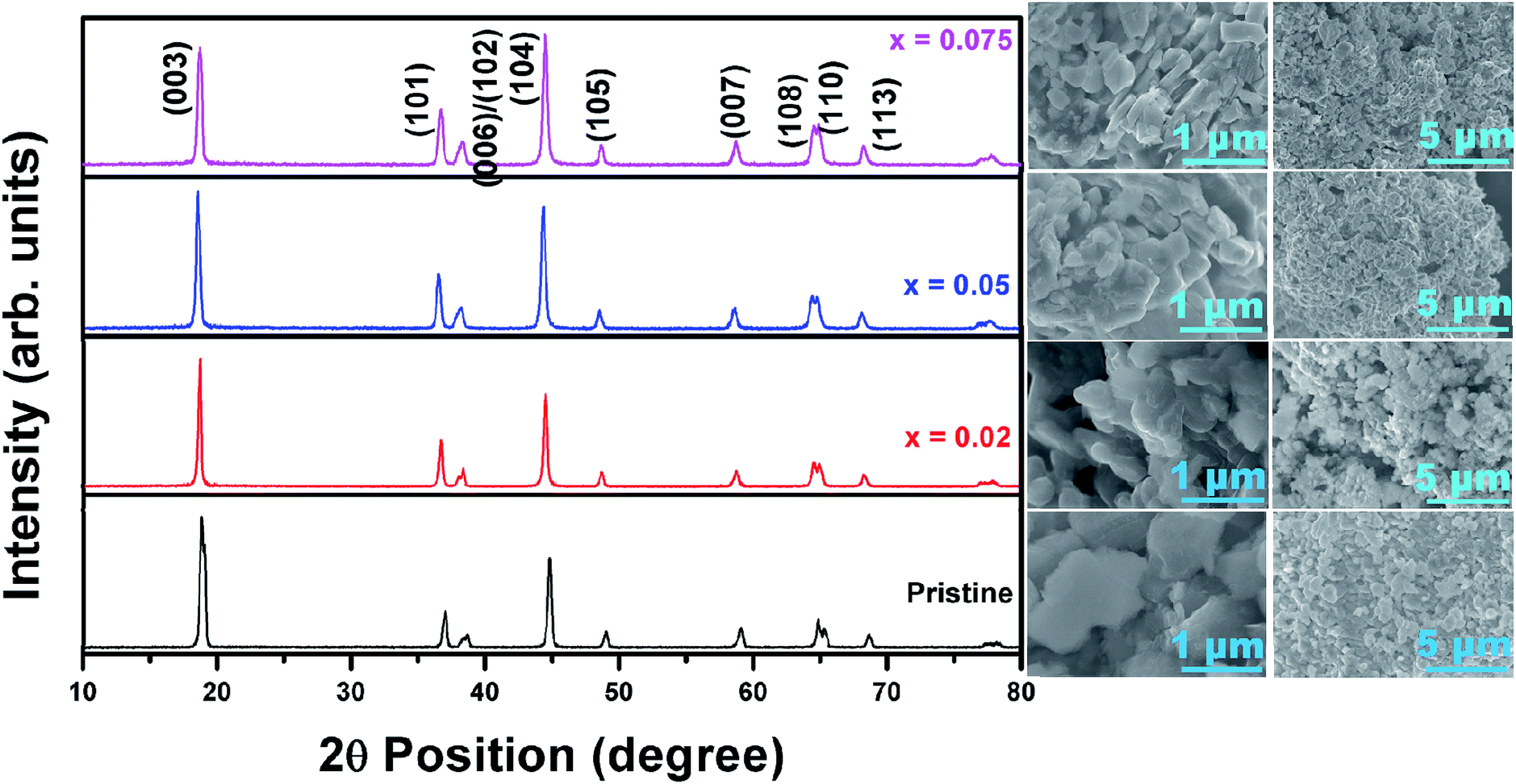 | ||
| Fig. 2 XRD patterns of pristine and fluorine-doped Ni-rich cathodes along with the corresponding FESEM images showing their morphologies. | ||
| Sample ID | Lattice parameters | |||
|---|---|---|---|---|
| a (Å) | c (Å) | c/a | V (Å3) | |
| Pristine | 2.8548 ± 0.0036 | 14.1219 ± 0.0194 | 4.9467 ± 0.0068 | 99.674 |
| x = 0.025 | 2.8697 ± 0.0026 | 14.1920 ± 0.0121 | 4.9455 ± 0.0042 | 101.217 |
| x = 0.050 | 2.8756 ± 0.0053 | 14.2109 ± 0.0294 | 4.9419 ± 0.0102 | 101.766 |
| x = 0.075 | 2.8684 ± 0.0031 | 14.1815 ± 0.0194 | 4.9440 ± 0.0068 | 101.051 |
The parameters a and c respectively indicate the interlayer metal–metal distance and cumulative of MO6 and LiO6 octahedron layers thickness in the LiMO2 layer structure.8 The parameters for the pristine sample were a = 2.8548 Å and c = 14.1219 Å. Upon increasing fluorine doping from x = 0.025 to 0.05, both a and c increased, while the c/a ratio decreased, accompanied by a decrease in slab thickness and an increase in the thickness of the interslab space. This may be because the occupancy of Ni2+, which has a small radius (0.69 Å), in the Li layer along with that of Li+, with the largest radius (0.76 Å), in the transition metal layer increased the slab thickness (a). In addition, an increase in c favors Li ion intercalation and deintercalation, further enhancing the electrochemical performance. Further increasing x beyond 0.075 led to reduced a and c values, which is unfavorable for electrochemical performance, as reflected by testing the electrodes against lithium.
The XPS spectra in Fig. 3 show the high-resolution spectra of (a) Li 1s, (b) F 1s and (c) Ni 2p. In panel (a), the Li 1s spectrum of the pristine sample (x = 0.00) was fitted to a single symmetric function with its peak centered at 55.2 eV, while the rest of the Li 1s spectra from the F-doped samples were fitted by two symmetric functions. The peaks at 55.2 eV, which dominate 75% to 85% of the total experimental Li peak area in the F-doped samples, are assigned to Li2CO3.38 The presence of Li2CO3 was confirmed with the detection of the strong carbonate C 1s peak at ∼290 eV (not shown). The stoichiometric concentration of the carbonate peak is in agreement with peak in the Li 1s spectrum at 55.2 eV. The smaller fitted peaks at 56.3 eV are assigned to LiF.38 Additional evidence for the detection of LiF is discussed below, focusing on the F 1s region (Fig. 3b). It is important to note that Li co-ordinated in the pristine or F-doped Ni-rich layered of LiNi0.7Co0.15Mn0.15O1.95F0.05 structure was not detected in any of the Li 1s peaks due to overall structure of the samples that consist of a top layer of excess LiF and Li2CO3. This top layer screened the signals from Li in the pristine and F-doped LiNi0.7Co0.15Mn0.15O1.95F0.05 structures. This observation is further discussed below.
 | ||
| Fig. 3 High-resolution XPS spectra of LiNi0.7Co0.15Mn0.15O1.95F0.05: (a) lithium, (b) fluorine, and (c) nickel regions. | ||
As expected, the spectrum of the pristine sample (x = 0.00) shows a flat line (with noise) in the F 1s region, while the F 1s spectra of the F-doped samples show broad, asymmetric peaks. Each of these peaks was adequately fitted to two symmetric functions. One of the fitted functions has a dominant peak area at 685.1 eV. Based on the binding energy value of this peak and its calculated concentration, which is in agreement with the fitted Li 1s peak at 56.3 eV, it is assigned to LiF.38 The second fitted F 1s peak decreases in binding energy as the concentration of doped fluorine (x) increases (686.6, 686.1, and 685.9 eV for x = 0.025, 0.05, 0.075, respectively). This chemical shift is trending toward a binding energy value where the F 1s binding energy in NiF2 was measured at 685.1 eV.38 Thus, the second fitted F 1s peak is assigned to doped fluorine from the ion-exchange reaction during the synthesis of the Ni-rich layered LiNi0.7Co0.15Mn0.15O1.95F0.05 materials. Additional evidence of the detection of doped fluorine is discussed below. A small shoulder peak at 687.2 eV, which is only detected in one sample (x = 0.075), was not identified. Based on its relatively small concentration, we assume that it plays an inactive role.
The pristine layered Ni-rich electrode material without fluorine has two oxidation states, Ni3+ and Ni2+, with a theoretical Ni3+/Ni2+ ratio of 3.7. In Fig. 3c, the Ni 2p1/2 and Ni 2p3/2 regions show a single asymmetric core transition peak for the pristine and F-doped samples. The spectra also show broad peaks near 862 and 880 eV. The latter peaks are assigned to satellite peak transitions of the Ni 2p region.34,35,37 A peak deconvolution procedure to determine the spectroscopic properties of the Ni3+ and Ni2+ states was unsuccessful since the binding energies of these states are separated by less than 1.2 eV.34,35,37 These Ni chemical states become convoluted into a single peak in both the 2p1/2 and 2p3/2 spin states. Another fitting complication is that there is a considerable overlap of the broad satellites peaks with the Ni chemical states. The electrons of the Ni3+/Ni2+ redox couple are commonly itinerant in Ni-rich oxides. To evaluate the effect(s) of fluorine doping, we chose a single fitting function for the Ni 2p3/2 peak to represent both the Ni3+ and Ni2+ chemical states. Using a Shirley function as a background account for the in-elastically photoelectron scattering, the Ni 2p2/3 peaks were adequately fitted with a single asymmetric Lorentzian function with full-width-half-maximum (FWHM) fixed at 2.22 eV, while the satellite peak of Ni 2p3/2 peak is fitted to a broader symmetric Gaussian–Lorentzian function. The fits of the Ni 2p3/2 peaks of the pristine and F-doped samples show nearly identical line shapes, indicating that the Ni3+/Ni2+ ratio was not strongly perturbed in any of the fluorine-doped samples. However, a small positive shift in the binding energy peak of 0.1 to 0.2 eV was observed for the F-doped samples compared to the pristine sample. This small shift in binding energy trends toward the binding energy of the Ni 2p peak of NiF2.37 This Ni 2p shift is also consistent with the trend observed for the small fitted F 1s peak near 686 eV. These observations are consistent with the XRD data and indicate that the ion-exchange reaction yielded an F-doped LiNi0.7Co0.15Mn0.15O1.95F0.05 structure.
The Ni 2p3/4 peak was used as a reference to calculate the peak ratios of the detected species on the surfaces of the pristine and F-doped Ni-rich layered samples. Table 2 summarizes the concentration ratios of Li2CO3, LiF, and doped F with respect to the Ni concentration. The LiF/Ni ratios are similar, while the doped F/Ni ratios increase with increasing x, as expected from the ion-exchange reaction of the doped F samples. Table 2 also shows that the doped F/Ni ratio from XPS correlates with the I(003)/I(104) ratios from XRD. This correlation between the XRD (i.e., bulk technique) and XPS (i.e., surface technique) results has not been reported in previous studies. The correlation characteristics will be further considered in a future study. It is desirable to explore such a study since the target ratios of fluorine-doped LiNi0.7Co0.15Mn0.15O2−xFx (x = 0.025, 0.05, and 0.075), on the one hand, are consistent with the XRD while, on the other hand, the XPS ratios are nearly 5 to 6 times larger for the x = 0.12, 0.20, and 0.50 than the theoretical values. Moreover, the surfaces of the Ni-rich layered materials have layers of excess Li2CO3 and LiF. Under similar acquisition parameters (e.g., pass energy of 20 eV), the 2p peaks of Mn and Co were extremely weak, indicating that the overlayer of un-reacted Li2CO3 and LiF effectively screened the signal from these elements, but not from nickel, suggesting that the top layer is approximately 4 to 6 nm in thickness. This top layer clearly originates from the 5% Li excess in the stoichiometric amount from a mixture of Li2CO3 and LiF, which was added to compensate for Li loss at elevated temperature. Without this excess Li, the pristine and LiNi0.7Co0.15Mn0.15O2−xFx samples would be deficient in Li,8,9,14,20,34,35,37 and battery performance would decrease. Furthermore, the removal of excess Li2CO3 and LiF after annealing is not feasible due to the insolubility of LiF and Li2CO3 in most solvents that would not alter the structure of the layered, nickel-rich cathode particles. In conclusion, the pristine and LiNi0.7Co0.15Mn0.15O2−xFx samples prepared by a solvothermal method followed by annealing consisted of particles with diameters of 3–4 microns and intrinsic top layers of Li2CO3 and LiF.
| Sample ID | LiF/Ni | Li in Li2CO3/Ni | F-Doped/Ni-rich layer | I (003)/I(104) |
|---|---|---|---|---|
| x = 0.000 | 0.00 | 5.26 | 0.00 | 1.83 |
| x = 0.025 | 0.422 | 8.72 | 0.12 | 1.19 |
| x = 0.050 | 0.55 | 4.10 | 0.20 | 1.02 |
| x = 0.075 | 0.47 | 4.00 | 0.53 | 0.90 |
The electrochemical performances of LiNi0.7Co0.15Mn0.15O2−xFx samples with x = 0, 0.025, 0.05 and 0.075 were tested in typical CR 2032 coin half-cells against lithium metal. Fig. 4a–c compare the long cycle stability and charge–discharge plots of the pristine and various fluorine-doped LiNi0.7Co0.15Mn0.15O2 electrodes obtained in the voltage range of 2.8–4.4 V at a current density of 200 mA g−1. The pristine sample delivered initial charge and discharge capacities of 261.3 and 162.6 mA h g−1, respectively, with an initial coulombic efficiency of 78.2%. The sample with x = 0.05 delivered reversible initial charge and discharge capacities of 302.6 and 197.6 mA h g−1, respectively, with 80.3% coulombic efficiency, making it the best among the pristine and fluorine-doped samples. The low initial coulombic efficiency (CE) in both the doped and undoped samples may be due to the (1) irreversible formation of a solid–electrolyte interface (SEI) over the electrode surface and (2) the top layer containing excess Li2CO3 and LiF on the surface of the Ni-rich layered materials, which could impede electrolyte impregnation into the electrode surface. The electrochemical performance increased with fluorine doping content until x = 0.05; further increasing the F concentration adversely affected the electrochemical performance. This might be due to the disordering of Li+ and Ni2+, as evidenced by the XRD pattern of the sample with x = 0.075. The sample with x = 0.05 exhibited negligible voltage fade compared to the pristine sample (Fig. 4b and c) and delivered a reversible capacity of 169.8 mA h g−1, even after 100 cycles; the reversible capacity of the pristine sample was only 131.8 mA h g−1. Moreover, the F-doped surface layer suppressed the surface reaction as it is anticipated that the anion doping of high-voltage cathode materials form a M–F bond that which directly modifies the anionic oxidation processes at high voltage as reported by Tarascon's group.30–33 Oxyfluoride-based high-voltage cathode materials show improved electrochemical voltage and rate performance.36,39–41 This is well-supported by the electrochemical impedance spectra of the pristine and fluorine-doped (x = 0.05) samples. The Nyquist plot consists of semicircles in the high- and medium-frequency regions and a straight line in the low-frequency region in both spectra. The first semicircle at high frequency and the other semicircle at medium frequency respectively correspond to the interfacial resistance of Li+ ions passing through the SEI layer and electrochemical charge-transfer resistance between the active material and the electrolyte, which agrees well with the charge–discharge plots. The straight line in the low-frequency region refers to the Warburg diffusion inside the active material. Accordingly, the fluorine-doped sample with x = 0.05 exhibited a lower charge-transfer resistance of 152.9 Ω than the pristine sample (751.2 Ω; Fig. 4d).
 | ||
| Fig. 4 (a) Cycle stability of pristine and F-doped LiNi0.7Co0.15Mn0.15O2 tested at 200 mA g−1. Charge–discharge curves at various cycles for the (b) pristine sample and (c) LiNi0.7Co0.15Mn0.15O1.95F0.05 obtained at 200 mA g−1. (d) Electrochemical impedance spectra of the pristine sample and LiNi0.7Co0.15Mn0.15O1.95F0.05. | ||
In this work, we successfully synthesized Ni-rich layered LiNi0.7Co0.15Mn0.15O2−xFx compounds (0 < x < 0.075) and studied the effect of fluorine doping on their electrochemical performances for reversible Li+ extraction in the potential window of 2.8–4.4 V. Increasing the fluorine content (x > 0.05) resulted in the disordering of Li+ and Ni2+ ions, which adversely affected their cycling performances. The sample with x = 0.05 exhibited the lowest charge-transfer resistance and delivered a remarkable reversible capacity of 169.8 mA h g−1 at 200 mA g−1 with negligible voltage drop, even after 100 cycles.
Acknowledgements
Chandrasekar M Subramaniyam, Prof. Hua Kun Liu and Prof. Shi Xue Dou are grateful to the Commonwealth of Australia, Excellerate Australia, and the Automotive Australia 2020 Cooperative Research Centre (CRC) for financial support under project code AutoCRC 1-111 and the Institute for Superconducting and Electronic Materials (ISEM) for the use of its infrastructure as in-kind support. Dr John B Goodenough thanks the Robert A. Welch Foundation of Houston, Texas, USA for financial support.Notes and references
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461–472 CrossRef CAS.
- M. M. Thackeray, P. J. Johnson, L. A. de Picciotto, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1984, 19, 179–187 CrossRef CAS.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- C. Fu, G. Li, D. Luo, Q. Li, J. Fan and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 15822–15831 CAS.
- A. Manthiram, J. C. Knight, S.-T. Myung, S.-M. Oh and Y.-K. Sun, Adv. Energy Mater., 2016, 6, 1501010 CrossRef.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783–789 CrossRef CAS.
- P. Oh, M. Ko, S. Myeong, Y. Kim and J. Cho, Adv. Energy Mater., 2014, 4, 1400631 CrossRef.
- T. Ohzuku and A. Ueda, J. Electrochem. Soc., 1994, 14, 2972–2977 CrossRef.
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091–2097 CrossRef CAS.
- J. Tian, Y. Su, F. Wu, S. Xu, F. Chen, R. Chen, Q. Li, J. Li, F. Sun and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 582–587 CAS.
- N. Yabuuchi, K. Yoshii, S.-T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS PubMed.
- Y.-K. Sun, D.-J. Lee, Y. J. Lee, Z. Chen and S.-T. Myung, ACS Appl. Mater. Interfaces, 2013, 5, 11434–11440 CAS.
- J. Xiao, N. A. Chernova and M. S. Whittingham, Chem. Mater., 2008, 20, 7454–7464 CrossRef CAS.
- J. Xiao, N. A. Chernova and M. S. Whittingham, Chem. Mater., 2010, 22, 1180–1185 CrossRef CAS.
- J. Zheng, W. H. Kan and A. Manthiram, ACS Appl. Mater. Interfaces, 2015, 7, 6926–6934 CAS.
- N. A. Chernova, M. Ma, J. Xiao, M. S. Whittingham, J. Breger and C. P. Grey, Chem. Mater., 2007, 19, 4682–4693 CrossRef CAS.
- X. Zeng, G.-L. Xu, Y. Li, X. Luo, F. Maglia, C. Bauer, S. F. Lux, O. Paschos, S.-J. Kim, P. Lamp, J. Lu, K. Amine and Z. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3446–3451 CAS.
- J. Yang, M. Hou, S. Haller, Y. Wang, C. Wang and Y. Xia, Electrochim. Acta, 2016, 189, 101–110 CrossRef CAS.
- J. Yang and Y. Xia, ACS Appl. Mater. Interfaces, 2016, 8, 1297–1308 CAS.
- H. Zhang, B. Li, J. Wang, B. Wu, T. Fu and J. Zhao, RSC Adv., 2016, 6, 22625–22632 RSC.
- W. Cho, S.-M. Kim, K.-W. Lee, J. H. Song, Y. N. Jo, T. Yim, H. Kim, J.-S. Kim and Y.-J. Kim, Electrochim. Acta, 2016, 198, 77–83 CrossRef CAS.
- D. Mohanty, K. Dahlberg, D. M. King, L. A. David, A. S. Sefat, D. L. Wood, C. Daniel, S. Dhar, V. Mahajan, M. Lee and F. Albano, Sci. Rep., 2016, 6, 26532 CrossRef CAS PubMed.
- K. Yang, L.-Z. Fan, J. Guo and X. Qu, Electrochim. Acta, 2012, 63, 363–368 CrossRef CAS.
- Y.-K. Sun, M.-J. Lee, C. S. Yoon, J. Hassoun, K. Amine and B. Scrosati, Adv. Mater., 2012, 24, 1192–1196 CrossRef CAS PubMed.
- Z. Chen, Y. Qin, K. Amine and Y. K. Sun, J. Mater. Chem., 2010, 20, 7606–7612 RSC.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Nat. Mater., 2013, 12, 827–835 CrossRef CAS PubMed.
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M.-L. Doublet, D. Gonbeau, P. Novák, G. Van Tendeloo, R. Dominko and J.-M. Tarascon, Science, 2015, 350, 1516 CrossRef CAS PubMed.
- M. Sathiya, A. M. Abakumov, D. Foix, G. Rousse, K. Ramesha, M. Saubanère, M. L. Doublet, H. Vezin, C. P. Laisa, A. S. Prakash, D. Gonbeau, G. VanTendeloo and J. M. Tarascon, Nat. Mater., 2015, 14, 230–238 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- G. Du, Y. NuLi, J. Yang and J. Wang, Mater. Res. Bull., 2008, 43, 3607–3613 CrossRef CAS.
- http://www.xpsfitting.com/search/label/Nickel .
- https://www.srdata.nist.gov/xps/Default.aspx .
- S. H. Kang and K. Amine, J. Power Sources, 2005, 146, 654–657 CrossRef CAS.
- S. H. Kang, I. Belharouak, Y. K. Sun and K. Amine, J. Power Sources, 2005, 146, 650–653 CrossRef CAS.
- X.-Z. Liao, Y.-S. He, Z.-F. Ma, X.-M. Zhang and L. Wang, J. Power Sources, 2007, 174, 720–725 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |