
Monolithic capillary columns consisting of poly(glycidyl methacrylate-co-ethylene glycol dimethacrylate) and their diol derivatives with incorporated hydroxyl functionalized multiwalled carbon nanotubes for reversed-phase capillary electrochromatography
Nisansala
Ganewatta
 and
Ziad
El Rassi
*
and
Ziad
El Rassi
*
Department of Chemistry, Oklahoma State University, Stillwater, OK 74078-3071, USA. E-mail: elrassi@okstate.edu; Fax: +(405) 744-6007
First published on 13th November 2017
Abstract
Two types of monolithic stationary phases with incorporated hydroxyl functionalized multiwalled carbon nanotubes (OH-MWCNTs) were introduced and evaluated, namely, the poly(glycidyl methacrylate-co-ethylene glycol dimethacrylate) monolith, denoted as poly(GMA-co-EDMA), and a diol derivative of the poly(GMA-co-EDMA) monolith. The diol derivative monolith was obtained by subjecting the poly(GMA-co-EDMA) monolith with physically incorporated OH-MWCNTs to an acid treatment with 0.1 M sulfuric acid at a moderate temperature of 50 °C for a total of 7.5 h. Also, the poly(GMA-co-EDMA) monolith with both physically and covalently incorporated OH-MWCNTs was prepared by subjecting the physically incorporated monolithic column to a Lewis acid catalyzed reaction in the presence of BF3 in order to react some of the OH-MWCNTs with the epoxy rings of the poly(GMA-co-EDMA) monolith. In all cases, the OH-MWCNTs were subjected to high power sonication at an output power of 10 W for 15 min with the aim of better dispersing the incorporated nanotubes into the monoliths under investigation. In fact, high power sonication yielded columns with a relatively higher plate count (∼2 fold increase) when compared to low power sonication. While the incorporation of OH-MWCNTs into the poly(GMA-co-EDMA) monolith acted as an amendment boosting the nonpolar character of the monolith and providing additional π–π interactions, the diol derivative monolith with its polar backbone character acted nearly as a support for the OH-MWCNT stationary phase giving rise to a carbon nanotube sorbent providing hydrophobic and π–π interactions via the incorporated OH-MWCNTs. These two kinds of columns were evaluated using alkylbenzenes, toluene derivatives, aniline compounds, phenols and polyaromatic hydrocarbons.
Introduction
In recent years, capillary electrochromatography (CEC) has evolved into a rapidly growing modern analytical separation technique mainly due to the introduction of monolithic stationary phases as the primary sorbents for CEC separations; for recent reviews on this topic, see ref. 1 and 2. In fact, monolithic capillary columns have demonstrated some advantages over their capillary column counterparts packed with microparticulate sorbents such as the absence of retaining frits, ease of column preparation, relatively high permeability, readily adjustable column porosity and tunable separation selectivity. These advantages have facilitated the expansion of monolithic columns in general and CEC in particular over the past few decades. However, although polymer-based monoliths can be easily fabricated and confined into columns of all sizes, these stationary phases have low surface areas which limit their ability to sufficiently retain small solutes due to the reduced amount of mesopores. To remedy this deficiency, a variety of nanoparticles (NPs) have been incorporated into polymeric monoliths in order to modulate the retention properties and selectivity of monolithic columns and to consequently improve the separation of small molecules; for a recent review on this topic, see ref. 3. The unique surface characteristics of NPs combined with their large surface-to-volume ratio allows the preparation of stationary phases with distinct retentive power and in turn enhanced separation selectivity toward chiral and achiral solutes.4,5After the brief initial study by Li et al.6 on the incorporation of single-walled carbon nanotubes (SWCNTs) into an organic polymer based monolithic stationary phase for CEC, other types of NPs have more recently been investigated in monolithic CEC including graphene oxide nanosheets,7 carboxylated SWCNTs with bonded chiral pepsin,8 gold NPs (GNPs),9 cyclodextrin-GNPs,10 silver NPs,11 iron oxide magnetic NPs,12 lanthanide metal–organic frameworks,13 core–shell silica NPs, hexagonal and wormlike mesoporous silica rods,14 silica NPs functionalized with chiral pepsin15 and zeolitic imidazolate frameworks.16 These monolithic columns incorporated with various NPs were among other things, demonstrated in the CEC separations of chiral basic drugs,8,15 selected chiral drugs,10 peptides,9 neutral compounds,11 organophosphorus pesticides,12 aromatic test solutes,7,13 organic acids,14 PAH and non-steroidal anti-inflammatory drugs,16 just to name a few.
Based on the above recent investigations, only a limited number of attempts have been made for developing nanocarbon-modified organic polymer-based monolithic stationary phases for CEC6–8 despite the remarkable characteristics of nanocarbon materials and the beneficial properties exhibited by polymeric monoliths as ideal media for these nano-entities.17 The unique properties of nanocarbon materials such as large specific surface area and capability of establishing non-covalent interactions with a variety of solutes, mainly via π–π stacking and hydrophobic interactions (for a typical reference, see ref. 17), should in principle allow the successful development of nanocarbonaceous stationary phases with improved separation performances. Therefore, the aim of this study is to further contribute to this field by developing suitable methacrylate based monolithic capillary columns for incorporating multiwalled carbon nanotubes (MWCNTs) for use in CEC separations. Due to their poor dispersion in solvents, bare MWCNTs are difficult to handle and covalent modifications of the nanotube skeleton are needed to prevent their aggregation in tight bundles; for a recent review, see ref. 18. Thus, hydroxyl functionalized carbon nanotubes, namely, OH-MWCNTs were used in this study. These OH-MWCNTs performed very well in monolithic HPLC columns with incorporated OH-MWCNTs.17 In the first part of this contribution, OH-MWCNTs were incorporated into the poly(GMA-co-EDMA) monolith either by physical entrapment or by covalent/physical entrapment. The second part of this investigation focused on the physical entrapment of OH-MWCNTs into a diol derivative of the poly(GMA-co-EDMA) monolithic stationary phase in order to further assess the contribution of the incorporated OH-MWCNTs to solute retention and the residual retention arising from the monolithic backbone. Furthermore, the effects of low power and probe tip high power sonication on the separation performance were also studied. The optimized columns thus obtained were tested under reversed-phase chromatography (RPC) conditions using CEC, which is a suitable technique for generating a constant nano-flow by electroosmotic flow (EOF) without the need for a mechanical pump.
Materials and methods
Instruments
All CEC experiments were performed on an in-house-assembled instrument from commercially available components. It comprised two high voltage power supplies of positive and negative polarity, Models MJ30P0400-11 and MJ30N0400-11, respectively, from Glassman High Voltage, Inc. (Whitehouse Station, NJ, USA) and a Model 200 UV-Vis variable wavelength detector (Vestec Corporation, TX, USA) equipped with a cell for on-capillary detection. The high voltage end of the system was enclosed in a plexiglass box with an interlock system which automatically cuts off high voltage when the plexiglass box is opened. Electrochromatograms were recorded using a Shimadzu computing integrator Model Chromatopac CR501 (Kyoto, Japan). The in situ polymerization of the monolithic capillary columns was carried out in a Model 105 Isotemp water bath from Fisher Scientific (Fairlawn, NJ, USA). A Model-45 solvent delivery system from Waters Associates (Milford, MA, USA) was used in conditioning the pre-polymerized capillary columns. An automated syringe pump from KD Scientific (Holliston, MA, USA) was used to pass the reagents through the fused-silica capillary when silanizing its inner walls and to condition the monolithic capillary column with a given mobile phase. An ultrasonic cleaner Model 1510R-MTH from Branson Ultrasonic Corporation (Danbury, CT, USA) was used for low power sonication and a Model F50 sonic dismembrator from Fisher Scientific (Waltham, MA, USA) was used for high power sonication. A Sigma 300 GC oven from PerkinElmer (Norwalk, CT, USA) was used for heating the columns during post-polymerization modifications.Reagents and materials
Fused silica capillaries with an internal diameter (i.d.) of 100 μm and an outer diameter (o.d.) of 375 μm were purchased from Polymicro Technologies (Phoenix, AZ, USA). OH-MWCNTs, batch number SN32547, with a specific surface area of >200 m2 g−1, an o.d. of 10–20 nm, an i.d. of 5–10 nm and a length of 10–30 μm, were purchased from Sun Innovations Inc. (Fremont, CA, USA); for more information, see the link: https://www.nanomaterialstore.com/oh-nt.php. Glycidyl methacrylate (GMA), ethylene glycol dimethacrylate (EDMA), 1-dodecanol, cyclohexanol, 3-(trimethoxysilyl)propylmethacrylate, 2,2′-azobis(isobutyronitrile) (AIBN), acetone, 1,4-dioxane, alkylbenzenes (ABs), p-ethylaniline, p-chloroaniline, p-bromoaniline, 3,4-dichloroaniline, p-nitrophenol, m-chlorophenol, 3,4,5-trichlorophenol, p-tolualdehyde, p-tolunitrile, m-tolualdehyde, m-tolunitrile, 2,3-dinitrotoluene, fluorenone and 1-naphthol were purchased from Sigma-Aldrich (Milwaukee, WI, USA). m-Toluidine was obtained from Fluka Chemie GmbH (Buchs, Switzerland). Naphthalene and p-toluidine were purchased from Eastman Kodak Co. (Rochester, NY, USA). Anhydrous Na2HPO4 and NaOH were obtained from EM Science (Gibbstown, NJ, USA). BF3 etherate was purchased from TCI America (Portland, OR, USA). Phenol, sulfuric acid, sodium acetate and aniline were obtained from Fisher Scientific (Fairlawn, NJ, USA). Acetonitrile (ACN) and methanol were purchased from Pharmco-Aaper (Brookfield, CT, USA).Silanization of the capillary walls
The inner walls of a 45 cm long, 100 μm i.d. fused silica capillary were pre-treated using an automated syringe pump, by first passing water for 2 min, then 1 M NaOH for 30 min followed by 0.1 M HCl for 30 min and water again for 30 min at a flow rate of 1 mL h−1. Subsequently, the inner walls of the fused silica capillary were silanized in the presence of a solution of 50% (v/v) 3-(trimethoxysilyl)propylmethacrylate in acetone for 6 h at room temperature at a flow rate of 33.3 μL h−1. Next, the capillary was rinsed with methanol for 10 min at a flow rate of 1 mL h−1 and was dried under a stream of nitrogen gas for another 10 min.In situ polymerization of monoliths for the covalent and/or physical entrapment of OH-MWCNTs
The polymerization solutions used in the preparation of monolithic capillary columns with the covalent and/or physical entrapment of OH-MWCNTs were prepared using glass vials and have a final weight of 1 g (see Fig. 1 for the polymerization reactions) containing 14.95 wt% GMA as the functional monomer, 9.97 wt% EDMA as the crosslinker, a binary porogen consisting of 37.38 wt% cyclohexanol and 37.38 wt% 1-dodecanol, 0.25 wt% AIBN and 0.07 wt% OH-MWCNTs. OH-MWCNTs were first added to the binary porogenic mixture consisting of cyclohexanol and 1-dodecanol, and then the mixture was subjected to high power sonication at an output power of 10 W for 15 min while keeping the glass vial in ice to prevent evaporation. This was followed by adding GMA, EDMA and AIBN to the vial. Thereafter, the prepared OH-MWCNTs containing the polymerization solution was thoroughly degassed using normal sonication for 20 min. The polymerization solution thus prepared was introduced into a pre-silanized fused silica capillary, which was filled up to a 28 cm demarcation line marked on the 45 cm long fused silica capillary by applying a negative pressure at the outlet end while the inlet end was immersed in the solution. Subsequently, the ends of the capillary were sealed using GC septa and were allowed to undergo polymerization in a heated water bath at 50 °C for a total of 24 h. After the completion of the polymerization step, the capillary was washed with 80% (v/v) ACN using the Waters solvent delivery system. The poly (GMA-co-EDMA) monolithic capillary column thus prepared had physically trapped OH-MWCNTs, and was then ready for post-polymerization modification in order to facilitate the covalent attachment of OH-MWCNTs to the monolith.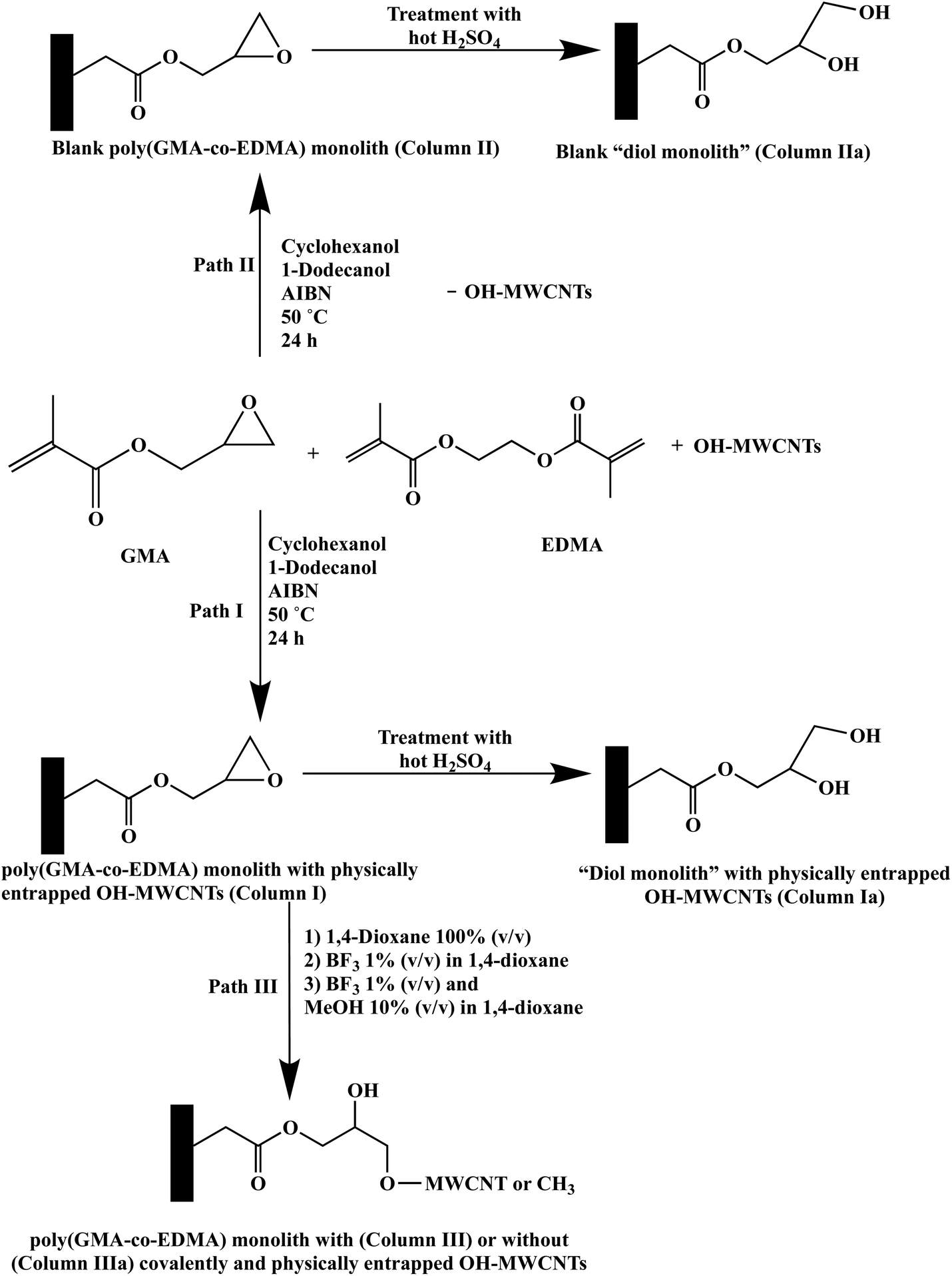 | ||
| Fig. 1 Reaction schemes showing different paths by which the poly(GMA-co-EDMA) monolith and its diol monolith derivative were prepared with and without incorporated OH-MWCNTs. | ||
After the above in situ polymerization, the porogen free poly(GMA-co-EDMA) monolithic capillary column with physically entrapped OH-MWCNTs was equilibrated with 1,4-dioxane for 4 h. Subsequently, 1% (v/v) BF3 in 1,4-dioxane was allowed to pass through the column for 24 h, and thereafter a mixture of 1% (v/v) BF3 and 10% (v/v) methanol in 1,4-dioxane was passed through the column for 4 h to scavenge the unreacted surface epoxy groups. All these reagent perfusion steps were achieved using an automated syringe pump whose flow rate was maintained constant at 0.03 mL h−1.
At the completion of the reaction time, it was anticipated to covalently attach the OH-MWCNTs to the monolithic support via the opening of the epoxy groups present on the functional monomer, GMA. The resultant column was equilibrated for 1 h with the desired mobile phase prior to CEC analyses.
In order to convert the unreacted epoxy rings to diol functional groups on the surface of the poly(GMA-co-EDMA) monolith with physically entrapped OH-MWCNTs or without entrapped OH-MWCNTs (i.e., a blank column), the monolithic column in question was treated with hot sulfuric acid.19,20 The acid catalyzed diol formation from the epoxy groups of GMA was carried out as follows: the monolithic capillary column was filled with a 0.1 M H2SO4 solution, and then heated in a GC oven at 50 °C for 2.5 h. This process was repeated two more times so that the sulfuric acid treatment lasted a total of 7.5 h. After the completion of the reaction period, the column was rinsed with water and was equilibrated with the desired mobile phase prior to the analyses.
Results and discussion
Poly(GMA-co-EDMA) monolithic columns with covalently and/or physically entrapped OH-MWCNTs
With a few trial and errors guided by the extensive experience in the design of these types of monoliths in our laboratories,20–22 the optimal polymerization solution composition was readily determined and it is given in the Materials and methods section. In fact, this optimal composition yielded a stable and reproducible column (see below) with good permeability. Once this optimal composition was identified, the amount of OH-MWCNTs that could be incorporated into this monolith was determined by preparing three monolithic columns at different amounts of incorporated OH-MWCNTs, namely, 0.05 wt%, 0.07 wt% and 0.10 wt% while keeping everything else constant including the polymerization temperature and duration (50 °C for 24 h) and high power sonication for 15 min. Using alkylbenzenes (ABs) as the test solutes, the average plate counts per meter were found to be 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 41000 and 20000 for the columns at 0.05 wt%, 0.07 wt% and 0.10 wt%, respectively. Thus, the column at 0.07 wt% OH-MWCNTs (or 5.45 × 10−5 mg cm−1) was chosen as the most efficient column that can be produced with incorporated OH-MWCNTs. This column is designated as Column I that is made using Path I, as shown in Fig. 1.
000, 41000 and 20000 for the columns at 0.05 wt%, 0.07 wt% and 0.10 wt%, respectively. Thus, the column at 0.07 wt% OH-MWCNTs (or 5.45 × 10−5 mg cm−1) was chosen as the most efficient column that can be produced with incorporated OH-MWCNTs. This column is designated as Column I that is made using Path I, as shown in Fig. 1.
As shown in Fig. 1, the poly(GMA-co-EDMA) monolith was prepared in various formats including (i) poly(GMA-co-EDMA) with physically incorporated OH-MWCNTs and its diol derivative monolith with physically incorporated OH-MWCNTs, designated as Column I and Column Ia, respectively (via Path I), (ii) the blank poly(GMA-co-EDMA) monolith (i.e., without incorporated OH-MWCNTs, designated as Column II) and its blank diol derivative monolith designated as Column IIa (both columns are made via Path II) and (iii) poly(GMA-co-EDMA) with physically and covalently incorporated (designated as column III) and without OH-MWCNTs designated as Column IIIa (via Path III). Path III involved an additional Lewis acid catalyzed reaction in the presence of BF323 between the epoxy-based poly(GMA-co-EDMA) monolith and the hydroxylated carbon nanotubes (OH-MWCNTs), which should in principle allow the covalent attachment of some of the physically entrapped OH-MWCNTs to the monolithic structure through its reactive epoxy groups.
In a recent investigation from our laboratory on incorporating MWCNTs in a monolithic column for HPLC,17 it was shown that a short duration of high power sonication, which is known to break up, disperse or de-bundle the carbon nanotube aggregates in solution,24,25 resulted in maximizing the homogeneity of the monolithic column with incorporated carbon nanotubes. Similarly, in this study, high power sonication was continuously applied for 15 min on the OH-MWCNT–porogen solution which was used in the preparation of the capillary monolithic column. The column thus prepared showed the highest average separation efficiency of 41000 plates per m for ABs, out of a series of prepared columns. This optimum duration and the type of sonication are in agreement with those reported earlier by Mayadunne and El Rassi17 concerning the incorporation of OH-MWCNTs in monolithic HPLC columns.
To assess the quality of the above monolith with physically entrapped and covalently attached OH-MWCNTs (Column III), the reproducibility of the analysis was investigated using ABs (from toluene to heptylbenzene) as model solutes. This was carried out by measuring the retention factor, k, values of ABs obtained on the monolithic column from run-to-run (n = 3), day-to-day (n = 3), and column-to-column (n = 2) using a mobile phase of 40% (v/v) ACN and 60% (v/v) of 2 mM Na2HPO4, pH 7.0. The day-to-day and column-to-column reproducibilities were calculated by averaging triplicate measurements of the k values on each day and for each column. In all cases, the reproducibility was determined by the % RSDs of the k values, which were found to be 1.75%, 2.59% and 6.47% for run-to-run, day-to-day and column-to-column, respectively.
Retention properties of Column III with covalently and physically entrapped OH-MWCNTs
000 plates per m vs. 18000 plates per m).
 | ||
| Fig. 2 Electrochromatograms of ABs obtained on the monolithic column with physically and covalently incorporated OH-MWCNTs (A) and its blank monolithic column (B) obtained via Path III (Column III and Column IIIa, respectively). Conditions: 40 cm (effective length 25 cm) × 100 μm i.d.; running voltage, 15 kV; sample injection, voltage at 15 kV for 7 s; hydro-organic mobile phase, 40% ACN:60% (v/v) of 2 mM Na2HPO4, pH 7.0. Solutes: (1) toluene, (2) ethylbenzene, (3) propylbenzene, (4) butylbenzene, (5) pentylbenzene, (6) hexylbenzene, and (7) heptylbenzene; EOF tracer indicating t0, uracil. | ||
| Solutes | Retention factor, k | η = kColumn III/kColumn IIIa | |
|---|---|---|---|
| Column III | Blank column (Column IIIa) | ||
| Toluene | 0.82 ± 0.02 | 0.69 ± 0.00 | 1.19 |
| Ethylbenzene | 1.06 ± 0.02 | 0.84 ± 0.00 | 1.26 |
| Propylbenzene | 1.43 ± 0.03 | 1.09 ± 0.02 | 1.31 |
| Butylbenzene | 1.96 ± 0.03 | 1.42 ± 0.04 | 1.38 |
| Pentylbenzene | 2.70 ± 0.04 | 1.81 ± 0.06 | 1.49 |
| Hexylbenzene | 3.58 ± 0.05 | 2.29 ± 0.09 | 1.56 |
| Heptylbenzene | 4.77 ± 0.07 | 2.85 ± 0.07 | 1.67 |
In the first set of measurements, five toluene derivatives, namely, p-toluidine (has a strong –NH2 and a weak –CH3 benzene ring activating group), p-tolualdehyde (has a weak activating group, –CH3, and a moderate deactivating group, –CHO), p-tolunitrile (has a weak activating group, –CH3, and a moderate deactivating group, –CN), toluene (has only a weak activating group, –CH3) and 2,3-dinitrotoluene (has two strong deactivating groups, –NO2) were used as model solutes. They eluted from Column III in the order of p-toluidine, p-tolualdehyde, p-tolunitrile, toluene and 2,3-dinitrotoluene with average k values (n = 3) of 2.17 ± 0.26, 3.18 ± 0.29, 4.60 ± 0.41, 7.75 ± 0.63 and 12.69 ± 0.13, respectively. This order parallels the order of increasing deactivation effect caused by the substituted functional groups on the benzene ring thus showing π–π interactions with the incorporated OH-MWCNTs in addition to nonpolar interactions with both the monolith and its incorporated OH-MWCNTs. The values of the selectivity factors (α values) were greater than 1.4 as follows: α2,1 = 1.47, α3,2 = 1.45, α4,3 = 1.68 and α5,4 = 1.64 (refer to Fig. 3A for the identity of solute pairs). An average separation efficiency of 37300 plates per m was obtained; see Fig. 3A for a typical electrochromatogram.
 | ||
| Fig. 3 Electrochromatograms of p-substituted (A) and m-substituted (B and C) toluene derivatives obtained on the monolithic column with physically and covalently incorporated OH-MWCNTs (A) and (B) obtained via Path III (Column III) and on the diol monolithic column derivative with physically incorporated OH-MWCNTs (C) obtained via Path I (Column Ia). Conditions: 40 cm (effective length 25 cm) × 100 μm i.d.; running voltage, 15 kV; sample injection, voltage at 15 kV for 5 s; hydro-organic mobile phase, 20% ACN:80% (v/v) of 2 mM Na2HPO4, pH 7.0. Solutes in (A), (1) p-toluidine, (2) p-tolualdehyde, (3) p-tolunitrile, (4) toluene, and (5) 2,3-dinitrotoluene; in (B) and (C), (1) m-toluidine, (2) m-tolualdehyde, (3) m-tolunitrile, and (4) toluene; EOF tracer indicating t0, uracil. | ||
In another set of experiments, a mixture of toluene and three m-substituted toluene compounds, consisting of m-toluidine, m-tolualdehyde, and m-tolunitrile was separated with an average separation efficiency of 6400 plates per m on the above Column III, as shown in Fig. 3B. The calculated k values were 2.18 ± 0.20, 3.51 ± 0.26, 4.75 ± 0.34 and 7.37 ± 0.52 for m-toluidine, m-tolualdehyde, m-tolunitrile and toluene, respectively. The α values were as follows: α2,1 = 1.61, α3,2 = 1.35 and α4,3 = 1.55 (see Fig. 3B for solute pair identities). In this case also, the activation/deactivation power of the corresponding substituted groups on the benzene ring played a significant role in determining the elution order which was similar to that of the p-substituted toluene derivatives. In addition, toluene, the most hydrophobic solute, eluted last.
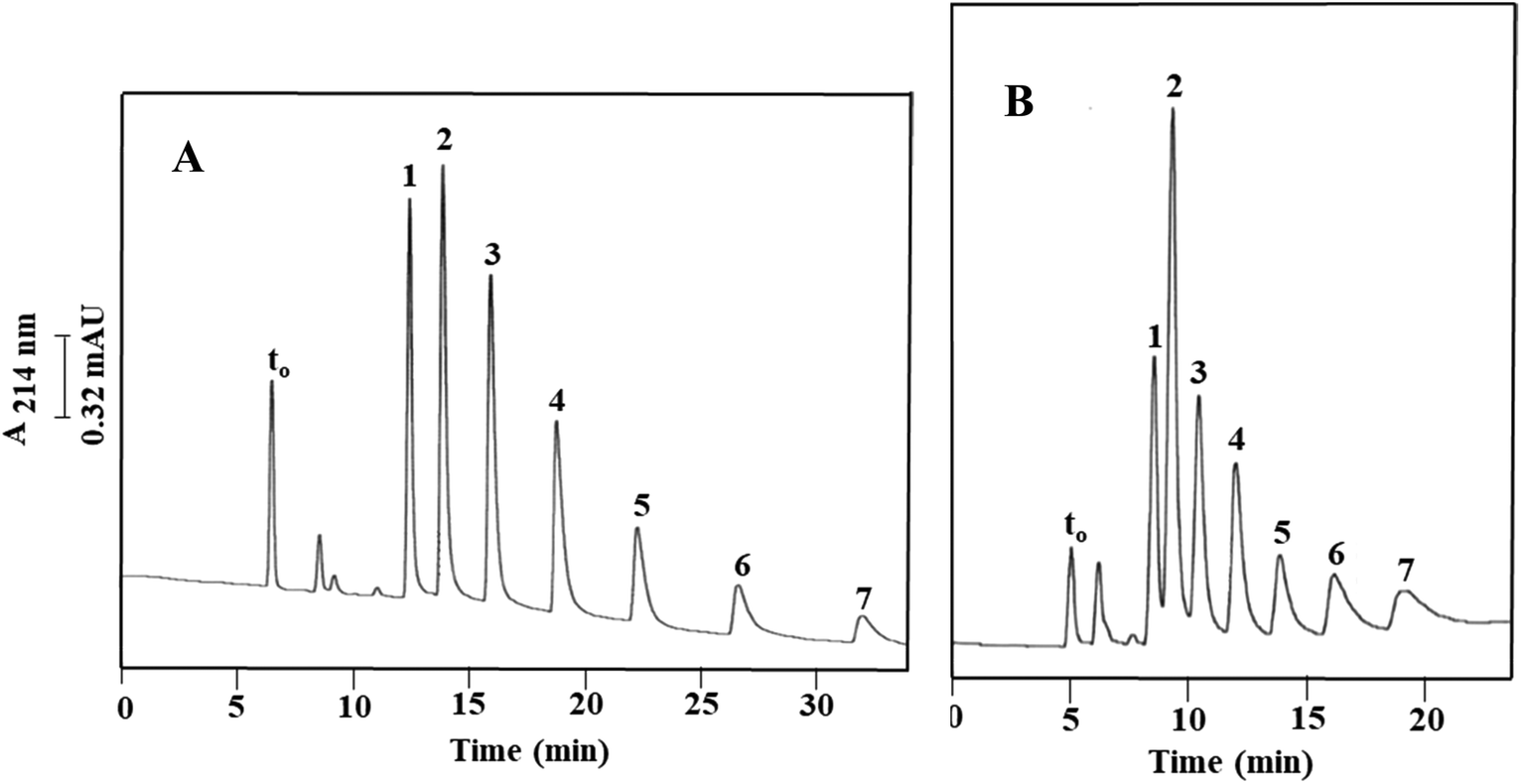
The retentive properties of Column III were further evaluated using a mixture of aniline (k = 0.42 ± 0.02) and four substituted aniline compounds, consisting of p-ethylaniline (k = 0.59 ± 0.02), p-chloroaniline (k = 1.09 ± 0.05), p-bromoaniline (k = 1.37 ± 0.04) and 3,4-dichloroaniline (k = 2.33 ± 0.04). They eluted from the column following the order in which they were just listed; see also Fig. 4A. Aniline and its derivatives yielded an average plate count of 25000 plates per m. The retention follows the typical RPC behavior where the halogenated anilines were more retained than the ethyl substituted anilines. 3,4-Dichloroaniline, which is a disubstituted compound with a weakly deactivating –Cl group, eluted last. This demonstrates that both hydrophobicity and π–π interactions determine the retention of anilines under investigation. The calculated α values for the solute pairs shown in Fig. 4A were α2,1 = 1.39, α3,2 = 1.86, α4,3 = 1.26 and α5,4 = 1.69.
 | ||
| Fig. 4 Electrochromatograms of aniline compounds obtained on the monolithic column with physically and covalently incorporated OH-MWCNTs (A) obtained via Path III (Column III) and on the diol monolithic column derivative with physically incorporated OH-MWCNTs (B) obtained via Path I (Column Ia). Conditions: 40 cm (effective length 25 cm) × 100 μm i.d.; running voltage, 15 kV; sample injection, voltage at 15 kV for 7 s; hydro-organic mobile phase, 40% ACN:60% (v/v) of 4 mM CH3COONa, pH 4.0. Solutes: (1) aniline, (2) p-ethylaniline, (3) p-chloroaniline, (4) p-bromoaniline, and (5) 3,4-dichloroaniline; EOF tracer indicating t0, uracil. | ||
Some phenols were electrochromatographed on Column III using a hydro-organic mobile phase composed of 40% (v/v) ACN and 60% (v/v) of 4 mM CH3COONa, pH 4.0, and the results are portrayed in Fig. 5A. Under these experimental conditions, the tested phenols were baseline separated and eluted in the following increasing order of phenol (k = 0.48 ± 0.00), p-nitrophenol (k = 0.73 ± 0.01), m-chlorophenol (k = 1.05 ± 0.01) and 3,4,5-trichlorophenol (k = 2.77 ± 0.05). These phenols yielded an average plate count of 44600 plates per m. Phenol eluted faster than the substituted phenols such as p-nitrophenol, m-chlorophenol and 3,4,5-trichlorophenol which were retained more strongly due to their hydrophobic nature and π–π interactions. Moreover, the degree of halogenation tends to control solute retention. Therefore, as per the RPC retention behavior, the trihalogenated phenol was retained for a longer time on the column than the monohalogenated phenol. The α values for the solute pairs shown in Fig. 5A were found to be α2,1 = 1.52, α3,2 = 1.44 and α4,3 = 2.64.
 | ||
| Fig. 5 Electrochromatograms of phenol compounds obtained on the monolithic column with physically and covalently incorporated OH-MWCNTs (A) prepared via Path III (Column III) and on the diol monolithic column derivative with physically incorporated OH-MWCNTs (B) prepared via Path I (Column Ia). Conditions: 40 cm (effective length 25 cm) × 100 μm i.d.; running voltage, 15 kV; sample injection, voltage at 15 kV for 10 s; hydro-organic mobile phase, 40% ACN:60% (v/v) of 4 mM CH3COONa, pH 4.0. Solutes: (1) phenol, (2) p-nitrophenol, (3) m-chlorophenol, and (4) 3,4,5-trichlorophenol; EOF tracer indicating t0, uracil. | ||
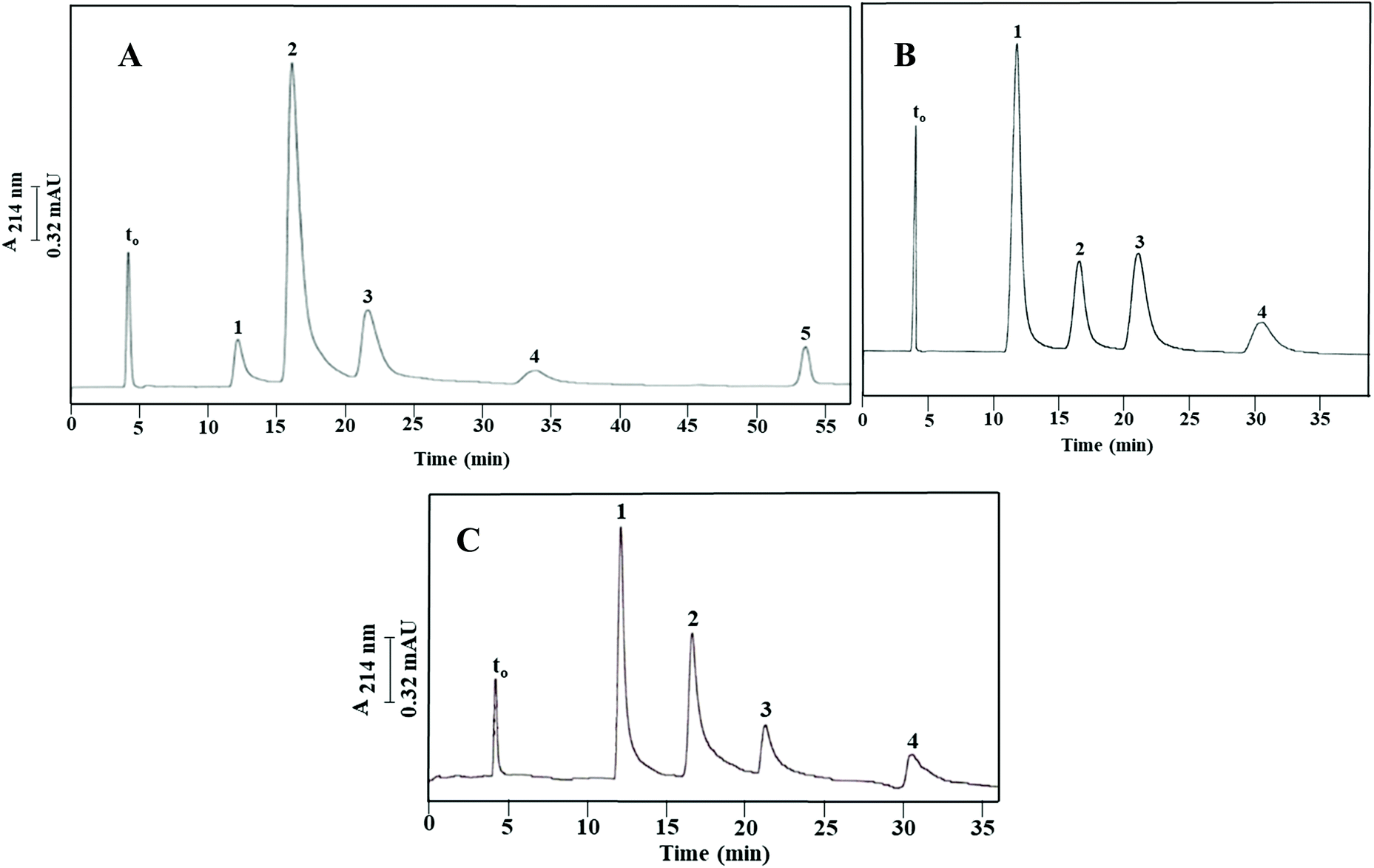
Finally, a series of four PAHs were injected on Column III and was baseline separated as shown in Fig. 6 with an average plate number of 20000 plates per m. PAHs are composed of multiple aromatic rings in which the electrons are delocalized. The less hydrophobic compound 1-naphthol (k = 0.79 ± 0.05) eluted faster than the other tested PAHs, due to its attached –OH group. Also, fluorenone (k = 1.06 ± 0.08) which has a carbonyl group between the two aromatic rings eluted faster than naphthalene (k = 1.46 ± 0.12) due to the added polarity to the molecule from the carbonyl group. The absence of a carbonyl group in fluorene (k = 2.51 ± 0.11) imparted this PAH with a more hydrophobic character and contributed to its higher k value. The calculated α values for the solute pairs shown in Fig. 6 were α2,1 = 1.34, α3,2 = 1.37 and α4,3 = 1.72.
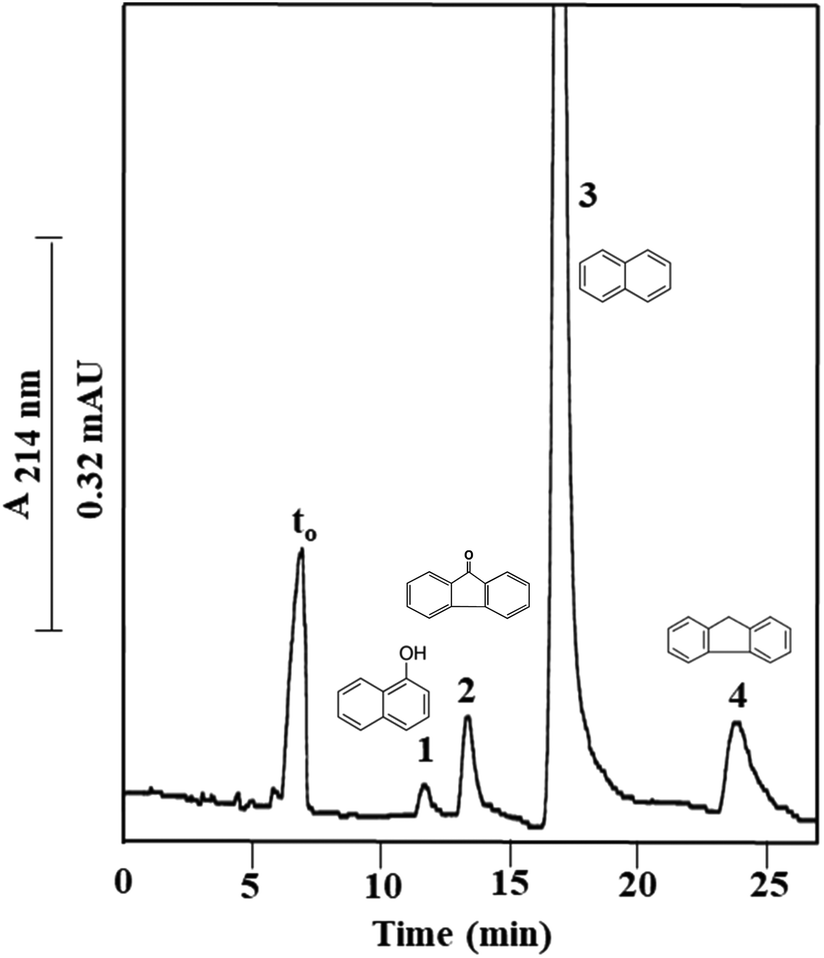 | ||
| Fig. 6 Electrochromatograms of PAHs obtained on the monolithic column with physically and covalently incorporated OH-MWCNTs prepared via Path III (Column III). Conditions: 40 cm (effective length 25 cm) × 100 μm i.d.; running voltage, 15 kV; sample injection, voltage at 15 kV for 10 s; hydro-organic mobile phase, 40% ACN:60% (v/v) of 2 mM Na2HPO4, pH 4.0. Solutes: (1) 1-naphthol, (2) fluorenone, (3) naphthalene, and (4) fluorene; EOF tracer indicating t0, uracil. | ||
Retention properties of Column Ia, the diol derivative of Column III with physically entrapped OH-MWCNTs
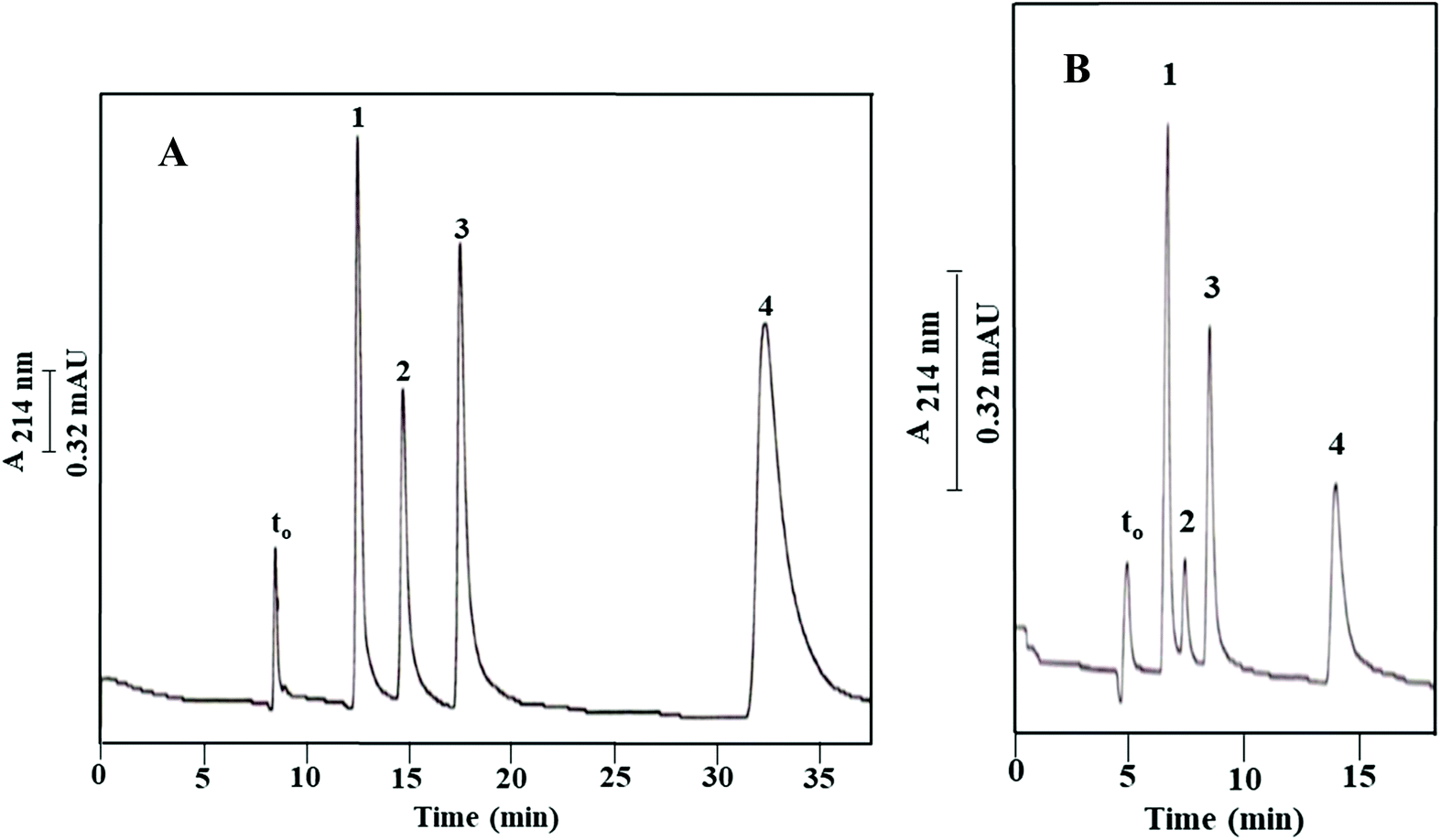
As shown in Fig. 2B and reported in Table 1, the blank column of poly(GMA-co-EDMA), i.e., Column IIIa, shows a significant residual hydrophobicity. This is not surprising since both GMA and EDMA possess a weakly nonpolar character, and also the scavenging of the epoxy groups (see Fig. 1) on the surface of the monolith with methanol would add nonpolar methyl groups. In order to provide a more pacific and neutral monolith to “support” the OH-MWCNTs, the GMA-based monolith was converted to a diol derivative by opening the epoxy ring via a strong acid treatment using sulfuric acid;19,20 see Materials and methods. The diol derivative monolithic capillary column thus obtained with physically incorporated OH-MWCNTs (Column Ia, see Path I in Fig. 1) was tested using ABs, and the measured k values were compared to those obtained on its blank column (Column IIa, see Path II in Fig. 1). The resulting electrochromatograms on the OH-MWCNT bearing column and its blank column are shown in Fig. 7A and B, respectively, and the calculated k values are reported in Table 2. The blank column (Column IIa) separated the ABs with an average plate count of only 10000 plates per m while the monolithic column containing OH-MWCNTs yielded an average of 33000 plates per m (Column Ia). As shown in Table 2, the modulus values, η, listed are significantly higher than those obtained on the poly(GMA-co-EDMA) base monolith, as shown in Table 1. The moduli are higher by 15% to 35% for ABs on the diol monolithic derivative (Column Ia, Table 2) than on its base monolith Column III (see Table 1). Also, there was a more than three-fold increase in the average plate count when comparing the 33000 plates per m obtained on the diol monolithic column with incorporated nanoparticles (Column Ia) to the 10000 plates per m obtained on its blank monolith (Column IIa). However, the plate count on the diol column with physically incorporated OH-MWCNTs is about 20% less than that on the base monolith with physically and covalently incorporated OH-MWCNTs, i.e., Column III (compare 41000 to 33000 plates per m).
 | ||
| Fig. 7 Electrochromatograms of ABs obtained on the monolithic column with incorporated OH-MWCNTs treated with acid (A) prepared via Path I (Column Ia) and its blank diol monolithic column derivative (B) prepared via Path II (Column IIa). Conditions: 40 cm (effective length 25 cm) × 100 μm i.d.; running voltage, 15 kV; sample injection, voltage at 15 kV for 7 s; hydro-organic mobile phase, 40% ACN:60% (v/v) of 2 mM Na2HPO4, pH 7.0. Solutes: (1) toluene, (2) ethylbenzene, (3) propylbenzene, (4) butylbenzene, (5) pentylbenzene, (6) hexylbenzene, and (7) heptylbenzene; EOF tracer indicating t0, uracil. | ||
| Solutes | Retention factor, k | η = kColumn Ia/kColumn IIa | |
|---|---|---|---|
| Column Ia | Blank column (Column IIa) | ||
| Toluene | 0.82 ± 0.06 | 0.51 ± 0.00 | 1.61 |
| Ethylbenzene | 1.01 ± 0.06 | 0.60 ± 0.00 | 1.68 |
| Propylbenzene | 1.26 ± 0.07 | 0.76 ± 0.05 | 1.66 |
| Butylbenzene | 1.60 ± 0.09 | 0.91 ± 0.02 | 1.76 |
| Pentylbenzene | 2.01 ± 0.01 | 1.15 ± 0.08 | 1.75 |
| Hexylbenzene | 2.49 ± 0.13 | 1.34 ± 0.01 | 1.86 |
| Heptylbenzene | 3.17 ± 0.16 | 1.65 ± 0.01 | 1.92 |
Based on the above results, the sulfuric acid treatment accomplished to a large extent its aim of rendering the monolithic backbone more hydrophilic by converting the epoxy groups to diols, and consequently reduced the hydrophobicity of the monolithic backbone yielding a monolithic column (Column Ia) with physically incorporated OH-MWCNTs whose retention properties toward nonpolar solutes (e.g., ABs) are due in major part to the incorporated carbon nanotubes. This can be noticed by just glancing the AB electrochromatograms in Fig. 7A and B. Thus, the diol derivative of the poly(GMA-co-EDMA) monolith can be viewed as nearly a “support” for the nano entities-based stationary phase, namely, the “OH-MWCNT stationary phase”.
The above diol monolithic column with physically incorporated OH-MWCNTs (Column Ia), or simply the “OH-MWCNT stationary phase”, was evaluated using some m-substituted toluene derivatives (see Fig. 3C), aniline compounds (see Fig. 4B) and phenol compounds (see Fig. 5B). As expected, the elution orders of the three sets of solutes on the diol derivative column (Column Ia) remained the same as those obtained on the original monolithic column (Column III). The k values of m-toluidine, m-tolualdehyde, m-tolunitrile and toluene obtained on Column Ia were 1.87 ± 0.00, 2.96 ± 0.00, 4.05 ± 0.00 and 6.24 ± 0.01, respectively, which were lower by about 14% to 16% than those obtained on the original poly(GMA-co-EDMA) monolithic column with physically and covalently incorporated OH-MWCNTs (Column III). However, the α values were about the same on both columns. In fact, these α values were 1.61, 1.35 and 1.55 for the solute pairs 2/1, 3/2 and 4/3, respectively, shown in Fig. 3B, compared to 1.58, 1.37 and 1.54 for the solute pairs 2/1, 3/2 and 4 /3, respectively, shown in Fig. 3C. Similarly, the other test solutes, namely, anilines and phenols yielded k values which were lower on the diol derivative monolithic column (Column Ia) than on the original poly(GMA-co-EDMA) column (Column III). In fact, the k values were 0.30 ± 0.01, 0.40 ± 0.01, 0.75 ± 0.02 and 1.45 ± 0.03 for aniline, p-ethylaniline, p-bromoaniline and 3,4-dichloroaniline, respectively, on Column Ia, and the k values for phenol, p-nitrophenol, m-chlorophenol and 3,4,5-trichlorophenol were 0.34 ± 0.01, 0.51 ± 0.01, 0.72 ± 0.01 and 1.85 ± 0.03, respectively, on Column Ia. Depending on the nature of the solute, the k values for anilines and phenols were anywhere from 28 to 38% lower on Column Ia than on Column III. The α values on the other hand are more or less the same for anilines and phenols on both columns. The tested m-substituted toluene derivatives, anilines and phenols yielded average plate counts of 25800 plates per m, 24000 plates per m and 24000 plates per m, respectively.
In short, and as expected, the diol based monolith with incorporated OH-MWCNTs (Column Ia) offered lower solute retention than Column III while exhibiting more or less the same α values under otherwise the same mobile phase composition. In Column III, the unreacted epoxy groups were scavenged with methanol, a process that introduced nonpolar methyl groups whereas in Column Ia, the unreacted epoxy groups were converted to polar diol groups. In addition, and because of the higher electroosmotic flow velocity obtained on Column Ia, the separations were achieved in shorter times especially when using a hydro-organic mobile phase consisting of 40% ACN:60% (v/v) of 4 mM CH3COONa, pH 4.0, and a running condition that is shown in Fig. 4 and 5 for aniline and phenol compounds at a running voltage of 15 kV. Under these conditions, the EOF velocity was 1.75-fold higher on Column Ia (0.84 ± 0.02 mm s−1) than on Column III (0.48 ± 0.00 mm s−1). The higher EOF velocity arises from the adsorption of more mobile phase ions to the more polar Column Ia thus leading to a higher surface charge density and in turn stronger EOF velocity. Also, the EOF velocity was 1.4-fold higher on Column Ia (0.89 ± 0.07 mm s−1) than on Column III (0.63 ± 0.01 mm s−1) using a hydro-organic mobile phase consisting of 40% ACN:60% (v/v) of 2 mM Na2HPO4, pH 7.0, and 15 kV as the running voltage. These are the running conditions for electrochromatograms in Fig. 7A and 2A, respectively.
The reproducibility of the analysis on Column Ia was investigated using ABs (from toluene to heptylbenzene) as model solutes. This was carried out by measuring the k values of ABs from run-to-run (n = 3) and day-to-day (n = 3) using a mobile phase of 40% (v/v) ACN and 60% (v/v) of 2 mM Na2HPO4, pH 7.0. The day-to-day reproducibilities were calculated by averaging the triplicate measurements of the k values on each day. In all cases, the reproducibility was determined by the % RSD of the k values which were 2.99% and 4.62% for run-to-run and day-to-day, respectively.
Conclusion
The poly(GMA-co-EDMA) monolith (Column III) and its diol derivative (Column Ia) proved to be useful for incorporating OH-MWCNTs physically and covalently. In the case of Column III, the incorporation of OH-MWCNTs provided an amendment to the chromatographic property of this monolith by increasing/modulating its hydrophobic interactions with nonpolar solutes and adding π–π interactions toward aromatic solutes, while the diol derivative monolithic column (Column Ia) acted as a “support” for the carbon nanoentities, and brought about the realization of nearly an OH-MWCNT stationary phase proper. The application of high-power sonication for a short period of time allowed the generation of polymerization solutions with well dispersed and shortened OH-MWCNTs which may have increased the specific surface area of the OH-MWCNTs. The separation selectivities and solute retention were controlled by hydrophobic and π–π interactions with the OH-MWCNTs incorporated into the organic polymer monoliths.Abbreviations
| ABs | Alkylbenzenes |
| ACN | Acetonitrile |
| AIBN | 2,2′-Azobis(isobutyronitrile) |
| EDMA | Ethylene glycol dimethacrylate |
| GMA | Glycidyl methacrylate |
| OH-MWCNTs | Hydroxyl functionalized multiwalled carbon nanotubes. |
Conflicts of interest
There are no conflicts to declare.References
- M. Jonnada, R. Rathnasekara and Z. El Rassi, Electrophoresis, 2015, 36, 76–100 CrossRef CAS PubMed.
- R. Rathnasekara, S. Khadka, M. Jonnada and Z. E. Rassi, Electrophoresis, 2017, 38, 60–79 CrossRef CAS PubMed.
- W. Hu, T. Hong, X. Gao and Y. Ji, TrAC, Trends Anal. Chem., 2014, 61, 29–39 CrossRef CAS.
- M. Ahmed, M. M. A. Yajadda, Z. J. Han, D. Su, G. Wang, K. K. Ostrikov and A. Ghanem, J. Chromatogr., A, 2014, 1360, 100–109 CrossRef CAS PubMed.
- S. D. Chambers, F. Svec and J. M. Fréchet, J. Chromatogr., A, 2011, 1218, 2546–2552 CrossRef CAS PubMed.
- Y. Li, Y. Chen, R. Xiang, D. Ciuparu, L. D. Pfefferle, C. Horváth and J. A. Wilkins, Anal. Chem., 2005, 77, 1398–1406 CrossRef CAS PubMed.
- Z. Lin, J. Wang, R. Yu, X. Yin and Y. He, Electrophoresis, 2015, 36, 596–606 CrossRef CAS PubMed.
- C. Miao, R. Bai, S. Xu, T. Hong and Y. Ji, J. Chromatogr., A, 2017, 1487, 227–234 CrossRef CAS PubMed.
- Q. Cao, Y. Xu, F. Liu, F. Svec and J. M. Frechet, Anal. Chem., 2010, 82, 7416–7421 CrossRef CAS PubMed.
- M. Li, M. Tarawally, X. Liu, X. Liu, L. Guo, L. Yang and G. Wang, Talanta, 2013, 109, 1–6 CrossRef CAS PubMed.
- M. Navarro-Pascual-Ahuir, M. J. Lerma-Garcia, G. Ramis-Ramos, E. F. Simo-Alfonso and J. M. Herrero-Martinez, Electrophoresis, 2013, 34, 925–934 CrossRef CAS PubMed.
- E. J. Carrasco-Correa, G. Ramis-Ramos and J. M. Herrero-Martinez, J. Chromatogr., A, 2015, 1385, 77–84 CrossRef CAS PubMed.
- L. S. Zhang, P.-Y. Du, W. Gu, Q.-L. Zhao and Y.-P. Huang, J. Chromatogr., A, 2016, 1461, 171–178 CrossRef CAS PubMed.
- W. Lei, L.-Y. Zhang, L. Wan, B.-F. Shi, Y.-Q. Wang and W.-B. Zhang, J. Chromatogr., A, 2012, 1239, 64–71 CrossRef CAS PubMed.
- S. Xu, R. Mo, C. Jin, X. Cui, R. Bai and Y. Ji, J. Pharm. Biomed. Anal., 2017, 140, 190–198 CrossRef CAS PubMed.
- E. J. Carrasco-Correa, A. Martínez-Vilata, J. M. Herrero-Martínez, J. B. Parra, F. Maya, V. Cerdà, C. P. Cabello, G. T. Palomino and F. Svec, Talanta, 2017, 164, 348–354 CrossRef CAS PubMed.
- E. Mayadunne and Z. El Rassi, Talanta, 2014, 129, 565–574 CrossRef CAS PubMed.
- S. W. Kim, T. Kim, Y. S. Kim, H. S. Choi, H. J. Lim, S. J. Yang and C. R. Park, Carbon, 2012, 50, 3–33 CrossRef CAS.
- F. Okanda and Z. El Rassi, Electrophoresis, 2006, 27, 1020–1030 CrossRef CAS PubMed.
- H. Zhong and Z. El Rassi, J. Sep. Sci., 2009, 32, 10–20 CrossRef CAS PubMed.
- Y. Jmeian and Z. El Rassi, J. Proteome Res., 2007, 6, 947–954 CrossRef CAS PubMed.
- S. Selvaraju and Z. El Rassi, J. Sep. Sci., 2012, 35, 1785–1795 CrossRef CAS PubMed.
- S. Khadka and Z. El Rassi, Electrophoresis, 2016, 37, 3178–3185 CrossRef PubMed.
- C. A. Hewitt, M. Craps, R. Czerw and D. L. Carroll, Synth. Met., 2013, 184, 68–72 CrossRef CAS.
- K. Yang, Z. Yi, Q. Jing, R. Yue, W. Jiang and D. Lin, Chin. Sci. Bull., 2013, 58, 2082–2092 CrossRef CAS.
- T. W. G. Solomons and C. B. Fryhle, Organic Chemistry, John Wiley & Sons, Inc., 2008 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |