
Mechanism and reactivity of catalytic propargylic substitution reactions via metal–allenylidene intermediates: a theoretical perspective
Ken
Sakata
 *a and
Yoshiaki
Nishibayashi
*b
*a and
Yoshiaki
Nishibayashi
*b
aFaculty of Pharmaceutical Sciences, Hoshi University, Ebara, Shinagawa-ku, Tokyo 142-8501, Japan. E-mail: sakata@hoshi.ac.jp
bDepartment of Systems Innovation, School of Engineering, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ynishiba@sys.t.u-tokyo.ac.jp
First published on 17th October 2017
Abstract
Development of transition metal-catalyzed propargylic substitution reactions is still in progress as a novel synthetic tool, while a variety of allylic substitution reactions have been well established and widely used in organic synthesis. In this review, we summarize the mechanism and reactivity of transition metal-catalyzed propargylic substitution reactions from a theoretical point of view. We classify the reaction mechanisms for transition metal-catalyzed propargylic substitution reactions which have been reported to date, and then discuss, in particular, the reactions via metal–allenylidene intermediates.
 Ken Sakata | Ken Sakata received his Bachelor degree in 1995 and Ph.D. degree in 2000 from Kyoto University under the supervision of Professor Hiroshi Fujimoto. He became a research associate in 2000 and then a lecturer in 2005 at Hoshi University. Since 2010, he has been an associate professor at Hoshi University. His current research interests are focused on theoretical investigations of organic and organometallic chemistry. |
 Yoshiaki Nishibayashi | Yoshiaki Nishibayashi received his Ph.D. in 1995 from Kyoto University under the supervision of Professor Sakae Uemura. He became an assistant professor at the University of Tokyo in 1995 and moved to Kyoto University in 2000. In 2005, he became an associate professor at the University of Tokyo as PI. Since 2016, he has been a full professor at the University of Tokyo. He received the CSJ Award for Distinguished Young Chemists in 2001, the JSPS Prize in 2012, the Green & Sustainable Chemistry Honorable Award in 2012, the SSOCJ Company Award in 2015, and the JSCC Award for Creative Work in 2017. |
1 Introduction
Transition metal-catalyzed allylic substitution reactions have been widely examined since the 1960s.1,2 A variety of allylic substitution reactions have been developed, and their reaction mechanism, in which the allylmetal intermediate plays an important role, has been understood in detail. At present, the allylic substitution reaction is one of the most important tools for both the carbon–carbon and carbon–heteroatom bond formations in organic synthesis.On the other hand, less attention has been paid to the corresponding propargylic substitution reaction until recently. The Nicholas reaction is known to be a useful propargylic substitution reaction.3,4 However, it requires a stoichiometric amount of [Co2(CO)8] and several steps are needed to obtain the propargylic substitution product. Catalytic propargylic substitution reactions had been fairly limited5–9 until ruthenium-catalyzed propargylic substitution reactions with a variety of nucleophiles were reported in 2000.10 Since then, transition metal-catalyzed propargylic substitution reactions have been gradually developed as novel transformation reactions.
Several reviews have already been published for catalytic propargylic substitution reactions.11–15 In contrast to previous reviews, in this review, we focus on the reaction mechanism and reactivity of transition metal-catalyzed propargylic substitution reactions mainly from a theoretical point of view. First, we classify transition metal-catalyzed propargylic substitution reactions in terms of their reaction mechanisms, and then discuss the mechanism and reactivity, especially for the reactions via metal–allenylidene intermediates.
2 Mechanisms for catalytic propargylic substitution reactions
Reaction mechanisms for the transition metal-catalyzed propargylic substitution reactions which have been reported to date are generally classified into six types as shown in Scheme 1.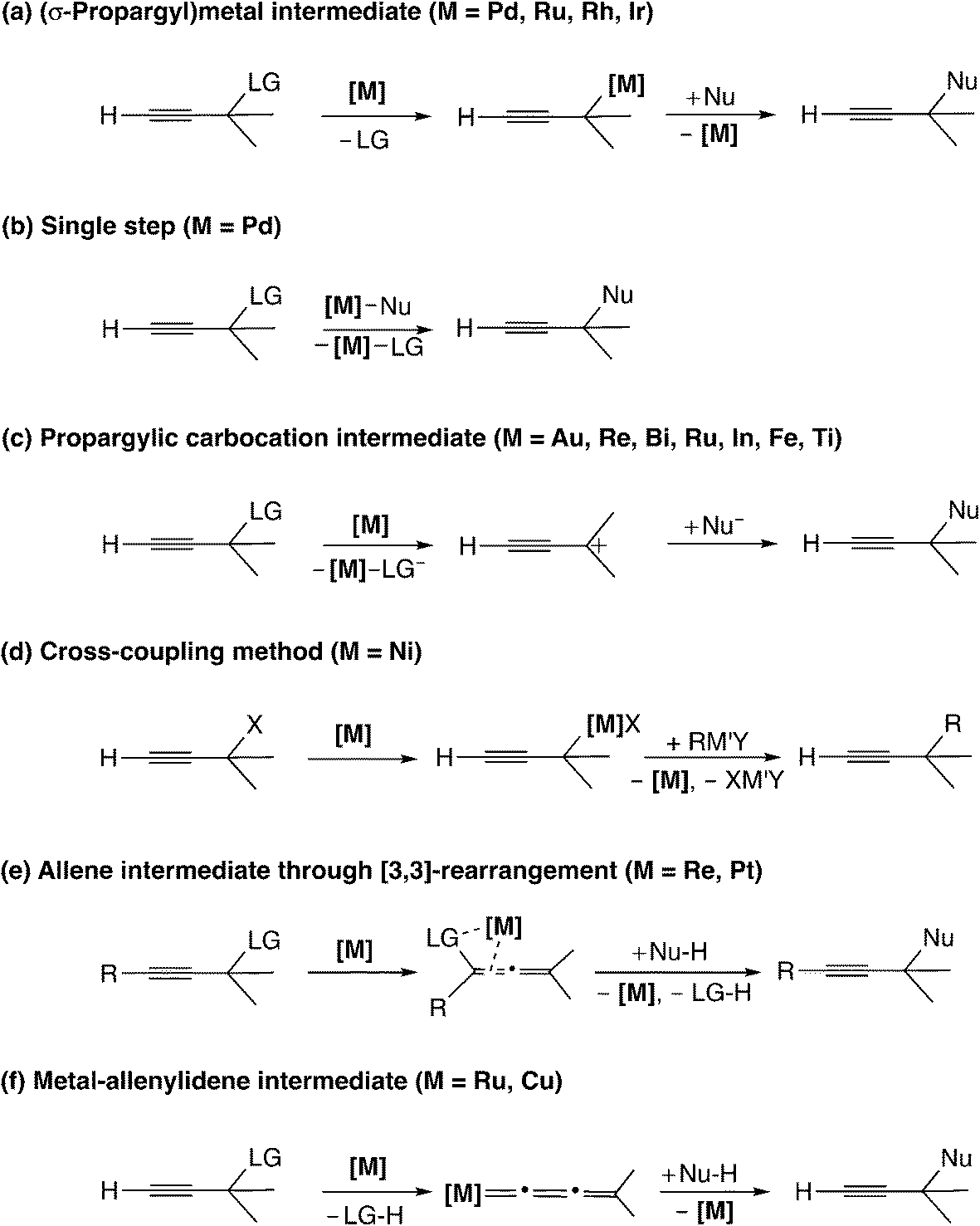 | ||
| Scheme 1 Reaction mechanisms for catalytic propargylic substitution reactions (LG = leaving group; Nu = nucleophile). | ||
Transition metal-catalyzed propargylic substitution reactions which have been known since early times proceed through a (σ-propargyl)metal intermediate (Scheme 1(a)).16 The detailed mechanism for this type of reaction is shown in Scheme 2. The reaction pathway involves an η3-allenylmetal intermediate (A), which gives a (σ-allenylic)metal complex (B) as well as a (σ-propargylic)metal complex (C). The attack of a nucleophile on complex B affords allene derivatives (D), while the attack on complex C provides the propargyl substitution products (E). Thus, the selectivity control is indispensable to yield predominantly the product E, because allene derivatives can also be formed (B → D) in this type of mechanism.
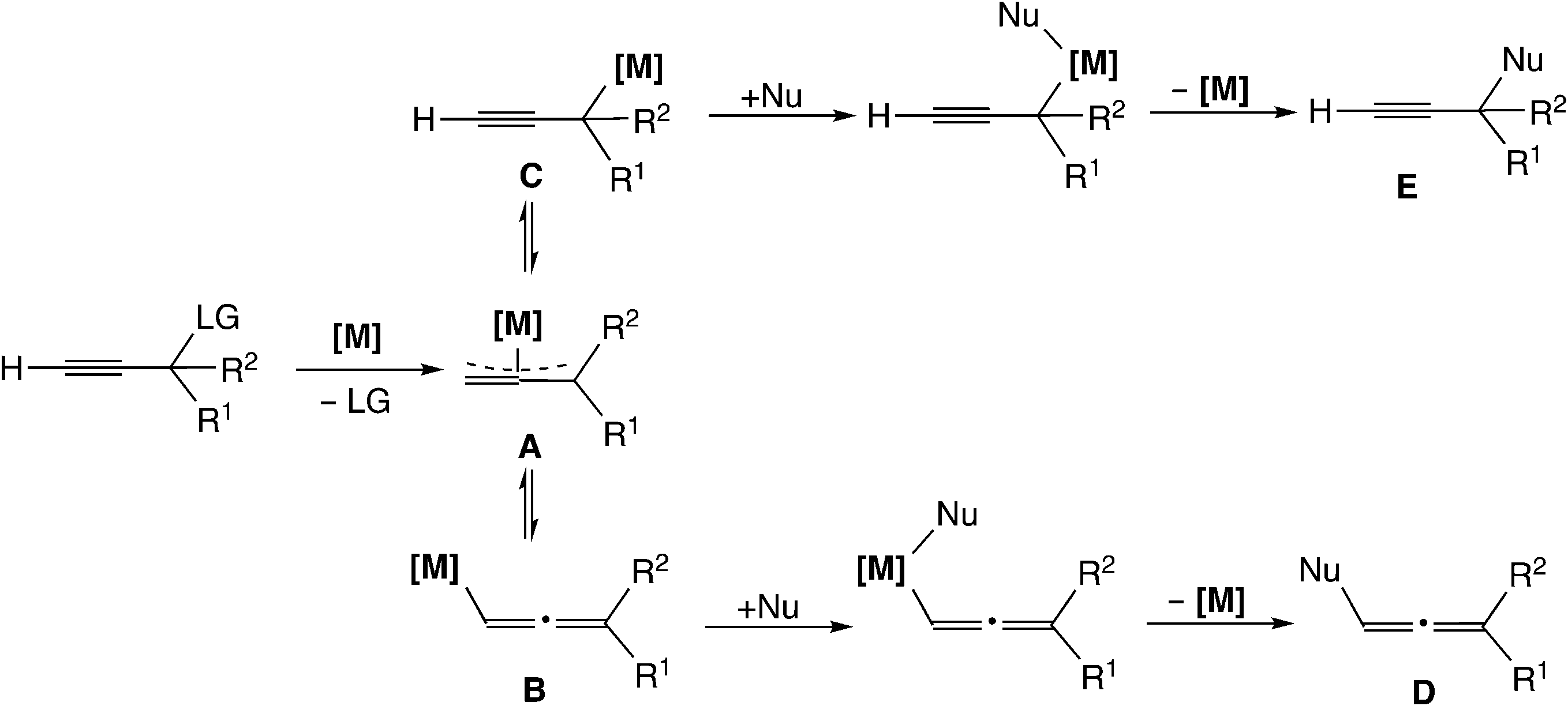 | ||
| Scheme 2 Reaction mechanism via (σ-propargyl)metal intermediates (LG = acetate, carbonate, halogen, and phosphate; Nu = nitrogen nucleophiles, sulfur nucleophiles, carbon nucleophiles, phosphorus nucleophiles, and hydride species). | ||
This mechanism corresponds to that for many reactions catalyzed by palladium complexes, which have been known to promote several types of transformations of propargylic compounds.6,16 Recently, Ma and co-workers reported the palladium-catalyzed propargylation reaction of tertiary propargylic carbonates with tri-substituted carbon nucleophiles to give propargyl substituted products.17 Reactions catalyzed by a mono-ruthenium complex18 have also been assumed to proceed through (σ-propargyl)metal intermediates.
Himo and co-workers performed a B3LYP-D level DFT study for this type of reaction mechanism.19 They examined whole reaction pathways for the [Pd(DPEPhos)]-catalyzed reaction of propargylic carbonate with phosphorus nucleophiles (Scheme 3), and compared the allenylphosphonate formation with the propargyl one.
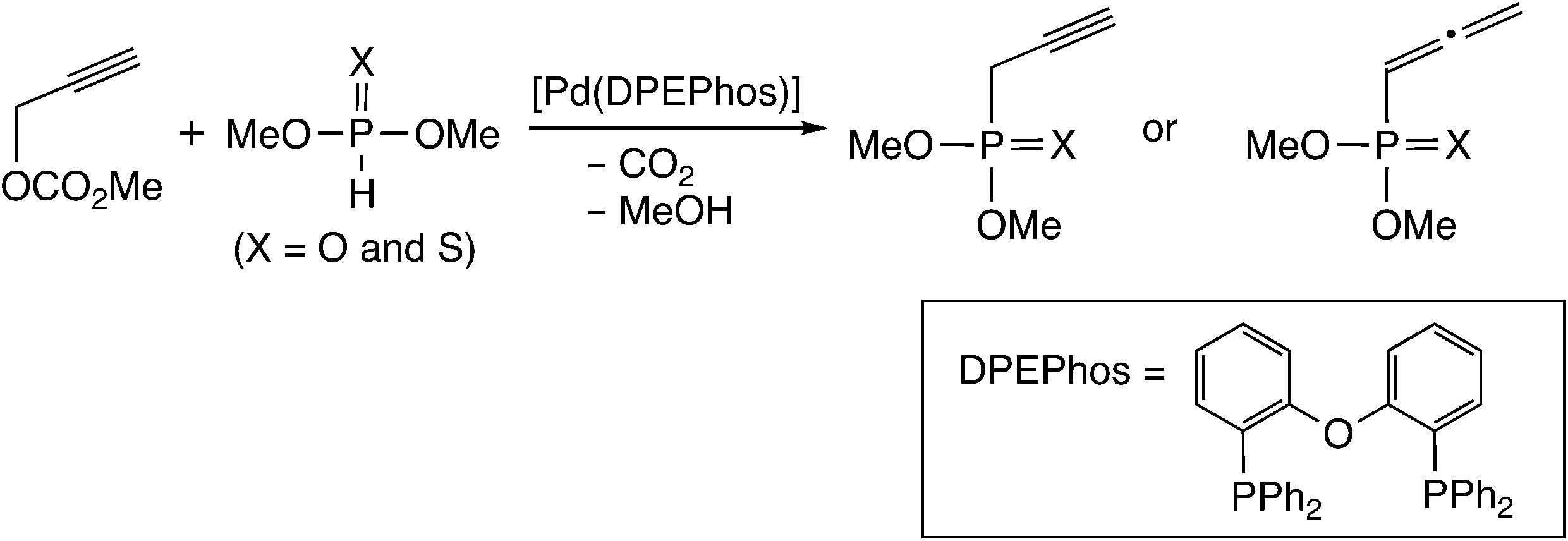 | ||
| Scheme 3 Model reaction system examined by Himo and co-workers.19 | ||
Wu, Zhao, and co-workers examined the reaction mechanism via allenylpalladium complexes for Pd(0)-catalyzed coupling of propargylic carbonates with N-tosylhydrazones by using ωB97X-D level DFT calculations.20
Another type of mechanism has been proposed for the palladium-catalyzed propargylic reaction.21 Szabó examined palladium pincer complex-catalyzed stannyl and silyl transfer to propargylic substrates (Scheme 4). They also performed B3PW91 level DFT calculations, and proposed the mechanism in which the trimethylstannyl group is transferred to the propargylic substrates in a single reaction step (Scheme 1(b)).21b In this pathway, the SN2′-like transition state for the allenyl product (I in Scheme 5) has been found to be preferred to the SN2-like transition state for the propargyl product (II in Scheme 5). They indicated that the palladium–tin σ-bond orbital plays an important role as a nucleophile.
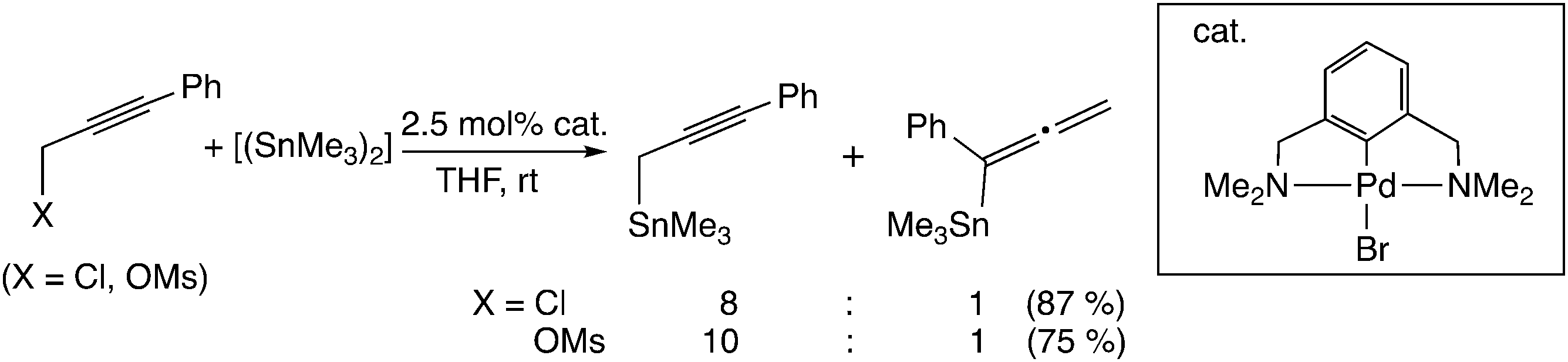 | ||
| Scheme 4 Single-step reaction mechanism. | ||
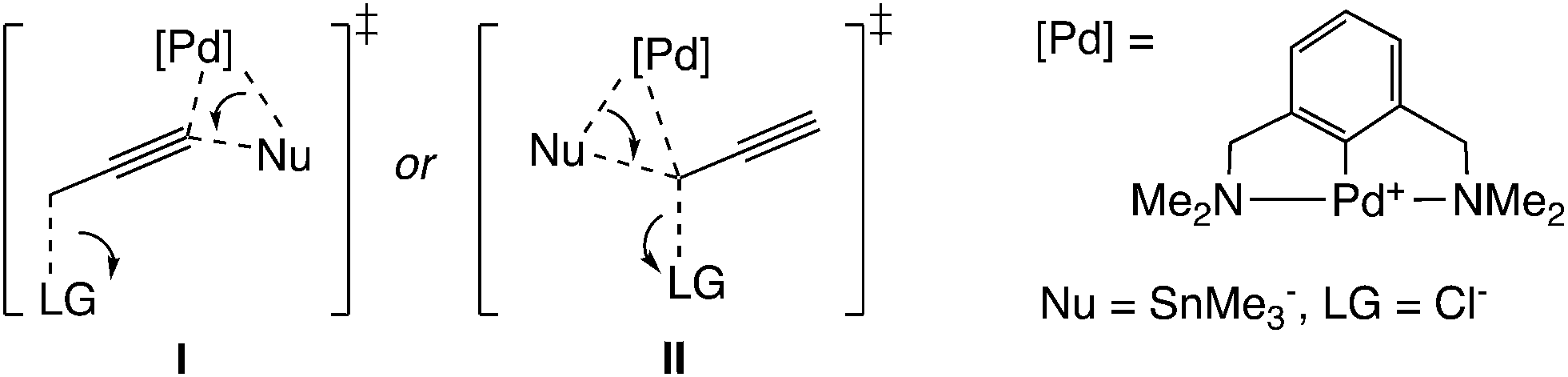 | ||
| Scheme 5 Transition states for single-step reactions. | ||
The third type of reaction is assumed to proceed through propargylic carbocation intermediates (Scheme 1(c)). The reaction using Lewis acid catalysts corresponds to this type of reaction (Scheme 6). Campagne and co-workers reported the gold-catalyzed propargylic substitution reaction, in which the gold catalyst acts as a propargylic alcohol-activating agent through π-coordination.22 Other gold complexes,23 rhenium complexes,24 bismuth complexes,25 ruthenium complexes,26 indium chloride,27,28 iron chloride,29 titanium complexes,8,30 and heterobimetallic iridium–tin complexes31 have been proposed to catalyze the propargylic substitution reaction via the propargylic carbocation intermediate. Reactions catalyzed by iridium32 and rhodium33 complexes may also proceed through propargylic carbocation intermediates.
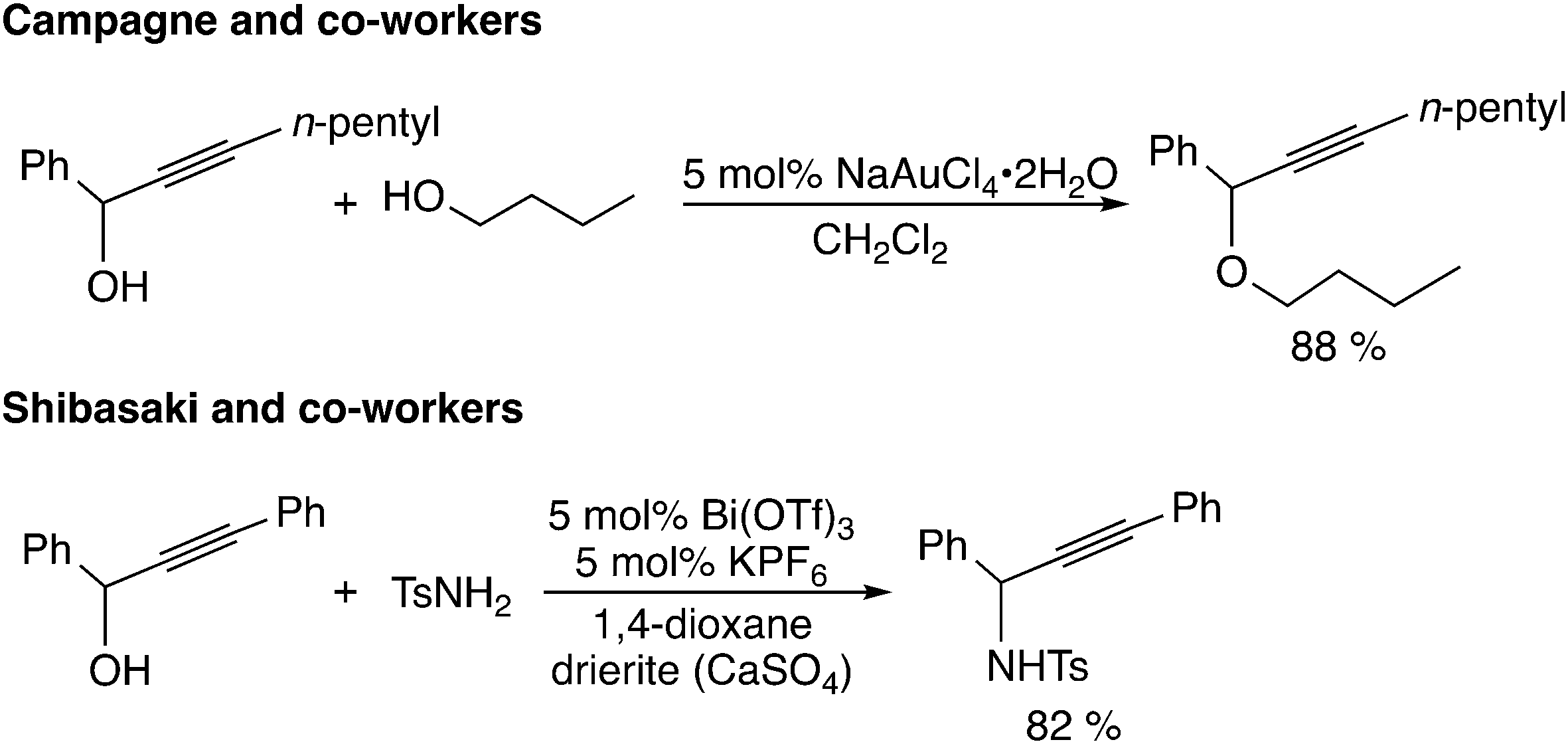 | ||
| Scheme 6 Reactions using Lewis acid catalysts. | ||
In this mechanism, the π-coordinated Lewis acid abstracts the hydroxyl group at the propargylic carbon atom. Thus, both the π- and σ-Lewis acidities are essential for this type of catalyst (Scheme 7). Recently, Olah, Bach, and co-workers examined the SN1-type diastereoselective substitution reactions of propargylic acetates, which bear a stereogenic center (–C*HXR2; X = Me, OMe, and OCH2Ar; R2 = tBu, iPr, and Et) adjacent to the electrophilic carbon atom, with silyl nucleophiles catalyzed by [Bi(OTf)3] (Scheme 8(a)), and concluded that the experimental results can be explained by assigning a chiral propargylic cation as an intermediate.34 The B3LYP and M06-2X(D3) level DFT calculations showed that the conformation in which the R2 group is almost perpendicular to the plane defined by the three substituents at the cationic carbon was preferred. It was also shown that the energy in the transition state IV is lower than that in the transition state III (Scheme 8(b)), reproducing the experimentally observed anti-selectivity. Furthermore, the NMR spectra for propargylic cations were obtained in order to examine the conformations.
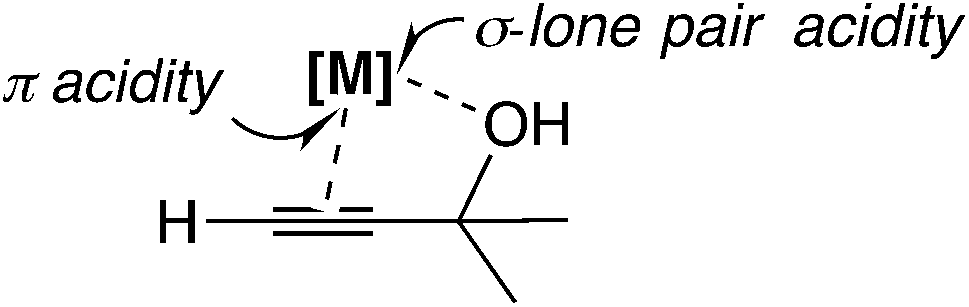 | ||
| Scheme 7 π-Acidity and σ-lone pair acidity in the Lewis acid. | ||
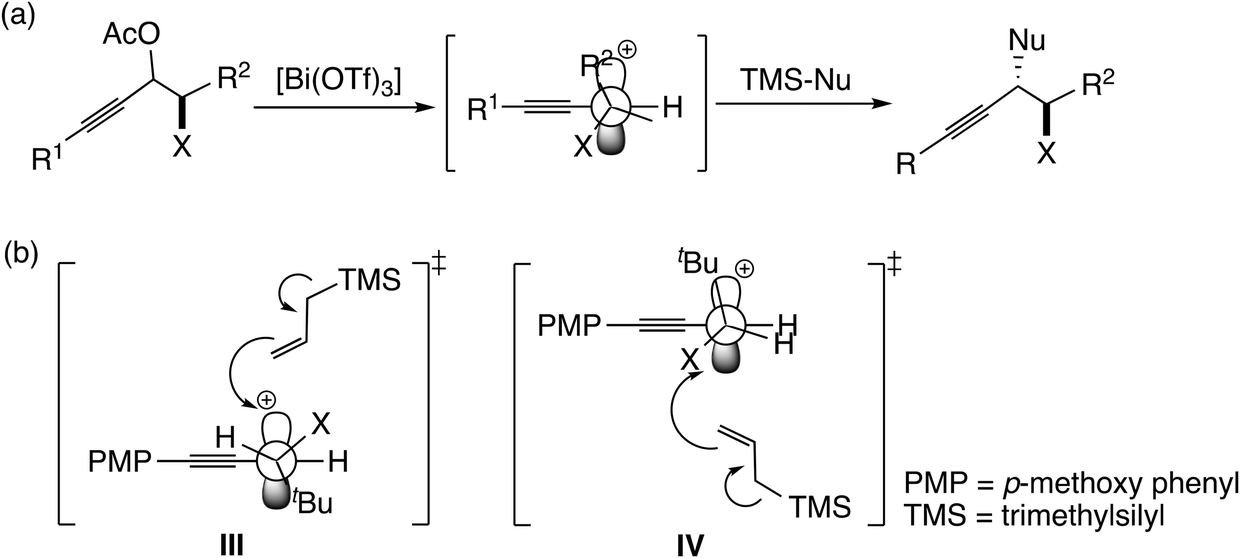 | ||
| Scheme 8 (a) SN1-type diastereoselective substitution reactions studied by Olah, Bach, and co-workers.34 (b) Examined transition states for the attack of allyltrimethylsilane to propargylic cation.34 | ||
Brønsted acids are known to catalyze propargylic substitution reactions in the same manner as Lewis acid catalysts. Sanz and co-workers reported the substitution reaction of the hydroxyl group in propargylic alcohols with 1,3-dicarbonyl compounds catalyzed by p-toluenesulfonic acid monohydrate (PTS) (Scheme 9(a)).35 The reaction of β-diketones bearing a substituent at the active methylene position with tertiary propargyl alcohol gave allene derivatives (Scheme 9(b)). It has been indicated that the regioselectivity depends on the cationic character of the propargylic cation (Scheme 9(c)).36 Zhan and co-workers examined the silver-catalyzed cycloaddition reactions of propargylic alcohols with thioamides, and proposed two pathways based on the resonance structures of the propargylic cation.37 Díez-González and co-workers reported that HBF4 catalyzes the nucleophilic substitution of propargylic alcohols.38
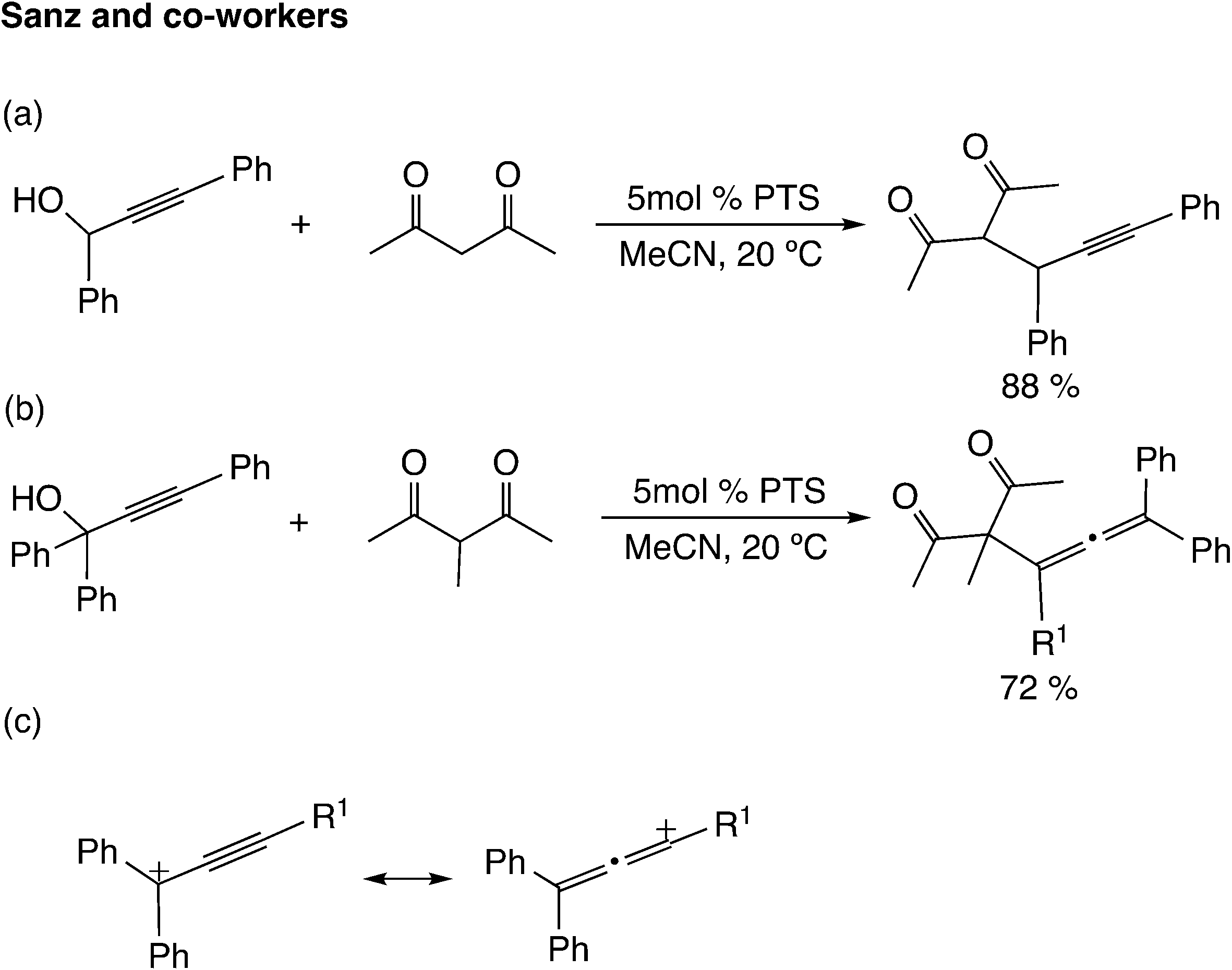 | ||
| Scheme 9 Reactions using a Brønsted acid catalyst. (a) Propargylation of 1,3-diketone with secondary propargylic alcohol. (b) Allenylation of substituted 1,3-diketone with tertiary propargylic alcohol. (c) Cationic character of the propargylic cation represented by resonance structures. | ||
The fourth type is the reaction utilizing the cross-coupling method (Scheme 1(d)). The nickel-catalyzed Negishi cross-coupling reactions of propargylic electrophiles with secondary alkyl nucleophiles have been reported by Fu and co-workers (Scheme 10).39 Later, asymmetric carbon–carbon bond-forming reactions have been reported.40
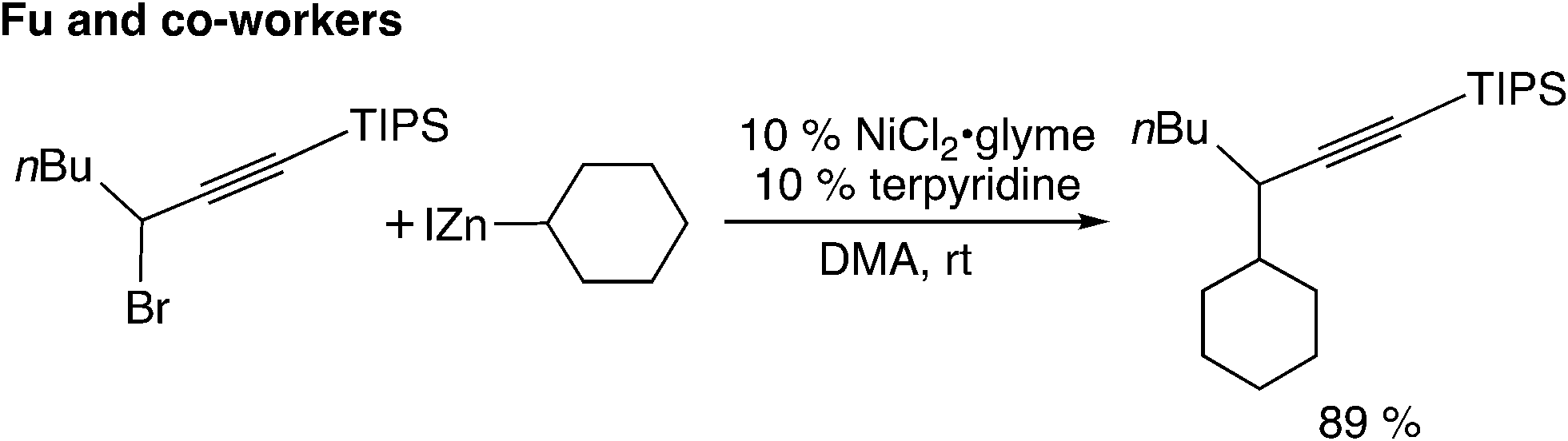 | ||
| Scheme 10 Cross-coupling reaction. | ||
The fifth type of reaction (Scheme 1(e)) proceeds through the allene intermediate generated by [3,3]-rearrangement of the leaving group. Toste and co-workers reported propargyl etherification catalyzed by a rhenium–oxo complex (Scheme 11).41 In this reaction, it has been postulated that the allenolate intermediate (V in Scheme 12), which is produced by the rearrangement of propargyl alcohol to an enone, undergoes SN2′ addition of a nucleophile. There have been no theoretical studies for the reaction, so the reaction may proceed via propargylic cation intermediates.
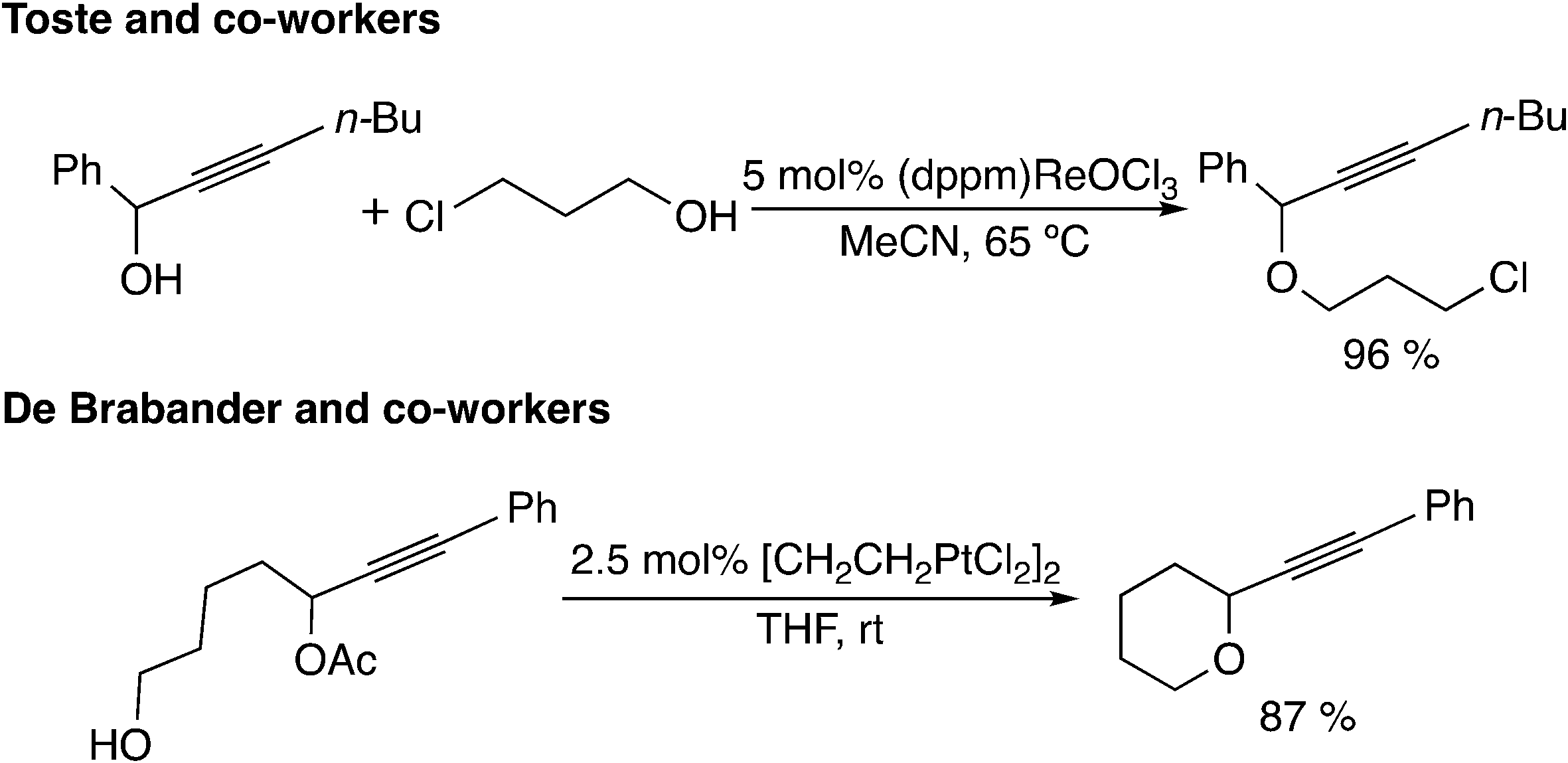 | ||
| Scheme 11 Reactions via allene intermediates. | ||
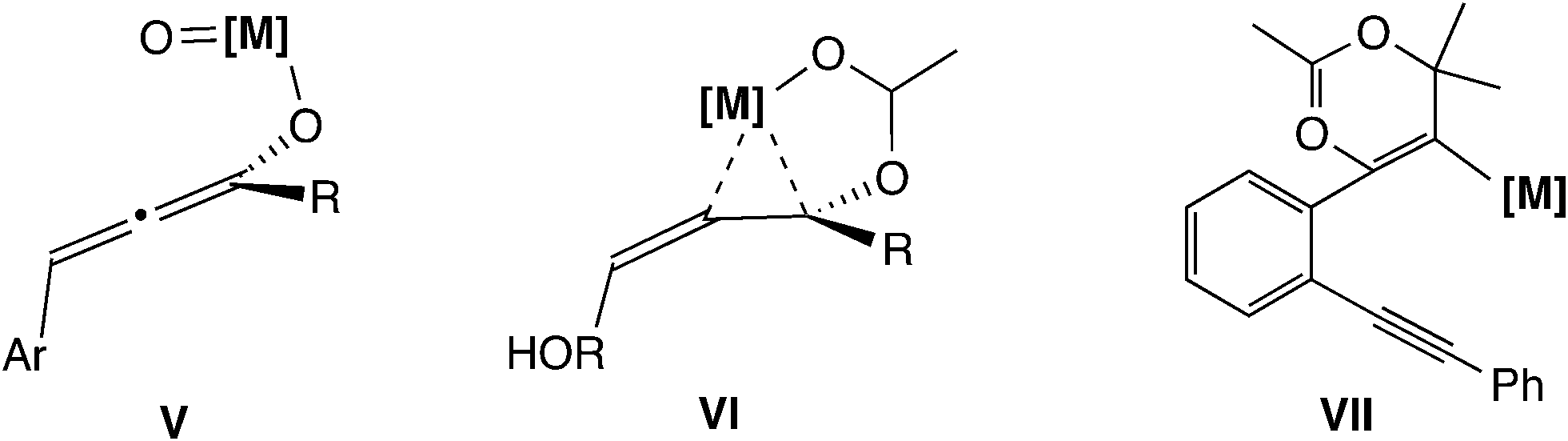 | ||
| Scheme 12 Proposed allene intermediates. | ||
Another example of this type was reported by De Brabander and co-workers.42 They found the cycloetherification of ω-hydroxypropargylic esters catalyzed by Zeise's dimer [CH2CH2PtCl2]2. In the key intermediate VI (Scheme 12) generated via [3,3]-rearrangement, the ester leaving group is activated by a cationic platinum(II) center. Thus, concerted SN2′-type cyclization has been proposed to occur easily. Theoretically, Fang, Yang and co-workers examined the reaction mechanisms via allene intermediate VII for gold(I)-catalyzed propargyl ester tandem cyclization reactions.43
A recently developed type of propargylic substitution reaction is the reaction via metal–allenylidene intermediates (Scheme 1(f)). In 2000, thiolate-bridged di-ruthenium complexes were found to work as effective catalysts toward propargylic substitution reactions (Scheme 13).10,44 A variety of nucleophiles, not only heteroatom-centered nucleophiles such as alcohols, amines, amides, thiols, and phosphite oxides but also carbon-centered nucleophiles such as β-diketones, silyl enol ethers, and ketones, are available for the propargylic substitution reactions.45 Enantioselective reactions have also been developed.46–48 Cadierno, Gimeno, and co-workers reported the propargylic substitution reaction catalyzed by a mononuclear 16-electron ruthenium(II) complex (2 in Scheme 13). This monoruthenium-catalyzed reaction has been proposed to proceed via a ruthenium–allenylidene intermediate.49 Haak and co-worker also assumed that ruthenium-catalyzed addition of carboxylic acids or cyclic 1,3-dicarbonyl compounds to propargyl alcohols proceeds through ruthenium–allenylidene or ruthenium–vinylidene complexes.50
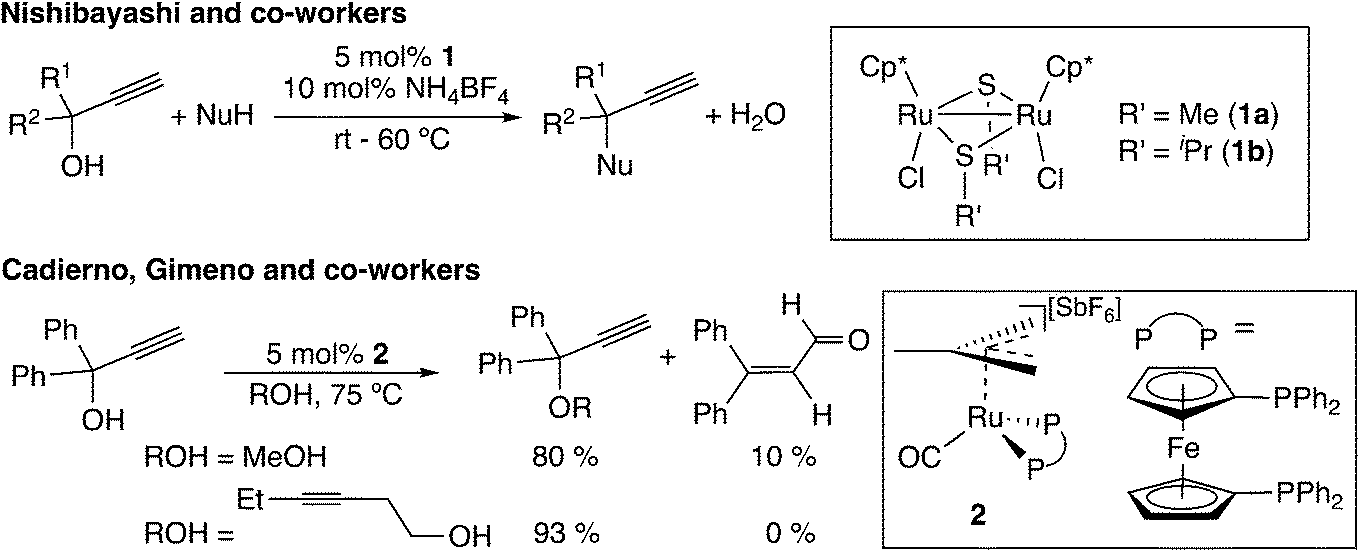 | ||
| Scheme 13 Ruthenium-catalyzed propargylic substitution reactions. | ||
Another example for this type of reaction is the copper-catalyzed reaction. In 1994, copper-catalyzed propargylic amination of propargylic esters with amines was reported independently by Murahashi's group7a and Caporusso's group.7b At the same time, Godfrey and co-workers reported the copper-catalyzed propargylic etherification of propargylic esters or chlorides with phenols.7c Later, in 2008, asymmetric amination reactions have been reported independently by two groups (Scheme 14).51,52 Recently, reactions with not only nitrogen-centered nucleophiles51a,52a–d,f,h,53 but also carbon-52e,i,54 and oxygen-centered52g,55 nucleophiles have been developed. These reactions have also been proposed to proceed via copper–allenylidene complexes. Detailed mechanisms are discussed in the following section.
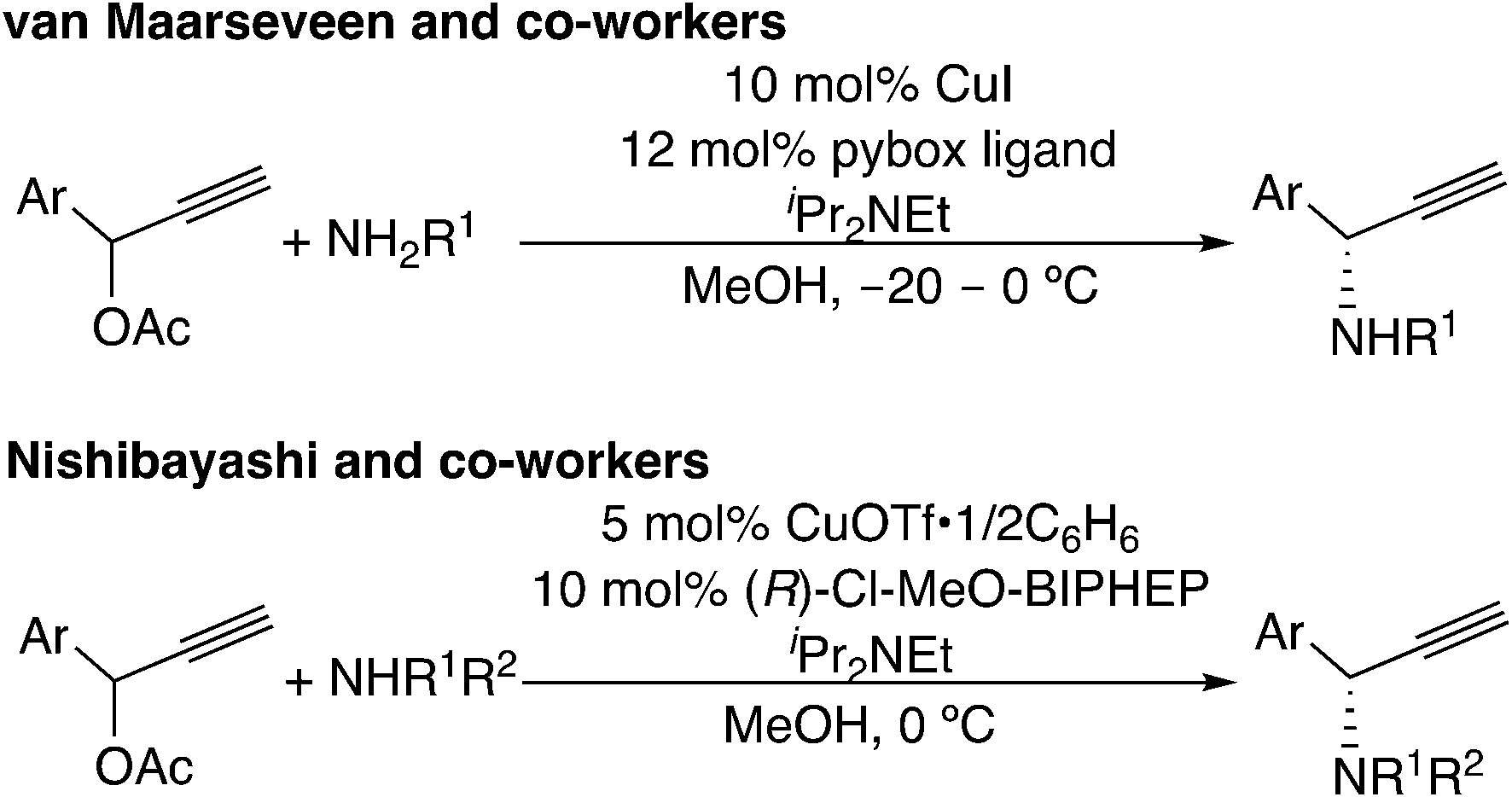 | ||
| Scheme 14 Copper-catalyzed propargylic substitution reactions. | ||
3 Reactions via metal–allenylidene intermediates
3.1 Mechanism
In the case of the thiolate-bridged diruthenium-catalyzed reaction, the detailed reaction mechanism has been examined based on stoichiometric and catalytic reactions.45 The proposed reaction pathway is shown in Scheme 15. The initial step is the formation of a vinylidene complex (C) via a π-alkyne complex (B). The vinylidene complex transforms into an allenylidene complex (D) through dehydration. Then, nucleophilic attack of a nucleophile on the γ-C atom in the allenylidene gives another vinylidene complex (E). The vinylidene complex transforms into another π-alkyne complex (F), which liberates the propargyl substituted product to regenerate the diruthenium active species (A).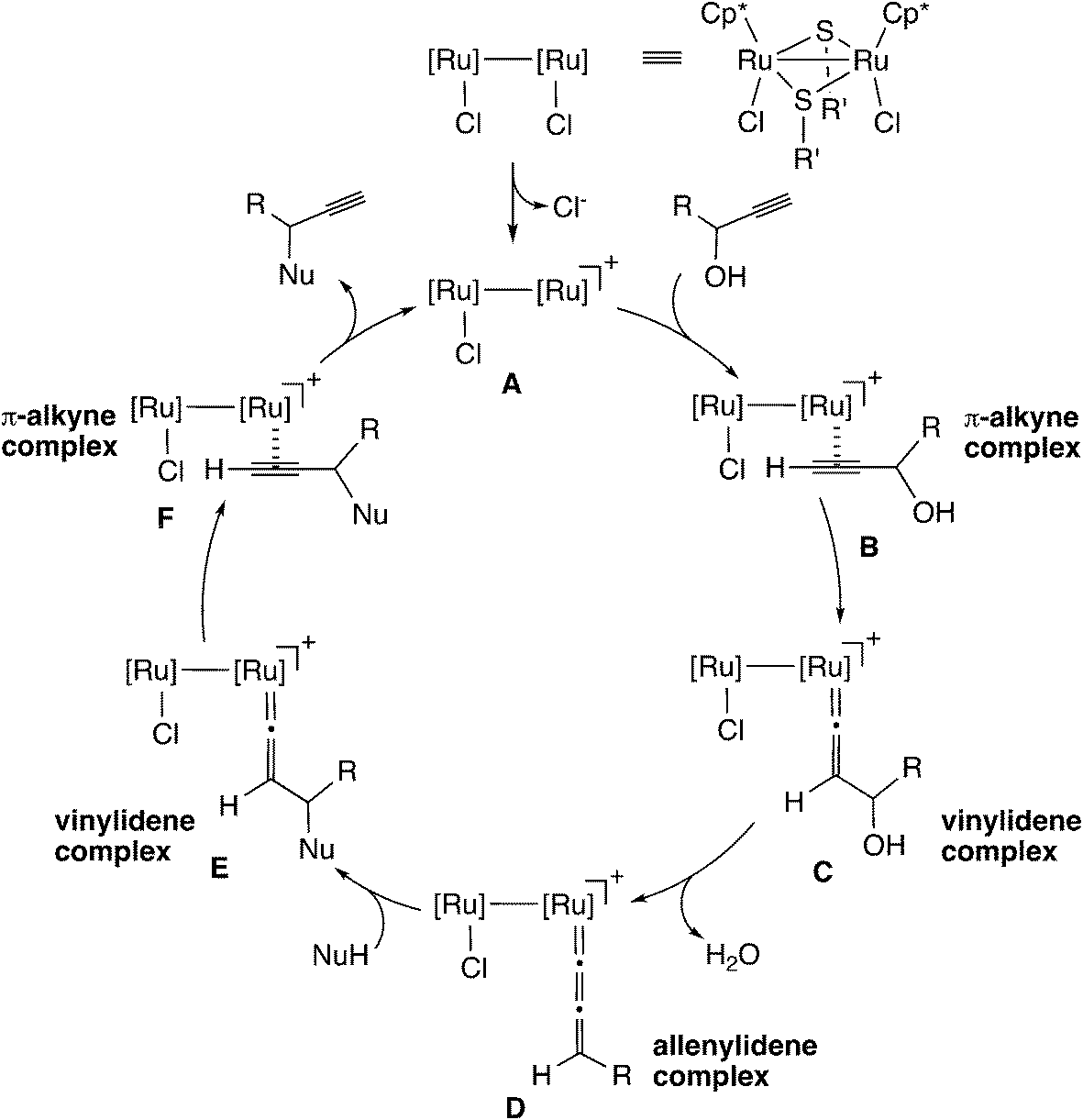 | ||
| Scheme 15 Proposed reaction pathway for the thiolate-bridged diruthenium-catalyzed reaction. | ||
Nakamura and co-workers performed a B3LYP level DFT study of the model reaction system as shown in Scheme 16.56 As shown in Scheme 15, the proposed catalytic cycle is symmetrical before and after the formation of the allenylidene complex. Therefore, the examined model reaction is not only the formation reaction of the ruthenium–allenylidene complex accompanied by dehydration (A → B → C → D in Scheme 15) but also the backward reaction for the nucleophilic attack of a water molecule, which is the oxygen-centered nucleophile, on the ruthenium–allenylidene complex (D → E → F → A in Scheme 15). Propargyl alcohol attached with zero to two methanol molecules, (MeOH)n (n = 0–2), was applied to the model substrate in methanol solvent. They compared the di-ruthenium catalyst with the mono-ruthenium one, and found that two factors are important for the catalytic efficiency of the diruthenium catalysts. One is the high stability of the coordinatively unsaturated complex, which provides smooth catalyst turnover. The other is the weaker back-donation, which makes the reaction steps easy by decreasing the reaction barriers in the reaction pathway.
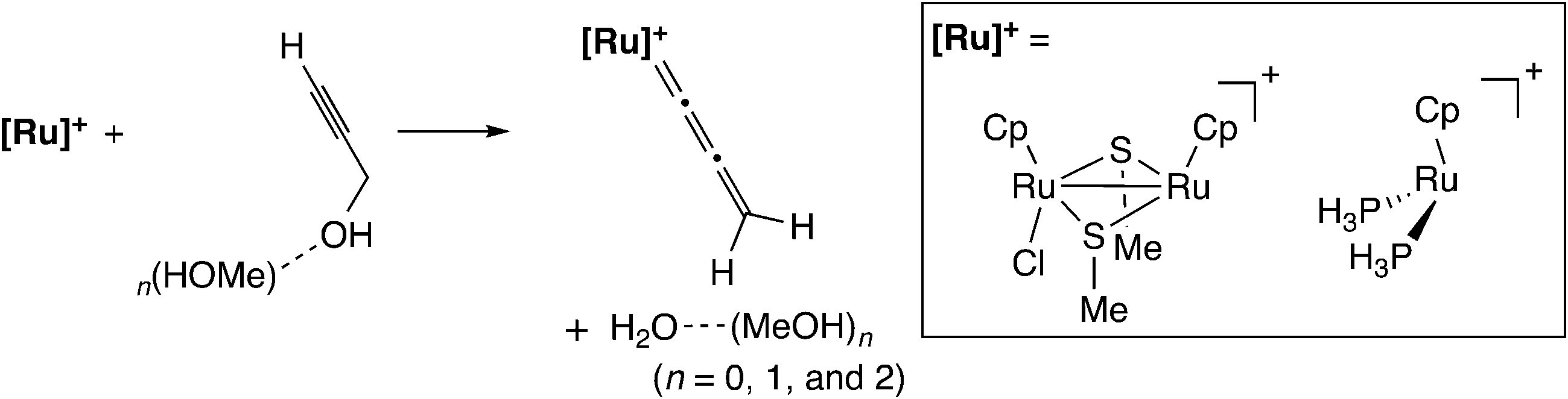 | ||
| Scheme 16 Model reaction system examined by Nakamura and co-workers.56 | ||
Based on the above results, Sakata and co-workers further examined the reaction of the ruthenium–allenylidene complex with carbon-centered nucleophiles by using B3LYP level DFT calculations.57 They investigated the reaction pathways for the reaction of propene and vinyl alcohol with the ruthenium–allenylidene complex (Scheme 17), and revealed that two reaction pathways are different after the attack of nucleophiles on the ruthenium–alkenyl complexes based on the results of IRC calculations. The carbon–carbon bond-forming reaction proceeds through a stepwise process in the case of propene, while it proceeds through a concerted process in the case of vinyl alcohol (Scheme 18). This is caused by the difference in the acidity of the hydrogen atom which is transferred to the β-C atom. They concluded that the C–C bond formation reactions have the same type of reaction mechanism as the reactions with heteroatom-centered nucleophiles.
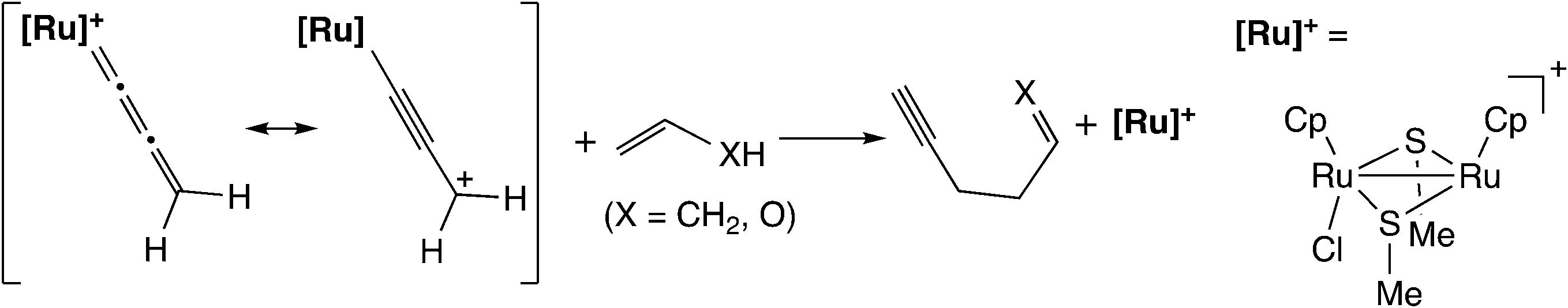 | ||
| Scheme 17 Model reaction system examined by Sakata and co-workers.57 | ||
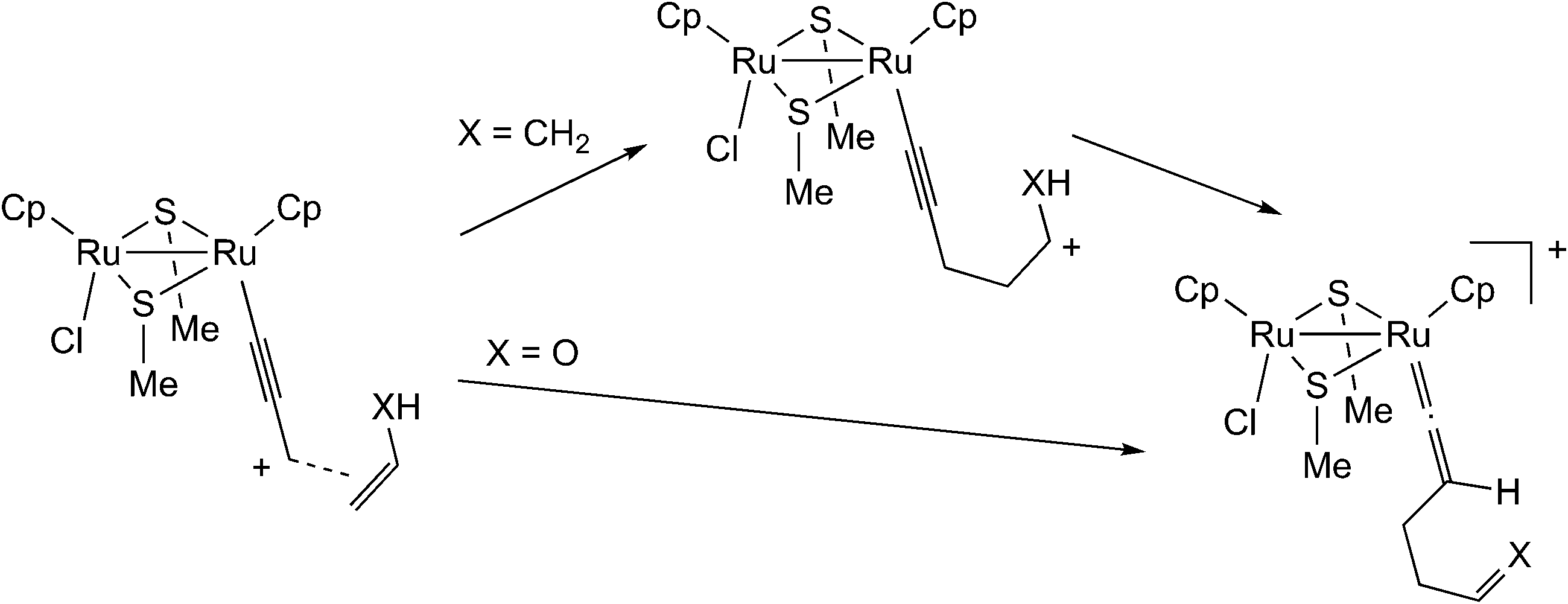 | ||
| Scheme 18 Reactions of the ruthenium–allenylidene complex with propene (X = CH2) and with vinyl alcohol (X = O).57 | ||
Their finding promoted better understanding of the reaction of carbon-centered nucleophiles. In the case of the reaction of 1-phenyl-2-propyn-1-ol with 2,4-dimethylpentadiene (Scheme 19), 2-methyl-4-methylene-6-phenyl-2-octen-7-yne was observed as the product.58 For this reaction, not the concerted mechanism but the stepwise mechanism was proposed, and the mechanism was confirmed to be reasonable by performing B3LYP/LANL2DZ level DFT calculations.58
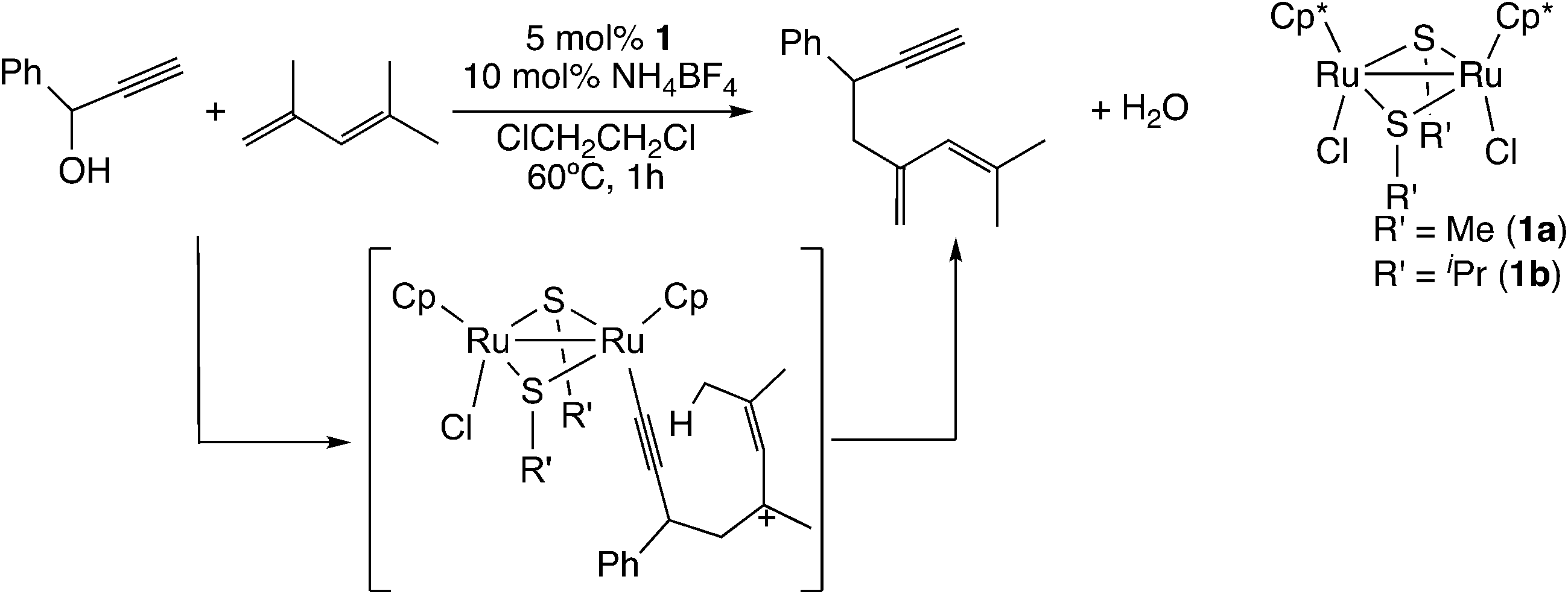 | ||
| Scheme 19 Reactions of ruthenium–allenylidene complexes with 1,3-dienes.58 | ||
Furthermore, Sakata and co-workers have examined other reactions via the ruthenium–allenylidene complex by using DFT calculations.59,60 In the diruthenium-catalyzed reaction of 1-cyclopropyl-2-propyn-1-ols with anilines and water (Scheme 20), the nucleophilic attack of aniline or water on the ε-C atom of cyclopropane of the allenylidene complex has been proposed as the key step in the reaction.59 The B3LYP/LANL2DZ level DFT calculations for the model reaction system showed that the nucleophilic attack of NH3 on the ε-C atom of a cyclopropane ring connected to an allenylidene moiety (VIII) occurs easily to give the corresponding ruthenium–alkenyl complex (IX) (Scheme 21). The subsequent proton transfer affords the vinylidene complex (X). The free energy barriers for these two steps were calculated to be relatively small (15.5 and 1.2 kcal mol−1).
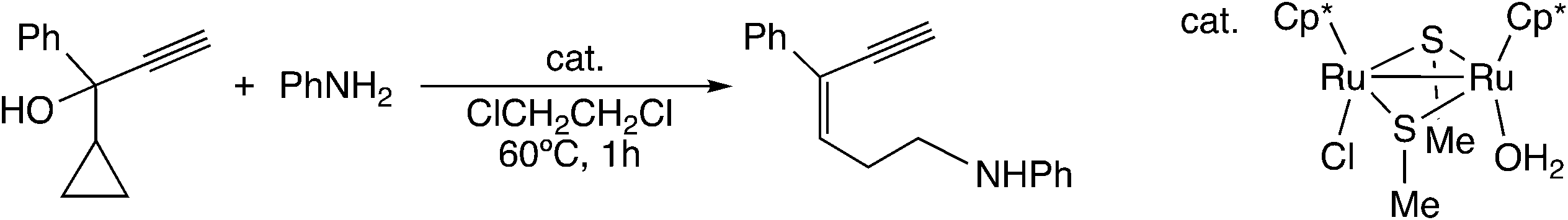 | ||
| Scheme 20 Reaction of 1-cyclopropyl-2-propyn-1-ols with anilines.59 | ||
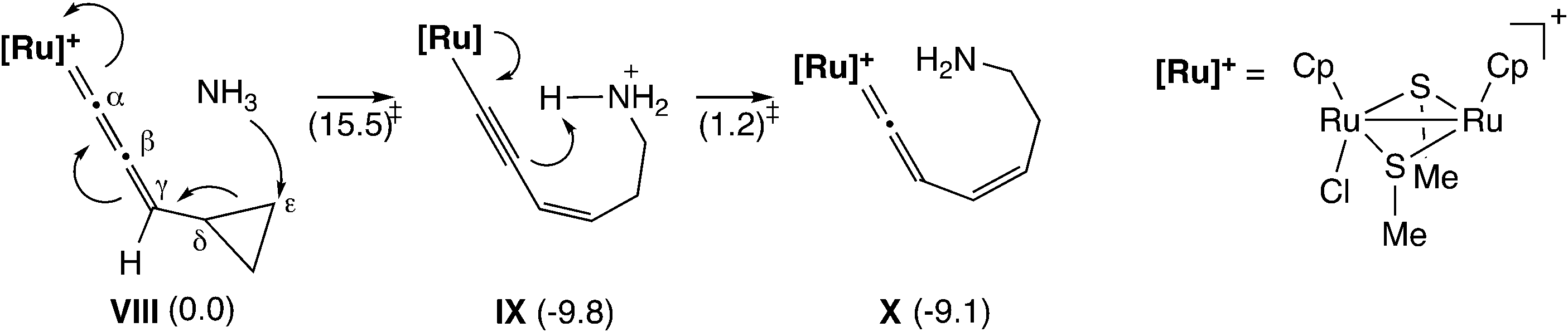 | ||
| Scheme 21 Model reaction pathway.59 Values in parentheses are relative Gibbs free energies at 298.15 K (kcal mol−1). | ||
In the [3 + 2] cycloaddition reaction of dimethyl 2-ethynyl-cyclopropane-1,1-dicarboxylate with aldehydes and aldimines (Scheme 22), the cyclopropyl vinylidene complex has been assumed to give the ruthenium–allenylidene complex.60 B3LYP level DFT calculations were performed for the model reaction system, and it was shown that the reaction proceeds smoothly along the proposed reaction pathway (Scheme 23). On the other hand, it was confirmed that the concerted cycloaddition pathway is disfavored.
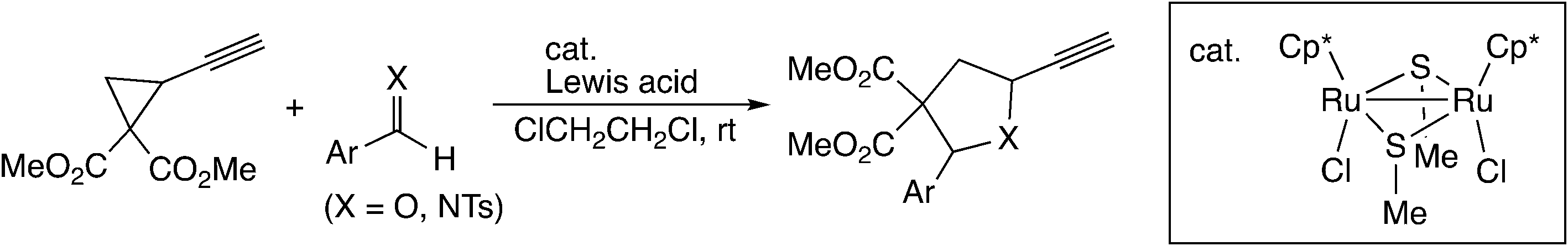 | ||
| Scheme 22 [3 + 2] cycloaddition reaction of dimethyl 2-ethynyl-cyclopropene-1,1-dicarboxylate with aldehydes and aldimines.60 | ||
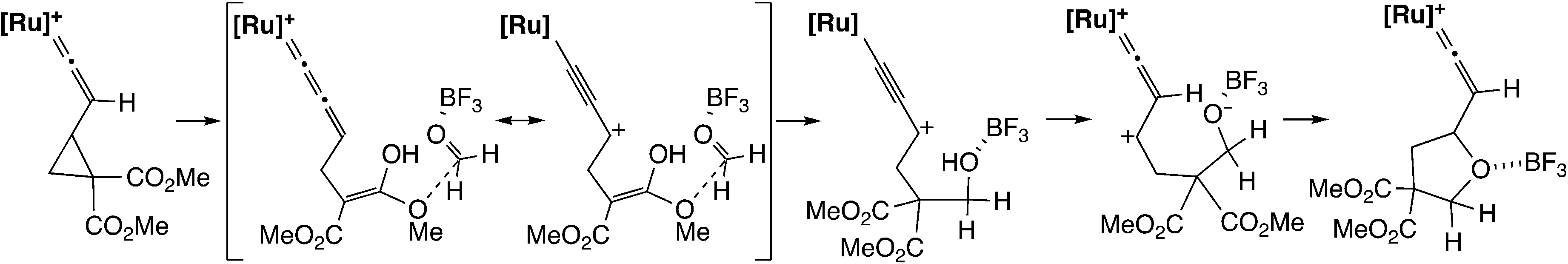 | ||
| Scheme 23 Model reaction pathway.60 | ||
Another typical example of the reaction via metal–allenylidene complexes is the copper-catalyzed reaction. It has been proposed that the reaction proceeds through the copper–allenylidene complex. The detailed reaction pathway proposed by Nishibayashi is shown in Scheme 24.52d The π-alkyne complex (A) transforms into a copper–acetylide complex (B).61 The elimination of acetic acid gives the copper–allenylidene complex (C). The nucleophilic attack of the amine on the γ-C atom provides another copper–acetylide complex (D), which is followed by another π-alkyne complex (E). The ligand exchange between the product and the substrate regenerates the initial π-alkyne complex. Although silver– and gold–allenylidene complexes have been experimentally isolated62–66 and also been theoretically examined,63–66 the copper–allenylidene complexes have not been isolated.
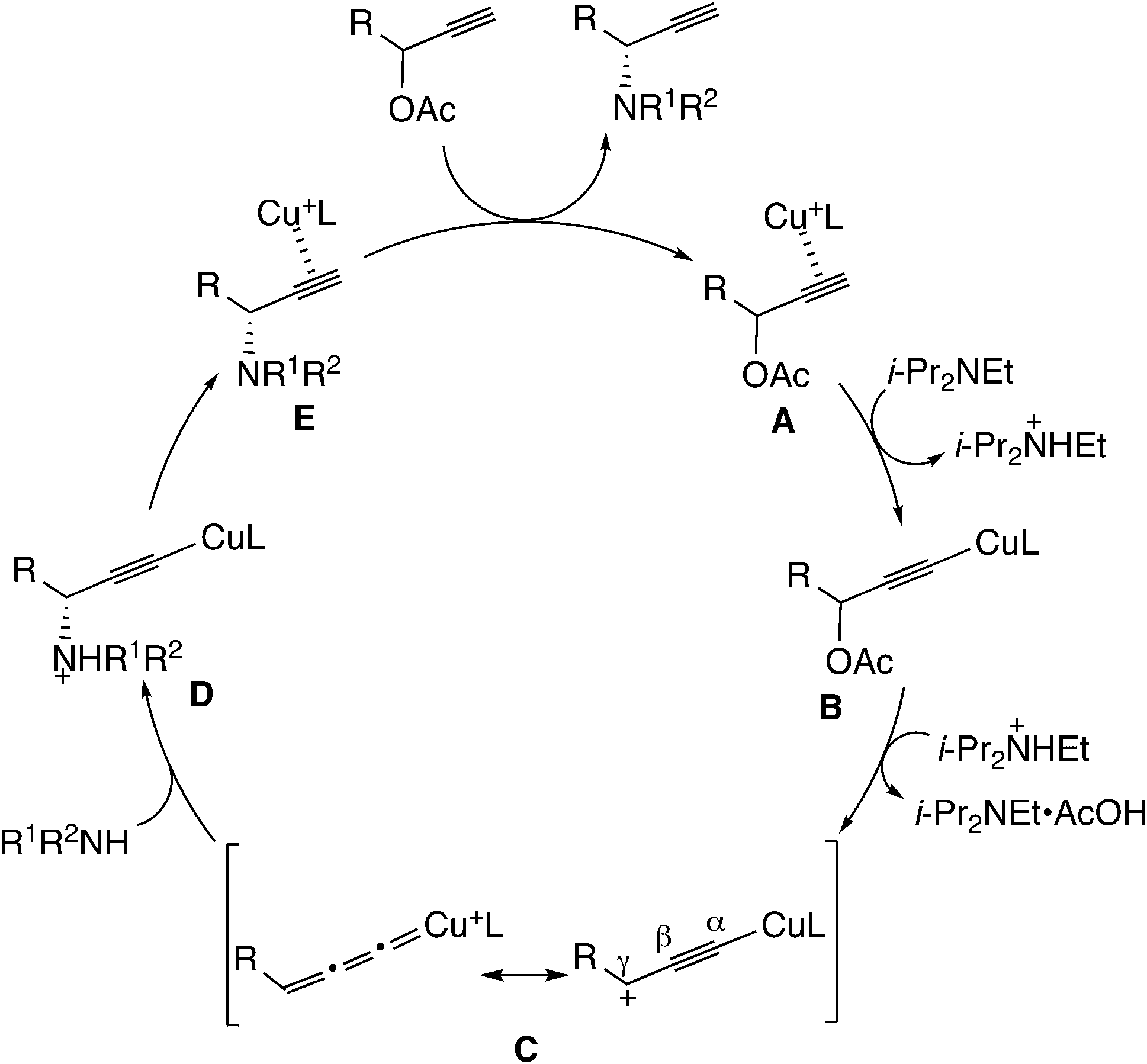 | ||
| Scheme 24 Proposed reaction pathway for the copper-catalyzed amination reaction. | ||
Sakata, Nishibayashi and co-workers performed a DFT study for the model reaction system between propargylic acetate (XI, XI′, or XI′′) and dimethylamine in the presence of [Cu(PH3)2(MeOH)]+ (Scheme 25)52d and revealed that the assistance of the Lewis base molecule, methanol or trimethylamine is an important factor to promote the propargylic amination. That is, trimethylamine used primarily for the neutralization of eliminated AcOH also makes the formation of the copper–acetylide complex easier.
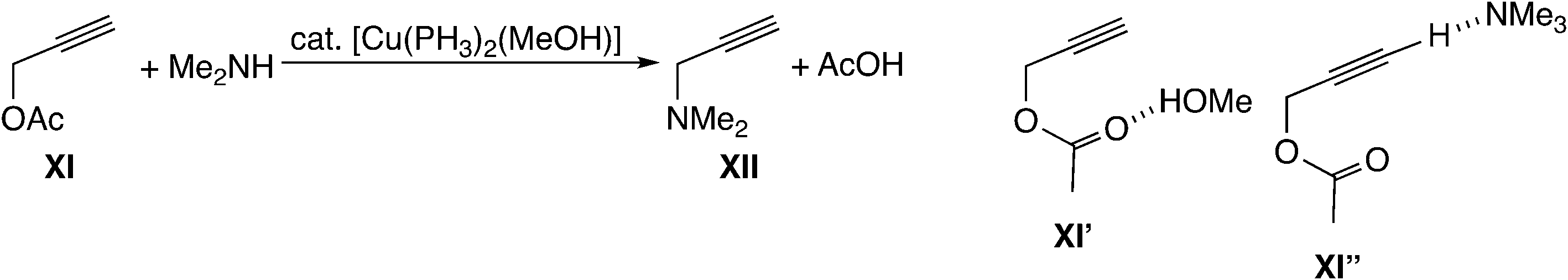 | ||
| Scheme 25 Model reaction system for copper-catalyzed amination.52d | ||
Experimentally, a linear relationship between the ligand enantiomeric excess (ee) and the product ee was observed in the case of the copper-catalyzed reaction using the BIPHEP ligand.52d This indicates that a monomeric copper complex works as the active species to promote the reaction. On the other hand, chiral amplification was observed in the case of the pybox ligand.52f,h This suggests that a dinuclear copper complex is the active species. In fact, the prepared dinuclear copper–pybox complex shows the same catalytic activity as the copper catalyst formed in situ from CuOTf·1/2C6H6 and pybox.52f Identification of the true active species in the reaction system provides a better understanding of the origin of the stereoselectivity. Very recently, by using M06-2X level DFT calculations, it has been shown that the stereo-chemical model of dinuclear copper–allenylidene complexes with 1,2-diols activated by borinic acid rationalizes the experimentally observed selectivity in the propargyl substitution reactions of polyols, where a nonlinear relationship was shown.55b Thus, further experimental and computational studies have been desired.
3.2 Structures and reactivities of metal–allenylidene intermediates
In the reaction via metal–allenylidene intermediates, the structure and reactivity of metal–allenylidene complexes play a crucial role. For metal–allenylidene complexes, some reviews including theoretical discussions have been published to date.67 In this section, we discuss the roles of metal–allenylidene complexes in catalytic reaction systems.We first compare the reaction free energies of the metal–allenylidene complex formation between propargyl alcohols or esters and the metal complexes, ΔG298K, as shown in Table 1. In the reaction of the mono-ruthenium complex [CpRu(PH3)2]+ with propargyl alcohol, the reaction free energy is largely exergonic (ΔG298K = −13.4 to −18.3 kcal mol−1). On the other hand, the reaction free energy is nearly zero (ΔG298K = 0.3 to −4.6 kcal mol−1) in the reaction of the di-ruthenium complex, [CpRuCl(μ2-SMe)2RuCp(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCCH2)]+, which shows the catalytic activity for propargylic substitution reactions. The reactions of the copper complex, [Cu(PH3)2(MeOH)]+, with propargylic acetate are largely endergonic (ΔG298K = 17.7–23.8 kcal mol−1). The copper complex also has catalytic activity as shown in the above discussion. These results indicate that the formation of energetically unstable metal–allenylidene complexes is favored for the catalytic reaction systems via metal–allenylidene complexes. In the case of the reaction of propargyl alcohol with the copper complex, however, the reaction energy, 26.1 kcal mol−1, is too endergonic. This may be the reason why the substitution reaction of propargyl alcohol catalyzed by copper catalysts has not been reported until now.
CCCH2)]+, which shows the catalytic activity for propargylic substitution reactions. The reactions of the copper complex, [Cu(PH3)2(MeOH)]+, with propargylic acetate are largely endergonic (ΔG298K = 17.7–23.8 kcal mol−1). The copper complex also has catalytic activity as shown in the above discussion. These results indicate that the formation of energetically unstable metal–allenylidene complexes is favored for the catalytic reaction systems via metal–allenylidene complexes. In the case of the reaction of propargyl alcohol with the copper complex, however, the reaction energy, 26.1 kcal mol−1, is too endergonic. This may be the reason why the substitution reaction of propargyl alcohol catalyzed by copper catalysts has not been reported until now.
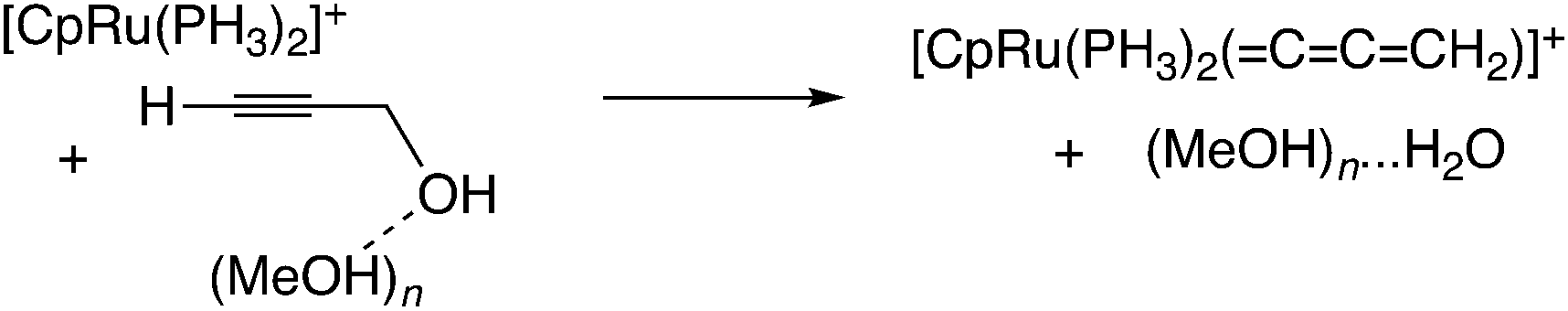
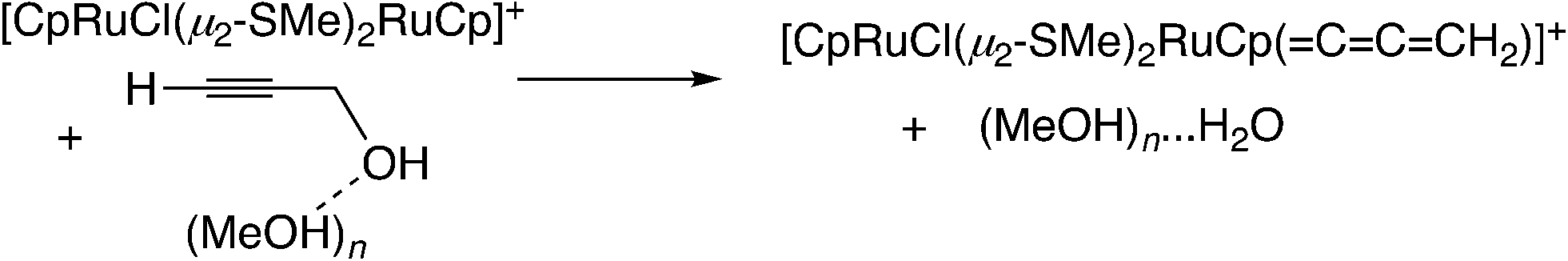
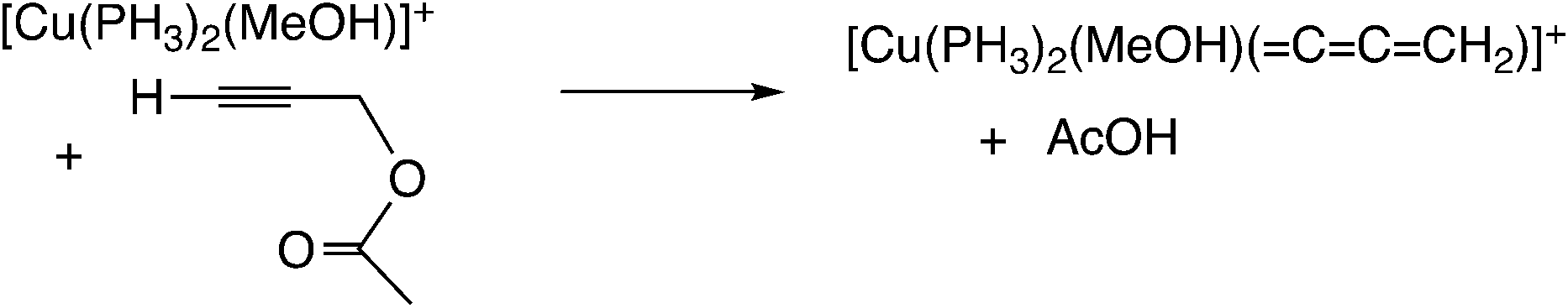
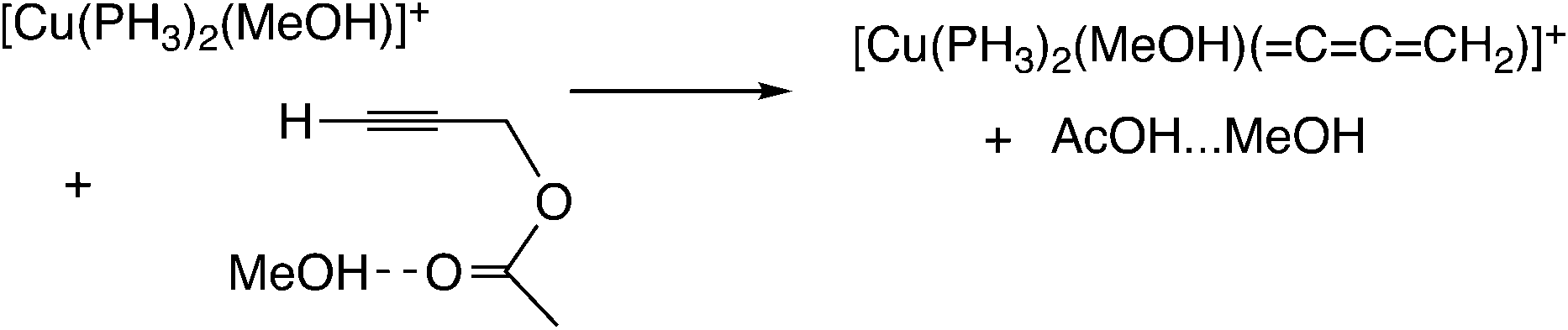
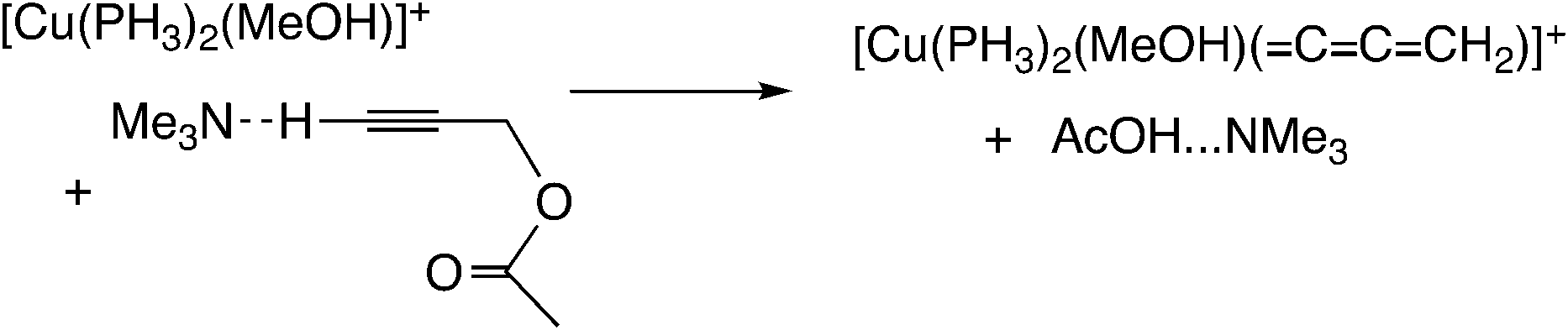
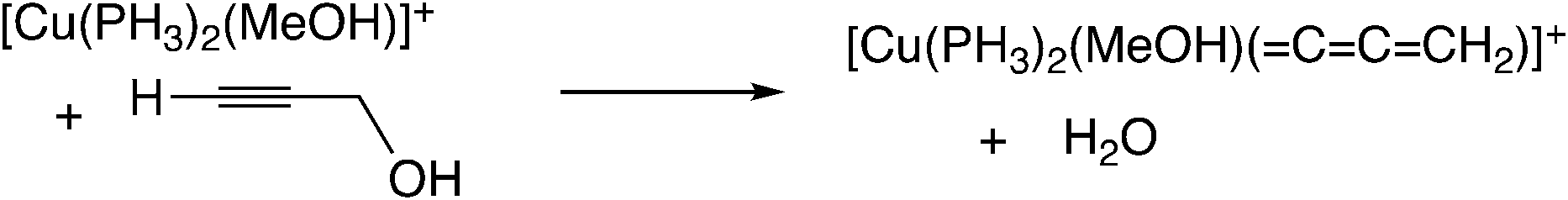
Nakamura and co-workers indicated that the difference in energetic stability between the monoruthenium– and diruthenium–allenylidene complexes is due to the strength of back-donation from the metal to the allenylidene ligand.56 Sakata and co-workers compared the interaction energy68 of the copper–allenylidene complex with that of the monoruthenium complex, and showed that the interaction between the metal complex and the allenylidene ligand in the former complex is much weaker than that in the latter complex.52d
In canonical MOs for the metal–allenylidene complex, the back-donation, that is, the electron delocalization from the occupied orbital in the metal complex to the unoccupied orbital in the allenylidene ligand, is represented by the orbital ψin-phase, which is generated by in-phase overlap between the occupied metal d-orbital χ in the metal fragment and the LUMO φ in the allenylidene ligand (Scheme 26). On the other hand, the out-of-phase overlap between χ and φ gives the unoccupied orbital in the metal–allenylidene complex, ψout-of-phase, which is responsible for the electrophilicity of α-C and/or γ-C in the metal–allenylidene complex.67g
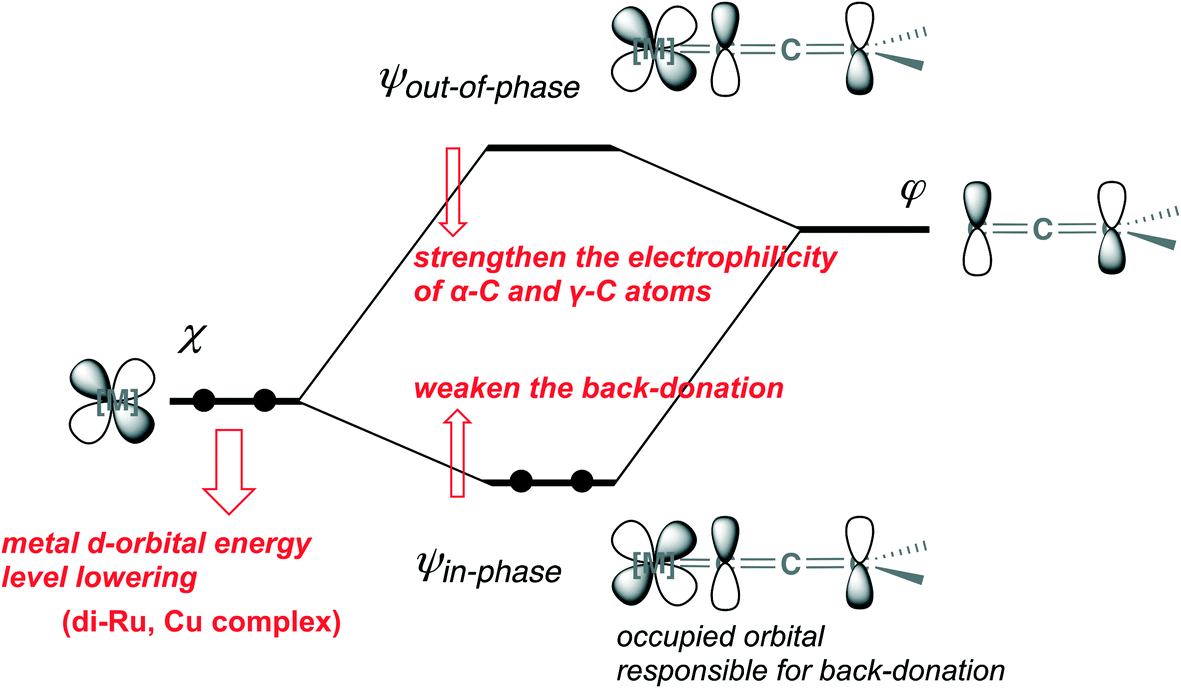 | ||
| Scheme 26 Orbital interaction between the metal d-orbital and the LUMO of the allenylidene ligand. | ||
Along with the lowering of the energy level in the orbital χ, the energy of the orbital ψin-phase is elevated while the energy of the orbital ψout-of-phase is lowered in terms of the orbital interaction scheme (Scheme 26).69 This means that the back-donation weakens and simultaneously the electrophilicity of α-C and/or γ-C in the metal–allenylidene complex strengthens. Thus, the energy of the metal d-orbital which interacts with the LUMO in the allenylidene ligand is predicted to affect both the back-donation and the electrophilicity. The orbitals which correspond to the orbitals ψin-phase and ψout-of-phase in the model metal–allenylidene complexes, [CpRu(PH3)2(CCCH2)]+, [CpRuCl(μ2-SMe)2RuCp(CCCH2)]+, and [Cu(PH3)2(MeOH)(CCCH2)]+, are shown in Fig. 1(a)–(c), respectively. In the [CpRu(PH3)2(CCCH2)]+ complex, the orbitals ψin-phase and ψout-of-phase correspond to the (HO − 2)MO and LUMO, which are mainly composed of the HOMO in the [CpRu(PH3)2]+ fragment and the LUMO in the allenylidene ligand. The HOMO in the [CpRu(PH3)2]+ fragment (−10.0 eV) and the LUMO in the allenylidene ligand (−4.1 eV) correspond to the orbitals χ and φ.
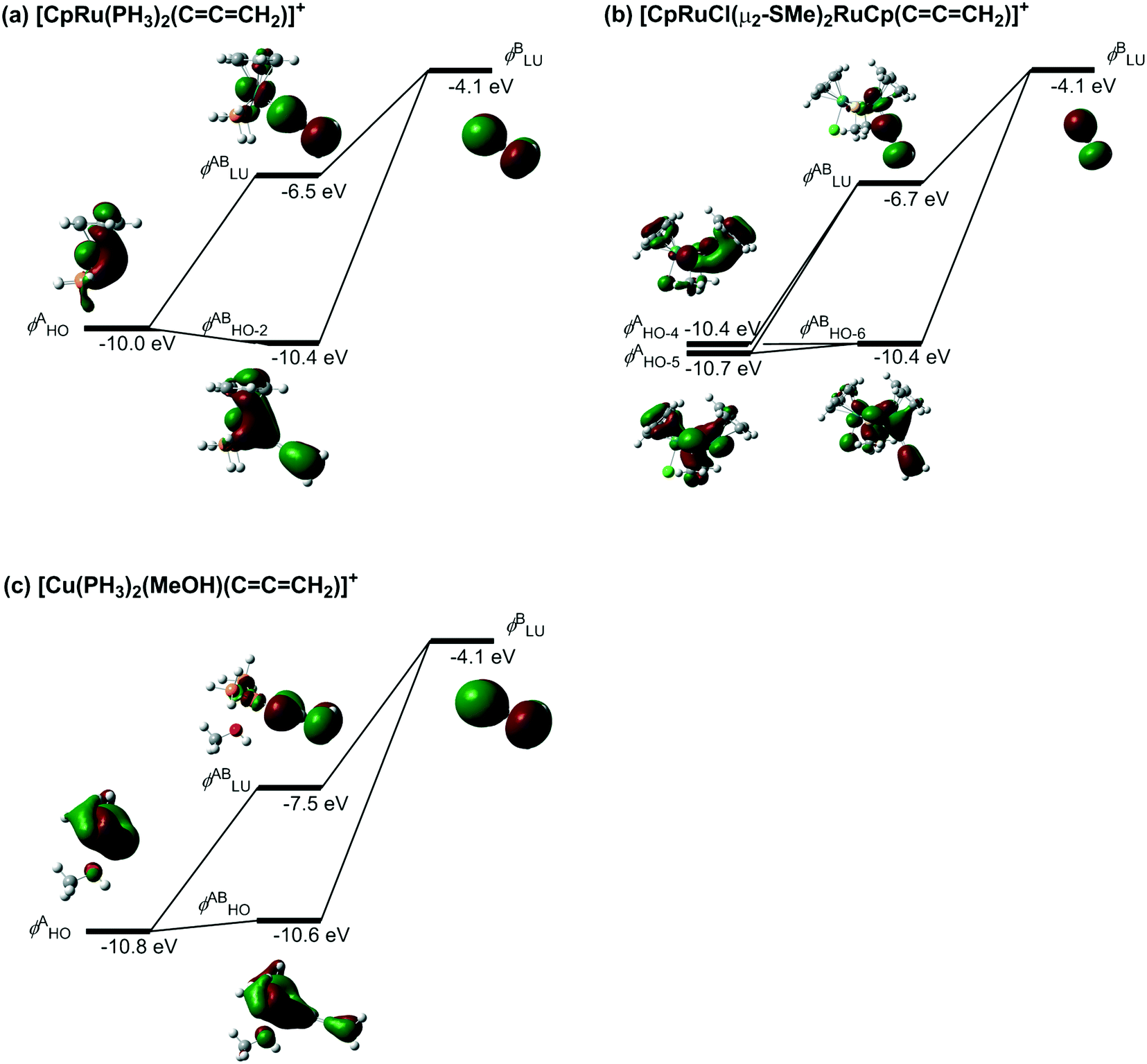 | ||
| Fig. 1 Orbital interactions between the metal d-orbital and the LUMO of the allenylidene ligand (B3LYP/(SDD, 6-311G(d,p)) level). | ||
In the [CpRuCl(μ2-SMe)2RuCp(CCCH2)]+ complex, the orbitals (HO − 6)MO and LUMO, which correspond to ψin-phase and ψout-of-phase, respectively, consist of the (HO − 4)MO and (HO − 5)MO in the diruthenium fragment and the LUMO in the allenylidene ligand. The energies of the (HO − 4)MO and (HO − 5)MO in the diruthenium fragment, −10.4 and −10.7 eV, are lower than that of the HOMO in the monoruthenium fragment (−10.0 eV). Therefore, the energy of the LUMO in the diruthenium–allenylidene complex (−6.7 eV) is lower than that of the LUMO in the monoruthenium–allenylidene complex (−6.5 eV). In the case of [Cu(PH3)2(MeOH)(CCCH2)]+, the HOMO in the [Cu(PH3)2(MeOH)]+ fragment, which corresponds to the orbital χ, is located at −10.8 eV energy, so the energy of the LUMO in the copper–allenylidene complex (−7.5 eV) is much lower than that of the LUMO in the monoruthenium–allenylidene complex (−6.5 eV). Thus, the energy levels of the metal d-orbitals, which are responsible for both the back-donation and electrophilicity, in the copper–allenylidene and diruthenium–allenylidene complexes are lower than that of the orbital in the monoruthenium–allenylidene complex. These perspectives suggest that an appropriate adjustment for the metal d-orbital energy levels is significant for the development of catalytic reaction systems via metal–allenylidene complexes.
4 Summary
In this review, we have summarized the reaction mechanisms and reactivities of transition metal-catalyzed propargylic substitution reactions. According to the experimental results, some types of mechanisms have been proposed, and a part of them have been examined theoretically. In particular, we focused on the reactions via metal–allenylidene complexes, which have recently been developed as a method for propargylic substitutions. Unfortunately, reactions of which the mechanisms are unknown remain.70 We believe that the combination of computational studies with experimental ones gives important clues for further development of catalytic propargylic substitution reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by CREST, JST (JPMJCR1541) and JSPS KAKENHI grants to K. S. (JP17K05795) and Y. N. (JP17H01201 and JP15H05798).Notes and references
- (a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed.
- J. F. Hartwig, Organotransition Metal Chemistry, University Science Books, California, chapter 20, 2010 Search PubMed.
- K. M. Nicholas, Acc. Chem. Res., 1987, 20, 207–214 CrossRef CAS.
- Recently, the enantioselective Nicholas reaction catalyzed by a chiral Brønsted acid was reported. See: M. Terada, Y. Ota, F. Li, Y. Toda and A. Kondo, J. Am. Chem. Soc., 2016, 138, 11038–11043 CrossRef CAS PubMed.
- C. Darcel, S. Bartsch, C. Bruneau and P. H. Dixneuf, Synlett, 1994, 457–458 CrossRef CAS.
- J. A. Marshall and M. A. Wolf, J. Org. Chem., 1996, 61, 3238–3239 CrossRef CAS.
- (a) Y. Imada, M. Yuasa, I. Nakamura and S.-I. Murahashi, J. Org. Chem., 1994, 59, 2282–2284 CrossRef CAS; (b) R. Geri, C. Polizzi, L. Lardicci and A. M. Caporusso, Gazz. Chim. Ital., 1994, 124, 241–248 CAS; (c) J. D. Godfrey Jr., R. H. Mueller, T. C. Sedergran, N. Soundararajan and V. J. Colandrea, Tetrahedron Lett., 1994, 35, 6405–6408 CrossRef.
- A. Bartels, R. Mahrwald and S. Quint, Tetrahedron Lett., 1999, 40, 5989–5990 CrossRef CAS.
- M. Hayashi, A. Inubushi and T. Mukaiyama, Chem. Lett., 1987, 1975–1978 CrossRef CAS.
- Y. Nishibayashi, I. Wakiji and M. Hidai, J. Am. Chem. Soc., 2000, 122, 11019–11020 CrossRef CAS.
- Y. Miyake, S. Uemura and Y. Nishibayashi, ChemCatChem, 2009, 1, 342–356 CrossRef CAS.
- N. Ljungdahl and N. Kann, Angew. Chem., Int. Ed., 2009, 48, 642–644 CrossRef CAS PubMed.
- R. J. Detz, H. H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2009, 6263–6276 CrossRef CAS.
- C.-H. Ding and X.-L. Hou, Chem. Rev., 2011, 111, 1914–1937 CrossRef CAS PubMed.
- For catalytic cycloaddition with terminal propargylic compounds, see: X.-H. Hu, Z.-T. Liu, L. Shao and X.-P. Hu, Synthesis, 2015, 47, 913–923 CrossRef CAS.
- J. Tsuji and T. Mandai, Angew. Chem., Int. Ed. Engl., 1995, 34, 2589–2612 CrossRef CAS.
- X. Huang, S. Xu, W. Wu, P. Li, C. Fu and S. Ma, Nat. Commun., 2016, 7, 12382 CrossRef PubMed.
- T. Kondo, Y. Kanda, A. Baba, K. Fukuda, A. Nakamura, K. Wada, Y. Morisaki and T. Mitsudo, J. Am. Chem. Soc., 2002, 124, 12960–12961 CrossRef CAS PubMed.
- J. O. C. Jiménez-Halla, M. Kalek, J. Stawinski and F. Himo, Chem. – Eur. J., 2012, 18, 12424–12436 CrossRef PubMed.
- W.-W. Ping, L. Jin, Y. Wu, X.-Y. Xue and X. Zhao, Tetrahedron, 2014, 70, 9373–9380 CrossRef CAS.
- (a) J. Kjellgren, H. Sunden and K. J. Szabó, J. Am. Chem. Soc., 2004, 126, 474–475 CrossRef CAS PubMed; (b) J. Kjellgren, H. Sundén and K. J. Szabó, J. Am. Chem. Soc., 2005, 127, 1787–1796 CrossRef CAS PubMed.
- (a) M. Georgy, V. Boucard and J.-M. Campagne, J. Am. Chem. Soc., 2005, 127, 14180–14181 CrossRef CAS PubMed; (b) M. Georgy, V. Boucard, O. Debleds, C. Dal Zotto and J.-M. Campagne, Tetrahedron, 2009, 65, 1758–1766 CrossRef CAS.
- (a) J. Liu, E. Muth, U. Florke, G. Henkel, K. Merz, J. Sauvageau, E. Schwake and G. Dyker, Adv. Synth. Catal., 2006, 348, 456–462 CrossRef CAS; (b) K. Y. Lee, H. S. Lee, H. S. Kim and J. N. Kim, Bull. Korean Chem. Soc., 2008, 29, 1441–1442 CrossRef CAS.
- Y. Kuninobu, E. Ishii and K. Takai, Angew. Chem., Int. Ed., 2007, 46, 3296–3299 CrossRef CAS PubMed.
- H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2007, 46, 409–413 CrossRef CAS PubMed.
- (a) Y. Inada, Y. Nishibayashi, M. Hidai and S. Uemura, J. Am. Chem. Soc., 2002, 124, 15172–15173 CrossRef CAS PubMed; (b) Y. Nishibayashi, Y. Inada, M. Yoshikawa, M. Hidai and S. Uemura, Angew. Chem., Int. Ed., 2003, 42, 1495–1498 CrossRef CAS PubMed; (c) Y. Nishibayashi, A. Shinoda, Y. Miyake, H. Matsuzawa and M. Sato, Angew. Chem., Int. Ed., 2006, 45, 4835–4839 CrossRef CAS PubMed; (d) E. Bustelo and P. H. Dixneuf, Adv. Synth. Catal., 2007, 349, 933–942 CrossRef CAS.
- K. Motoyama, M. Ikeda, Y. Miyake and Y. Nishibayashi, Eur. J. Org. Chem., 2011, 2239–2246 CrossRef CAS.
- In the InBr3-catalyzed deacetoxylation of propargylic acetate with triethylsilane, indium radical species were assumed to mediate the reaction. See: N. Sakai, M. Hirasawa and T. Konakahara, Tetrahedron Lett., 2005, 46, 6407–6409 CrossRef CAS.
- (a) Z.-P. Zhan, J.-L. Yu, H.-J. Liu, Y.-Y. Cui, R.-F. Yang, W.-Z. Yang and J.-P. Li, J. Org. Chem., 2006, 47, 8298–8301 CrossRef PubMed; (b) Z.-P. Zhan, Y.-Y. Cui and H.-J. Liu, Tetrahedron Lett., 2006, 47, 9143–9146 CrossRef CAS; (c) Z.-P. Zhan and H.-J. Liu, Synlett, 2006, 2278–2280 CrossRef CAS; (d) Z.-P. Zhan, X.-B. Cai, S.-P. Wang, J.-L. Yu, H.-J. Liu and Y.-Y. Cui, J. Org. Chem., 2007, 72, 9838–9841 CrossRef CAS PubMed.
- (a) R. Mahrwald, S. Quint and S. Scholtis, Tetrahedron, 2002, 58, 9847–9851 CrossRef CAS; (b) G. V. Karunakar and M. Periasamy, J. Org. Chem., 2006, 71, 7463–7466 CrossRef CAS PubMed.
- P. N. Chatterjee and S. Roy, J. Org. Chem., 2010, 75, 4413–4423 CrossRef CAS PubMed.
- P. A. Evans and M. J. Lawler, Angew. Chem., Int. Ed., 2006, 45, 4970–4972 CrossRef CAS PubMed.
- I. Matsuda, K. Komori and K. Itoh, J. Am. Chem. Soc., 2002, 124, 9072–9073 CrossRef CAS PubMed.
- D. Nitsch, S. M. Huber, A. Pöthig, A. Narayanan, G. A. Olah, G. K. S. Prakash and T. Bach, J. Am. Chem. Soc., 2014, 136, 2851–2857 CrossRef CAS PubMed.
- R. Sanz, D. Miguel, A. Martínez, J. M. Álvarez-Gutiérrez and F. Rodríguez, Org. Lett., 2007, 9, 727–730 CrossRef CAS PubMed.
- T. Ishikawa, T. Aikawa, Y. Mori and S. Saito, Org. Lett., 2003, 5, 51–54 CrossRef CAS PubMed.
- X. Gao, Y.-M. Pan, M. Lin, L. Chen and Z.-M. Zhan, Org. Biomol. Chem., 2010, 8, 3259–3266 CAS.
- E. Barreiro, A. Sanz-Vidal, E. Tan, S.-H. Lau, T. D. Sheppard and S. Díez-González, Eur. J. Org. Chem., 2015, 7544–7549 CrossRef CAS PubMed.
- S. W. Smith and G. C. Fu, Angew. Chem., Int. Ed., 2008, 47, 9334–9336 CrossRef CAS PubMed.
- A. J. Oelke, J. Sun and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 2966–2969 CrossRef CAS PubMed.
- B. D. Sherry, A. T. Radosevich and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 6076–6077 CrossRef CAS PubMed.
- J. K. De Brabander, B. Liu and M. Qian, Org. Lett., 2008, 10, 2533–2536 CrossRef CAS PubMed.
- Y. Li, A. M. Kirillov, R. Fang and L. Yang, Organometallics, 2017, 36, 1164–1172 CrossRef CAS.
- Hidai and co-workers originally prepared a series of thiolate-bridged diruthenium complexes [Cp*RuCl(SR)]2 (R = alkyl and aryl) and investigated their reactivity in detail. See: (a) S. Dev, K. Imagawa, Y. Mizobe, G. Cheng, Y. Wakatsuki, H. Yamazaki and M. Hidai, Organometallics, 1989, 8, 1232–1237 CrossRef CAS; (b) M. Hidai and Y. Mizobe, Can. J. Chem., 2005, 83, 358–378 CrossRef CAS and references therein.
- For reviews, see (a) Y. Nishibayashi, Curr. Org. Chem., 2006, 10, 135–150 CrossRef CAS; (b) Y. Nishibayashi and S. Uemura, in Metal Vinylidenes and Allenylidenes in Catalysis, ed. C. Bruneau and P. H. Dixneuf, Wiley-VCH, Weinheim, 2008, ch. 7, p. 217 Search PubMed.
- Y. Nishibayashi, G. Onodera, Y. Inada, M. Hidai and S. Uemura, Organometallics, 2003, 22, 873–876 CrossRef CAS.
- Y. Inada, Y. Nishibayashi and S. Uemura, Angew. Chem., Int. Ed., 2005, 44, 7715–7717 CrossRef CAS PubMed.
- For review: Y. Nishibayashi, Synthesis, 2012, 44, 489–503 CrossRef CAS.
- (a) V. Cadierno, J. Díez, S. E. García-Garrido and J. Gimeno, Chem. Commun., 2004, 2716–2717 RSC; (b) V. Cadierno, J. Gimeno and N. Nebra, Adv. Synth. Catal., 2007, 349, 382–394 CrossRef CAS; (c) V. Cadierno, S. E. García-Garrido, J. Gimeno and N. Nebra, Inorg. Chim. Acta, 2010, 363, 1912–1934 CrossRef CAS.
- S. Berger and E. Haak, Tetrahedron Lett., 2010, 51, 6630–6634 CrossRef CAS.
- (a) R. J. Detz, M. M. E. Delville, H. Hiemstra and J. H. van Maarseveen, Angew. Chem., Int. Ed., 2008, 47, 3777–3780 CrossRef CAS PubMed; (b) R. J. Detz, Z. Abiri, R. le Griel, H. Hiemstra and J. H. van Maarseveen, Chem. – Eur. J., 2011, 17, 5921–5930 CrossRef CAS PubMed.
- (a) G. Hattori, H. Matsuzawa, Y. Miyake and Y. Nishibayashi, Angew. Chem., Int. Ed., 2008, 47, 3781–3783 CrossRef CAS PubMed; (b) G. Hattori, A. Yoshida, Y. Miyake and Y. Nishibayashi, J. Org. Chem., 2009, 74, 7603–7607 CrossRef CAS PubMed; (c) G. Hattori, Y. Miyake and Y. Nishibayashi, ChemCatChem, 2010, 2, 155–158 CrossRef CAS; (d) G. Hattori, K. Sakata, H. Matsuzawa, Y. Tanabe, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2010, 132, 10592–10608 CrossRef CAS PubMed; (e) A. Yoshida, M. Ikeda, G. Hattori, Y. Miyake and Y. Nishibayashi, Org. Lett., 2011, 13, 592–595 CrossRef CAS PubMed; (f) A. Yoshida, G. Hattori, Y. Miyake and Y. Nishibayashi, Org. Lett., 2011, 13, 2460–2463 CrossRef CAS PubMed; (g) K. Nakajima, M. Shibata and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 2472–2475 CrossRef CAS PubMed; (h) M. Shibata, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2014, 50, 7874–7877 RSC; (i) K. Tsuchida, Y. Senda, K. Nakajima and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 9728–9732 CrossRef CAS PubMed.
- For other nitrogen-centered nucleophiles, see: (a) C. Zhang, Y.-H. Wang, X.-H. Hu, Z. Zheng, J. Xu and X.-P. Hu, Adv. Synth. Catal., 2012, 354, 2854–2858 CrossRef CAS; (b) Z.-T. Liu, Y.-H. Wang, F.-L. Zhu and X.-P. Hu, Org. Lett., 2016, 18, 1190–1193 CrossRef CAS PubMed; (c) Z. Zheng, G. Deng and Y. Liang, RSC Adv., 2016, 6, 103478–103481 RSC; (d) C. Zhang, Y.-Z. Hui, D.-Y. Zhang and X.-P. Hu, RSC Adv., 2016, 6, 14763–14767 RSC; (e) L.-J. Cheng, A. P. N. Brown and C. J. Cordier, Chem. Sci., 2017, 8, 4299–4305 RSC.
- For carbon-centered nucleophiles, see: (a) P. Fang and X.-L. Hou, Org. Lett., 2009, 11, 4612–4615 CrossRef CAS PubMed; (b) F.-L. Zhu, Y. Zou, D.-Y. Zhang, Y.-H. Wang, X.-H. Hu, S. Chen, J. Xu and X.-P. Hu, Angew. Chem., Int. Ed., 2014, 53, 1410–1414 CrossRef CAS PubMed; (c) F.-L. Zhu, Y.-H. Wang, D.-Y. Zhang and X.-P. Hu, Angew. Chem., Int. Ed., 2014, 53, 10223–10227 CrossRef CAS PubMed; (d) G. Huang, C. Cheng, L. Ge, B. Guo, L. Zhao and X. Wu, Org. Lett., 2015, 17, 4894–4897 CrossRef CAS PubMed; (e) Q. Wang, T.-R. Li, L.-Q. Lu, M.-M. Li, K. Zhang and W.-J. Xiao, J. Am. Chem. Soc., 2016, 138, 8360–8363 CrossRef CAS PubMed; (f) J. Song, Z.-J. Zhang and L.-Z. Gong, Angew. Chem., Int. Ed., 2017, 56, 5212–5216 CrossRef CAS PubMed.
- For oxygen-centered nucleophiles, see: (a) L. Shao, D.-Y. Zhang, Y.-H. Wang and X.-P. Hu, Adv. Synth. Catal., 2016, 358, 2558–2563 CrossRef CAS; (b) R.-Z. Li, H. Tang, K. R. Yang, L.-Q. Wan, X. Zhang, J. Liu, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2017, 56, 7213–7217 CrossRef CAS PubMed.
- S. C. Ammal, N. Yoshikai, Y. Inada, Y. Nishibayashi and E. Nakamura, J. Am. Chem. Soc., 2005, 127, 9428–19438 CrossRef CAS PubMed.
- K. Sakata, Y. Miyake and Y. Nishibayashi, Chem. – Asian J., 2009, 4, 81–88 CrossRef CAS PubMed.
- M. Daini, M. Yoshikawa, Y. Inada, S. Uemura, K. Sakata, K. Kanao, Y. Miyake and Y. Nishibayashi, Organometallics, 2008, 27, 2046–2051 CrossRef CAS.
- Y. Yamauchi, G. Onodera, K. Sakata, M. Yuki, Y. Miyake, S. Uemura and Y. Nishibayashi, J. Am. Chem. Soc., 2007, 129, 5175–5179 CrossRef CAS PubMed.
- Y. Miyake, S. Endo, T. Moriyama, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2013, 52, 1758–1762 CrossRef CAS PubMed.
- For the reactivity of the copper–acetylide complex, see: A. F. Adeleke, A. P. N. Brown, L.-J. Cheng, K. A. M. Mosleh and C. J. Cordier, Synthesis, 2017, 49, 790–801 CrossRef CAS.
- M. Asay, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 4796–4799 CrossRef CAS PubMed.
- M. M. Hansmann, F. Rominger and A. S. K. Hashmi, Chem. Sci., 2013, 4, 1552–1559 RSC.
- X.-S. Xiao, W.-L. Kwong, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem. – Eur. J., 2013, 19, 9457–9462 CrossRef CAS PubMed.
- X.-S. Xiao, C. Zou, X. Guan, C. Yang, W. Lu and C.-M. Che, Chem. Commun., 2016, 52, 4983–4986 RSC.
- L. Jin, M. Melaimi, A. Kostenko, M. Karni, Y. Apeloig, C. E. Moore, A. L. Rheingold and G. Bertrand, Chem. Sci., 2016, 7, 150–154 RSC.
- (a) M. I. Bruce, Chem. Rev., 1991, 91, 197–257 CrossRef CAS; (b) M. I. Bruce, Chem. Rev., 1998, 98, 2797–2858 CrossRef CAS PubMed; (c) V. Cadierno, M. P. Gamasa and J. Gimeno, Eur. J. Inorg. Chem., 2001, 571–591 CrossRef CAS; (d) R. F. Winter and S. Záliš, Coord. Chem. Rev., 2004, 248, 1565–1583 CrossRef CAS; (e) S. Rigaut, D. Touchard and P. H. Dixneuf, Coord. Chem. Rev., 2004, 248, 1585–1601 CrossRef CAS; (f) M. I. Bruce, Coord. Chem. Rev., 2004, 248, 1603–1625 CrossRef CAS; (g) H. Fischer and N. Szesni, Coord. Chem. Rev., 2004, 248, 1659–1677 CrossRef CAS; (h) J. Zhu and Z. Lin, in Metal Vinylidenes and Allenylidenes in Catalysis, ed. C. Bruneau and P. H. Dixneuf, Wiley-VCH, Weinheim, 2008, ch. 4, p. 129 Search PubMed; (i) V. Cadierno and J. Gimeno, Chem. Rev., 2009, 109, 3512–3560 CrossRef CAS PubMed; (j) C. Coletti, A. Marrone and N. Re, Acc. Chem. Res., 2012, 45, 139–149 CrossRef CAS PubMed.
- Interaction energy is defined as the difference between the energy of the metal–allenylidene complex and the sum of energies of two fragment systems, the metal complex and the allenylidene ligand. Here, geometries of the two fragments are fixed in the structure of the metal–allenylidene complex.
- Frontier Orbitals and Reaction Paths, ed. K. Fukui and H. Fujimoto, World Scientific Publishing, Singapore, 1997 Search PubMed.
- M. Guisán-Ceinos, V. Martín-Heras and M. Tortosa, J. Am. Chem. Soc., 2017, 139, 8448–8451 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |