
Eco-friendly direct (hetero)-arylation polymerization: scope and limitation
Simiao
Yu
a,
Fuchuan
Liu
a,
Jianwei
Yu
a,
Shiming
Zhang
*a,
Clement
Cabanetos
*b,
Yongqian
Gao
a and
Wei
Huang
*a
aKey Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University (NanjingTech), 30 South Puzhu Road, Nanjing 211816, China. E-mail: iamsmzhang@njtech.edu.cn
bCNRS UMR 6200, MOLTECH-Anjou, University of Angers, 2 Bd Lavoisier, 49045 Angers, France
First published on 16th November 2016
Abstract
Polymer semiconductors have recently attracted considerable attention owing to their (i) excellent optical properties, (ii) processability, (iii) inherent tunability of the energetics, and (iv) synthetic versatility. Consequently, researchers have shown great interest in developing eco-friendly polymerization methods to reduce the synthetic cost of such macromolecular materials, including the so-called direct (hetero)-arylation polymerization (DHAP). In addition to reducing the number of synthetic steps, required in conventional cross-coupling polymerizations, DHAP avoids the use of lithiated and/or stannylated intermediates that are highly toxic and/or dangerous. In this contribution, we reviewed a number of conjugated polymers prepared by DHAP for applications in organic electronics, and more precisely for organic photovoltaics and field-effect transistors. Moreover, emphasis has been put on polymerization reaction conditions (i.e., the nature of the catalysts, ligands and solvents) and their impact on the properties of the material. Even though some optimizations still remain, regarding the current trends, it is obvious that DHAP will play a larger role in the design and synthesis of polymer semiconductors.
 Simiao Yu | Simiao Yu was born in Dalian, China in 1993. She received her BE degree from the Department of Chemical Engineering at Nanjing Tech University in 2015. And she continued her master’s degree under the supervision of Prof. Shiming Zhang and Yongqian Gao in the Nanjing Tech University in the same year. She is currently pursuing master’s studies in the area of synthesis of specific molecules applied in organic photovoltaics and field effect transistors. |
 Shiming Zhang | Shiming Zhang received his PhD in 2009 from the Institute of Chemistry, Chinese Academy of Sciences, and subsequently began two years of postdoctoral research within the Department of Chemistry at Northwestern University under the supervision of Prof. Tobin J. Marks and Antonio Facchetti. He worked at KAUST (Saudi Arabia) as a postdoctoral fellow from 2011 to 2012, after which he moved to Singapore to work in Silecs International as a senior chemist from 2012 to 2014 and WinTech Nano as a quality lead/principal engineer from 2014 to 2015. In 2015 he joined the Institute of Advanced Materials (IAM) and began his professorship at Nanjing Tech University (NanjingTech) as a professor. His research interests include organic/flexible electronics and microelectronics-packaging materials. |
 Clement Cabanetos | After graduation in 2008, Clement Cabanetos undertook a PhD at the CEISAM laboratory, University of Nantes (France), focusing on the design and synthesis of new crosslinkable macromolecular materials for nonlinear optical applications. Further to completion, he joined in 2011 the KAUST (Saudi Arabia) as a postdoctoral fellow to prepare macromolecular materials for organic photovoltaic (OPV) applications. In 2013, he joined the “Systemes Conjugués Linaires” group in Angers (France) as a CNRS researcher to develop new molecular systems for organic electronics. |
 Wei Huang | Wei Huang received his PhD degree from Peking University in 1992. In 1993, he began his postdoctoral research with the Department of Chemistry under the supervision of Prof. HUANG Hsing Hua in the National University of Singapore (NUS) and then taught at NUS. In the mean time, he took part in the foundation of the Institute of Materials and Engineering (IMRE), Singapore. In 2002, he moved to Fudan University where he founded the Institute of Advanced Materials (IAM). In 2006, he was appointed as the Deputy President of the Nanjing University of Posts and Telecommunications (NUPT). He was elected as Academician of the Chinese Academy of Sciences in 2011. In 2012, he assumed his duty as the President of the Nanjing Tech University (NanjingTech). He was elected as Foreign Academician of the Russia Academy of Sciences in 2016. His research interests include organic/plastic/flexible (opto) electronics, nanomaterials and nanotechnology. |
1. Introduction
Transition-metal-catalyzed C–C coupling reactions have been, and continue to be, some of the most principal reactions in organic chemistry, widely applied to the synthesis of natural products, pharmaceutical intermediates or even organic functional materials. Until recently, traditional cross-coupling reactions, namely, Suzuki–Miyaura, Stille, Negishi, Sonogashira and Kumada, have been the primary means of forming carbon–carbon (C–C) bonds.1–3 However, these methods typically involve halogenated and organometallic reagents, thus generating stoichiometric amounts of undesirable and toxic by-products. Moreover, additional synthetic steps, including group-protection and/or activation of aromatic rings, are required to prepare these intermediates. It is noteworthy that such synthetic and environmental problems have plagued researchers for a long time.4 Furthermore, within the current context of the increasing demand for atomic economy reactions and green chemistry,5 researchers have more recently focused their efforts on the exploration of new cross-coupling methods.Considering that the carbon–hydrogen (C–H) bond is one of the most common chemical bonds among organic compounds, the formation of a C–C bond by coupling an aromatic hydrogen activated compound directly to a halogenated aromatic reagent would provide a powerful tool to fulfill the atomic economy requirement since the only by-product generated within the media is a hydrogen halide (H–X).6 This synthetic method, called direct (hetero)-arylation (DHA), has shown great potential over the last few years in synthesizing compounds without organometallic reagents (such as Grignard reagents, organozinc or organotin).7 Additionally, this method is not only a good way to resolve problems inherent within traditional cross-coupling reactions but also a particularly appealing strategy for generating C–C bonds as well as C–X bonds.8 Many researchers have been eyeing this emerging field in recent decades. In 2002, Lemaire et al.1 summarized the results obtained in the field of aryl–aryl bond formation using various kinds of reactions and technologies. To some extent they drew a relatively complete picture of the current efforts in this area. More recently, Leclerc and fellow researchers8 systematically and comprehensively summed up the present studies on direct (hetero)-arylation polymerization (DHAP). They discussed general and adaptable reaction conditions for the synthesis of defect-free as well as high-molecular-weight conjugated polymers; a discussion which constructed a bright outlook about the field.
Thus, DHA represents an efficient strategy for the preparation of a large number of aromatic blocks or monomers. Particularly, it offers a highly valuable and economical synthetic method for large-scale production, and commercially viable preparations of various compounds for organic electronics4 including applications such as organic photovoltaics (OPV),9–13 organic field-effect transistors (OFET)14–19 and/or organic light-emitting diodes (OLED);20,21 for instance, polymer solar cells (PSCs) and polymer photovoltaic materials have benefited from significant and remarkable advances over the last decades.22 New photovoltaic materials (donors and acceptors) and device structures are reported with each passing day, and peak power conversion efficiency (PCE) is regularly being increased.23–26
As a C–H activation method, DHAP can simplify and shorten the synthetic process, minimize the presence of difficult-to-remove by-products, and at the same time afford new compounds that can be achieved with relatively less cost.27 Consequently, this method will undoubtedly contribute in removing technological and practical barriers. In this context, the aim of this review is to summarize selected examples of conjugated polymers prepared via DHAP and to discuss the added value, scope and limitation inherent in this method with a particular focus on the structure–property relationships.
2. Comparison with traditional cross-coupling reactions
Recently, DHAP has emerged to provide efficient access to new conjugated macromolecular materials. Since then, a significant amount of effort has gone into developing, refining, and popularizing this new method. Indeed, among its advantages recent literature has reported DHAP as a cheap and atom-economical tool when compared to conventional organometallic cross-coupling reactions.28–31Allard et al.32 described for the first time the preparation of thieno[3,4-d]thiazole-based (TTz) alternating copolymers synthesized via Stille, Suzuki or DHAP methods (Scheme 1). First, polymer P1-1 was synthesized via conventional Stille cross-coupling between 2-octylthieno[3,4-d]thiazole and 2,6-dibromo-4,8-di(ethylhexyl-oxyl)benzo[1,2-b:4,5-b′]dithiophene (BDT) and compared to its analogous P1-2 synthesized via DHAP. Both polymers showed a comparable molecular weight (Mn) of ca. 32 kDa,33 demonstrating that direct arylation is a possible method to obtain Mn as high as those achieved by conventional coupling reactions.
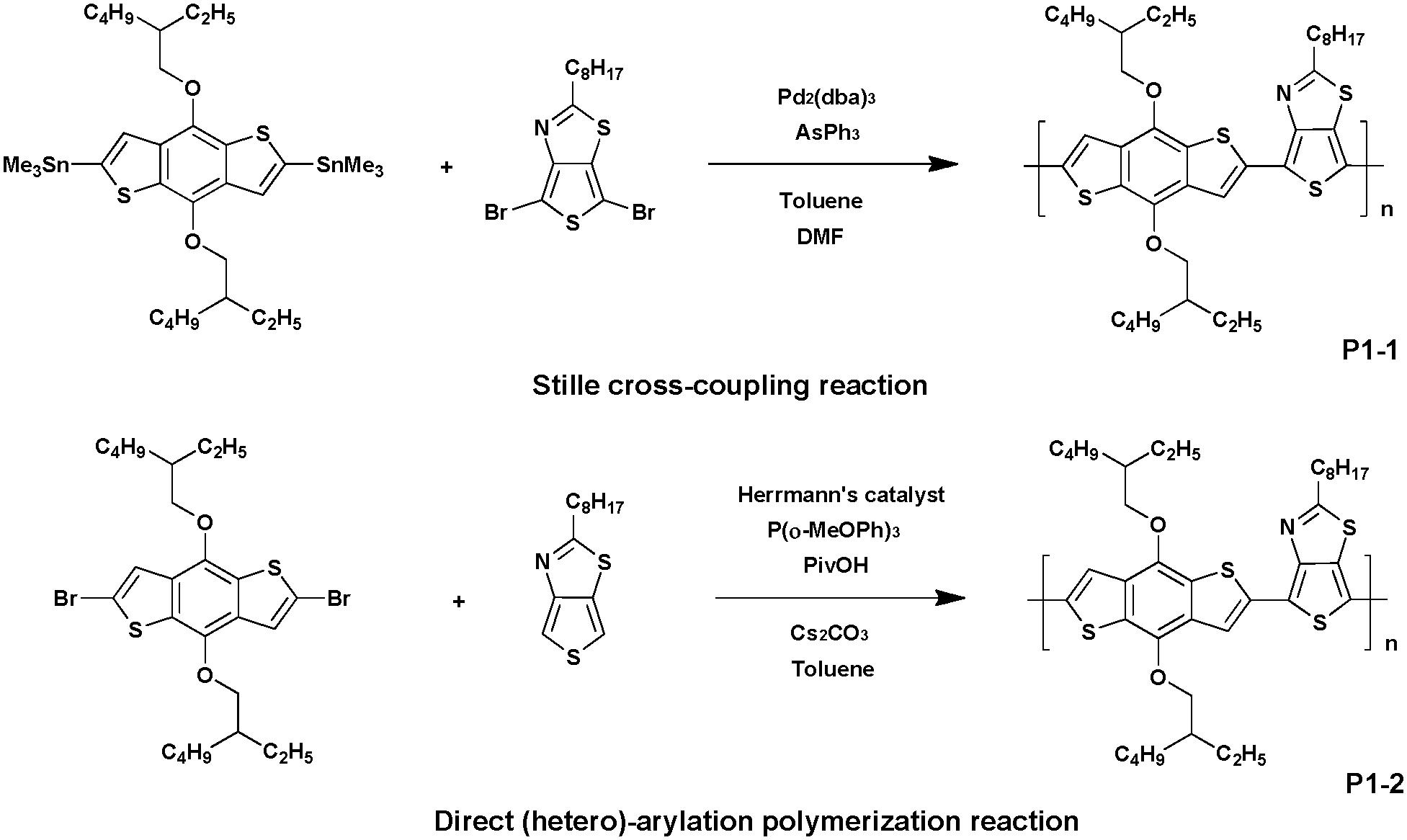 | ||
| Scheme 1 Preparation of P1-1via Stille coupling and P1-2via DHAP. | ||
In parallel, both polymer P2 and polymer P3 were synthesized via DHAP (Scheme 2). For P2, 5-(2-ethylhexyl)thieno[3,4-c]pyrrole-4,6-dione (TPD) was coupled with 4,6-dibromo-2-octylthieno[3,4-d]thiazole. A low molecular weight of 8 kDa was determined and was correlated with the strong rigidity of the polymer backbone resulting from the interaction between the thiophene ring of the TTz unit and the ketone borne by the TPD unit, resulting in the reduction of the solubility of this material.34 However, it turns out that DHAP is probably the only available method to afford this polymer. Indeed, all attempts performed in conventional conditions failed. Polymer P3 has also been synthesized under the same conditions used for P2 by polymerizing the 3,6-bis(5-bromothiophen-2-yl)-2,5-bis(2-octyldodecyl)pyrrolo-[3,4-c]pyrrole-1,4(2H,5H)-dione and 2-octylthieno[3,4-d]thiazole monomers. In this case, a molecular weight of ca. 17 kDa was measured. It is noteworthy that P3 could also be prepared via Suzuki coupling since diketopyrrolopyrrole (DPP) units bearing pinacol borane groups have already been reported.35 However, the synthesis cost of P3via Suzuki coupling is quite high, which leads to DHAP being the method of choice.
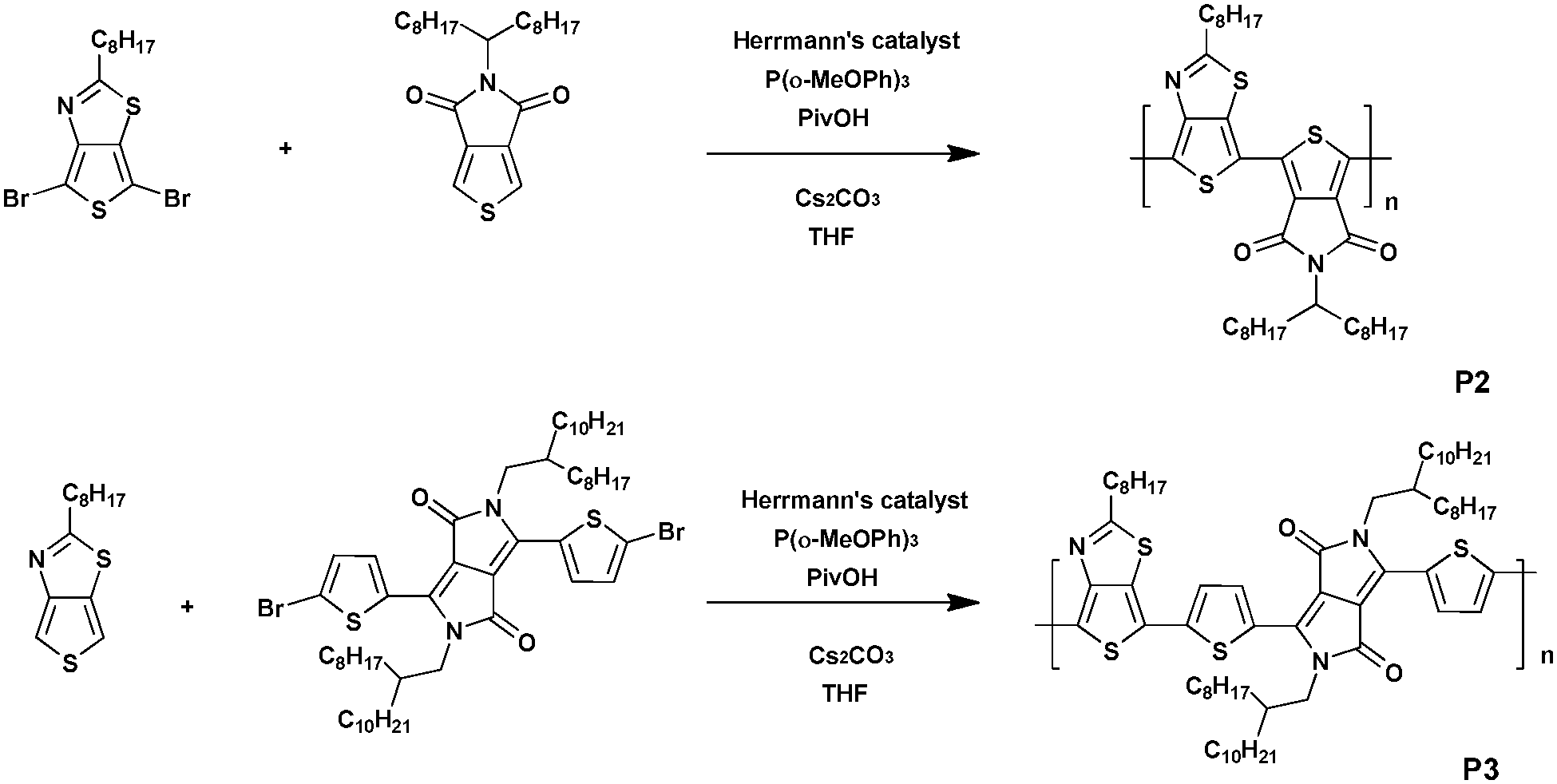 | ||
| Scheme 2 Synthesis of P2 and P3via DHAP. | ||
In 2015, Marzano et al.36 reported a new random copolymer prepared via both Stille polymerization (P4-1) and DHAP (P4-2), and which contained two different acceptor units, namely benzo[c][1,2,5]thiadiazole and benzo[d][1,2,3]triazole along with one donor moiety, namely benzo[1,2-b;4,5-b′]dithiophene (Scheme 3). The DHAP afforded the polymer P4-2 in lower yields (70% vs. 85% for P4-1) but with longer chains (Mn = 10.3 kDa for P4-2vs. Mn = 20 kDa for P4-1). All polymers synthesized were tested in bulk heterojunction (BHJ) solar cells with [6,6]-phenyl C71 butyric acid methyl ester (PC71BM). The device based on P4-1 showed a modest power conversion (PCE) of 2.5%. However, after adding 2% (v/v) of 1-chloronaphthalene (CN), which was reported to improve the PCEs,37 the device performance was significantly increased to 4.8%. Meanwhile, without the additive P4-2, it exhibited a comparable PCE of ca. 1.9%. Upon CN processing the PCE of P4-2 was barely improved to 2.8%, indicating that the polymer P4-2 is almost insensitive to this treatment, probably due to the structural defects of DHAP polymers.38
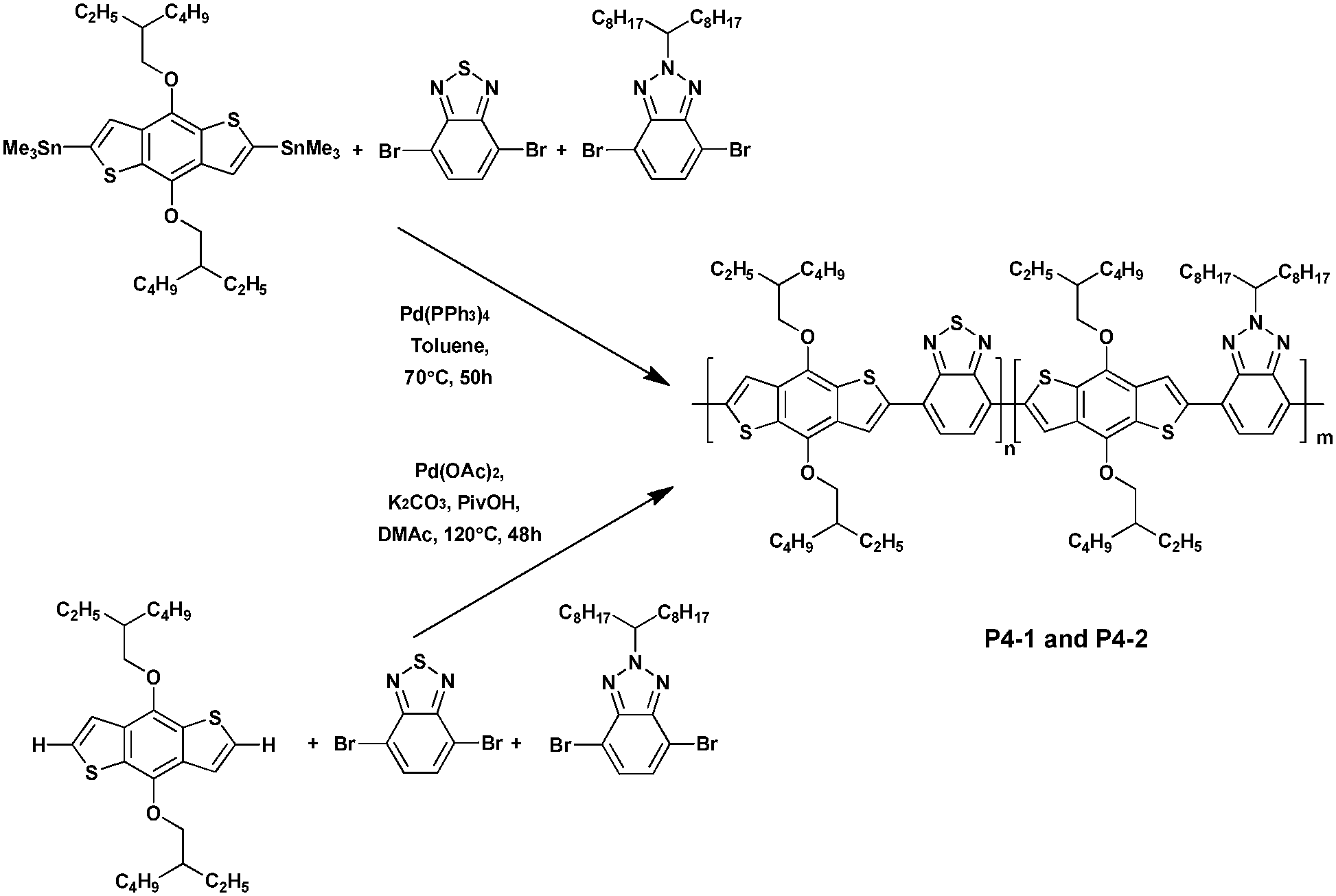 | ||
| Scheme 3 Preparation of P4-1via Stille coupling and P4-2via DHAP. | ||
3. Homopolymers synthesized via DHAP
Polythiophenes are considered to be part of the most promising material family in both conductive polymers and photovoltaic materials. However, without substituents, the latter are neither soluble nor feasible. To solve this solubility issue, alkyl-substitution turns out to be the most effective shortcut. For instance, poly(3-alkylthiophene) (P3AT) appears to be the most common polythiophene bearing alkyl-substituted side chains, and the well-known poly-3-hexylthiophene (P3HT) can be cited as a key example.39–41 For the most part, polymerization typically occurs in the 2-position and the 5-position of thiophene. If the 2-position is chosen as the “head” (simply as “H”) of the polythiophene units and the 5-position as the “tail” (simply as “T”), the proportion of the units with “head-to-tail” (HT–HT) structures in the poly(3-alkylthiophene) is called regioregularity.42 Due to the small steric hindrance among the repeated units, such a process is more likely to achieve better planarity and stronger interchain interactions. Thus, compared with irregular polythiophene, head-to-tail regioregular (RR) P3ATs have not only elevated the effective conjugated chain length, but also higher charge mobility.43In 2010, Ozawa et al.44 reported the palladium-catalyzed direct arylation polymerization of 2-halo-3-hexylthiophene affording RR-P3HT (Scheme 4). The use of Herrmann's catalyst and Tris(2-dimethylaminophenyl)phosphine as the ligand lead to the preparation of P5 characterized by a high molecular weight (Mn = 30.6 kDa) and a promising regioregularity of ca. 98% in an almost quantitative yield (99%). Interestingly, the regioregularity improved significantly with increasing molecular weight, probably because of the cross-coupling reaction between C–H and C–Br, which dominates the polymerization at the later stage. From these experimental data higher regioregularity leads to higher charge mobility and conductivity, therefore improving the PCEs when the polymer is used in BHJ solar cells.
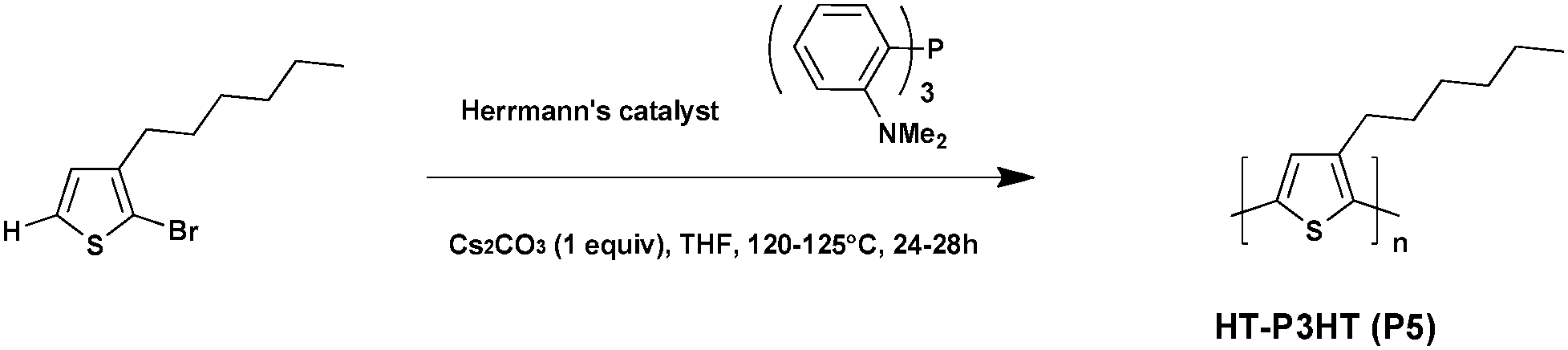 | ||
| Scheme 4 Synthesis of regioregular polymer P3HTvia DHAP. | ||
Then in 2015 the DHAP method, which can selectively synthesize unprotected thiophene units under an appropriated catalytic system, was first reported by Bura et al.45 The use of the Herrmann–Beller catalyst and P(o-NMe2Ph)3 in the solvent of dioxane with an acidic additive was quite useful to achieve well-defined thiophene–thiophene couplings. A high molecular weight of poly(3,3′′′-didodecyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene) (PQT12) (P6) (43 kDa) was achieved by synthesizing 5-bromo-3,3′′′-didodecyl 2,2′:5′,2′′:5′′,2′′′-quaterthiophene (monomer A) and 5-bromo-3′,4′′-didodecyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene (monomer B) (Scheme 5). The experiments undertaken highlighted that adding steric hindrance protection around the β-positions of the brominated thiophene unit is of great importance to improve the selectivity of the cross-couplings at the α-positions. Plus, these experiments also proved that DHAP could be a practicable synthetic tool to obtain various polythiophene derivatives applied in both organic electronics and in the fabrication of photovoltaic devices.
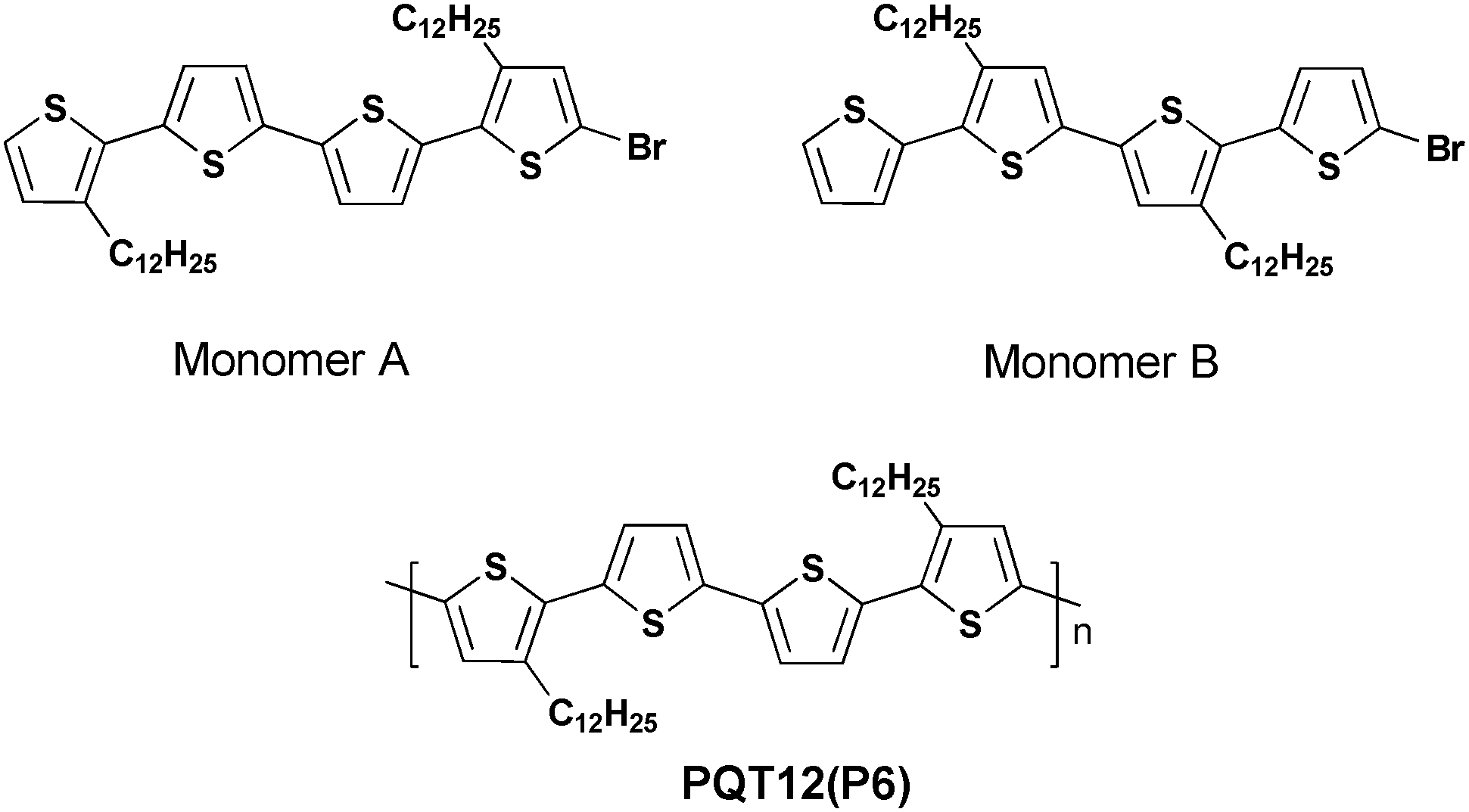 | ||
| Scheme 5 Synthesis of polythiophene P6via DHAP. | ||
To date, polythiophene is one of only a few homopolymers that can be synthesized via DHAP. Beyond this a wide variety of homopolymers, based on different monomers prepared via DHAP, have been attempted and studied for years. DPP, which is naturally regarded as an acceptor-type building block, is a suitable option. In 2013, Guo et al.46 prepared the dithienyldiketopyrrolopyrrole (DTDPP)-based homopolymer P7, which is traditionally synthesized by nickel-mediated Yamamoto-type polycondensation,47 using the concise and highly efficient new method, DHAP (Scheme 6). Under the optimized reaction conditions of 5 mol% of Pd(OAc)2, 10 mol% of PCy3·HBF4 (Cy = cyclohexyl), 2.5 equivalents of K2CO3 and 1.0 equivalent of pivalic acid in the mixed solvent of N,N-dimethylacetamide (DMAc)/xylene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the unsymmetrical DTDPP–DTDPP copolymer P7 with different alkyl substituents on each DPP unit48 was obtained as a black solid in a moderate yield of 43% with an Mn of 30.2 kDa and a polymer dispersity index (PDI) of 3.56. The optical and electrochemical properties were also characterized. The UV-vis-NIR absorption spectrum of P7 in a film on the quartz plate was tested and the absorption band edge (λonset) was 1014 nm, which showed strong and broad NIR absorption bands. In addition, P7 exhibited a remarkable optical low bandgap (Eoptg) down to 1.22 eV, which estimated from the absorption band edge in film (λonset) was probably due to the common features of ideal planarity and good π-conjugation. Such structural features may make it a promising feature in OPV materials.
:1), the unsymmetrical DTDPP–DTDPP copolymer P7 with different alkyl substituents on each DPP unit48 was obtained as a black solid in a moderate yield of 43% with an Mn of 30.2 kDa and a polymer dispersity index (PDI) of 3.56. The optical and electrochemical properties were also characterized. The UV-vis-NIR absorption spectrum of P7 in a film on the quartz plate was tested and the absorption band edge (λonset) was 1014 nm, which showed strong and broad NIR absorption bands. In addition, P7 exhibited a remarkable optical low bandgap (Eoptg) down to 1.22 eV, which estimated from the absorption band edge in film (λonset) was probably due to the common features of ideal planarity and good π-conjugation. Such structural features may make it a promising feature in OPV materials.
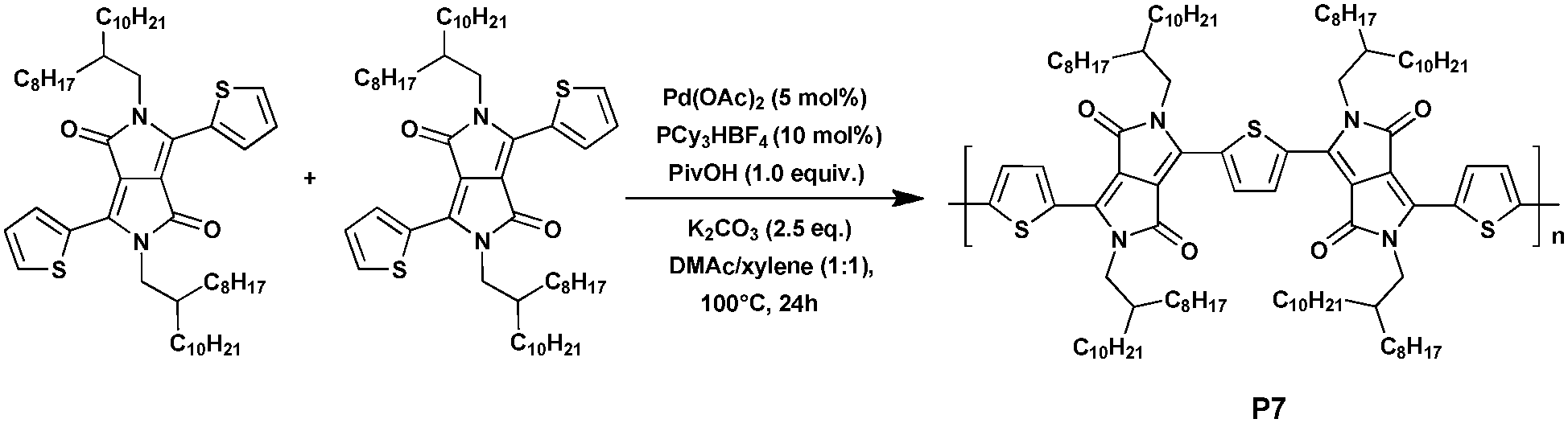 | ||
| Scheme 6 Synthesis of homopolymer P7via DHAP. | ||
4. D–A conjugated polymers synthesized via DHAP
4.1 Polymers containing benzothiadiazole (BT)
Benzothiadiazole (BT) is an altogether applicable acceptor unit exceptionally suited for photovoltaic materials owing to its appropriate electron-withdrawing ability. Retaining a relative low highest occupied molecular orbital (HOMO) energy level in the copolymers, which is advantageous for the chemical stability of the copolymers as well as for attaining a high open-circuit voltage (Voc) in the final organic photovoltaics, makes it stand out among the numerous acceptor groups (such as thienothiadiazole, thienopyrazine, etc.).49–51 Consequently, BT has become an extremely important building block utilized in efficient photovoltaic materials. And the utilization of a BT unit in push–pull conjugated polymers has shown a high PCE of up to 9.1% in dye-sensitized solar cells.52Wang et al.53 recently reported the synthesis of small molecules containing bare thiophene, alkyl-substituted thiophene and 4,7-dibromo-2,1,3-benzothiadiazole (DBrBT) (Scheme 7). Under optimized conditions, Pd2(dba)3-catalyzed DHAP produced the corresponding small molecules, i.e., a thiophene-flanked benzothiadiazole derivative (DTBT) and 4,7-bis(4-hexylthiophen-2-yl)-2,1,3-benzothiadiazole (DHTBT), with a comparable yield to that of the reference Stille or Suzuki coupling reactions.
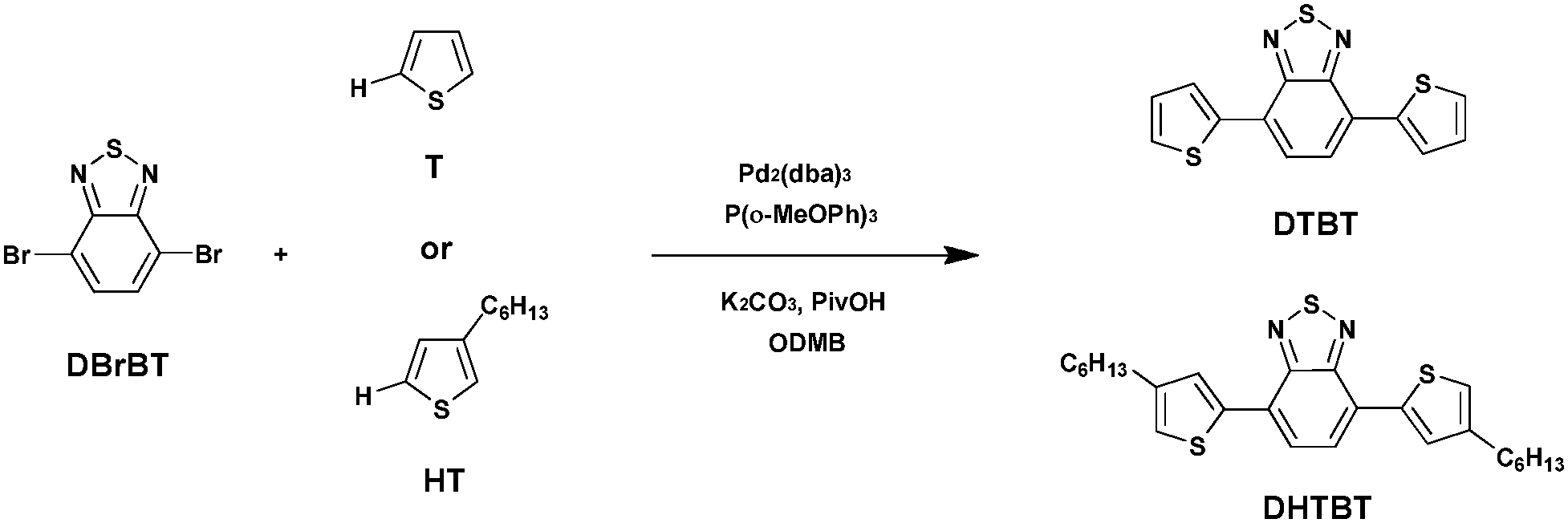 | ||
| Scheme 7 Synthesis of the small molecules DTBT and DHTBT. | ||
DTBT and DHTBT were then used respectively to synthesize conjugated polymers PFTBT (P8) and PFHTBT (P9) with 9,9-dioctyl-2,7-dibromofluorene (DBrF) still via DHAP in high yield, i.e., 82% and 91%, respectively. In addition, P8 was characterized by an Mn of 13.8 kDa while P9 was characterized by a slightly higher Mn of 17.6 kDa. Due to the presence of hexyl side chains on the thiophene units the solubility of P9 was remarkably improved compared to P8. According to the high-temperature NMR, this method of DHAP catalyzed with Pd2(dba)3 allows the synthesis of almost defect-free polymers with good C–H selectivity. Furthermore, it is worth noting that P9 exhibits a hypsochromic-shifted absorption spectrum compared with a P8 of ca. 35 nm, attributed to the steric hindrance of the hexyl groups on the thiophene rings. Indeed, it is supposed that the π-conjugation along the backbone in P9 could impede by the increasing dihedral angles between the thiophene and the fluorene unit (Scheme 8).54
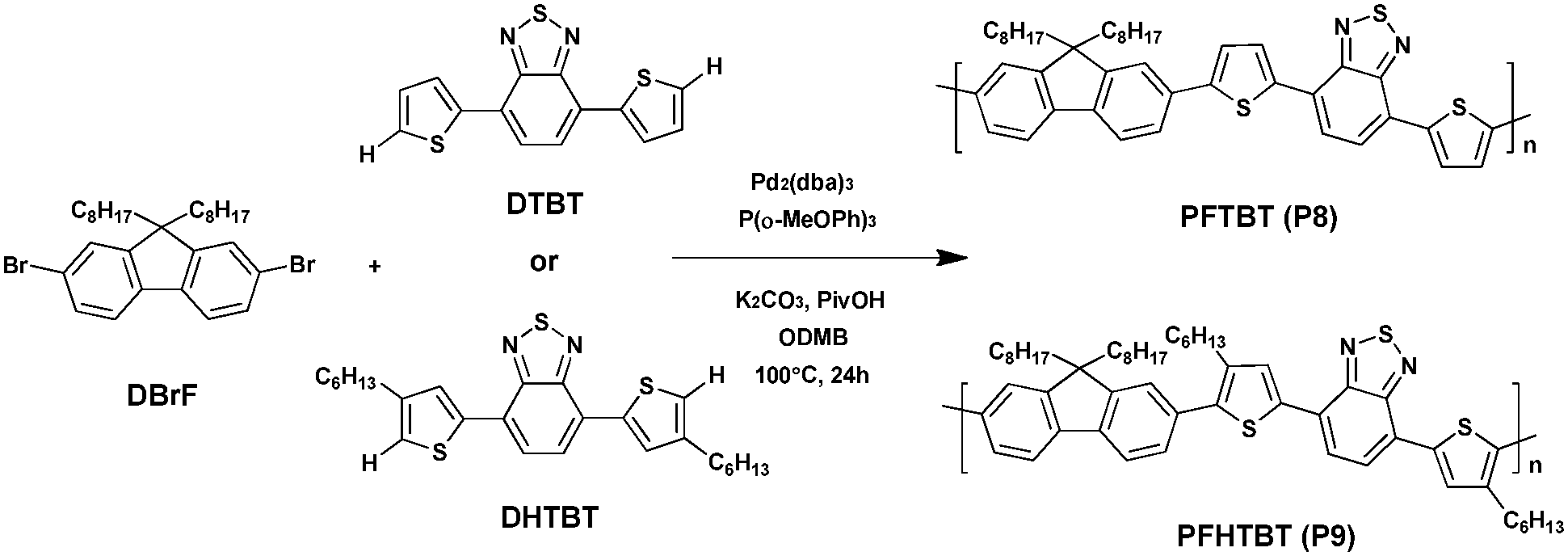 | ||
| Scheme 8 Synthesis of P8 and P9via DHAP under the same conditions. | ||
In 2016, Tomar et al.55 reported four donor–acceptor type polymers based on benzothiadiazole and thiophene–benzothiadiazole–thiophene (TBTT) synthesized via DHAP (Scheme 9). Under the optimal conditions of Pd(OAc)2, pivalic acid, DMAc at 80 °C and in a nitrogen atmosphere, alternate polymers P10 and P11 were achieved by reacting thiophene–benzothiadiazole–thiophene with quinoline and biquinoline, respectively; a random polymer P12 was achieved by reacting benzothiadiazole, cyclopentadithiophene (CPDT) and quinoline; and P13 was obtained using biquinoline instead of quinoline. All polymers exhibited good solubility in ordinary organic solvents and the number-average molecular weights of 3.6 kDa, 5.03 kDa, 5.3 kDa and 7.6 kDa were obtained by the measurements of gel permeation chromatography against polystyrene standards. The UV/Vis absorption spectra of the four polymers were then investigated, which revealed that P10 (494 nm) and P12 (664 nm) showed a higher value of absorbance maxima compared to P11 (481 nm) and P13 (649 nm) on account of more efficient electron delocalization caused by the more planar structure.56,57 In addition, the deep-lying HOMO energy levels of P10, P11, P12 and P13, which were −5.82 eV, −5.75 eV, −5.19 eV, and −5.03 eV, respectively, showed improved oxidative stability for this variety of polymers. Furthermore, OFET characteristics were also studied. P12 and P13 could be observed exhibiting a hole-type transport of 11 × 10−3 cm2 V−1 s−1 and 6 × 10−3 cm2 V−1 s−1, respectively, whereas P10 and P11 did not show any OFET characteristics due to the existence of the dominant contact resistance in the P10 and P11 systems.
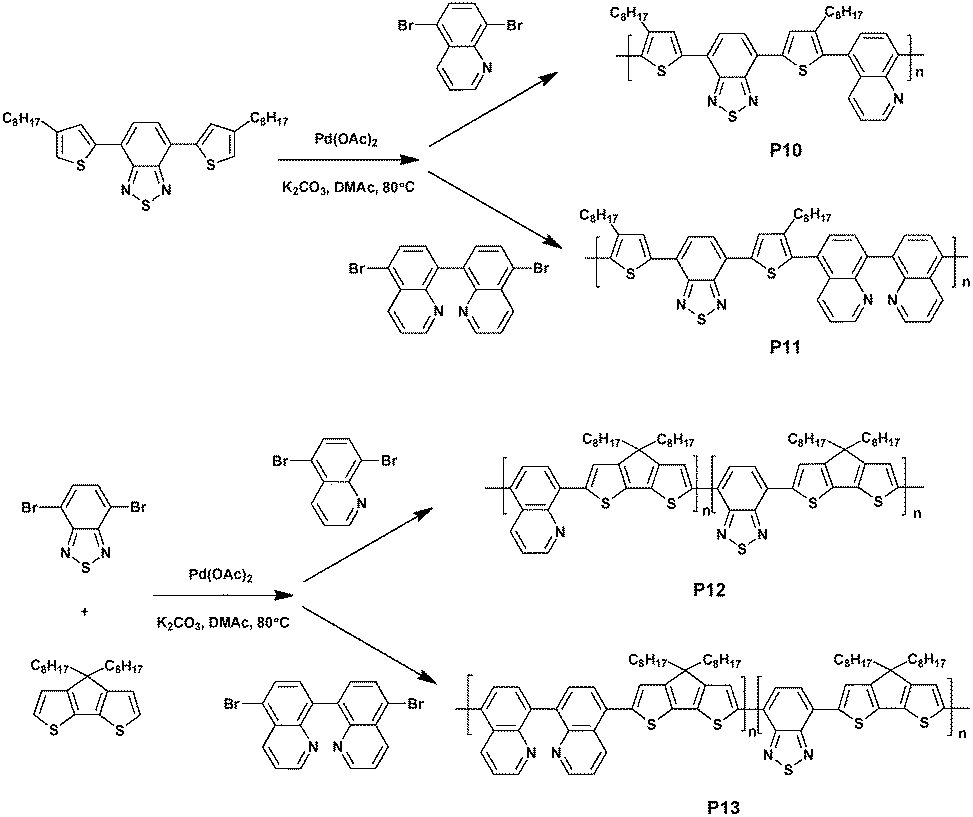 | ||
| Scheme 9 Synthesis of P10–P13via DHAP. | ||
4.2 Polymers containing thieno[3,4-c]pyrrole-4,6-dione (TPD)
The TPD unit is a good electron-withdrawing co-monomer, which has great potential for photovoltaic applications. Indeed, recent studies reported that some highly efficient TPD-based PSCs exhibit a PCE of up to 8.5% when fabricated and tested under an inert atmosphere.58–61 Its relatively planar structure could prove to be beneficial for electron delocalization when incorporated into conjugated polymers promoting intra-molecular/intermolecular interactions. In addition, its strong electron-withdrawing effect could lead to low HOMO and lowest unoccupied molecular orbital (LUMO) energy levels.22 For these reasons the TPD based materials were shown to be good electron donors when synthesizing donor–acceptor conjugated polymers, developing specific interactions between the imide group and the fullerene derivatives.62 Moreover, the imide group in the TPD moiety could also be used as a directional activating group so that C–H activation could typically occur in the 2-position and the 5-position of thiophene.63In 2012, Leclerc et al.64 synthesized a TPD-bithiophene based polymer via DHAP in high yield (96%). Under optimal conditions, using Tris(3-methoxyphenyl)phosphine as the ligand and Pd(OAc)(o-Tol) as the catalyst, high molecular weights of ca. 60 kDa were achieved. In parallel, the Stille polymerization was carried out using Pd2(dba)3 as the catalyst and Tris(3-methoxyphenyl)phosphine as the ligand. The resulting polymer (P14-2) showed a lower yield (71%) and molecular weight (Mn of 9 kDa). The UV/Vis absorption spectra of the two analogues were then investigated and revealed that both P14-1 and P14-2 exhibit comparable features in chloroform solutions and in the solid-state. However, compared to P14-2 the absorption maximum of P14-1 is slightly red shifted, by ca. 18 nm in film, probably because of the different molecular weights and morphologies/packing in the solid state. Additionally, X-ray and thermal analyses were performed to study the structural regularity of these polymers, showing that both the enthalpies of crystallization and melting were higher for P14-1 than for P14-2 (Scheme 10).
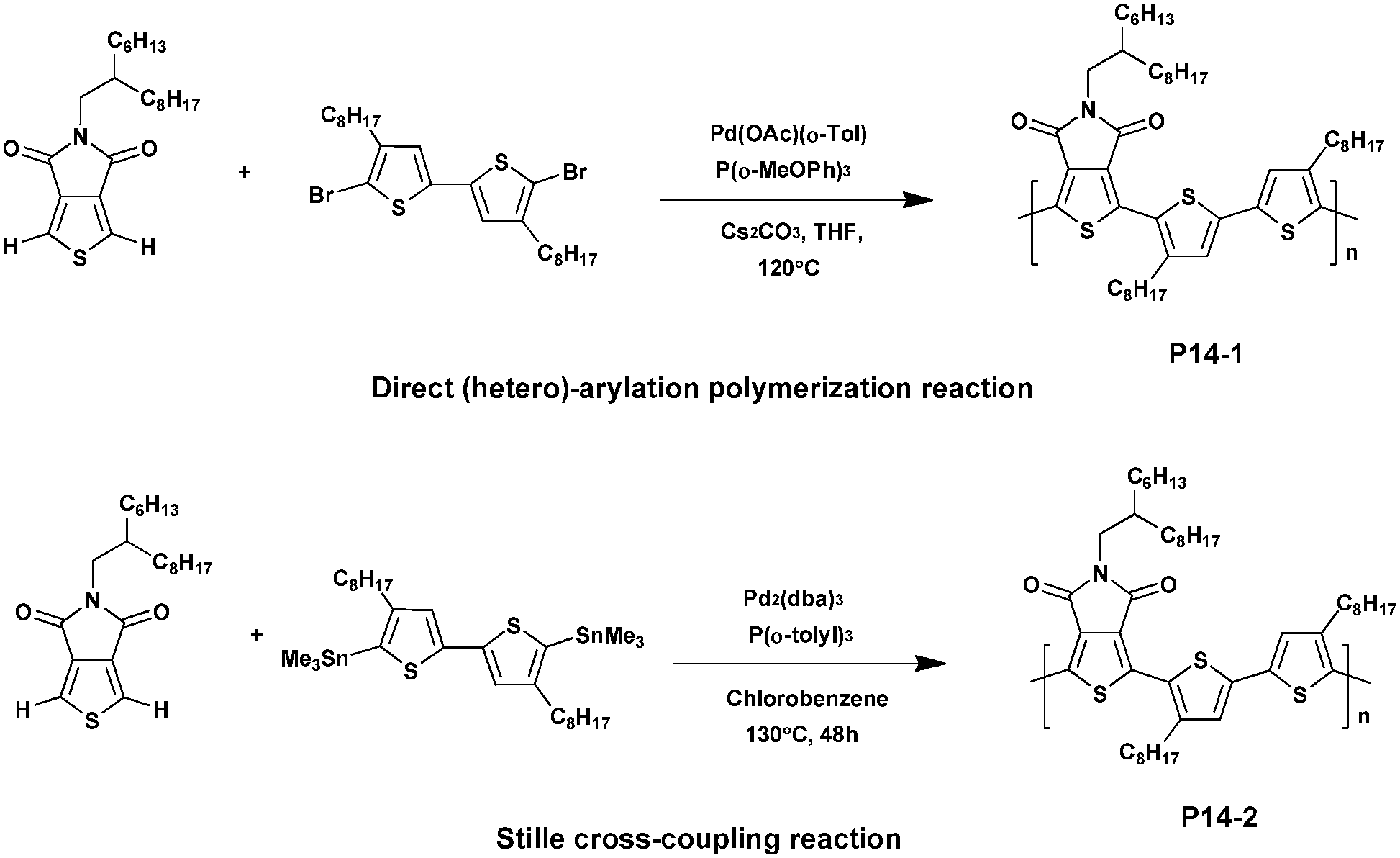 | ||
| Scheme 10 Preparation of P14-1via DHAP and P14-2via Stille coupling. | ||
In 2012, two TPD based polymers, namely P15 and P16, containing bithiophene or terthiophene as electron-rich moieties, respectively, were prepared by Jo et al.65via a direct heteroarylation procedure in the presence of trans-di(μ-acetato)bis[o-(dio-tolyl-phosphino)benzyl]dipalladium(II) and Tris(o-methoxyphenyl)phosphine (Scheme 11). The bithiophene-containing polymer P15 achieved 94% yield and was characterized by a Mn of 50 kDa while P16 barely reached a Mn of 41 kDa with a similar yield (92%). Furthermore, it is worth noting that these polymers exhibit HOMO energy levels of ca. −5.66 eV. From this, BHJ solar cells based on photoactive films made of a blend of P15 or P16 with [6,6]-phenyl C71-butyric acid methyl ester (PC70BM) were fabricated. Upon optimization, PCEs of 1.90% and 6.10% were obtained from the P15 and P16 based devices, respectively. These differences are probably induced by the terthiophene unit favouring better conjugation along the backbone, higher electron mobility and better π–π stacking.66 Indeed, it is noteworthy that the absorption spectrum of P16 exhibits a vibronic shoulder at 600 nm, indicating that the polymer chains were already aggregated in solution. Moreover, this peculiar aggregation shows that integrating the polymers into field-effect transistors led to a better charge carrier mobility (1.3 × 10−2 cm2 V−1 s−1 for P16vs. 10−5 cm2 V−1 s−1 for P15).
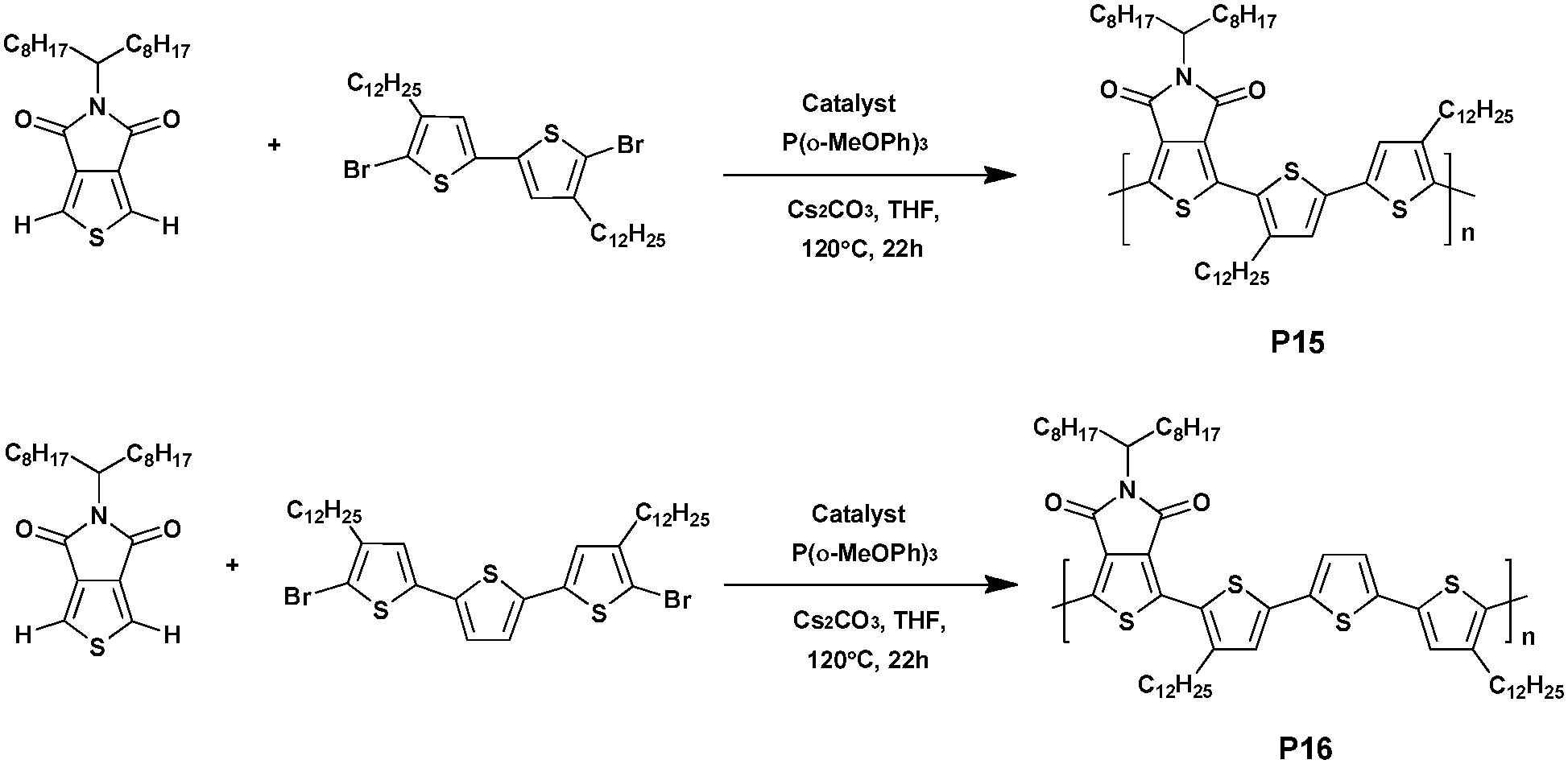 | ||
| Scheme 11 Synthesis of P15 and P16via DHAP. | ||
Despite its multiple advantages, including simplified steps and no need for preprocessing the organometallic monomers, DHAP still suffers from two main limitations: the first one concerns the homocoupling reaction leading to structural defects of the polymer chains,67–70 and the second deals with the lack of selectivity when different C–H bonds are present causing branching as well as cross-linking and generally leading to insoluble materials.71–73
However, in 2016, Ozawa et al.74 studied these two questions by exploring an original strategy (Scheme 12). When preparing P17, composed of a dithienopyrrole unit as the donor and the TPD unit as the acceptor, they found that using a blend of ligands in palladium catalyzed direct arylation polymerizations may avoid side reactions effectively and favor high yield. Thus, the combined use of Tris(2-methoxyphenyl)phosphine and tetramethylethylenediamine in the presence of Pd2(dba)3 led to a high yield, reduced defect formation and no insoluble materials.
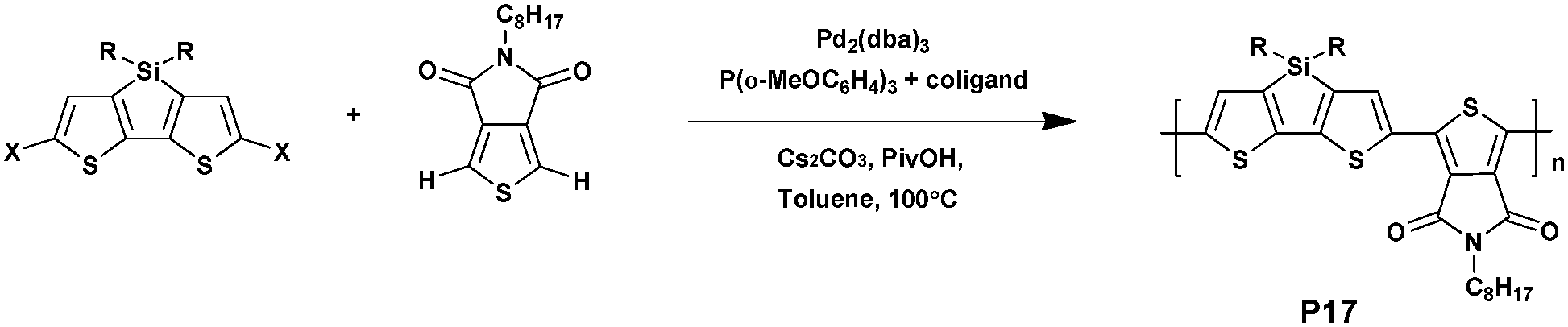 | ||
| Scheme 12 Preparation of P17via DHAP. | ||
4.3 Polymers containing isoindigo
Isoindigo is an ideal building block for synthesizing D–A conjugated polymers. Indeed, the latter displays many advantages such as a strong electron-withdrawing character, outstanding stability, a highly fused structure, outstanding absorption properties, and so on.75–77 As a result, polymers of isoindigo generally show low bandgaps, strong π–π interactions and high charge carrier mobility, suggesting a potential compatibility in building D–A polymers for efficient organic photovoltaic materials.78–81 High performance BHJ solar cells based on isoindigo polymers have already been fabricated with PCEs of up to 7%,82–84 and field-effect transistors with a mobility as high as 3.62 cm2 V−1 s−1.85–87Moreover, while generally used as a p-type material, only a few studies have been devoted to afford n-type isoindigo-based materials. In 2013, Grenier et al.88 combined three different electron-withdrawing co-monomers with a low steric hindrance, namely TPD, the 5,5′-dioctyl-1,1′-4H-bithieno[3,4-c]pyrrole-4,4′,6,6′(5H,5′H)-tetrone (BTPD) or DPP with the isoindigo to prepare n-type copolymers characterized by good charge mobility, low bandgaps and low energy levels. At first, the three copolymers were synthesized via Suzuki–Miyaura cross-coupling reaction. However, only P18 could be isolated with an acceptable yield of ca. 70% and a Mn of 44 kDa (Scheme 13).
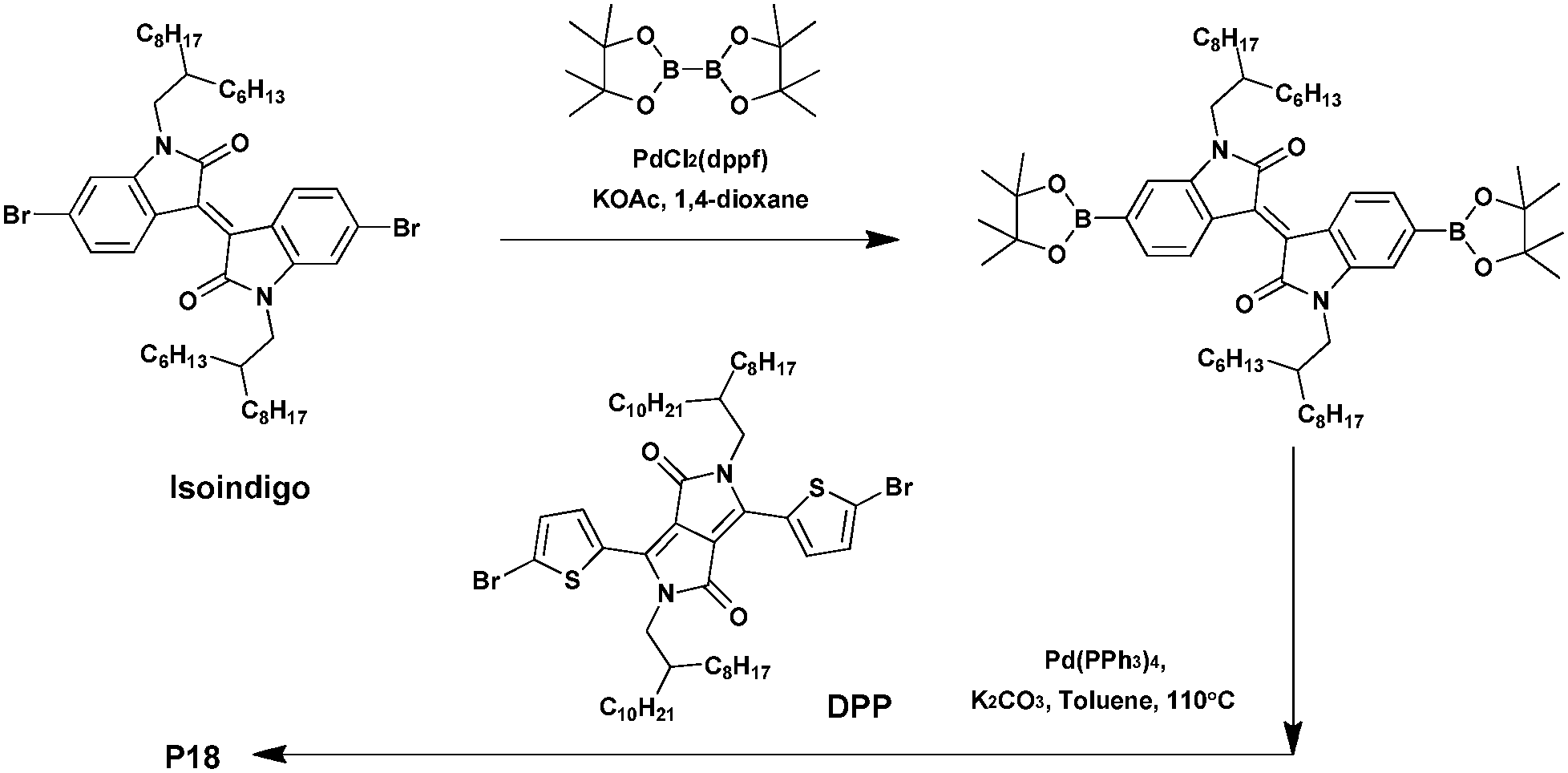 | ||
| Scheme 13 Synthesis of P18via Suzuki–Miyaura coupling. | ||
From these observations, the synthesis of the two other copolymers was carried out via DHAP (Scheme 14). After optimization, P19 and P20 were isolated in 77% and 87% yields, respectively, and exhibit a Mn of 24 kDa and 20 kDa, respectively. From the respective cyclic voltammograms (CV) and UV-abs, the energy levels of the copolymers P19 and P20 were found to be relatively stabilized with LUMO levels of around −4.2 eV, i.e., close to that of PC61BM.89 Moreover, while these two polymers show no electrochemical reversibility in the oxidation process, polymer P18 shows reversibility in its oxidation as well as reduction. In addition, the latter is characterized by a higher LUMO energy level of −4.0 eV and a HOMO energy level of −5.3 eV. As a result, P19 and P20 could be ideal candidates as n-type polymers and can possibly be used in all-polymer solar cells. In parallel, thin film transistor properties were investigated. P19, P20 and P18 exhibit electron mobilities of ca. 2.0 × 10−4 cm2 V−1 s−1, 2.5 × 10−3 cm2 V−1 s−1 and 1.6 × 10−4 cm2 V−1 s−1, respectively, suggesting that using a BTPD unit instead of a TPD unit contributes to increase the electron mobility due to the centrosymmetric structure of the BTPD unit. Indeed, because of the improved packing properties, copolymers containing isoindigo and centrosymmetric co-monomers may have higher charge transport properties.90 Finally, these results show an almost comparable electron mobility of that characterizing PC61BM in the thin film (∼10−3 cm2 V−1 s−1)91,92 meaning that they can have promising features and great potential as n-type materials in organic photovoltaics.
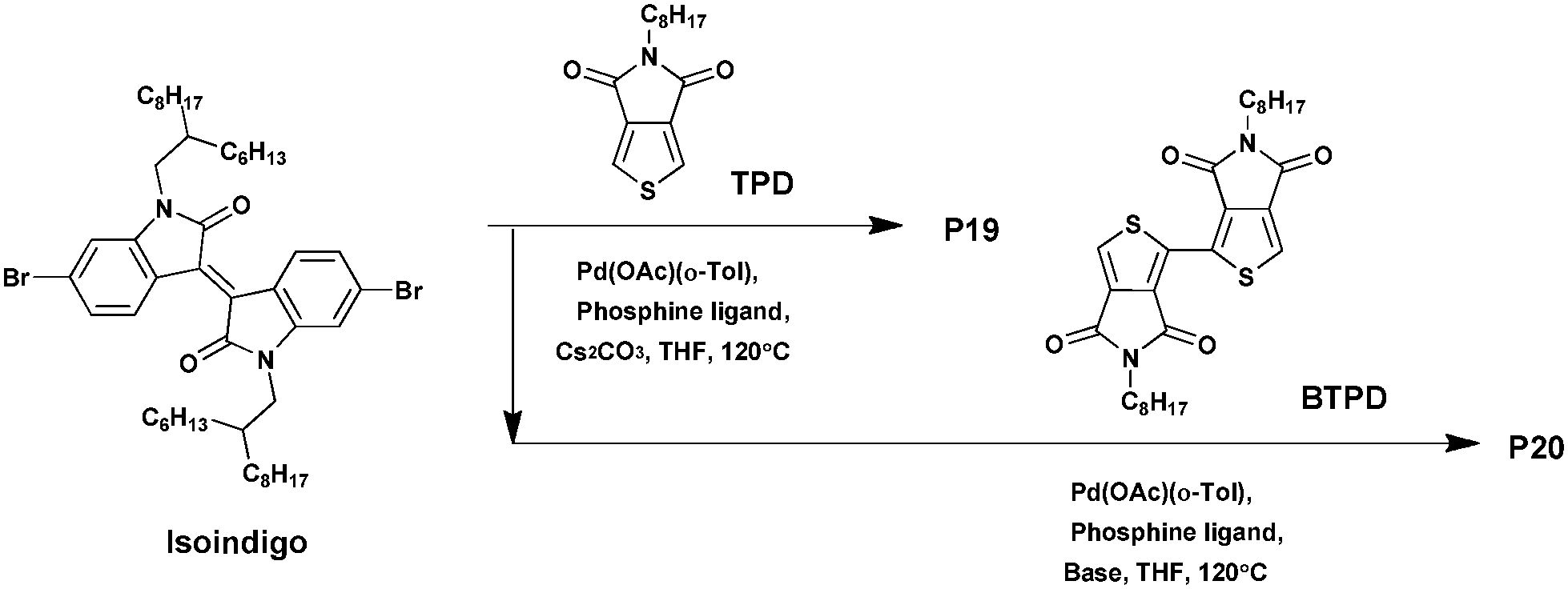 | ||
| Scheme 14 Synthesis of P19 and P20via DHAP. | ||
Furthermore, Elsawy et al.93 recently reported the synthesis and characterization of a series of D–A–D–A′ copolymers incorporating different strong acceptors, i.e., the isoindigo (A) unit and 4,7-dibromo[c][1,2,5]-(oxa, thia, or selena)diazole (A′) with 3,4-ethylenedioxythiophene as the donor (D) via DHAP (Scheme 15). In this study, the effects of different heteroatoms namely the oxygen, sulfur and selenium borne by the benzimidazole unit on the photovoltaic properties were investigated. At first, the authors prepared the 6,6′-bis(2,3-dihydrothieno[3,4-b][1,4]-dioxin-5-yl)1,1′-bis-(2-octyldodecyl)-[3,3′-biindolinylidene]-2,2′-dione (IDED) polymer via conventional Stille coupling reaction with a high yield of ca. 80%. Then the preparation of three polymers, namely PIDEDO (P21), PIDES (P22) and PIDEDSe (P23), was carried out under CH-arylation activation in high yields (>80%). Molecular weights ranging from 15.2 kDa to 17.3 kDa were estimated using gel permeation chromatography. Thin-film UV/Vis spectra exhibit red-shifted absorption profiles demonstrating more π–π stacking and ordering in the solid state. In addition, moving from O to Se results in a bathochromic shift with a difference of ca. 68 nm (λmax) between P21 and P23.
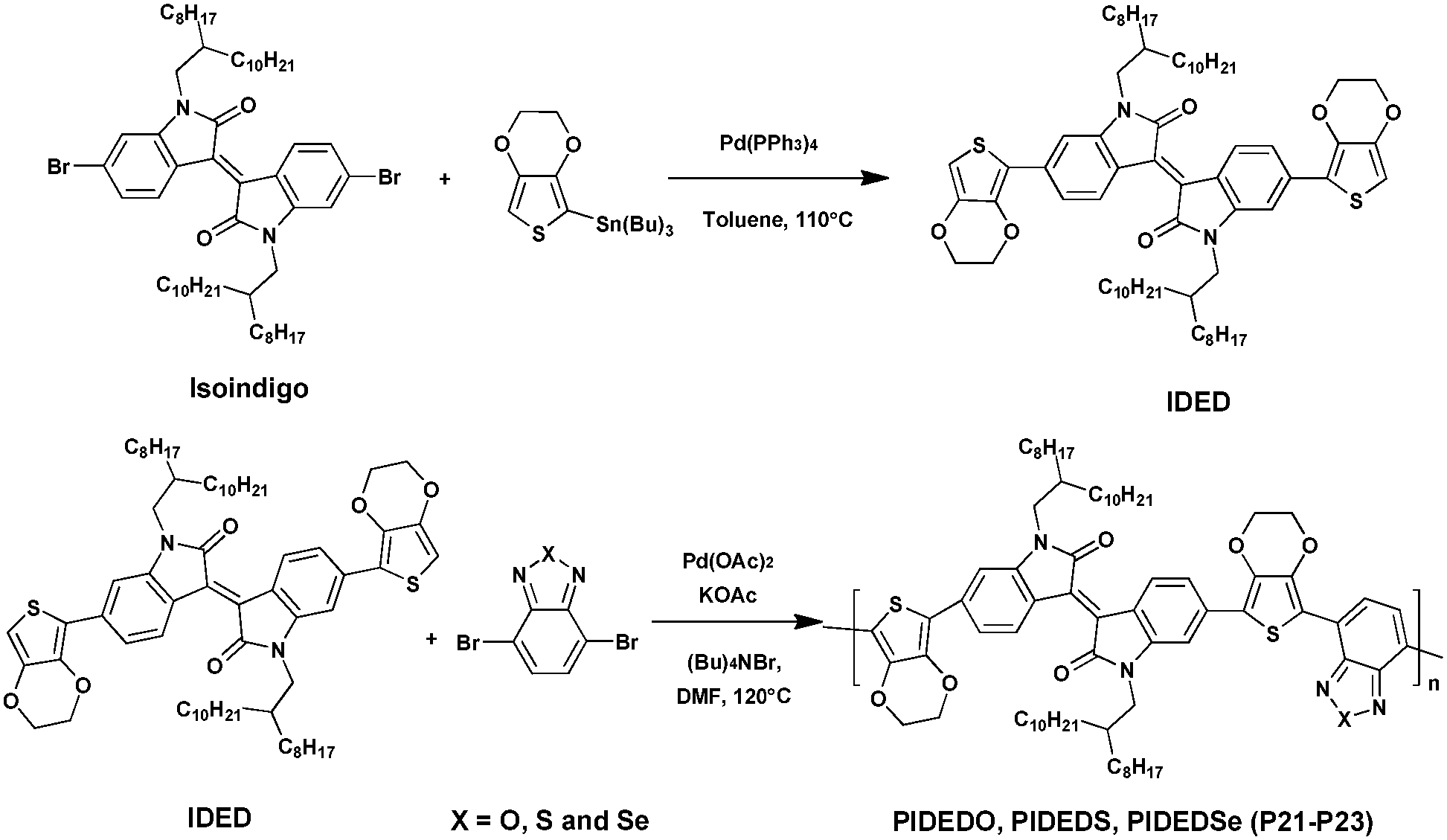 | ||
| Scheme 15 Preparation of P21–P23via DHAP. | ||
Next, the photovoltaic performances of three polymers as donor materials were assessed through the preparation of PC61BM based BHJ. The device performances are summarized in Table 1. Compared with the P21-based device, the PCEs of the two other devices were significantly higher, due to the well-improved short-circuit current density (Jsc) parameters. This enhancement can be attributed to the higher electron densities and the appropriate morphology of the films.94 Moreover, a comparison of thiophene and selenophene based devices revealed a superior charge balance of the P22:PC61BM blend resulting in its higher Jsc.
| Active layer | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) |
|---|---|---|---|---|
| P21 | 2.27 | 0.58 | 46.0 | 0.61 |
| P22 | 8.10 | 0.56 | 35.0 | 1.60 |
| P23 | 7.13 | 0.56 | 34.0 | 1.36 |
The charge transport properties of each material were estimated using OFETs and the hole mobilities of 1.9 × 10−4, 4.0 × 10−4 and 3.5 × 10−4 cm2 V−1 s−1 were measured for P21, P22 and P23, respectively. From Table 2, the blend of P22:PC61BM exhibits the highest electron mobility and a good charge balance due to its appropriate film morphology.
| Active layer | μ e (cm2 V−1 s−1) | μ h (cm2 V−1 s−1) |
|---|---|---|
| P21:PC61BM | 1.85 × 10−6 | 5.41 × 10−4 |
| P22:PC61BM | 2.31 × 10−4 | 4.46 × 10−4 |
| P23:PC61BM | 7.45 × 10−5 | 2.25 × 10−4 |
Although high yields and high molecular weights of isoindigo-based copolymers are reachable via DHAP, the biggest challenge still remains, that being its reproducibility. In this context, Leclerc et al.6 reported for the first time continuous flow methods applied to DHAP. This inexpensive technology uses a reactor under fixed and constant reaction conditions.95,96 From this set up a new D–A polymer containing a strong electron-donating unit, namely 3,4-ethylenedioxythiophene (EDOT), and isoindigo (iI) as the acceptor (A), was synthesized and characterized with an Mn ranging from 34 to 42 kDa (Scheme 16). In addition, OFET and BHJ solar cells were fabricated to estimate the charge carrier mobility and organic photovoltaic performance, respectively. A comparison of PiIEDOT (P24) synthesized in a conventional flask reveals a similar hole mobility of ca. 2 × 10−3 cm2 V−1 s−1 and a comparable PCE of ca. 1.74% and 1.80%, respectively, when blended with PC71BM. Although the Voc and fill factor (FF) are nearly identical, the main limitation, impacting the PCEs, comes from the low Jsc recorded in both cases (5.0 mA cm−2). However, it is noteworthy that both polymers exhibit comparable molecular weights. Consequently, these promising results confirm the great potential of the continuous flow methods.
 | ||
| Scheme 16 Polymerization of P24via DHAP. | ||
4.4 Polymers containing diketopyrrolopyrrole (DPP)
Due to the strong electron-withdrawing nature, intermolecular hydrogen bonding, planar backbone and strong π–π stacking interactions, the DPP moiety has been widely used for the preparation of active macromolecules.97–99 For instance, high performance DPP-based polymers with PCEs of up to 8% were reported.100–102Recently, Kuwabara et al.103 discussed the optimization of reaction conditions for the synthesis of height diketopyrrolopyrrole-based conjugated polymers via DHAP (Scheme 17). In this study, Kuwabara et al. report on the importance of reaction time to afford high-molecular-weight polymers in DHAP while avoiding overreactions in unexpected C–H bonds. Thus, polymer P25 (copolymerized by 2,5-di-(2-ethylhexyl)-3,6-bis(4-bromophenyl)pyrrolo[3,4-c]pyrrole-1,4-dione and 3,3′,4,4′-tetramethylbithiophene) was synthesized with a reaction time of 6 h, isolated in good yield, and characterized by a high molecular weight (Mn of 18.1 kDa). Moreover, since the polymer shows wide absorption in the visible region, as well as the low-lying HOMO energy level, it was investigated as a potential donor in BHJ solar cells.89,104 When blended with PC61BM, a Voc of 1.01 V, a Jsc of 2.93 mA cm−2, a FF of 0.30 and a resulting PCE of 0.89% were achieved for P25. The device exhibited high Voc attributed to the low-lying HOMO energy level and an optimized morphology. The low Jsc and FF may be due to the twisted structure of the polymer, which is not suitable for carrier transport.105,106 Even though the PCE value of the device is much lower than those typically recorded using P3AT-based devices,107–109 the results of this study point out that DHAP can be used commendably to synthesize polymers for PSCs.110
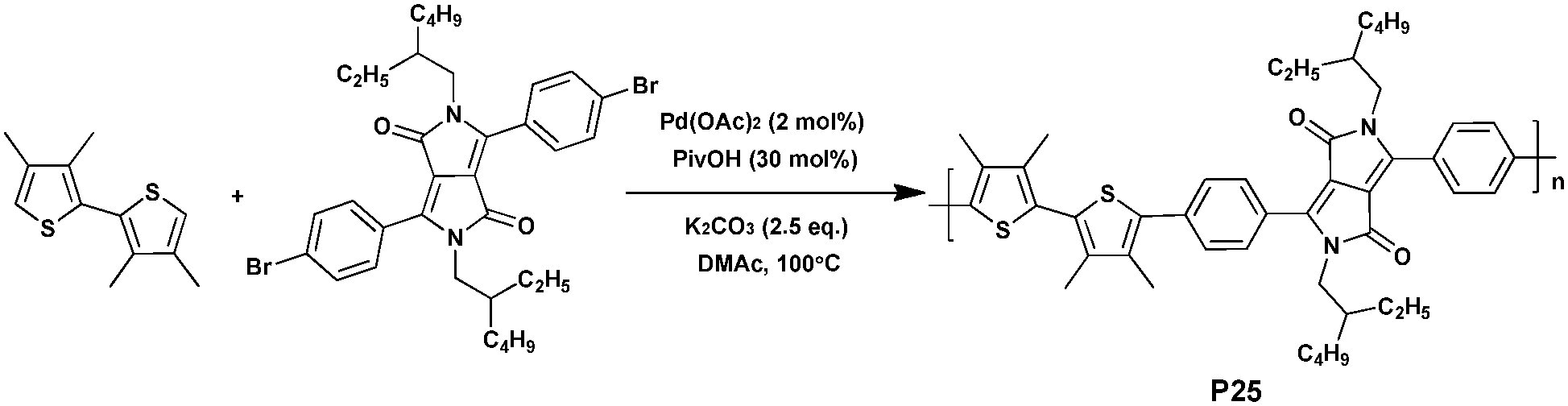 | ||
| Scheme 17 Synthesis of P25via DHAP. | ||
Guo et al.46 conducted a series of studies on DTDPP-based polymers synthesized via DHAP in 2013 (Scheme 18). The DTDPP-based copolymers exhibited outstanding photoelectronic properties, such as a low bandgap and high hole mobilities, making them key prospects for application in optoelectronic materials.111 Under the optimized reaction conditions of 5 mol% of Pd(OAc)2, 10 mol% of PCy3·HBF4 (Cy = cyclohexyl), 2.5 equivalents of K2CO3 and 1.0 equivalents of pivalic acid in the mixed solvent of DMAc/xylene (1:1), both electron-donating units (P28, P29, P30 and P31) and electron-withdrawing units (P26 and P27) were successfully achieved with high yields of up to 94% and comparable molecular weights of up to 45 kDa (summarized in Table 3).
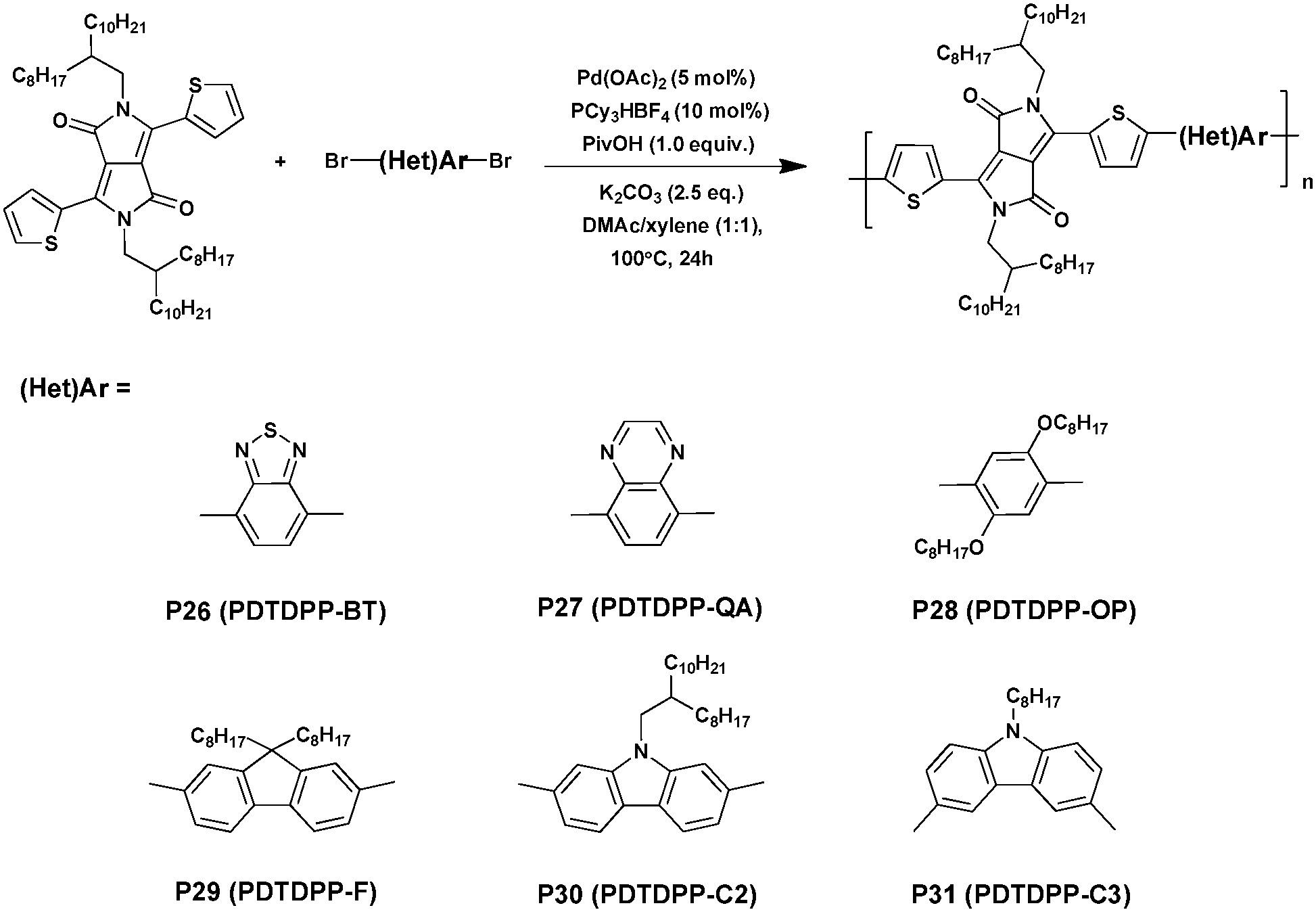 | ||
| Scheme 18 Synthesis of P26–P31via DHAP. | ||
| Polymer | M n (kDa) | PDI | Yield (%) |
|---|---|---|---|
| P26 | 23.5 | 4.13 | 90 |
| P27 | 23.5 | 4.36 | 92 |
| P28 | 36.7 | 3.46 | 94 |
| P29 | 27.7 | 1.72 | 35 |
| P30 | 45.0 | 3.23 | 90 |
| P31 | 10.2 | 1.73 | 91 |
What’s more, the optical and electrochemical properties of the polymers are characterized and summarized in Table 4.
| Polymer | UV-vis absorption spectra | Cyclic voltammetry | |||
|---|---|---|---|---|---|
| λ onset (nm) | E optg (eV) | HOMO (eV) | LUMO (eV) | E CVg (eV) | |
| P26 | 1015 | 1.22 | −5.29 | −3.70 | 1.59 |
| P27 | 1010 | 1.23 | −5.17 | −3.64 | 1.53 |
| P28 | 932 | 1.33 | −5.11 | −3.68 | 1.43 |
| P29 | 885 | 1.40 | −5.38 | −3.56 | 1.82 |
| P30 | 923 | 1.34 | −5.10 | −3.62 | 1.48 |
| P31 | 909 | 1.36 | −5.00 | −3.64 | 1.36 |
Compared with the analogues synthesized through Suzuki or Stille coupling reported previously,112–115 polymers based on DHAP showed a difference of about 10–40 nm in the absorption maxima, which may be interpreted as the diverse Mn, branching structures from the side reactions and morphology in the solid state. In comparison with the three polymers P28, P29 and P30, P26 and P27 exhibited broader absorption bands (up to 1000 nm) and bathochromic shifted absorption maxima. These phenomena imply that a donor–acceptor–donor–acceptor (D–A–D–A) sequence involving DTDPP (regarded as an inherent D–A–D unit) and an acceptor unit offers advantages for the red shift of the absorption band.113 In addition, P26 and P27 exhibited remarkable optical bandgaps (Eoptg) down to 1.22 eV, which were estimated from the absorption band edge in film (λonset). Additionally, the bandgaps (ECVg) estimated using CV were somewhat larger than Eoptg, which can be attributed to the exciton binding energy of polymers between the polymer film and the electrode.115,116 So DHAP can not only offer an approach to select low-bandgap and NIR absorbing polymers, but can also accelerate the discovery of high-performance organic photovoltaics.
In 2015, Homyak et al.117 used a synthetic method of DHAP to prepare four DPP-based polymers (Scheme 19). High molecular weight (Mn) polymers ranging from 10 to 30 kDa were obtained; the polymers were applied in OPV as well as OFET devices. While PDPPTTT (P32) and PDPPTPT (P33) achieved an average PCE of 3.8–3.9%, their fluorinated analogues (PDPPTTfT (P34) and PDPPTPfT (P35)) show much lower efficiencies mainly due to an inappropriate energy gap between the LUMO energy levels of the polymers and the fullerene derivative. However, although OFET devices revealed that all materials showed high hole mobilities within the same order of magnitude (ca. 10−2 cm2 V−1 s−1), values obtained for the fluorinated derivatives P34 and P35 materials are 2–3 times higher than that for the P32 and P33 materials.
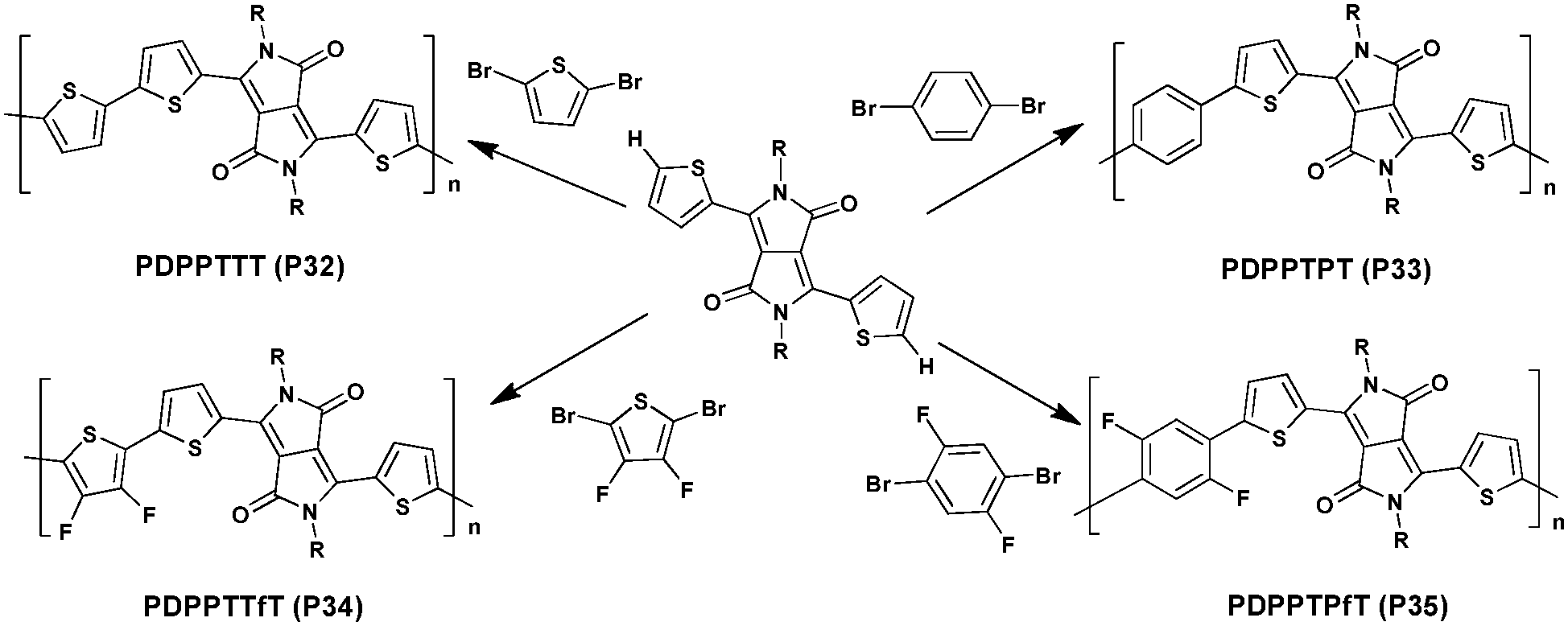 | ||
| Scheme 19 Synthesis of P32–P35via DHAP. | ||
To further improve the charge transport properties, the use of oligothiophene derivatives appears to be an effective option.118 However, the latter often display low reactivity when involved in direct arylation,119 and the multiple C–H bonds borne by the aromatic rings may lead to undesired cross-linked defects.72,120,121 Consequently, to solve these problems fluorine-atoms were introduced in the β-positions of thiophene rings (position 3 and 4) and a new DPP-based high-mobility conjugated polymer containing (E)-1,2-bis(3,4-difluorothien-2-yl)ethene (4FTVT) was synthesized via DHAP in high yield (93%) by Gao et al.122 (Scheme 20). Under optimal conditions using Herrmann's catalyst, a polymer with an Mn of 60 kDa was obtained.
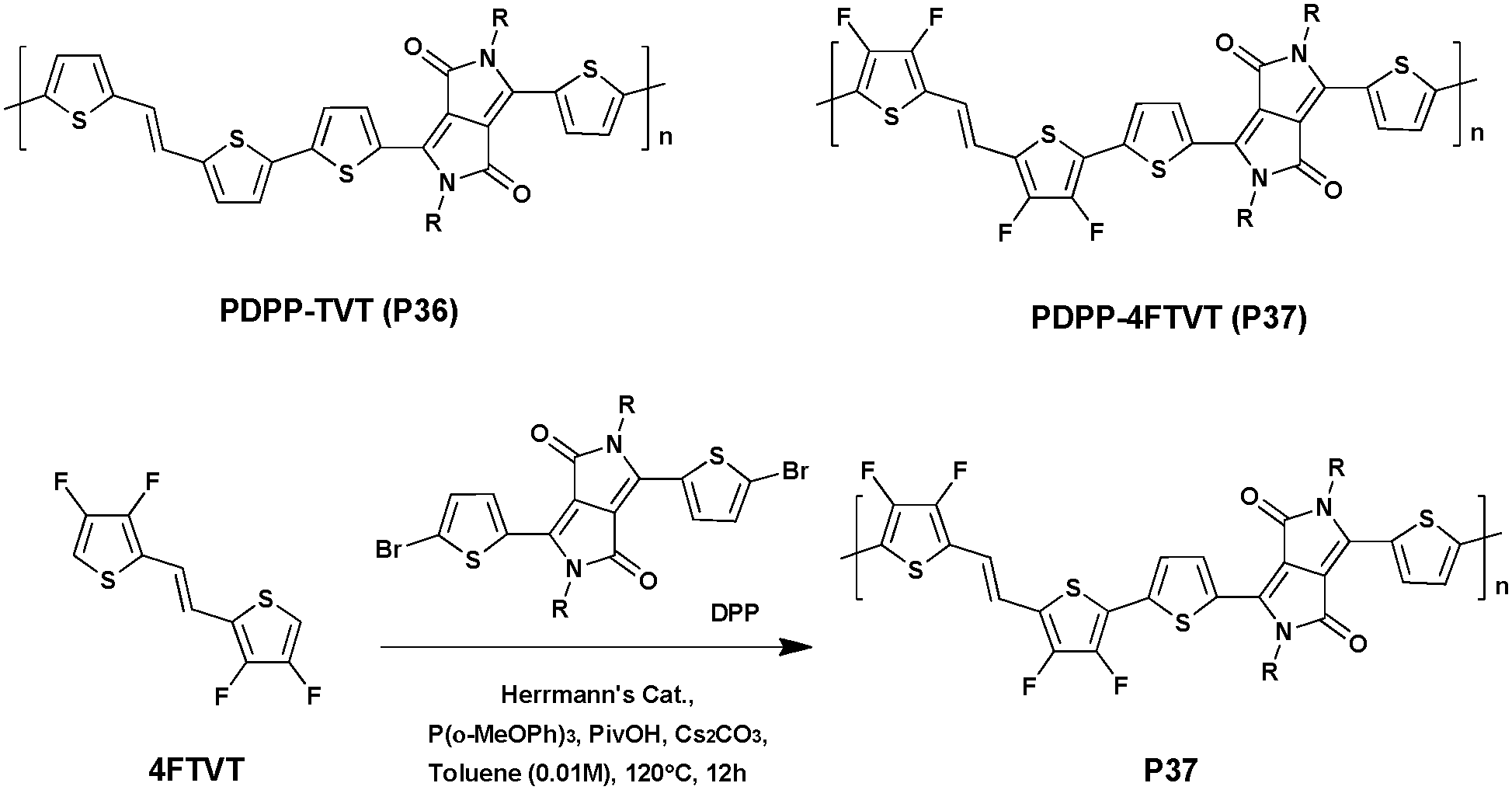 | ||
| Scheme 20 Polymerization of P36 and P37via DHAP. | ||
The HOMO and LUMO energy levels of PDPP–4FTVT (P37) were estimated using CV and UV-abs. The incorporation of a fluorine-atom resulted in a significant stabilization of both the HOMO and LUMO energy levels (−5.36 eV and −3.50 eV vs. 5.17 eV and −3.35 eV for the reference material PDPP–TVT (P36)). From bottom-gate and top-contact OFETs P37 shows ambipolar characteristics and a hole mobility (μh) of ca. 3.40 cm2 V−1 s−1vs. an electron mobility (μe) of ca. 5.86 cm2 V−1 s−1. It is noteworthy that these values are comparable to those reached by analogous polymers synthesized via the Stille cross coupling reaction.123–127
5. Summary and outlook
This review summarizes and gathers data on several donor–acceptor conjugated polymers synthesized via direct(hetero)-arylation polymerization with applications in organic electronics. The use of DHAP as a route toward the formation of the specific C–C bond has been a great challenge in organic chemistry over the past twenty years, but the results reported herein clearly demonstrate that the way has been paved. According to the recent literature, DHAP offers a powerful approach for the synthesis of efficient active materials, and is a strong substitute for conventional cross-coupling reactions. However, there are still shortcomings that cannot be ignored. For some monomers the reactions are actually controllable with difficulties generating branched, cross-linked polymers and/or by-products due to the activation of more than one C–H bond. Additionally, the effects of the steric hindrance of the monomers may have an impact on the polymerization and should also be taken into consideration. Furthermore, it is worth noting that the reaction conditions require specific optimization for each monomer. There are actually, to date, no universal catalytic systems. Under palladium catalysis many parameters including the nature of the additive(s), solvent, ligand(s), reaction temperature and time have a great impact on the conversion. Therefore, the optimum reaction conditions of this non-stationary catalytic system should be achieved by repeated experiments and data analysis. Many questions still remain unanswered and much further work needs to be conducted. Nevertheless, comparisons of materials obtained via DHAP with their analogues prepared via conventional cross-coupling reactions clearly show that DHAP is a method of choice. Indeed, efficient materials prepared using DHAP for polymer solar cells and OFETs have already been reported and this is hopefully just the beginning.Acknowledgements
The authors acknowledge financial support from the National Key R&D Program of “Strategic Advanced Electronic Materials” (No. 2016YFB0401100), the National Natural Science Foundation of China (Grant No. 61574077), and the Natural Science Foundation of Jiangsu (No. SBK2016022423).Notes and references
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed.
- A. Suzuki, Chem. Commun., 2005, 4759 RSC.
- C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS PubMed.
- P. Morin, T. Bura, B. Sun, S. I. Gorelsky, Y. Li and M. Leclerc, ACS Macro Lett., 2015, 4, 21 CrossRef CAS.
- M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807 CrossRef CAS PubMed.
- F. Grenier, B. R. Aïch, Y.-Y. Lai, M. Guérette, A. B. Holmes, Y. Tao, W. W. H. Wong and M. Leclerc, Chem. Mater., 2015, 27, 2137 CrossRef CAS.
- A. E. Rudenko and B. C. Thompson, Macromolecules, 2015, 48, 569 CrossRef CAS.
- T. Bura, J. T. Blaskovits and M. Leclerc, J. Am. Chem. Soc., 2016, 138, 10056 CrossRef CAS PubMed.
- D. H. Wang, A. Pron, M. Leclerc and A. J. Heeger, Adv. Funct. Mater., 2013, 23, 1297 CrossRef CAS.
- S.-Y. Liu, W.-Q. Liu, J.-Q. Xu, C.-C. Fan, W.-F. Fu, J. Ling, J.-Y. Wu, M.-M. Shi, A. K. Y. Jen and H.-Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 6765 CAS.
- P. D. Homyak, Y. Liu, J. D. Harris, F. Liu, K. R. Carter, T. P. Russell and E. B. Coughlin, Macromolecules, 2016, 49, 3028 CrossRef CAS.
- A. E. Rudenko, P. P. Khlyabich and B. C. Thompson, ACS Macro Lett., 2014, 3, 387 CrossRef CAS.
- M. Shaker, C. K. Trinh, W. Kim, H. Kim, K. Lee and J.-S. Lee, New J. Chem., 2015, 39, 4957 RSC.
- S.-W. Chang, H. Waters, J. Kettle and M. Horie, Org. Electron., 2012, 13, 2967 CrossRef CAS.
- S. Wang, J. Yang, K. Broch, J. Novák, X. Cao, J. Shaw, Y. Tao, Y. Hu and W. Huang, RSC Adv., 2016, 6, 57163 RSC.
- J.-R. Pouliot, B. Sun, M. Leduc, A. Najari, Y. Li and M. Leclerc, Polym. Chem., 2015, 6, 278 RSC.
- P. Sonar, T. R. Foong and A. Dodabalapur, Phys. Chem. Chem. Phys., 2014, 16, 4275 RSC.
- A. D. Hendsbee, J.-P. Sun, L. R. Rutledge, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 4198 CAS.
- M. K. Poduval, P. M. Burrezo, J. Casado, J. T. López Navarrete, R. P. Ortiz and T.-H. Kim, Macromolecules, 2013, 46, 9220 CrossRef CAS.
- W. Lu, J. Kuwabara, T. Iijima, H. Higashimura, H. Hayashi and T. Kanbara, Macromolecules, 2012, 45, 4128 CrossRef CAS.
- M.-L. Sun, W.-S. Zhu, Z.-S. Zhang, C.-J. Ou, L.-H. Xie, Y. Yang, Y. Qian, Y. Zhao and W. Huang, J. Mater. Chem. C, 2015, 3, 94 RSC.
- Y. Zou, A. Najari, P. Berrouard, S. Beaupre, B. Reda Aich, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330 CrossRef CAS PubMed.
- C. Liu, C. Yi, K. Wang, Y. Yang, R. S. Bhatta, M. Tsige, S. Xiao and X. Gong, ACS Appl. Mater. Interfaces, 2015, 7, 4928 CAS.
- J. Zhang, Y. Zhang, J. Fang, K. Lu, Z. Wang, W. Ma and Z. Wei, J. Am. Chem. Soc., 2015, 137, 8176 CrossRef CAS PubMed.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- Z. B. Henson, K. Müllen and G. C. Bazan, Nat. Chem., 2012, 4, 699 CrossRef CAS PubMed.
- E. Iizuka, M. Wakioka and F. Ozawa, Macromolecules, 2015, 48, 2989 CrossRef CAS.
- M. Wakioka, N. Ichihara, Y. Kitano and F. Ozawa, Macromolecules, 2014, 47, 626 CrossRef CAS.
- K. Okamoto, J. Zhang, J. B. Housekeeper, S. R. Marder and C. K. Luscombe, Macromolecules, 2013, 46, 8059 CrossRef CAS.
- J. Kuwabara, T. Yasuda, S. J. Choi, W. Lu, K. Yamazaki, S. Kagaya, L. Han and T. Kanbara, Adv. Funct. Mater., 2014, 24, 3226 CrossRef CAS.
- N. Allard, A. Najari, J. Pouliot, A. Pron, F. Grenier and M. Leclerc, Polym. Chem., 2012, 3, 2875 RSC.
- N. Allard, S. Beaupre, B. R. Aich, A. Najari, Y. Tao and M. Leclerc, Macromolecules, 2011, 44, 7184 CrossRef CAS.
- M. Pomerantz, Tetrahedron Lett., 2003, 44, 1563 CrossRef CAS.
- H. Burckstummer, A. Weissenstein, D. Bialas and F. Wurthner, J. Org. Chem., 2011, 76, 2426 CrossRef PubMed.
- G. Marzano, D. Kotowski, F. Babudri, R. Musio, A. Pellegrino, S. Luzzati, R. Po and G. M. Farinola, Macromolecules, 2015, 48, 7039 CrossRef CAS.
- D. Kotowski, S. Luzzati, G. Bianchi, A. Calabrese, A. Pellegrino, R. Po, G. Schimperna and A. Tacca, J. Mater. Chem. A, 2013, 1, 10736 CAS.
- A. E. Rudenko and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 135 CrossRef CAS.
- A. E. Rudenko and B. C. Thompson, Macromolecules, 2015, 48, 569 CrossRef CAS.
- Q. Wang, M. Wakioka and F. Ozawa, Macromol. Rapid Commun., 2012, 33, 1203 CrossRef CAS PubMed.
- A. E. Rudenko, C. A. Wiley, J. F. Tannaci and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2660 CrossRef CAS.
- C. H. Woo, B. C. Thompson, B. J. Kim, M. F. Toney and J. M. J. Frechet, J. Am. Chem. Soc., 2008, 130, 16324 CrossRef CAS PubMed.
- R. D. Mccullough, Adv. Mater., 1998, 2, 93 CrossRef.
- Q. Wang, R. Takita, Y. Kikuzaki and F. Ozawa, J. Am. Chem. Soc., 2010, 132, 11420 CrossRef CAS PubMed.
- T. Bura, P. O. Morin and M. Leclerc, Macromolecules, 2015, 48, 5614 CrossRef CAS.
- Q. Guo, J. Dong, D. Wan, D. Wu and J. You, Macromol. Rapid Commun., 2013, 34, 522 CrossRef CAS PubMed.
- A. P. Zoombelt, S. G. J. Mathijssen, M. G. R. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2010, 20, 2240 RSC.
- C. Kanimozhi, N. Yaacobi-Gross, K. W. Chou, A. Amassian, T. D. Anthopoulos and S. Patil, J. Am. Chem. Soc., 2012, 134, 16532 CrossRef CAS PubMed.
- C. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416 RSC.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. N. Plesu, M. Belletete, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS PubMed.
- J. Hou, M. Park, S. Zhang, Y. Yao, L. Chen, J. Li and Y. Yang, Macromolecules, 2008, 41, 6012 CrossRef CAS.
- X. Kang, J. Zhang, D. O’Neil, A. J. Rojas, W. Chen, P. Szymanski, S. R. Marder and M. A. El-Sayed, Chem. Mater., 2014, 26, 4486 CrossRef CAS.
- X. Wang, K. Wang and M. Wang, Polym. Chem., 2015, 6, 1846 RSC.
- X. Wang, Z.-G. Zhang, H. Luo, S. Chen, S. Yu, H. Wang, X. Li, G. Yu and Y. Li, Polym. Chem., 2014, 5, 502 RSC.
- M. Tomar, A. Z. Ashar, K. S. Narayan, K. Müllen and J. Jacob, J. Polym. Res, 2016 DOI:10.1007/s10965-016-0945-1.
- M. Tomar, N. T. Lucas, H. Kim, F. Laquai, K. Müllen and J. Jacob, Polym. Int., 2012, 61, 1318 CrossRef CAS.
- M. Tomar, N. T. Lucas, M. G. Gardiner, K. Muellen and J. Jacob, Tetrahedron Lett., 2012, 53, 285 CrossRef CAS.
- C. Piliego, T. W. Holcombe, J. D. Douglas, C. H. Woo, P. M. Beaujuge and J. M. J. Frechet, J. Am. Chem. Soc., 2010, 132, 7595 CrossRef CAS PubMed.
- C. Cabanetos, A. E. Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. J. Frechet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656 CrossRef CAS PubMed.
- G. Zhang, Y. Fu, Q. Zhang and Z. Xie, Chem. Commun., 2010, 46, 4997 RSC.
- X. Guo, N. Zhou, S. J. Lou, J. Smith, D. B. Tice, J. W. Hennek, R. P. Ortiz, J. T. L. Navarrete, S. Li, J. Strzalka, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Nat. Photonics, 2013, 7, 825 CrossRef CAS.
- K. R. Graham, C. Cabanetos, J. P. Jahnke, N. M. Idso, A. E. Labban, G. O. N. Ndjawa, T. Heumueller, K. Vandewal, A. Salleo, B. F. Chmelka, A. Amassian, P. M. Beaujuge and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 9608 CrossRef CAS PubMed.
- L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597 CrossRef CAS PubMed.
- P. Berrouard, A. Najari, A. Pron, D. Gendron, P. Morin, J. Pouliot, J. Veilleux and M. Leclerc, Angew. Chem., Int. Ed., 2012, 51, 2068 CrossRef CAS PubMed.
- J. Jo, A. Pron, P. Berrouard, W. L. Leong, J. D. Yuen, J. S. Moon, M. Leclerc and A. J. Heeger, Adv. Energy Mater., 2012, 2, 1397 CrossRef CAS.
- Y. Li and Y. Zou, Adv. Mater., 2008, 20, 2952 CrossRef CAS.
- F. Lombeck, H. Komber, S. I. Gorelsky and M. Sommer, ACS Macro Lett., 2014, 3, 819 CrossRef CAS.
- S. Broll, F. Nübling, A. Luzio, D. Lentzas, H. Komber, M. Caironi and M. Sommer, Macromolecules, 2015, 48, 7481 CrossRef CAS.
- S. Kowalski, S. Allard and U. Scherf, Macromol. Rapid Commun., 2015, 36, 1061 CrossRef CAS PubMed.
- K. Wang, G. Wang and M. Wang, Macromol. Rapid Commun., 2015, 36, 2162 CrossRef CAS PubMed.
- W. Lu, J. Kuwabara and T. Kanbara, Macromolecules, 2011, 44, 1252 CrossRef CAS.
- Y. Fujinami, J. Kuwabara, W. Lu, H. Hayashi and T. Kanbara, ACS Macro Lett., 2012, 1, 67 CrossRef CAS.
- L. G. Mercier, B. R. Aïch, A. Najari, S. Beaupré, P. Berrouard, A. Pron, A. Robitaille, Y. Tao and M. Leclerc, Polym. Chem., 2013, 4, 5252 RSC.
- E. Iizuka, M. Wakioka and F. Ozawa, Macromolecules, 2016, 49, 3310 CrossRef CAS.
- T. Lei, Y. Cao, Y. Fan, C. Liu, S. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099 CrossRef CAS PubMed.
- G. Zhang, Y. Fu, Z. Xie and Q. Zhang, Macromolecules, 2011, 44, 1414 CrossRef CAS.
- R. Stalder, J. Mei and J. R. Reynolds, Macromolecules, 2010, 43, 8348 CrossRef CAS.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303 CrossRef CAS.
- Z. Ma, E. Wang, M. E. Jarvid, P. Henriksson, O. Inganas, F. Zhang and M. R. Andersson, J. Mater. Chem., 2012, 22, 2306 RSC.
- K. Mahmood, Z.-P. Liu, C. Li, Z. Lu, T. Fang, X. Liu, J. Zhou, T. Lei, J. Pei and Z. Bo, Polym. Chem., 2013, 4, 3563 RSC.
- P. Sonar, H.-S. Tan, S. Sun, Y. M. Lam and A. Dodabalapur, Polym. Chem., 2013, 4, 1983 RSC.
- Y. Deng, J. Liu, J. Wang, L. Liu, W. Li, H. Tian, X. Zhang, Z. Xie, Y. Geng and F. Wang, Adv. Mater., 2014, 26, 471 CrossRef CAS PubMed.
- L. Fang, Y. Zhou, Y.-X. Yao, Y. Diao, W.-Y. Lee, A. L. Appleton, R. Allen, J. Reinspach, S. C. B. Mannsfeld and Z. Bao, Chem. Mater., 2013, 25, 4874 CrossRef CAS.
- Z. Ma, D. Dang, Z. Tang, D. Gedefaw, J. Bergqvist, W. Zhu, W. Mammo, M. R. Andersson, O. Inganäs, F. Zhang and E. Wang, Adv. Energy Mater., 2014, 4, 1301455 CrossRef.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2012, 134, 20025 CrossRef CAS PubMed.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457 CrossRef CAS PubMed.
- F. Grenier, P. Berrouard, J. Pouliot, H.-R. Tseng, A. J. Heeger and M. Leclerc, Polym. Chem., 2013, 4, 1836 RSC.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS.
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762 CrossRef CAS.
- E. von Hauff, V. Dyakonov and J. Parisi, Sol. Energy Mater. Sol. Cells, 2005, 87, 149 CrossRef CAS.
- C. Waldauf, P. Schilinsky, M. Perisutti, J. Hauch and C. J. Brabec, Adv. Mater., 2003, 15, 2084 CrossRef CAS.
- W. Elsawy, H. Kang, K. Yu, A. Elbarbary, K. Lee and J.-S. Lee, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2926 CrossRef CAS.
- D. Gebeyehua, C. J. Brabeca, F. Padingerb, T. Fromherzb, J. C. Hummelenc, D. Badtd, H. Schindlerd and N. S. Sariciftcia, Synth. Met., 2001, 118, 1 CrossRef.
- K. Geyer, J. D. C. Codée and P. H. Seeberger, Chem. – Eur. J., 2006, 12, 8434 CrossRef CAS PubMed.
- G. Jas and A. Kirschning, Chem. – Eur. J., 2003, 9, 5708 CrossRef CAS PubMed.
- Y. S. Kwon, J. Lim, H. Yun, Y. Kim and T. Park, Energy Environ. Sci., 2014, 7, 1454 CAS.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Energy Environ. Sci., 2012, 5, 6857 CAS.
- W. Li, W. S. C. Roelofs, M. Turbiez, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2014, 26, 3304 CrossRef CAS PubMed.
- L. Dou, J. You, J. Yang, C.-C. Chen, Y. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180 CrossRef CAS.
- W. Li, A. Furlan, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 5529 CrossRef CAS PubMed.
- K. H. Hendriks, G. H. L. Heintges, V. S. Gevaerts, M. M. Wienk and R. A. J. Janssen, Angew. Chem., Int. Ed., 2013, 52, 8341 CrossRef CAS PubMed.
- J. Kuwabara, Y. Nohara, S. J. Choi, Y. Fujinami, W. Lu, K. Yoshimura, J. Oguma, K. Suenobu and T. Kanbara, Polym. Chem., 2013, 4, 947 RSC.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- H. Zhou, L. Yang, S. Xiao, S. Liu and W. You, Macromolecules, 2010, 43, 811 CrossRef CAS.
- Q. Shi, H. Fan, Y. Liu, J. Chen, L. Ma, W. Hu, Z. Shuai, Y. Li and X. Zhan, Macromolecules, 2011, 44, 4230 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
- P. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456 CrossRef CAS.
- A. Facchetti, Chem. Mater., 2011, 23, 733 CrossRef CAS.
- S.-W. Chang, H. Waters, J. Kettle, Z.-R. Kuo, C.-H. Li, C.-Y. Yu and M. Horie, Macromol. Rapid Commun., 2012, 33, 1927 CrossRef CAS PubMed.
- Y. Zou, D. Gendron, R. Neagu-Plesu and M. Leclerc, Macromolecules, 2009, 42, 6361 CrossRef CAS.
- E. Zhou, S. Yamakawa, K. Tajima, C. Yang and K. Hashimoto, Chem. Mater., 2009, 21, 4055 CrossRef CAS.
- S. Cho, J. Lee, M. Tong, J. H. Seo and C. Yang, Adv. Funct. Mater., 2011, 21, 1910 CrossRef CAS.
- S. Qu and H. Tian, Chem. Commun., 2012, 48, 3039 RSC.
- Y. Li, S. P. Singh and P. Sonar, Adv. Mater., 2010, 22, 4862 CrossRef CAS PubMed.
- L. Dou, W.-H. Chang, J. Gao, C.-C. Chen, J. You and Y. Yang, Adv. Mater., 2013, 25, 825 CrossRef CAS PubMed.
- P. Homyak, Y. Liu, F. Liu, T. P. Russel and E. B. Coughlin, Macromolecules, 2015, 48, 6978 CrossRef CAS.
- Z. Yi, S. Wang and Y. Liu, Adv. Mater., 2015, 27, 3589 CrossRef CAS PubMed.
- J. Kuwabara, K. Yamazaki, T. Yamagata, W. Tsuchida and T. Kanbara, Polym. Chem., 2015, 6, 891 RSC.
- A. E. Rudenko, C. A. Wiley, S. M. Stone, J. F. Tannaci and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3691 CrossRef CAS.
- A. E. Rudenko and B. C. Thompson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 135 CrossRef CAS.
- Y. Gao, X. Zhang, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2015, 27, 6753 CrossRef CAS PubMed.
- J. Lee, A. Han, J. Kim, Y. Kim, J. H. Oh and C. Yang, J. Am. Chem. Soc., 2012, 134, 20713 CrossRef CAS PubMed.
- I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896 CrossRef CAS PubMed.
- B. Sun, W. Hong, Z. Yan, H. Aziz and Y. Li, Adv. Mater., 2014, 26, 2636 CrossRef CAS PubMed.
- K.-H. Kim, S. Park, H. Yu, H. Kang, I. Song, J. H. Oh and B. J. Kim, Chem. Mater., 2014, 26, 6963 CrossRef CAS.
- H.-J. Yun, S.-J. Kang, Y. Xu, S. O. Kim, Y.-H. Kim, Y.-Y. Noh and S.-K. Kwon, Adv. Mater., 2014, 26, 7300 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |