
Biomass-derived interconnected carbon nanoring electrochemical capacitors with high performance in both strongly acidic and alkaline electrolytes†
Xianjun
Wei
*,
Yongbin
Li
and
Shuyan
Gao
*
Collaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, Key Laboratory of Green Chemical Media and Reactions (Ministry of Education), School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, China. E-mail: xj_wei@163.com; shuyangao@htu.cn
First published on 7th November 2016
Abstract
Besides a lack of low-cost electrode materials, the match between the electrode materials and electrolytes is one of the top issues to achieve high-rate electrochemical supercapacitors. Herein, we report an environmentally friendly strategy to prepare three-dimensionally interconnected carbon nanorings (TDICNs) from biomass waste, batata leaves and stalks (BLS). The microstructure of TDICNs matches well with acidic and alkaline electrolytes, making them exhibit high specific capacitances of 532.5 F g−1 (1 A g−1) and 264.0 F g−1 (30 A g−1), excellent capacitance retention (95.1% and 91.7% after 1000 cycles at 1 A g−1 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 30 A g−1, respectively) and a high energy density of 25.8–11.9 W h kg−1 with a power density of 249.5–13068.0 W kg−1 in 1 mol L−1 H2SO4 aqueous electrolyte. In 6 mol L−1 KOH electrolyte, they still exhibit high specific capacitances of 350.0 F g−1 (1 A g−1) and 246.9 F g−1 (30 A g−1), excellent cycling stability (95.06% and 91.1% of capacitance retention after 1000 (1 A g−1) and 10000 (30 A g−1) cycles, respectively), a high energy density of 24.5 W h kg−1 and a power density of 12918.0 W kg−1.
000 cycles at 30 A g−1, respectively) and a high energy density of 25.8–11.9 W h kg−1 with a power density of 249.5–13068.0 W kg−1 in 1 mol L−1 H2SO4 aqueous electrolyte. In 6 mol L−1 KOH electrolyte, they still exhibit high specific capacitances of 350.0 F g−1 (1 A g−1) and 246.9 F g−1 (30 A g−1), excellent cycling stability (95.06% and 91.1% of capacitance retention after 1000 (1 A g−1) and 10000 (30 A g−1) cycles, respectively), a high energy density of 24.5 W h kg−1 and a power density of 12918.0 W kg−1.
Introduction
Electrical double layer capacitors (EDLCs), also known as supercapacitors, with the characteristics of outstanding power density, long cycling stability, environmental friendliness and fast charge/discharge, have been widely applied as electrical sources for high-power electronic devices, digital devices, and so on.1–3 Compared with batteries, EDLCs usually suffer from low energy density (around 5 W h kg−1), which greatly hampers their potential applications.4 Based on eqn (1), the real voltage and specific capacitance are key to enhance the energy density,5 both of which substantially rely on the electrolyte and the EDLC electrode material, the two main components of EDLCs. Practically, the development of high energy density always sacrifices power density.6 This is expected to be overcome by achieving a good match between the electrolytes and electrode materials, which includes the match between the electrolyte ion size and the pore size of the electrode material, between electrolyte conductivity and wettability of the electrode material, between the pseudocapacitance and the functional electrode material, between the pseudocapacitance and the nature of the used electrolyte, and so on.The electrolyte is one of the basic components of EDLCs. Appropriate selection of the electrolyte can facilitate obtaining high energy densities. Aqueous electrolytes have been reported extensively over the last few years.7 This is mainly attributable to their low cost, safety, environmental friendliness and ease of manufacture.8 In general, 1 mol L−1 H2SO4 and 6 mol L−1 KOH are the most frequently used aqueous electrolytes due to their high ionic conductivity (the maximum values of 0.8 S cm−1 (ref. 7, 9 and 10) and 0.6 S cm−1 at 25 °C (ref. 9 and 11)) and low viscosity (about 1.12 mPa s and less than 2.32 mPa s at 20 °C),11,12 which are beneficial to achieving low equivalent series resistance and high power densities.13
For electrode materials, carbonaceous materials, such as activated carbons (ACs), carbon nanotubes (CNTs), graphene-based materials, and carbon aerogels,14–17 are considered as the most potential EDLC electrode candidates due to their high electrical conductivity, outstanding chemical stability, environmental friendliness, and wide availability.18–21 Among them, ACs with tailored porosity have been attractive candidates due to their three-dimensional (3D) nanostructures, hierarchical pores, large specific surface areas and surface heteroatom doping (N, O, B, S, P, etc.).22–26 Abundant micropores and mesopores provide a high accessible surface area, and play an essential role in high energy storage resulting in a large capacitance and high energy density, whereas interconnected mesopores and macropores facilitate rapid ion transport by supplying ion-buffering reservoirs and ion-transport pathways, which ensure high rate capability and high power density.27 The nitrogen or oxygen groups in the carbon can provide an additional pseudocapacitance from the redox reaction occurring on the heteroatom surface, improve the wettability of the carbon materials and enhance their electrochemical performance.28 Traditionally, ACs are prepared from costly and non-renewable coal and petroleum,29 and their synthesis procedure is complicated, costly and always involves highly toxic chemicals.30 Therefore, there is an urgent need to design advanced ACs via a renewable route. In quite recent years, biomass-based approaches are becoming more and more attractive and promising in that biomass, a natural renewable resource, features uniform porosity and rich heteroatoms such as N and O27 and carbon materials derived from biomass resources usually possess optimal hierarchical structures with naturally perfect interconnection and in situ heteroatom doping.23 Sweet potatoes (Ipomoea batatas) as agricultural byproducts represent the sixth most important food crop in the world, and in China, the planting area and output of sweet potatoes stand first in the world. Nevertheless, for a long time, people have been eating only sweet potato tuberous roots, and 5 billion kg sweet potato leaves and stalks, 48 billion kg sweet potato leaves and 47 billion kg sweet potato vines are discarded or burned to produce heat in most regions except in some regions where they are used as feed, which certainly causes serious social and environmental problems.31–33 The leaves and stalks of sweet potato are rich in proteins, amino acids, polyphenols, polysaccharose, dietary fibers, minerals, and vitamins such as carotene, vitamin B2, vitamin C and vitamin E.31,34 So, in principle, BLS should be easily pyrolyzed and the resulting functional carbon materials would be suitable for the electrodes of electrochemical supercapacitors.
Until now, no combination of biomass-waste-based three-dimensional hierarchically porous heteroatom-doped carbons and the relevant match with electrolytes has been reported to achieve high performance EDLC devices.9 This herein motivates us to develop TDICNs from BSL. The TDICNs exhibit high specific capacitances of 532.5 F g−1 and 350.0 F g−1, excellent cycling stabilities (95.1% and 95.06% retention after 1000), high energy densities (25.8 W h kg−1 and 24.5 W h kg−1) and high power densities (13068.0 W kg−1 and 12918.0 W kg−1) in 1 mol L−1 H2SO4 and 6 mol L−1 KOH, respectively. This work integrates biomass resource utilization with efficient high-rate EDLC performance in both strongly acidic and alkaline electrolytes via a good match between the electrode materials and electrolytes.
Results and discussion
The mechanism of the preparation of carbon materials by pyrolysis of sweet potato stems and leaves has not been reported in the literature. However, the structures and chemical properties of lignin, cellulose, hemicellulose, β-carotene, flavonoids and amino acids in sweet potato stems and leaves are similar to those of other plants;32–35 see Fig. S1.† So we can speculate the pyrolysis mechanism of sweet potato stems and leaves according to the relevant literature and our results. Lignin is a complex three-dimensional amorphous polymer composed of three units: p-coumaryl alcohol (H), coniferyl alcohol (G) and sinapyl alcohol (S) as presented in Fig. S1a.† The ratio of the monomer units is highly variable and mainly depends on the different plant species.36 Cellulose is a linear biopolymer composed of two β-glucopyranose units as shown in Fig. S1b.† The two glucan units are connected by 1,4-glycosidic linkages. The frequent intermolecular hydrogen bonds between different layers of the polysaccharides result in a lamellar structure.37,38 Hemicelluloses are not uniform-polysaccharides, but hetero-polysaccharides, whose composition varies with different shaws. The basic units in hemicelluloses are pentoses, hexoses, and sugar acids. Arabinose, xylose, glucose, galactose, mannose and glucuronic acid are the main constituents of the hemicelluloses in agricultural residues,38,39 see Fig. S1c.† Flavonoids are one of the most abundant secondary metabolites in plants and the most significant natural phenolic compounds, which usually exist in the form of glycosides in sweet potato stems and leaves, see Fig. S1d.†32,40 Pyrolysis is a partial depolymerization process of biomass to form volatile gases (CO, CO2, H2, CH4, and C2H4), liquid bio-oils (carbohydrates, organic acids, organic aldehydes, and mixed phenolic substances) and char under the conditions of free oxygen and the suitable temperature (300–700 °C).37 Biomass is mainly composed of cellulose, hemicellulose and lignin, and its pyrolysis behavior can be considered as the comprehensive effect of the pyrolysis of the three major components.38Fig. 1A shows the proposed mechanism of sweet potato stem and leaf pyrolysis. According to the literature,38,41–48 lignin, cellulose and hemicellulose are dehydrated and depolymerized to form a lot of oxygenated organic compounds such as 1, 2, 3, 4 and 5 as shown in Fig. 1A at about 300 °C temperature, which are commonly referred to as the main conversion products. The reactions responsible for the resulting compounds are mostly due to the instability of the alkyl chains, of some linkages between monomer units and of the methoxy substituents of the aromatic rings, and are generally accompanied by the release of water or gas. These main products contain two or even three active points, so that a polycyclic polymer 12 may be formed through intermolecular cross-linking reactions among them. On the other hand, the main products are further decomposed into secondary products 6, 7, 8, 9, 10 and 11 at even higher temperature. A lot of intermolecular condensation reactions could happen among the secondary products and the polycyclic polymer 12, which result in the formation of aromatic polycyclic structures. All these rearrangement reactions are also usually associated with the release of water or incondensable gas. The mechanism is in good coincidence with the SEM and TEM analysis. The TEM micrographs in Fig. 1B and C and SEM images in Fig. S2† clearly show that TDICNs feature 3D interconnected nanorings consisting of multilayer graphenes (from 3 to 16 layers).49 The lattice fringes with a distance of 0.34 nm correspond to the interlayer distance of graphite. Fig. 1D and E show the monolayer and multilayer structure models of TDICNs, which clearly display the possible microstructure of TDICNs.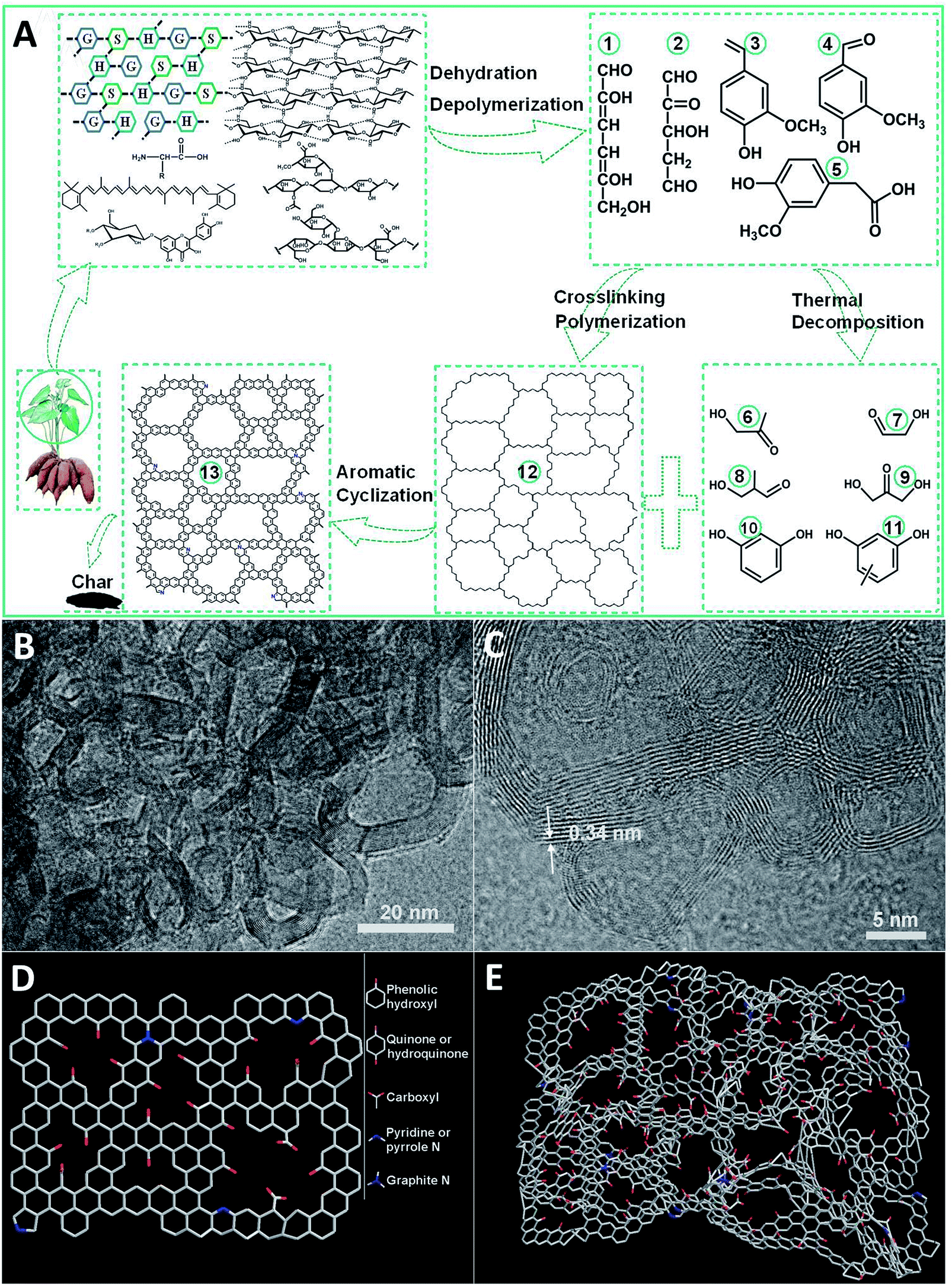 | ||
| Fig. 1 The proposed formation mechanism (A) of TDICN, TEM images (B and C); and monolayer (D) and multilayer (E) structure models of TDICNs. Compounds 1–13: 2,3-anhydro-D-mannose; 2-carbonyl, 3-hydroxyl, 1,5-glutaraldehyde; 2-methoxy, 3-vinylphenol; 2-methox, 3-carbonylphenol; 2-methoxy, 3-carboxyethylphenol; 1-hydroxylacetone; 2-glycolaldehyde; 2-methyl, 3-hydroxylpropionaldehyde; 1,3-dihydroxyacetone; catechol; methyl resorcinol; polycyclic polymer; aromatic polycyclic structure. | ||
Raman results are shown in Fig. 2A, exhibiting two bands located at 1320 cm−1 (D band) and 1590 cm−1 (G band), probably originating from heteroatom doping and KOH activation.50 The G band is attributed to the vibration of sp2-hybridized carbon atoms in a planar hexagonal crystal lattice. The G/D intensity ratio is often used to indicate the degree of graphitization. And 1.03 is obtained in our case, suggesting a weakly ordered graphitic microstructure with a relatively good electronic conductivity.51 As compiled in Fig. 2B and C and S2C,† the XPS results of TDICNs show C1s as the main element at 282.6 eV accompanied by weak signals of N1s and O1s at binding energies of 397.7 and 530.3 eV. The N1s could be deconvoluted into four peaks, i.e. nitrogen-containing functional groups: pyridinic (N-6, 398.2 eV), pyrrolic and/or pyridonic (N-5, 399.6 eV), quaternary nitrogen (N-Q, 401.2 eV), and oxidized N (N–O, 406.5 eV). N-6 and N-5 are beneficial to create pseudocapacitance.52 The N-Q would effectively improve the conductivity of electrode materials and contribute to the cycling performance of supercapacitors. Additionally, the nitrogen-containing functional groups could improve the wettability.53,54 The O1s could also be resolved into four peaks, quinone (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 531.3 eV), hydroxyl (C–OH, 532.2 eV), ether (C–O–C, 533.4 eV), and carboxyl (OC–OH, 535.5 eV), all of which are beneficial to electrochemical redox activity and wettability.55–58
O, 531.3 eV), hydroxyl (C–OH, 532.2 eV), ether (C–O–C, 533.4 eV), and carboxyl (OC–OH, 535.5 eV), all of which are beneficial to electrochemical redox activity and wettability.55–58
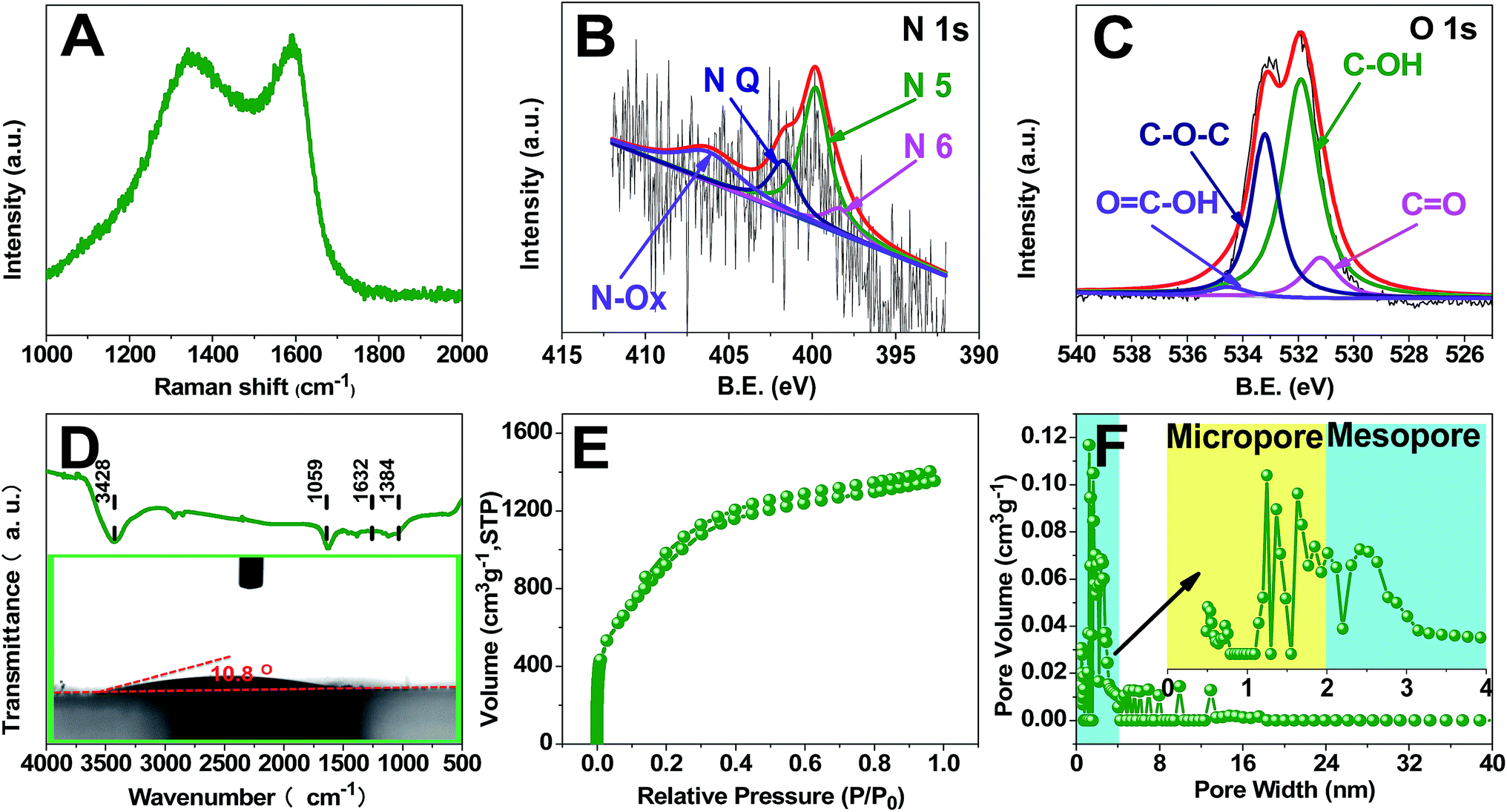 | ||
| Fig. 2 Raman spectrum (A); N1s XPS spectrum (B); O1s XPS spectrum, FT-IR spectrum (D); Ar adsorption/desorption isotherms (E); and pore size distribution (F) of TDICNs; the inset of (D) is the water contact angle measurement. | ||
The chemical nature of the as-synthesized TDICNs is further characterized by FTIR in the 500–4000 cm−1 region. As shown in Fig. 2D, the strong peak at 3428 cm−1 is typically attributed to the O–H stretching vibration (hydroxyl groups and chemisorbed water) and/or N–H symmetric stretching vibration.59,60 The band at 1632 cm−1 is designated to the stretching vibration of CO (COOH functional groups).61–63 The spectrum at 1384 cm−1 corresponds to the C–N stretching vibration, together with the deformation vibration of C–H and stretching vibration of C–O.64 The signal located at 1059 cm−1 is assigned to C–O–C (symmetric angular deformation of ethers).65,66 Additionally, the contact angle is 10.8° (the inset of Fig. 2D), demonstrating good wettability. The Ar adsorption/desorption isotherms and pore size distributions of TDICNs in Fig. 2E and F indicate hybrid type-I and type-IV (IUPAC) adsorption isotherms, suggesting the coexistence of micropores (centered at about 1.25 nm) and mesopores (centered at about 2.51 nm) The wider micropore size distribution provides a higher ion accessible surface area and plays an important role in achieving higher energy storage.67 The BET surface area and the t-plot micropore area are 3114.74 and 2644.96 m2 g−1, respectively.
Altogether, several important morphological and structural features should be noted. First, TDICNs have a graphitic structure with 3D interconnected nanoring networks, resulting in appropriate pore size matching well with electrolyte ions. Second, the high surface area ensures more storage sites and hence higher energy density. Last but not least, O, N in situ doping and related surface functional groups contribute to good electronic conductivity and wettability. Therefore, it is reasonable to anticipate that TDICNs may have great performance in electrochemical energy storage, especially for pH-universal high-rate supercapacitors.
We first note that the CV profiles of TDICNs within scan rates from 2 to 200 mV s−1 between −1 and 0 V in 1 mol L−1 H2SO4 aqueous electrolyte show that the plateau current increases along with scan rate (Fig. 3A). The quasi-rectangular shape can be maintained even at a high scan rate of 200 mV s−1, indicating quick and efficient charge transfer and excellent capacitive behavior.68 In order to further study the capacitive performance, the galvanostatic charge–discharge tests of TDICNs are carried out in 1 mol L−1 H2SO4 aqueous electrolyte at loading potentials between −1.0 and 0 V at different current densities from 0.5 to 30.0 A g−1. All the galvanostatic charging–discharging curves display a symmetric triangular shape without obvious IR drop, indicating the small internal resistance and good capacitive performance (Fig. 3B).69,70 At a current density of 1.0 A g−1, a specific capacitance (calculated from eqn (2)) of 532.5 F g−1 is obtained, which is much higher than those of previously published biomass/biowaste-based activated carbons (Fig. 3C and Table S1†). The specific capacitances decrease gradually with the increase of discharging current density possibly due to inadequate time for electrolyte ion diffusion into the entire pores.71–73 However, they still retain 264 F g−1 even at 30 A g−1, indicating excellent rate capability.
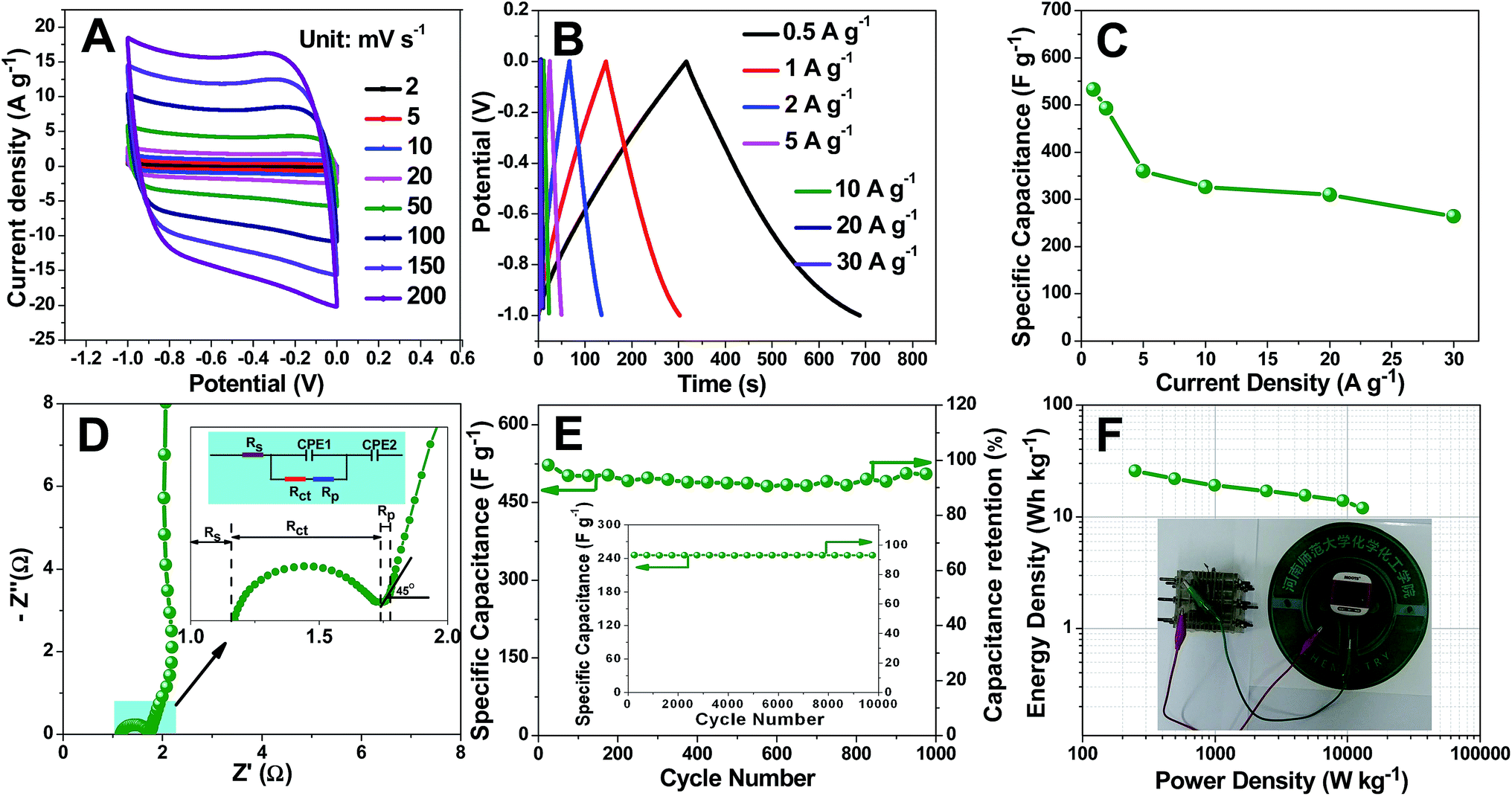 | ||
| Fig. 3 CV curves of a TDICN-based supercapacitor at various scan rates in 1 mol L−1 H2SO4 aqueous electrolyte (A); galvanostatic charge–discharge curves of the TDICN-based supercapacitor at different current densities from 0.5 to 30 A g−1 (B); specific capacitances of the TDICN-based supercapacitor at different current densities (C); Nyquist plot for the TDICN electrode (D); specific capacitance and capacitance retention of the TDICN-based supercapacitor at 1 A g−1 for 1000 cycles (E); Ragone plot of the TDICN-based supercapacitor (F); the inset of (D) shows the equivalent circuit model and the magnified high frequency range; the inset of (E) is the cycle test of TDICNs at 30 A g−1 for 10000 cycles; the inset of (F) demonstrates that three assembled symmetric TDICN-based supercapacitors can efficiently drive a commercial pedometer. | ||
EIS in 1 mol L−1 H2SO4 (three-electrode system) over the range from 10 mHz to 10 kHz (Fig. 3D) shows an equivalent series resistance (Rs, the sum of the intrinsic resistance of the material, ionic resistance of H2SO4 electrolyte and the contact resistance between the stainless steel grid and material)74–76 of about 1.2 Ω, suggesting the good conductivity and low internal resistance.77 At high frequency, the Nyquist plot exhibits a semicircle, which represents Rct charge transfer resistance between the electrolyte and electrode.61,74,78 At intermediate frequency, there is a slope of 45° between the semicircle and perpendicular line corresponding to Warburg impedance (Rp), representing the diffusion rate of electrolyte ions into the micropore surface of electrode materials.78–82 As shown in the inset of Fig. 3D, TDICNs exhibit small Rct (0.567 Ω) and Rp (0.347 Ω), which may be attributed to the 3D interconnected ring-like structure, high hydrophilicity and doping.83,84 At low frequency, the vertical line indicates an ideal capacitive behavior.80,81
Considering cycling stability is an important parameter, cycling performance is evaluated in a three-electrode system by continuous charging–discharging technology at current densities of 1 and 30 A g−1 in the potential range of −0.8 to 0.2 V (Hg/HgSO4) in 1 mol L−1 H2SO4 electrolyte. 95.1 and 91.7% of the initial capacitances at current densities of 1 and 30 A g−1 after 1000 and 10000 cycles were obtained, indicating the excellent capacitance retention. To further investigate the electrochemical performance, the energy density and power density are calculated using eqn (1), (3) and (4). The Ragone plot is illustrated in Fig. 3F. The TDICN-based capacitor presents energy densities of 25.8–11.9 W h kg−1 at power densities of 249.5–13068.0 W kg−1, superior to those of most reported biomass derived carbon electrodes (Table S1†), which indicates that TDICNs are a promising electrode material for supercapacitors and it is demonstrated here that three tandem symmetric supercapacitor devices could easily drive a commercial pedometer as shown in the inset of Fig. 3F and the video in the ESI.†
For pH-universal application, the energy storage test is carried out in 6 mol L−1 KOH aqueous electrolyte. As shown in Fig. 4A, the TDICN-based supercapacitor still exhibits the rectangular voltammetry characteristic of EDLCs in the alkaline electrolyte. Its specific capacitance can reach 350 F g−1 at 1.0 A g−1. When increasing the current density to 30 A g−1, a specific capacitance of 246.9 F g−1 is obtained (Fig. 4B). The Rct and Rp of TDICN-based supercapacitors in 6 mol L−1 KOH electrolyte are 0.587 and 0.376 Ω, manifesting the fast charge transfer and ion diffusion. The near vertical line indicates ideal capacitive behavior (Fig. 4C). Fig. 4D demonstrates 95.0% capacitance retention at 1 A g−1 after 1000 cycles. The capacitive retention is still up to 91.1% even after 10000 charging–discharging cycles at 30 A g−1 (inset of Fig. 4D), suggesting excellent electrochemical stability. The Ragone plot (the inset of Fig. 4B) shows energy densities of 24.5–10.8 W h kg−1 at power densities of 249.0–12918.0 W kg−1, higher than those of previously reported biomass/biowaste-based activated carbons (Table S1†). Note that the capacitance values in 6 mol L−1 KOH aqueous electrolyte for TDICNs are smaller than those in 1 mol L−1 H2SO4. This phenomenon can be interpreted by the principle of Andreas and Conway,85 where the pseudocapacitive contribution of oxygenated functional groups (e.g., quinone-type functionalities) is less efficient in KOH electrolyte than in H2SO4 electrolyte.
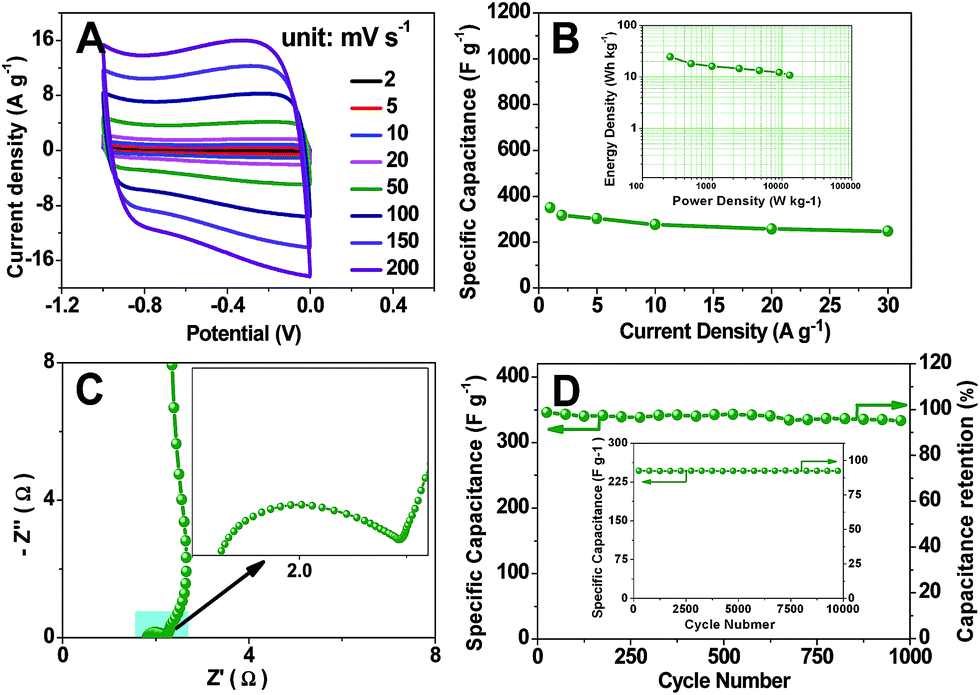 | ||
| Fig. 4 CV curves of a TDICN-based supercapacitor in 6 mol L−1 KOH aqueous electrolyte at various scan rates (A); specific capacitances of the supercapacitor at different current densities (B); Nyquist plot and the magnified high frequency range in the inset (C); and plot of specific capacitance and capacitance retention at 1 A g−1 for 1000 cycles (D); the inset of (B) is the Ragone plot; the inset of (D) is the cycle test of the TDICN-based supercapacitor at 30 A g−1 for 10000 cycles. | ||
Conclusions
In summary, novel TDICNs have been prepared via facile carbonization followed by a KOH activation process. The as-obtained TDICNs consisted of 3D interconnected carbon nanorings with high BET surface area (3114.7 m2 g−1), appropriate pore size (centered at about 1.25 nm) and O–N doping. These characteristics show excellent multiple synergistic effects and are beneficial for matching very well with H2SO4 and KOH electrolytes. That is to say, TDICNs can show high performance in the full pH range: as high as 532.5 F g−1 and 350.0 F g−1 specific capacitance, 25.8 W h kg−1 and 24.5 W h kg−1 energy density, and 13068.0 W kg−1 and 12918.0 W kg−1 power density in 1 mol L−1 H2SO4 and 6 mol L−1 KOH. Such a supercapacitor with high power density up to 25.8 W h kg−1 has enough energy to power some electronic equipment such as a commercial pedometer. Therefore, our work not only describes a low-cost, environmentally friendly and facile preparation method to produce a carbon electrode material for high-performance supercapacitors in strongly alkaline/acidic environments, but also inspires a promising and potential application in energy conversion devices.
Experimental
Materials
The disposable BSL is used as the starting material and recycled from the farmland of Henan, China. Potassium hydroxide (KOH) was purchased from Sino pharm Chemical Reagent Co., Ltd.Preparation of TDICNs
Typically, BSL is firstly rinsed with deionized water and dried in an oven at 65 °C for 10 hours to remove all the intrinsic moisture. The dry BSL is crushed and then ground at 25000 rpm (revolutions per minute) using a grinder. After that, BSL is subjected to carbonization in a tube furnace at 600 °C for 2 hours with a heating rate of 5 °C min−1 under a continuous flow of nitrogen and cooled down to room temperature to get a pre-carbon (marked as BSL-600). To prepare the porous carbon material, the BSL-600 is mixed with KOH at a BSL-600/KOH weight ratio of 1:4 by grinding using an agate mortar followed by activation in a tube furnace at 700 °C for 2 hours with a heating rate of 10 °C min−1 under a continuous flow of nitrogen and cooled down to room temperature, which are washed with 2 mol L−1 HCl and distilled water until neutral, and then dried in an oven at 75 °C to obtain the TDICN carbon material.
Electrochemical measurements
Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed on a CHI660E electrochemical workstation (Shanghai Chen Instrument Co. Ltd., China), the galvanostatic charge–discharge curves were determined using a Land (CT2001A, Land, Wuhan, China). The electrochemical performance was determined using a two-electrode or three-electrode cell under ambient conditions. To prepare the working electrode, TDICNs are mixed with acetylene black and poly(tetrafluoroethylene) binder at a weight ratio of 75:20:5, and the mixture is then pasted on the stainless steel grid (1 cm2), followed by drying at 85 °C in an electric thermostatic drying oven. For the three-electrode system, the TDICNs are used as the working electrode, stainless steel grid as the counter electrode and Hg/Hg2SO4 electrode (in acidic medium) as the reference. The two-electrode system is assembled using two equal TDICN electrodes. The CV test is performed in a two-electrode system with 1 mol L−1 H2SO4 or 6 mol L−1 KOH electrolyte under ambient conditions; the potential range of CV was −1–0 V at various sweep rates. The EIS test is performed in a three-electrode system with 1 mol L−1 H2SO4 or 6 mol L−1 KOH electrolytes under ambient conditions in the frequency range of 0.01–10 kHz at the open circuit voltage. The specific capacitance is calculated by charge/discharge experiments in a three-electrode system with 1 mol L−1 H2SO4 or 6 mol L−1 KOH electrolytes under ambient conditions. The power and energy densities are calculated by charge/discharge experiments in a two-electrode system with 1 mol L−1 H2SO4 or 6 mol L−1 KOH electrolyte under ambient conditions.
The specific capacitance is calculated through eqn (1) and (2)as follows:
| C1 = 2 × IP × t(two-electrode system) | (1) |
| C2 = IP × t(three-electrode system) | (2) |
The stored specific energy is obtained by eqn (3):
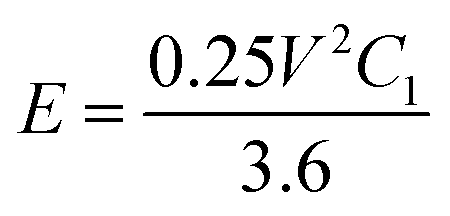 | (3) |
The power density is given by eqn (4):
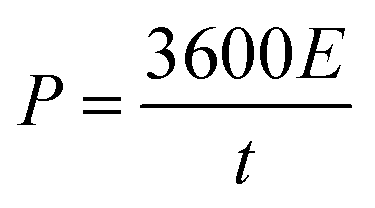 | (4) |
Characterization
The morphology of TDICNs is characterized using field-emission scanning electron microscopy (FESEM, Zeiss Supra 40, 5 kV) and transmission electron microscopy (TEM, JEOL JEM-2100). X-ray diffraction (XRD) measurements are carried out using a Bruker D8 instrument with Cu-Kα radiation (λ = 0.154 nm). X-ray photoelectron spectroscopy (XPS) was performed on an ESCALab220i-XL instrument with monochromatic Al-Kα radiation (300.0 eV). Raman spectroscopy is carried out using a Renishaw in Via unit with a 514 nm excitation laser. Argon adsorption/desorption is performed at −186.15 °C using a Micromeritics ASAP-2020. Before measurements, the materials are outgassed at 190 °C for 3 hours. The specific surface area is calculated according to the Brunauer–Emmett–Teller (BET) method, whereas the micropore surface area and mesopore surface area are determined using the t-plot theory. The pore size distribution is calculated by the density functional theory (DFT) method. FTIR spectra are collected on a NEXUS (Thermo Nicolet Co., USA) spectrophotometer in the wavenumber region from 4000 to 400 cm−1. The wettability is measured by using a water contact angle technique (Kruss, FM 40).Acknowledgements
Profs. Haibo Zeng, Yu Gu, and Xiaoming Li from Nanjing University of Science and Technology are greatly appreciated for their kind advice and help. This work is supported by NSFC (21471048), the Research Project of Chinese Ministry of Education (No. 213023A), NCET-11-0944, the Excellent Youth Foundation of Henan Scientific Committee (124100510004), and the Program for Innovative Research Team in University of Henan Province (No. 14IRTSTHN005).Notes and references
- S. Bose, T. Kuila, A. K. Mishra, R. Rajasekar, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 767–784 RSC.
- L. Zhou, H. Cao, S. Zhu, L. Hou and C. Yuan, Green Chem., 2015, 17, 2373–2382 RSC.
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- Y. S. Yun, G. Yoon, K. Kang and H.-J. Jin, Carbon, 2014, 80, 246–254 CrossRef CAS.
- Q. Liang, L. Ye, Z.-H. Huang, Q. Xu, Y. Bai, F. Kang and Q.-H. Yang, Nanoscale, 2014, 6, 13831–13837 RSC.
- L. Zhu, Q. Gao, Y. Tan, W. Tian, J. Xu, K. Yang and C. Yang, Microporous Mesoporous Mater., 2015, 210, 1–9 CrossRef CAS.
- C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- V. Khomenko, E. Raymundo-Piñero and F. Béguin, J. Power Sources, 2010, 195, 4234–4241 CrossRef CAS.
- D. Jiménez-Cordero, F. Heras, M. A. Gilarranz and E. Raymundo-Piñero, Carbon, 2014, 71, 127–138 CrossRef.
- H. Wu, X. Wang, L. Jiang, C. Wu, Q. Zhao, X. Liu, B. Hu and L. Yi, J. Power Sources, 2013, 226, 202–209 CrossRef CAS.
- S. I. Fletcher, F. B. Sillars, N. E. Hudson and P. J. Hall, J. Chem. Eng. Data, 2010, 55, 778–782 CrossRef CAS.
- G. Liu, L. Ma and J. Liu, The Physical Properties Data of Chemical Enginerring, Chemical Industry Press, Beijing, 2002, ch. 3 Search PubMed.
- P. J. Hall, M. Mirzaeian, S. I. Fletcher, F. B. Sillars, A. J. R. Rennie, G. O. Shitta-Bey, G. Wilson, A. Cruden and R. Carter, Energy Environ. Sci., 2010, 3, 1238–1251 CAS.
- A. Alabadi, X. Yang, Z. Dong, Z. Li and B. Tan, J. Mater. Chem. A, 2014, 2, 11697–11705 CAS.
- D. Ge, L. Yang, L. Fan, C. Zhang, X. Xiao, Y. Gogotsi and S. Yang, Nano Energy, 2015, 11, 568–578 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- H. An, Y. Wang, X. Wang, L. Zheng, X. Wang, L. Yi, L. Bai and X. Zhang, J. Power Sources, 2010, 195, 6964–6969 CrossRef CAS.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. P. Marco-Lozar, M. Kunowsky, F. Suárez-García, J. D. Carruthers and A. Linares-Solano, Energy Environ. Sci., 2012, 5, 9833–9842 CAS.
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- Y. Zhao, W. Ran, J. He, Y. Song, C. Zhang, D.-B. Xiong, F. Gao, J. Wu and Y. Xia, ACS Appl. Mater. Interfaces, 2015, 7, 1132–1139 CAS.
- S. Kondrat, C. R. Pérez, V. Presser, Y. Gogotsi and A. A. Kornyshev, Energy Environ. Sci., 2012, 5, 6474–6479 CAS.
- W. Huang, H. Zhang, Y. Huang, W. Wang and S. Wei, Carbon, 2011, 49, 838–843 CrossRef CAS.
- S. Gao, H. Fan, X. Wei, L. Li, Y. Bando and D. Golberg, Part. Part. Syst. Charact., 2013, 30, 864–872 CAS.
- S. Dutta, A. Bhaumik and C.-W. Kevin, Energy Environ. Sci., 2014, 7, 3574–3592 CAS.
- W. Kim, J. B. Joo, N. Kim, S. Oh, P. Kim and J. Yi, Carbon, 2009, 47, 1407–1411 CrossRef CAS.
- M.-M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 Search PubMed.
- C. Long, L. Jiang, X. Wu, Y. Jiang, D. Yang, C. Wang, T. Wei and Z. Fan, Carbon, 2015, 93, 412–420 CrossRef CAS.
- Y. Zhang, Y.-N. Wu, L. Chen, Y.-Y. Gao and J.-B. Zhu, Food Sci., 2006, 27, 252–256 CAS.
- H. Ishida, H. Suzuno, N. Sugiyama, S. Innami, T. Tadokoro and A. Maekawa, Food Chem., 2000, 68, 359–367 CrossRef CAS.
- S. Gao, H. Fan, Y. Chen, L. Li, Y. Bando and D. Golberg, Nano Energy, 2013, 2, 1261–1270 CrossRef CAS.
- C. C. Teow, V.-D. Truong, R. F. McFeeters, R. L. Thompson, K. V. Pecota and G. C. Yencho, Food Chem., 2007, 103, 829–838 CrossRef CAS.
- K. Sasaki, T. Oki, Y. Kai, Y. Nishiba and S. Okuno, Biosci., Biotechnol., Biochem., 2015, 79, 1308–1314 CrossRef CAS PubMed.
- L. Xi, T. Mu and H. Sun, Food Chem., 2015, 172, 166–174 CrossRef CAS PubMed.
- W. M. Walter, A. E. Purcell and G. K. McCollum, J. Agric. Food Chem., 1978, 26, 1222–1226 CrossRef CAS.
- E. M. Rubin, Nature, 2008, 454, 841–845 CrossRef CAS PubMed.
- C. Falco, N. Baccile and M.-M. Titirici, Green Chem., 2011, 13, 3273–3281 RSC.
- F.-X. Collard and J. Blin, Renewable Sustainable Energy Rev., 2014, 38, 594–608 CrossRef CAS.
- K. Werner, L. Pommer and M. Broström, J. Anal. Appl. Pyrolysis, 2014, 110, 130–137 CrossRef CAS.
- C. S. Winefield, D. H. Lewis, E. E. Swinny, H. Zhang, H. S. Arathoon, T. C. Fischer, H. Halbwirth, K. Stich, C. Gosch, G. Forkmann and K. M. Davies, Physiol. Plant., 2005, 124, 419–430 CrossRef CAS.
- D. K. Shen, S. Gu and A. V. Bridgwater, J. Anal. Appl. Pyrolysis, 2010, 87, 199–206 CrossRef CAS.
- W. Chaiwat, I. Hasegawa, T. Tani, K. Sunagawa and K. Mae, Energy Fuels, 2009, 23, 5765–5772 CrossRef CAS.
- J. E. White, W. J. Catallo and B. L. Legendre, J. Anal. Appl. Pyrolysis, 2011, 91, 1–33 CrossRef CAS.
- Y.-C. Lin, J. Cho, G. A. Tompsett, P. R. Westmoreland and G. W. Huber, J. Phys. Chem. C, 2009, 113, 20097–20107 CAS.
- A. Dieguez-Alonso, A. Anca-Couce, N. Zobel and F. Behrendt, Fuel, 2015, 153, 102–109 CrossRef CAS.
- D. K. Shen and S. Gu, Bioresour. Technol., 2009, 100, 6496–6504 CrossRef CAS PubMed.
- S. Gao, L. Li, K. Geng, X. Wei and S. Zhang, Nano Energy, 2015, 16, 408–418 CrossRef CAS.
- L. Sun, C. Tian, M. Li, X. Meng, L. Wang, R. Wang, J. Yin and H. Fu, J. Mater. Chem. A, 2013, 1, 6462–6470 CAS.
- J. H. Kaufman and S. Metin, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 13053–13060 CrossRef CAS.
- S. Gao, K. Geng, H. Liu, X. Wei, M. Zhang, P. Wang and J. Wang, Energy Environ. Sci., 2015, 8, 221–229 CAS.
- G. Xu, J. Han, B. Ding, P. Nie, J. Pan, H. Dou, H. Li and X. Zhang, Green Chem., 2015, 17, 1668–1674 RSC.
- J. Tan, H. Chen, Y. Gao and H. Li, Electrochim. Acta, 2015, 178, 144–152 CrossRef CAS.
- L. Hao, X. Li and L. Zhi, Adv. Mater., 2013, 25, 3899–3904 CrossRef CAS PubMed.
- D. Usachov, O. Vilkov, A. Grüneis, D. Haberer, A. Fedorov, V. K. Adamchuk, A. B. Preobrajenski, P. Dudin, A. Barinov, M. Oehzelt, C. Laubschat and D. V. Vyalikh, Nano Lett., 2011, 11, 5401–5407 CrossRef CAS PubMed.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS.
- X. Wei, S. Wan and S. Gao, Nano Energy, 2016, 28, 206–215 CrossRef CAS.
- W.-H. Qu, Y.-Y. Xu, A.-H. Lu, X.-Q. Zhang and W.-C. Li, Bioresour. Technol., 2015, 189, 285–291 CrossRef CAS PubMed.
- M. Wahid, G. Parte, D. Phase and S. Ogale, J. Mater. Chem. A, 2015, 3, 1208–1215 CAS.
- Y. Guo and D. A. Rockstraw, Microporous Mesoporous Mater., 2007, 100, 12–19 CrossRef CAS.
- X. Li, E. Xin and J. Zhang, Electron. Mater. Lett., 2015, 11, 143–148 CrossRef CAS.
- Z. Lei, J. Zhang and X. S Zhao, J. Mater. Chem., 2012, 22, 153–160 RSC.
- H. Denga, G. Li, H. Yang, J. Tang and J. Tang, Chem. Eng. J., 2010, 163, 373–381 CrossRef.
- J. Zhang, L. Jin, J. Cheng and H. Hu, Carbon, 2013, 55, 221–232 CrossRef CAS.
- S. Fajardo and G. S. Frankel, Electrochim. Acta, 2015, 166, 255–267 CrossRef.
- W. Tian, Q. Gao, Y. Tan, Y. Zhang, J. Xu, Z. Li, K. Yang, L. Zhu and Z. Liu, Carbon, 2015, 85, 351–362 CrossRef CAS.
- T.-H. Liou and S.-J. Wu, J. Hazard. Mater., 2009, 171, 693–703 CrossRef CAS PubMed.
- X. Wei, X. Jiang, J. Wei and S. Gao, Chem. Mater., 2016, 28, 445–458 CrossRef CAS.
- S. Gao, Y. Chen, H. Fan, X. Wei, C. Hu, H. Luo and L. Qu, J. Mater. Chem. A, 2014, 2, 3317–3324 CAS.
- C. Long, X. Chen, L. Jiang, L. Zhi and Z. Fan, Nano Energy, 2015, 12, 141–151 CrossRef CAS.
- Y. Gao, W. Zhang, Q. Yue, B. Gao, Y. Sun, J. Kong and P. Zhao, J. Power Sources, 2014, 270, 403–410 CrossRef CAS.
- J. Zhou, Z. Zhang, W. Xing, J. Yu, G. Han, W. Si and S. Zhuo, Electrochim. Acta, 2015, 153, 68–75 CrossRef CAS.
- W. Qian, F. Sun, Y. Xu, L. Qiu, C. Liu, S. Wang and F. Yan, Energy Environ. Sci., 2014, 7, 379–386 CAS.
- W. Xing, C. C. Huang, S. P. Zhuo, X. Yuan, G. Q. Wang, D. Hulicova-Jurcakovac, Z. F. Yan and G. Q. Lu, Carbon, 2009, 47, 1715–1722 CrossRef CAS.
- D. Puthusseri, V. Aravindan, S. Madhavi and S. Ogale, Energy Environ. Sci., 2014, 7, 728–735 CAS.
- J.-G. Wang, Y. Yang, Z.-H. Huang and F. Kang, Carbon, 2013, 61, 190–199 CrossRef CAS.
- L. Sun, C. Tian, Y. Fu, Y. Yang, J. Yin, L. Wang and H. Fu, Chem.–Eur. J., 2014, 20, 564–574 CrossRef CAS PubMed.
- J. Chang, Z. Gao, X. Wang, D. Wu, F. Xu, X. Wang, Y. Guo and K. Jiang, Electrochim. Acta, 2015, 157, 290–298 CrossRef CAS.
- P. Tammela, H. Olsson, M. Strømme and L. Nyholm, J. Power Sources, 2014, 272, 468–475 CrossRef CAS.
- X. Wei, S. Wan, X. Jiang, Z. Wang and S. Gao, ACS Appl. Mater. Interfaces, 2015, 7, 22238–22245 CAS.
- X. Wu, X. Hong, Z. Luo, K. S. Hui, H. Chen, J. Wu, K. N. Hui, L. Li, J. Nan and Q. Zhang, Electrochim. Acta, 2013, 89, 40–406 CrossRef.
- X. Wang, X. Fan, G. Li, M. Li, X. Xiao, A. Yu and Z. Chen, Carbon, 2015, 93, 258–265 CrossRef CAS.
- M. Biswal, A. Banerjee, M. Deoab and S. Ogale, Energy Environ. Sci., 2013, 6, 1249–1259 CAS.
- X. Deng, B. Zhao, L. Zhu and Z. Shao, Carbon, 2015, 93, 48–58 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta07826e |
| This journal is © The Royal Society of Chemistry 2017 |