
AB3-loaded and tumor-targeted unimolecular micelles for medullary thyroid cancer treatment†
Renata
Jaskula-Sztul‡
a,
Guojun
Chen‡
bc,
Ajitha
Dammalapati
d,
April
Harrison
d,
Weiping
Tang
e,
Shaoqin
Gong
*bcf and
Herbert
Chen
*a
aDepartment of Surgery, School of Medicine University of Alabama at Birmingham, Birmingham, AL 35233, USA. E-mail: herbchen@uab.edu
bDepartment of Materials Science and Engineering, University of Wisconsin–Madison, Madison, WI 53715, USA. E-mail: shaoqingong@wisc.edu
cWisconsin Institute for Discovery, University of Wisconsin–Madison, Madison, WI 53715, USA
dDepartment of Surgery, School of Medicine and Public Health, University of Wisconsin–Madison, Madison, WI 53706, USA
eSchool of Pharmacy, University of Wisconsin–Madison, Madison, WI 53706, USA
fDepartment of Biomedical Engineering, University of Wisconsin–Madison, Madison, WI 53706, USA
First published on 2nd December 2016
Abstract
Medullary thyroid cancer (MTC) is often resistant to standard therapies, emphasizing the need for the development of other treatments. A new histone deacetylase inhibitor, AB3, can effectively inhibit MTC cell proliferation in vitro. However, its poor aqueous solubility and stability, fast clearance, and lack of tumor targeting ability limit its in vivo application. Therefore, multifunctional unimolecular micelles were developed for targeted delivery of AB3 for MTC therapy. The unimolecular micelles exhibited a spherical core–shell structure, uniform size distribution, and excellent stability. AB3 was encapsulated into the hydrophobic core of the unimolecular micelles, thus significantly enhancing its aqueous solubility and stability. KE108, a somatostatin analog possessing high affinity to all five subtypes of SSTR, was used as an MTC-targeting ligand. In vitro cellular uptake analyses demonstrated that the KE108 exhibited superior targeting ability in MTC cells compared to octreotide, the first clinically used somatostatin analog. Moreover, the AB3-loaded and KE108-conjugated unimolecular micelles exhibited the best efficacy in suppressing MTC cell growth and tumor marker expression in vitro. Furthermore, AB3-loaded, KE108-conjugated micelles demonstrated the best anticancer efficacy in vivo without any apparent systemic toxicity, thereby offering a promising approach for targeted MTC therapy.
1. Introduction
Medullary thyroid cancer (MTC) is a neuroendocrine tumor that arises from the calcitonin-producing C-cells of the thyroid. While MTC accounts for only 5% of all thyroid cancers, it causes approximately 14% of all deaths from thyroid cancers.1,2 Surgery at a relatively early stage remains the only potential cure for MTC.3,4 Despite complete surgical resection, more than 50% of patients with MTC will have persistent or recurrent disease.5 More importantly, MTC liver metastases are almost always small and widely distributed throughout the liver, precluding curative surgical resection.6 Unfortunately, there are no effective treatments for patients with MTC liver metastases and/or widely metastatic diseases.7 Thus, there is an urgent need for new treatment modalities for metastatic diseases.Our previous studies showed that the inhibition of histone deacetylases (HDACs) triggered activation of Notch signaling and induced cell cycle arrest and apoptosis, leading to MTC cell growth suppression.7 HDAC inhibitors have emerged as a new type of anticancer agent. Up to now, there have been three FDA-approved HDAC inhibitors, including SAHA,8 PXD-101,9 and natural product FK228.10 AB3, a new synthetic HDAC inhibitor identified by Tang et al.,11 was easily prepared by condensing commercially available para-methoxybenzaldehyde with a known bifunctional reagent, which has a six methylene carbon tether between a hydrazide and a hydroxamic acid. We have demonstrated that AB3 can effectively inhibit MTC proliferation and suppress the expression of tumor markers, thereby making it a promising candidate as an anticancer drug for MTC treatment.7 However, AB3, like most of the anticancer drugs, has poor aqueous solubility and stability, experiences fast in vivo clearance, and lacks in vivo tumor-targeting capabilities when administered systemically, thus limiting its application.
Drug nanocarriers have been wildly explored to overcome the limitations of conventional drugs, including poor solubility in aqueous media, inadequate stability, and lack of selectivity for tissues/cells.12–14 Particularly, drug nanocarriers are attractive for targeted cancer therapy due to their passive (via the enhanced permeability and retention (EPR) effect) and active (via cell-specific ligand conjugation) tumor-targeting capabilities.13–15 Among the various drug nanocarriers, polymer micelles, exhibiting a core–shell structure, have been extensively studied. Hydrophobic payloads can be readily encapsulated into the hydrophobic core of the micelles through hydrophobic interactions as well as hydrogen bonding, while the hydrophilic shell provides micelles with excellent aqueous dispersibility.16–18 Typically, polymer micelles are formed by the self-assembly of multiple linear amphiphilic block copolymers. However, one major concern with these conventional polymer micelles is their stability due to the dynamic nature of the self-assembly process. The in vivo stability of these self-assembled micelles can be affected by the concentration of the amphiphilic block copolymer molecules (critical micelle concentration), pH, ionic strength, temperature, or their interaction with serum proteins.19–21 Premature rupture of these drug nanocarriers during circulation can cause a burst release of payloads into the bloodstream, which can cause potential systemic toxicity and surrender their tumor-targeting ability, thereby limiting their in vivo applications. Unimolecular micelles—formed by individual multi-arm star amphiphilic block copolymers—have been investigated to overcome this problem.22–27 Because of their covalent nature and unique chemical structure, properly engineered unimolecular micelles can possess excellent in vivo stability. Moreover, due to their excellent chemical versatility, these unique unimolecular micelles have been successfully functionalized with different targeting ligands (e.g. small molecules, peptides, antibodies, and aptamers) and imaging probes (e.g., dyes, radioisotopes, etc.).27–30
The majority of MTCs overexpress five subtypes of somatostatin receptors (SSTR 1–5).24,31–33 KE108 peptide is a somatostatin analog displaying strong binding affinities to all five subtypes of SSTRs. Our recent report demonstrated, for the first time, that KE108, a true pansomatostatin synthetic nonapeptide, can be used as an efficient SSTR-targeting ligand for targeted carcinoid cancer therapy.34 In this study, we aimed to develop multifunctional unimolecular micelles for targeted MTC treatment (Fig. 1(A)). KE108 or octreotide (OCT) as an MTC-targeting ligand was conjugated to the unimolecular micelles. OCT peptide is the first somatostatin analog used in clinic.35 However, it only displays a high binding affinity to SSTR2, and a moderate affinity to SSTR5, but has very little affinities to the other three SSTR subtypes (i.e., SSTR1, SSTR3, and SSTR4).36–38 Our studies demonstrate that, in comparison to OCT, KE108 was much more effective in enhancing the cellular uptake of micelles in MTC cells. AB3 was encapsulated into the hydrophobic core of the unimolecular micelles. AB3-loaded and KE108-conjugated unimolecular micelles were much more effective at suppressing MTC cell growth and tumor marker production. Moreover, in vivo studies demonstrated that the AB3-loaded and KE108-conjugated micelles exhibited the best antitumor efficacy, thus offering a promising approach for targeted MTC therapy.
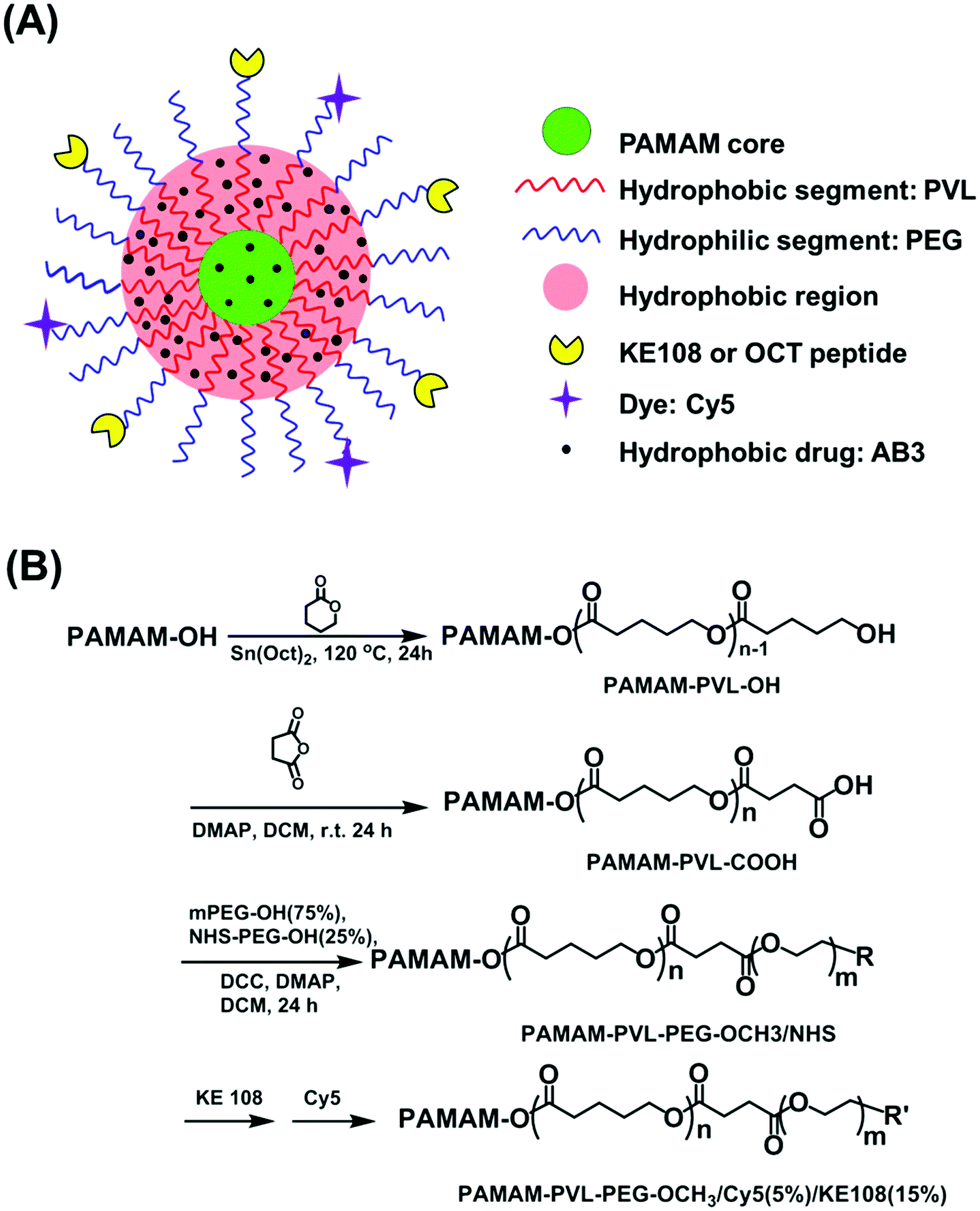 | ||
| Fig. 1 (A) A schematic illustration of a multifunctional unimolecular micelle formed by multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–OCH3/Cy5/KE108 for targeted delivery of AB3 for MTC treatment. (B) A synthetic scheme of the multi-arm star amphiphilic block copolymers PAMAM–PVL–PEG–OCH3/Cy5/KE108 or PAMAM–PVL–PEG–OCH3/Cy5/OCT. | ||
2. Methods
2.1 Synthesis of PAMAM–PVL–PEG–OCH3/Cy5/KE108 and PAMAM–PVL–PEG–OCH3/Cy5/OCT
PAMAM–PVL–PEG–OCH3/Cy5/KE108 and PAMAM–PVL–OCH3/Cy5/OCT polymers were synthesized following the scheme shown in Fig. 1(B). Detailed experimental procedures can be found in the ESI.† All of the intermediates and final polymer products were fully characterized by 1H NMR (Fig. S1 and S2 in the ESI†), Fourier transform infrared (FT-IR) spectroscopy (Fig. S1, ESI†), and gel permeation chromatography (GPC, Table S1, ESI†).2.2 Preparation of AB3-loaded unimolecular micelles
To prepare the AB3-loaded targeted (i.e., KE108-conjugated) unimolecular micelles, AB3 (5 mg) and PAMAM–PVL–PEG–OCH3/KE108 (20 mg) were dissolved in acetonitrile (3 mL). Thereafter, DI water (9 mL) was added dropwise into the solution. The resulting mixture was stirred for 4 h at room temperature before acetonitrile was fully removed by rotary evaporation. The final AB3-loaded targeted unimolecular micelles were obtained after freeze-drying. The AB3-loaded non-targeted unimolecular micelles were prepared following a similar method using the PAMAM–PVL–PEG–OCH3 polymer instead. The AB3 drug loading level, defined as the weight percentage of AB3 in the AB3-loaded unimolecular micelles, was measured by UV-vis spectroscopy at 293 nm.2.3 In vitro release of AB3 from AB3-loaded unimolecular micelles
The release profiles of AB3 from AB3-loaded micelles were studied in a glass apparatus at 37 °C in a release medium at pH 5.3 or 7.4.39 Briefly, AB3-loaded unimolecular micelles (5 mg) dispersed in a medium (5 mL) were enclosed in a dialysis bag with a molecular weight cut-off of 2 kDa. The dialysis bag was immersed in 50 mL of the release medium and kept at 37 °C under a horizontal laboratory shaker at 100 rpm (Thermo Scientific MaxQ Shaker, USA). At specific time points, 3 mL of release media were collected and replaced by the same volume of fresh media. The amount of released AB3 was analyzed by a UV-Vis-NIR spectrophotometer at 293 nm.2.4 Quantitative real-time PCR of SSTR 1–5 receptors in TT cells
The mRNA expression levels of SSTRs in TT cells were measured by qRT-PCR. RNA was isolated and transcribed into cDNA following a protocol published previously.40 A qRT-PCR reaction was performed using a CFX Thermal Cycler and SsoFast EvaGreen labeling system (Bio-Rad) at conditions described earlier41 on three biological replicates. The SSTR 1–5 primer sequences were as follows: SSTR1 forward, 5′-ATGGTGGCCCTCAAGGCCGG-3′ and reverse, 5′-CGCGGTGGCGTAATAGTCAA-3′; SSTR2 forward, 5′-CCAACACCTCAAACCAGAC-3′ and reverse, 5′-CATAGCCAAGAAAGGCAGAC-3′; SSTR3 forward, 5′-TCATCTGCCTCTGCTACCTG-3′ and reverse, 5′-GAGCCCAAAGAAGGCAGGCT-3′; SSTR4 forward, 5′-ATCTTCGCAGACACCAGACC-3′ and reverse, 5′-ATCAAGGCTGGTCACGACGA-3′; SSTR5 forward, 5′-CCGTCTTCATCATCTAACACGG-3′ and reverse, 5′-GGCCAGGTTGACGATGTTGA-3′. The s27 primer sequences were as follows: forward, 5′-TCTTTAGCCATGCACAAACG-3′ and reverse, 5′-TTTCAGTGCTGCTTCCTCCT-3′. The mRNA expression levels of the SSTRs in MZ-CRC-1 cells were measured following a similar procedure.34 Both TT and MZ-CRC-1 are human MTC cell lines.The cycle numbers obtained at the log-linear phase of the reactions for target genes were normalized to housekeeping gene s27 from the same sample measured concurrently. Finally, the expression ratios were calculated using the comparative cycle threshold (ΔCt) method and presented as the average ± standard error of the mean (SEM).
2.5 Cellular uptake analyses
The cellular uptake behaviors of the micelles in a human MTC cell line (i.e., TT) were analyzed using both flow cytometry and confocal laser scanning microscopy (CLSM). Multi-arm star amphiphilic block copolymers PAMAM–PVL–PEG–OCH3/Cy5/KE108, PAMAM–PVL–PEG–OCH3/Cy5/OCT, and PAMAM–PVL–PEG–OCH3/Cy5 were dissolved in the cell culture medium directly to form the KE108-conjugated, OCT-conjugated, and non-targeted micelles, respectively. For flow cytometry analyses, cells were seeded in 6-well plates (3 × 105 cells per mL) and incubated overnight. Cells were treated with KE108-conjugated, OCT-conjugated, or non-targeted micelles (100 μg mL−1). The culture medium was used as a blank control. After 2 h of incubation, the cells were washed with PBS and lifted using a non-enzymatic cell dissociation solution. The cellular uptake of the micelles based on Cy5 intensity was analyzed using a FACS Calibur four-color analysis cytometer (Becton Dickinson, San Jose, CA) and FlowJo analysis software (Tree Star, Inc., Ashland, OR). A minimal of 1 × 105 cells were analyzed for each sample. The cellular uptake of the micelles in WI-38 (pulmonary fibroblasts), a cell line that does not overexpress SSTRs, was studied following similar procedures using flow cytometry and was used as a negative control. For the CLSM studies, cells were seeded (1 × 105 cells per mL) onto 96-well high-optical-quality plates and incubated overnight. Cells were treated with either KE108-conjugated (i.e., targeted) or non-targeted micelles (100 μg mL−1). After 2 h of incubation, the cells were washed with PBS, fixed with 1.5% formaldehyde for 15 min, and washed with DI water. Then the cells on the optical plate were mounted with Prolong Gold anti-fade reagent with DAPI (Life Technologies, Carlsbad, CA) and the images were taken under a Nikon A1R-Si high-speed spectral laser scanning confocal inverted microscope (Nikon, Melville, NY) with NIS-Elements BR Software.2.6 Cell proliferation assays
The cytotoxicity of free AB3 and AB3-loaded micelles with or without KE108-targeting ligands on two human MTC cell lines (i.e., TT and MZ-CRC-1) was assessed using MTT assays. Cells were plated on 96-well plates and incubated overnight. Cells were treated with pure medium, free AB3, AB3-loaded targeted and non-targeted micelles, and empty targeted and non-targeted micelles (AB3-free micelles). The equivalent AB3 concentration for all AB3-loaded micelles was 3 μM.42 The cells were then incubated for 24, 48, and 96 h, and the MTT assay was performed at each time point using a standard protocol.26 The plates were then measured at 570 nm using a spectrophotometer (Quant, Bio-Tek Instruments, Winooski, VT). The percent of cell viability relative to the control (pure medium) was calculated.2.7 Tumor marker expression assays
MTC marker expressions, including achaete–scute complex-like 1 (ASCL-1) and chromogranin A (CgA), were assessed using Western blot analyses. Cells were seeded in 100 mm culture plates and incubated overnight. Cells were then treated with pure medium, free AB3 (3 μM), or AB3-loaded, KE108-conjugated targeted and non-targeted micelles. The equivalent AB3 concentration for all AB3-loaded micelles was 3 μM. After incubation for 48 h, proteins were harvested according to a previously described protocol.31 Protein concentrations were determined by a bicinchoninic acid assay (Thermo Scientific, Waltham, MA). The protein lysates (20 μg) were denatured and resolved on 10% SDS-PAGE gels. Protein bands in the gel were then transferred to a nitrocellulose membrane and incubated with the appropriate primary antibody with dilutions as follows: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2000 for ASCL-1, 1:1000 for CgA, and 1:1000 for β-actin. Membranes were incubated overnight at 4 °C and then washed with buffer (1 × PBS, 0.05% Tween 20). Horseradish peroxidase-conjugated goat, anti-rabbit, or anti-mouse secondary antibodies were used for secondary incubation. Proteins were then visualized with chemiluminescent substrates Immunstar (CgA and β-actin) or Femto (ASCL-1). The quantitative analyses of the protein expression of ASCL1 and CgA were done using ImageJ bundled with Java 1.8.0_77 (http://imagej.nih.gov/ij/).
:2000 for ASCL-1, 1:1000 for CgA, and 1:1000 for β-actin. Membranes were incubated overnight at 4 °C and then washed with buffer (1 × PBS, 0.05% Tween 20). Horseradish peroxidase-conjugated goat, anti-rabbit, or anti-mouse secondary antibodies were used for secondary incubation. Proteins were then visualized with chemiluminescent substrates Immunstar (CgA and β-actin) or Femto (ASCL-1). The quantitative analyses of the protein expression of ASCL1 and CgA were done using ImageJ bundled with Java 1.8.0_77 (http://imagej.nih.gov/ij/).
2.8 In vivo anticancer study
Four-week-old male athymic nude mice were obtained from Charles River (Wilmington, Maryland, USA). A subcutaneous MTC tumor xenograft model was established by subcutaneously implanting 1 × 107 TT cells in 200 μL of Hanks balanced salt solution (Mediatech, Inc., Manassas, Virginia, USA) into the left flank of each mouse. Ten days after inoculation, TT-tumor-bearing mice were randomly divided into six treatment groups (6 mice per group). The six groups of mice were treated with saline (control), free AB3, AB3-loaded non-targeted micelles, AB3-loaded KE108-conjugated targeted micelles, empty non-targeted micelles, and empty KE108-conjugated targeted micelles at the equivalent AB3 dose of 20 mg kg−1 BW. Each treatment group received five intravenous injections every five days. Tumor volumes were measured by a caliper and then calculated by the modified ellipsoidal formula: Tumor volume = (length × width2)/2. At the end of the experiment, mice were sacrificed and postmortem examination of the lungs, liver, heart, and spleen were performed to confirm that there was no evidence of metastases or tumor growth outside of the inoculation site. All major organs, including the liver, brain, heart, and leg muscles, of the mice treated with AB3-loaded targeted micelles were collected and hematoxylin and eosin (H&E)-stained sections were prepared for pathological assessment. All experimental procedures were carried out in compliance with our animal care protocol (M01474-0-01-09), which was approved by the Animal Care and Use Committee of the University of Wisconsin–Madison.2.9 Statistical analysis
Weights and volumes were compared between groups using an analysis of variance (ANOVA) with pair-wise comparisons performed using Fisher's protected least significant difference tests. Data were log-transformed prior to analyses to better meet the assumptions of the ANOVA. P-Values less than 0.05 were considered to be statistically significant.3. Results
The synthesis of multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–OCH3/Cy5/KE108 or PAMAM–PVL–PEG–OCH3/Cy5/OCT was carried out following Fig. 1(B). First, PAMAM–PVL–OH was prepared by the ring-opening polymerization of valerolactone (VL) using PAMAM–OH (G4, 64 –OH terminal groups) as a macroinitiator. The terminal –OH groups of PAMAM–PVL–OH were then converted into –COOH by reacting with succinic anhydride to form PAMAM–PVL–COOH. Thereafter, two types of PEG (namely, OH–PEG–OCH3 and OH–PEG–NHS) were conjugated onto the PAMAM–PVL–COOH via an esterification reaction. The NHS functional groups were used subsequently to conjugate the targeting ligand (KE108 or OCT) and Cy5 dye. All of the intermediates and final products were fully characterized by 1H NMR, FT-IR and GPC analyses as shown in ESI.†The resulting multi-arm star amphiphilic block copolymers formed stable unimolecular micelles in an aqueous solution. The hydrophobic core of the micelles allowed for the encapsulation of the hydrophobic drug AB3 through hydrophobic interactions and hydrogen bonding, thereby enhancing the water solubility and stability of AB3. Peptides KE108 or OCT on the surface of the unimolecular micelles provided the micelles with specific SSTR targeting abilities. The effects of KE108 and OCT targeting ligands on the cellular uptake of the micelles were studied and will be discussed later. The Cy5 dye conjugated onto the micelles allowed for the visualization of the micelles. The average hydrodynamic size of the unimolecular micelles measured by dynamic light scattering (DLS, Fig. S3(A), ESI†) was 58 nm (PDI = 0.114). The unimolecular micelles showed a spherical morphology with a clear core–shell structure under transmission electron microscopy (TEM, Fig. S3(B), ESI†). The drug-loading level and drug-loading efficiency of AB3-loaded unimolecular micelles were 15.1% and 76.6%, respectively.
The in vitro drug release profiles are shown in Fig. 2. A clear pH-dependent drug release behavior was observed. At pH = 7.4, only 15% of AB3 was released after 5 days. In contrast, at pH = 5.3, a relatively rapid drug release rate was observed with 59% of AB3 being released after 5 days. The pH-dependent drug release behavior could be possibly due to the fact that the hydrophobic core of the micelle, within which AB3 was encapsulated, was made of PVL, a member of the polyester family, which can be hydrolyzed faster under acidic conditions, thereby leading to a faster drug release.43–46 Swelling of the PAMAM in an acidic condition due to the protonation of the 62 tertiary amine groups in an acidic condition could be another possible reason.47–49
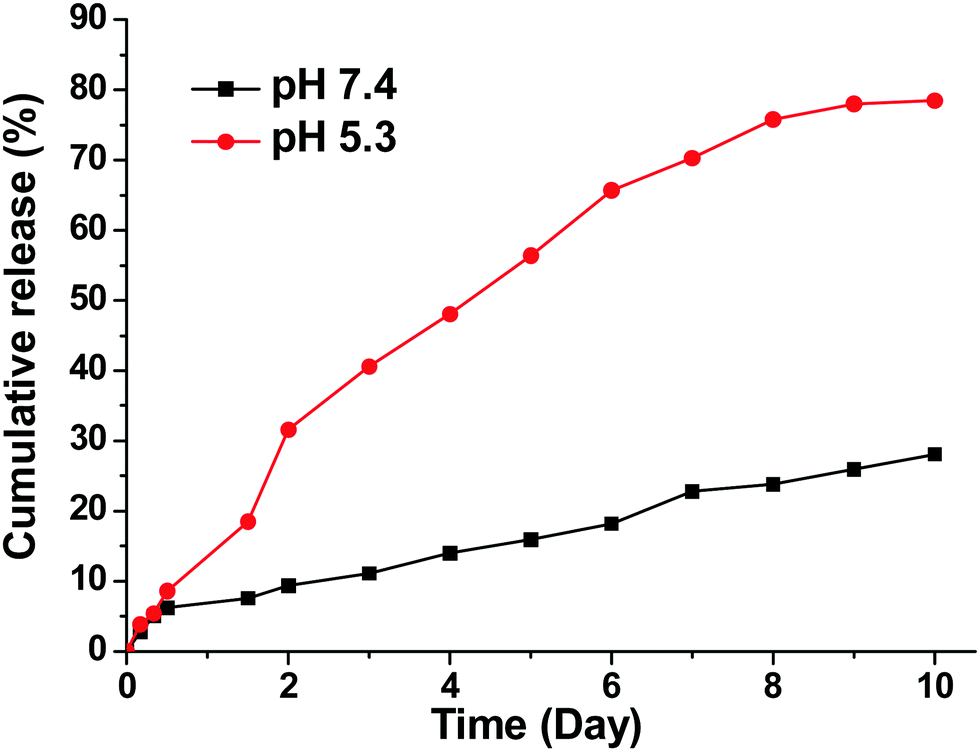 | ||
| Fig. 2 In vitro release of AB3 from AB3-loaded unimolecular micelles. | ||
The efficacy of KE108 as an SSTR-targeting ligand for MTC cells was evaluated first in vitro. As shown in Fig. 3(A), two MTC cell lines (TT and MZ-CRC-1) have high expression of all five SSTRs except for SSTR4. To test whether KE108 has a superior targeting compared to OCT, TT cells were treated with either non-targeted (without any targeting ligand) unimolecular micelles or unimolecular micelles conjugated with either KE108 or OCT. Based on the fluorescent intensity of the Cy5 dye conjugated on the micelles, the cellular uptake of the micelles was measured by flow cytometry. As shown in Fig. 3(B), OCT—which has a high affinity for SSTR2, a moderate affinity for SSTR5, and very little affinity to SSTR1, SSTR3, and SSTR4—increased the cellular uptake of the micelles 4-fold compared with non-targeted micelles. The KE108 peptide, on the other hand, has a high affinity for all 5 subtypes of SSTRs (SSTR 1–5) and thus was expected to display much better targeting efficacy than OCT. Indeed, the intracellular uptake of the KE108-conjugated micelles was 50 and 15 times higher than that of the non-targeted micelles and OCT-conjugated micelles, respectively. To further demonstrate the specific SSTR targeting ability of KE108, the cellular uptake behavior of the unimolecular micelles in a WI-38 cell line (pulmonary fibroblasts), that does not overexpress SSTRs, was also studied and no differences were observed in the level of cellular uptake between the KE108-conjugated, OCT-conjugated, or non-targeted micelles (Fig. 3(C)). These findings indicate that MTC cells overexpressing SSTRs on their surface exhibited an increased intracellular uptake of KE108-conjugated micelles through the SSTR-mediated endocytosis process. Furthermore, KE108 peptides exhibited a much more superior SSTR-targeting ability than OCT due to its high affinity for all five SSTR subtypes (SSTR 1–5).
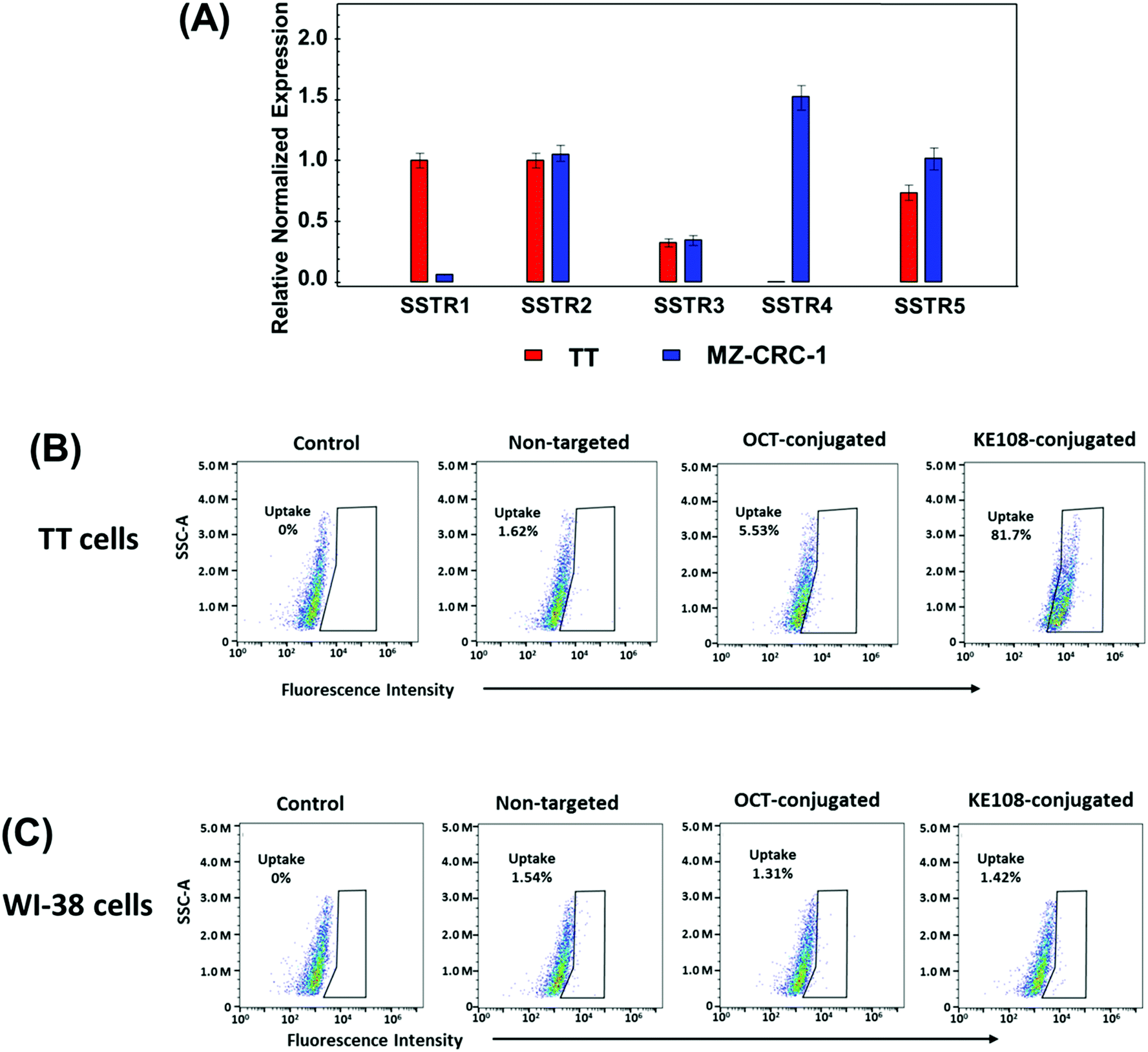 | ||
| Fig. 3 (A) The expression levels of the five subtypes of SSTR in TT and MZ-CRC-1 cells. Flow cytometry analysis of the (B) TT and (C) WI-38 cells treated with pure medium (control), non-targeted, OCT-conjugated or KE108-conjugated unimolecular micelles (100 μg mL−1) for 2 h at 37 °C. | ||
The targeting ability of KE108 was further confirmed by CLSM analyses (Fig. 4). As expected, targeted (KE108-conjugated) unimolecular micelles exhibited significantly higher cellular uptake than non-targeted micelles, manifested by much stronger red Cy5 fluorescence. Moreover, both non-targeted and targeted micelles accumulated preferentially in the cytoplasm of the cells because nanoparticles were taken up via an endocytosis process.23 Overall, the greater cellular uptake of the targeted micelles compared to the non-targeted ones revealed that the KE108-targeting ligand was effective at promoting SSTR-mediated endocytosis. In contrast, non-targeted micelles were taken up through non-specific endocytosis.
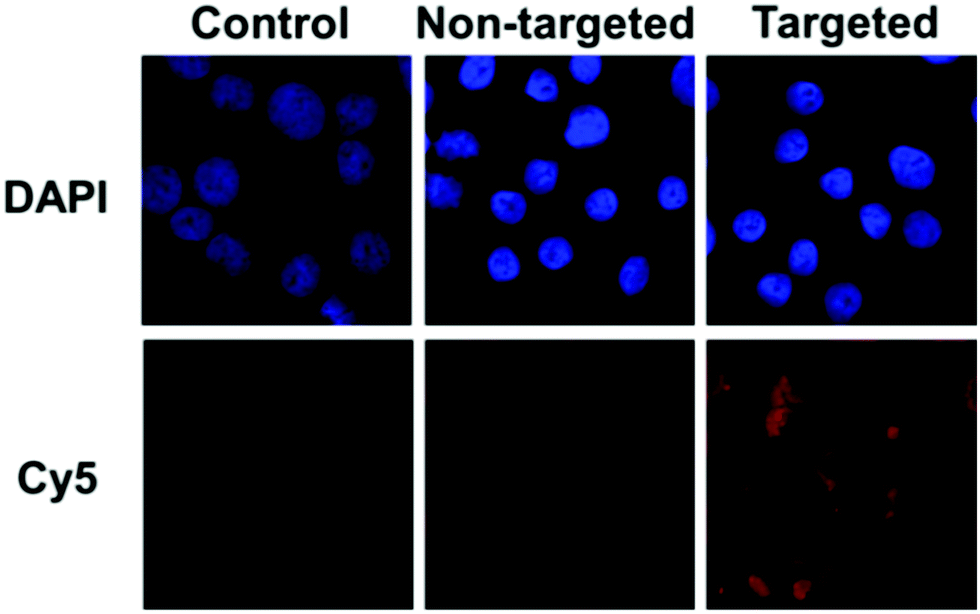 | ||
| Fig. 4 CLSM images of TT cells treated with pure medium (control), or non-targeted or targeted (KE108-conjugated) unimolecular micelles (100 μg mL−1) for 2 h at 37 °C. | ||
To further demonstrate the superior targeting ability of KE108, the anti-proliferative efficacies of AB3-loaded micelles were evaluated in MTC cells (i.e., TT and MZ-CRC-1). As shown in Fig. 5(A) and (B), at each time point (24, 48, or 96 h), AB3-loaded targeted (i.e., KE108-conjugated) micelles (i.e., AB3-T) exhibited the best efficacy of suppressing proliferation in both TT and MZ-CRC-1 cells in comparison with other treatment groups. The enhanced cytotoxicity of AB3-T over non-targeted ones (i.e., AB3-NT) was attributed to their enhanced cellular uptake as discussed above. Moreover, neither the empty non-targeted (i.e., Empty NT) micelles nor targeted (i.e., Empty T) ones exhibited any apparent cytotoxicity. Notably, it was also observed that cells treated with AB3-T were more sensitive than free AB3. To further investigate the effect of KE108 conjugation on the anticancer efficacy of AB3-loaded targeted micelles, Western blot analyses on MTC marker expression in MTC cells were carried out. ASCL-1 and CgA are well established MTC markers, and high levels of ASCL-1 and CgA are linked to the poor prognosis of MTC.50,51 As shown in Fig. 5(C) and (D), the AB3-T were much more effective in reducing the expression of both ASCL-1 and CgA than AB3-NT as well as free AB3, which is in good agreement with the MTT results. The quantitative analyses of Western blot data are shown in the Fig. S4 (ESI†).
 | ||
| Fig. 5 MTT assays of the (A) TT and (B) MZ-CRC-1 cells (AB3 concentration: 3 μM) treated with cell culture medium (control), free AB3, and AB3-loaded non-targeted (AB3-NT) and AB3-loaded targeted (AB3-T) micelles. AB3-loaded targeted micelles (AB3-T) were much more effective at suppressing tumor cell proliferation than other treatment groups. All values are presented as a mean ± SD (n = 4). *: P < 0.05; **: P < 0.01; ***: P < 0.005; NS: not significant. Western blot analyses for ASCL-1 and CgA of (C) TT and (D) MZ-CRC-1 cells (AB3 concentration: 3 μM) treated with Control, free AB3, and AB3-NT and AB3-T micelles. AB3-T exhibited the highest efficacy at reducing the MTC markers (i.e., ASCL-1 and CgA). | ||
The antitumor study of the AB3-loaded micelles in vivo was then performed. TT-tumor-bearing mice were intravenously treated with saline (control), pure AB3, empty non-targeted micelles (Empty NT), empty targeted (i.e., KE108-conjugated) micelles (Empty T), AB3-loaded non-targeted micelles (AB3-NT), or AB3-loaded targeted micelles (AB3-T) at an equivalent AB3 dosage of 20 mg kg−1 BW every five days for a total of five treatments. As shown in Fig. 6(A), neither Empty NT nor Empty T induced any antitumor effect, while all drug treatment groups significantly decreased the tumor burden as compared to the saline control group. However, AB3-T induced the highest antitumor efficacy among all treatment groups. No noticeable changes in body weight or survival were observed in the AB3-NT or AB3-T groups. In contrast, while free AB3 also exhibited inhibition of tumor growth when compared to the control, its severe toxicity in mice was demonstrated by decreased survival (3 out of the 6 mice died during the experiment). Pathological assessment of H&E-stained sections of different organs (i.e., brain, heart, liver and leg muscles) of mice treated with AB-T micelles (Fig. 6(B)) did not indicate any signs of acute or chronic inflammation, or apoptotic or necrotic regions, suggesting that the AB3-loaded targeted unimolecular micelle delivery system was safe for organs other than the NE cancerous tissues, and that the potential off-target uptake of micelles by normal tissues did not cause any detectable damage. These findings may support the development of AB3-loaded, KE108-conjugated tumor-targeting unimolecular micelles in the treatment and palliation of patients with MTC cancers. This approach can significantly enhance the therapeutic outcome of cancer therapy while minimizing undesirable side-effects.
 | ||
| Fig. 6 Therapeutic efficacy of free AB3 and AB3-loaded micelles in subcutaneous TT tumor xenografts. Each mouse received five intravenous injections spaced 5 days apart indicated by the arrows. (A) In vivo anticancer efficacy of different AB3 formulations in TT-tumor xenografts. All values are presented as a mean ± SD (n = 6). *: P < 0.05; **: P < 0.01; NS: not significant. #: three mice died in the AB3 group due to its severe toxicity. (B) Representative H&E-stained sections of brain, heart, liver, and leg muscles of a mouse treated with AB3-T. | ||
4. Discussion
MTC, derived from the parafollicular cells (C-cells) of the thyroid, is an aggressive subtype of thyroid cancers.1,2 Like most thyroid cancers, surgical resection is the predominant treatment modality and can be curative in selected patients. However, unlike well-differentiated thyroid cancers which can be treated with surgery and/or radioactive iodine even when metastases develop, patients with MTC metastases have limited options. Hence, it is of great importance to developing other effective treatment modalities. We have recently reported that AB3, a new HDAC inhibitor, can effectively inhibit MTC proliferation and suppress the expression of tumor markers.7 In order to further increase its therapeutic indexes and minimize its off-target toxicities, in this study, we have successfully developed a multifunctional unimolecular micelle for targeted delivery of AB3 for MTC treatment. Unimolecular micelle was formed by individual multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–OCH3/KE108/Cy5. All of the components of the unimolecular micelles were covalently linked, thereby conferring outstanding stability. Moreover, the average hydrodynamic size of the micelles was 58 nm, which is desirable for tumor-targeted drug delivery.52 AB3 was physically loaded into the hydrophobic core of the unimolecular micelles via hydrophobic interaction as well as hydrogen bonding. The in vitro drug release study showed that AB3 was released in a pH-dependent manner. This pH-dependent release behavior is of particular interest for drug delivery systems for cancer therapies. It is expected that AB3 is sufficiently stable in micelles with minimal drug leakage during blood circulation. After the AB3-loaded micelles are taken up by cancer cells through endocytosis, where acidic endosomes or lysosomes are involved, AB3 will be released relatively quickly to effectively kill the cancer cells.Somatostatin analogs, KE108 and OCT, were used as the targeting ligand. Through quantitative flow cytometry analyses, we demonstrated that both KE108-conjugated and OCT-conjugated micelles were taken up to a greater extent than non-targeted micelles in MTC cells. Meanwhile, the cellular uptake of the KE108-conjugated micelles exhibited superior targeting ability compared to OCT-conjugated micelles, which is due to the fact that KE108 peptide possesses high affinity for all five subtypes of somatostatin receptors (SSTR 1–5), while OCT, the clinically used somatostatin analog, only displays high affinity to SSTR2. It is worth mentioning that other than MTCs, these KE108-conjugated micelles can also be potentially used as targeted drug nanocarriers for several other types of cancers that overexpress SSTRs, including melanomas53 and breast cancers.54
The KE108-functionalized AB3-loaded unimolecular micelles were able to suppress cell growth and tumor marker expression in MTC cells to a significantly greater extent compared to non-targeted ones, further demonstrating the targeting ability of KE108. It is noted that the KE108 functionalized AB3-loaded unimolecular micelles also exhibited better anticancer effect than free AB3 in vitro. These differences may be attributable to two possible reasons. First, it may be due to the different mechanisms of cellular uptake for free drug versus drug encapsulated in targeted micelles. The cellular uptake of free AB3 occurs through a passive diffusion mechanism and the drug may be expelled out through the P-glycoprotein (P-gp) pump,55 while AB3-T were taken up by receptor-mediated endocytosis, which may overcome the P-gp pumping action.56 Second, it might be due to the poor solubility and stability of free AB3. The stability and solubility of AB3 loaded inside of the targeted micelles can be greatly enhanced. In addition to highlighting the ability of KE108-conjugated micelles to effectively target and deliver AB3 to MTC cells in vitro, we studied the in vivo anti-tumor efficacy. AB3-loaded targeted micelles demonstrated a much higher anti-tumor effect statistically compared to other treatments groups. H&E analyses on the major organs did not show any indication of inflammation, or apoptotic or necrotic regions. However, the pure AB3 treatment group showed a low survival rate (3 out of the 6 mice died).
Currently, the majority of drug discovery and development efforts for MTCs have been limited to somatostatin analogs and repurposing various existing anticancer drugs. Both approaches have significant shortcomings. First, somatostatin analogs lack adequate anti-proliferative efficacy against MTCs and patients develop resistance to the therapy over time. Second, systemic non-targeted delivery of chemotherapeutics can lead to profound side effects. Our findings address these major issues by demonstrating that (1) SSTRs can serve as targets for patients with MTCs, (2) KE108 peptide can be used as a superior targeting ligand for SSTRs in comparison with other common somatostatin analogs, such as OCT, and (3) unimolecular micelles conjugated with KE108 can serve as drug delivery vehicles for any hydrophobic anticancer drugs including therapeutic HDAC inhibitors such as AB3, which will reduce systemic side-effects.
5. Conclusions
The multifunctional unimolecular micelles formed by multi-arm star amphiphilic block copolymer PAMAM–PVL–PEG–OCH3/KE108/Cy5 were developed for targeted MTC therapy. KE108 peptide, displaying high affinity to all five subtypes of SSTRs (SSTR 1–5), was employed as a novel targeting moiety for MTCs overexpressing SSTRs. The cellular uptake of the targeted unimolecular micelles in the MTC cells was dramatically enhanced compared to that of either OCT-conjugated ones or non-targeted ones. Moreover, AB3-loaded and KE108-conjugated unimolecular micelles were much more effective at suppressing MTC cell growth and hormone production than other types of micelles or free AB3. Finally, in vivo studies demonstrated that the AB3-loaded and KE108-conjugated micelles exhibited the best antitumor efficacy. Thus, these AB3-loaded and KE108-conjugated micelles offer a promising approach for targeted MTC therapy.Acknowledgements
This project was financially supported by grants from the NIH (R01 CA121115 to H. Chen, 1K25CA166178 and R21CA196653 to S. Gong), the Aly Wolff Memorial Foundation (S. Gong and H. Chen), the American Cancer Society (MEN2 Thyroid Cancer Professorship 120319-RPM-11-080-01-TBG to H. Chen and Research Scholar Award RSGM TBE–121413 to H. Chen), the Caring for Carcinoids Foundation, and the AACR. The authors would like to thank Dr Ricardo Lloyd for the pathological assessment of the mice tissues and Dr Glen Leverson for the statistical analysis.References
- M. Schlumberger, F. Carlomagno, E. Baudin, J. M. Bidart and M. Santoro, Nat. Clin. Pract. Endocrinol. Metab., 2008, 4, 22–32 CrossRef CAS PubMed.
- M. Papotti, U. Kumar, M. Volante, C. Pecchioni and Y. C. Patel, Clin. Endocrinol., 2001, 54, 641–649 CrossRef CAS PubMed.
- D. Y. Greenblatt, A. M. Vaccaro, R. Jaskula-Sztul, L. Ning, M. Haymart, M. Kunnimalaiyaan and H. Chen, Oncologist, 2007, 12, 942–951 CrossRef CAS PubMed.
- R. S. Sippel, M. Kunnimalaiyaan and H. Chen, Oncologist, 2008, 13, 539–547 CrossRef PubMed.
- H. R. Hahm, M. S. Lee, Y. K. Min, M. K. Lee, K. W. Kim, S. J. Nam, J. H. Yang and J. H. Chung, Thyroid, 2004, 11, 73–80 CrossRef PubMed.
- H. Chen, J. R. Robert, D. W. Ball, D. W. Eisele, S. B. Baylin, R. Udelsman and G. Bulkley, Ann. Surg., 1998, 227, 887–895 CrossRef CAS PubMed.
- R. Jaskula-Sztul, J. Eide, S. Tesfazghi, A. Dammalapati, A. D. Harrison, X. M. Yu, C. Scheinebeck, G. Winston-McPherson, K. R. Kupcho, M. B. Robers, A. K. Hundal, W. Tang and H. Chen, Mol. Cancer Ther., 2015, 14, 499–512 CrossRef CAS PubMed.
- M. Duvic, R. Talpur, X. Ni, C. Zhang, P. Hazarika, C. Kelly, J. H. Chiao, J. F. Reilly, J. L. Ricker, V. M. Richon and S. R. Frankel, Blood, 2007, 109, 31–39 CrossRef CAS PubMed.
- H. Z. Lee, V. E. Kwitkowski, P. L. Del Valle, M. S. Ricci, H. Saber, B. A. Habtemariam, J. Bullock, E. Bloomquist, Y. Li Shen, X. H. Chen, J. Brown, N. Mehrotra, S. Dorff, R. Charlab, R. C. Kane, E. Kaminskas, R. Justice, A. T. Farrell and R. Pazdur, Clin. Cancer Res., 2015, 21, 2666–2670 CrossRef CAS PubMed.
- K. M. VanderMolen, W. McCulloch, C. J. Pearce and N. H. Oberlies, J. Antibiot., 2011, 64, 525–531 CrossRef CAS PubMed.
- W. Tang, T. Luo, E. F. Greenberg, J. E. Bradner and S. L. Schreiber, Bioorg. Med. Chem. Lett., 2011, 21, 2601–2605 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS PubMed.
- J. M. K. D. Pan, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef PubMed.
- A. Z. Wang, R. Langer and O. C. Farokhzad, Annu. Rev. Med., 2012, 63, 185–198 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- V. P. Torchilin, Pharm. Res., 2007, 24, 1–16 CrossRef CAS PubMed.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomedicine, 2010, 6, 714–729 CAS.
- A. Heise, J. L. Hedrick, C. W. Frank and R. D. Miller, J. Am. Chem. Soc., 1999, 121, 8647–8648 CrossRef CAS.
- M. J. Lawrence, Chem. Soc. Rev., 1994, 23, 417–424 RSC.
- S. Kim, Y. Shi, J. Kim, K. Park and J. Cheng, Expert Opin. Drug Delivery, 2010, 7, 49–62 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 3009–3019 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Q. Gong, Biomaterials, 2009, 30, 5757–5766 CrossRef CAS PubMed.
- W. Xu, J. F. Burke, S. Pilla, H. Chen, R. Jaskula-Sztul and S. Gong, Nanoscale, 2013, 5, 9924–9933 RSC.
- J. Guo, H. Hong, G. Chen, S. Shi, T. R. Nayak, C. P. Theuer, T. E. Barnhart, W. Cai and S. Gong, ACS Appl. Mater. Interfaces, 2014, 6, 21769–21779 CAS.
- G. Chen, L. Wang, T. Cordie, C. Vokoun, K. W. Eliceiri and S. Gong, Biomaterials, 2015, 47, 41–50 CrossRef CAS PubMed.
- Y. Xiao, H. Hong, A. Javadi, J. W. Engle, W. Xu, Y. Yang, Y. Zhang, T. E. Barnhart, W. Cai and S. Gong, Biomaterials, 2012, 33, 3071–3082 CrossRef CAS PubMed.
- J. Guo, H. Hong, G. Chen, S. Shi, Q. Zheng, Y. Zhang, C. P. Theuer, T. E. Barnhart, W. Cai and S. Gong, Biomaterials, 2013, 34, 8323–8332 CrossRef CAS PubMed.
- W. Xu, I. A. Siddiqui, M. Nihal, S. Pilla, K. Rosenthal, H. Mukhtar and S. Gong, Biomaterials, 2013, 34, 5244–5253 CrossRef CAS PubMed.
- R. Jaskula-Sztul, W. Xu, G. Chen, A. Harrison, A. Dammalapati, R. Nair, Y. Cheng, S. Gong and H. Chen, Biomaterials, 2016, 91, 1–10 CrossRef CAS PubMed.
- Y. Xiao, R. Jaskula-Sztul, A. Javadi, W. Xu, J. Eide, A. Dammalapati, M. Kunnimalaiyaan, H. Chen and S. Gong, Nanoscale, 2012, 4, 7185–7193 RSC.
- L.-C. Sun and D. H. Coy, Curr. Drug Delivery, 2011, 8, 2–10 CrossRef CAS.
- S. N. Pinchot, K. Holen, R. S. Sippel and H. Chen, Oncologist, 2008, 13, 1255–1269 CrossRef CAS PubMed.
- G. Chen, R. Jaskula–Sztul, A. Harrison, A. Dammalapati, W. Xu, Y. Cheng, H. Chen and S. Gong, Biomaterials, 2016, 97, 22–33 CrossRef CAS PubMed.
- S. W. Lamberts, A.-J. Van der Lely, W. W. de Herder and L. J. Hofland, N. Engl. J. Med., 1996, 334, 246–254 CrossRef CAS PubMed.
- Y. Zou, X. Xiao, Y. Li and T. Zhou, Oncol. Rep., 2009, 21, 379–386 CAS.
- H. Jiang, X. F. Deng, C. M. Duan, C. Chen, J. L. Xiang, Y. Lu and Q. F. Ma, Lymphology, 2011, 44, 21–28 CAS.
- L. Shimon, J. E. Taylor, J. Z. Dong, R. A. Bitonte, S. Kim, B. Morgan, D. H. Coy, M. D. Culler and S. Melmed, J. Clin. Invest., 1997, 99, 789–798 CrossRef PubMed.
- Y. Bao, M. Yin, X. Hu, X. Zhuang, Y. Sun, Y. Guo, S. Tan and Z. Zhang, J. Controlled Release, 2016, 235, 182–194 CrossRef CAS PubMed.
- T. P. Wyche, A. Dammalapati, H. Cho, A. D. Harrison, G. S. Kwon, H. Chen, T. S. Bugni and R. Jaskula-Sztul, Cancer Gene Ther., 2014, 21, 518–525 CrossRef CAS PubMed.
- R. Jaskula-Sztul, P. Pisarnturakit, M. Landowski, H. Chen and M. Kunnimalaiyaan, J. Surg. Res., 2011, 171, 23–27 CrossRef CAS PubMed.
- R. Jaskula-Sztul, J. Eide, S. Tesfazghi, A. Dammalapati, A. D. Harrison, X.-M. Yu, C. Scheinebeck, G. Winston-McPherson, K. R. Kupcho and M. B. Robers, Mol. Cancer Ther., 2015, 14, 499–512 CrossRef CAS PubMed.
- X. Shuai, H. Ai, N. Nasongkla, S. Kim and J. Gao, J. Controlled Release, 2004, 98, 415–426 CrossRef CAS PubMed.
- S. R. Abulateefeh, S. G. Spain, K. J. Thurecht, J. W. Aylott, W. C. Chan, M. C. Garnett and C. Alexander, Biomater. Sci., 2013, 1, 434–442 RSC.
- D. Li, J. X. Ding, Z. H. Tang, H. Sun, X. L. Zhuang, J. Z. Xu and X. S. Chen, Int. J. Nanomed., 2012, 7, 2687 CAS.
- N. Arya and D. S. Katti, Int. J. Nanomed., 2015, 10, 2997 CAS.
- M. A. van Dongen, B. G. Orr and M. M. Banaszak Holl, J. Phys. Chem. B, 2014, 118, 7195–7202 CrossRef CAS PubMed.
- M. H. Kleinman, J. H. Flory, D. A. Tomalia and N. J. Turro, J. Phys. Chem. B, 2000, 104, 11472–11479 CrossRef CAS.
- P. K. Maiti, T. Cagin, S.-T. Lin and W. A. Goddard, Macromolecules, 2005, 38, 979–991 CrossRef CAS.
- K. Oberg, E. T. Janson and B. Eriksson, Ital. J. Gastroenterol. Hepatol, 1999, 31, S160–S162 Search PubMed.
- H. Chen, R. Udelsman, M. A. Zeiger and D. W. Ball, Oncol. Rep., 1997, 4, 775–778 CAS.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed.
- M. Martinez-Alonso, N. Llecha, M. Mayorga, A. Sorolla, X. Dolcet, V. Sanmartin, L. Abal, J. Casanova, M. Bardad, A. E. Yeramian, R. S. Puig, R. Vilella, X. Matias-Guiu and R. Marti, J. Int. Med. Res., 2009, 37, 1813–1822 CrossRef CAS PubMed.
- M. Huo, A. Zou, C. Yao, Y. Zhang, J. Zhou, J. Wang, Q. Zhu, J. Li and Q. Zhang, Biomaterials, 2012, 33, 6393–6407 CrossRef CAS PubMed.
- P. Liang, D. Zhao, C. Q. Wang, J. Y. Zong, R. X. Zhuo and S. X. Cheng, Colloids Surf., B, 2013, 102, 783–788 CrossRef CAS PubMed.
- D. W. Miller, E. V. Batrakova, T. O. Waltner, V. Y. Alakhov and A. V. Kabanov, Bioconjugate Chem., 1997, 8, 649–657 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb02530g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |