
Featuring Xantphos
Piet W. N. M.
van Leeuwen
 *a and
Paul C. J.
Kamer
*b
*a and
Paul C. J.
Kamer
*b
aLaboratoire de Physique et Chimie des Nano-Objets, INSA-Toulouse, 135 Avenue de Rangueil, 31077 Toulouse, Cedex 4, France. E-mail: vanleeuw@insa-toulouse.fr
bLeibniz-Institut für Katalyse e. V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany
First published on 7th November 2017
Abstract
This review highlights the use of a bisphosphine ligand group in homogeneous catalysis. This ligand group is known as the Xantphos-like ligands, which are based on the generic formula of 2,2′-bis(phosphino)diaryl ether. The synthesis of the main members is summarized and emphasis is given to ligands that can be easily synthesized, also on large scale. The catalytic applications are not comprehensively presented, but instead, about 20% of the examples from literature have been extracted, aiming at the demonstration of diversity, peculiar examples, historical development, design vs. trial and error, and new reactions. Metals ranging from group 7 to 11 have been used in a wide variety of reactions: hydroformylation, alkoxycarbonylation, hydrocyanation, cross-coupling reactions for many C–C and C–X bond forming reactions, carbonylative coupling reactions, phosphinations, etc.
1. Introduction
Phosphines are widely used as ligand components in homogeneous catalysts. Large-scale processes include rhodium-catalysed hydroformylation of propene, butene, and heptene, oligomerization of ethene, cobalt-catalysed hydroformylation of internal higher alkenes, ethene methoxycarbonylation towards acrylates, and butadiene dimerization and telomerization. Small-scale operations include asymmetric hydrogenation of enamides and substituted acrylic acids, asymmetric isomerization to make menthol, alkoxycarbonylation (ibuprofen), and group 10 metal-catalysed C–C and C–N bond formation reactions (Heck reaction, Suzuki reaction, Buchwald–Hartwig reaction).1 In the early years, the prototypical monophosphines were triphenylphosphine and tributylphosphine. Triphenylphosphine was used by Reppe in nickel-catalysed trimerization and tetramerization of alkynes and in carbonylation chemistry, as reported in 1948; Bu3P was briefly mentioned.2,3 Ph3P is the most widely used ligand in large-scale homogeneous catalysis due to the study reported by Wilkinson and co-workers in the 1960s using Rh and Ru,4–6 whereas the more sensitive and therefore less popular Bu3P plays an important role in the development of Shell's cobalt-catalysed hydroformylation process.7 The first bisphosphines made were probably 1,n-bisdicyclohexylphosphino(CH2)n (n = 3–5) from dichloroalkanes and metal phosphides, reported by Issleib in 1959.8 For n = 2, ethene and Cy4P2 (diphosphine) were obtained via this procedure. When the less basic phenyl analogs PPh2M and dibromoethane or dichloromethane were used, the bisphosphines dppe and dppm were obtained. Dppe was rapidly incorporated in organometallic chemistry under the name diphos as the most popular bisphosphine for several years after the first explorations by Chatt's group.9 For catalysis, this was not the best choice since dppe often provided the least active catalyst among the bisphosphines, as was discovered later and explained by Thorn and Hoffmann; according to their calculations, the rigidity of the complex disfavoured migratory insertions.10 The length of the backbone influenced catalysis enormously, as was reported as early as 1966 by Iwamoto and Yuguchi for the iron-catalysed codimerization of butadiene and ethene, for which dppp provided the fastest and most selective formation of 1,4-hexadiene.11 The popularity of bisphosphines increased enormously with the invention of chiral bidentate ligands, such as DIPAMP12 and DIOP,13 for asymmetric hydrogenation that were much more selective than their monophosphine homologs. Until the arrival of MonoPhos,14 it was thought that bidentate ligands were a prerequisite for enantioselective catalysis, and the paradigm changed in 2000 when MonoPhos and related ligands were introduced. An endless list and a variety of bidentate ligands, not just bisphosphines, have been published since. Typically, the ligands synthesized were all designed for coordination in a cis fashion in square-planar complexes, i.e. P–M–P bite angles (vide infra, section 3.1.1) in the range of 80–100 degrees. In the mid 1980s the quest for ligands having wider bite angles was growing,15 initially in the area of rhodium hydroformylation, vide infra. Successful members of this new group were BISBI16 and Xantphos.Xantphos was first published slightly more than 20 years ago,17–20 and today there are more than 2900 research publications that use Xantphos 1 or a close relative as a ligand in metal-complex catalysed reactions. Over 3700 compounds can be found in Scifinder containing the substructure of 2,2′-bis(X2phosphino)diphenyl ether, the generic structure for the Xantphos-like ligands. Haenel and co-workers synthesized Xantphos as part of their project on heteroaromatic phosphines such as 4,6-bis(diphenylphosphino)dibenzofuran (DBFphos, 2) and 4,6-bis(diphenylphosphino)dibenzothiophene. Their work included an X-ray structure of Xantphos,21,22 which proved the large and fixed P–P distance as compared to the common, cis-coordinating bisphosphines. They discovered that DBFphos forms bimetallic complexes with Co2(CO)6 as wide-bite angle ligands tend to do,23 but also tridentate, pincer-like complexes of Ru were found.24 In 1990, we started a project on bidentate bisphosphines that would form P–M–P bite angles larger than the common 90° found abundantly in square planar and octahedral complexes, for their use in catalysis. The first reaction in mind was rhodium catalysed hydroformylation; we were searching for ligands that would coordinate in a bis-equatorial fashion in the trigonal bipyramidal rhodium catalyst, RhH(bisphos)(CO)2. The best synthetically accessible design involved the Xantphos-type ligands,17 while the terphenyl based designs, for example, posed more difficulties at the time because the cross-coupling methods were still less developed.25 The generic structure is shown in Scheme 1 and by changing the atom(s) X in position 9, we subtly varied the P–P distance and thus, the ligand-preferred P–M–P angles.26 The Xantphos-like backbones available in 2000 are depicted in Scheme 1 and it seems that apart from methyl, t-butyl, carboxylate or other functional groups added to the backbone, no new backbones have been developed since then.27
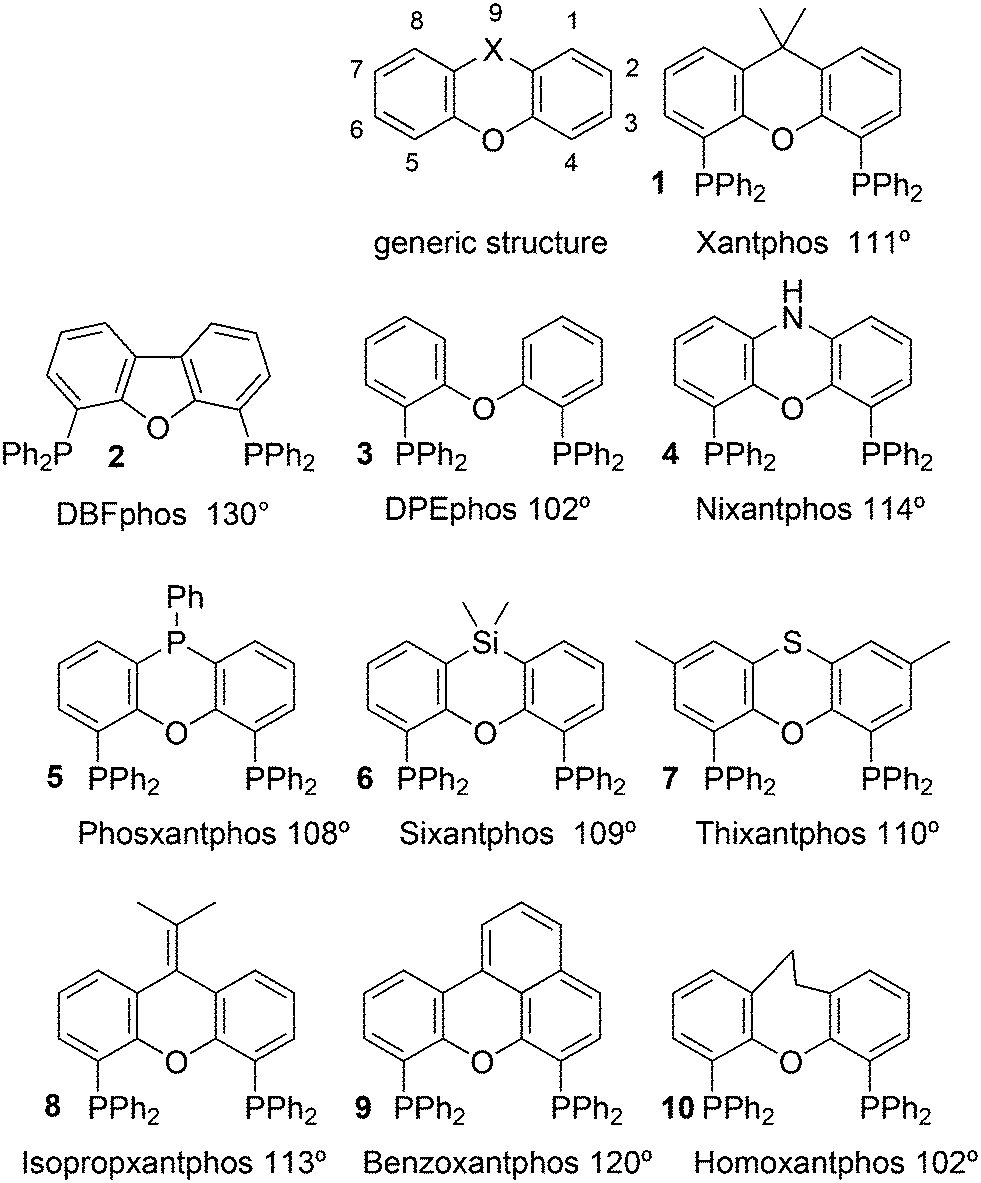 | ||
| Scheme 1 Xantphos type ligands. | ||
The most used ligands in literature are Xantphos, DPEphos, and Nixantphos; the reason being that they are readily commercially available on the mg to kg scale and several providers offer ton-scale quantities on their websites.28 However, the number of Xantphos related backbones and compounds explored carrying substituents other than the phenyl group at phosphorus is small, compared with BINAP for instance, of which the axial chiral motif has been translated into a large number of bisphosphines as was reviewed by Lemaire and co-authors in 2005.29 They focused on the synthesis of axially chiral ligands and presented an example of an enantioselective catalytic reaction for each ligand. Here, we will review the Xantphos derivatives that have been reported; most of them were initially synthesized in the framework of hydroformylation studies.
Ligands with properties similar to the Xantphos group are 11 and 12, and BISBI 13 (Scheme 2),16 but these will only be mentioned in passing where appropriate. Triptycene based ligand 11 (ref. 30) behaves very much the same as Xantphos in several reactions, while both ligands 11 (ref. 31) and 12 (ref. 32) were studied by Gelman as trans-coordinating bidentates. Note that Xantphos also occurs as a trans coordinating ligand, although in most of these instances it acts as a tridentate as the oxygen is involved in a weak interaction with the metal.33,34
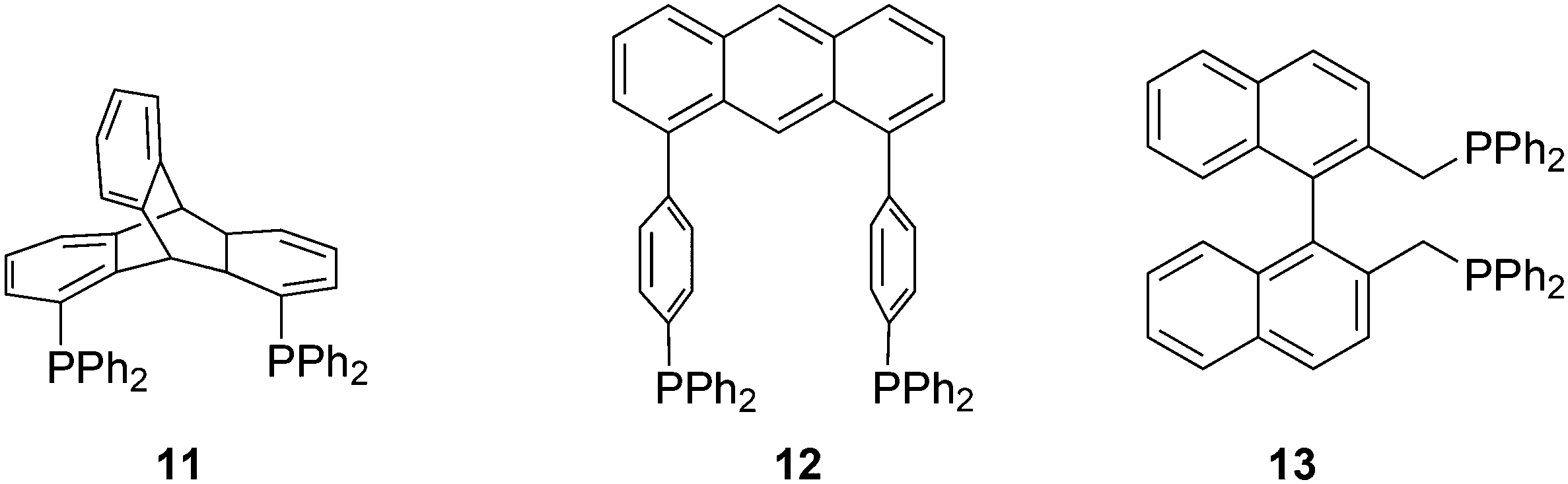 | ||
| Scheme 2 Wide bite angle ligands. | ||
We have structured the review by reaction type and since it is neither useful nor possible to review all articles featuring Xantphos, we have selected those in which this ligand group plays a particular role, usually where it leads to an exceptional selectivity; however, failures may also be instructive for a better understanding. The variety of reactions is huge and we do not expect that a single explication will be revealed. For a few popular reactions, mechanistic studies and DFT calculations leading to explanations have been reported, but for most reactions, these data are not available. We will keep the description of the bite angle concept to a minimum as this has been reviewed many times for broader groups of ligands35–37 and for certain reaction types38,39 or ligands.40
2. Synthesis
2.1. Standard ligands
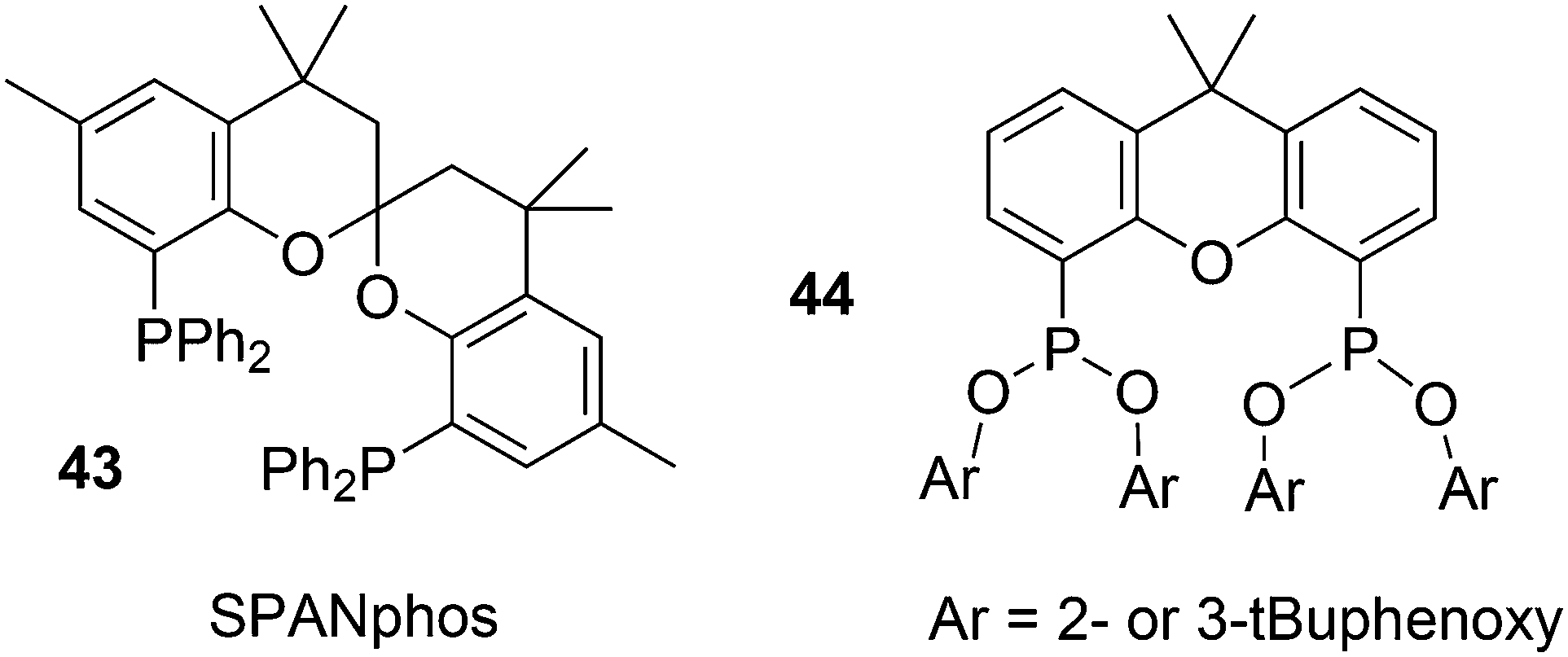
The simple Xantphos type ligands with bis(diphenylphosphino) substituents are shown in Scheme 1. The general synthesis involves the dilithiation of the free backbone, with either “free” n-BuLi, or s-BuLi at room temperature or with TMEDA modified n-BuLi at lower temperatures. Thanks to the oxygen atom in the 5-position, lithiation takes place exclusively at the 4 and 6-positions in high yield. Reaction with Ph2PCl gives the ligands in 60–80% isolated yields. For 9,9-dimethylxanthene, the lithiation reaction was already extensively explored for the building of supramolecular scaffolds by Rebek via coupling with DMF or CO2.41 One can obtain 9,9-dimethylxanthene from the reaction of xanthone with AlMe3 in quantitative yield.42 Xanthone is the product of a thermolysis of phenyl salicylate or phenol and salicylic acid at 275 °C.43 Carbon-9, carrying two methyl groups, can also be introduced by acid catalysed alkylation with acetone, but in this instance di-p-tolyl ether has to be used to block the para position and avoid the formation of bisphenol-A. This reaction requires long reaction times, large volumes of solvents, and gives 4,4,4′,4′,6,6′-hexamethyl-2,2′-spirobi[chromane] as the co-product, the starting material for SPANphos.44 Eventually, this route leads to 2,7-dimethyl-Xantphos, also cited as Xantphos, 1. DBFphos 2 has too wide a bite angle for mononuclear complexes and will not be considered here.DPEphos 3 is made from cheap diphenyl ether via the lithiation route; di-p-tolyl ether can also be used. The bite angle of DPEphos (also written as dpe-phos)45 is only slightly larger than that of dppf and the behaviour of the two ligands in catalysis is often similar; however, for commercial production, 3 is much cheaper. Both central atoms, Fe and O, can interact with the coordinated metal. For the production of Nixantphos 4, the backbone 10-H-phenoxazine is used, which is also widely available as a component in dye synthesis. Before the lithiation, the NH group was protected by a bulky alkylsilyl group.27
The next row of ligands, 5–7, can also be made in high yields from convenient starting materials, but they have so far found limited application. When lithiation of diphenyl ether is followed by reaction of the respective dichlorides, PhPCl2 and Me2SiCl2, ligands 5 (ref. 27) and 6 (ref. 17) are obtained. In a second lithiation step, the phosphines are introduced. The Phosxantphos backbone exchanges phenyl for butyl during the lithiation and thus, in this case, the use of PhLi was recommended.27 The phenoxathiine backbone for 7 can be conveniently obtained from a Friedel–Crafts reaction of di-p-tolyl ether with sulfur and AlCl3 as the catalyst, yielding the 2,7-dimethyl derivative of 7 in modest yield.46 It has also been used extensively as a scaffold for aminoacids and in macrocycles.47
The backbone synthesis for 8–10 is more tedious and these ligands are not suited for broad application. Only benzoxantphos 9, having a curious bite angle of 120°, might be worth synthesizing. 2-(Naphthalen-1-yl)phenol obtained via a C–C cross-coupling reaction was converted to benzo[kl]xanthene over Pd in 30% isolated yield after column chromatography;27 however, better methods have since become available.48 The backbone 9-(propan-2-ylidene)-9H-xanthene can be generated via a McMurry reaction of xanthone and acetone in 60% yield and the lithiation/phosphorylation gave a similarly modest yield in this instance.
Electronic variations of the PPh2 group have been reported; thus, for Thixantphos 7 several 4-phenyl substituted homologs have been synthesized, 7a–7g, which have the same steric properties as 7, but have either more electron-donating or withdrawing character.49
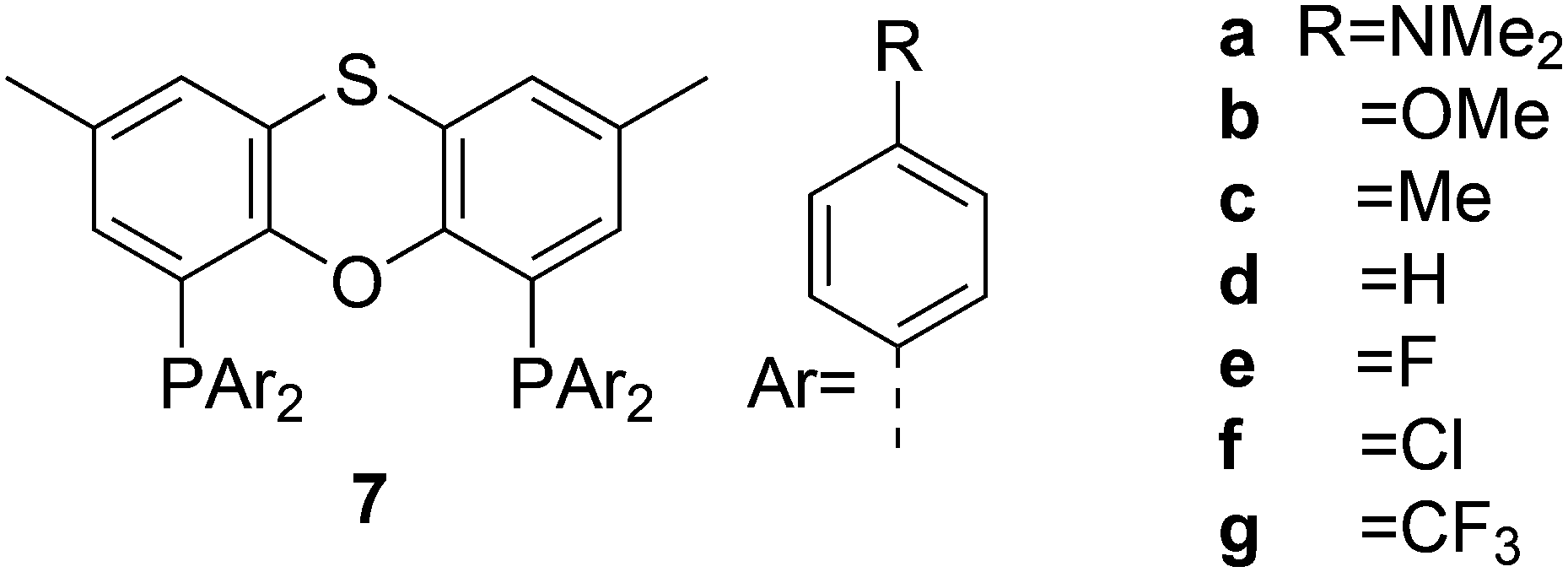
For Xantphos, Nixantphos, and DPEphos, a few alkyl (Me, Et, tBu, Cy) derivatives are available.28,33 DuBois and co-workers reported the synthesis of Et2P Xantphos and DPEphos, named EtXantphos and depPE. Their stronger donor properties and wider bite angles were used to modify the hydricity of bis(diphosphine) Pd–H+ complexes.50
2.2. Functionalized ligands
Replacement of the phenyl groups of Xantphos by 3-phenylpropoxyphenyl groups, which can be conveniently sulfonated at the para position of the phenyl group (Scheme 3), gives tetrasulfonate derivative 15. This molecule gives vesicles in water, where water-insoluble substrates can be captured to undergo a catalytic reaction with the metal inside.59
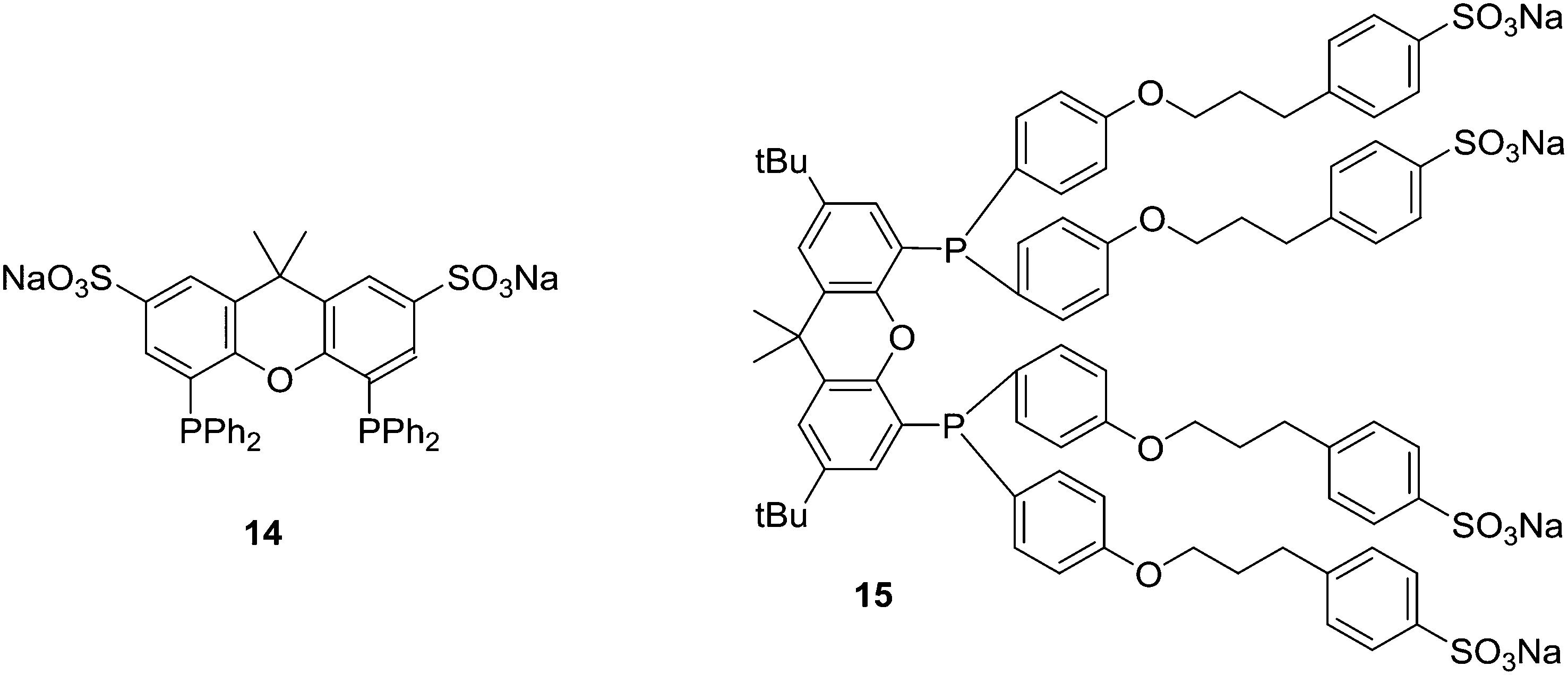 | ||
| Scheme 3 Water-soluble ligands. | ||
 | ||
| Scheme 4 Amphiphilic ligands. | ||
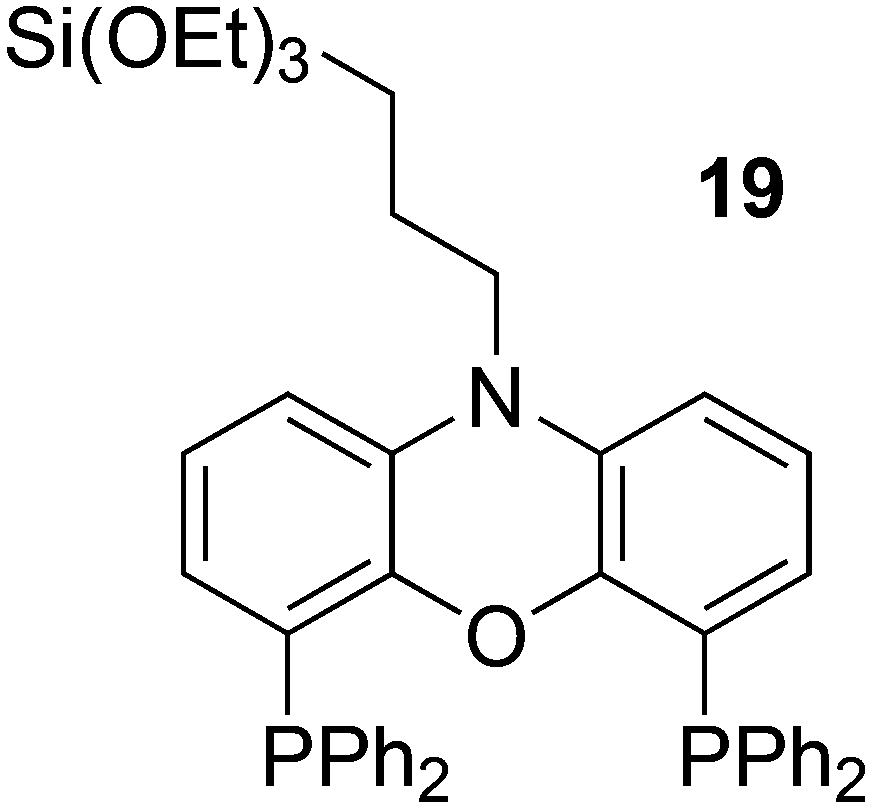
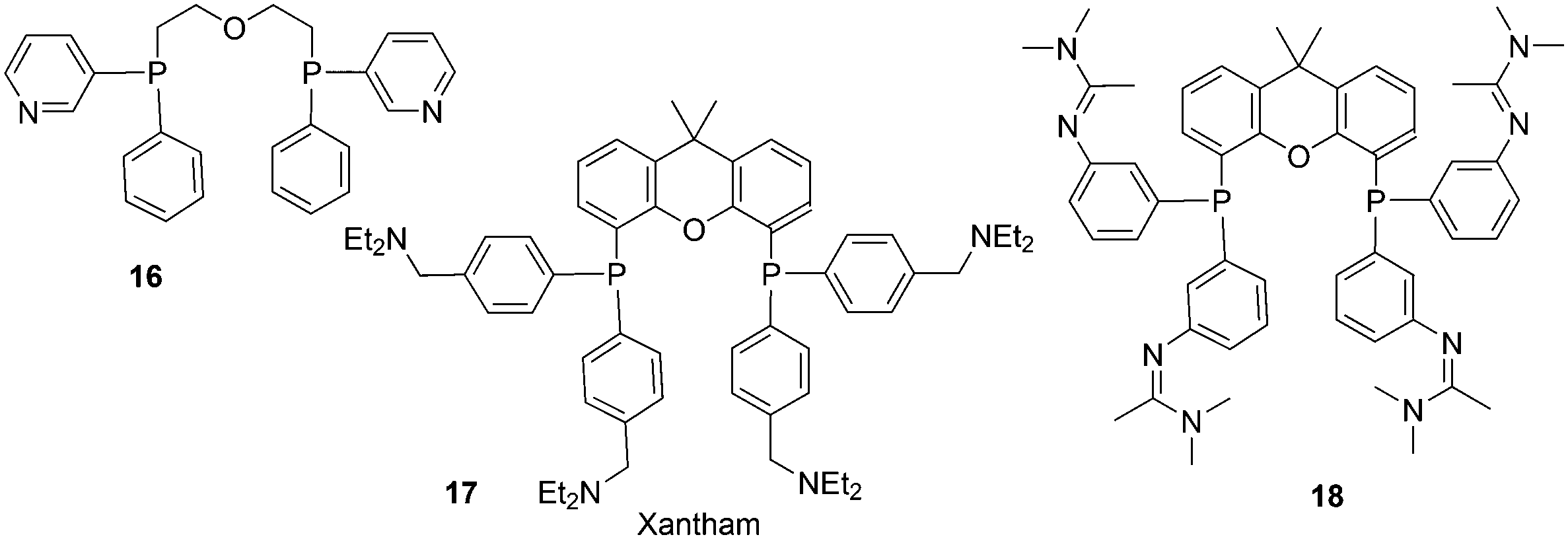
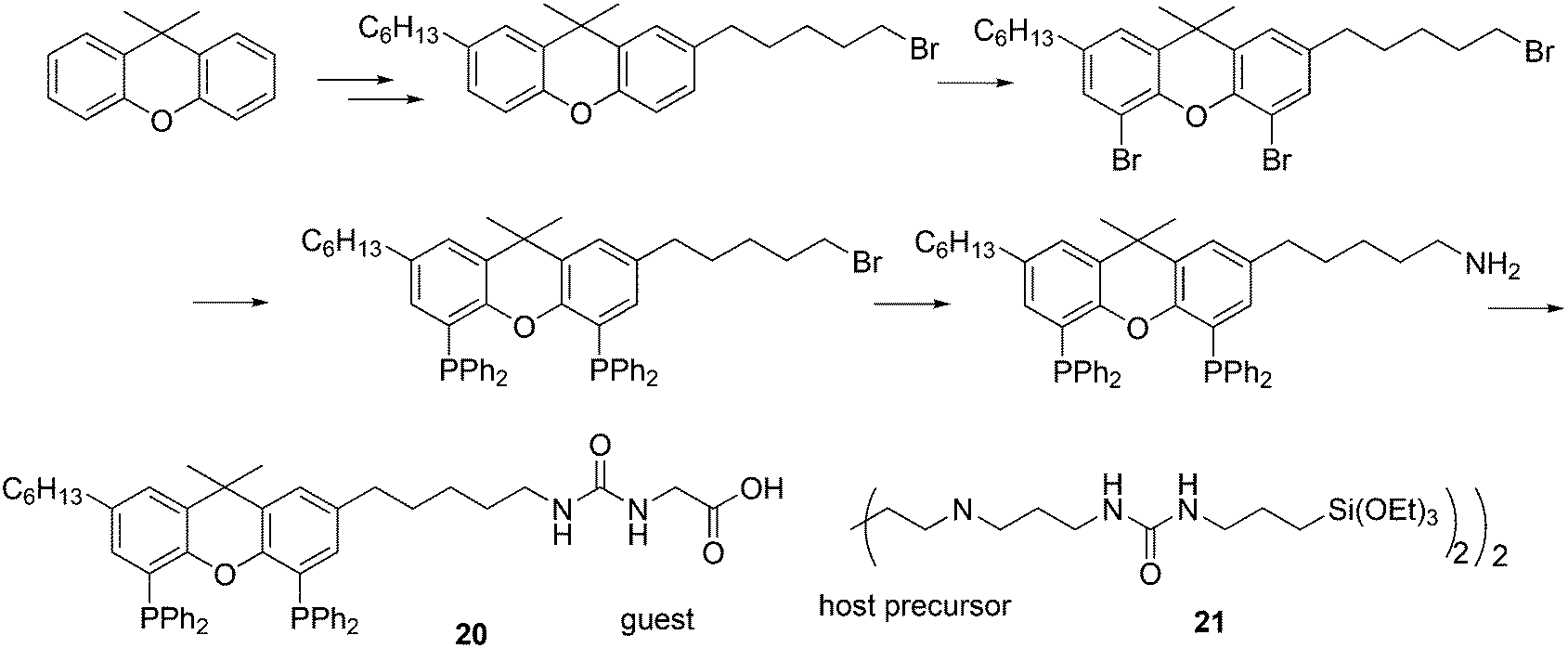
In a different approach, the ligands were connected to dendrimers and silica via non-covalent interactions. Phosphine ligand 20 equipped with a glycinylurea tail acts as the guest for a bisurea–propyl amine, as introduced by Meijer and co-workers, first for polypropylamine dendrimers and triphenylphosphine as the guest ligand.68 The synthesis from the respective isocyanates is straightforward. The amine precursor for 20 was made by two acylation steps via a Friedel–Crafts reaction, followed by chlorodimethyl silane reduction of the acyl groups and replacement of the bromide in the chain by ammonia. All steps are high yielding, except the phosphination in this instance. The amine obtained after introduction of the desired phosphine can be reacted with glycinyl isocyanate to obtain 20, or with triethoxysilylpropylisocyanate for immobilization of the ligand on silicagel.69 Complexation in the non-covalent immobilization involves two urea groups of the host and the basic amine, and one urea of the guest and the carboxylic acid. Chen et al. used Xantphos derivative 20 and 21 immobilized on silica (Scheme 5).70 The binding is reversible and in methanol, the ligand guest and metal complex can be washed out.
 | ||
| Scheme 5 Non-covalent immobilization and synthesis of 20. | ||
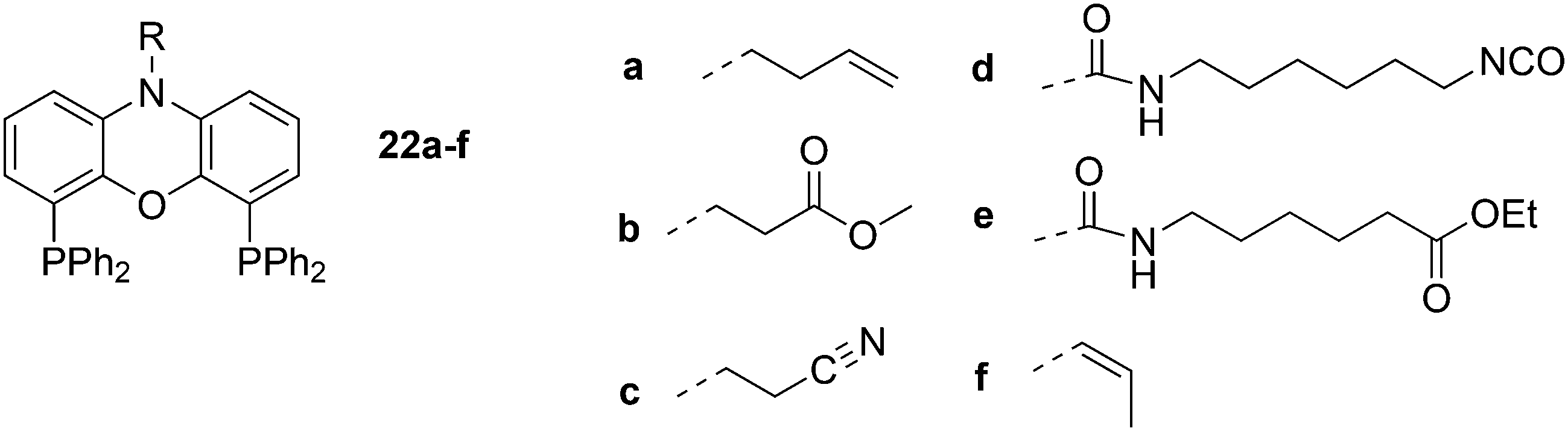
Dendrimers can be used as carriers for catalysts and are separated from the reaction medium by nanofiltration. The ligands are covalently attached to the dendrimer in this approach (Scheme 6). The catalyst can be located in the core of the dendrimer or on the surface of the dendrimer, which allows for more catalysts to be connected.71 The general experience is that rhodium is well retained by the Xantphos ligands in a continuous flow reactor with a nanofilter, down to the ppm level, although for an industrial application this is not good enough. Eilbracht and Haag et al. immobilized Nixantphos in a variety of ways on hyperbranched polyglycerol support after modification of the nitrogen function by Michael addition to methyl acrylate and acrylonitrile, or by addition to isocyanates, and related reactions.72 The amide anion of Nixantphos is not a strong nucleophile and prolonged reaction times were needed, unless one compromises for lower yields. To obtain 22d in high yield, a large excess of the diisocyanate should be used. Once the linker is on the backbone, the newly introduced functional group shows efficient coupling.
 | ||
| Scheme 6 Dendrimeric immobilizations. | ||
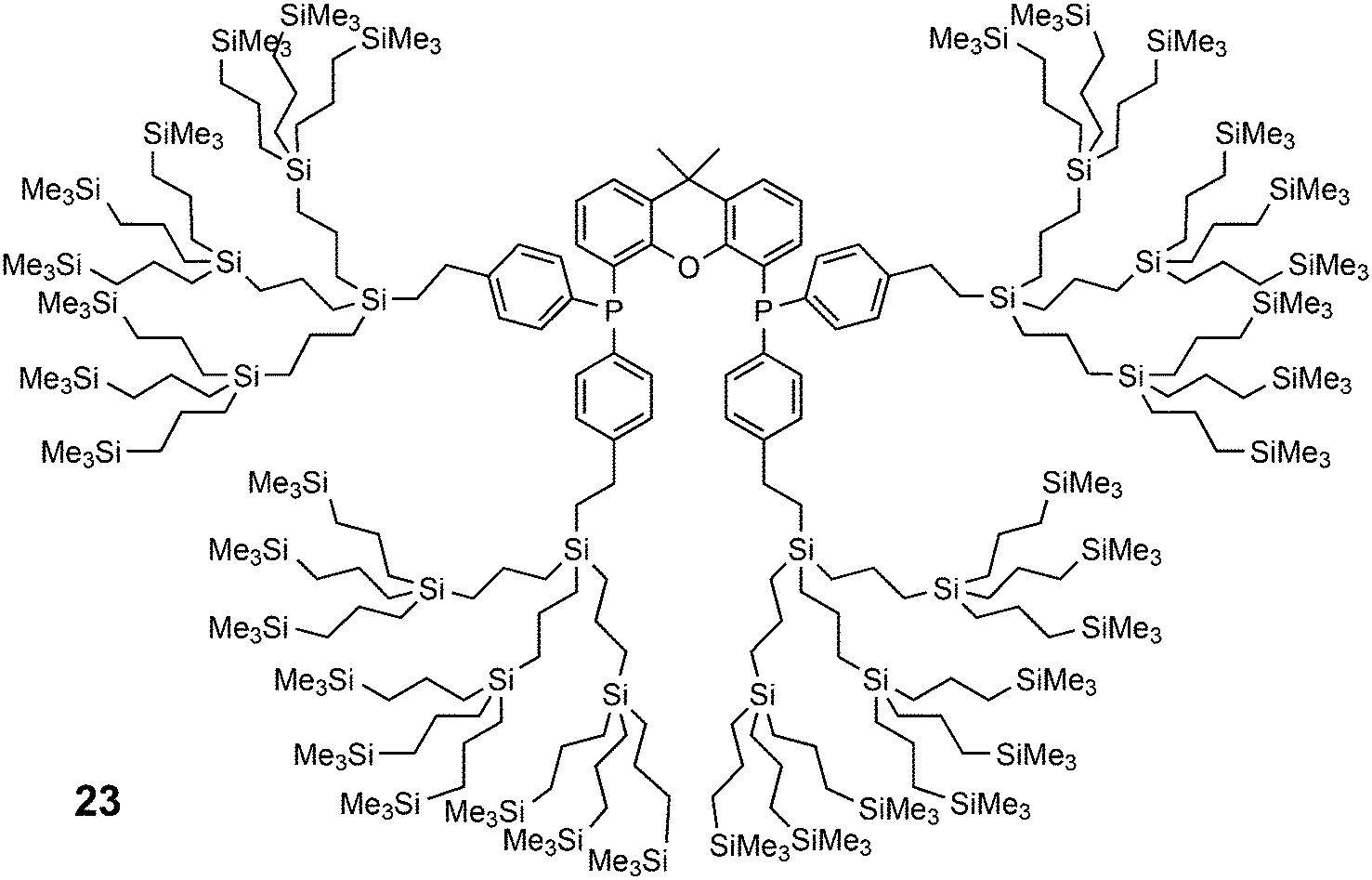
Core-substituted dendrimeric ligand 23 was made by substitution of chloride in (9,9-dimethyl-9H-xanthene-4,5-diyl)bis(dichlorophosphane) by the lithiated aryl dendrimers.73 The dendrimeric silane units were prepared via a sequence of hydrosilylation of allyl groups and substitution of the chlorides in –SiCl3 by the allyl Grignard reagent. The first hydrosilylation took place on p-bromostyrene. The system was applied in palladium catalysed allylic alkylation in a continuous flow reactor, but retention is usually low for palladium in this reaction.69,74 The catalytic reaction continues in the effluent in the absence of phosphine ligand.
Sulfoxantphos 14 has been used in several ways to facilitate convenient product-catalyst separation. For instance, Fehrmann and co-workers reported continuous fixed-bed gas-phase hydroformylation using supported ionic liquid-phase (SILP) Rh catalysts.57 The ionic liquids used were 1-n-butyl-3-methylimidazolium hexafluorophosphate and n-octyl sulfate. The ligand rhodium ratio needed was higher than in organic solvents, but after this was taken care of, the same high selectivity was obtained. It was proposed by Bell and co-workers that the sulfonic salts groups of 14 interacted with the silanol groups on the silica surface.75
Ligand 24 [1,1′-((4,5-bis(2,8-dimethyl-10H-phenoxaphosphinin-10-yl)-9,9-dimethyl-9H-xanthene-2,7-diyl)bis(pentane-5,1-diyl))bis(3-methyl-1H-imidazol-3-ium)] represents a combination of favourable properties for, i.a., hydroformylation in ionic liquids.53 It was synthesized from 9,9-dimethylxanthene. First, the backbone was acylated with 5-bromovaleric chloride in 90% yield. With Lewis acidic catalysts, the ketones were reduced by chlorodimethylsilane. Then, the POP group was introduced via lithiation of the backbone and reaction with the product of lithiated ditolyl ether and phosphorus trichloride. In most cases, this is a reaction producing only 50% yield, most likely due to the reactivity of the bromopentyl groups. The slow reaction with N-methylimidazole gives the salt, used as hexafluorophosphate.76 The ligand combines the solubility in ILs, a wide bite angle ensuring good l/b selectivity, and the relatively electron-withdrawing POP group giving high rates of reaction. Note that the POP group differs considerably from diphenylphosphino sterically, as it is completely flat.
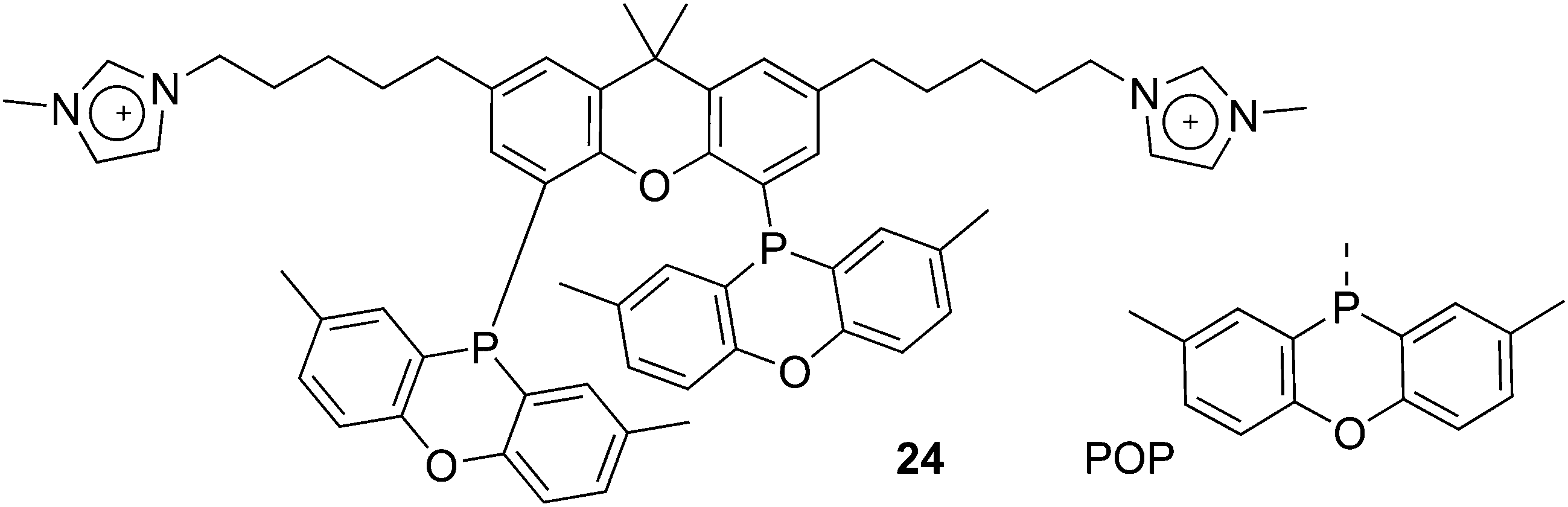
Optimization of various reaction parameters led to an unprecedentedly active (TOF > 6200 mol mol−1 h−1, T = 100 °C), selective (linear/branched ratio >40) and stable hydroformylation procedure. Catalyst losses were extremely low, but not quite in the desired ppb region for Rh in the production of bulk chemicals, i.e. low value aldehydes.
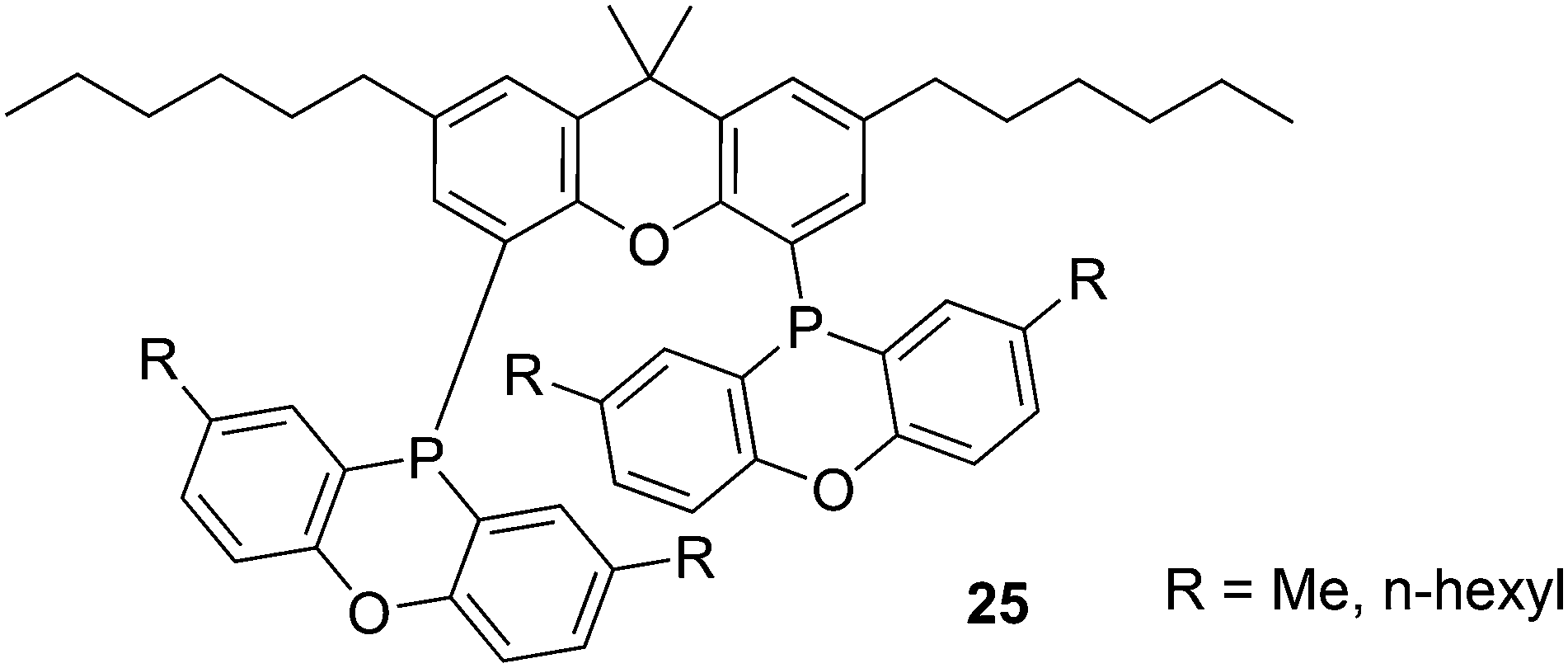
A range of alkyl substituted Xantphos ligands 25 was prepared in a similar way to 24. For application of POP-Xantphos (1 equipped with POP groups instead of PPh2) in a rhodium catalysed hydroformylation by Celanese Chemicals Europe, it was noted that the solubility of POP-Xantphos in organic solvents, or a mixture of the hydroformylation product and the starting alkene, was too low for the efficient operation of the hydroformylation reaction. The solubility of POP-Xantphos could be raised from 2.6 mM to 300 mM in toluene by alkylating it at the 2 and 7 positions with an n-hexyl group. The synthesis involved the sequence of acylation and reduction of the ketones, as mentioned above. Backbone and POP syntheses gave excellent yields, but the non-optimized phosphination step gave on average 70%, and on occasion much less. When also the methyl groups of POP were replaced by n-hexyl groups, now totalling 6, the solubility in toluene at room temperature further increased to >500 mM. Surprisingly, linear alkyl groups have a more favourable effect than branched alkyl groups.
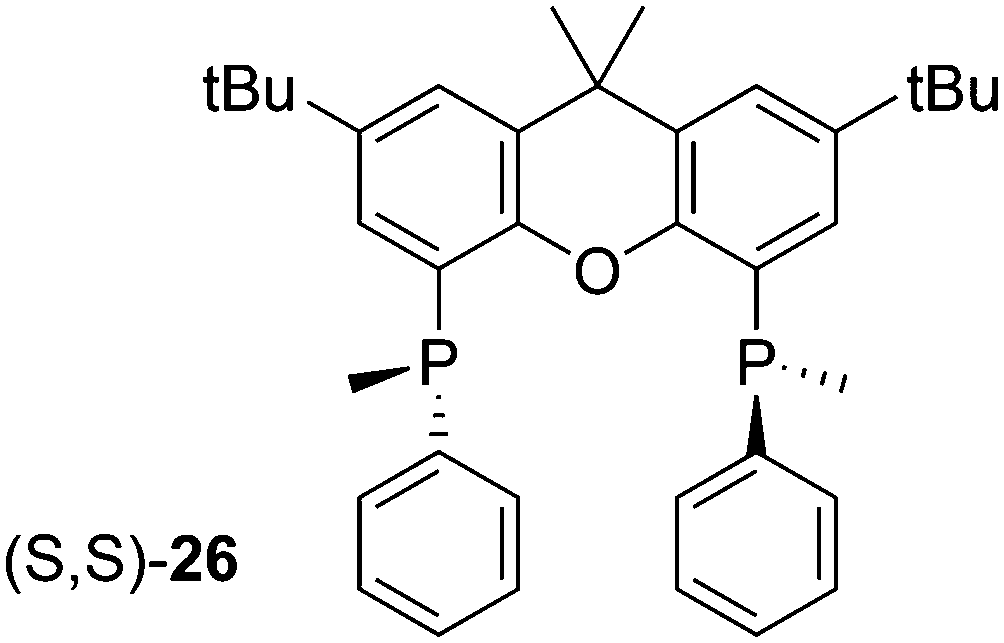
The first chiral Xantphos derivative was reported by Hamada and co-workers, the bis(methylphenylphosphino) of tBu substituted xanthene, 26.82 The synthesis involved the reaction of dilithioxanthene with MePhPCl to give the racemic mixture (two rac forms and meso) in good yield, although the undesired meso was in slight excess. However, this could be epimerized with LiAlH4 in THF at room temperature after oxidation. The rac oxides were separated by diastereomeric salt formation with (R,R)-(−)-dibenzoyltartaric acid. The enantiopure ligands, obtained by reduction with PMHS and Ti(OiPr)4, were used in Pd catalysed asymmetric allylic alkylation (AAA) with good ees. It is well known for AAA that wide bite angles cause more steric interaction between ligand and allyl group, thus giving higher ees.83
Recently, Börner and co-workers developed an efficient asymmetric synthetic protocol for P-stereogenic Xantphos and related ligands via an adaptation of the well-established Jugé method.84 The removal of the borane protection before the coupling reaction proved to be crucial. The ligands induced high enantioselectivities in the Rh-catalysed hydrogenation of industrially relevant enones.
The successful efforts of Duphos and Xantphos were brought together by Dierkes and co-workers.85 The synthesis follows the usual steps for Duphos ligands, as introduced by Burk, shown in Scheme 7.86 Duxantphos 27 and Duthixantphos 28 were used in Pd catalysed AAA and interestingly, they performed well for a variety of substrates, for which otherwise different ligands would be needed (O-acetylcyclohex-2-enol, 93% ee; O-acetylpent-2-en-3-ol, 82% ee; O-acetyl-1,3-diphenylprop-1-en-2-ol, 97% ee).87 It is known that substitution of the methyl substituents by more bulky groups in the phospholene ring can have a large effect on the ees;88 unfortunately, only the dimethyl derivatives were prepared in this study.
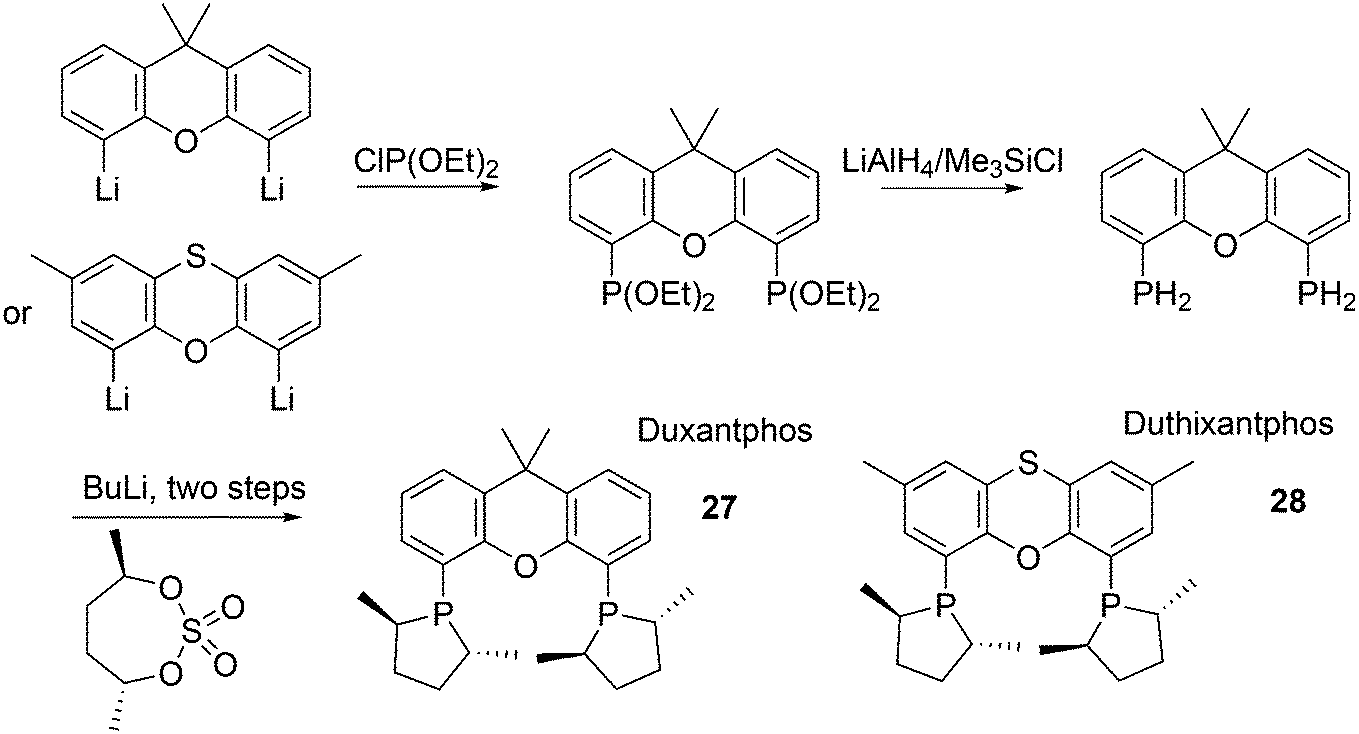 | ||
| Scheme 7 Synthesis of Duphos derivatives of Xantphos. | ||
Kamer and co-workers synthesized chiral DPEphos derivatives 29a–d in good to high yields (29d) using standard phosphine ligand synthesis (Scheme 8).89
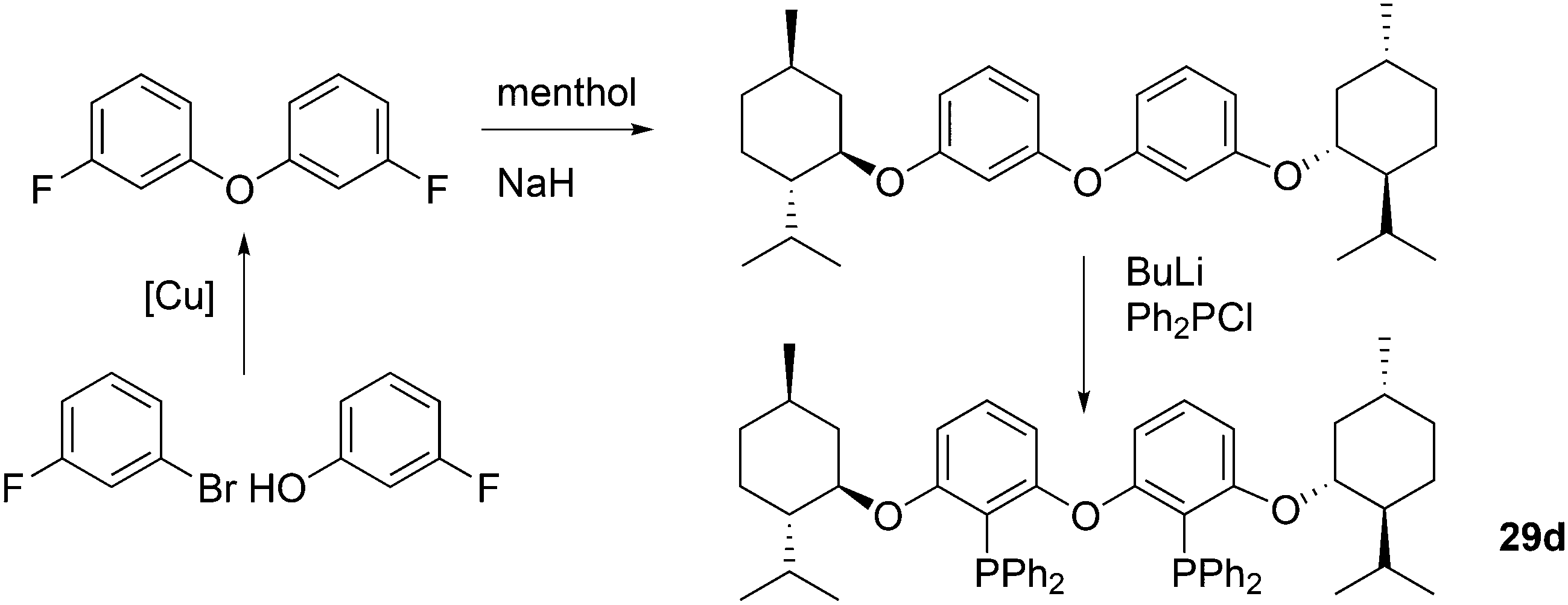
 | ||
| Scheme 8 Synthesis of chiral ether derivatives of DPEphos 29d. | ||
The ester derivatives proved not to be very effective ligands in AAA, but the menthol ether derivative 29d was rather effective for a first try of easily accessible, modular ligands. Fig. 1 shows the effect of the proximity and size of the substituents on the ee. Surprisingly, the ees for O-acetylcyclohex-2-enol exceeded those of O-acetyl-1,3-diphenylprop-1-en-2-ol; usually, the converse is true.
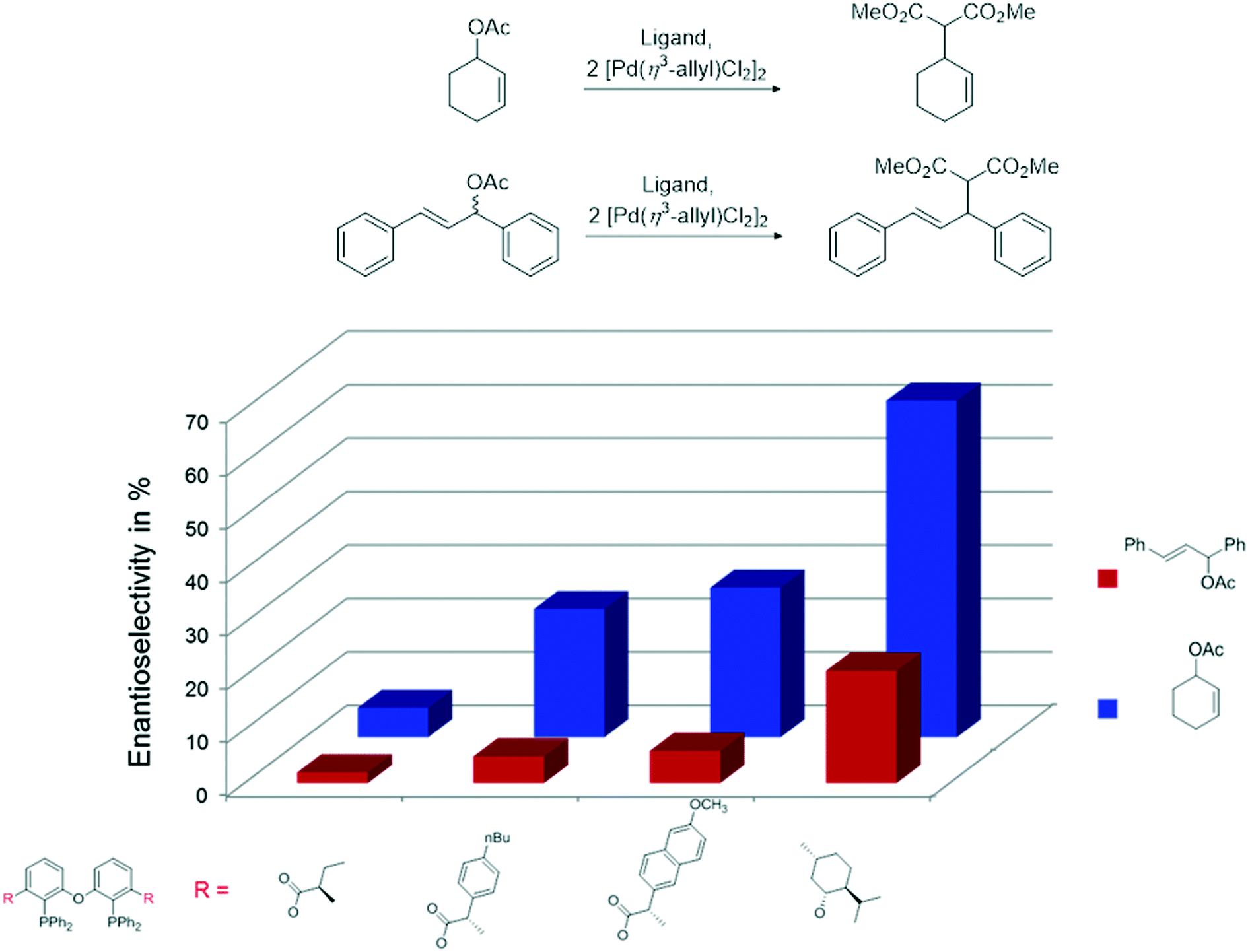 | ||
| Fig. 1 Pd-Catalysed AAA for modular, chiral DPEphos ligands. Reprinted with permission from C. F. Czauderna, A. G. Jarvis, F. J. L. Heutz, D. B. Cordes, A. M. Z. Slawin, J. I. van der Vlugt and P. C. J. Kamer, Organometallics, 2015, 34, 1608. Copyright 2015 American Chemical Society. | ||
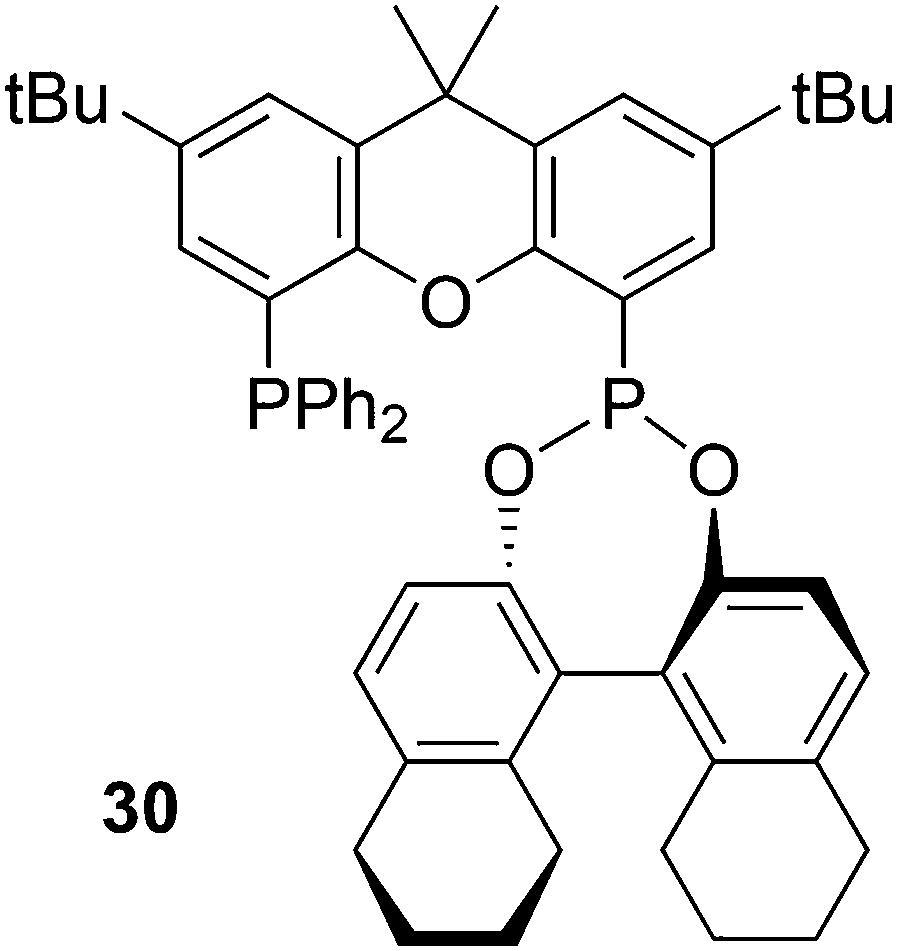
Inspired by Xantphos, Binaphos and other phosphine–phosphite or phosphonites90 Reek and co-workers prepared ligand 30 by substitution of bromide in (5-bromo-2,7-di-tert-butyl-9,9-dimethyl-9H-xanthen-4-yl)diphenylphosphine by ClP(NEt2)2.91 The phosphoramidite was converted by refluxing with the desired binol and tetrazole as the catalyst. The “half” product bromo/diphenyl-phosphinoxanthene was prepared from 4,5-dibromoxanthene and just one equivalent of n-BuLi and PPh2Cl in modest yield (45%).92 Note that the substitution of xanthene at the 4 and 5 position by different groups (via bromination, lithiation, cross-coupling, etc.) usually gives statistical distributions, and overall yields of 45% of the mixed compound are typical. Ligand 30 stands out among other ligands in the asymmetric hydrogenation of demanding dihydrofurans,93 giving ees > 90%. Note that Binaphos also has 5 atoms in the ligand backbone, but the coordination behaviour of the two is completely different; 30 occupies two equatorial positions in the rhodium trigonal bipyramid and Binaphos coordinates predominantly equatorially–apically (vide infra, 3.1.1).94 The use of hybrid diphosphorus ligands in rhodium catalysed asymmetric hydroformylation has recently been reviewed by Reek et al.95
3. Reactions
3.1. Hydroformylation and carbonylation
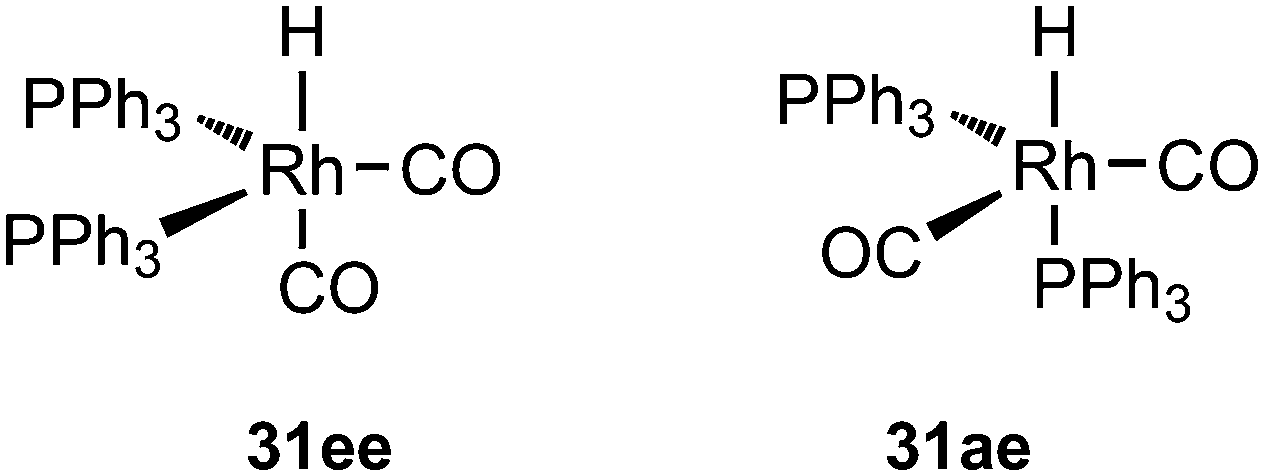
Wilkinson's hydroformylation catalyst HRh(CO)(PPh3)3 (or a suitable precursor) was commercialized in the 1970s.101 At low arylphosphine concentration, the selectivity is of the order of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 for linear/branched aldehydes, but at the cost of rates that are an order of magnitude lower it can be raised to 92:8 (120 °C, 30 bar) at high phosphine concentrations for propene to butanal. Studies of the rhodium species present in the hydroformylation mixture, especially those performed by by Brown and Kent,102 led to the conclusion that two species were responsible for the reaction, having the composition HRh(CO)2(PPh3)2, and two bipyramidal structures were assigned to them, 31ee and 31ae. There were indications that 31ee was the species that induced higher selectivity to linear product: (1) enforced apical–equatorial species with a ligand such as dppe gave low l/b ratios (1–2); (2) dppf103 and DIOP,104 which bind more easily in an equatorial–equatorial fashion, gave somewhat higher l/b ratios than PPh3; (3) dissociation of one phosphine from HRh(CO)(PPh3)3 might give an intermediate resembling species 31ee after the loss of one CO to start the cycle. In the mid 1980s, one of the authors started the design of wide bite angle bisphosphines in the Shell labs of Amsterdam, and the predecessor of Xantphos 2,2′-bis(diphenylphosphino)ethyl ether was published.15 Since Shell at this point had no interest in rhodium catalysed hydroformylation, the project was continued at the University of Amsterdam with the study of more rigid backbones with this structure. Simple models and molecular mechanics showed that a 5 or 6 atom link between the two P-atoms would favour eq–eq coordination; however, if the middle atom would be a C-atom carrying hydrogen, metalation might occur, as found by Shaw for 1,5-bis(dialkylphosphino)pentane ligands.105 Hence, we chose oxygen in the middle of the chain to solve this issue. Simultaneously, in the mid 1980s, a ligand screening at Texas Eastman by Devon and co-workers led to the discovery of BISBI (13) as a highly selective catalyst for linear aldehyde formation.16 BISBI catalysts in hydroformylation catalysis at Eastman Chemical were reviewed by Puckette.106
:30 for linear/branched aldehydes, but at the cost of rates that are an order of magnitude lower it can be raised to 92:8 (120 °C, 30 bar) at high phosphine concentrations for propene to butanal. Studies of the rhodium species present in the hydroformylation mixture, especially those performed by by Brown and Kent,102 led to the conclusion that two species were responsible for the reaction, having the composition HRh(CO)2(PPh3)2, and two bipyramidal structures were assigned to them, 31ee and 31ae. There were indications that 31ee was the species that induced higher selectivity to linear product: (1) enforced apical–equatorial species with a ligand such as dppe gave low l/b ratios (1–2); (2) dppf103 and DIOP,104 which bind more easily in an equatorial–equatorial fashion, gave somewhat higher l/b ratios than PPh3; (3) dissociation of one phosphine from HRh(CO)(PPh3)3 might give an intermediate resembling species 31ee after the loss of one CO to start the cycle. In the mid 1980s, one of the authors started the design of wide bite angle bisphosphines in the Shell labs of Amsterdam, and the predecessor of Xantphos 2,2′-bis(diphenylphosphino)ethyl ether was published.15 Since Shell at this point had no interest in rhodium catalysed hydroformylation, the project was continued at the University of Amsterdam with the study of more rigid backbones with this structure. Simple models and molecular mechanics showed that a 5 or 6 atom link between the two P-atoms would favour eq–eq coordination; however, if the middle atom would be a C-atom carrying hydrogen, metalation might occur, as found by Shaw for 1,5-bis(dialkylphosphino)pentane ligands.105 Hence, we chose oxygen in the middle of the chain to solve this issue. Simultaneously, in the mid 1980s, a ligand screening at Texas Eastman by Devon and co-workers led to the discovery of BISBI (13) as a highly selective catalyst for linear aldehyde formation.16 BISBI catalysts in hydroformylation catalysis at Eastman Chemical were reviewed by Puckette.106
Casey and Whiteker studied the rhodium carbonyl hydride species of BISBI that forms a purely eq–eq isomer and along with this they introduced the natural bite angle (βn) concept.107 The natural bite angle is the angle that the P–M–P would adopt, given a certain P–M distance, if the metal had no preference electronically and in the absence of steric interactions with additional ligands in the complex. It is calculated by MM2 using a dummy metal atom and setting bending force constants to zero.35–37
In addition to the developments in the bisphosphine area, the discovery of highly selective, fast, bulky diphosphites took place at about the same time by the Bryant group at UCC.108 Out of the hundreds of diphosphites reported, a few were studied by in situ NMR and IR and the studies showed that the successful bidentate ligands all contained a 5 or 6 atom bridge and coordinated in the eq–eq fashion of 31ee.109
Returning to Xantphos ligands, as mentioned above, ligands 1–10 were developed in the first place for the rhodium catalysed hydroformylation of terminal alkenes and were subsequently applied in other reactions as well before a broad patent was submitted. Ligands of the second group 14–24 were also synthesized in the framework of hydroformylation, but except for Sulfoxantphos 14, their use for other reactions has remained limited; availability is the key to wider applications.
We will summarize here a few general characteristics of terminal and internal alkene hydroformylation with Xantphos-like ligands, since the early data have been reviewed in the past.110,111
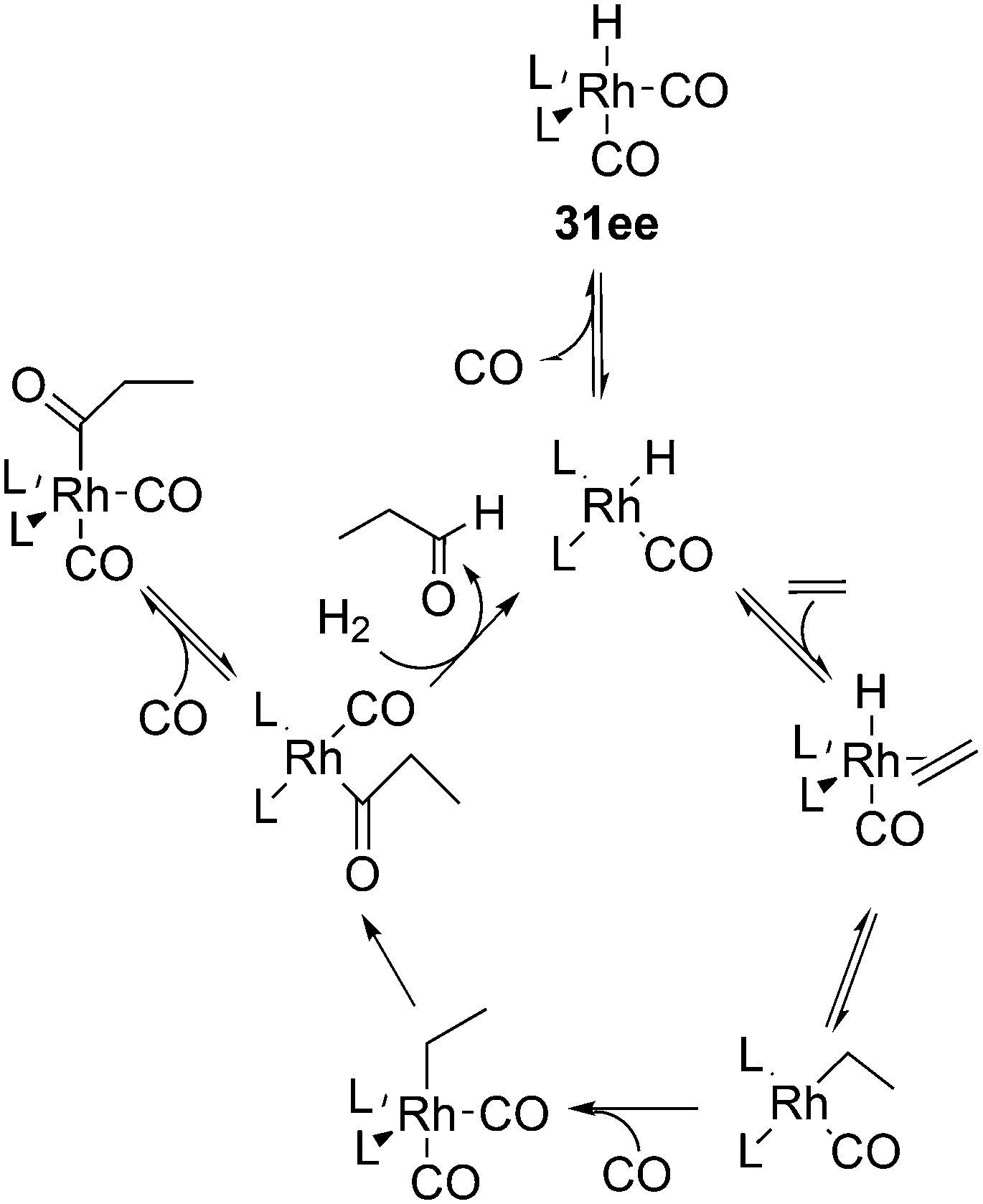
The kinetic studies of 1-alkene hydroformylation showed, as in most rhodium catalysts, that the reaction is first order in rhodium (except when rhodium carbonyl dimers are formed, a tendency especially noted for Nixantphos), a positive to first order in alkene concentration, and a negative order in CO, up to −1. The dependency on CO and ligand concentrations can be complicated as the two ligands compete with one another. For instance a 1:1 L/Rh ratio never leads to the typical high l/b ratios under the routine laboratory conditions (80–100 °C, 10–20 bar, 0.5–1 mM Rh), somewhat milder than the industrial conditions. At these conditions, the Xantphos concentration should be kept at 2.5 mM. These kinetics are in accord with two possible rate-limiting steps, alkene coordination or alkene migratory insertion, or an in between situation of the two (Scheme 9). Thus, in Scheme 9, we have drawn the CO coordination to the unsaturated alkylrhodium species as irreversible.
 | ||
| Scheme 9 Simplified mechanism of Rh-catalysed hydroformylation. | ||
To distinguish between the two steps for one ligand (1), a 12C, 13C kinetic isotope study at natural abundance was conducted and also the rates of H2 and D2 hydroformylation were compared.112 The outcome for this ligand under the conditions chosen was inconclusive as the experimental outcome of the isotope effect was between the calculated values for the two steps. CO exchange was studied for three ligands and it was found to be a dissociative process and two orders of magnitude faster than the overall rate of hydroformylation.113 Therefore, the two CO exchange reactions in Scheme 9 are thought to be fast equilibria.
Changes in product l/b distribution seem “spectacular”, as the common ratio is 2:1 and a small change in the free energy of activation between the two products can shift the ratio to 50:1. A smooth relation between l/b ratio and natural bite angle within this ligand group was obtained, which gives some confidence in the bite angles calculated by simple MM2 methods. Absolute values may not represent actual angles in complexes 31, but the trend seems correct.37 Depending on the electronic properties of the two diarylphosphino groups, the l/b ratio increases with βn and reaches a plateau for diphenylphosphino at 111°. The development of in situ IR measurements at practical conditions and concentrations has greatly contributed to our knowledge of the catalyst systems, as the two species 31 can be clearly distinguished both for phosphine and phosphite ligands.17,114 NMR measurements were not performed on reacting systems, but thanks to the abundance of atoms with a nuclear spin of ½, large amounts of data can be collected for the characterization of the intermediates. Also, cobalt hydroformylation underwent some rejuvenation, thanks to in situ spectroscopy.114c The interesting outcome for Xantphos ligands is that both structures 31ee and 31ae are observed in the spectra, although the proportion of 31ee increases with bite angle. However, even when 31ae is the major species, l/b ratios are high. Electron withdrawing substituents on the diarylphosphino groups increase the proportion of eq–eq, a characteristic of a trigonal bipyramid, and both the rate and the l/b ratio increase. The higher rate is due to a faster dissociation of CO, or more precisely a higher proportion of unsaturated species.49 Thus, formation of an eq–eq isomer is not a prerequisite for high l/b ratios, and 31ee as well as 31ae can lead to the same intermediate that in the next step(s) favours the formation of a linear rhodium alkyl species. Thus, while BISBI occurs nicely as 100% eq–eq, Xantphos ligands with only a partial eq–eq occurrence perform just as well. It is interesting to note that several bisphosphines have been reported that have an ideal βn for equatorial–equatorial coordination, but yet give low l/b ratios in 1-alkene hydroformylation. Two examples of wide bite angle ligands are 32 (ref. 105) and 33 (Scheme 10);115 it was argued that 32 did not give stable chelates in spite of the calculated βn of 126°, and a low l/b ratio resulted,116 while 33 is sterically too demanding and needs to convert to a monodentate ligand in the catalytic cycle, although it exclusively forms an eq–eq intermediate. Steric hindrance also spoiled the performance of electron poor ligand 34.117
 | ||
| Scheme 10 Various wide bite angle bisphosphines. | ||
In the hydroformylation of 4-pentenal to adipic aldehyde, Xantphos 1 does not produce a fast catalyst, but among a group of selected bidentate diphosphites, it is the most selective catalyst (98% linear).118
The known, strong electron withdrawing properties of pyrrole groups on phosphorus were exploited in 35 (ref. 119) (PyXANT). Initial rates for ethene hydroformylation (80 °C, 20 bar ethene/CO/H2 = 1:1:1) were as high as 20300 mol mol−1 h−1.120 For 1-octene under these conditions l/b was 88 and it increased to 104 at 40 bar, while the rate remained the same. Pyrrolyl phosphoramidites of bisphenol gave the same high linearities, but much higher rates, probably because they are even better π-acceptors.121,122 Ligand 35 is stable in water and base, since hydrolysis is catalysed by acids.
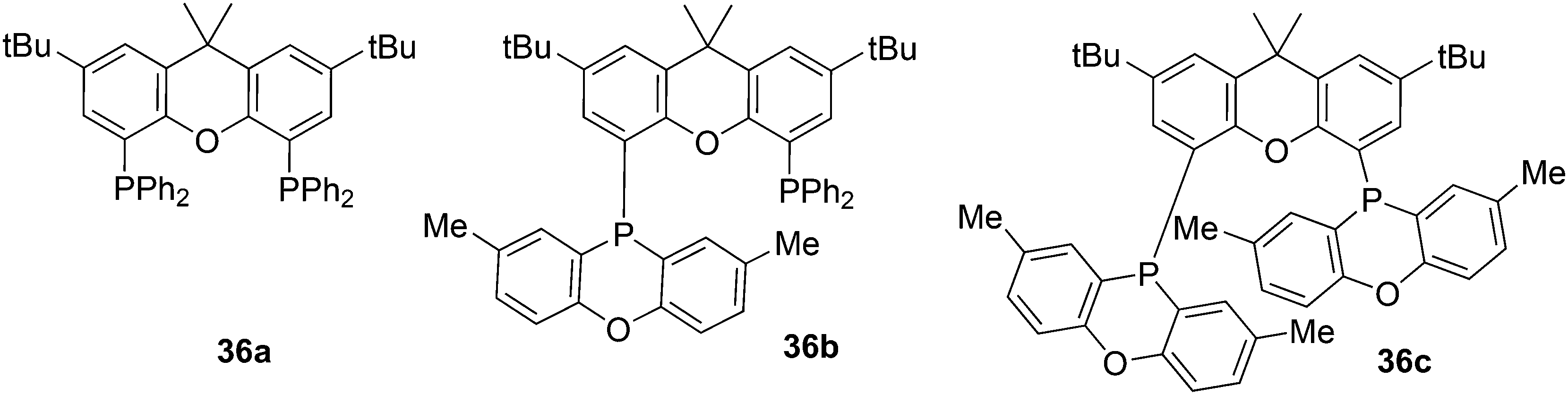
In the series 36a–c the rate of 1-octene hydroformylation increases by a factor of 5, but the selectivity to linear aldehyde goes down (l/b 44, 31, 22). The POP group (2,8-dimethylphenoxaphosphinine) is a weaker donor than PPh2, and from crystal structures it can be seen that the flat POP group is bent away from the metal centre, thus giving a lower barrier for branched alkyl formation.123 Yamakawa and co-workers hydroformylated 3,3,3-trifluoropropene with the use of various Xantphos ligands, Nixantphos and DPEphos, which resulted in l/b ratios of 99 at complete conversion and mild conditions (80 °C).124 At 100 °C, the l/b ratio was 999, but at 50 °C, after incubation at 80 °C, the l/b dropped to 0.6. Conversion increased linearly with reaction time and thus the kinetics were different for this electron poor alkene, compared to those of normal alkenes; the strong changes with temperature suggest different kinetics for the linear and branched pathways.
A remarkable ligand effect was reported for the hydroformylation reaction of substituted alkylidenecyclopropanes by Simaan and Marek, Scheme 11.125 Under very mild conditions, complete conversion was obtained to give two products, the ring-opened linear product 37 and the expected product 38. Notably, the stereochemistry of the quaterny stereogenic C-centre was retained. In all three instances, Rh reacted with the exocyclic, benzylic C-atom, and the ring opening and CO insertion compete for the subsequent step; there is no simple explanation for the ligand effect.
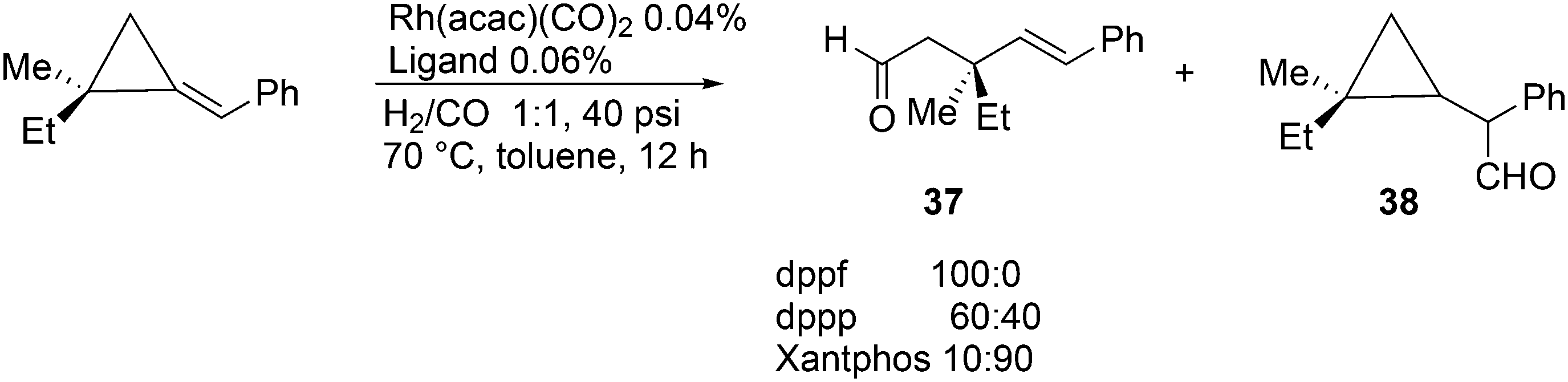 | ||
| Scheme 11 Hydroformylation of alkylidenecyclopropanes. | ||
The first DFT studies on xanthene ligands in Rh hydroformylation were conducted by Carbó et al.75,126 All results point to the decisive role of non-bonding interactions and thus the full ligands have to be taken into account; consequently, a QM/MM method was used. The migratory insertion starting with the rotation of the alkene in a favourable position was taken as the rate limiting step. A trend to slightly higher l/b values was found when the bite angle was increased. Electronic changes in the ligand do not directly change the l/b ratio, but they do change the geometry of the complex; electron withdrawing ligands prefer a trigonal bipyramidal structure, while strong donor ligands cause a distortion towards a square bipyramid.127 These structural changes influence the energetics of the two pathways. Generally, electron withdrawing ligands lead to higher l/b values and thus, in a trigonal bipyramid the steric influence is most effective. Steric effects on l/b ratios, keeping electronics and backbone the same, have not been conducted. Landis and Uddin conducted similar ONIUM studies on Xantphos/propene hydroformylation, exploring 56 independent isomers and although they could reproduce the l/b value nicely, they concluded that it was still too early to draw conclusions because the computed activation barrier was strongly overestimated.128 Kumar, Jackson and co-workers studied the regioselectivity of as many as seven Xantphos ligands (1, 4–9) with M06-L-based quantum chemical calculations.129 In addition, they calculated the stabilities of the eq–eq and eq–ap conformers of 16 Xantphos ligands (1–10, and the electronic variations 7a–7g). They were able to reproduce (i) the correct ordering of the equatorial–equatorial and equatorial–axial conformers of the resting state of the Rh catalysts, HRh(Xantphos)(CO)2; (ii) the dissociation energy for removing the CO ligand; (iii) the bite angle ranges in the HRh(Xantphos)(olefin)(CO) complexes and (iv) the experimental regioselectivities for the HRh(Xantphos)(CO)2-catalysed octene hydroformylation. The experimental l/b ratio vs. βn was reproduced, but interestingly, the differences in bite angles in the transition states for the various ligands was not more than 1°. The conclusion remains that non-bonding ligand–ligand and ligand–substrate interactions determine the regioselectivity (in the absence of isomerization). Butyl acrylate was hydroformylated by Jianliang Xiao et al. to the linear product and they found that the rate increased in the following order: dppe (118 mol h−1) < dppp (236 mol h−1) < Binap (177 mol h−1) < dppf (861 mol h−1) < dppb (2038 mol h−1) < Xanthphos (3052 mol h−1), which shows that unmodified xantphos is the bisphosphine with highest rate in this case (80 °C, 20 bar syngas, toluene), in a series coinciding with the order of the bite angle.130 Dppb modified with electron withdrawing p-CF3 groups gave an even faster catalyst (4700 mol h−1).
Kégl recently reviewed computational studies in hydroformylation.131 Kiprof and co-workers focused on the stabilities of intermediates in hydroformylation for Thixantphos, BISBI, and dppe.132 DFT-ONIUM studies of the isomerization of internal alkenes (see next section) have been reported.133 Octenes show a slightly higher rate than butenes. Entropy corrections were neglected. The large difference in the isomerization rate of Xantphos (slow) and POP-Xantphos (fast) rhodium complexes was confirmed, without including the CO dissociation step, although a difference of 8 kcal mol−1 would seem overestimated. The difference mainly arises from the stability of the initial alkene complexes caused by non-bonding ligand–alkene interactions.
Rhodium hydroformylation catalysts become isomerization catalysts at higher temperatures and lower CO pressures. As mentioned above, electron withdrawing phosphorus ligands will enhance isomerization compared to hydroformylation. The tandem isomerization/hydroformylation was recently reviewed by Boerner and co-workers.134 Catalytic isomerizing ω functionalization of fatty acids via hydroformylation and other reactions was recently reviewed by Mecking et al.135 Phosphites are good ligands for this tandem reaction and aromatic diphosphites with the correct bridge and steric properties, as introduced by Union Carbide, were the first preferred ligands reported, but nowadays there is a large body of suitable diphosphite or diphosphonite ligands.95 Regarding phosphines, the same ligands derived from BISBI and Xantphos that gives high l/b ratios for 1-alkenes can be used for the tandem process. Highly effective Xantphos ligands for tandem isomerization–hydroformylation were obtained for the phenoxaphosphinine substituted Xantphos or Nixantphos.136,137 With the use of ligand 25, a l/b ratio of 93:7 was achieved for a butene mixture (Raffinate II) at 115 °C and 25 bar. Earlier, it had been shown that 4-octene gave 86:14 as l/b ratio with an unsubstituted ligand 25 (i.e. lacking the alkyl groups added for an increase in the ligand's solubility).138
Tandem reactions, in which hydroformylation is followed by another reaction, have recently been reviewed by Bondzic.139 As mentioned above, hydrogenation of the aldehyde products to alcohols is often a desired subsequent reaction and it can be carried out by the same metal if the metal is a phosphine modified Co or Pd.140 For Rh catalysts, this is less common as Wilkinson's rhodium hydride carbonyl arylphosphine complexes do not hydrogenate aldehydes. If a different rhodium species that is ionic can be generated and co-exist with the hydroformylation catalyst, one obtains a tandem system. For Xantphos, one such example was reported by Sandee et al.63 Thus, when Nixantphos 19 was attached to silica via a sol–gel method, during rhodium hydroformylation, an ionic hydrogenation catalyst could co-exist in the solid, depending on the solvent. This way 1-octene could be directly converted to 1-nonanol in high selectivity as the linearity of the product was retained. The addition of water at high temperature (160 °C) to a Xantphos catalyst in polar solvents produces alcohols directly from 1-octene, with the usual high linearity. As most components are insoluble in water, the reaction was called “on water catalysis”.141 [Rh(cod)2]BF4 and Xantphos or tris(2,4-di-tert-butylphenyl)phosphite in alcoholic solvents gave the acetals directly without added acid catalysts, as probably some cationic Rh(I) was retained in solution.142
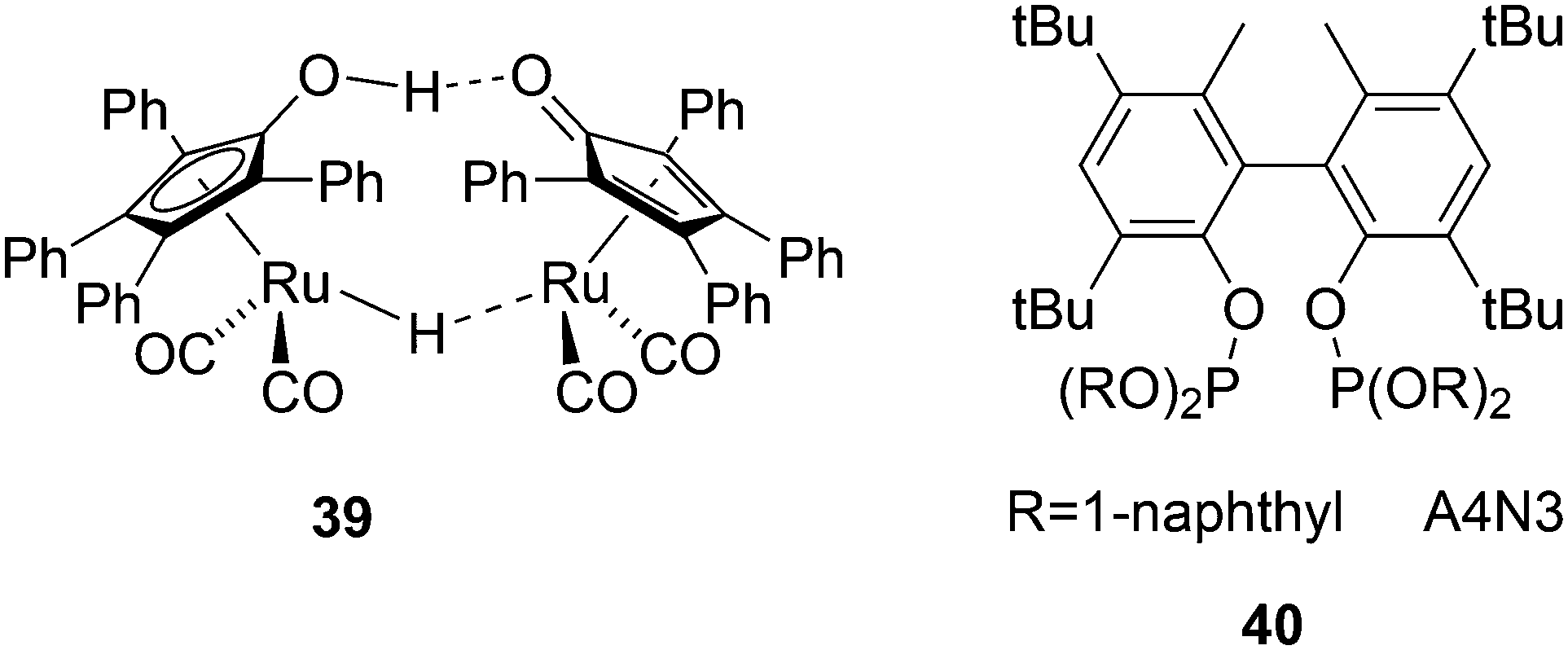
Another solution for obtaining alcohols from a rhodium hydroformylation catalyst was reported by Nozaki and co-workers who added Shvo's ruthenium based aldehyde hydrogenation catalyst 39 to a Xantphos rhodium hydroformylation system and both catalysts maintained their properties under these conditions.143 An l/b of 22 was obtained for 1-decene to 1-undecanol for three ligands considered, 40, a diphosphite type ligand, BISBI and Xantphos.144 For higher internal alkenes the rates were considerably lower and phosphites are now much more effective than Xantphos, likely due to the π-acidity of the former.145
Another important tandem sequence of reactions is called hydroaminomethylation; the aldehyde product reacts with an amine present and the resultant enamine is hydrogenated by the same catalyst. The reaction requires two equivalents of hydrogen and water is a co-product (Scheme 12). The linear products of simple alkenes are important large scale chemicals, while the branched products are valuable chemical intermediates in the laboratory.
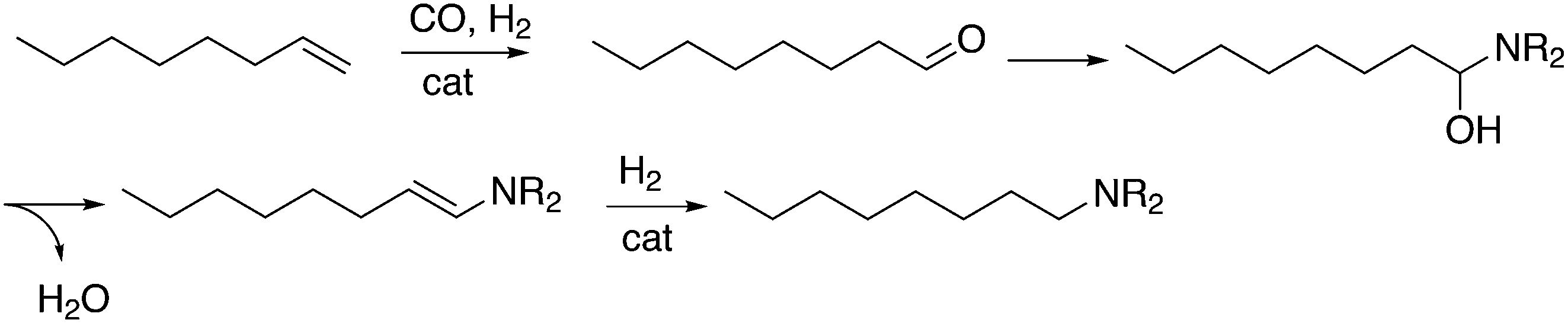 | ||
| Scheme 12 Aminomethylation of 1-octene. | ||
For 1-alkenes, a highly chemoselective and regioselective hydroaminomethylation using a cationic rhodium pre-catalyst together with Xantphos as ligand has been reported.146 Linear amines from internal alkenes were the more demanding targets; eight Xantphos ligands were equipped with POP moieties to raise activity for isomerization and the POP derivative of 8 gave full conversion of 2-pentene with 68% linear product with piperidine as the amine (CO, 7 bar; H2, 33 bar; toluene/methanol (1:1); 125 °C).147
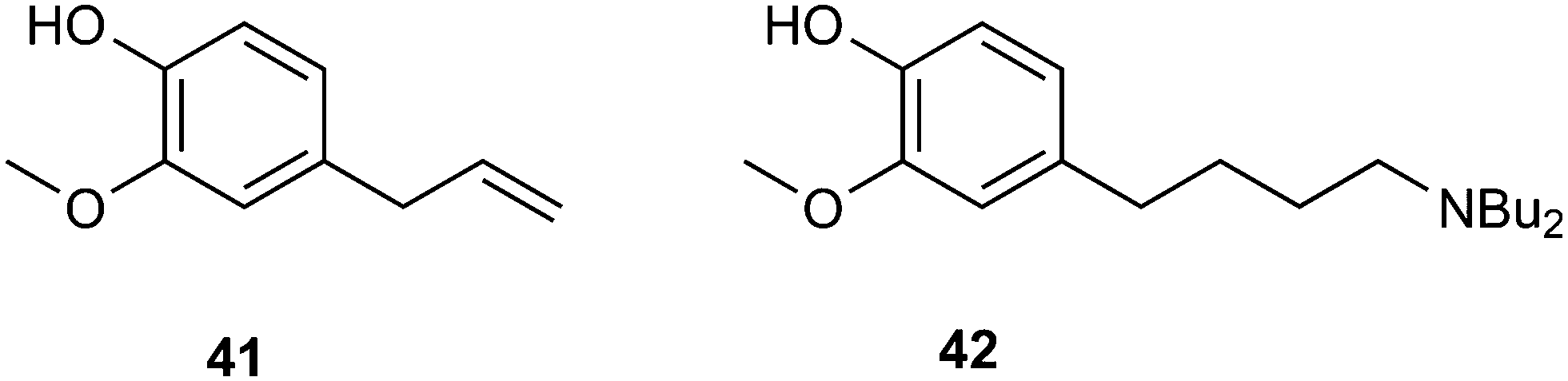
As an example of an application in fine chemicals carried out by Dos Santos and co-workers, we show the aminomethylation of eugenol 41 with dibutylamine as the amine to obtain 42.148 Ligands such as BISBI raised the preference for linear aldehydes, but the hydrogenation step is sluggish compared to the ligand-free system. The addition of acid to this system restored the hydrogenation activity and the linear amine was obtained in good yield. Hydrogenation activity requires the presence of a Rh(I) salt species, the formation of which is promoted by acid. In a normal hydroformylation system, such large quantities of acid (20% on substrate) would stop the hydroformylation reaction, but now there is a large amount of base present.
Vogt and co-workers have shown that Sulfoxantphos 14 dissolved in ionic liquids in a two-phase system can also be used for an efficient and selective aminomethylation tandem reaction, with piperidine as the amine. Phase separation was good and rates as high as 16000 h−1 at 125 °C and 35 bar, were recorded.149 The same group reported that pyXANT ligand 39 gave an excellent catalyst for aminomethylation of terminal alkenes with very high l/b ratios (6200 h−1, 12/24 bar CO/H2, 110 °C; substrates piperidine and 1-octene).117
Aminomethylation is more or less restricted to the use of dialkylamines, since monoalkylamines or ammonia give multiple substitutions in addition to mono-substitution. Bouwman and co-workers investigated the use of mono-substituted N-atoms and focused on amides as the nitrogen source, starting with acetamide.150 Triphenylphosphine gave only 3% of the desired alkylamide, which was not very encouraging. They managed to develop a catalyst system for this new reaction with the use of ligands 1–3, 6, 9, 10 and anisyl and t-butyl substituted Xantphos. Ligands 1, 3, and 10 performed best, but the solvent composition was equally important (diglyme, substituted phenols, acids, fluoroalcohols). For acetamide, a selectivity of 61% for the desired linear product was obtained, and interestingly, valeramide gave a higher selectivity of 83%; the amide reacts preferentially with the linear aldehyde.
Substituted phenylhydrazines can be reacted with aldehydes or ketones to give hydrazones. The latter are converted with acid to indoles, the Fischer indole synthesis. The aldehyde can be generated in situ, as shown by Eilbracht and co-workers, and the three steps of the reaction are compatible, thus turning it into a tandem reaction.151 A selective hydroformylation catalyst will selectively give one product and hydroformylation is not sensitive to functional groups. A simple reaction scheme is presented in Scheme 13. The broad scope of the reaction was shown with the use of Sulfoxantphos 14 in water.152
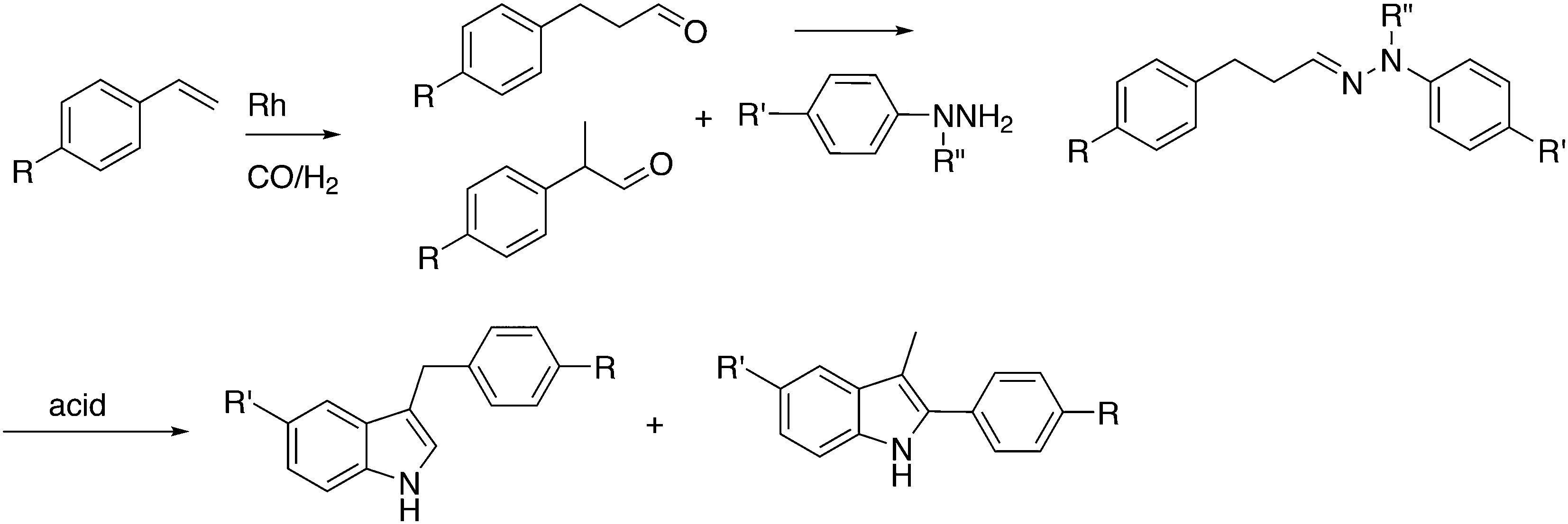 | ||
| Scheme 13 Hydroformylation and Fischer indole synthesis. | ||
Octanol was used recently as the surrogate for CO with the use of Rh/DPEphos in a Pauson–Khand reaction with alkynes or allenes, while Xantphos was inactive under the conditions tested.155
A related reaction is the transferhydroformylation discovered by Dong and co-workers.156 As in hydroacylation, the C–H (aldehyde) bond breakage occurs on a Rh(I) salt (rhodium benzoate in this instance) via oxidative addition (Scheme 14). Elimination of benzoic acid gives an acyl intermediate of the catalytic cycle of hydroformylation, and both the backward and the forward reactions can take place to give the new aldehyde. Interestingly, the proton on the new acylrhodium species must be delivered by benzoic acid in order to arrive at a catalytic process. One could also imagine the formation of dihydrogen as an intermediate, but experiments in deuterated methanol indicated that this was not the case.
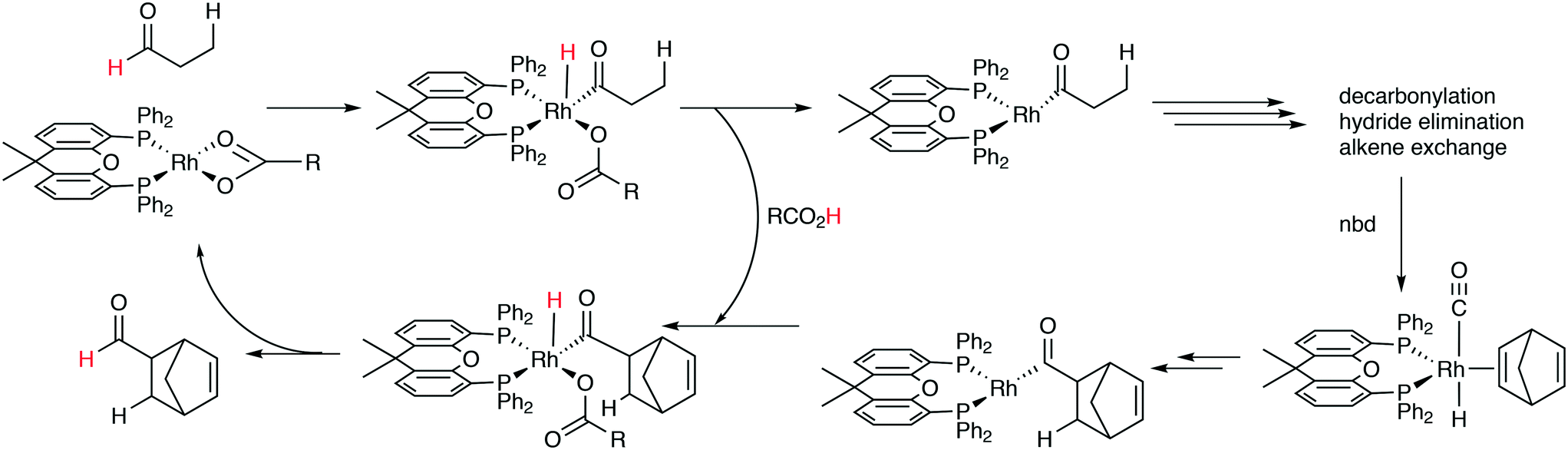 | ||
| Scheme 14 Transfer hydroformylation. | ||
Arsine ligands are not very common in homogeneous catalysis, but the arsine homologues of 4,7-di-t-butyl Xantphos provide active catalysts in the Pt–Sn hydroformylation of 1-octene. The mixed arsine phosphine ligand (βn = 111°) is 20 times faster than Xantphos, and the bisarsine ligand more than 10 times, but both are slower than Homoxantphos (40 times faster than Xantphos) (60 °C, 40 bar);167 l/b ratios >250 were obtained and isomerization with the mixed ligand was <4%. In rhodium-catalysed hydroformylation, substitution of arsine for phosphine slows the catalysis.
Vogt and co-workers studied the isomerization–hydroformylation of terminal and internal octenes with a catalyst composed of Sixantphos with PtCl2 and SnCl2.168 This catalyst is as fast as Homoxantphos, using 1-octene as the substrate. Regarding internal octenes, isomerization apparently favours a slightly wider bite angle. The isomerization activity of the catalyst and the l/b ratio increased with temperature, as does the formation of alkane. The same research group investigated the Pt/Sn-catalysed isomerization–hydroformylation of methyl 3-pentenoate. In this instance, a catalyst based on Thixantphos (βn = 107°) gave the desired aldehyde exclusively.155
Alkoxycarbonylation of internal alkynes was reported by Tsuji et al. with the use of aryl formates as the source for carbon monoxide and the arylalcohol.180 Hitherto, the reaction had to be carried out at high temperatures (180 °C), but a ligand effect study revealed that with the use of wide bite angle ligands such as Xantphos and BINAP the reaction could be carried out at 100 °C. It was shown that with Xantphos as the ligand the reaction proceeds via conversion of aryl formates to phenols and carbon monoxide, not via direct addition of aryl formates to alkynes. The reaction can also be done with alkyne, phenol, and 1 bar of CO (Scheme 15).
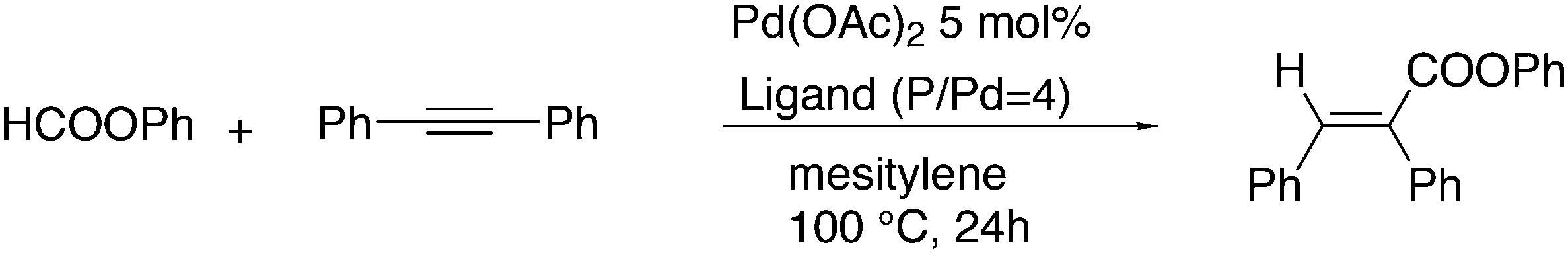 | ||
| Scheme 15 Addition of phenyl formate to an alkyne. | ||
Amadio and co-workers studied the effect of phosphine ligands in the palladium catalysed oxidative carbonylation of alcohols.185 When 1,4-benzoquinone is used as the oxidant the main products formed are oxalates, and carbonates are the by-products. The anion has a major effect on rate and selectivity; tosylate gives the highest rate and mainly oxalate as the product. In the dppe series, dppp, dppb, dppf, DPEphos and Xantphos, the rate strongly increases for dppf and its derivatives. This is typical of systems in which the reductive elimination is rate-determining. Oxalate forms via reductive elimination from (diphos)Pd(CO2R)2 and this reaction is enhanced by wider bite angles via increasing steric hindrance; for small bite angles these complexes are stable. The mono-alkoxycarbonyl-Pd complexes react with alkoxides to give the carbonates. The ether-based ligands probably give inactive resting states with κ3-POP as the coordination mode as we have seen above for Rh.
In the methoxycarbonylation of alkenes, the catalytic species is usually cationic as well and the absence of Xantphos in this area will also be due to the formation of κ3-POP species. By far, the best ligand is mostly 1,2-bis-di-tBu-phosphinomethylbenzene 45, the “INEOS” ligand, especially for non-functionalized alkenes. Behr and Vorholt reported a few exceptions. Hydroesterification of methyl 10-undecenoate in thermomorphic solvent systems yielded C12 diesters with the use of a Pd mesylate catalyst; Xantphos gave the fastest catalyst, but it was less selective toward the linear product (72%) than 1,2-(tBu2PCH2)2benzene (94%).186 Methoxycarbonylation of 2,7-octadien-1-ol 46, however, gave very low yields when 45 was used, as mainly acetal products were obtained in low yield.187 In this instance, Xantphos was the best ligand tested and it gave 64% of the diester 47 (Scheme 16).
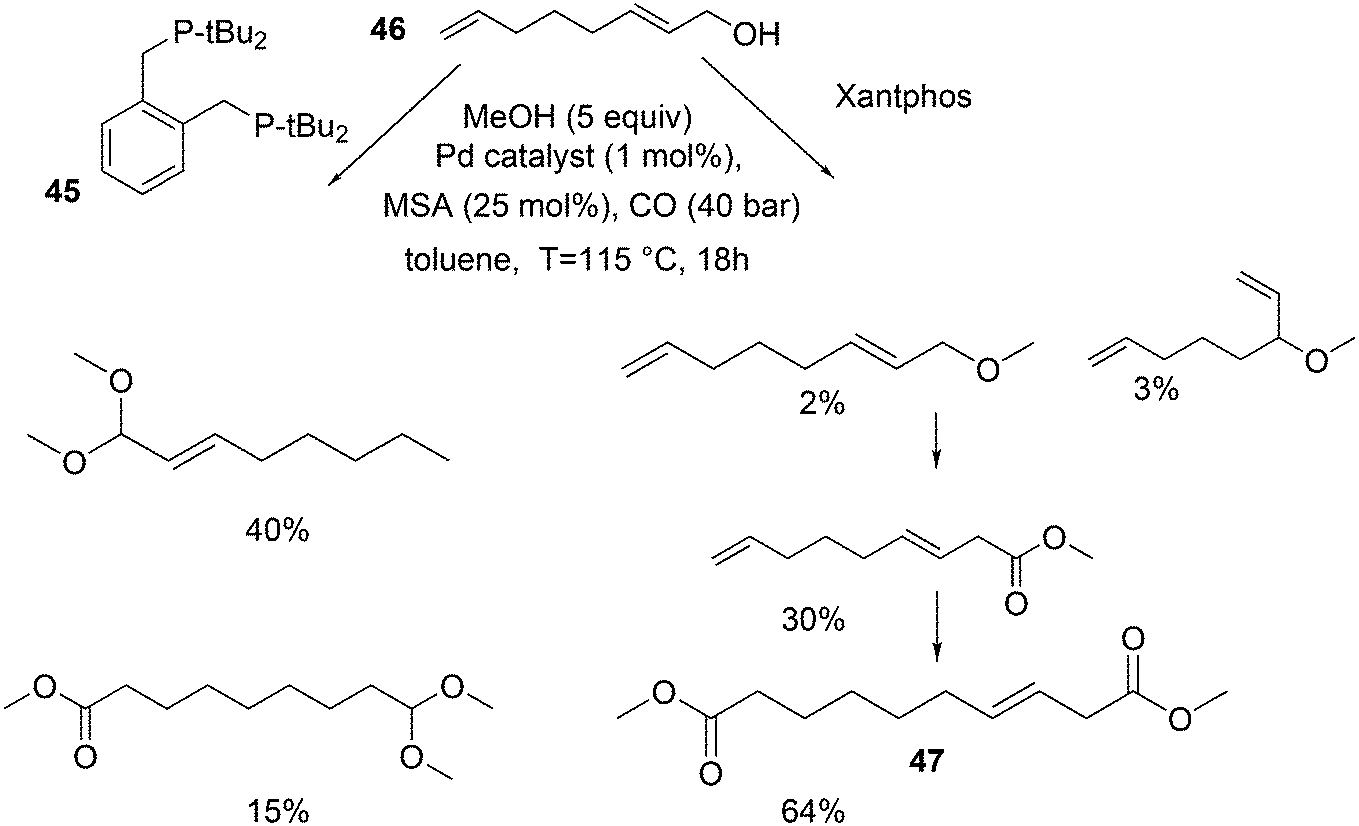 | ||
| Scheme 16 Hydroesterification of methyl 10-undecenoate with ligands 45 and 11 (Xantphos). | ||
3.2. Hydrocyanation
Vogt and co-workers combined all ligand properties that might enhance reductive elimination in one ligand, 44, and variations thereof:193 a) a wide bite angle, b) electron withdrawing aryloxy groups on phosphorus, c) steric bulk due to tBu groups.194 The nickel-catalysed isomerization of 2-methyl-3-butenenitrile to 3-pentenenitrile was studied, a key-step in the industrial hydrocyanation of butadiene, which involves a reversible hydrocyanation. Good activities and selectivities to the desired 3-pentenenitrile were obtained for the meta-tBu derivatives, while the ortho-tBu derivatives yielded slower catalysts. Triptycene based bisphosphines 11 have also been reported as good ligands in the nickel catalysed hydrocyanation of alkenes.195,196
As stated above (3.2.1), we began the hydrocyanation work using the Xantphos ligands bearing in mind electronic bite angle effects that were based on EH calculations on very simple models by Otsuka,200 and calculations by Whitesides et al. from the 1980s, rather than the steric effects brought forward so far.201 Before returning to reductive eliminations, we will mention two occasions that support an electronic orbital effect brought about by bite angle. DFT calculations on bisphosphine copper hydride show that the hydridic character changes under the influence of the bite angle.202 An electronic effect of βn was observed in the hydricity of hydrides in metal hydride bisphosphine complexes.203 However, the authors pointed out that steric changes, brought about by changes in backbone or substituents, could have a much larger effect on the measured physical properties than the electronically angle induced energy differences.204 The formation of ruthenium hydride and dihydrogen complexes also was influenced by the bite angle of the ligand.205 Kégl and co-workers recently published their results from DFT calculations on CoH(CO)L2, in which the ligands were two molecules of triphenylphosphine or Xantphos.206 The angle between the monophosphines was varied in small steps and the energy minimum was found at 113°, close to the value of Xantphos. The Co–H bond increased in strength with the bite angle, while the reverse took place for the Co–C bond. Thus, we tentatively conclude that the static properties of metal hydrides experience an electronic bite angle influence.
Zuidema et al. investigated the reductive elimination of MeCN from various palladium complexes with the help of DFT and QM/MM strategies.207 Various saturated and unsaturated backbones C1–4 and H, Me, and Ph substituents at phosphorus were included. They confirmed the initial oversimplified idea that the square planar complexes were destabilized by wider bite angles and that not only the final states were stabilized, but the barrier was also lower for the widest bite angle systems. From their QM/MM analysis they concluded that electronic effects dominated. Anstaett and Schoenebeck compared the reductive elimination of ArCF3 from complexes with Xantphos and dppe as ligands in DFT calculations.208 They also varied the substituents on phosphorus (Ph, Me, H) and could clearly show that the steric effect of these substituents led to a fast reductive elimination reaction for Xantphos, while the dppe complex was unreactive, merely because the small backbone causes only a limited “embracing” effect of the ligand in the metal complex. Bickelhaupt and co-workers extensively analysed the reverse reaction of reductive elimination, viz. oxidative addition of, i.a., MeCl to the group 10 zerovalent metal, Pd, coordinated by two ligands at the initial point of the reaction.209,210 Their analyses showed that the rate of reaction is mainly determined by steric interactions. This means, for example, that due to smaller steric repulsions between substrate and bisligand complex, oxidative addition is faster for dppe than for the complexes of the higher homologues. For the same reason, the final energies of the square planar complexes are lower (i.e. more stable) for the small bite angle complexes, and for those with the smaller substituents. The reverse reaction, reductive elimination, which is the rate limiting step in the more reactive cross-coupling catalyst systems as we will see later, is faster in wide bite angle bisphosphine catalysts. However, all calculations show that this has mainly a steric origin. In a tutorial review, Fernández and Bickelhaupt give a complete account for the interested readers.211 Real life catalysis is more complicated as we reported above; the reaction rate “peaks” at an angle of 106° and at wider angles the trend breaks down.212 Other processes may be promoted or another reaction step may become rate limiting.
Keith, Daly and co-workers evaluated, with the use of K-edge X-ray absorption spectroscopy (XAS), how M–P orbital mixing in late transition metal complexes is affected by changes in coordination geometry and bisphosphine structure; here we are particularly interested in group 10 metal complexes.213 The intensity of the XAS features provides an estimate of ground-state metal–ligand covalency for bonds involving p-orbitals on the ligands; its application for phosphine ligands is relatively unexplored. LUMO energies were found to be highly dependent on P–M–P bite angles, but M–P covalency varied very little. It was concluded that other structural parameters may be more important when evaluating overlap contributions to covalency in bisphosphine complexes of Ni and Pd. M–P–C angles and dihedral angles would be distorted to obtain maximum overlap between P and M orbitals.35,214
In conclusion, the initial idea of an electronic bite angle effect on conversions between square planar divalent and tetrahedral zerovalent complexes was not a valid design element. So far, only in metal complex hydrides do charges and complex structures experience an electronic bite angle effect. Nevertheless, the “wrong” idea has led to the first phosphine ligand catalysing hydrocyanations. Wide angle diphosphites were later introduced into the field of Ni-catalysed hydrocyanation, which outperformed the monophosphites used since the 1970s.215 A second result of this was the recognition that the ligand bite angle behaviour in C–C cross-coupling reactions followed the same trend. At first, we regarded the results of cross-coupling as somewhat capricious, but this comparison convinced us early on that reductive elimination was the rate limiting step in the process, provided that the barriers of other steps, in particular oxidative addition, had been eliminated first.
3.3. C–C bond formation
3.3.1.1. Introduction. The first C–C cross-coupling examples using Xantphos ligands were reported in a patent submitted in 1994 as a result of a collaboration between the University of Amsterdam and DSM.20 The results became more widely available several years later in 1998.216 In the same year, both Buchwald and Hartwig successfully applied Xantphos and DPEphos in the now so-called Buchwald–Hartwig C–N cross-coupling reaction or amination reaction.217,218 The DPEphos based system is effective in coupling reactions involving a variety of substrates, including electron-poor anilines or electron-rich aryl bromides. It tolerates a high degree of steric congestion at both the aniline and the aryl bromide. Hamann and Hartwig did a systematic study of bidentate phosphines including DPEphos and Xantphos and found that for the amination reactions in this report (alkyl amines), the reductive elimination is apparently not a decisive step, since BINAP performed better than the wide bite angle ligands. The latter ligands promoted side reactions such as β-hydrogen elimination, which was perhaps, as was suggested, via de-coordination of one phosphorus donor in the more flexible bidentates. Guari and co-workers studied the arylation of anilines with preformed complexes ArPdBr(P–P-ligand); this led to a highly active catalyst with turnovers up to 140 h−1 at room temperature, with wider bite angle ligands giving faster catalysts.219 Hartwig et al. reported tBu3P and NHC based catalysts that are also active at room temperature.220
An interesting recent finding that belongs to the sub-heading C–H arylation is included in the general introduction of C–C bond formation. Eastgate, Blackmond, and co-workers reported recently on the use of Xantphos monoxide as the preferred ligand in C–C bond formation.221 Preliminary results showed that the monoxide of BINAP also gave good results. Xantphos monoxide and Xantphos gave the same results for the reaction shown in Scheme 17, but kinetics demonstrated that the bisphosphine was oxidized in situ to the monoxide 48 by palladium(II) acting as catalyst/oxidant and water as the source of oxygen.222 The monoxide was formed during a short incubation period and the reaction carried out with the preformed intermediate ArPdBr(Xantphos-oxide) was not faster than the reaction of the in situ formed catalyst. Carboxylates play a crucial role in the abstraction of a proton from the heteroaromatic ring (vide infra) and other bases were less effective or not effective at all. Catalyst decomposition seems to be related to the formation of the dioxide of Xantphos. This is a highly interesting finding that is worthy of attention in other catalytic reactions with bidentate phosphine ligands.
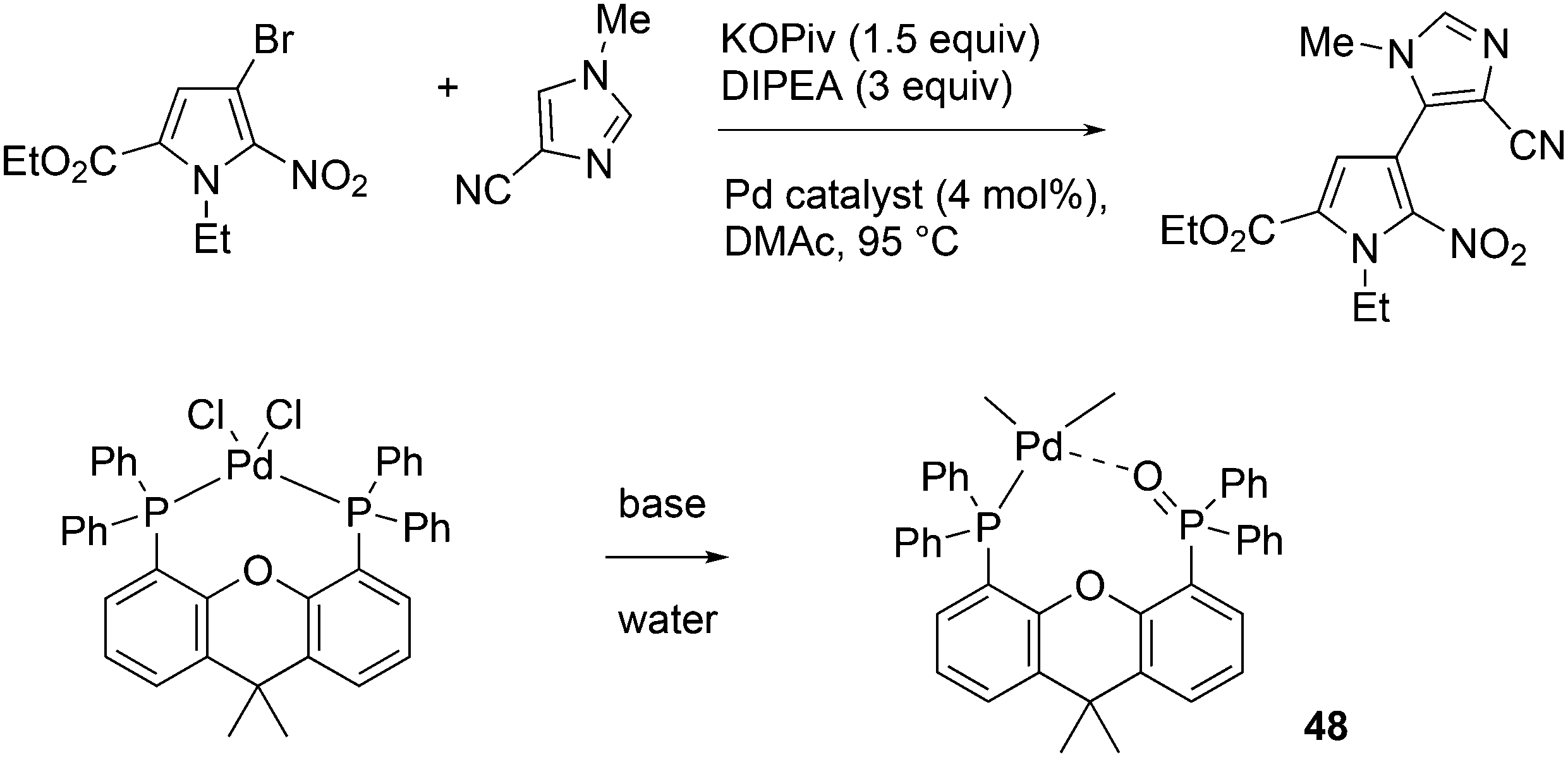 | ||
| Scheme 17 Formation of Xantphos monoxide during C–C coupling. | ||
3.3.1.2. Decarboxylative and decarbonylative coupling. The classic C–C cross-coupling reactions involve a hydrocarbyl metal compound and an organo halide or pseudohalide. The synthesis of the metal compound often starts with the metalation of an organo halide, such as the Grignard formation, unless direct metalation can be applied (organo lithium compounds, or catalytic metalation for borate esters). A direct C–H metalation by the palladium catalyst is even more attractive (vide infra). The metal halide procedure gives a large amount of salts as co-products and attempts have been reported to reduce the amounts of salt. For instance, De Vries et al. used carboxylic acid anhydrides as the metal hydrocarbyl precursors that only give CO as the co-product, since the carboxylic acid can be recycled.223 A recent example of a decarboxylative coupling was reported by Messaoudi, Alami, et al.224 In a system of this type, one starts with a metal carboxylate that is transferred to Pd and decarboxylated (or the reverse order) to produce, for example, an arylpalladium intermediate. The reaction shown in Scheme 18 afforded up to 76% yield of 49, a model for a wide range of pharmaceutically active compounds, with the use of a Pd/DPEphos catalyst. The stoichiometric use of silver carbonate offsets the advantageous use of the carboxylic acid.
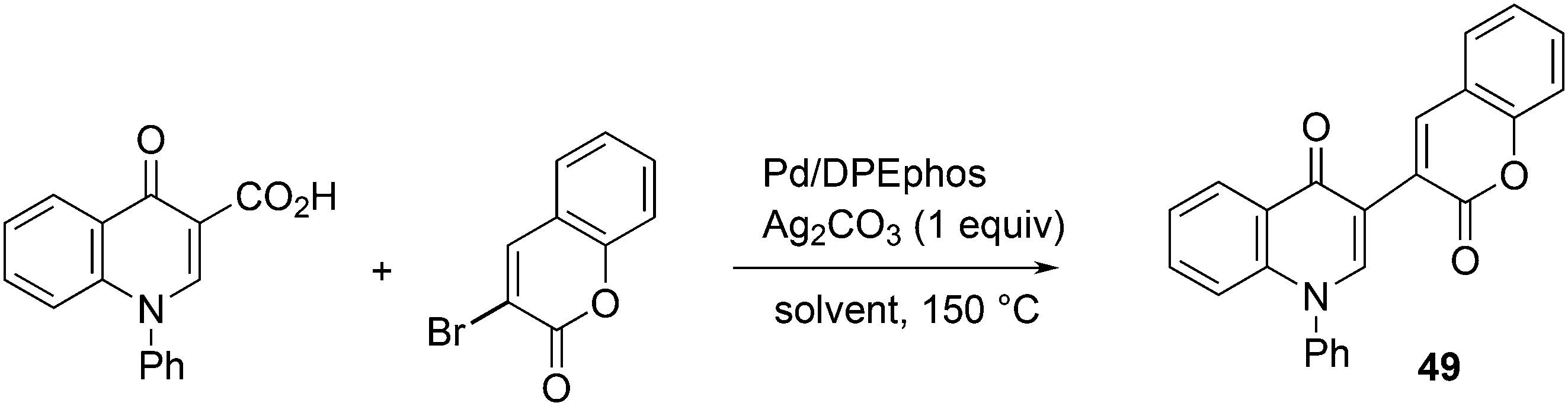 | ||
| Scheme 18 Decarboxylative C–C coupling. | ||
Mitchell and co-workers discovered a practical synthesis method for biaryls via a thermal decarboxylative Pd-catalysed cross-coupling reaction operating at moderate temperature.225 Importantly, the potassium carboxylate gave an excellent yield of the product with dppf as the ligand, DMF as the solvent, and tetra-n-butylammonium bromide as an additive (Scheme 19).
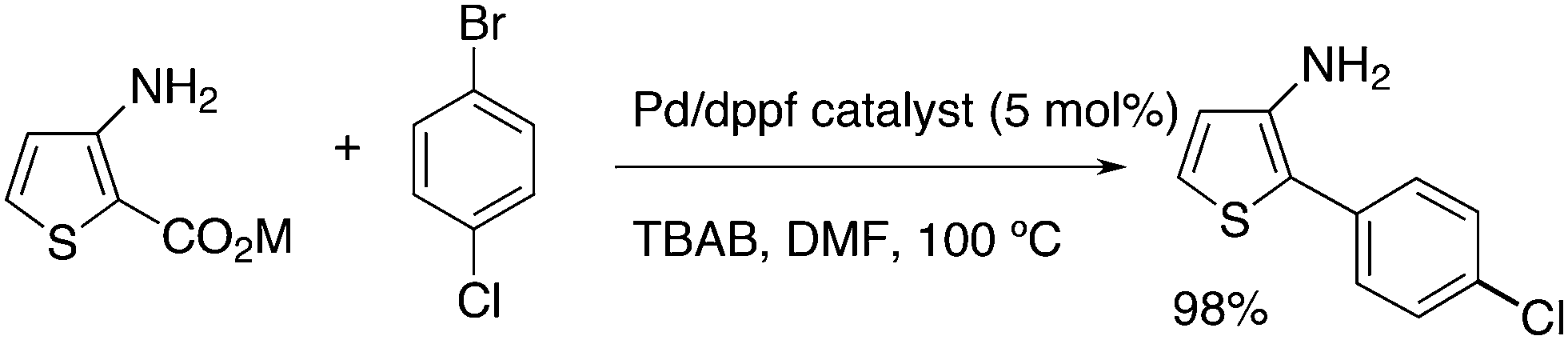 | ||
| Scheme 19 Decarboxylative C–C coupling of carboxylate. | ||
3.3.1.3. The Mizoroki–Heck reaction. The Mizoroki–Heck reaction can be successfully carried out with a variety of monodentate phosphine ligands, the more bulky ones often leading to the best result. Joshi and Pigge reported an intramolecular reaction (Scheme 20) in which Xantphos (99%) was superior to monodentates such as P(o-tolyl)3 and t-Bu3P (7 and 35%), and bidentates such as dppf (63%).226 R-BINAP gave a much slower reaction with the methyl substituted analogue, which required direct loss of the carboethoxy group, but under optimized conditions and with the use of AgOTf, the product was obtained in 93% yield and 94% ee.
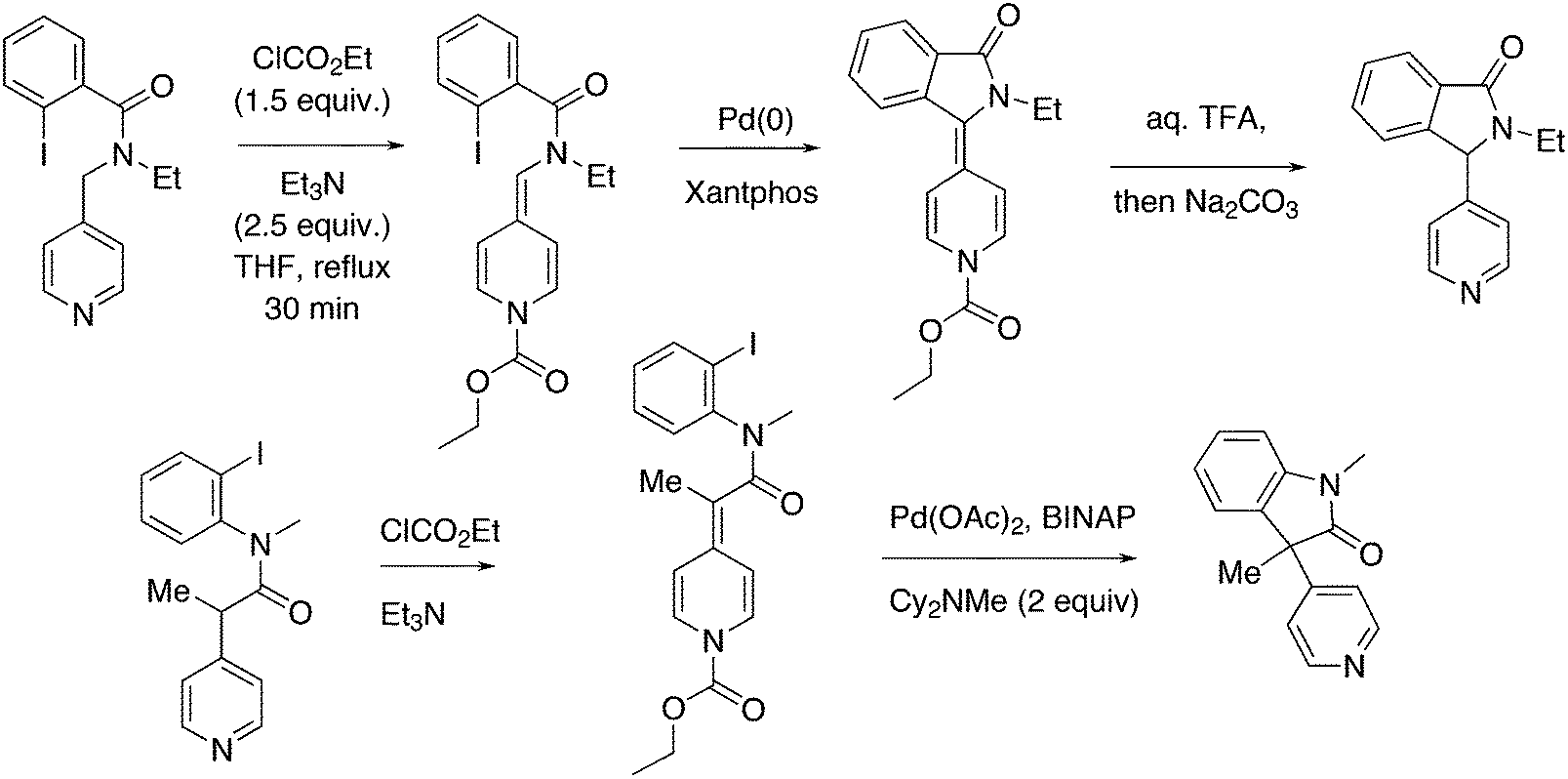 | ||
| Scheme 20 Intramolecular Mizoroki–Heck reaction. | ||
3.3.1.4. The Sonogashira reaction. Huang and co-workers explored the Sonogoshira reaction using the two common metals Pd and Cu and found that (Xantphos)CuI and unligated Pd salts gave the best results.227 Electron poor aryl chlorides also showed activity in the coupling reaction. As in previous work, the authors assumed that the phosphine remained on CuI.228 Another report on cooperation between CuXantphos and Pd concerns C–H arylation, but we will discuss it here since it concerns the same metals as the previous item.229 The authors found a profound effect of ligands on Cu, with Xantphos at the top of the list, while keeping the ligand on Pd the same (ClP(tBu)2, P-tBu3 and Xantphos could also be used on Pd). The authors proposed a mechanism (Scheme 21) in which CuX carries out the metalation at benzothiazole and the aromatic anion is transferred to Pd, in much the same way as the copper acetylide formation in the Sonogashira coupling reaction; yields up to 98% were obtained.
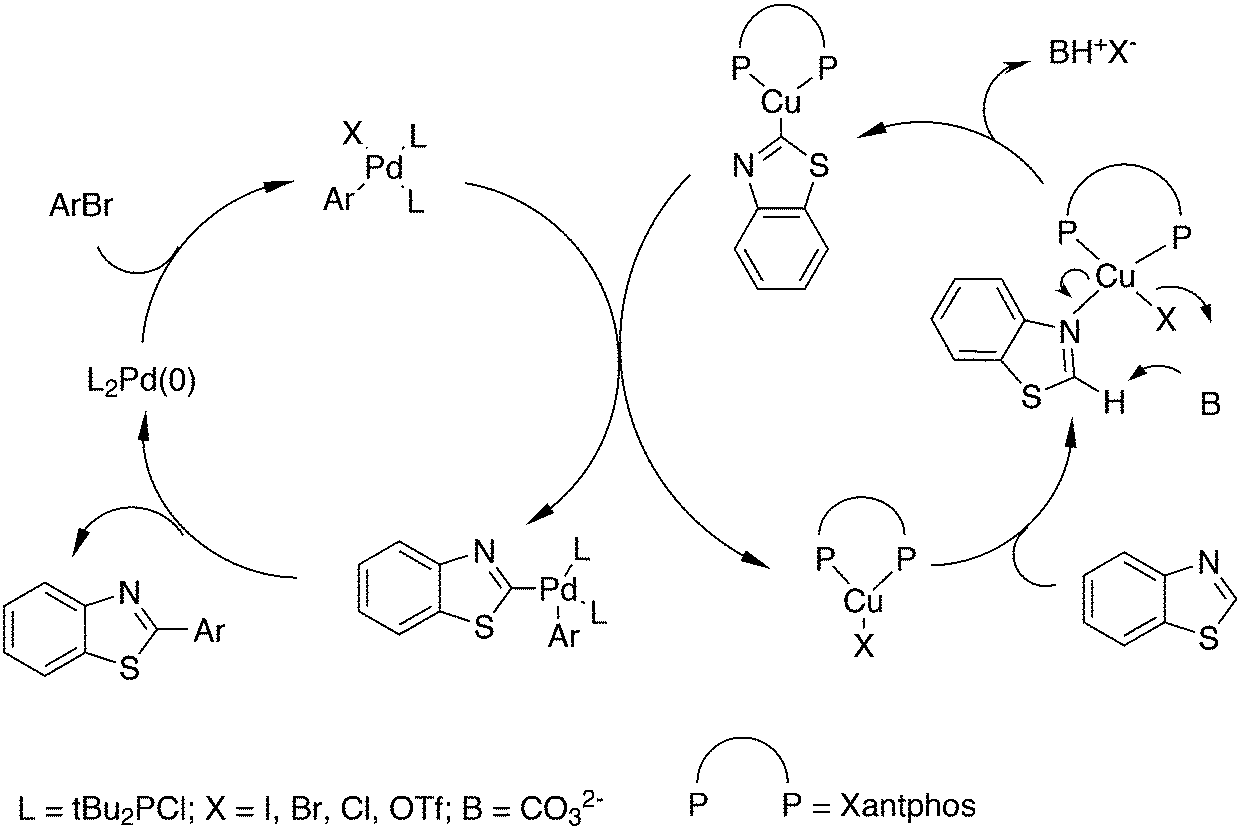 | ||
| Scheme 21 Combined Cu and Pd cross-coupling. | ||
3.3.1.5. The Kumada–Corriu and Negishi coupling reactions. As is known from simple model reactions,214 yield and selectivity for the Kumada–Corriu and Negishi type reactions increase with increasing bite angle for PPh2 substituted bidentate ligands until a maximum is reached, and further increasing the angle decreases the yield or selectivity. Carretero and co-workers developed a mild palladium-catalysed reaction of secondary benzylic bromides with aryl and alkenyl Grignard reagents. The use of Xantphos ensured that the undesired elimination pathway was minimized. The reaction proceeds with inversion of the configuration.230
Shi and Negishi reported a favourable result for DPEphos and tBu3P in the arylation of 1,1-dihalo-1-octene 50 (Scheme 22); wide angle bidentates gave a high preference (up to 99%) for the monosubstitution product 51, while tBu3P largely showed the disubstitution product 52.231 Alkynylation can also be achieved this way.232
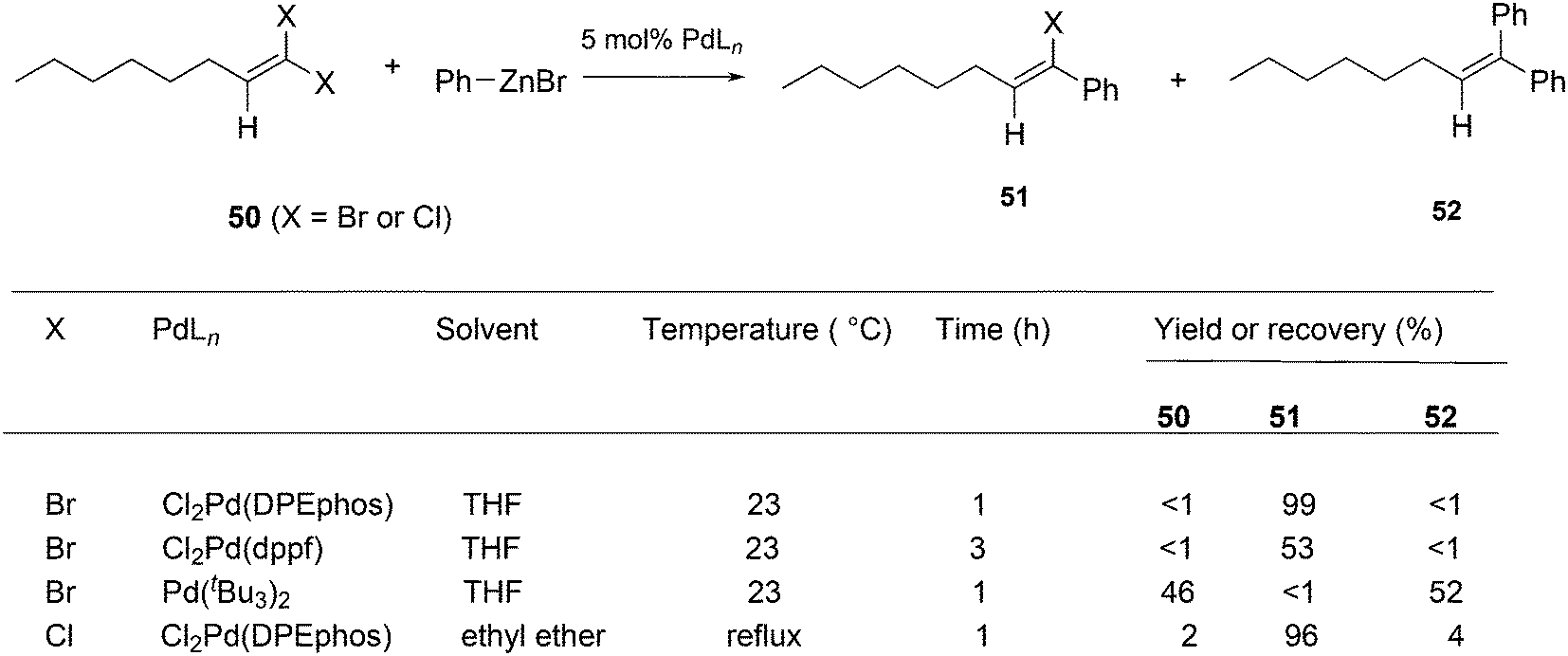 | ||
| Scheme 22 Mono- and diarylation of 1,1-dihalo-1-octene. | ||
Results along this line were reported by Roulland and co-workers for a Suzuki–Miyaura coupling.233 In the reaction (Scheme 23), they found a similar ligand effect with Xantphos giving the highest yield of monosubstitution product.
 | ||
| Scheme 23 Alkylation of 1,1-dichlorostyrene. | ||
In a subsequent publication, Negishi and co-workers achieved several million turnovers at still acceptable yields for the palladium-catalysed reactions of unsaturated organozincs with aryl or alkenyl iodides.234 Dppf and DPEphos were the preferred ligands.
Martin and Buchwald found ways to use the substituted Grignard reagents, as pioneered by Knochel, in order to avoid the change from Grignard to the milder zinc reagents, for example.235 At temperatures from −60 to −20 °C, they achieved cross-coupling of aryl triflates and iodides with Grignard reagents, either one of them containing reactive groups such as esters. Dicyclohexyl(diaryl)phosphines gave the best results (up to 79% for triflates) while Xantphos yielded only 41%. See Scheme 24 for an example.
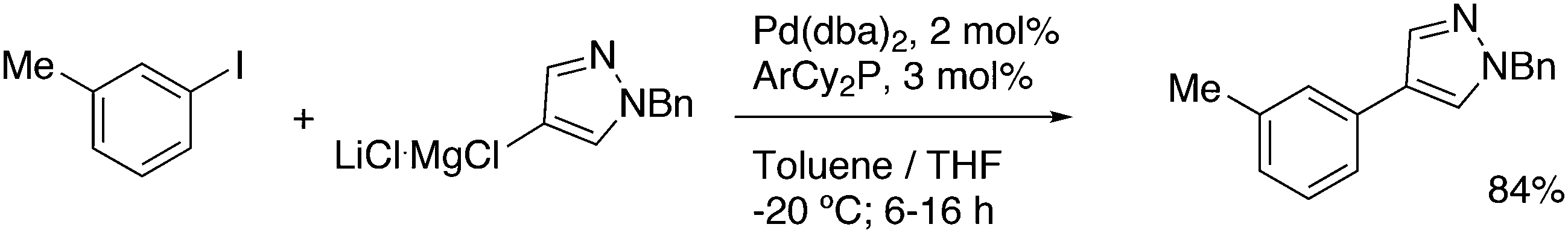 | ||
| Scheme 24 A mild method for Grignard vinylation. | ||
3.3.1.6. The Hiyama reaction. The Hiyama coupling reaction utilizes silyl compounds as the hydrocarbylmetal component in the cross-coupling (Scheme 25).
 | ||
| Scheme 25 Hiyama coupling reaction. | ||
For this reaction, mono-bulky phosphines and Xantphos gave the highest yields and selectivities and thus, this seems to be a well-behaved cross-coupling reaction.236
3.3.1.7. The addition of electrophiles to dienes. For coupling reactions of dienes, both Rh and Pd seem to give the highest selectivity when Xantphos ligands are used.
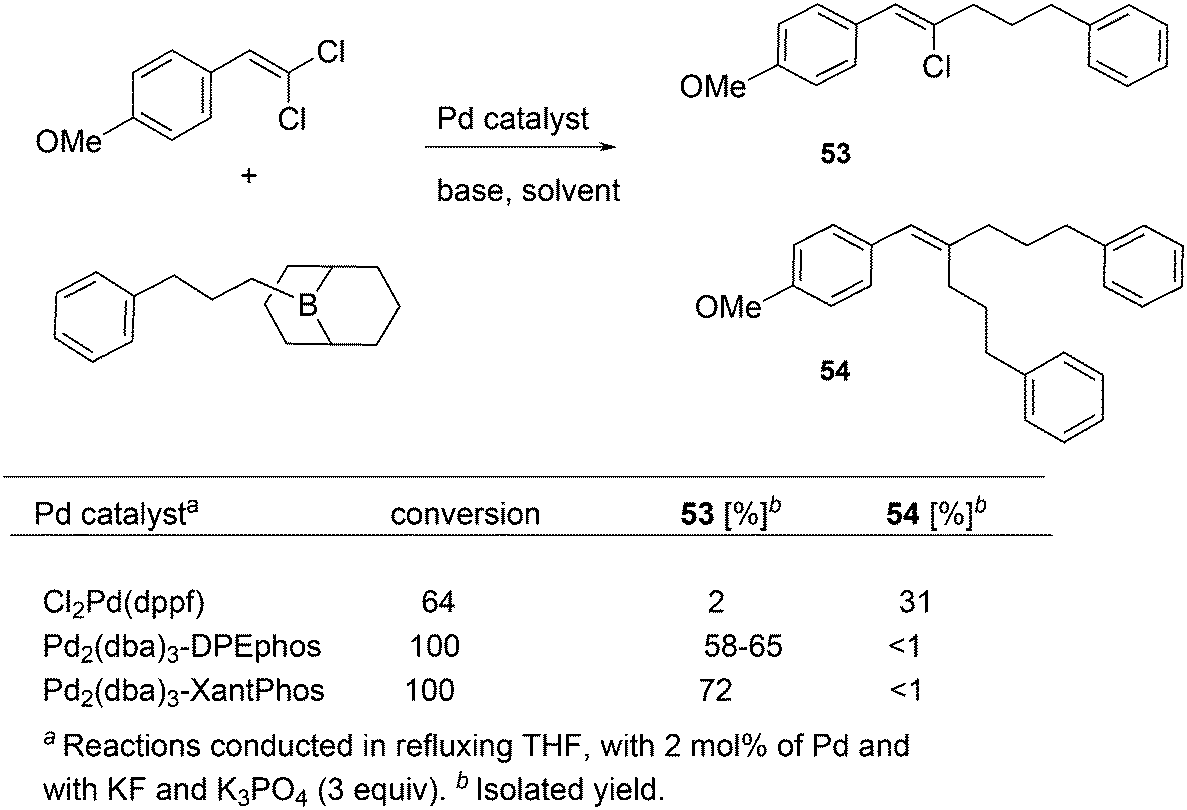
For the Pd catalysed coupling of dienes and aldehydes, and alkylboranes as the Lewis acids, Kimura and co-workers found that the yield increased in the ligand series PPh3 (0%), dppf (16%), DPEphos (33%), BINAP (64%), and Xantphos (99%) (Scheme 26).237 The series is not an exact bite angle sequence, since BINAP's angle (92°) is usually considered to be smaller than that of dppf (96°). Several metals catalyse this three-component reaction and in this case, β-hydride elimination takes place to give diene 55 and ethane, before the ethyl group moves to the allyl group, which results in an overall ene-type reaction.
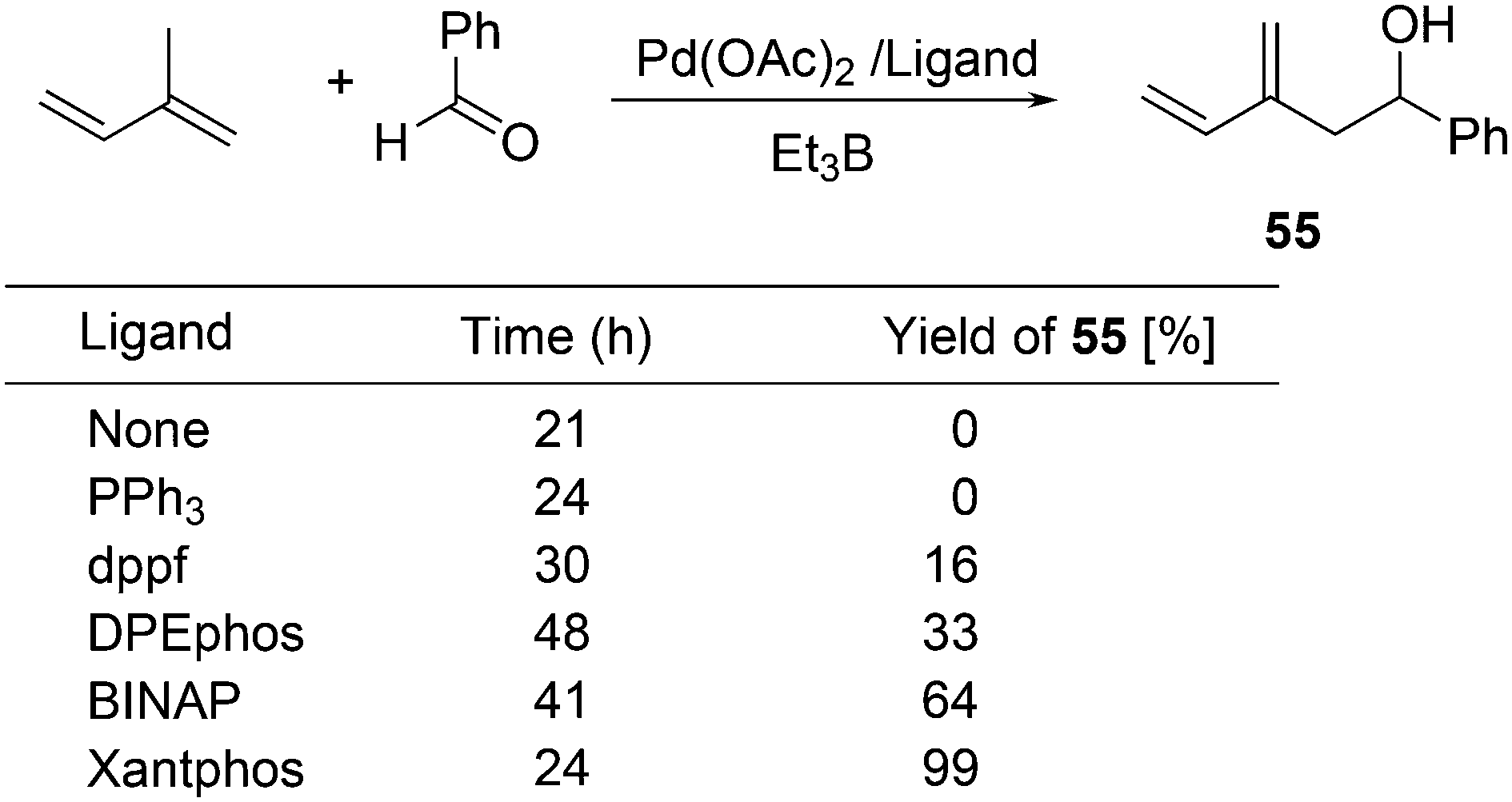 | ||
| Scheme 26 Allylation of aldehydes. | ||
3.3.1.8. C–H arylation. α-Arylation represented the first examples238 for the formation of Csp2–Csp3 bonds. The α-arylation of aldehydes with aryl bromides was successfully accomplished by Martín and Buchwald who showed that among many ligands, Xantphos (up to 76%) was best, closely followed by SPhos.239 Conditions were shown to be critical for obtaining good results, which were improved compared to previous results for BINAP (Scheme 27).240 For aryl chlorides, the use of SPhos and the like is mandatory.
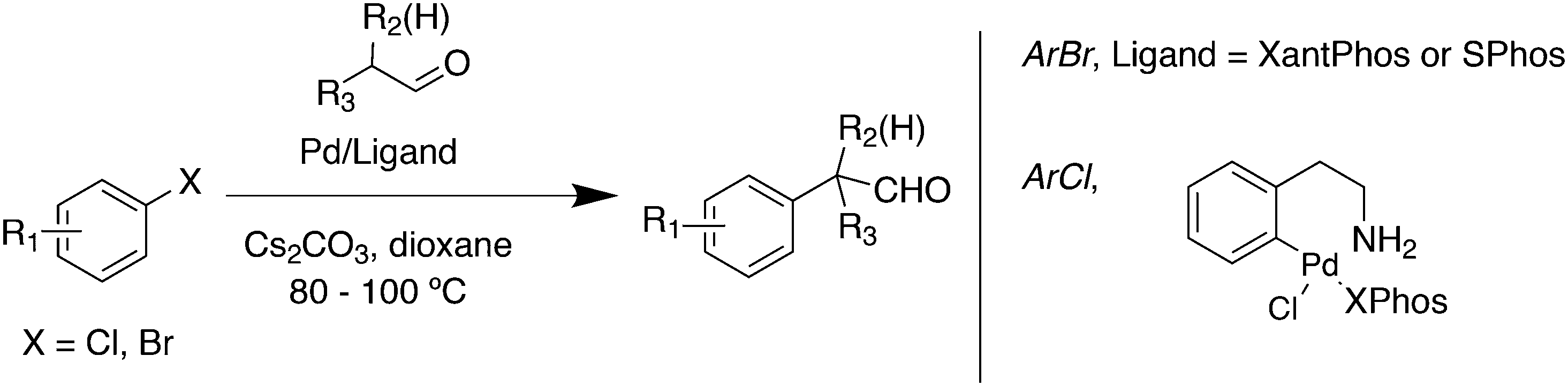 | ||
| Scheme 27 α-Arylation. | ||
An application of α-arylation involves the synthesis of Trileptal, a drug for the treatment of epilepsy, for which Xantphos is the preferred ligand (Scheme 28, protection, deprotection steps omitted).241 For the second coupling step, N-arylation, BINAP had to be used since Xantphos was completely inactive for this step.
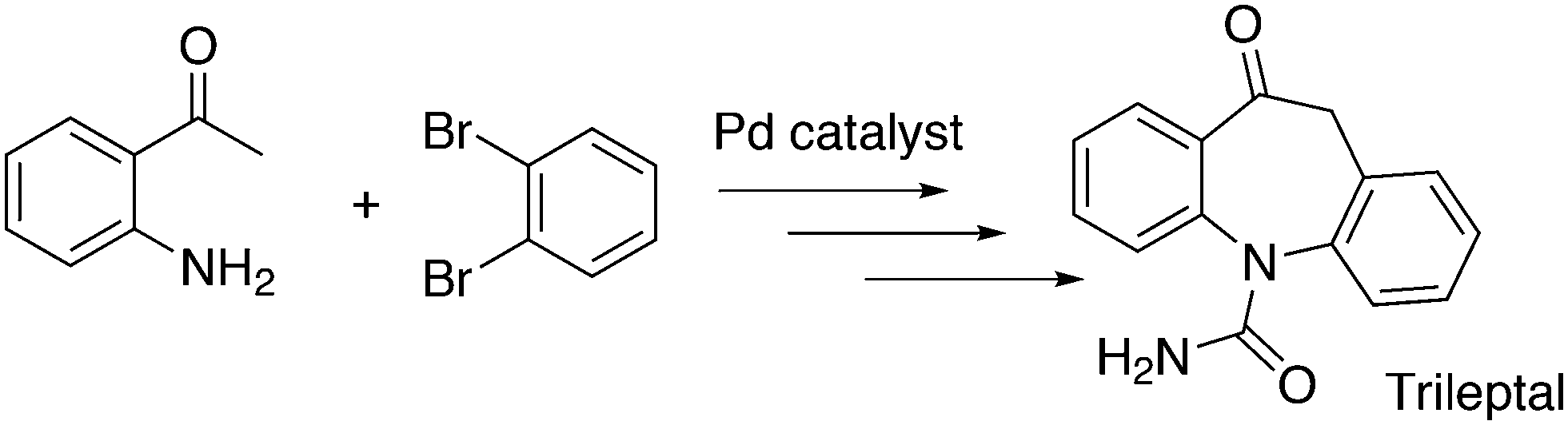 | ||
| Scheme 28 Sequence of α-arylation and N–C bond formation. | ||
Aliphatic ketone arylation as studied by Ackermann and Mehta focused on mono-arylation of simple ketones such as acetone (Scheme 29).242 For aryl imidazolylsulfonate 56 and acetone, monophosphines gave mostly mixtures of products. Bidentates such as dppe gave no conversion, while BINAP (80%), DPEphos (98%) and Xantphos (98%) worked very well. Competition experiments with tosylates and mesylates showed that imidazolylsulfonate was by far the most reactive of the three.
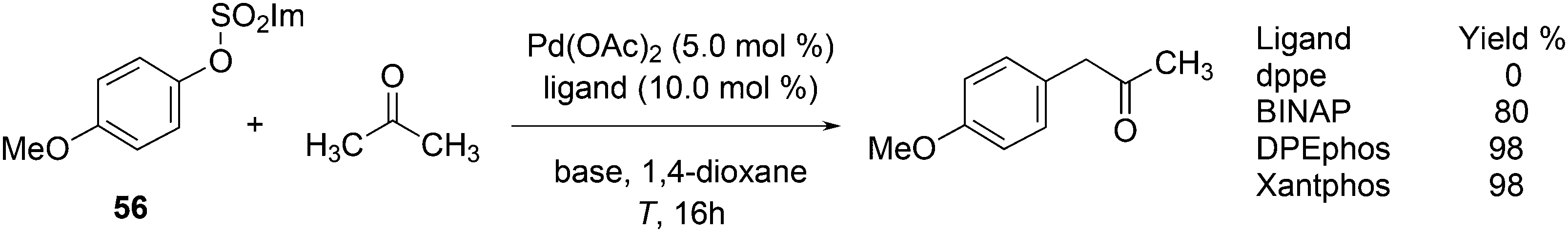 | ||
| Scheme 29 Aliphatic ketone arylation. | ||
α-Arylation of nitriles was reported by Wu and Hartwig (similar to Scheme 29) and both monophosphines and bisphosphines were investigated.243 Several bulky, electron-rich monophosphines gave good results, but Xantphos gave the highest conversion (95%) and the highest selectivity (100%) to monoarylation, a key problem for earlier catalytic systems.
Activation of sp3 C–H bonds was achieved by Martin et al. in o-tBu aryl bromides.244 It was assumed that oxidative addition of C–Br to Pd(0) takes place first and subsequently, C–H palladation occurs with the aid of pivalate on the nearby t-butyl group by Pd(II). The obtained metallacycle can undergo reductive elimination giving a benzocyclobutane, or the reaction can be terminated by the insertion of a carbene generated from precursor 57 and reductive elimination of a benzocyclopentane (Scheme 30).
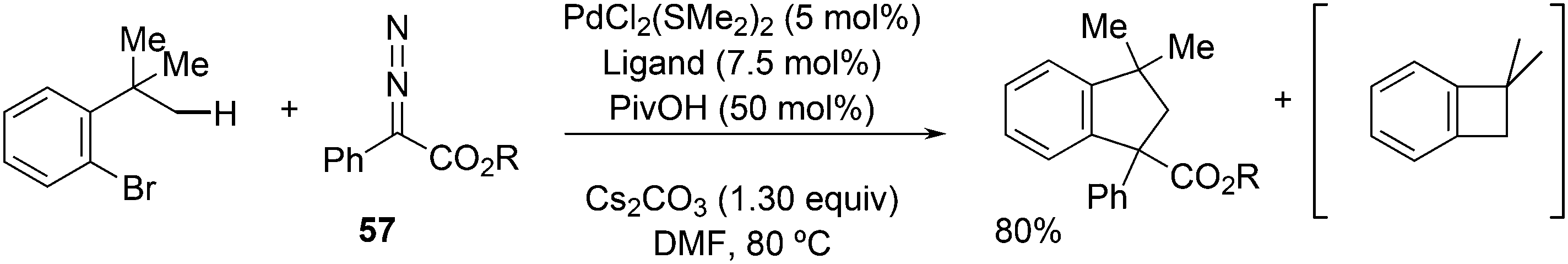 | ||
| Scheme 30 Three reactions in one. | ||
Bisphosphines such as Phanephos and the cyclohexyl analogue of DPEphos, which was better than DPEphos, were the preferred ligands, but surprisingly, Xantphos did not give any reaction under the circumstances used; thus, the reaction is susceptible to very small changes in the ligand.
Wolfe developed a new transformation for the conversion of 58 to cyclopentane-fused benzocyclobutenes 59via an unprecedented sequence of heteroatom-directed carbopalladation followed by intramolecular aryl C–H bond activation. DPEphos was the ligand of choice.245 The reaction started off as a Heck–Mizoroki reaction (Scheme 31); the source of side products was explained by an N–C cross-coupling reaction that started the sequence. The latter can be steered to high selectivity when NaOtBu is applied as a base, which generates the required amido-palladium intermediate.
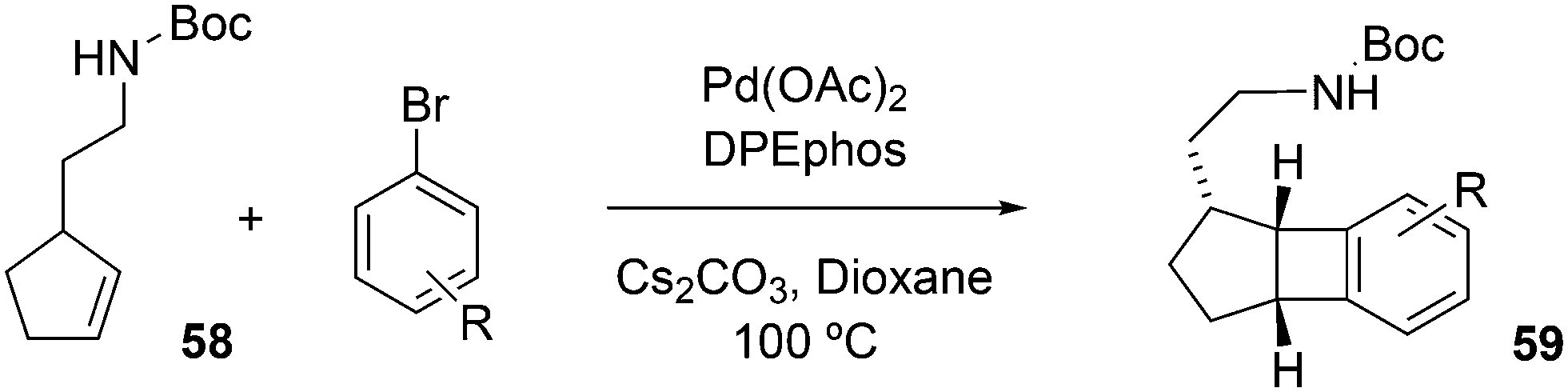 | ||
| Scheme 31 Heck–Mizoroki reaction followed by ring closure. | ||
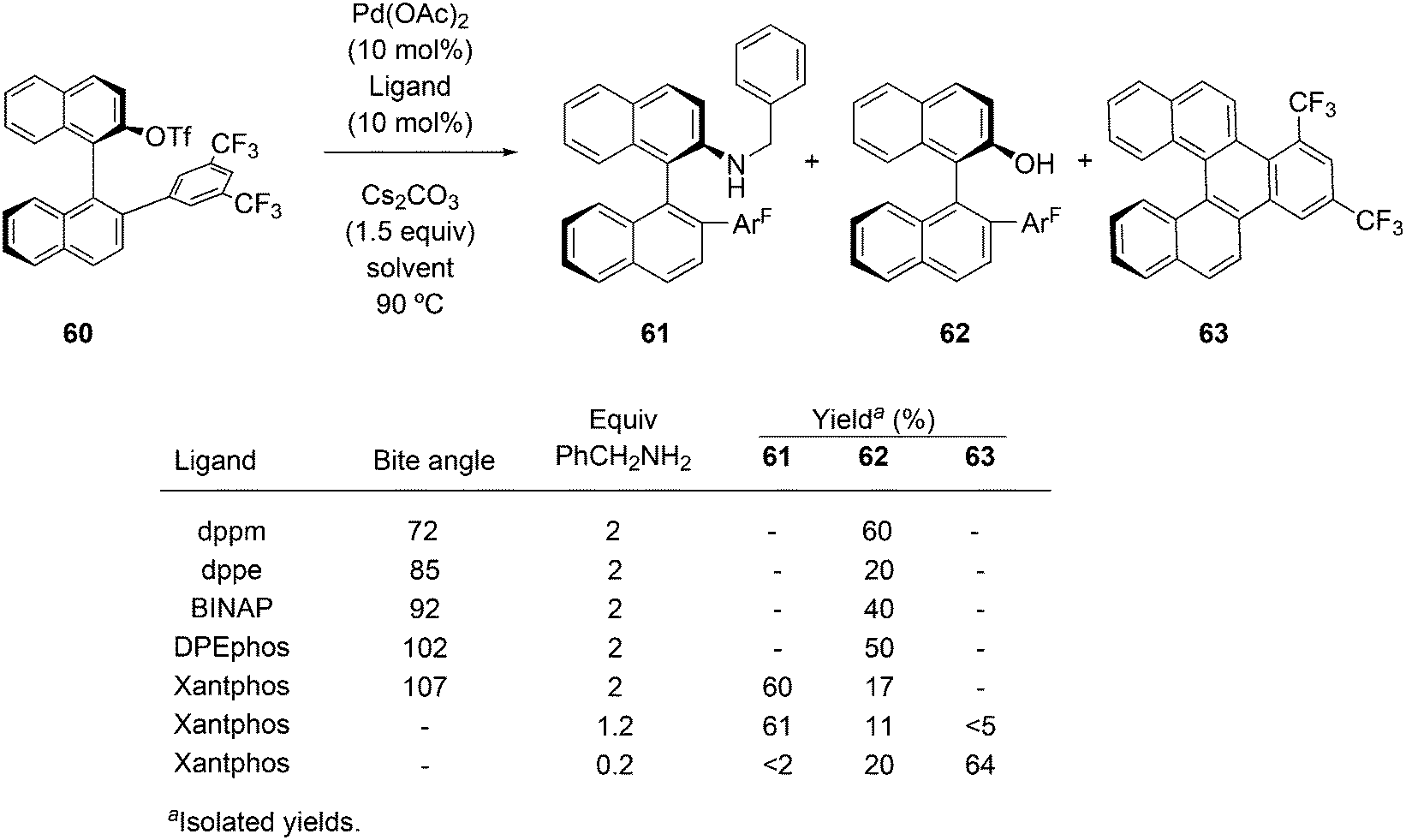
Of five bidentate ligands, Xantphos was the only one that gave the benzylamination (C–N bond formation) of a highly sterically hindered aryl triflate 60 to give 61 (Scheme 32).246 A binaphthyl triflate derivative with no 2′-substituent was effectively aminated using DPEphos in combination with Pd(OAc)2 and thus, the use of Xantphos is not necessary in the absence of steric bulk at the 2′-position. By lowering the amount of benzyl amine and changing the reaction conditions, the reaction could be directed to C–H substitution and C–C bond formation. By changing the base, the yield of 61 could be raised to 85%.
 | ||
| Scheme 32 Amination of a hindered binaphthyl triflate. | ||
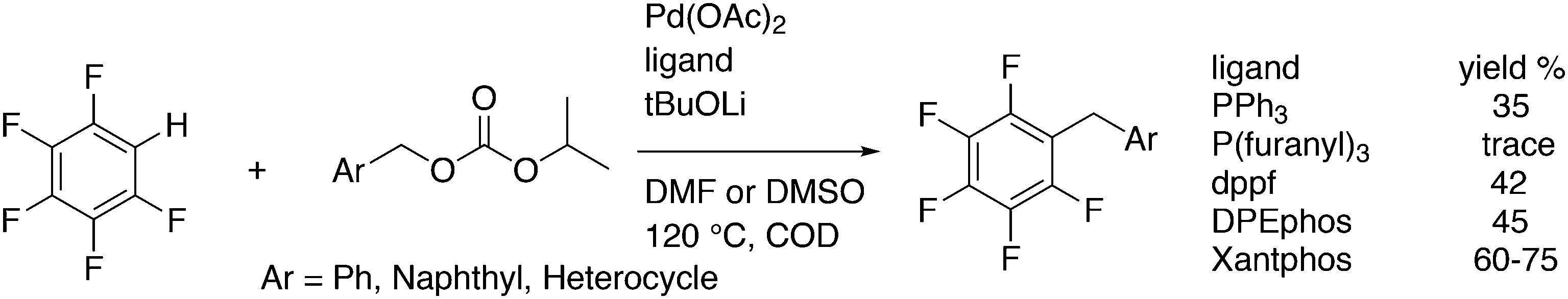
Yu and co-workers reported on a more demanding benzylation reaction, viz. the direct C–H substitution of polyfluoroaromatics, Scheme 33.247 Xantphos was found to be the best ligand, although 5% catalyst and 120 °C for 8 h were still needed to obtain yields up to 85%. Methoxy substituents on the benzyl group gave the highest yield.
 | ||
| Scheme 33 Benzylation of fluoroaromatics. | ||
3.3.1.9. Fluoroalkyl introduction. Addition of trifluoromethyl via organometallic catalysts has in recent years become an important topic of study.248 In palladium catalysed cross-coupling chemistry, the reductive elimination is extremely slow because of the exceptional strength and inertness of the Pd–CF3 bond. In sharp contrast to this, analogous methyl complexes give toluene via reductive elimination, even below room temperature. In 2006, a breakthrough was achieved by Grushin and Marshall when they showed that [(1)Pd(Ph)(CF3)] undergoes facile CF3Ph elimination at 50 °C.249 Buchwald et al. reported the first catalytic trifluoromethylation of chloroarenes with palladium and the monodentate ligands RuPhos and BrettPhos.250 Anstaett and Schoenebeck looked at the Xantphos system with DFT, while also comparing it with a small bite angle ligand such as dppe, and their PH2 and PMe2 analogues.251 They concluded that steric interaction in the starting Pd(II) complex raised the energy and that in the transition state the interaction was relatively lower than that in the small bite angle ligands. For ligands with small substituents on phosphorus, they showed that there was no predictable bite angle effect; e.g. for PH2 substituted Xantphos and dppe, nearly identical barriers were calculated. The elimination of CH3Ph is much faster for both ligands, with the barriers being some 10 kcal mol−1 lower, which was assigned to the lower costs of reorganization before reductive elimination can take place for the more weakly bound methyl groups. Grushin and co-workers studied the kinetics of their system in detail and also performed DFT calculations.252 Their study included the P-iPr2 analogue of Xantphos. The experimentally determined activation parameters (ΔH≠ = 25.9 kcal mol−1; ΔS≠ = 6.4 e.u.) are in excellent agreement with the computed data (ΔH≠calc = 24.8 kcal mol−1; ΔG≠calc = 25.0 kcal mol−1). The i-Pr derivative is slightly more stable, but decomposes less selectively than Xantphos and also forms a trans complex, which has to rearrange to the cis form before reductive elimination can take place. They considered more mechanisms than in the study mentioned above and for complexes containing two ligands, but the conclusions from the DFT studies are basically the same. The analogous Ni complex did not undergo reductive elimination of CF3Ph, and related complexes with other ligands underwent α-F elimination.253 Schoenebeck and co-workers extended their work in an interesting direction as they took an electron-poor ligand with a small bite angle, 1,2-bis(trifluoromethylphosphino)ethane.254 Computations had shown that this ligand should also facilitate the reductive elimination of PhCF3 in spite of the small bite angle. Not unexpectedly perhaps, since oxidative addition and reductive elimination are the best “behaved” organometallic elementary steps, regarding electronic and steric effects, cf. hydrocyanation, for instance.255 Thus, having made the ligand and complex in excellent yields, they observed a clean formation of CF3Ph at 80 °C, as predicted. The calculations showed that not only the electron-withdrawing power of P–CF3 was important, but also the electrostatic repulsion of the ligand and the fragments of the product to be formed.
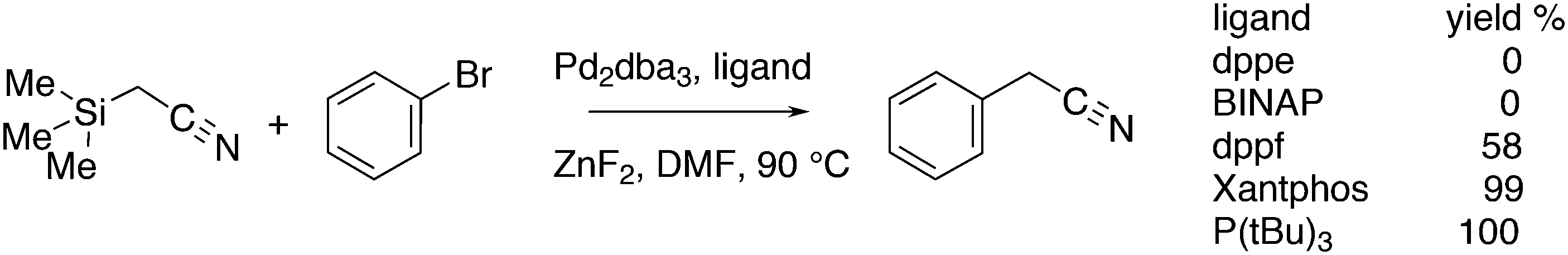
Introduction of CF2H groups via cross-coupling is, similar to CF3 introduction, not an easy process. Zhang and co-workers made a contribution to solve this, using BrCF2CO2K as the difluoromethyl source and an arylborylate as the acceptor.256 Thus, a complicated reaction mixture gave as high as 85% isolated yield for the substitution of the borate ester for the CF2H group, see Scheme 34.
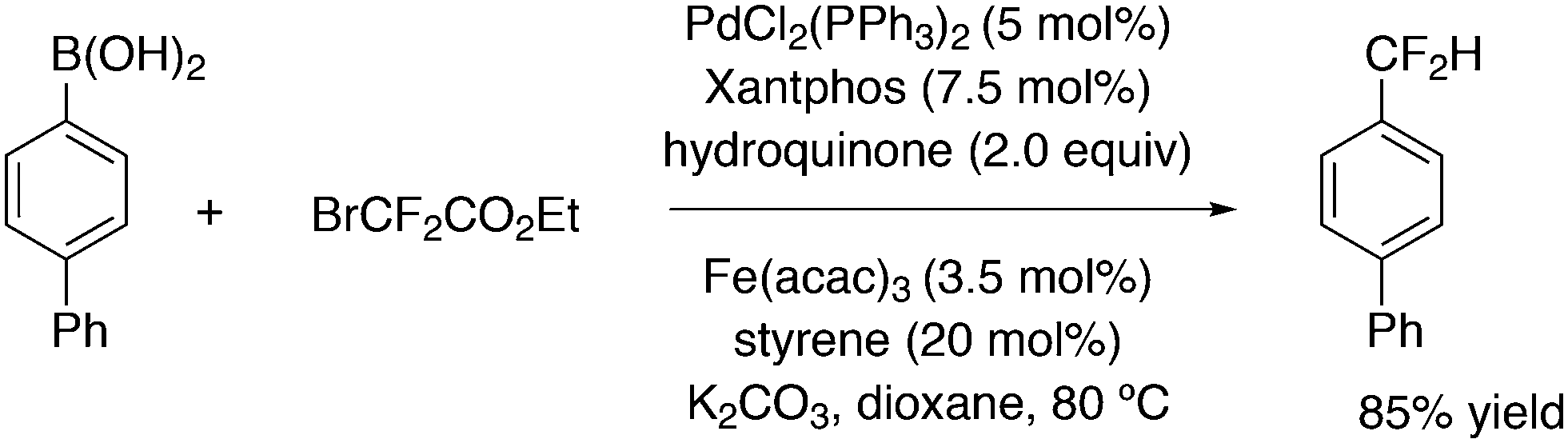 | ||
| Scheme 34 Introduction of CF2H groups. | ||
Scheme 35 shows the role of the additives in this ingenious system and two possible pathways; hydroquinone provides the proton that is needed and quinone reoxidizes Pd(0) to Pd(II). The role of the catalyst components could not be explained in detail, but apparently, only Xantphos led to active systems.
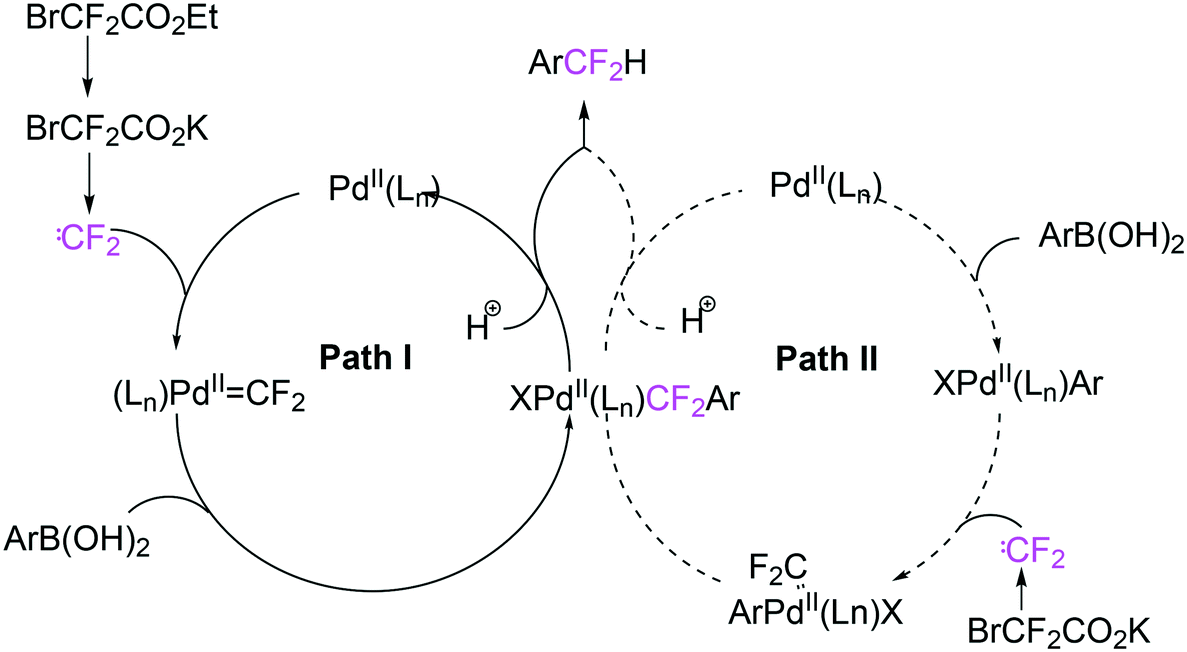 | ||
| Scheme 35 The role of the additives in the introduction of CF2H groups. | ||
Zhang also reported a radical addition reaction with this substrate and Xantphos/Pd as the catalyst (Scheme 36).257 The only ligand used was Xantphos, but palladium precursors containing several ligands, including the phosphines tpp or dppf, were used; in the presence of tpp the highest yields were obtained.
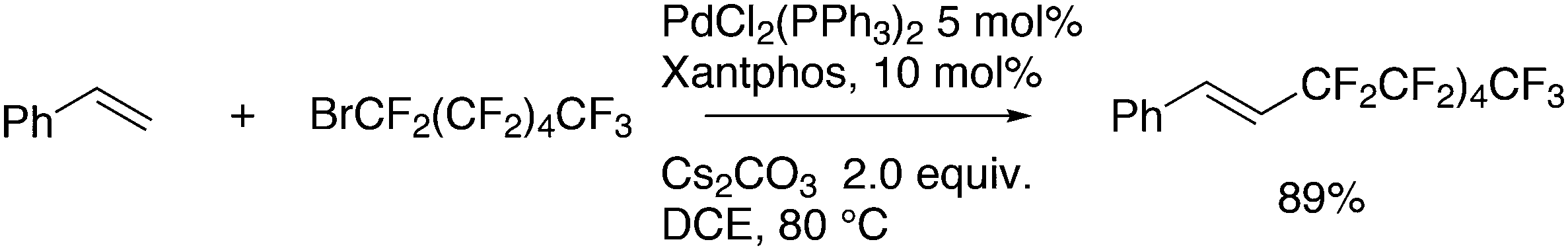 | ||
| Scheme 36 Alkene H-substitution by BrC6F11. | ||
Wang and co-workers described another addition reaction utilizing BrCF2CO2Et, which in this case also remains intact (Scheme 37).258 DPEphos, PdCl2(PPh3)2 and K2CO3 gave the best result. When TEMPO was added as a radical scavenger the yield dropped to zero and thus, this metal catalysed reaction involves a radical step, which might be an atom transfer of Br to give a Pd(I) intermediate, as Pd(0) was supposed to be the catalyst.
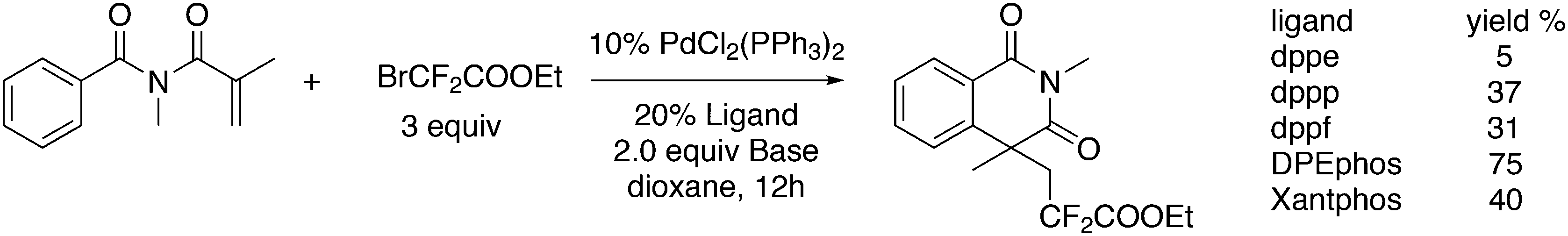 | ||
| Scheme 37 Ring closure by CF2COOEt radicals. | ||
A recent example of a radical addition of an iododifluorocarboxylic ester is shown in Scheme 38.259 The best catalyst system was based on PdCl2(PPh3)2, Xantphos, Cs2CO3 in dioxane (78%), while DPEphos comes second (58%). For pentafluoro-iodoethane, BINAP resulted as the ligand giving the highest yield. Palladium(0) can act as a catalyst by accepting an iodide radical; the organofluoro radical attacks the alkene system, and the resulting radical abstracts iodide from Pd(I)I, similar to other known catalysts based on Ru, Ni, etc. Pd(0) can also act as the radical initiator. It is not clear why this reaction depends so strongly on the ligand used.
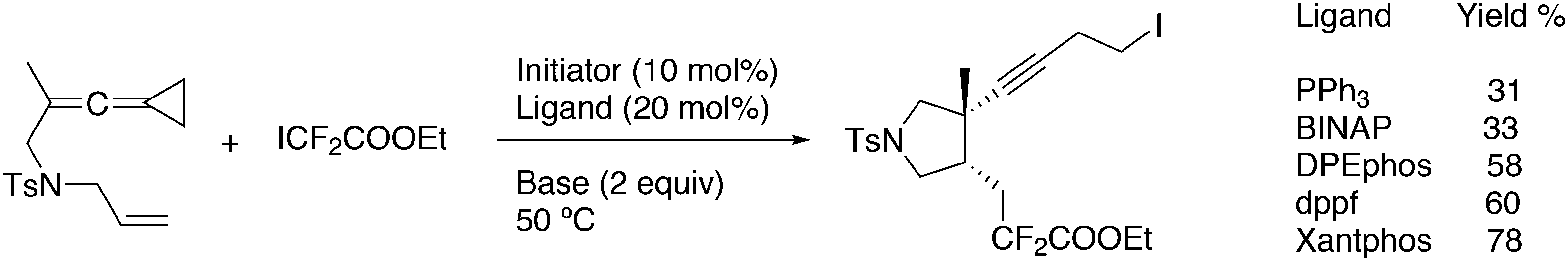 | ||
| Scheme 38 Radical addition of ICF2COOEt. | ||
3.3.1.10. Introduction of cyanide. Conversion of aryl halides into nitriles is an important reaction that received a lot of attention in the last few decades and efficient protocols are now available.260
For the industrial synthesis of a pharmaceutical reagent (citalopram, Scheme 39), Pitts and co-workers optimised the microwave enhanced palladium catalysed cyanation procedure for the final step of a production method for citalopram.261 Using Xantphos as the ligand (0.1%) yields of 99% were reported. The process cut the reaction time from 24 h to 2 min.
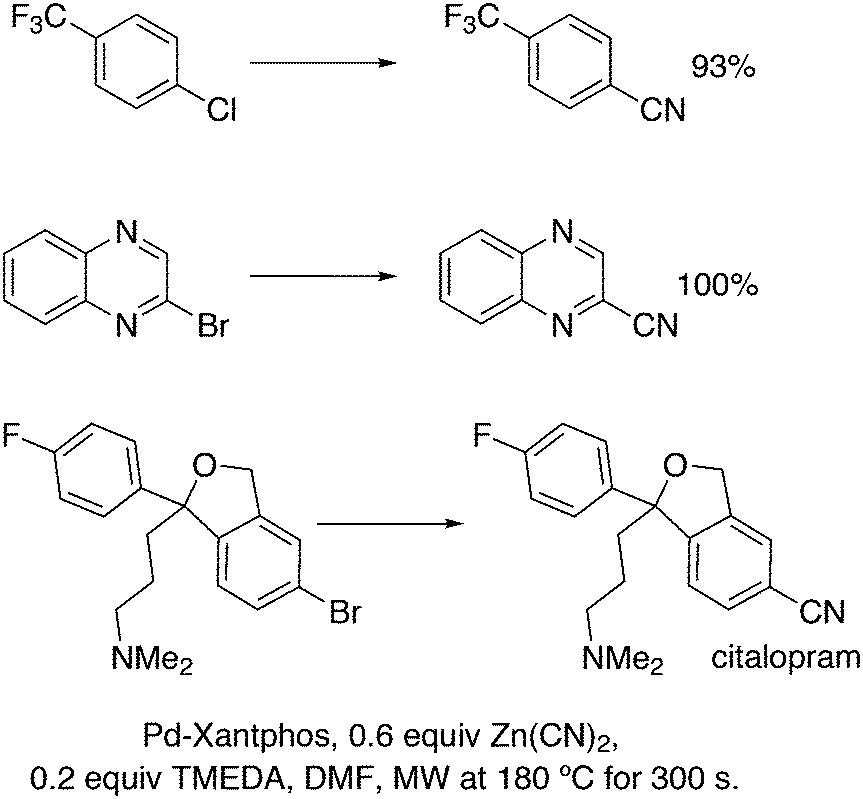 | ||
| Scheme 39 Cyanation of aryl halides. | ||
Intramolecular oxycyanation of alkenes was achieved by palladium and BPh3 catalysts through the cleavage of O–CN bonds and the subsequent insertion of double bonds (Scheme 40). The use of Xantphos as a ligand for palladium is essential for obtaining a transformation with high chemo- and regioselectivity.262 The cleavage of the O–C bond of the cyanate is a key step in this new reaction.263
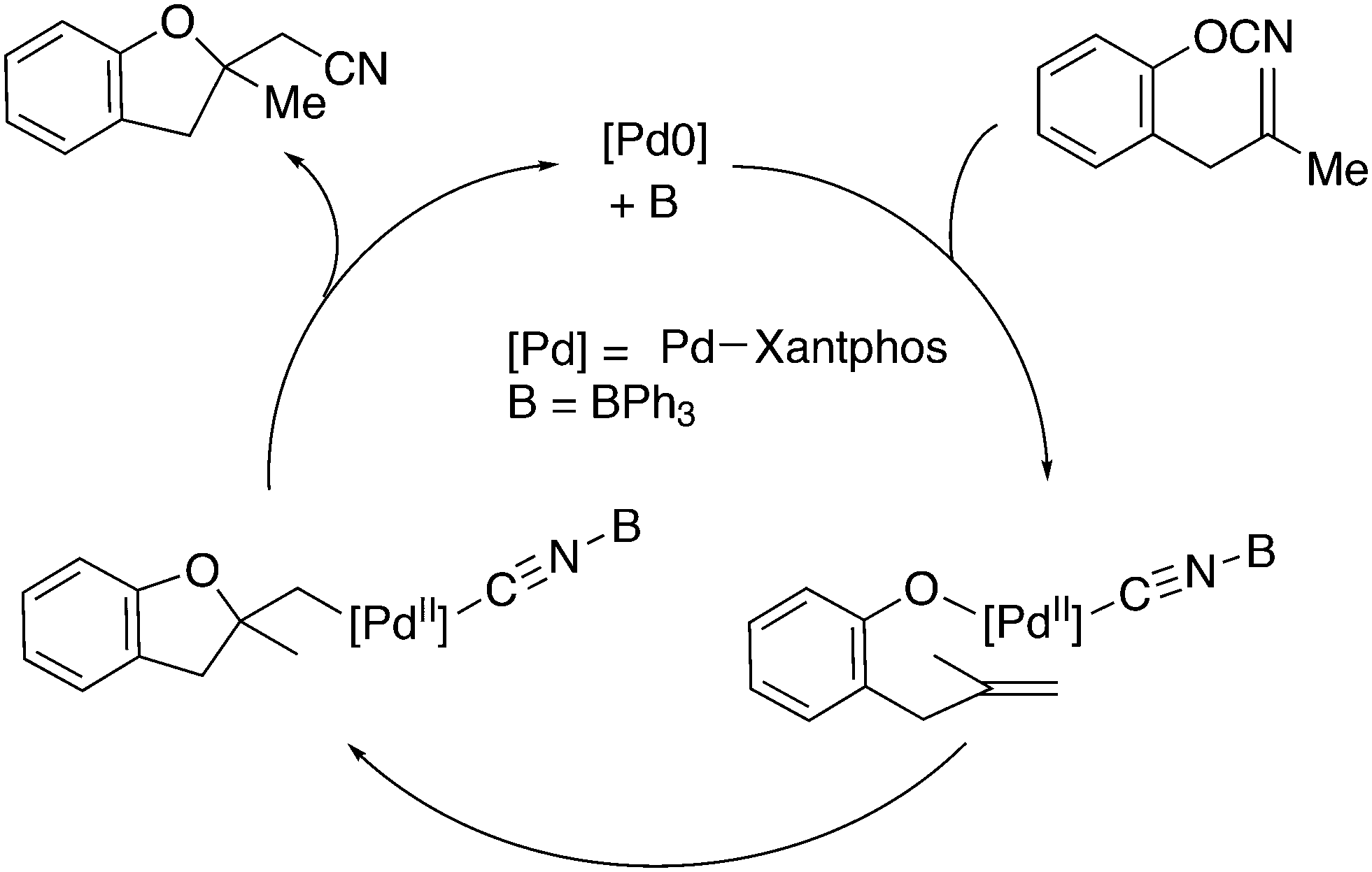 | ||
| Scheme 40 Oxycyanation of alkenes. | ||
An interesting analogue of this reaction is the aminocyanation of alkenes and alkynes reported by Nakao et al. (Scheme 41).264 The reaction was carried out with the same catalyst system and this time the sequence started with an oxidative addition of the cyanamide to Pd(0). For the BOC protected amine the yield was quantitative.
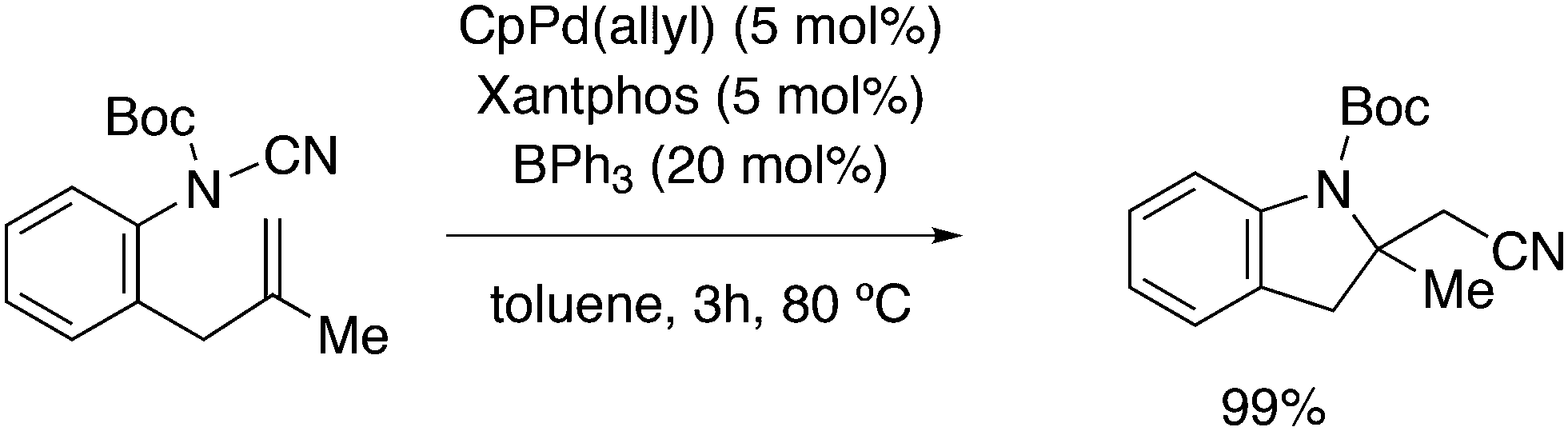 | ||
| Scheme 41 Aminocyanation of an alkene. | ||
3.3.1.11. Reactions of aminals. Palladium complexes are often reactive toward vinyl ethers, acetals, aminals, imines, and enamines, but selectivity for desired conversion is often an issue. Huang and co-workers introduced a novel, highly selective palladium-catalysed vinylation reaction for the direct synthesis of allylic amines from styrenes and aminals.265 Mechanistic studies showed that the reaction is an aminomethylation and thus involves a C–C bond formation, with an amine as a co-product. The mechanism is illustrated in Scheme 42. First a reduction of Pd(triflate)2 by isopropylalcohol takes place, followed by an oxidative addition of the iminium ion formed by the release of amine from the protonated aminal. The iminium ion undergoes an Umpolung to an aminomethyl group and for this palladacycle an X-ray structure was reported. Bidentate phosphines are needed for this reaction and Xantphos (up to 93%) outperformed dppf (17%) and BINAP (30%).
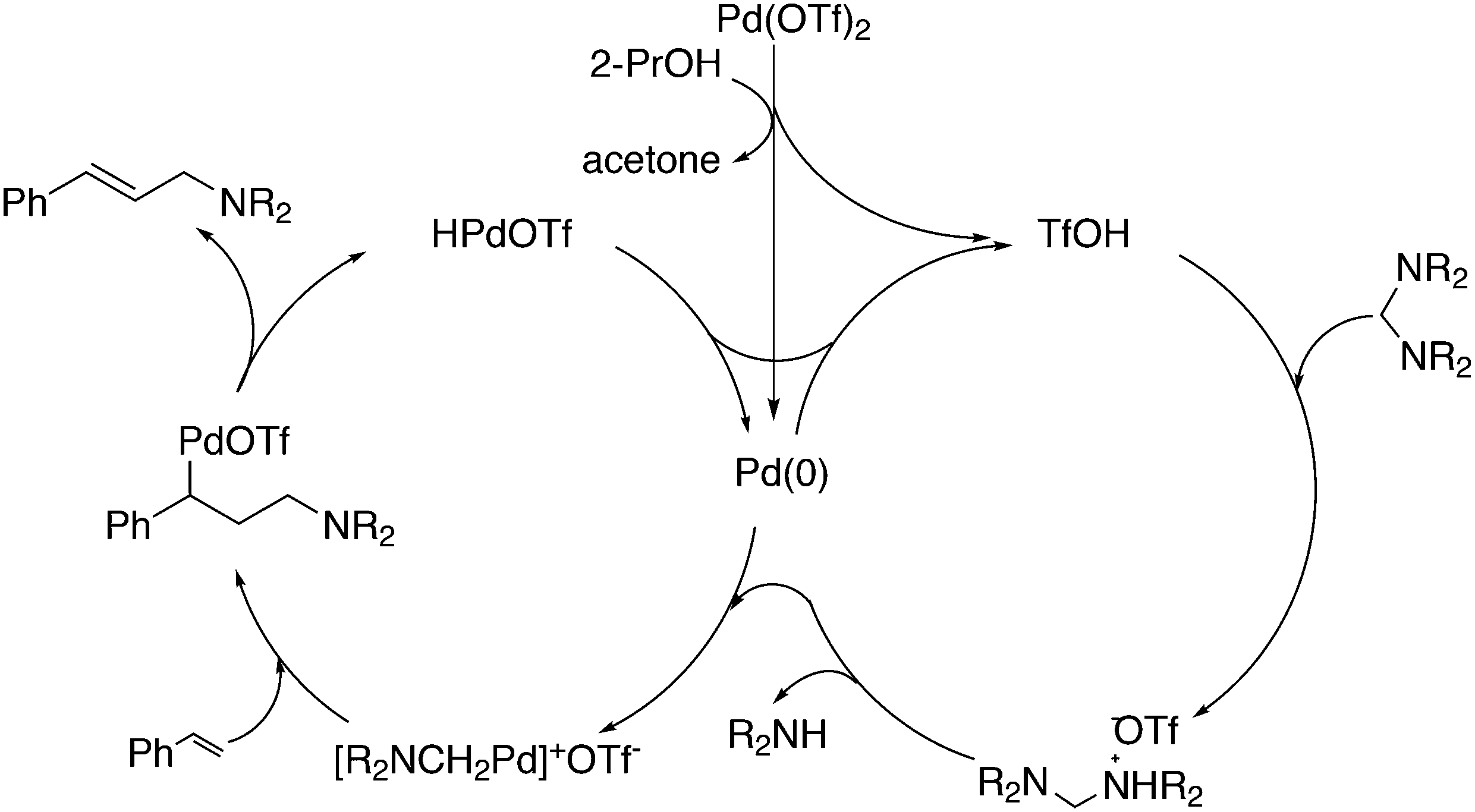 | ||
| Scheme 42 Mechanism of aminomethylation with aminals. | ||
Huang and co-workers also developed a method for the palladium catalysed addition of iminium ions to vinyl ethers by studying a range of phosphorus ligands.266 The basic reaction scheme is presented in Scheme 43. Some typical results from the ligand screening are PPh3 (traces), BINAP (59%), dppe (50%), DPEphos (55%), dppf (56%), and Xantphos (85%). The ligands and Pd were administered as the ligand complex of PdCl2. The scope of the reaction was developed using Xantphos. The key intermediate of palladium is the η2-bonded aminomethyl-Pd(II) complex or iminium-Pd(0) complex 64. Frontier orbital analysis says that the latter description is more appropriate.267 The computational results revealed that Xantphos aids in forming iminium-coordinated palladium complexes, promotes the reductive elimination step of aminomethylation, and stabilizes Pd(0) species.
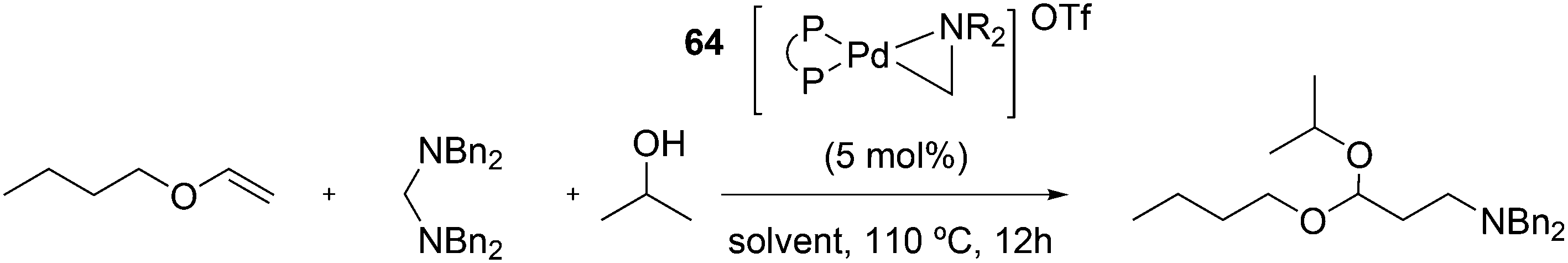 | ||
| Scheme 43 Aminomethylation of vinyl ethers. | ||
The subsequent invention by Huang using Pd/aminals involves the formation of a C–C bond and a C–N bond (section 3.4), but it is preferred to include it in this context. The reaction is a palladium catalysed methylamination and amination of an allene 65, and now the complete aminal molecule is incorporated in the product (66, Scheme 44).268 Most ligands gave only traces of the product and only Xantphos/Pd applied as the triflate gave good yields, although on average 20% of an allylamine 67 was formed as well. Many allenes reacted this way. A palladium π-allyl complex is the crucial intermediate, which undergoes an allylic substitution.
 | ||
| Scheme 44 Methylamination/amination of an allene. | ||
3.3.1.12. Tandem reactions. The formation of an aryl–C bond and a C–O bond in one reaction sequence was reported by Hay and Wolfe; Scheme 45 shows the stereoselective synthesis of isoxazolidines through the Pd-catalysed carboetherification of n-butenylhydroxylamines.269,270 DPEphos is the preferred ligand. Similar reactions for Pd-alkoxy compounds have been reported.
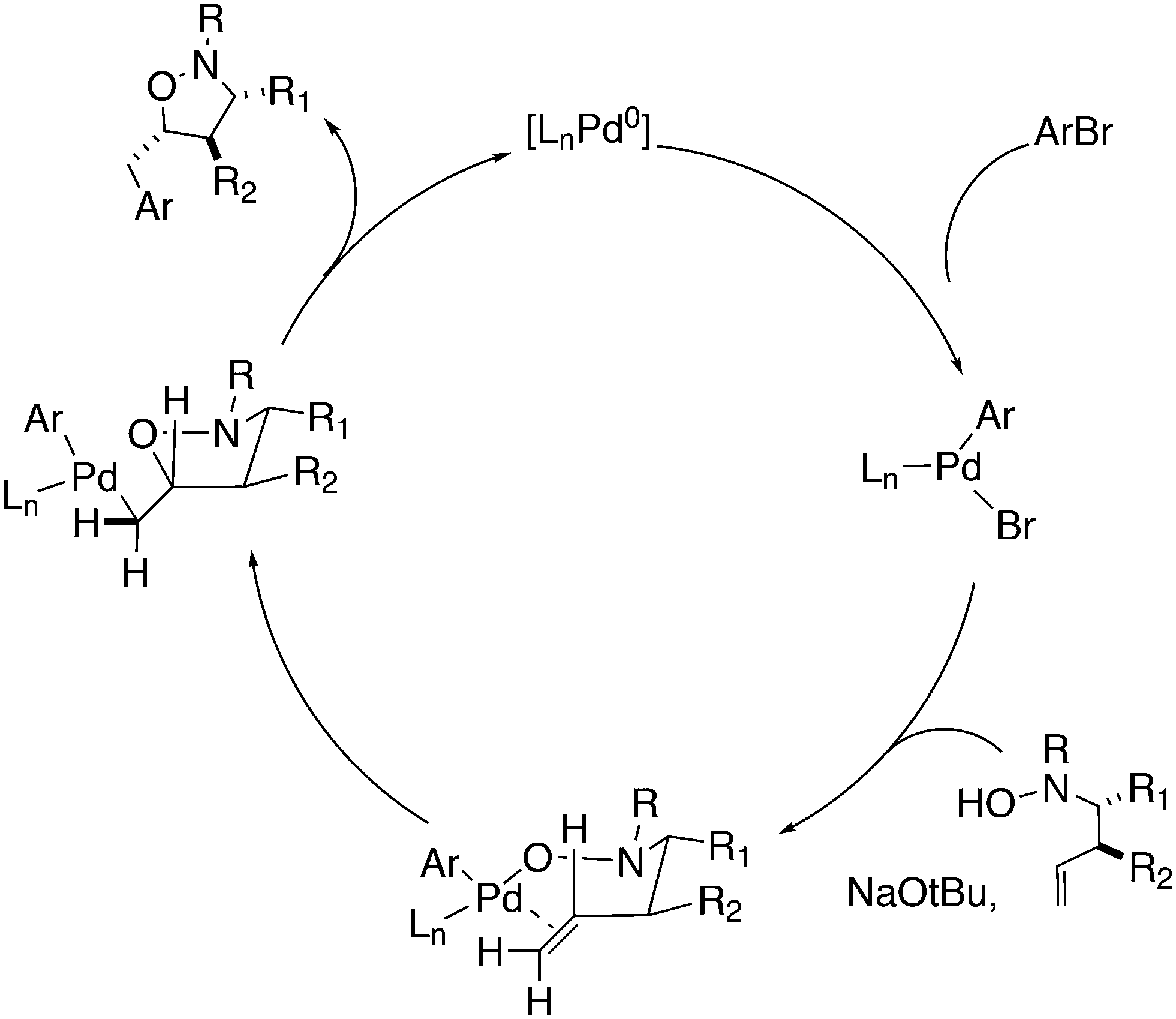 | ||
| Scheme 45 Tandem formation of isoxazolidines. | ||
Chen et al. extended this reaction to oxims, Scheme 46.271 The ligand effect was the same for this reaction. The isoxazolines were obtained in good yields. The use of Xantphos as ligand is crucial for the transformation. The carboetherification products can be further converted to hydroxy ketones in the presence of Fe powder and NH4Cl.
 | ||
| Scheme 46 Carboetherification of n-butenylhydroxylamines. | ||
Sharma and Van der Eycken reported a remarkable many component reaction with a palladium catalyst and a variety of ligands.272 They reacted a mixture of a 1,2-haloaromatic aldehyde, an amine, an isonitrile, and a substituted propiolic acid in one pot to give functionalized spiro[indoline-3,2′-pyrrole]-2,5′-diones in what they called a Post-Ugi domino Buchwald–Hartwig/Michael reaction, see Scheme 47. The alkynoic acids were also replaced with α,β-unsaturated acids leading to substituted spirooxindoles. There is a variety of pharmaceutically interesting compounds that contain a spirooxindole framework. The well-known monophosphines and bisphosphines such as dppf and DPEphos gave no or low yield and only Xantphos gave yields up to 65%. A plausible mechanism was presented, but like the authors we also refrain from speculations on the ligand effect.
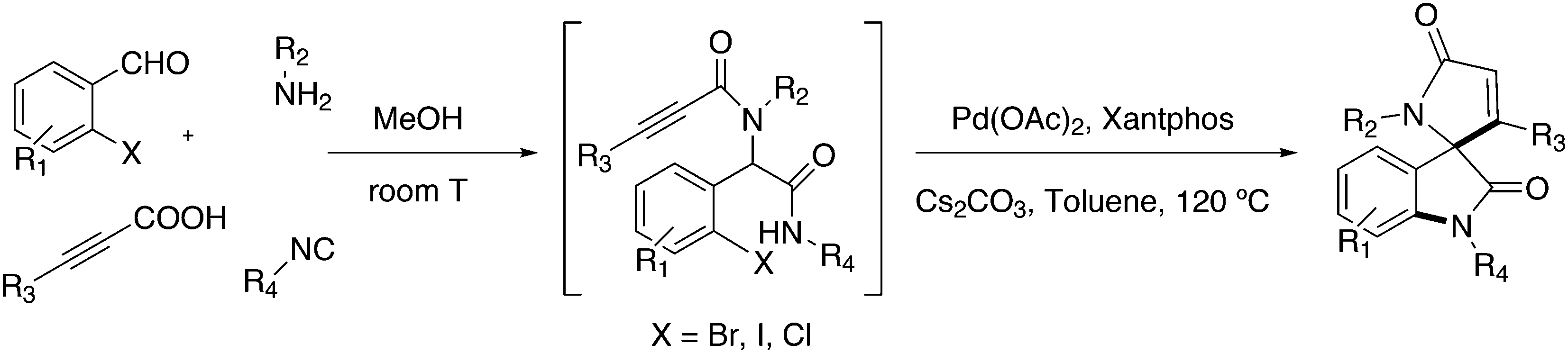 | ||
| Scheme 47 Multicomponent condensation. | ||
Ichikawa and co-workers reported on a tandem Tsuji–Trost and Heck–Mizoroki cyclization reaction with the introduction of a CF2 fragment into a heteroaromatic compound (Scheme 48).273 An excellent regioselectivity for the first step can be obtained with Pd/Xantphos and LiHMDS as the base. For the ring closure, palladium acetate was added to the isomeric mixture obtained after aqueous workup with the separation of the isomers. The methyleneindoline products can be easily derivatized further.
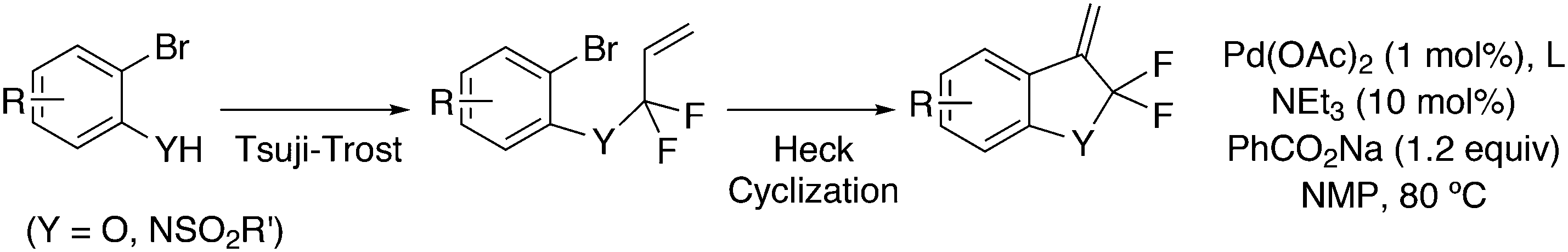 | ||
| Scheme 48 Tandem Tsuji–Trost and Heck–Mizoroki cyclization. | ||
Tandem reactions using two metals have also been reported, for example a rhodium-BINAP catalysed hydroarylation of an alkyne followed by a C–N bond formation catalysed by palladium and Xphos.274 In this instance BINAP is the exceptional ligand for the Rh/hydroarylation, dppp gave modest conversion, but Xantphos 0%.
In the last two decades Wolfe and his group have developed a large body of catalytic reactions centred around Pd/DPEphos, and Scheme 49 shows a recent example of a stereo-controlled synthesis of polycyclic heterocycles via a cascade Pd-catalysed alkene carboamination/Diels–Alder reaction sequence. The transformations effect the formation of 4 bonds, 3 rings, and 3–5 stereocentres.275
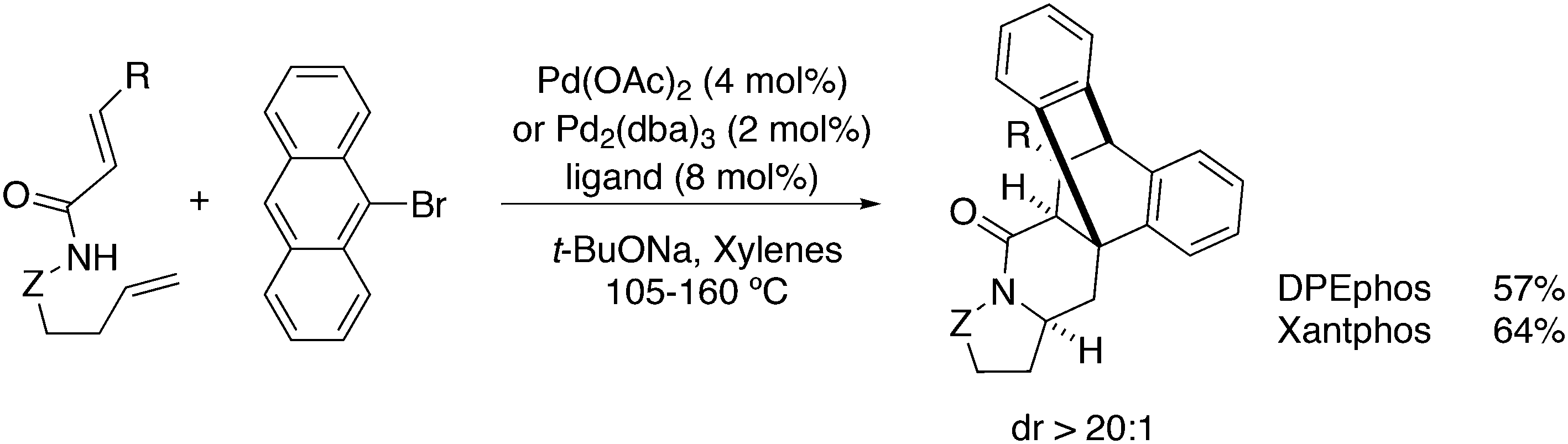 | ||
| Scheme 49 Multistep reaction sequences using DPEphos and Xantphos. | ||
Spiroindole formation can be accomplished by ring closure reactions of bromoarenes connected to furans and completed by alkoxy groups as shown in Scheme 50. As demonstrated by Yin et al., the yield increased for the intramolecular alkoxylation in the series dppp (40%), dppf (47%), DPEphos (55%) and Xantphos (84%).276 The authors suggest that this protocol likely involves an intramolecular dearomatizing Heck-type α-arylation of the furan ring to produce a cyclic allylic palladium intermediate and the subsequent intra- or intermolecular introduction of an alkyloxyl group at the other α-position of the furan ring, with opposite facial selectivities.
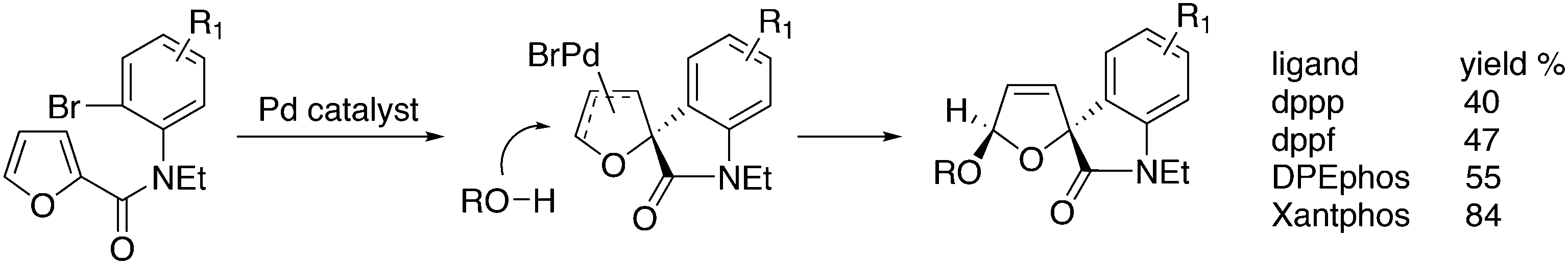 | ||
| Scheme 50 Spiroindole formation by cross-coupling. | ||
3.3.1.13. Nixantphos reactions. Nixantphos was first reported in 2000 by Van der Veen et al. and thus it was not included in the initial screening of C–C (>1993) and C–N cross-coupling reactions (>1998) in the 1990s. The first direct 2-arylation of benzoxazoles by Pd/Nixantphos was reported by Walsh and co-workers.277 In 2014, Walsh and his group discovered an important new feature of Nixantphos for reactions in the presence of strong bases, the C–H arylation of diarylmethanes.278 Hitherto, none of the many monophosphines or bisphosphines containing only aryl groups yielded an active catalyst for reactions with aryl chlorides. The less reactive chloride substrates needed ligands with strongly donating i-propyl or t-butyl groups to facilitate the oxidative addition of the aryl chloride to the ligand-Pd(0) intermediate. They discovered that Nixantphos under certain cross-coupling reaction conditions would be deprotonated and in this anionic form, the Pd complex undergoes oxidative addition of unactivated aryl chlorides, even at room temperature. Evidently, the deprotonated phenoxazine backbone (pKa = 22) might give rise to a highly electron-rich bisphosphine capable of inducing oxidative addition of aryl chlorides at Pd(0). The first reaction they reported involved the deprotonative cross-coupling process (DCCP), shown in Scheme 51, in which KN(SiMe3)2 serves as the base deprotonating the diphenylmethane substrate, and importantly, also the phenoxazine ligand backbone. However, DFT calculations showed that Nixantphos gained hardly any electron donicity upon deprotonation and that alkyl phosphines were more electron-rich than deprotonated Nixantphos. It was suggested that perhaps the neighbouring K–N ion pair catalyses the cleavage of the C–Cl bond. The resting state of the catalyst is the Pd-bis-Nixantphos dianionic species and thus, either the dissociation of one ligand or the oxidative addition is rate-limiting.
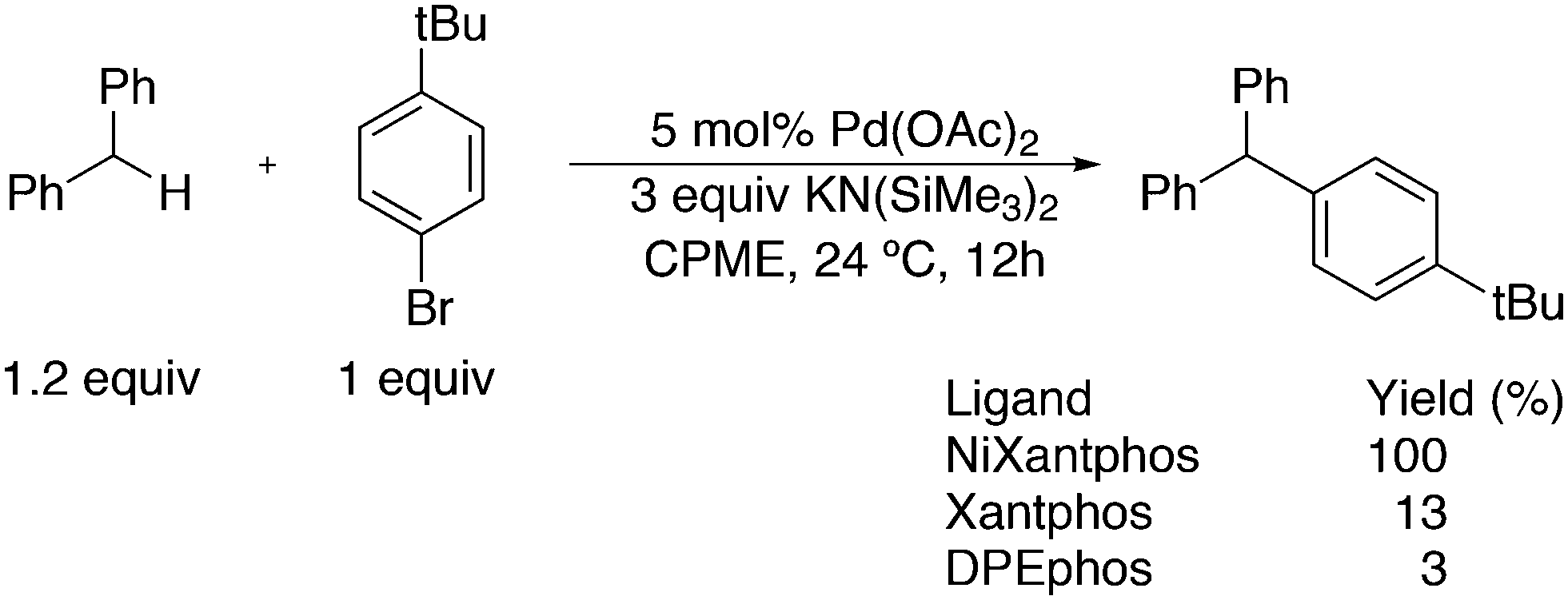 | ||
| Scheme 51 Deprotonative cross-coupling. | ||
The second example concerns the palladium-catalysed cross-coupling of aryl bromides and 2-aryl-1,3-dithianes, an Umpolung version of arylaldehydes, by Schmink and co-workers (Scheme 52).279 As in Walsh's work, neutral DPEphos or Xantphos gave very low or no conversion.
 | ||
| Scheme 52 C–H arylation of 1,3-dithianes. | ||
A room temperature Suzuki–Miyaura reaction can be carried out between aryl borates and a large variety of aryl chlorides with the new catalyst system, as was reported by Gao and co-workers.280Scheme 53 gives the conditions and yields for 68a–c.
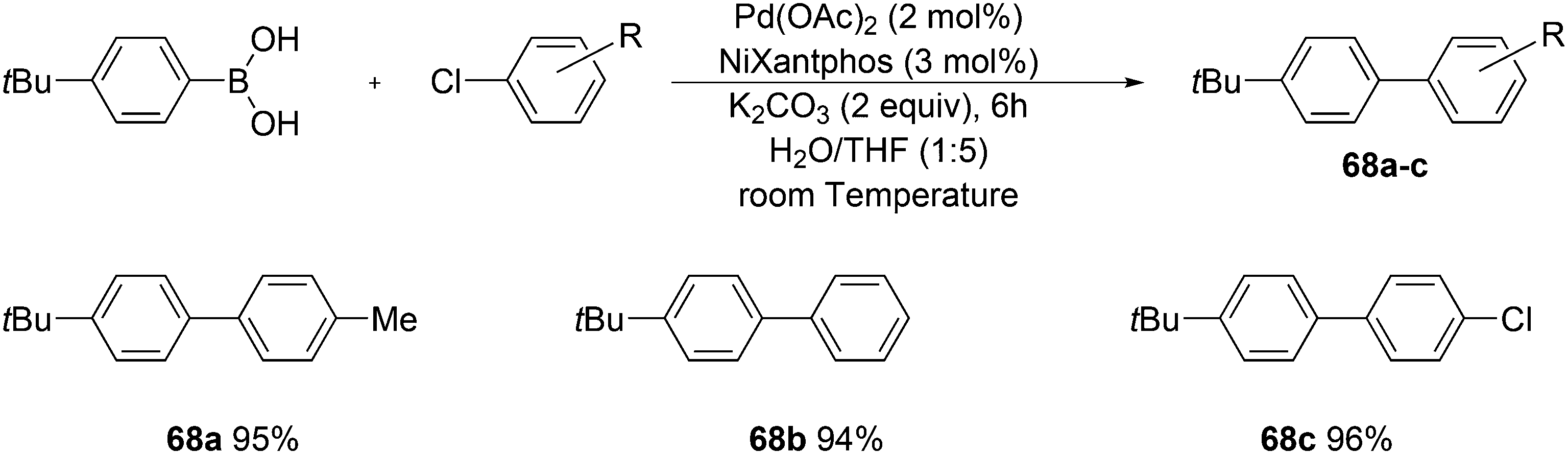 | ||
| Scheme 53 Suzuki–Miyaura reaction with Pd–Nixantphos. | ||
Direct C–H functionalization of azaarylmethylamines to produce aryl(azaaryl)methylamines without directing groups was described with the use of a Pd(OAc)2/Nixantphos-based catalyst that couples the resulting carbanions with many aryl halides (Scheme 54).281
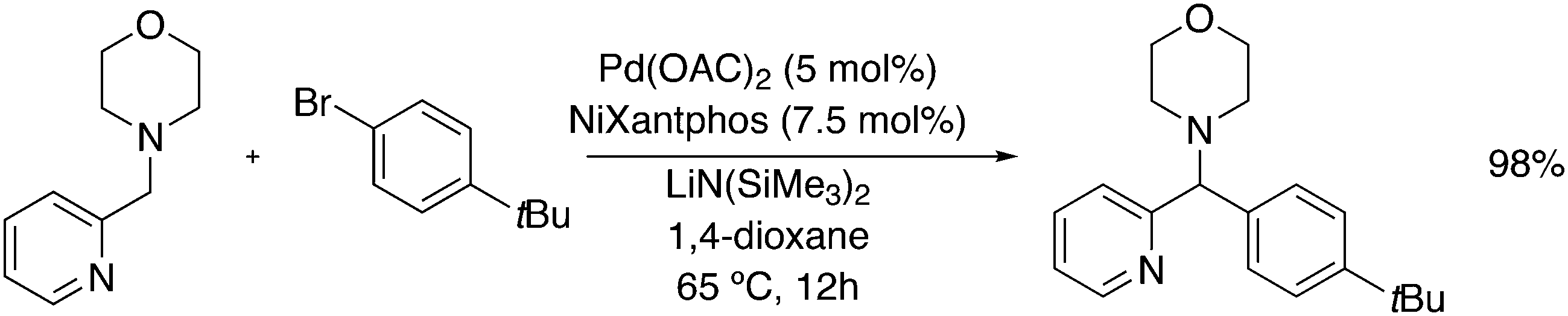 | ||
| Scheme 54 C–H functionalization of azaarylmethylamines. | ||
Pyridylmethyl silyl ethers undergo the DCCP reaction with this catalyst system (Scheme 55).282 A convenient one-pot cross-coupling/desilylation sequence was demonstrated to directly obtain arylated secondary alcohols. Vinyl bromides were also successfully used in the palladium-catalysed selective α-alkenylation of pyridylmethyl ethers.283
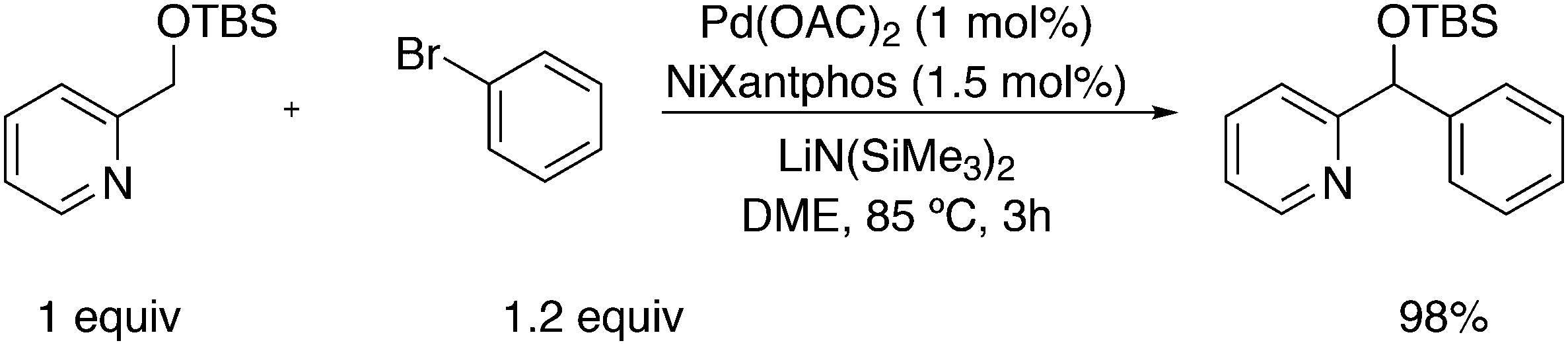 | ||
| Scheme 55 Pyridylmethyl silyl ethers in DCCP reaction. | ||
N-Benzyl-1,1-diphenylmethanimines can be arylated at the methylene group with aryl bromides with basically the same catalyst system, Scheme 56.284 The reaction takes place via 1,1,3-triaryl-2-azaallyl anions, which are selectively arylated at the 3-position. Aryl chlorides were not reactive under these conditions. After hydrolysis, diarylmethylamines are obtained while benzophenone can be recycled.
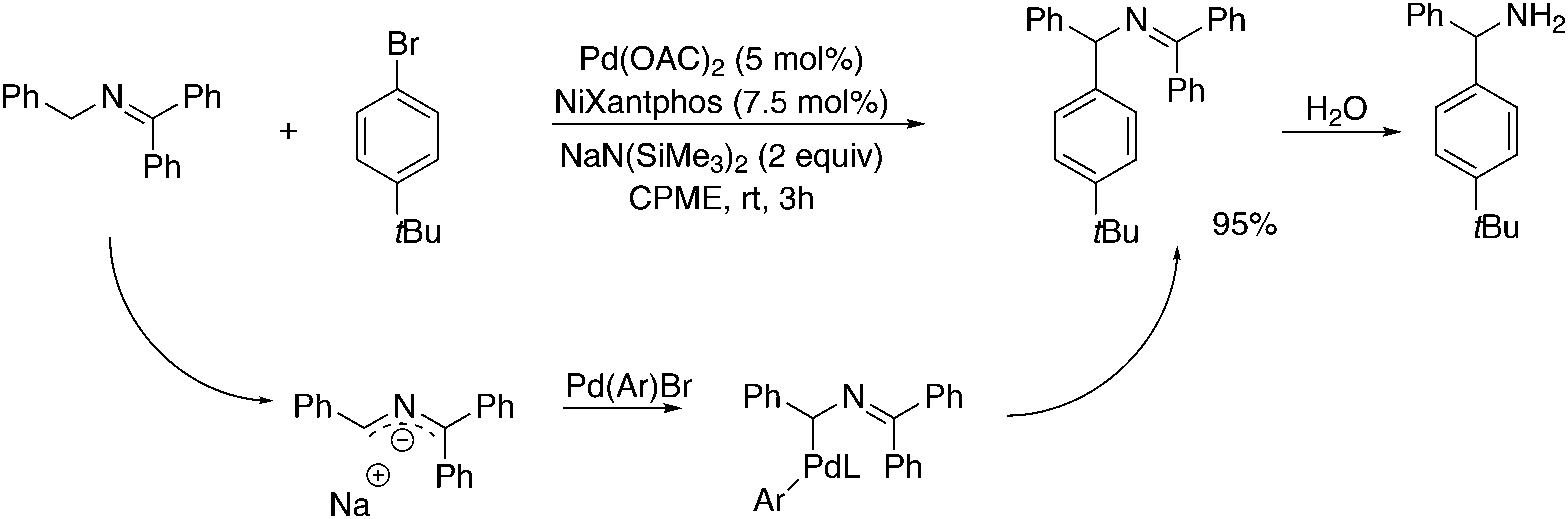 | ||
| Scheme 56 C-Arylation of benzylamines via imines. | ||
N-Benzhydryl-3-phenylprop-2-en-1-imines can be arylated either in the α-position or in the γ-position, depending on the catalyst system (Scheme 57).285 Monodentate PtBu3 based catalysts give α-arylation, 69, and Nixant-based catalysts give γ-arylation, 70. The authors proposed that the high selectivity observed for γ-arylation is the result of a cation–π interaction between the heterobimetallic Pd/Na/NiXantphos-based catalyst and the substrate, since the 22 other ligands that were explored all preferentially gave α-arylation. Xantphos and Bn-Nixantphos (N-benzyl-Nixantphos) gave equal amounts of 69 and 70.
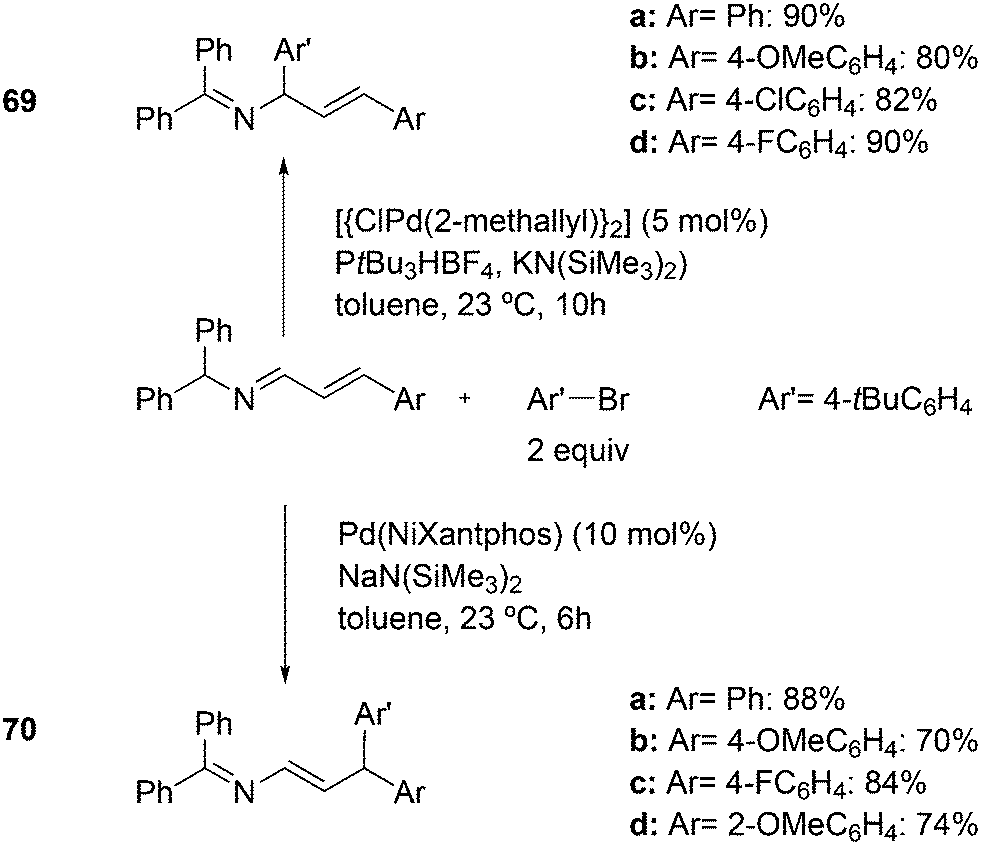 | ||
| Scheme 57 Regioselective arylation of allylamines via their imines. | ||
3.3.1.14. Theoretical studies. Maseras, Asensio and co-workers studied the competition between sp2 and sp3 C–Br bonds in Suzuki–Miyaura reactions, Scheme 58.286 Their study showed that small phosphines and Xantphos gave substitution of the sp3 C–Br halide, both in internal and external competition reactions, while bulky monodentates gave substitution at sp2 C–Br. DFT studies explained that for bulky ligands, dissociation of one ligand precedes oxidative addition of sp2 C–Br. Less bulky ligands gave an SN2 like substitution at sp3 carbon atoms. It was not elucidated why only Xantphos was active among the bidentate ligands investigated.
 | ||
| Scheme 58 Internal competition experiments for cross-coupling of sp2 and sp3 C–Br bonds. | ||
A DFT study of the full catalytic cycle of the Suzuki–Miyaura cross-coupling of vinyl bromide and a vinyl borate model system by Maseras et al. showed that there is not a single, general pathway for this reaction and that each system has to be separately studied in detail; for instance, both monophosphine and bisphosphine catalysts gave feasible pathways.287 An account by the same group illustrates the diversity for various cross-coupling reactions, such as Suzuki–Miyaura, Negishi, Stille, Sonagoshira, etc., and for the three main steps involved, oxidative addition, transmetallation, and reductive elimination.288
Huang et al. reported a successful new precursor (Xantphos)Pd(η2-CH2NBn2)+64 (Schemes 43 and 44) for aminomethylation of several reagents, which was studied by DFT by Yu Lan and co-workers.289 The complex can be described as a square-planar, nitrogen-coordinated aminomethyl-Pd(II) or a triangular η2-iminium-coordinated Pd(0). Frontier molecular orbital analysis favours the latter. Xantphos aids in forming iminium-coordinated palladium complexes, it promotes the aminomethylation, which can be described as a reductive elimination step and it stabilizes Pd(0) species. The reaction with allenes is shown in Scheme 44 (vide infra 3.3.1.11).
Scheme 59 shows a reaction that clearly is a radical reaction that starts with a SET forming the Rf radical (e.g. C6F13) that attacks the N-allyl group. Both Xantphos and DPEphos are good ligands for Pd and yields of benzazepine were up to 86%.290
 | ||
| Scheme 59 Radical initiated benzazepine formation. | ||
Formic acid can be used as the source of CO for the reaction of aryl iodides and phenols to give aryl arylcarboxylates in yields ranging from 60–88% when Xantphos is used as the ligand.292 Other bidentates and monodentates gave lower yields, but the difference between the ligands was less pronounced for this reaction than the examples we will see for carbonylative C–N coupling reactions (3.4.2).
Metal carbonyls have also been used as the source for CO, e.g. Co2(CO)8.293 Aryl triflates and the like were carbonylated with a Pd catalyst giving esters with the alcohol solvents. Co also accelerates the reaction. Various bisphosphines such as dppf, Josiphos, BINAP, dppb and Xantphos yielded good catalysts.
For the palladium-catalysed phthalazinone synthesis using paraformaldehyde as the carbon source (Scheme 60) Pd(tfa)2/Xantphos was reported as the best catalyst, DPEphos and dppf gave active catalysts as well.294 The insertion of CH2O into a Pd–C bond is a formylation reaction, rather than a carbon monoxide based insertion reaction.
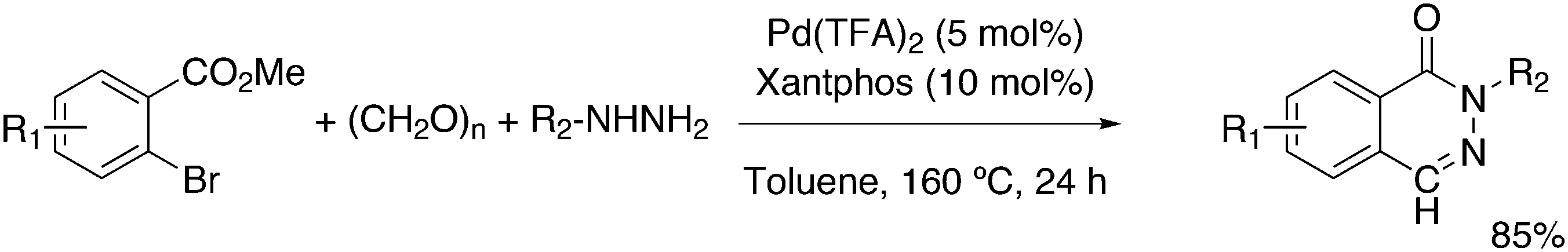 | ||
| Scheme 60 Phthalazinone synthesis. | ||
Furanones are present in several bio-active compounds. Huang and Alper reported a catalytic synthesis from aryl halides, two molecules of CO and substituted alkynes with yields ranging from 40–88%, as early as 1991 (Scheme 61).295 The scope was extended by Beller and co-workers by replacing PPh3 with other ligands, notably BuPAd2, dppf, and Xantphos.296 The latter ligand allowed the use of a variety of substituents and heteroaromatic rings.
 | ||
| Scheme 61 Furanone synthesis via carbonylative cross-coupling. | ||
Analogous to the fluoroalkylation discussed before, this can be carried out in a carbonylative way as was published recently and simultaneously by the groups of Skrydstrup and Zhang.297,298 The schematic reaction is shown in Scheme 62. A “classic” mechanism of oxidative addition etc. can be imagined, but both Skrydstrup and Zhang found indications of a radical pathway most likely involving Pd(I)Br intermediates.
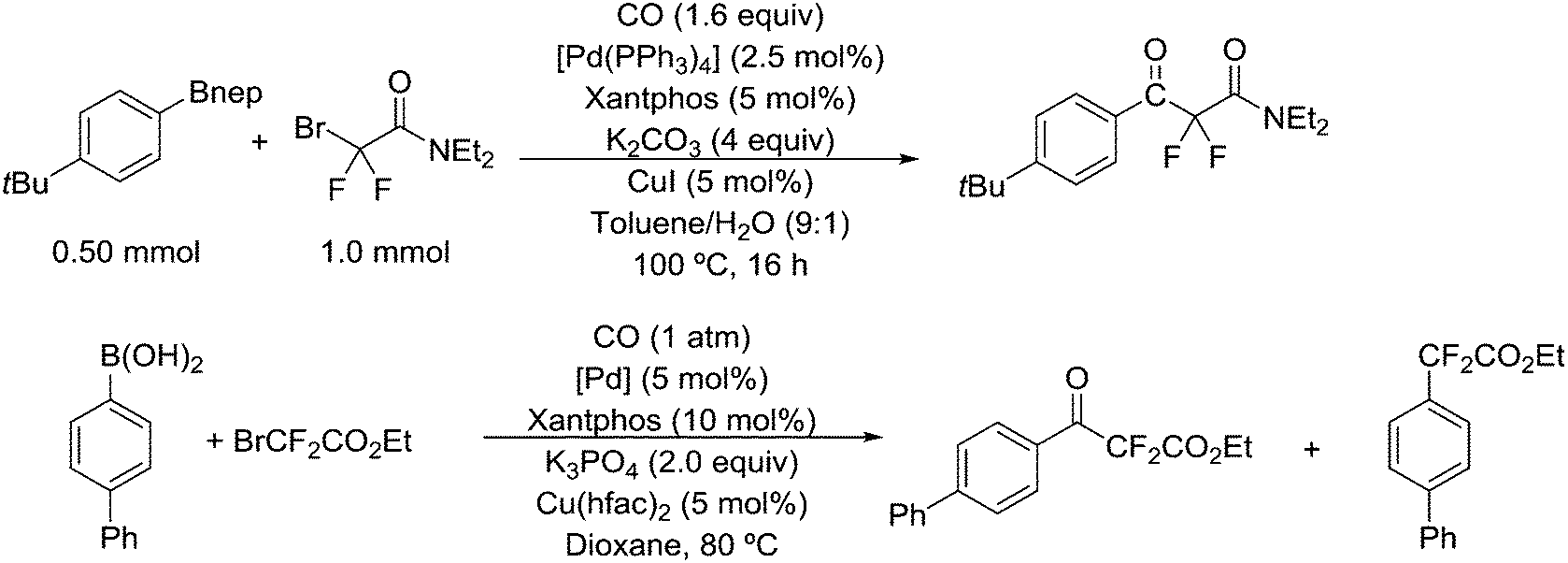 | ||
| Scheme 62 Radical initiated cross-coupling. | ||
A related reaction that does not involve cross-coupling was reported by Liang et al.Scheme 63.299 The preferred ligand in this instance is DPEphos. The reaction also starts with the transfer of a halide radical to Pd(0) and addition of the organic radical to the alkyne, which combines with Pd(I). After carbonylation, a nucleophile (alcohol or amine) completes the four-component reaction.
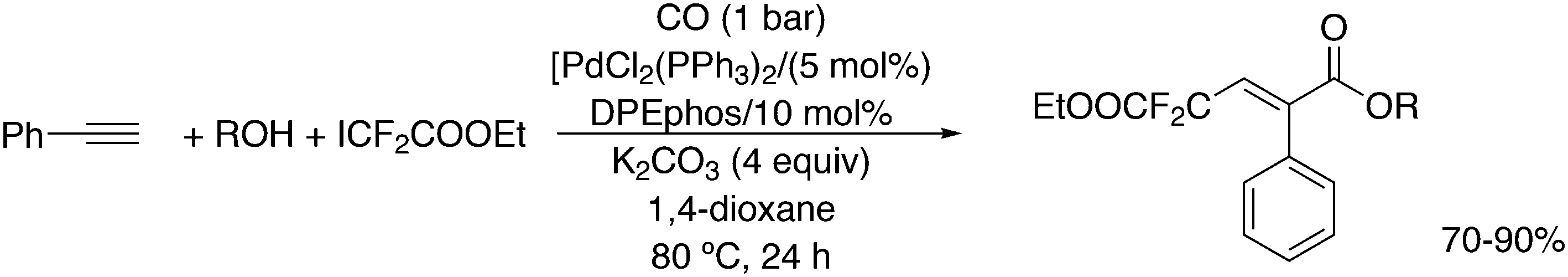 | ||
| Scheme 63 Radical addition and alkoxycarbonylation of alkynes. | ||
Carbonylative cross-coupling is, however, not unique for Xantphos, and apparently other ligands with similar properties can do the job. Gelman and co-workers described the use of a trans-chelated palladium complex derived from 1,8-bis-(4-(diphenylphosphino)phenyl)anthracene 12 as a catalyst in carbonylative Suzuki coupling and methoxycarbonylation of aryl iodides and bromides (Scheme 64).32 The selectivity was attributed to the unique structural features of the trans-chelating ligands (note that arylpalladium halides also give trans structures with several Xantphos-like ligands).33 Yields are excellent and side reactions and catalyst decomposition do not play a role, since catalyst loading could be reduced to 0.01%. The high temperature required would suggest that, as in Xantphos, a transient cis complex should form before reductive elimination can take place.
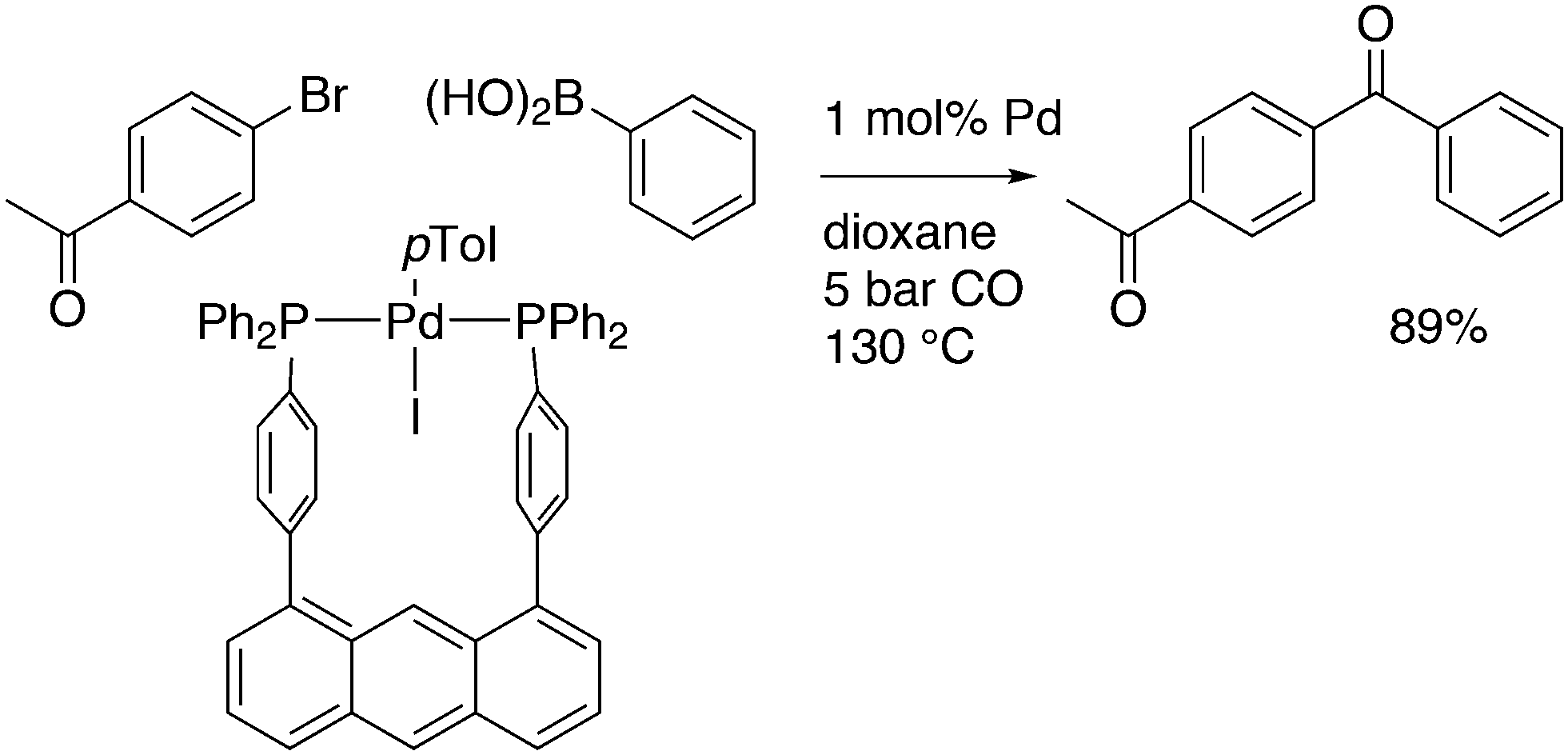 | ||
| Scheme 64 Carbonylative Suzuki reaction with a trans complex precursor. | ||
An alternative for aryl ketone formation that avoids carbonylative coupling, i.e. the use of CO, was reported by Takemiya and Hartwig, see Scheme 65.300 DPEphos and Xantphos stand out and gave the ketone in high yield after hydrolysis. The tBu group at N of the hydrazone plays an important role, since N-arylation was obtained when the phenyl derivative was used, especially when PPh3 was used as the ligand. The hydrazones are readily available from the aldehydes.
 | ||
| Scheme 65 Hydrazones as aldehyde mimics. | ||
 | ||
| Scheme 66 Alkyne coupling. | ||
Stereospecific nickel-catalysed cross-coupling using an inexpensive and sustainable nickel catalyst was reported by Dawson and Jarvo (Scheme 67).302 They reacted benzylic ethers in a Kumada reaction with Grignard reagents which proceeded with inversion at the benzylic position. The ligand used was achiral and potentially cheap DPEphos.
 | ||
| Scheme 67 Cross-coupling of benzyl ether with inversion. | ||
Louie and co-workers developed a cyclization reaction for diynes and nitriles to give substituted pyridine derivatives (Scheme 68).303 Xantphos outperformed all other ligands investigated. They developed for these reactions a stable yet reactive Ni(0) precursor (Xantphos)Ni(alkyne) that could also be used for Kumada cross-coupling chemistry.304
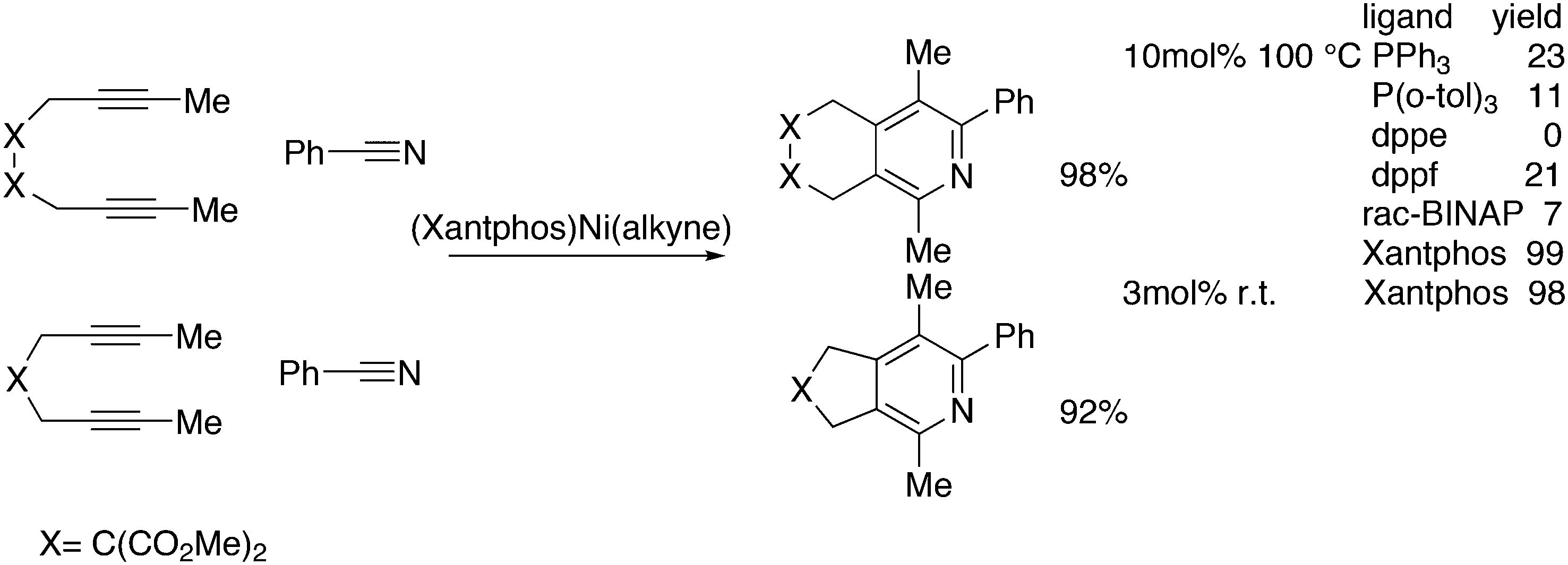 | ||
| Scheme 68 Cyclization reaction of diynes and nitriles. | ||
A computational screening of 42 bidentate phosphines by Maseras, Grushin and co-workers has yielded promising candidates for Ph–CF3 reductive elimination from Ni(II) complexes of the type [(P–P)Ni(Ph)(CF3)] (see also Pd, 3.3.1.9).305 Two electron rich ferrocene bisphosphines were identified as good candidates, but unfortunately the synthesis of the intermediates with these ligands failed, i.e. other lower energy decomposition pathways exist. Xantphos derived Ni–CF3 complexes led to the stable Ni–F bond formation.
Also, nickel based catalysts benefitted from the “deprotonated Nixantphos” effect; replacing Pd by Ni is attractive of course.306 Thus, for the nickel-catalysed arylation of heteroaryl-containing diarylmethanes an exceptional reactivity of the Ni(Nixantphos)-based catalyst was found. Scheme 69 shows the DCCP reaction studied. Substrates with pKa values up to 30 could be arylated, i.e. somewhat lower than the value for Pd catalysts. Out of the 37 mono- and bidentate cross-coupling ligands examined with Ni(COD)2, Nixantphos again showed the highest reactivity, Xantphos being second and ten times slower.
 | ||
| Scheme 69 Ni/Nixantphos catalysed DCCP. | ||
 | ||
| Scheme 70 Alkylylation of ketones by Cu/Xantphos or Cu/phenanthroline. | ||
Sawamura and co-workers have exploited many wide bite angle systems over the years,308 one example being the alkynylation of aldehydes with Cu, similar to the previous reaction (Scheme 71).309 They noted that wide bite angle ligands such as the TRAP ligands and Xantphos are able to solubilize alkynylcopper species, first used for C–C coupling in a stoichiometric way by Stephens and Castro in the reaction that carries their names.310 None of the many common chiral bisphosphines (bite angles 85–95°) gave any conversion and therefore they continued with the TRAP ligands that gave good conversions and modest ees up to 58%.
 | ||
| Scheme 71 Alkynylation of aldehydes. | ||
Diethylzinc triggers a condensation reaction of itaconate esters (Scheme 72) that can be catalysed by copper, giving modest yields without ligands or with dppp (38%).311 Very high yields were obtained with the use of Xantphos, and DPEphos was slightly behind in yield. No control of the stereochemistry of the substituted cyclopentanone was achieved.
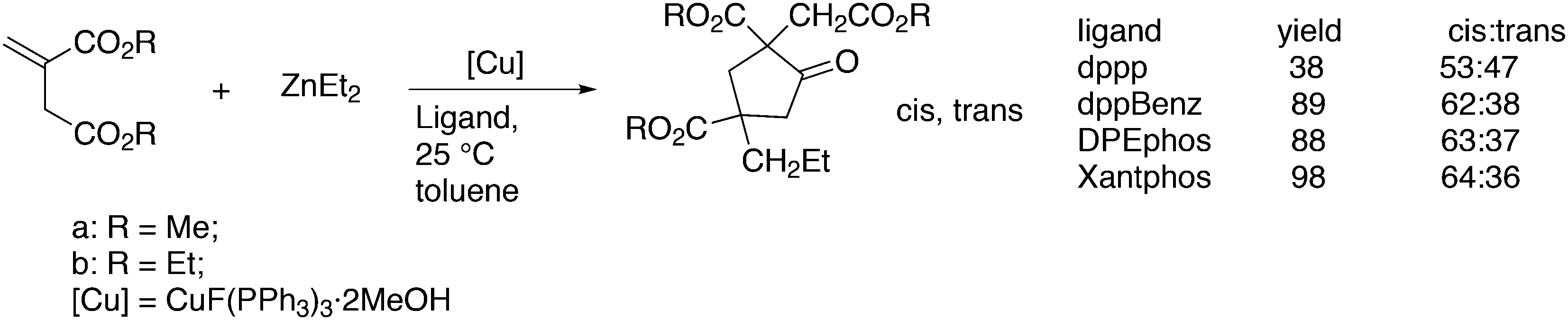 | ||
| Scheme 72 Alkylative dimerization/ring closure of itaconates. | ||
Alkynyl-3H-imidazo[4,5-b]pyridines 72 (Scheme 73) can be obtained by a Sonogashira reaction, but Piguel and co-workers reported a coupling route with the use of gem-dibromoalkenes as the alkyne source.312 Yields ranged from 33 to 77%. The reaction uses an inexpensive copper catalyst (CuBr·SMe2 or Cu(OAc)2), and a base in 1,4-dioxane at 120 °C; the preferred ligand is DPEphos, potentially the cheapest wide bite angle ligand.
 | ||
| Scheme 73 A disguised Sonogashira reaction. | ||
 | ||
| Scheme 74 Acylation via aldehyde C–H cleavage. | ||
Subsequently, the same authors published their studies involving a wider range of ligands, Scheme 75.314 The results were surprising; DPEphos and dppe gave high selectivity to the desired addition, but dpp-pentane showed a fast reaction to the Tishchenko product, the ester formed from two molecules of aldehydes. Intermediate selectivities were obtained for the POP ligand, Xantphos, dppp and dppb.
 | ||
| Scheme 75 Ligand effect in hydroacylation. | ||
In a series of publications, Breit and co-workers reported on Rh-catalysed additions to allenes for which several bidentate bisphosphines with somewhat wider bite angles were used as ligands.315 The intermediacy of π-allyl Rh species probably explains the preference for wide bite angles.316 Next, they argued that an isomeric analogue containing an alkyne (CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) would synthetically be much more accessible. They found that in the presence of Rh(DPEphos) and a benzoic acid, 1,3-diketones could be added regioselectively, depending on the substituents, under formation of a C–C bond (Scheme 76).317
CH) would synthetically be much more accessible. They found that in the presence of Rh(DPEphos) and a benzoic acid, 1,3-diketones could be added regioselectively, depending on the substituents, under formation of a C–C bond (Scheme 76).317
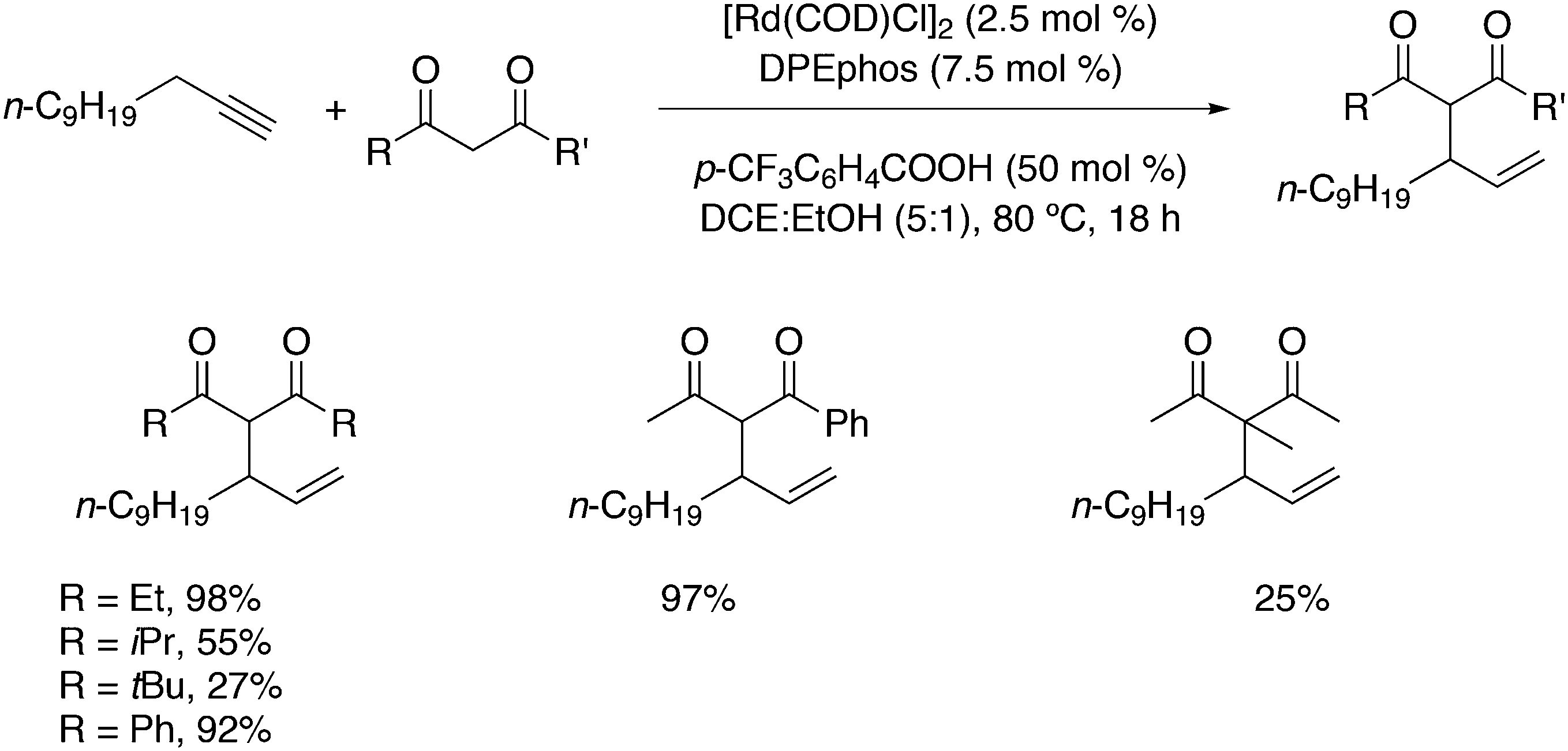 | ||
| Scheme 76 Addition of alkynes to diketones. | ||
Dong et al. reported Rh-catalysed C–C coupling reactions in which the ligand effect strongly “peaks” as we have seen occasionally in this study (Scheme 77).318 In this reaction, β-ketocarboxylic acids are coupled decarboxylatively to 2-alkynes using Rh and DPEphos (97%). Note the sharp decrease in yield for closely related ligands dppf (18%) and Xantphos (<5%). The tandem system is closely related to the Breit system reported above, as the 2-alkyne is first isomerized to the terminal allene and then coupling takes place as mentioned above and decarboxylation completes the scheme.
 | ||
| Scheme 77 Ligand effect in Rh catalysed C–C bond formation. | ||
Another recent, very neat example of Rh-DPEphos catalysed alkyne coupling was reported by Manan and Zhao.319 The reaction includes the formation of a C–C bond and a C–N bond, after C–H activation assisted by the X group as the “tug-in” ligand (Scheme 78). The reaction was designed to be redox neutral and produce 77 as the main product. Related oxidative reactions had been reported. In this instance the reaction was also sensitive to the Rh precursor, more so than in the previous reactions, which must be due to the C–H activation that must take place.
 | ||
| Scheme 78 Rh catalysed alkyne coupling followed by C–H activation and ring closure. | ||
3.4. C–N bond formation
Monoamination of 2,6-dibromotoluene (Scheme 79) was achieved with BINAP, DPEphos and dppf, while Xantphos gave no reaction.325 NHC ligands gave disubstitution. Mixed bromo-iodoarenes were monoaminated better by Xantphos than by BINAP based catalysts.
 | ||
| Scheme 79 Monoamination of aryl dibromides. | ||
Double N-arylation with t-butyl carbamate as the amine and ammonia analog provided a useful tool for the synthesis of unsymmetrically substituted carbazole derivatives (Scheme 80).326 Both diaryl phosphine ligands and Xantphos gave good results. The starting biphenyl derivative was made via a Suzuki–Miyaura coupling with Pd/PPh3 as the catalyst.
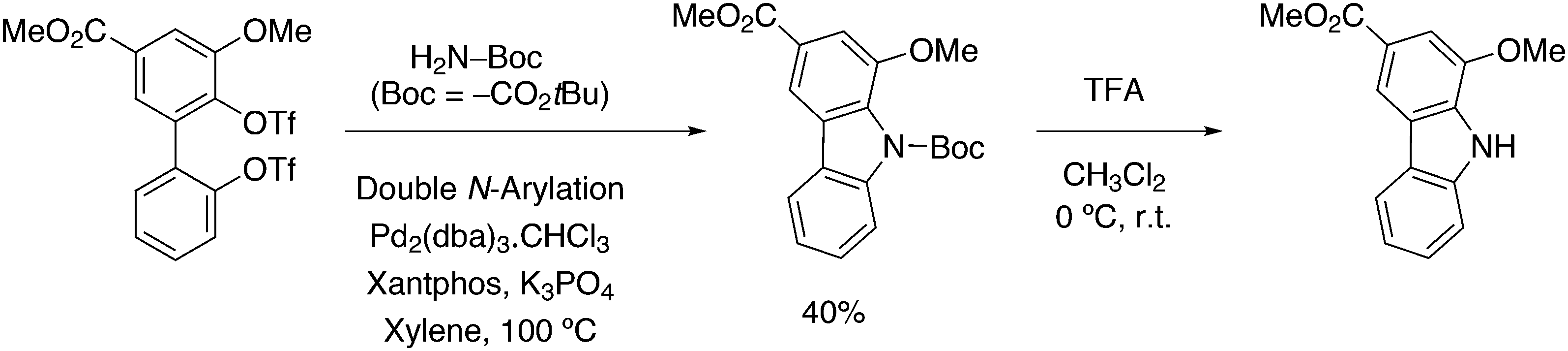 | ||
| Scheme 80 Double N-arylation with an ammonia analog. | ||
Willis, Frost and co-workers observed an interesting change in the selectivity of a dipeptide in a cross-coupling reaction (Scheme 81) when the ligand was changed.327
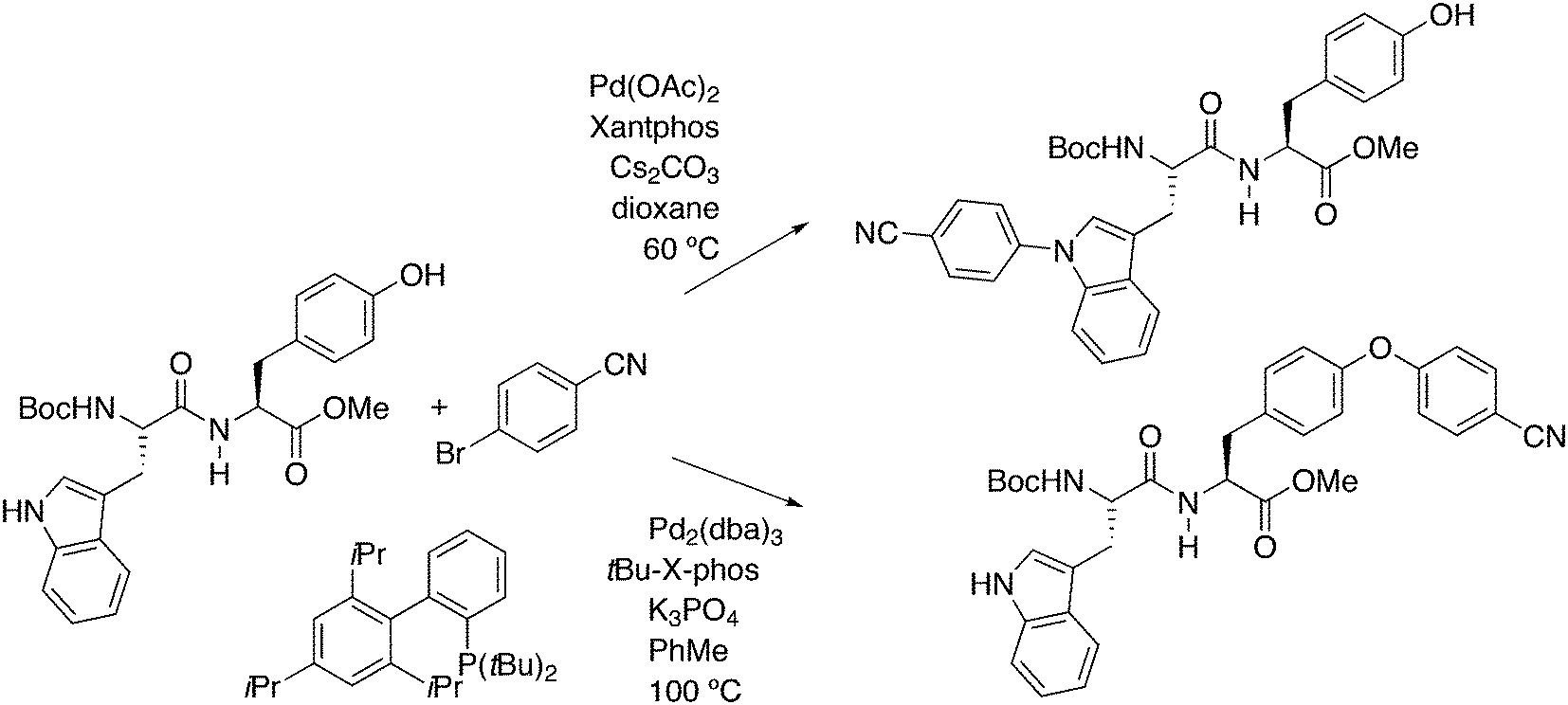 | ||
| Scheme 81 Ligand selective N- or O-arylation. | ||
Lakshman and co-workers studied the N-arylation of nucleosides, and Scheme 82 shows an example. Among the popular ligands, Xantphos gave the best results.328,329 This N-arylation was also carried out for a fully protected 2′-deoxyguanosine.330
 | ||
| Scheme 82 N-Arylation of protected 2′-deoxyadenosine. | ||
The same products were obtained in good yield with the use of Xantphos in a “reverse” coupling of chloronucleosides and anilines, Scheme 83, by the same group.331,332
 | ||
| Scheme 83 N-Arylation on chloronucleosides. | ||
An extensive study of the use of aryl nonaflates instead of aryl halides with primary and secondary amines was reported by Buchwald et al.333 Three ligand groups gave excellent yields: diaryl monophosphines, BINAP, and Xantphos. The latter also gave good results for even 2-carboxymethyl aryl nonaflate, which was effectively coupled with a primary alkylamine. Aryl nonaflates containing halides reacted preferentially with the nonaflate. A full account of the diarylphosphine ligands including an analysis by DFT has been presented.334 The reaction can be sped up by microwave irradiation.335 A kinetic study showed that phosphine dissociation from a bis(xantphos)Pd(0) complex can have a detrimental effect on the rate when using a ratio of >1.5:1 L:Pd, where L = Xantphos, in C–N bond forming reactions.336
The first general intermolecular C–N bond-forming reactions between aryl halides and amides were realized by Yin and Buchwald, using a palladium catalyst with Xantphos as the ligand.337 Aryl triflates, carbamates, and sulfonamides are also viable substrates for the amidations (Scheme 84), which proceed at 45–110 °C with 1–4 mol% of Pd catalyst in 66–99% yields and exhibit good functional group compatibility. A full account has appeared in which it was also shown that ArPdX complexes of Xantphos form trans intermediates.34
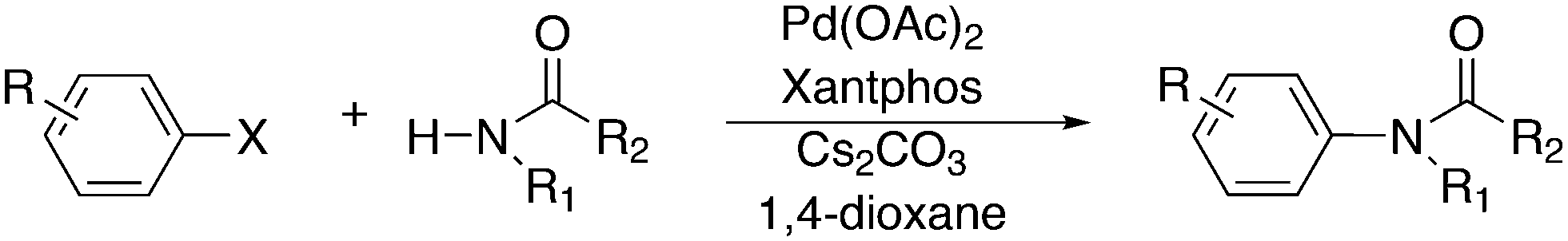 | ||
| Scheme 84 Amidation of aryl halides and pseudohalides. | ||
Selective amidation of 2,6-dihalogenopurines similar to the N-arylation shown in Scheme 83 employing Xantphos was reported by Piguel and Legraverend.338
Quinazolin-4(3H)-ones were prepared via Pd/Xantphos catalysed amidine N-arylation in moderate to high yields (Scheme 85).339 Other mechanisms can be envisaged, but it was shown to proceed via N–C coupling of the imine and the aryl group. Amidine coupling and ring closure with an ester has also been applied to nucleoside analogues of pharmaceutical interest.340
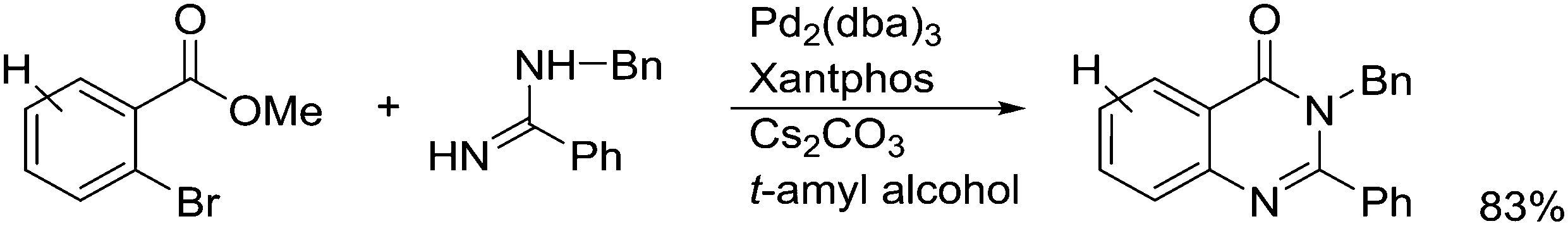 | ||
| Scheme 85 Quinazolin-4(3H)-ones via amidine N-arylation. | ||
N-Nucleophiles such as amidates that are only weakly nucleophilic take advantage of Xantphos ligands in the cross-coupling chemistry. Hartwig and co-workers showed that in small bite angle bidentates the amidate binding is κ2 and this greatly retards the reductive elimination (Scheme 86).341 Both dppf and the ferrocene based ligand show low conversion while Xantphos gives high yield.
 | ||
| Scheme 86 Weak N-nucleophiles in cross-coupling. | ||
Beletskaya reported that ureas require the same catalyst system as amides.342,343 Thus, urea arylation with aryl bromides in the presence of a Pd catalyst precursor, Xantphos and Cs2CO3 in dioxane at 100 °C afforded symmetrical N,N′-diarylureas in 64–92% yield. With the same catalytic system, the reaction between N-phenylurea and aryl bromides containing electron-withdrawing substituents in the para-position provided N-aryl-N′-phenylureas in 64–91% yield. The results were further refined by a series of Xantphos-based ligands containing various substituents in the diphenylphosphino groups.344
N,N-diaryl benzophenone hydrazones 78 are intermediates for the preparation of N-arylindoles. Buchwald et al. explored the Pd-catalysed cross-coupling reaction of benzophenone hydrazone 78 with aryl bromides (Scheme 87).345 The choice of ligand was crucial to the success; of the Pd/mono- and bis-phosphine catalysts that they surveyed, the Pd/Xantphos combination was the only catalyst system that effected the complete conversion of an N-aryl benzophenone hydrazone and an aryl bromide to the N,N-diarylhydrazone 79. Monoarylation was obtained with only 0.1% catalyst, much less than needed for the also active BINAP.
 | ||
| Scheme 87 N-Arylation of hydrazones. | ||
Barluenga and co-workers reported an interesting new double C–N bond formation, the addition of azide anions to a vinyl bromide that ring-closes to give 1H-1,2,3-triazoles, while the intermediate vinyl azides were never detected (Scheme 88).346 Monodentate phosphines gave no active Pd catalyst, neither did BINAP and again a very narrow range of ligands yielded highly active catalysts. The conventional route to triazoles is the Huisgen dipolar cycloaddition of alkynes and organic azides. Palladium catalysed cyclization of vinyl azides was disgarded as the mechanism, since independently prepared vinyl azides did not undergo cyclization with Pd/Xantphos. Instead, the authors proposed a 3+2 addition of azide and vinylpalladium as the crucial step, which makes the ligand effect even more peculiar.
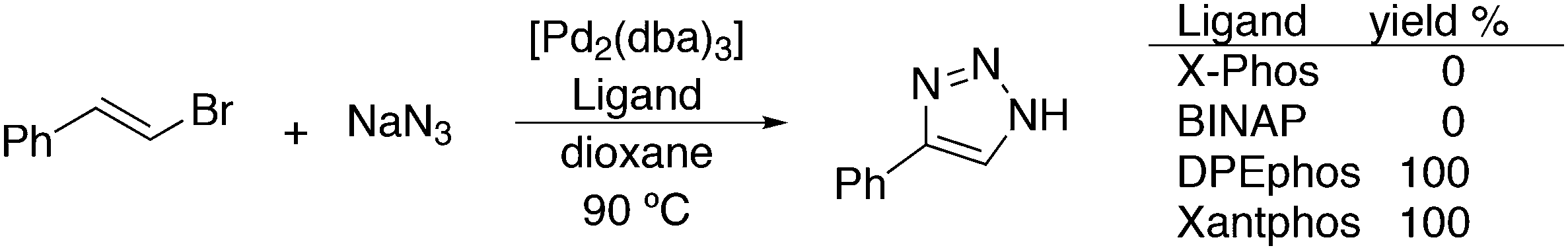 | ||
| Scheme 88 Triazole formation with sodium azide. | ||
Substituted piperazines and many related N-heterocycles were prepared from diamines and propargyl carbonates and a Pd catalyst (Scheme 89).347 The propargyl carbonates are masked sources of bis-electrophiles for palladium-catalysed reactions. DPEphos is the ligand of choice and a wide variety of products was obtained.
 | ||
| Scheme 89 Piperazine formation from propargyl carbonates and 1,n-diamines. | ||
Hydroamination of alkenes is a reaction that has been on the wishlist of synthetic chemists for a long time because unlike reactions involving halides, it does not produce salts as by-products. Unfortunately, the thermodynamics are borderline and several combinations of substituted alkenes and amines will not yield product. Thanks to the effective Pd catalysts developed by Hartwig and co-workers, the rates of reactions in both directions could be measured and they obtained thermodynamic data for several substituted styrenes and anilines.348 The ligands used were dppf and tBuXantphos (Scheme 90).349
 | ||
| Scheme 90 Reversible hydroamination of arylalkenes. | ||
Hydroamination of dienes was carried out with the same catalyst system based on tBuXantphos and palladium triflate (Scheme 91).350 The reaction closely resembles allylic substitution and mechanistically the hydroamination could start with a hydride addition to the diene providing the π-allylic intermediate; the strength of allylic alkylation is that there are numerous catalysts giving enantioselectivity, while chiral Xantphos ligands are scarce. Studies of substituted unsymmetrical and unsubstituted symmetrical model π-allyl complexes showed that nucleophilic attack on complexes ligated by Xantphos was faster than on complexes bearing ligands with smaller bite angles, and that nucleophilic attack on unsymmetrical allyl complexes with larger bite angle ligands was faster than attack on unsymmetrical allyl complexes with smaller bite angle ligands. Earlier, Van Haaren et al. reported on the rate and regioselectivity of allylic substitution in Pd complexes; they found the rate enhancing effect of wider bite angles, which was confirmed by PM3™ calculations.351
 | ||
| Scheme 91 Hydroamination of dienes. | ||
Instead of dienes, substituted allylic carbonates could also be used, which gave the same selectivity for substitution at the most substituted carbon, thus favouring the formation of the branched N-allyl products.352
Regioselective Pd-catalysed hydroamination of substituted dienes was further investigated by Van der Vlugt et al.353 As an example, chloroprene is shown in Scheme 92 with benzophenone hydrazone as the N-nucleophile. Only Xantphos gave conversion and even closely related ligands like Nixantphos, DPEphos, Josiphos and BINAP gave no conversion.
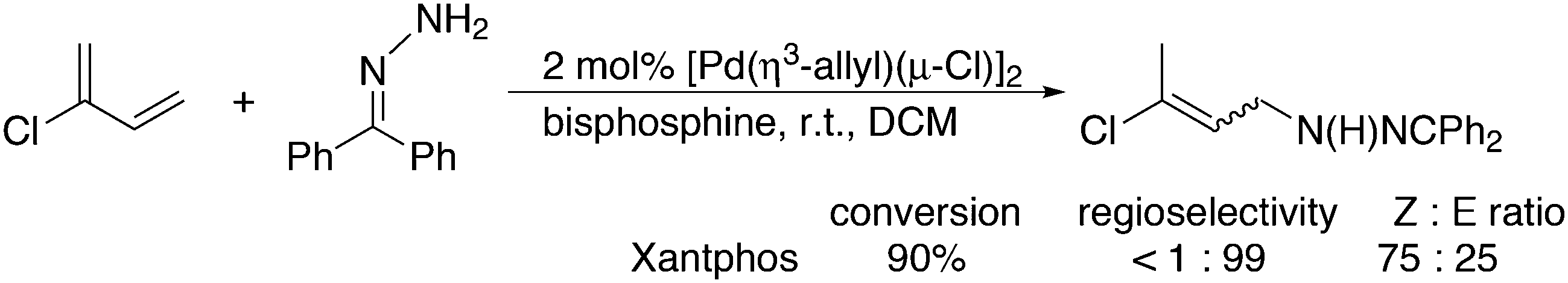 | ||
| Scheme 92 Hydroamination of chloroprene. | ||
Xantphos is also the preferred ligand for hydroamination of cycloheptatriene with aniline to yield tropene and cyclopent[b]quinoline derivatives as by-products (Scheme 93).354 Benzylamines reacted similarly. The reaction is a tandem sequential hydroamination. The quinolone side product does not result from a direct reaction of the catalyst with aryl-N-tropene; perhaps cycloheptatriene rearranges with catalyst and acid to a five-membered ring compound rather than a more common norcaradiene derivative.
 | ||
| Scheme 93 Hydroamination of cycloheptatriene. | ||
Carboamination of alkenes was studied by Wolfe and co-workers. The intramolecular alkenylamines gave cyclic products such as substituted pyrrolidines (Scheme 94).355 The reaction involves an insertion of the alkene into the Pd–N bond, i.e. a syn addition is obtained rather than the anti addition, which is the product of a Wacker type reaction of an alkene. Extensive kinetic studies were conducted on the stoichiometric reaction of the Pd intermediate for a range of ligands. The reaction rate increased in the order dppf, DPEphos, Xantphos, Nixantphos. Ligands with smaller bite angles gave an unstable intermediate or no product at all. Electronic studies on substituted dppf variants showed that the rate effect is a bite angle effect and not an electronic effect. Allylureas and Pd/Xantphos undergo this reaction to give cyclic ureas.356 Allylguanidines can be cyclized with Pd/Nixantphos to give substituted imidazolidin-2-imines. This method allows access to a number of cyclic guanidine derivatives in only two steps, from readily available allylic amines.357
 | ||
| Scheme 94 Carboamination of alkenes. | ||
A novel carboamination of alkynes was discovered by Hsung and co-workers.358 The reaction begins with an N–C bond breaking of an allylamine (a retro-allylic amination), Scheme 95. The ynamido-Pd complex rearranges as shown and the addition of an amine and reductive elimination completes the reaction sequence. The reaction outcome and rate strongly depend on the ligand, and high selectivities were obtained for PPh3, XPhos and Xantphos (all >95%), with the latter giving the fastest reaction. No reaction or slow reactions were obtained with e.g. dppp and BINAP. A broad scope was presented. The authors outlined that fast reductive elimination was key to the high selectivity for allylated amidines, which explains the results for XPhos and Xantphos; for a discussion on reductive elimination see the report by Hartwig et al.339
 | ||
| Scheme 95 Carboamination of alkynyl allylamines. | ||
The palladium-catalysed sequential one-pot N-arylation–carbo-amination–C-arylation of O-homoallylhydroxylamines and aryl bromides provided rapid entry to arylated N-aryl-3-arylmethylisoxazolidines with excellent diastereoselectivity (Scheme 96).359 The obtained isoxazolidines can be reductively cleaved to cis-N-aryl-β-amino alcohols in high yields at room temperature. The first N-arylation can be carried out with a diaryl monophosphine ligand and by adding Xantphos the reaction can be continued with a different aryl bromide, thus differentiating the two aryl groups. Other bidentate phosphines gave lower yields.
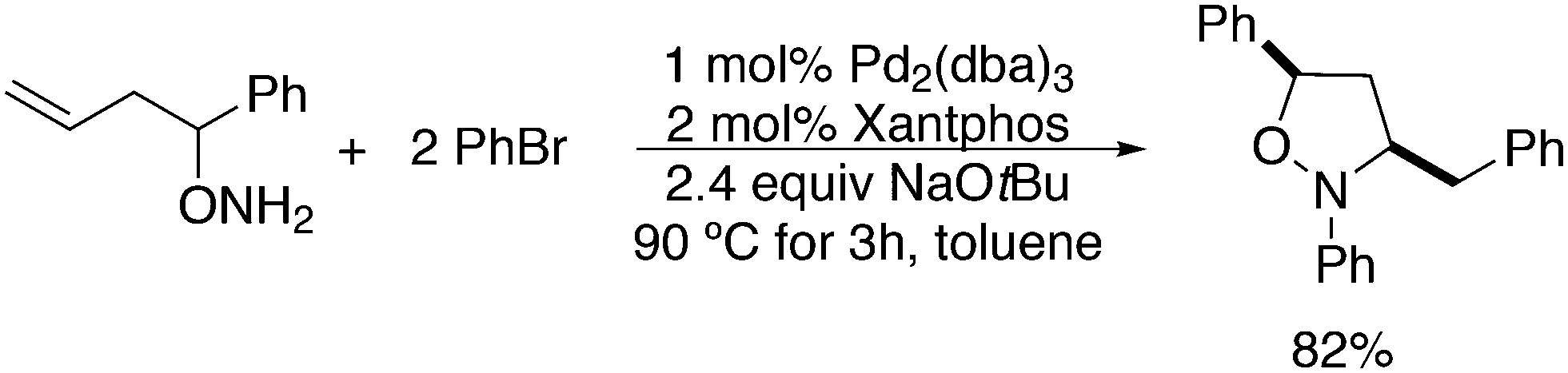 | ||
| Scheme 96 One-pot three bond forming reaction. | ||
Isoindolin-1-ones are attractive as pharmaceutical intermediates and a convenient new synthetic route was published by Kahn, Pardasani and co-workers (Scheme 97).360 Isocyanides readily insert into Pd–C bonds and in the presence of water the intermediate forms an amide; the reaction is an alternative for carbonylative C–N coupling, mentioned in section 3.4.2. In this case, the C–N bond is formed in the next step when the amide adds to the ortho alkynyl group catalysed by the same Pd catalyst. The starting (ortho-haloaryl)alkynes were synthesized by a Pd-catalysed Sonogashira reaction. The reaction conditions employed for their preparation and those of the subsequent reactions were very similar; thus, the authors developed a one-pot Pd-catalysed protocol for the synthesis of isoindolin-1-ones from 2-bromoiodobenzene. This is a robust reaction that was used for a wide range of substrates.
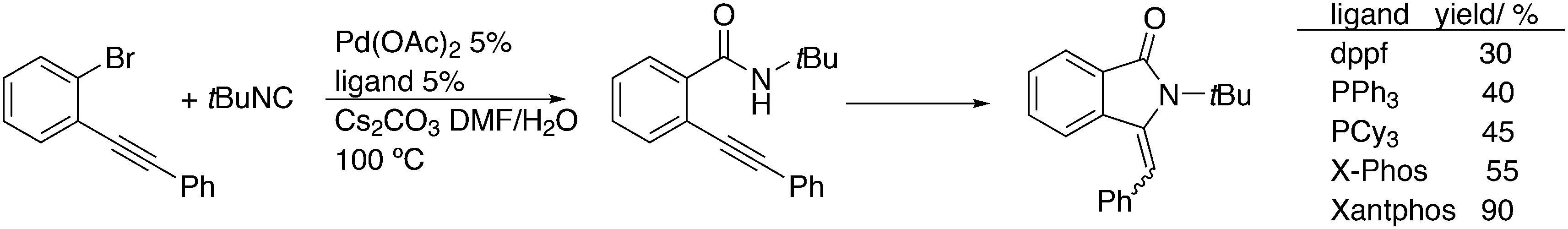 | ||
| Scheme 97 Isocyanide as an amide surrogate. | ||
Aryl chlorides do not react when the Xantphos based system is used and 1,3-bis(dicyclohexylphosphino)propane must be used as the ligand in order to facilitate the oxidative addition of the chloride to Pd(0).361
Buchwald and co-workers found a successful route to the so-called Weinreb amides with the use of N,O-dimethylhydroxylamine as the N-nucleophile (Scheme 98).362,363 The product N-methoxy-N-methyl amides (Weinreb amides) are well-established acylating agents. They were made with large functional group tolerance in 65–99% yield and only 1 bar of CO. Monophosphines, such as Ph3P and SPhos, and bisphosphines such as dppe, dppp, and dppb gave no conversion at all, but more surprising was that BINAP and DPEphos, often showing behaviour similar to that of Xantphos, gave no conversion either. Also, other amides could be made this way in high yield with secondary aliphatic amines, aniline, etc.364 Aminocarbonylation with this catalyst system was applied in a microflow system under elevated pressures.365 More examples of Weinreb amides were reported by Prandi et al. using Buchwald's catalyst system and vinyl triflates.366
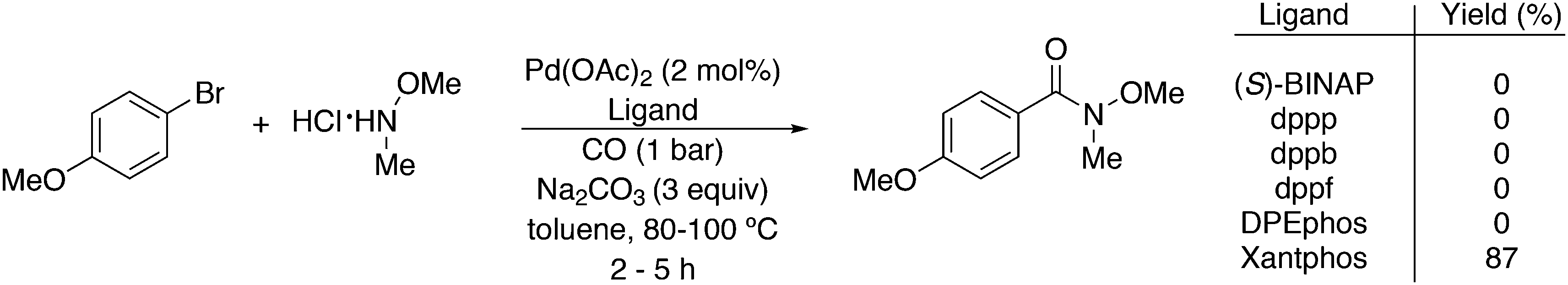 | ||
| Scheme 98 Weinreb amide synthesis via carbonylative coupling. | ||
The flexibility of Xantphos in forming both cis and trans complexes, the formation of pentacoordinated intermediates, and the less likely dissociation of the ligand compared with monodentates have been brought forward as important properties, but a detailed explanation is lacking.
Ley and co-workers developed a flow system for the application of Buchwald's catalyst system.367
Skrydstrup and co-workers pursued an application of carbonylative cross-coupling with Xantphos/Pd in the synthesis of radiolabelled 11C raclopride and olaparib (Scheme 99) for PET studies on this pharmaceutical.368 To this end, they prepared stoichiometric amounts of the Pd aryl intermediate, which then had to be quickly reacted with11CO, trapped and isolated. Much to their surprise, the phenyl derivative was isolated in large amounts, apparently formed by the known aryl exchange on arylphosphine arylpalladium complexes.369 Thus, in this case PtBu3 was the preferred ligand, which worked well in the stoichiometric reaction.
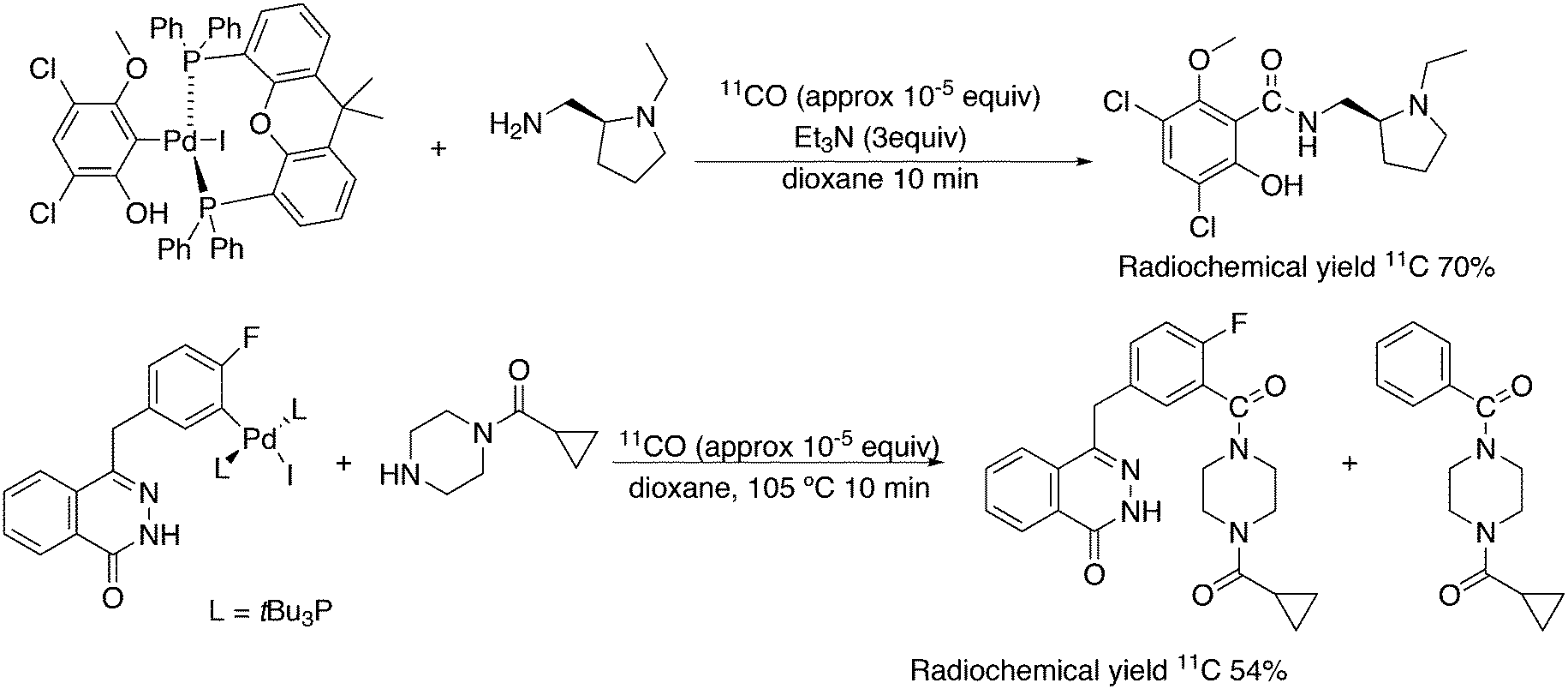 | ||
| Scheme 99 Stoichiometric carbonylative coupling. | ||
Hocek et al. applied Buchwald's aminocarbonylation catalyst to synthesize a range of modified C-aryl nucleosides using the typical mild conditions of this reaction (Scheme 100).370
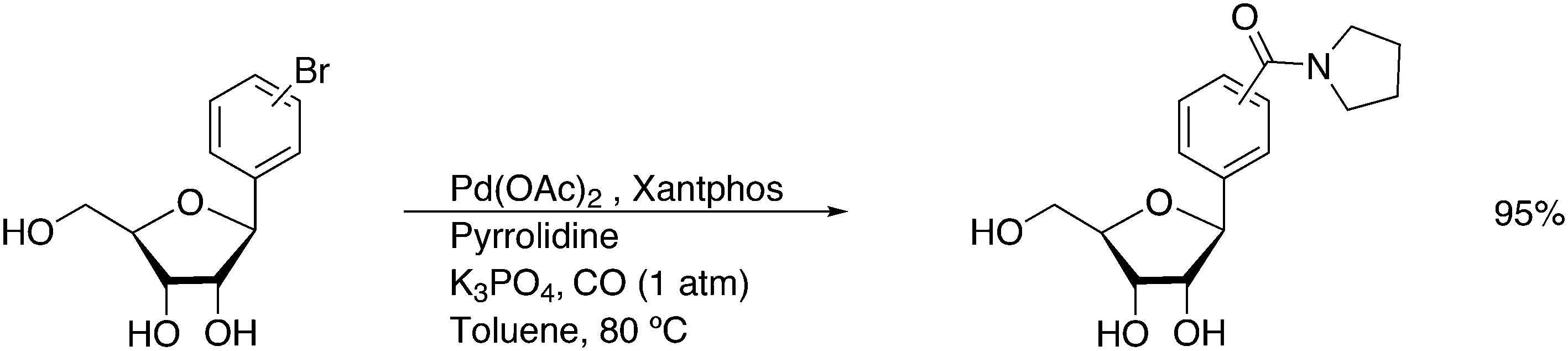 | ||
| Scheme 100 Nucleotide example of carbonylative coupling (silyl protection/deprotection omitted). | ||
Indol-3-yl aryl ketones were prepared from 1-methylindole and aryl formates as the source of the aryl group and CO with a Pd/Xantphos combination as the most effective catalyst.371 This way the use of CO pressures can be avoided, but the reaction uses 1.5 equivalents of C4F9SO2F and 3 equivalents of DBU.
Azidocarbonylation of aryl iodides was a challenge addressed by Macgregor, Grushin and co-workers.372 A careful study of the mechanism and deactivation led to the optimal procedure for the desired reaction, Scheme 101. The catalyst deactivation product is PdI2, which can be reduced in situ to Pd(0) by PMHS. Of numerous ligands screened in combination with Pd(OAc)2 or Pd2(dba)3 precursors, only Xantphos gave rise to an active catalyst. With the other ligands, the yield of PhCON3 from PhI never exceeded 6% under a variety of conditions. The catalyst shows a broad tolerance to functional groups in the aromatic ring.
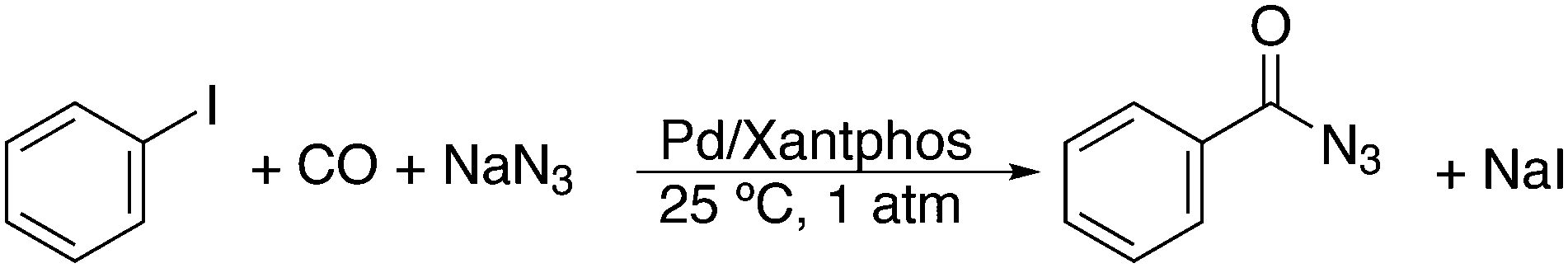 | ||
| Scheme 101 Azidocarbonylation. | ||
Above, we reviewed the hydroamidation of dienes and carbonylative C–C bond formation and we learnt that both reactions proceed quickly with Xantphos as the ligand. Thus, it may be no surprise that carbonylative hydroamidation of dienes also works well with the use of Xantphos, as was reported by the Beller group (Scheme 102).373 Thus, isoprene could be reacted with CO and amides to give a wide variety of β,γ-unsaturated imides.
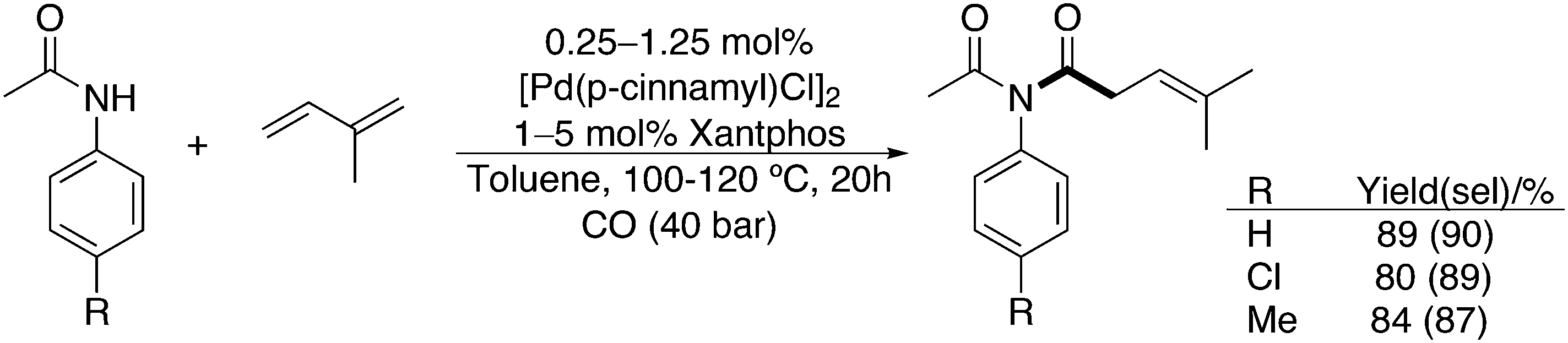 | ||
| Scheme 102 Carbonylative hydroamidation of dienes. | ||
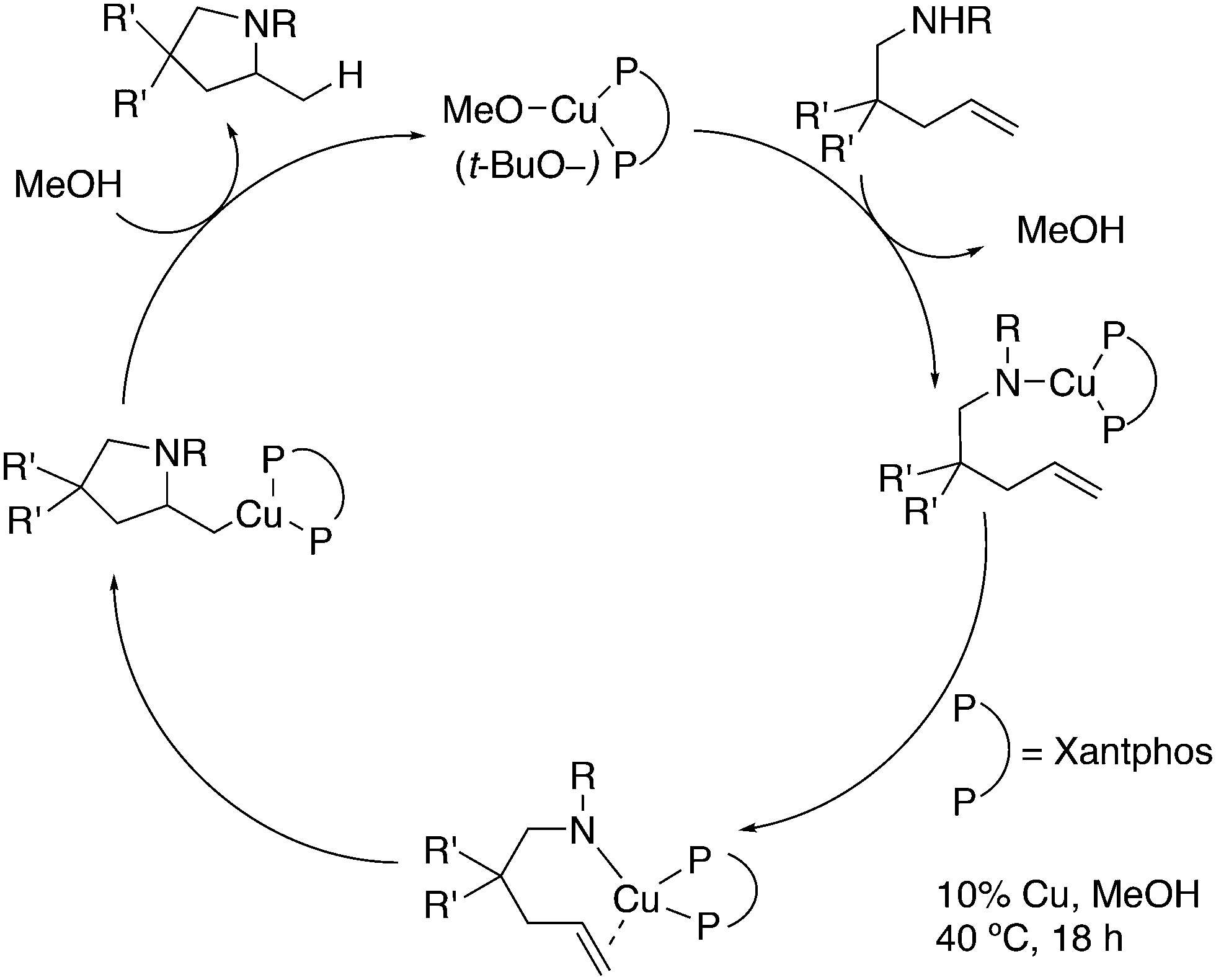 | ||
| Scheme 103 Cu/Xantphos hydroamination. | ||
Cu catalysed C–C and C–N coupling reactions have been known for a long time, starting with the Ullmann coupling carried out with stoichiometric copper bronze at high temperatures. After the discovery of the Buchwald–Hartwig reaction 20 years ago, ligand effects have also been used for Cu catalysed reactions and both N-based and P-based ligands provided good results. About 10 years ago, the first examples using Xantphos were reported. Maes and co-workers reported a tandem double C–N bond formation catalysed by Xantphos with either Cu or Pd (Scheme 104) with the use of 80, giving 81.375 For chloro-iodide derivatives, 82, only Pd was required and both BINAP and Xantphos gave 97% yield of isomer 83.
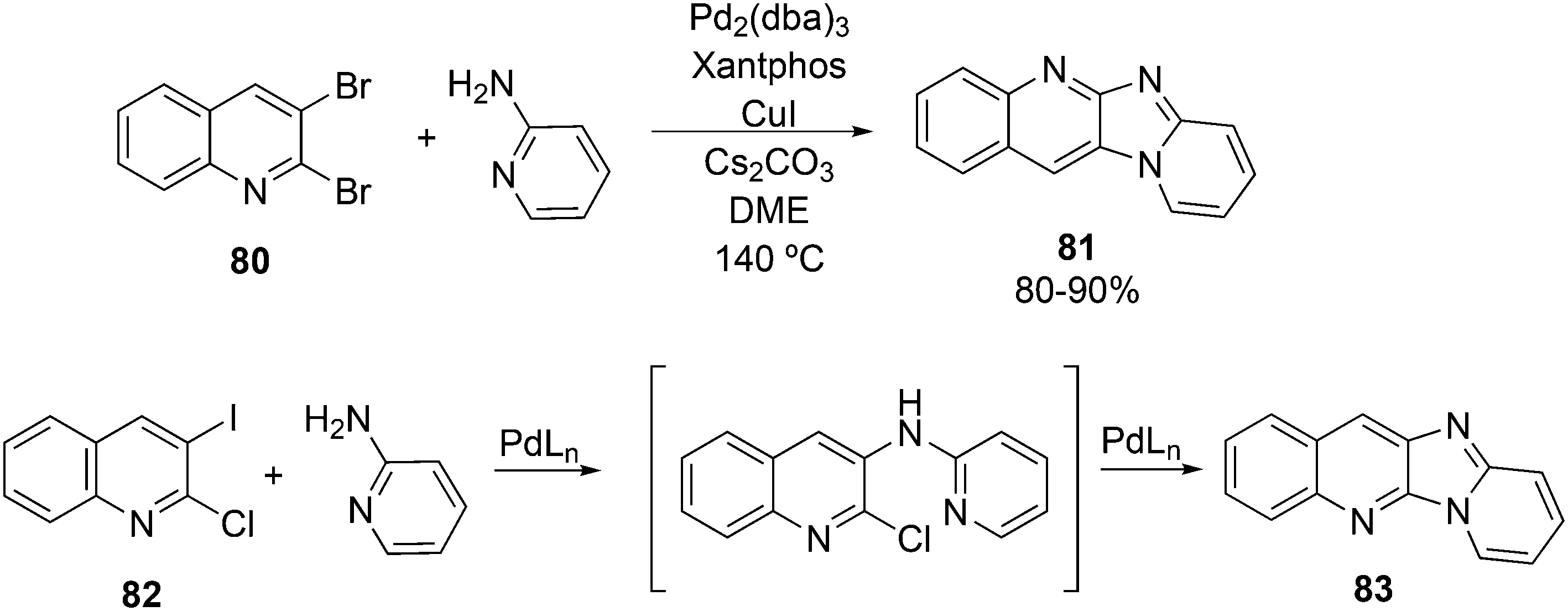 | ||
| Scheme 104 Double C–N bond formation. | ||
Aryl boronate esters can be coupled with benzoyloxyamines to give arylamines for which a protocol based on Cu/Xantphos was developed by Lalic et al.376 For the example shown in Scheme 105, Xantphos was the best ligand (99%) and the NHC ligand IMes gave only 72% yield, but for other combinations, IMes was sometimes preferred. Highly sterically hindered anilines can be prepared this way. The optimized reaction conditions proved to be remarkably general. The reaction involves transmetalation from boron to copper, with subsequent electrophilic amination of the aryl copper intermediate. Finally, the reactive copper alkoxide is regenerated with lithium alkoxide.
 | ||
| Scheme 105 Aryl boronate esters in amination reactions. | ||
More recently, Lalic's group improved this protocol while considerably expanding the scope with the use of Cy-DPEphos instead of Xantphos (all yields of hindered anilines > 88%).377
 | ||
| Scheme 106 Hydroamination of styrene catalysed by Rh(I)DPEphos. | ||
The anti-Markovnikov addition of the amine to styrenes was subjected to DFT studies by Ujaque et al.380 A variety of mechanisms were analyzed. According to the authors, the mechanism can be described by alkene coordination, amine nucleophilic addition, proton transfer via the metal center and reductive elimination, i.e. addition of N–H to Rh was excluded by the calculations. Attack of the amine cannot be charge controlled since that would always lead to the Markovnikov addition product; instead, the authors concluded that it is orbital controlled. A slippage of η2 coordination to η1 coordination is needed to enable the migratory insertion, as already proposed by Eisenstein and Hoffman in 1981.381 This process favours the anti-Markovnikov addition of the amine.
3.5. C–X bond formation
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)H fragment are much more attractive as starting materials. The first breakthroughs were reported by Tanaka in 1996 for (RO)2P(O)H (R = Me, Et) and R2P(O)H (R = Ph), which were added with Pd and Pt catalysts to terminal alkynes, giving 2-phosphinylalkenes.382,383 Several monodentate phosphine ligands were used and TON of 30 was obtained in several hours. Potential intermediates were identified by 31P NMR.
O)H fragment are much more attractive as starting materials. The first breakthroughs were reported by Tanaka in 1996 for (RO)2P(O)H (R = Me, Et) and R2P(O)H (R = Ph), which were added with Pd and Pt catalysts to terminal alkynes, giving 2-phosphinylalkenes.382,383 Several monodentate phosphine ligands were used and TON of 30 was obtained in several hours. Potential intermediates were identified by 31P NMR.
Wenbin Lin and co-workers extended this reaction to bisphosphinylation of alkynes, thus obtaining bisphosphines after reduction with silanes (Scheme 107).384 The TON for bisphosphine oxides was 20 after 20 h of reaction at 110 °C.
 | ||
| Scheme 107 Synthesis of bisphosphines via bishydrophosphinylation of alkynes. | ||
Montchamp and co-workers started an important variant of the reaction, both by widening the scope and the catalyst activity. The scope was widened by using hypophosphorous derivatives that could subsequently be converted to many other derivatives; thus, the catalyst needed to be optimized for only one P(O)H starting material, viz. hypophosphorous acid or ester (Scheme 108).385 Initially, hypophosphorous compounds also acted as hydrogen transfer agents (reducing/hydrogenating alkenes and alkynes), but modification of the catalyst led to complete suppression. In earlier work on the monophosphines, PPh3 was the ligand of choice, but as happened more often, Xantphos became the favorite ligand in hydrophosphinylation; in cis-(PPh3)2Pd complexes, the two phosphines occupy a similar space to Xantphos, with the added advantage that trans complexes are less easily accessible. Dppf and PPh3 afforded slower catalysts than Xantphos and DPEphos. TONs up to 200 were reported in 12–16 h.
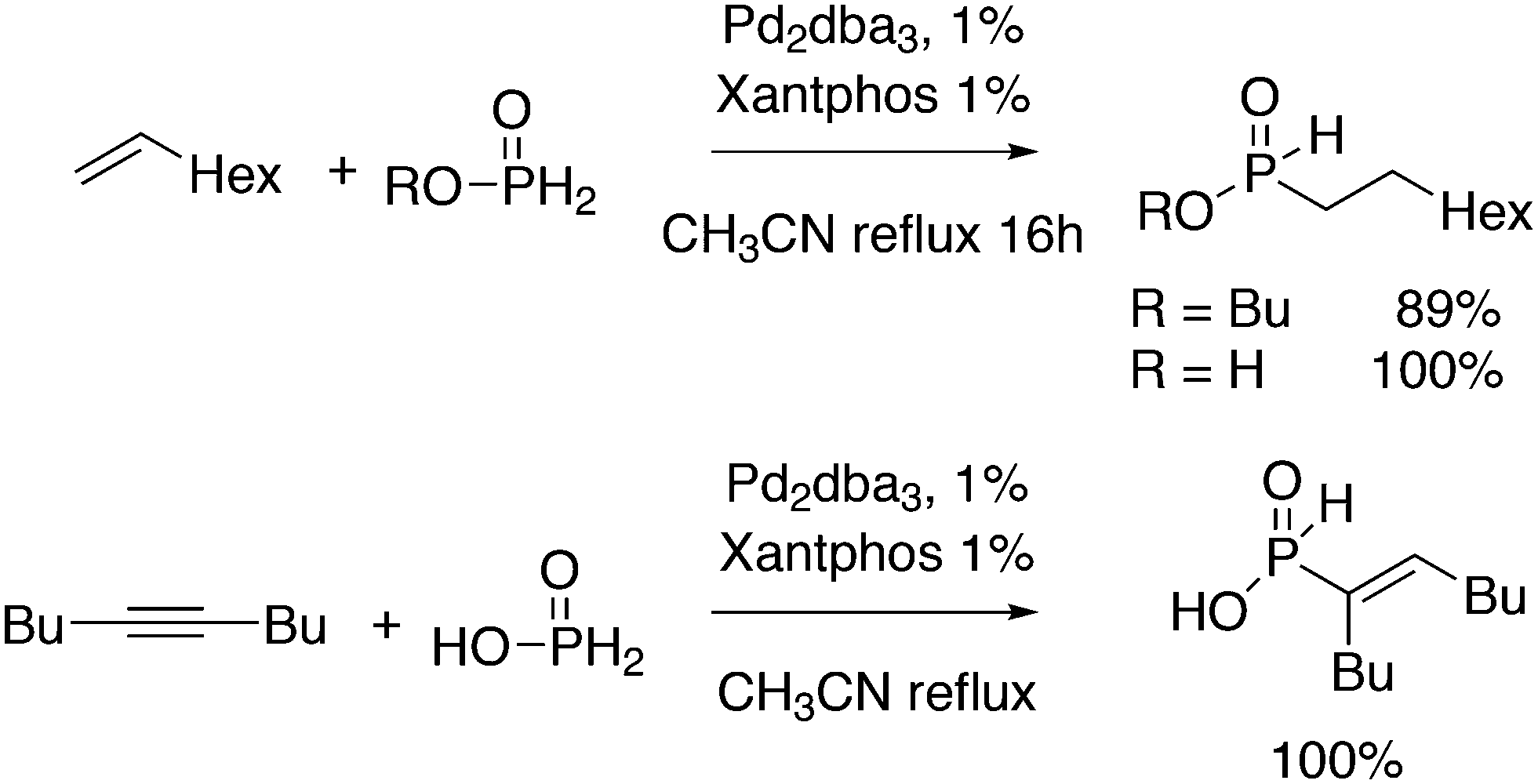 | ||
| Scheme 108 Addition of hypophosphorous compounds to alkynes and alkenes. | ||
In a comparison of Pd(0), with both Xantphos and dppf, and NiCl2 catalysts, the latter metal gave better results for internal alkynes, even in the absence of ligands, and the former Pd for alkenes.386 Presumably, the SPO substrate functions both as a ligand and reducing agent for Ni(II). Interestingly, commercially available aqueous H3PO2 can also be used and a polystyrene immobilized Nixantphos based catalyst also catalyses the reactions discussed here.387 In a tandem reaction in one-pot, the phosphinic acids can be oxidised to phosphonic acids by air.388
The regioselectivity in terminal alkyne phosphinylation could be steered by ligand and conditions for the Pd catalysed reaction (Scheme 109).389 With Pd/Xantphos in acetonitrile, the linear isomer was generally obtained with good selectivity and E-stereospecificity. On the other hand, by using Pd/dppf in the non-polar solvent toluene, good selectivity for the branched alkenyl-H-phosphinate was typically observed. Pd/Xantphos in toluene also gives the branched isomer as the major product (3/1) and thus the solvent plays an important role. The more electron-donating substituents on the terminal alkyne favor the formation of the branched isomer, whereas electron-withdrawing substituents favour the linear product.
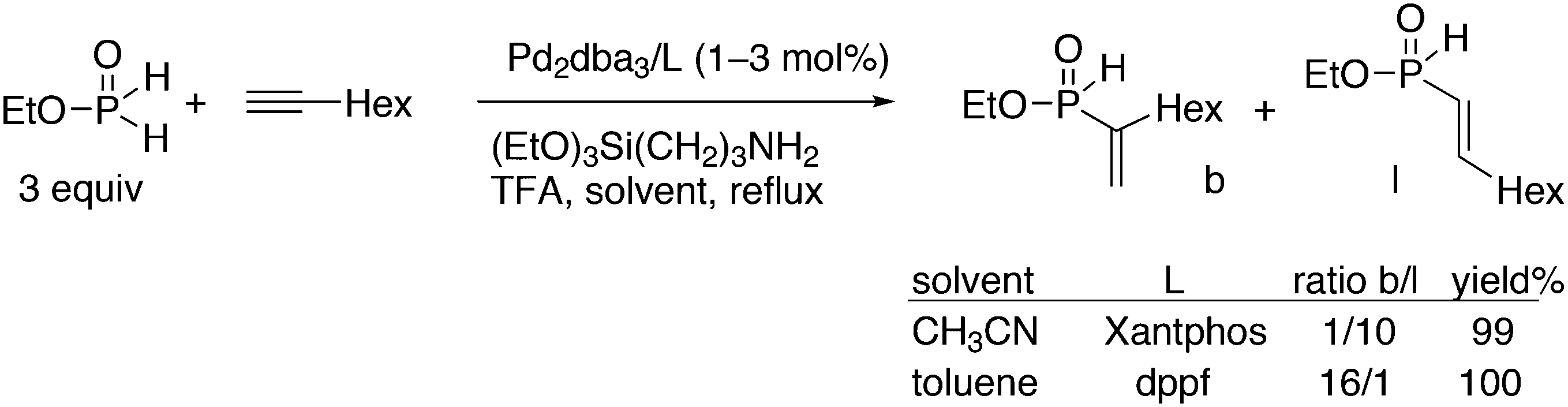 | ||
| Scheme 109 Regioselectivity in terminal alkyne phosphinylation. | ||
The desymmetrization of hypophosphorous esters was investigated using various strategies.390 Many chiral alcohols were tried in the reaction of Scheme 109 (see above, replacing ethoxy by chiral alcohols), followed by the Pd/Xantphos catalysed phosphinylation of 1-octene, but the results were modest. The use of chiral bisphosphines such as BINAP did not yield high ees, and thus work remains to be done here.
Allylic alcohols can be converted into allylphosphinites with hypophosphorous acid as shown in Scheme 110.391 First, the ester is formed by dehydration and the allyl ester is catalytically converted by Pd/Xantphos to the allylic-H-phosphinic acid. Mechanistically, this may not be akin to the hydrophosphinylation, but Xantphos behaves well in many allylic conversions (see 3.6). Likewise, benzylic alcohols cross-couple directly with concentrated H3PO2 by using Pd/Xantphos (1 mol%).392 Many examples have been reported and in several cases, the results were improved by a Dean–Stark distillation of t-amylalcohol. Chiral arylvinyl alcohols gave up to 77% retention of ee. Allylic alcohols were successfully replaced by 1,2 and 1,3-dienes, as is not uncommon in palladium-allyl chemistry.393 The catalytic system Pd2dba3/Xantphos is crucial to avoid or minimize the competitive reductive transfer hydrogenation pathway available to hypophosphorous acid derivatives.
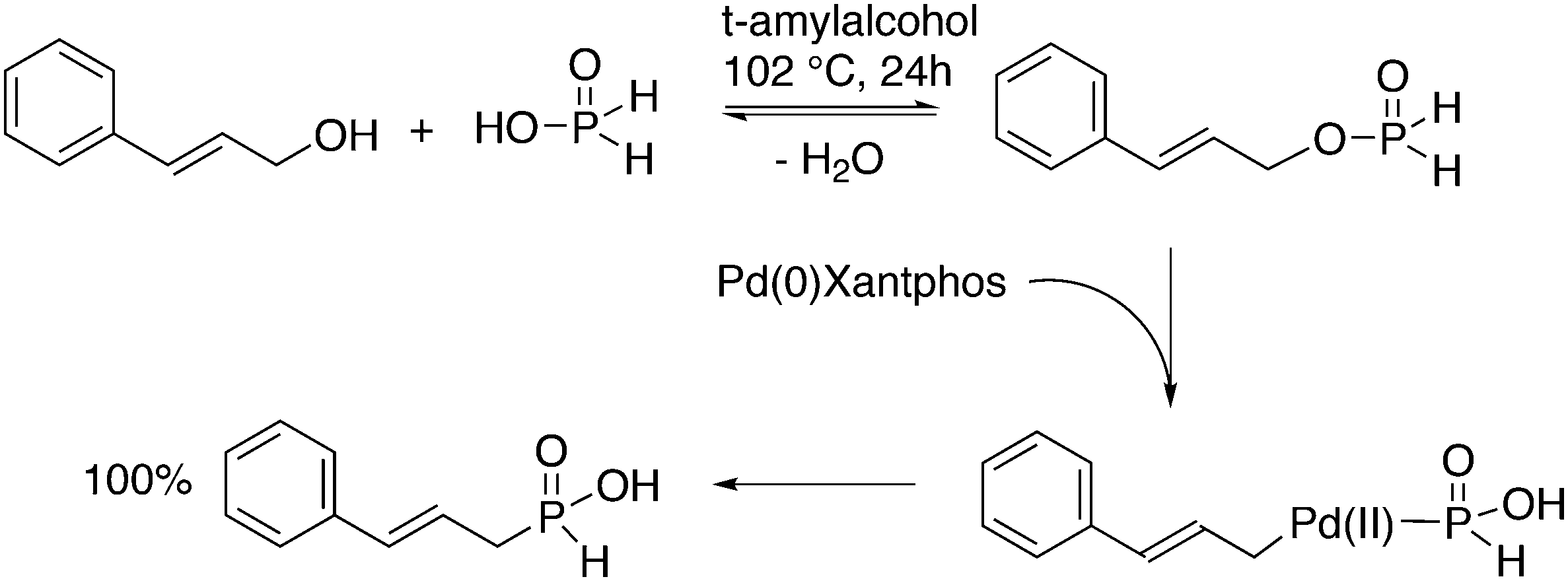 | ||
| Scheme 110 Formation of allylic-H-phosphinic acids. | ||
H-Phenylphosphinic acids and allyl alcohols were reacted with the same catalyst system to give allylphenyl phosphinic acids, which again demonstrates the versatility of this method (Scheme 111).394 Mol sieves can be used to remove water or a Dean–Stark trap can be applied.395
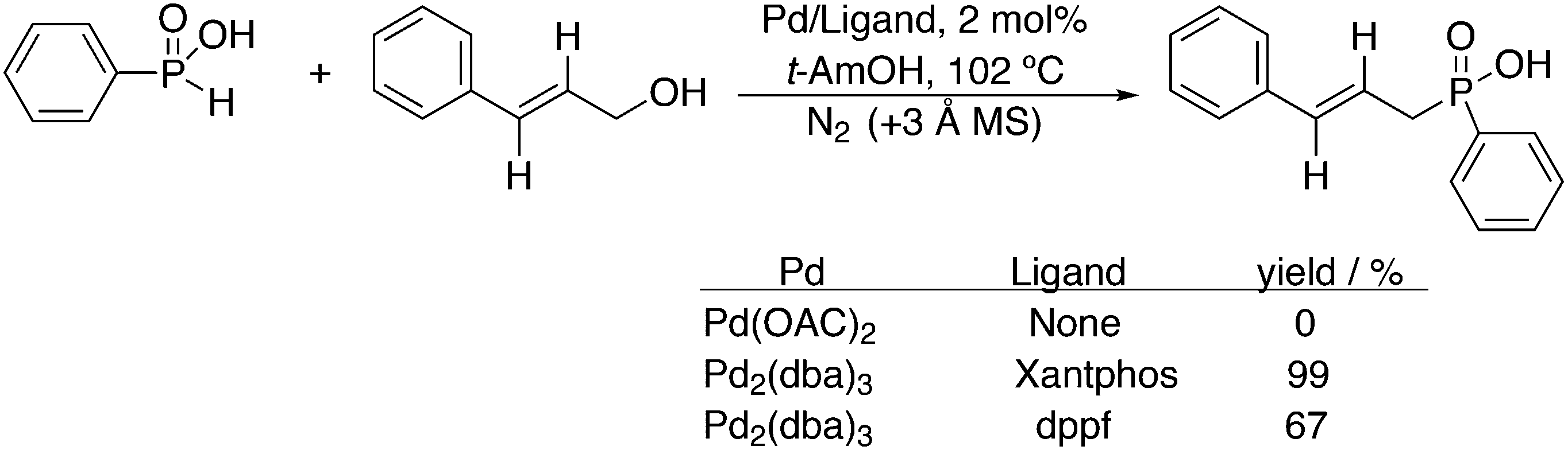 | ||
| Scheme 111 Allylation of H-phenylphosphinic acids. | ||
The rich palette of phosphinations with hypophosphorous acid (and salts) as developed by Montchamp and others was utilized by Sicken and co-workers at Clariant for the preparation of a wide variety of functionalized hydrocarbylphosphinic acid derivatives used in the production of fireproofing agents. This was reported in more than 20 patents, which mention tppts, Xantphos and Sulfoxantphos as the favoured ligands.396
In addition to the above reactions of hypophosphorous compounds, the latter can also be used in cross-coupling reactions with aryl halides (Scheme 112).397 Microwave heating improved the results. Reaction of the H-phosphinic acid with another molecule of aryl bromide afforded diarylphosphinic acids in good yields.
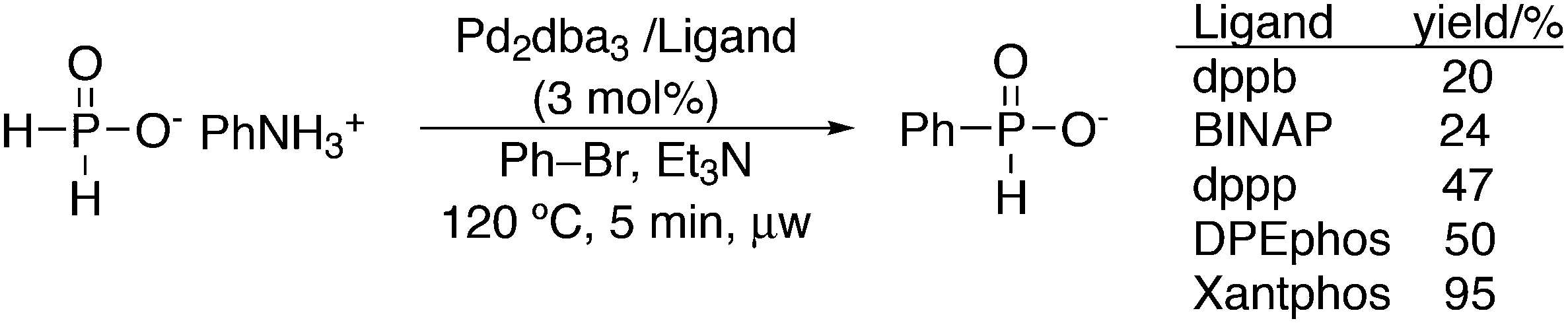 | ||
| Scheme 112 Cross-coupling of hypophosphorous acid (or ester, salt) to dihydrocarbylphosphinic acid. | ||
Benzyl halides could also be used as halide coupling agents with diethyl H-phosphinates with the same Pd/Xantphos catalyst.398 Mechanistic studies were conducted for the last reaction, which showed that benzyl halides oxidatively added to Pd(0) and that most likely the phosphinate anion replaced the halide on Pd to give the product by reductive elimination, disgarding a mechanism of outer-sphere attack. Chlorides and bromides reacted with similar rates, but for chlorides the o.a. was rate limiting, while for bromides the combination of r.e./ligand replacement was rate limiting. PPh3 underwent benzylphosphonium formation and showed a slower oxidative addition, thus explaining why it gives a poor catalyst.
Deelman and co-workers used sodium phosphinite (or sodium phosphinate) as the starting material to make diarylphosphinic acids (or sodium diarylphosphinate) via a cross-coupling protocol similar to the one above.399 The effect of the bidentate phosphine ligands followed the same trend and toluene at reflux was found to be the best solvent.
Bloomfield and Herzon reported the smooth reaction of SPOs with aryl iodides for the syntheses of trihydrocarbylphosphine oxides with Pd/Xantphos in yields of 70–97% (Scheme 113).400 For the reaction of Me2PH(O) and iodobenzene, a range of ligands were screened and the conversion increased with bite angle, as the reported by the authors (BINAP 2%; JosiPhos 0%; dppf 85%; Xantphos 97%). The reaction is shown to be amenable to diaryl, dialkyl, and alkylaryl SPOs as well as dialkylphosphites and in its current form, it is limited to iodides, albeit a broad scope of aryl iodides. The reaction was shown to proceed with retention of stereochemistry at phosphorus.
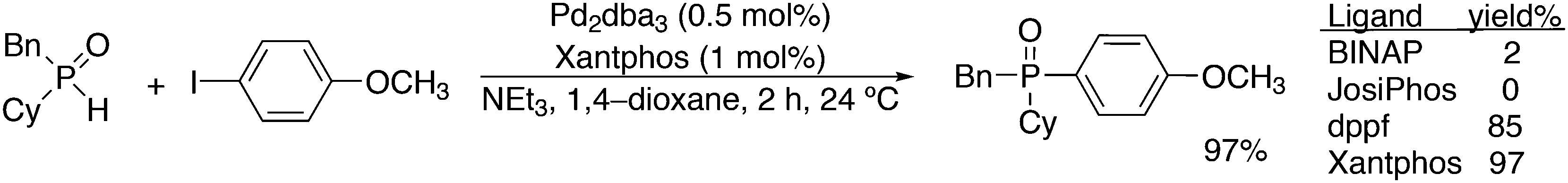 | ||
| Scheme 113 Cross-coupling of SPOs with aryl iodides. | ||
Aryl iodides and aryl thiols were effectively coupled by Schopfer and Schlapbach with a Pd2(dba)3 and DPEphos catalyst system and KOtBu as the base at 100 °C in toluene.403 Heteroaromatic iodides and heteroaromatic thiols were also efficiently converted to the coupling products with this catalyst. Murata and Buchwald reported the system Pd(OAc)2/DiPPF (1,1′-bis(diisopropylphosphino)ferrocene) for the coupling of C–S and C–P bonds (Scheme 114).404 A broad substrate scope was found, since both aliphatic and aromatic thiols can be coupled to many aryl bromides and chlorides. DPEphos was only effective for aryl bromides, since aryl chlorides do not oxidatively add to Pd(0) ligated by arylphosphines. Bidentate phosphines were needed for a stable catalyst since Buchwald's diarylphosphines gave no conversion. Using cesium carbonate as the base, Zhang and co-workers found that meso-brominated porphyrins could be coupled with alkyl and aryl thiols in the presence of both diarylphosphines and DPEphos (and the like).405
 | ||
| Scheme 114 Pd-Catalysed C–S bond formation. | ||
Catalysts based on Josiphos 84 gave rates up to two orders of magnitude higher than DiPPF as was reported by Hartwig et al.406 Substrate scope is very broad; for the system shown above and in almost all cases, chlorides could be used as the halide precursor. Homocoupling products were occasionally obtained as by-products, the amounts of which could be suppressed by using KOtBu, Pd(dba)2 and toluene.
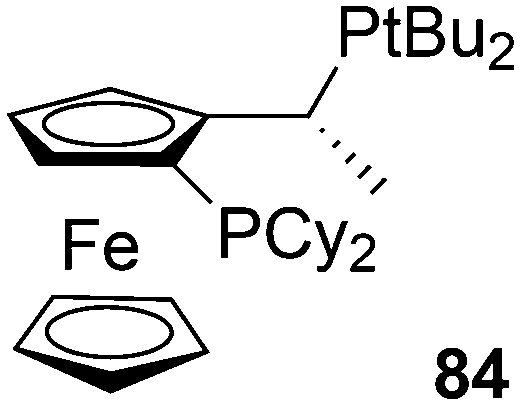
The evaluation of various catalytic systems for cross-coupling of n-BuSH and PhOTf with bidentates such as BINAP, dppp, Xantphos and DPEphos indicated that BINAP and Xantphos were the best ligands under the conditions tested (tBuONa as the base, Pd2(dba)3).407 Xantphos containing tBu groups was tried, but this catalyst gave no reaction; apparently, this electron-rich ligand is too bulky to undergo oxidative addition to its Pd complex. In a study with i-Pr2NEt as the base in dioxane, Xantphos performed the best, although it was orders of magnitude slower than the Josiphos based system mentioned above.408 Bromides as well as aryl triflates could be used.
Messaoudi and co-workers used the third generation of aminobiphenyl palladacycle pre-catalyst G3-Xantphos for the functionalization of peptides containing cysteine in high yields.409 The conjugation (bioconjugation) occurs chemoselectively at room temperature under biocompatible conditions. Scheme 115 shows a model reaction and Buchwald's Pd–G3-Xantphos.
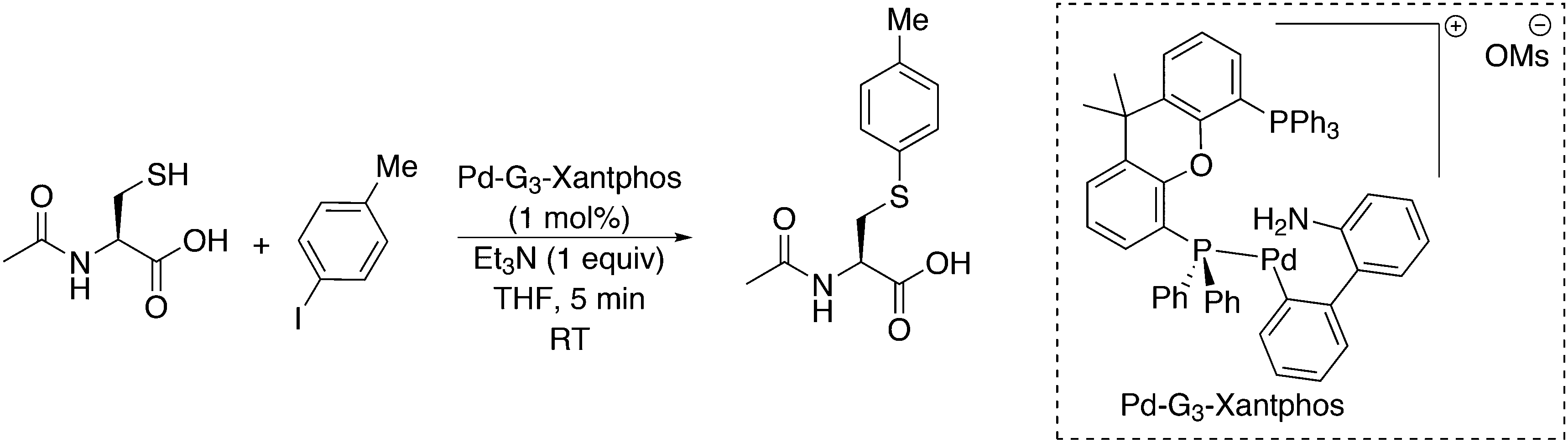 | ||
| Scheme 115 Model reaction of biocompatible C–S coupling. | ||
The application of C–S coupling for a pharmaceutical product increases the requirements for the purity and practicality of the methods and reagents used. Detailed analyses of the product may show traces of homocoupling, traces of phenyl coupled product due to phenyl(phosphine) and aryl(Pd) exchange. The introduction of sulfur in one of the components should occur via an odour free route. Products and side products must be separated by crystallisation rather than column chromatography and catalyst residues must be reduced to ppm levels. An interesting example can be found in the work by Yu et al. from Pfizer (Scheme 116) for the formation of PF-04191834, made on a scale of 20 kg.410 In this instance Xantphos was the ligand of choice.
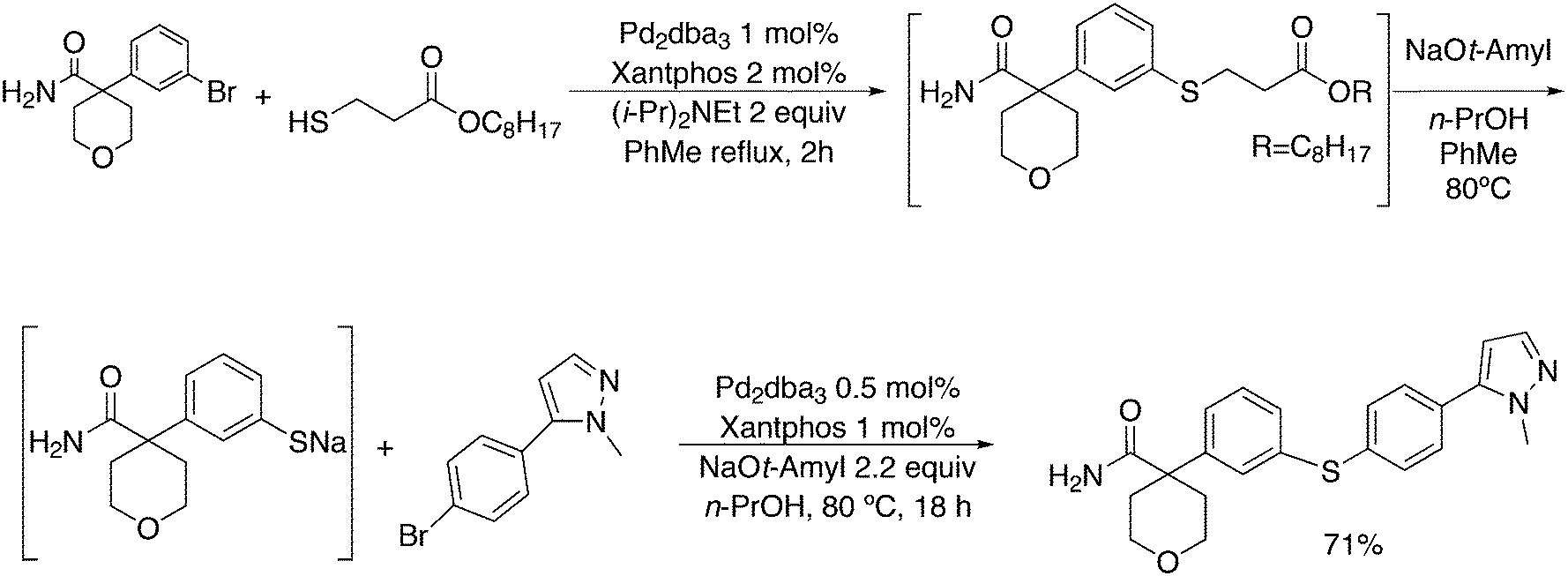 | ||
| Scheme 116 Pfizer's route for the formation of PF-04191834. | ||
Both Schmink411 and Walsh412 reported that the well-known Umpolung effect of 2-aryl-1,3-dithianes could be used for the Pd/Nixantphos catalysed arylation at C-2 (Scheme 117, reaction b) to eventually yield diarylketones or diarylmethanes. When applied to 2-benzyl-1,3-dithianes, a ring-opening took place (reaction a), and a new C–S bond was formed.413 Extension of the reaction to 2-diphenyl-1,3-oxathiolane led to a further surprise (reaction c) because instead of the expected 2,2-diaryl-1,3-oxathiolane, the products found were vinyl aryl sulfides.414 The system with Nixantphos as the preferred ligand was further improved by using 2,2-dimethyl-1,3-oxathiolane, which produced acetone as the co-product. The vinyl sulphides thus obtained can be used as precursors for thiols in coupling reactions, as has also been reported by Walsh for benzyl sulphides.415 The Pd/Xantphos systems therefore show reversible C–S bond formation, which leads to scrambling.
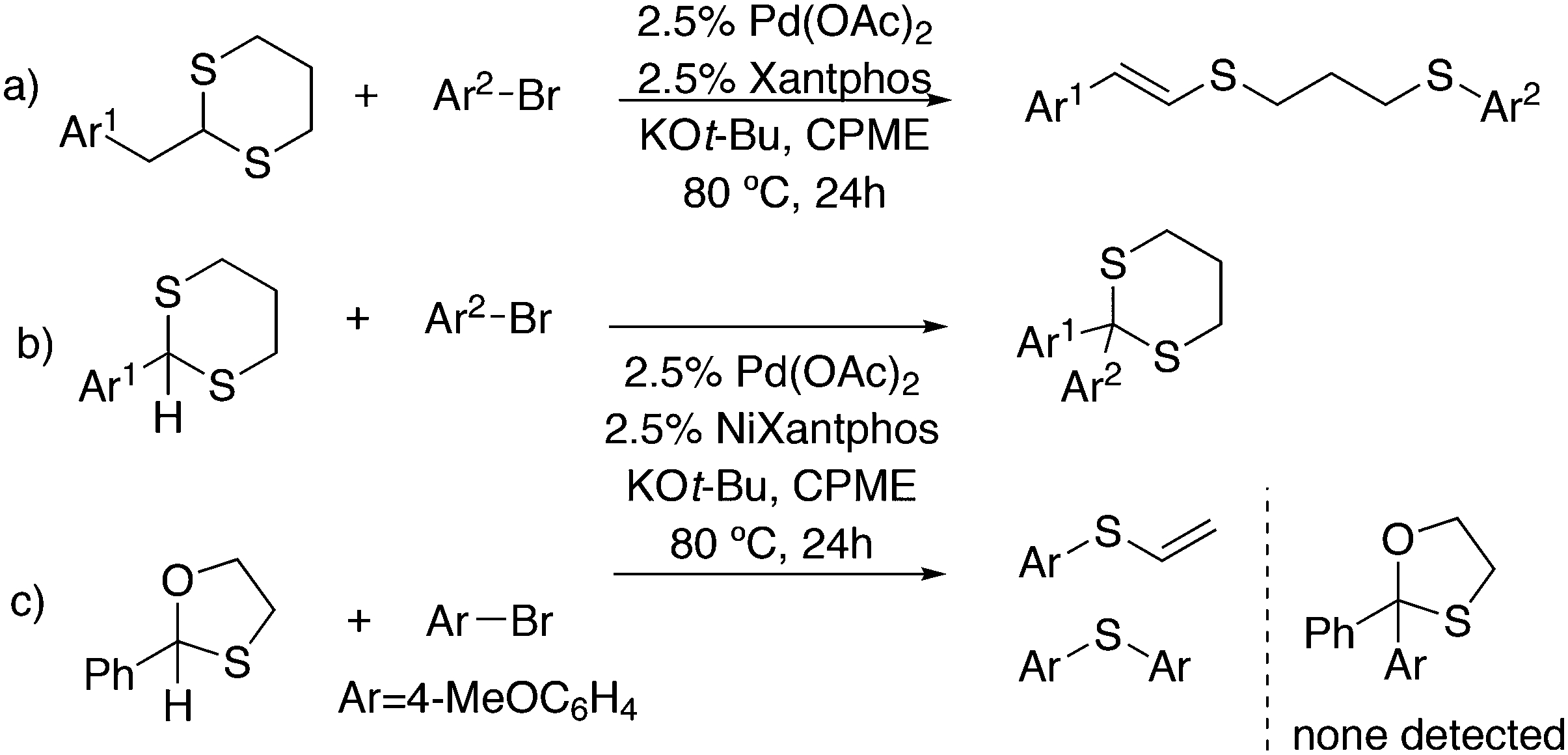 | ||
| Scheme 117 1,3-Dithianes (a and b) and 2,2-dimethyl-1,3-oxathiolane (c) as precursors. | ||
Easily accessible benzyl thioacetates can be used as precursors for benzyl thiol as was shown by Wager and Daniels.416 This system of Pd/Xantphos was particularly prone to Ph/Aryl exchange (Scheme 118) as mentioned above, which can sometimes be reduced by using the right base and solvent. These authors have shown that in this instance, Xphos was an effective ligand in suppressing the scrambling.
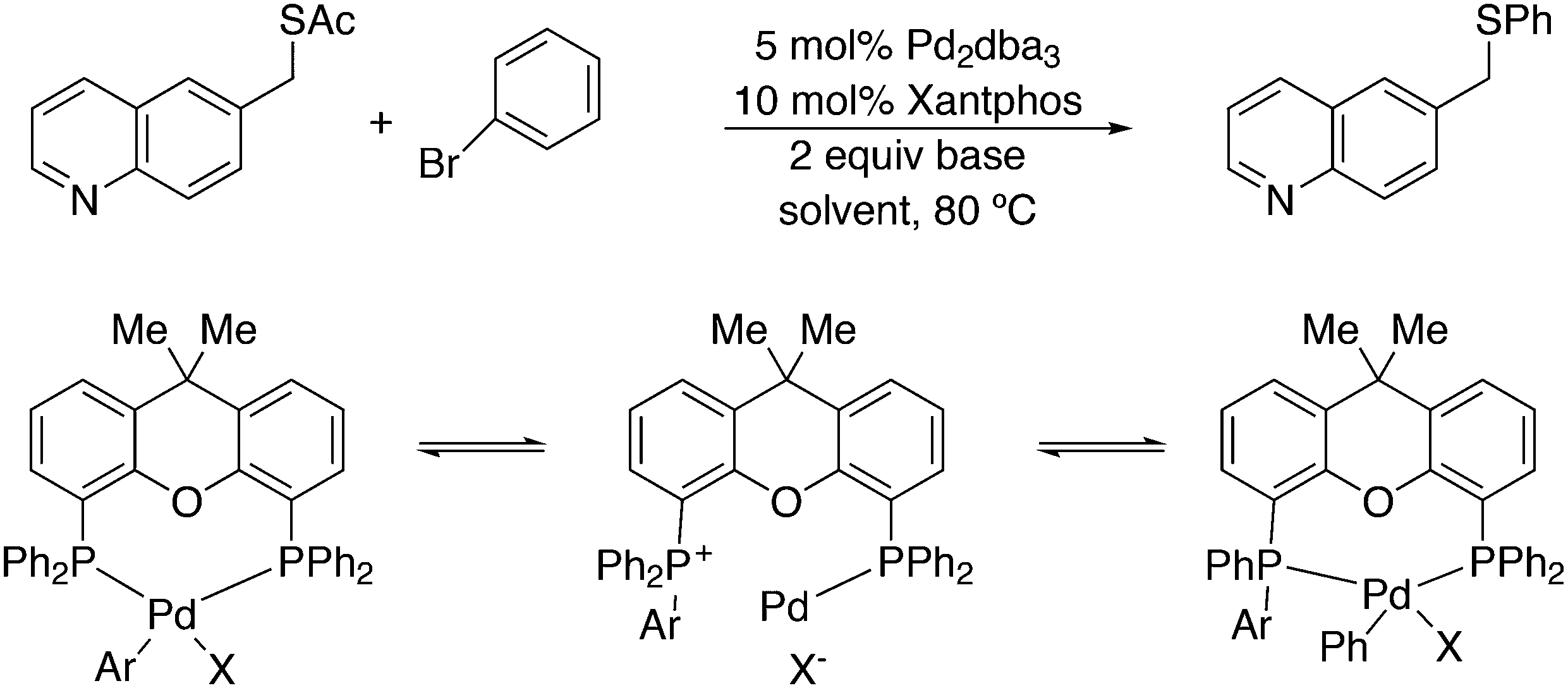 | ||
| Scheme 118 Benzyl thioacetates as precursors, and aryl/phenyl scrambling. | ||
Kleij and co-workers reported a method for the stereoselective preparation of highly functionalized allylic thioethers. This procedure uses a Pd-catalysed thiolation of vinyl cyclic carbonate substrates and features high (Z)-selectivity, good yields, minimal waste, and ample product scope as all three R groups can be varied (Scheme 119).417 DPEphos and Xantphos gave high yields and dppf gave no reaction.
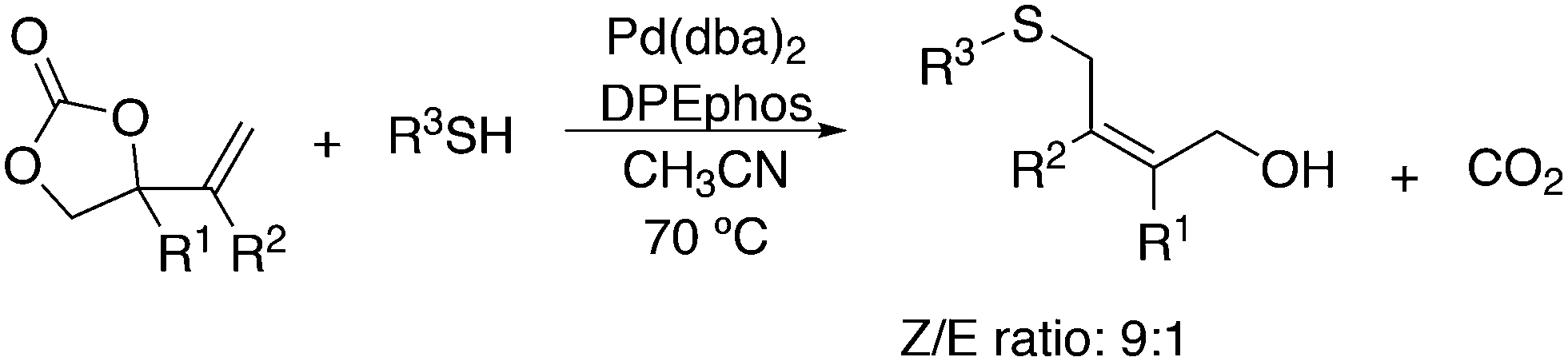 | ||
| Scheme 119 Decarboxylative functionalization of vinyl carbonates with thiols. | ||
The palladium-catalysed reaction of sulfinic acid salts with a wide variety of aryl and vinyl halides or triflates provides unsymmetrical diaryl sulfones and aryl vinyl sulfones in good to excellent yields (Scheme 120).418 The reaction is strongly influenced by the presence of nBu4NCl, and the use of Xantphos was found to be crucial for the success of the reaction. The example shown in Scheme 120 can be used for a subsequent coupling reaction.
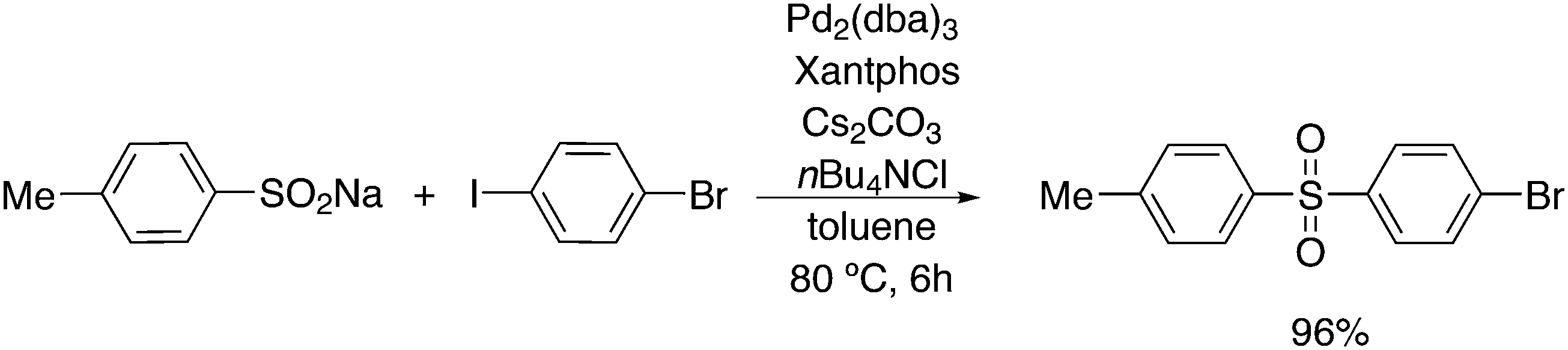 | ||
| Scheme 120 Synthesis of diaryl sulfones. | ||
Willis et al. used the DABCO adduct of SO2 (DABSO) as the source for generating sulfinic acid salts from aryllithium compounds.419 The cross-coupling with aryl halides (Scheme 121) follows the procedure described above. Since substantial incorporation of phenyl groups in the products was observed, the authors studied the ligand effect on this aryl exchange and they found that by using EWG-modified aryl groups on Xantphos (3,5-(CF3)2C6H4), the scrambling could be largely suppressed. Over 90% of the 37 examples given of diaryl sulfones were new compounds, which underscores the power of the new method.
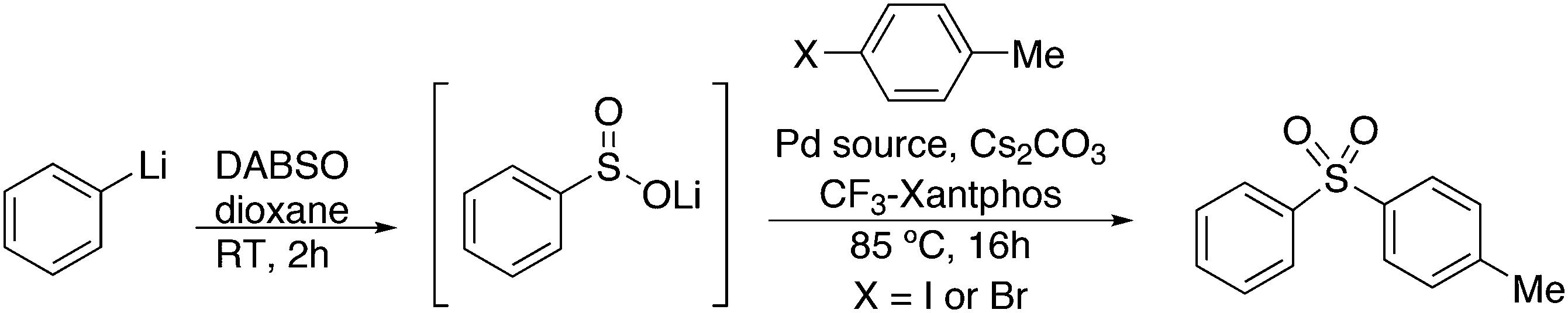 | ||
| Scheme 121 DABSO based sulfone synthesis. | ||
While the use of sulfur-based nucleophiles such as sulfinates and thiolates allows the easy generation of aryl sulfones and aryl thioethers, no related method allowing the generation of aryl sulfoxides via arylation of a sulfenate was available because the synthesis is more difficult. To this end, Madec, Poli and co-workers developed a method based on the in situ generation of the sulfenate in a retro-Michael reaction (Scheme 122);420 aryl/phenyl exchange with the ligand was also observed. The yields were modest to high, depending on the electronic properties of the aryl group (EWGs giving high yields). PPh3, dppe and BINAP gave no conversion when used as the ligand. Aryl bromides were not reactive and apparently, the reagents present inhibited the oxidative addition of the bromides. For heteroaromatics, the bromides were found to be more reactive.421
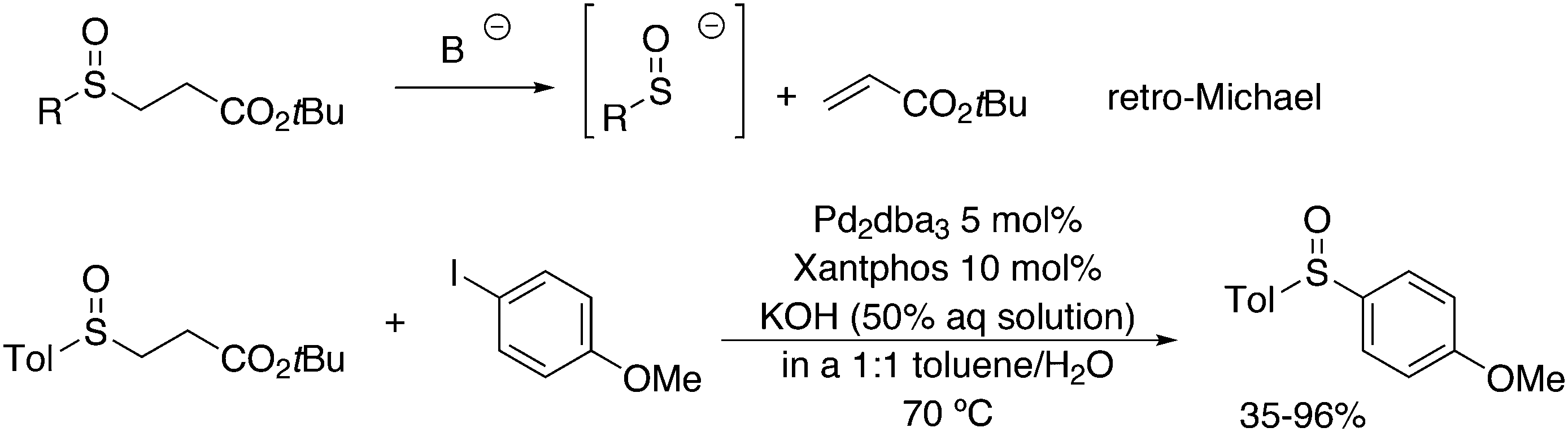 | ||
| Scheme 122 Synthesis of sulfoxides with sulfenates generated by retro-Michael reaction. | ||
During their studies on C–H arylation of benzyl sulfoxides Walsh and co-workers discovered that in addition to the expected product, diaryl sulfoxides were also obtained (Scheme 123).422,423 The reaction was further optimized to give diaryl sulfoxides in high yields. Nixantphos was the only ligand that gave excellent results. As the scheme shows, two equivalents of aryl halide are consumed. The catalyst is involved in three reactions, C–H arylation, sulfenate formation, and cross-coupling of the latter with aryl halide. The key step is S-arylation of a sulfenate anion.
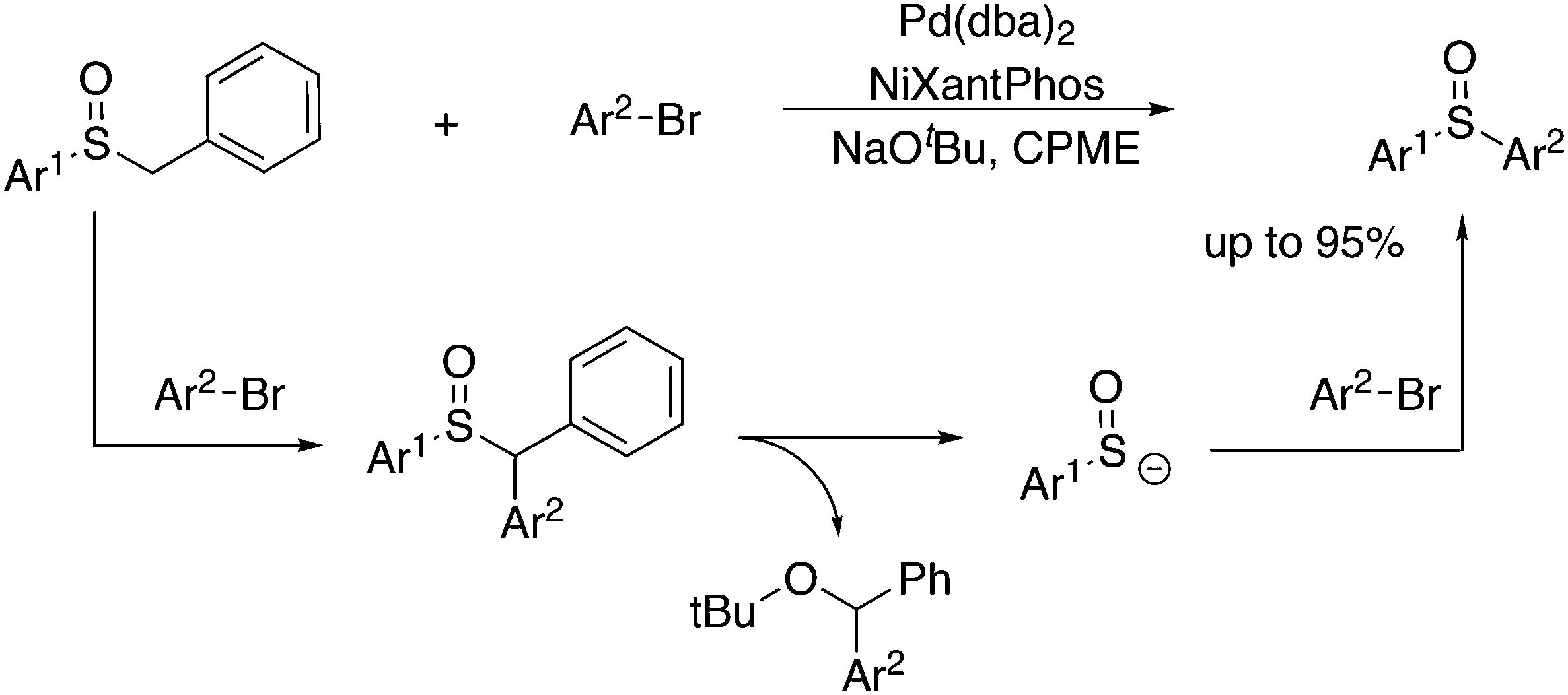 | ||
| Scheme 123 Sulfoxide synthesis with Pd/Nixantphos. | ||
A method for the preparation of S-aryl thioacetates was developed by Lai and Backes, using palladium-mediated couplings of aryl bromides and aryl triflates with potassium thioacetate as an inexpensive thiol source (Scheme 124).424 The resulting S-aryl thioacetates are versatile intermediates from which a number of sulfur-containing functional groups including arylthiols, aryl sulfenyl chlorides, aryl sulfinyl chlorides, and aryl alkyl sulfides can be accessed.
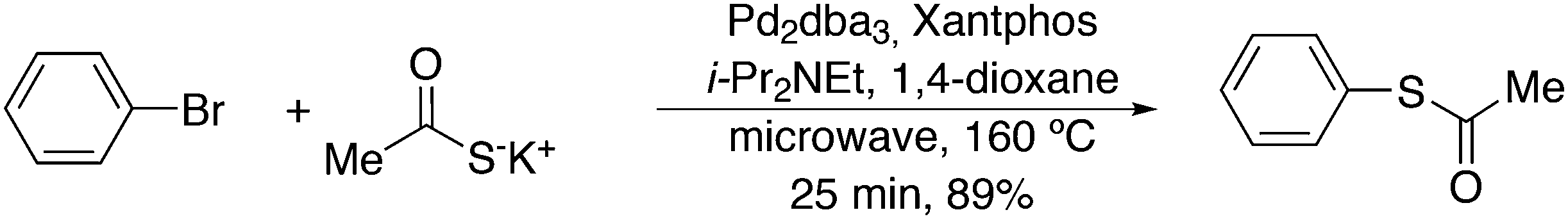 | ||
| Scheme 124 S-Aryl thioacetate synthesis. | ||
Regarding C–O bond forming reactions, the formation of vinyl esters is a desirable reaction in industry. The addition of a carboxylic acid to acetylene is a useful reaction for which zinc salts are used as catalysts, which replaced the now abandoned mercury catalysts, but neither are very active. Kimmich and co-workers reported a platinum acetylacetonate precursor and Xantphos as a good catalyst selected from a wide range of transition metal salts and complexes and phosphine ligands (Scheme 125).425
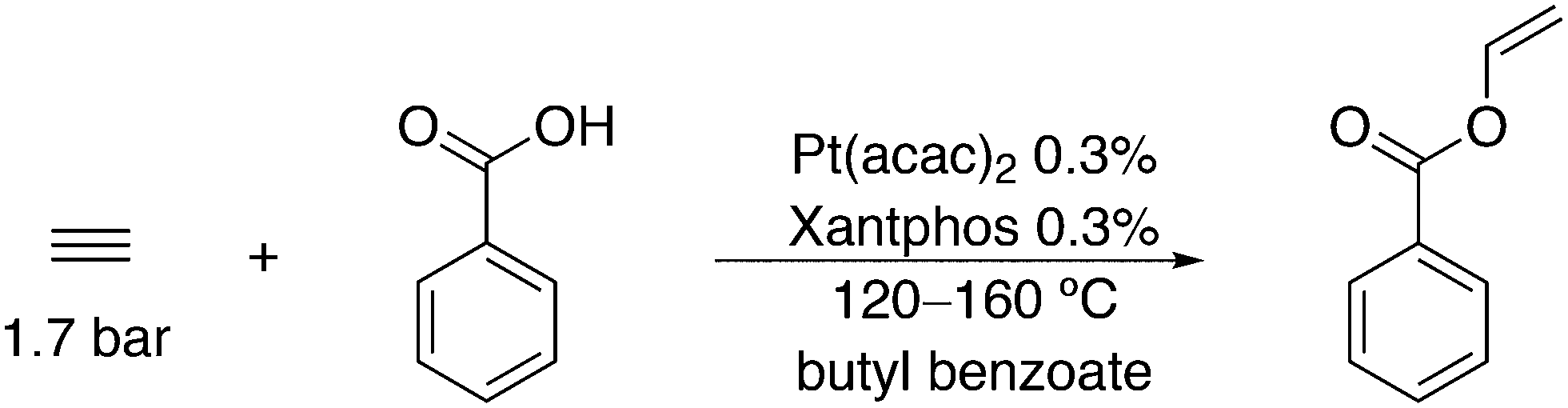 | ||
| Scheme 125 Vinyl benzoate synthesis. | ||
Tuning the properties of arylpalladium intermediates by the phosphine ligands and anions enables control of the catalytic arylative 5-exo and 6-endo cyclizations of alkynols (Scheme 126).426 The two modes of cyclizations represent a rare example of controllable, regioselective difunctionalization of alkynes and the most selective ligands are Xantphos for the endo coupling and Xphos for the syn–exo coupling, the latter being the common reaction product. The authors suggest that electronic differences change the coordination mode in the reaction intermediates as shown in Scheme 126.
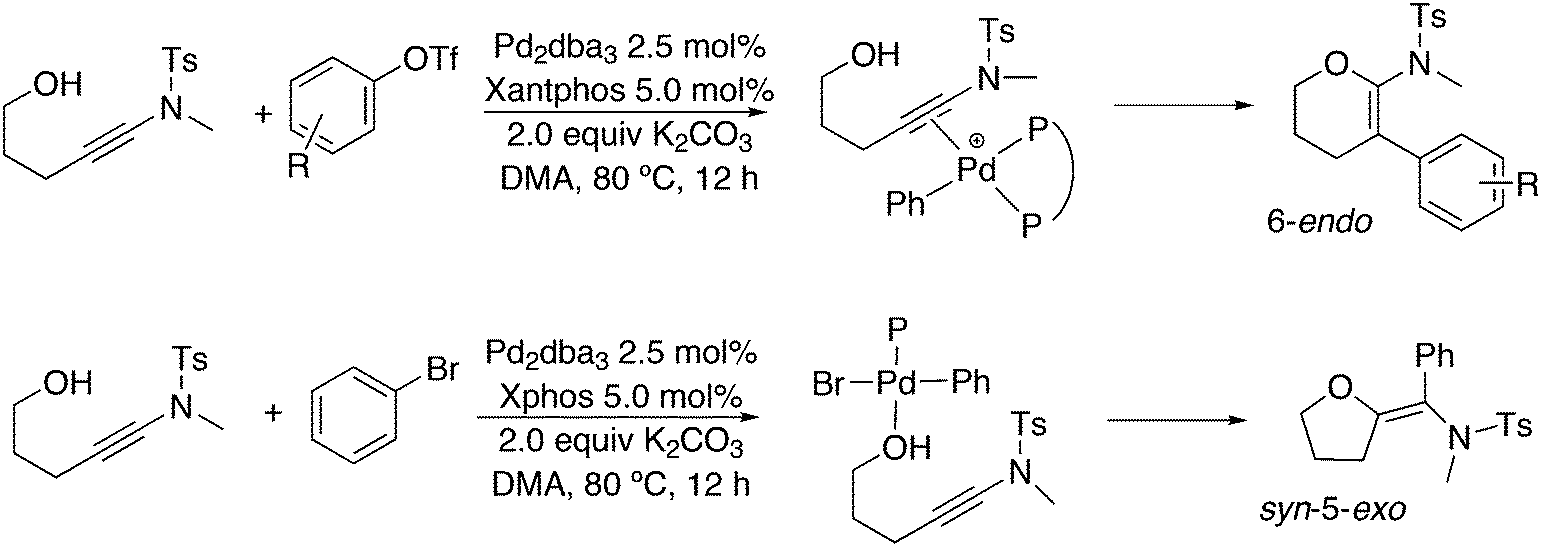 | ||
| Scheme 126 Ligand controlled ring-closure of alkynols. | ||
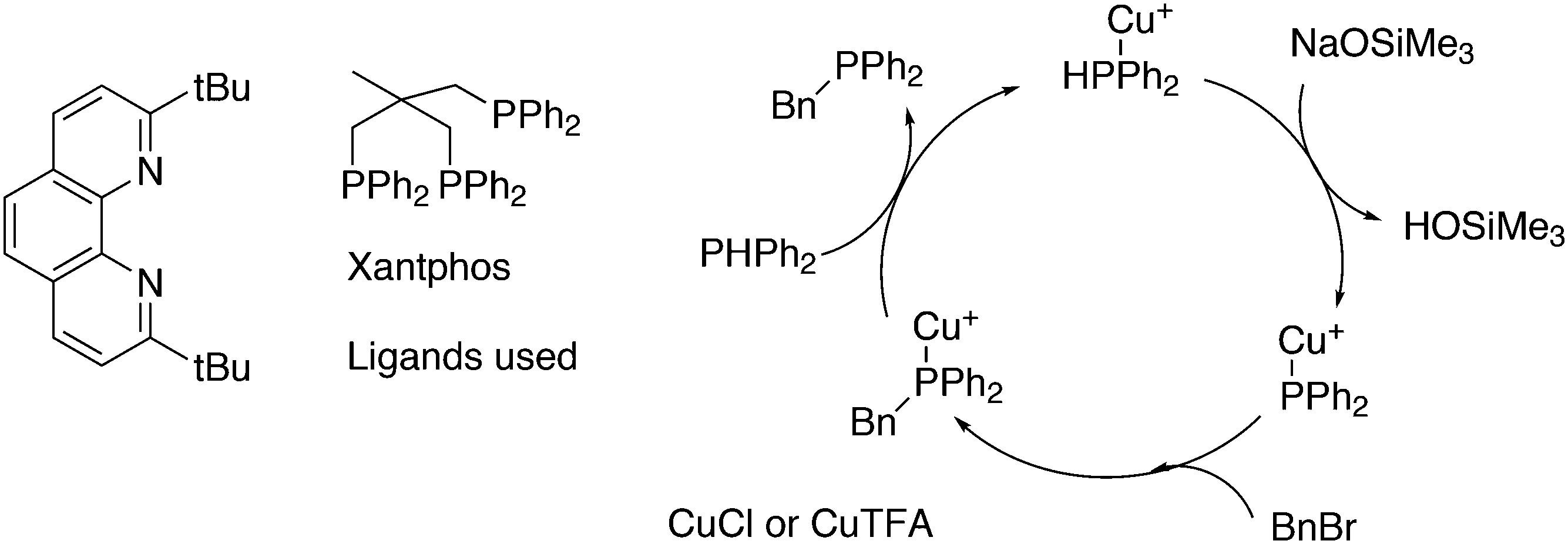 | ||
| Scheme 127 Cu catalysed alkylation of PPh2H. | ||
α,β-Alkynoates can be borylated by B2pin2 in alcohols, usually in a stereoselective manner, by Cu(I) catalysts (Scheme 128).428 Preferred ligands are DPEphos and Xantphos, followed at close distance by dppf, which is slower and/or less selective. The proposed mechanism involves reaction of Cu alkoxide with the bisboranate, giving a Cu–Bpin species and MeOBpin co-product. Then, insertion of the alkyne takes place, or the copper boryl complex conjugatively adds to the alkyne, followed by Cu–C cleavage by the alcohol. Further examples have been publishedby the Yun group.429
 | ||
| Scheme 128 Borylation of alkynoates. | ||
Likewise, Bpin can be added to silylallenes with high selectivity to the alkene isomer, as shown in Scheme 129.430 Monodentate phosphines of various electronic and steric natures all gave <13% conversion. DPEphos and Xantphos gave high conversions, but only Xantphos showed high stereoselectivity.
 | ||
| Scheme 129 Addition of Bpin to silylallenes. | ||
Instead of adding B2pin2/MeOH to allenes, one can also do a more Bpin efficient hydroboration reaction with HBpin as demonstrated by Tsuji and co-workers (Scheme 130).431 A very subtle ligand effect was observed for the p-methylphenyl and p-CF3-phenyl substituted Xantphos, which gave higher yields than Xantphos, but showed a slightly lower E/Z selectivity. NHC ligands gave less satisfactory results.
 | ||
| Scheme 130 Hydroboration with HBpin. | ||
More recently, Lee, Yun and co-workers reported highly selective hydroboration of terminal alkynes, for which the stereoselectivity was controlled by the ligand, with impressive high values (Scheme 131) and high yields based on HBdan.432 Mechanistically, CuH adds to the alkyne and the Cu-vinyl intermediate can rapidly isomerize; the σ-bond metathesis reaction with danB–H strongly depends on the ligand in regard to the stereo outcome.
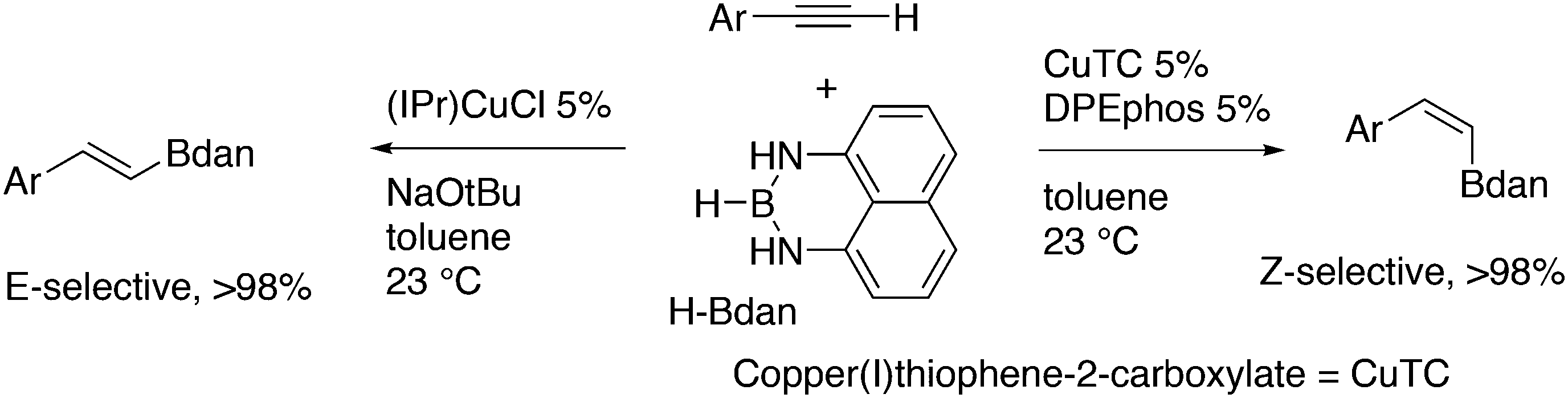 | ||
| Scheme 131 Stereoselective hydroborylation of terminal alkynes. | ||
Propargylic carbonates were borylated as shown in Scheme 132 in high yields.433 The preferred ligand stems from earlier work of the group with related Cu catalysis.
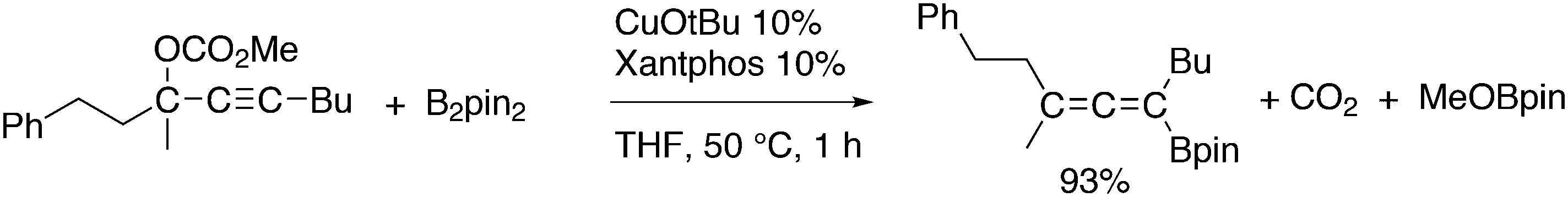 | ||
| Scheme 132 Reaction of propargylic carbonates with Cu–Bpin. | ||
When basically the same reaction was carried out with propargylic alcohols, allyl alcohols, and benzyl alcohols, the oxygen atom was efficiently replaced by Bpin, provided that the alcohol was stoichiometrically converted to the titanium alkoxide (Scheme 133).434 No addition to the alkyne occurred as was observed in the systems shown above in Scheme 132.
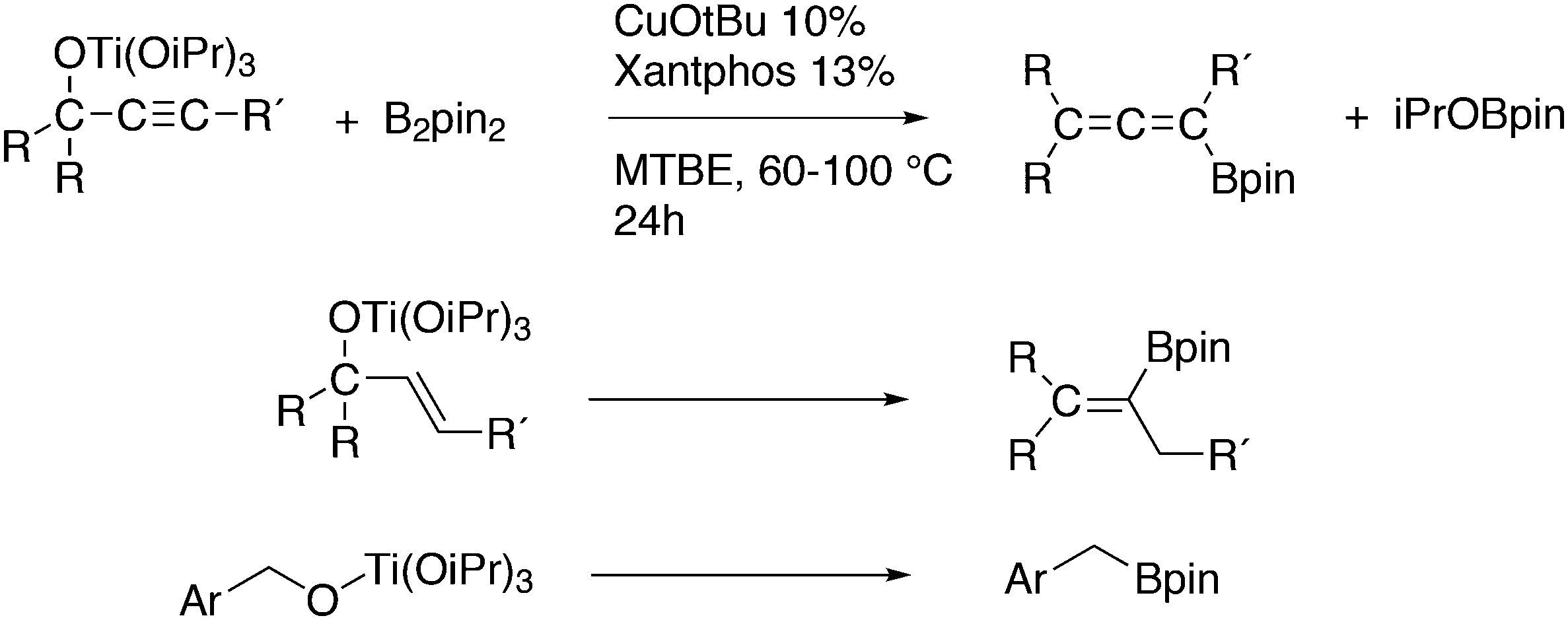 | ||
| Scheme 133 Borylation of alcohols with the aid of Ti alkoxides. | ||
Allyl carbonates can be borylated in high yields by B2pin2 and Cu/Xantphos catalysts and base (Scheme 134).435 The main product is E product 85.
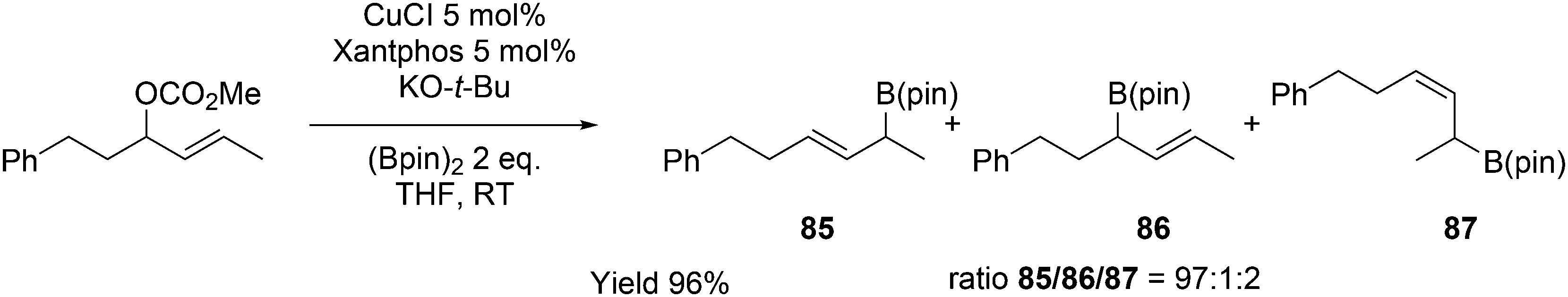 | ||
| Scheme 134 Borylation of allyl carbonates. | ||
Optically active carbonates retained high chirality, and in prochiral precursors high enantioselectivities could be induced with R,R-QuinoxP* 88 as the chiral ligand.
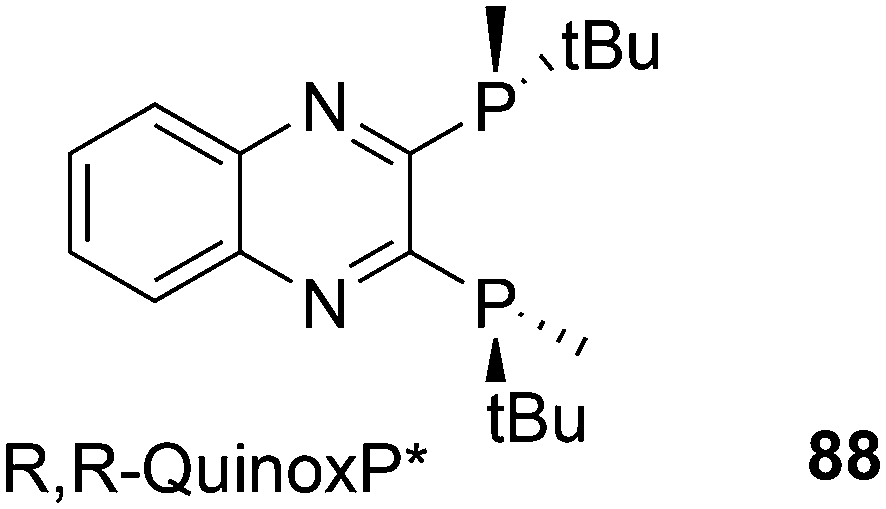
In the course of these investigations Ito and Sawamura, et al. discovered an interesting reaction when silyl substituted allyl precursors were used (Scheme 135).436 The tentative explanation given is that the Cu–B fragment adds to the double bond with Cu at the γ-position of the allyl bond, geminal to Si. Elimination of MeOBpin and CO2 leads to the cyclopropane product.
 | ||
| Scheme 135 Cyclopropane formation from silyl substituted allyl carbonates. | ||
So far, we have seen addition of Bpin/H to unsaturates, but interestingly, Xiao and Fu, et al. developed a protocol for the addition of Bpin/alkyl to a double bond (Scheme 136).437 In this approach, alkyl copper intermediates are generated in situ by the migratory insertion of CuBpin complexes across alkenes; these then react with alkyl electrophiles. Dppf gave less than 5% yield and Xantphos gave 57% to the Markovnikov adduct, but cyclohexyl substituted Xantphos gave yields up to 96% and anti-Markovnikov product selectivity of 94%. The DMF solvent played an important role; in toluene and THF there was hardly any reaction.
 | ||
| Scheme 136 Alkylation/boranation of alkenes. | ||
 | ||
| Scheme 137 Observed intermediate in Rh(Xantphos) B–N coupling, 89, and the proposed intermediate, 90, for dehydrogenation. | ||
The [Rh(Xantphos)]+ fragment was found to be an effective catalyst for the hydroboration of tBu-ethene using the amine–borane H3B·NMe3 at low (0.5 mol%) catalyst loadings. Investigations into the mechanism using the initial rate method and labelling studies showed that reductive elimination of the linear hydroboration product is likely the rate-limiting step.440
Chatanie and co-workers found that aryl cyanides could be made to react with (Bpin)2 and Rh–Xantphos catalyst to produce arylboronates in good to high yields (Scheme 138).441 Monophosphines and several bisphosphines gave lower conversion, but PPh3 and BINAP were good catalysts, coming in behind Xantphos. The authors report that the reaction involves an unprecedented C–CN bond activation that is promoted by a borylrhodium complex. The step-by-step mechanism was investigated by Esteruelas et al. and all intermediates were identified when i-propyl Xantphos was used.442
 | ||
| Scheme 138 Borylation of aryl cyanides. | ||
Rh–H complex 91 activates a variety of X–H bonds, including B–H in BpinH (Scheme 139)443 and C–H.444,445 This is a key intermediate in iPr-Xantphos hydride chemistry.
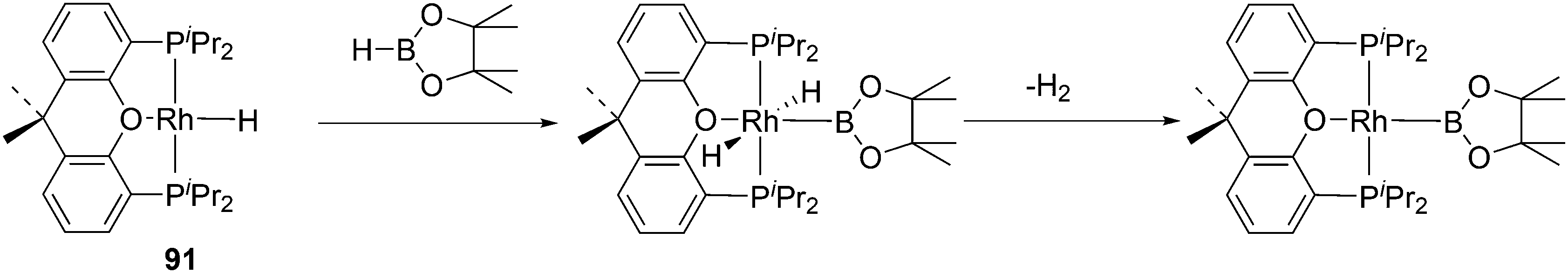 | ||
| Scheme 139 Intermediates in Rh/Xantphos hydroboration. | ||
Another C–X bond forming reaction catalysed by Rh is the formation of branched allylic esters from alkynes and carboxylic esters as reported by Breit and co-workers.446 The isomer distribution of the products is ligand dependent and DPEphos had the highest selectivity for the branched product (Scheme 140). An intermediate allene was proposed to be invoked in the mechanism after oxidative addition of the acid to Rh(I) to provide a RhH species, but after a thorough investigation, protonation of the alkyne was proposed as the crucial step (Scheme 141). The intermediate shown isomerizes to an allyl species via an allene complex.
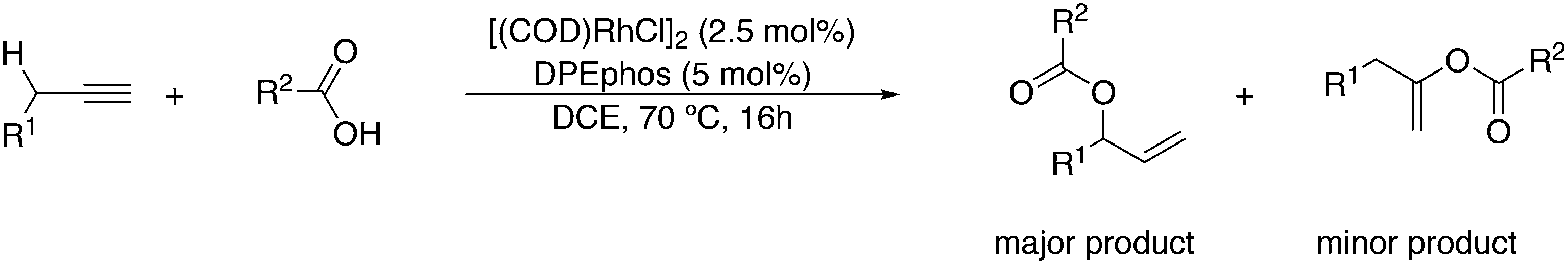 | ||
| Scheme 140 Reaction of alkynes and carboxylic acids. | ||
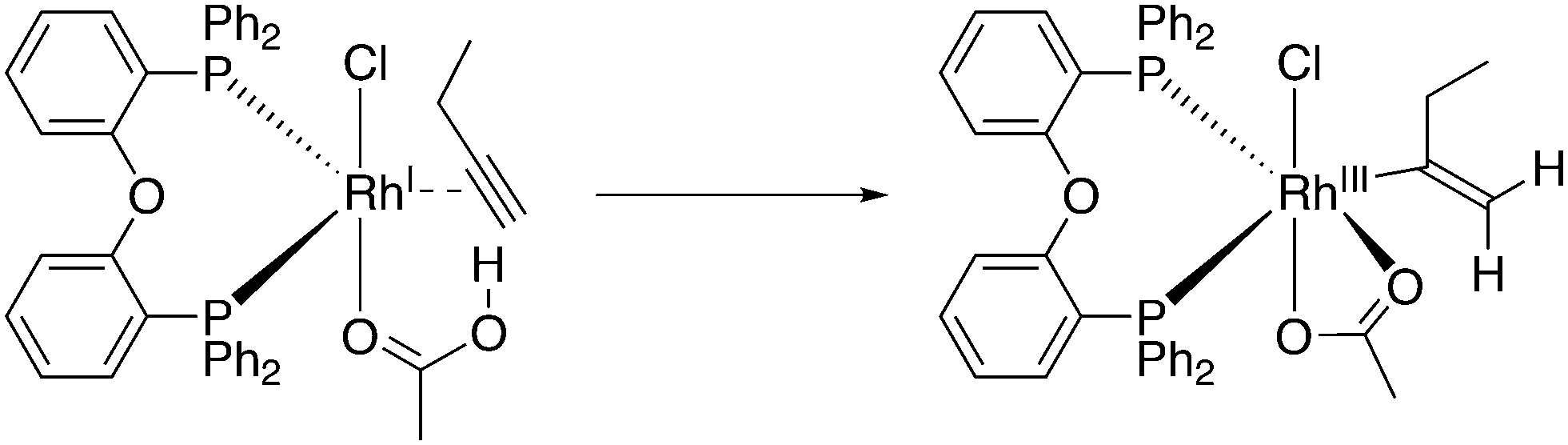 | ||
| Scheme 141 Alkyne protonation. | ||
3.6. Allylic fragments
The effect of Xantphos-like ligands in allylic alkylation and amination was discussed shortly after the introduction of the ligands, both chiral (see section 2.2.5, ligands 27 and 28, Scheme 7) and achiral.349 It was not unexpected that the wide bite angle ligands would have a strong influence on the outcome of the reaction, since the Trost ligands were known to give high enantioselectivities thanks to the “embracing” effect of the substituents on phosphorus caused by the wide bite angle.447 Indeed, for achiral reactive systems, the cone angle gave a better correlation with the selectivities for branched/linear product than the bite angle, both obtained from X-ray structures. In butenylpalladium complexes, small cone angle ligands (dppe) gave linear products, while the Xantphos group gave predominantly branched products.448 Back-bonding to the substituted site of the allyl moiety decreases when the cone angle is large, which enhances the electronic preference for nucleophilic attack on the branched position. The monosulphides of the bidentate bisphosphines were studied by Milheiro and Faller and they also found an increased preference for branched products from crotyl ethyl carbonate, using ligands with wider bite angles.449 In addition, they noted a higher rate of reaction for the more flexible ligands and to some extent, a memory effect for 1- or 3-substituted starting materials for most ligands except Xantphos-S and DPEphos-S (the memory effect refers to the observation that nucleophilic attack is more apt to occur at the position of the leaving group).Phosphole containing Xantphos ligands 92 and 93 were prepared by Le Floch et al. from the P-cyano substituted precursors as shown in Scheme 142.450 The ligands were reacted with [Pd(allyl)Cl]2 to afford the corresponding cationic complexes. The diphenyl analog 92 gave high yields in the catalytic allylation of anilines, which was attributed to the bite angle, steric bulk, and good π-acceptor properties of the ligand.
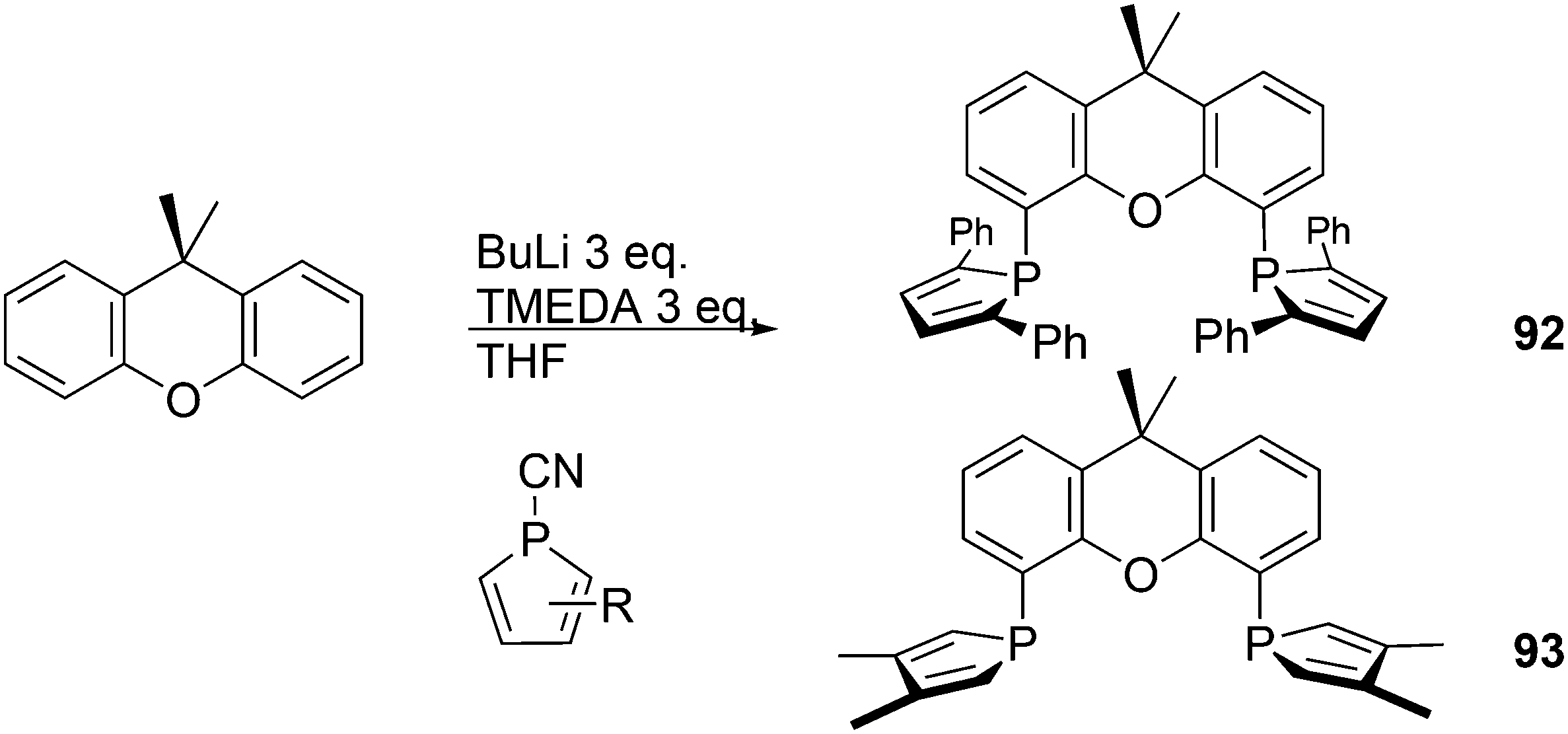 | ||
| Scheme 142 Preparation of phosphole Xantphos ligands. | ||
An intramolecular allylic amination converted the enantiopure propargylic carbonates/amines 94 into five-membered heterocyclic compounds 95 and six-membered compounds 96 (Scheme 143).451 DPEphos was the most promising ligand. Addition of a Lewis acid increased the selectivity to products 95, but the ee was somewhat lower. The detailed explanation was supported by DFT calculations.
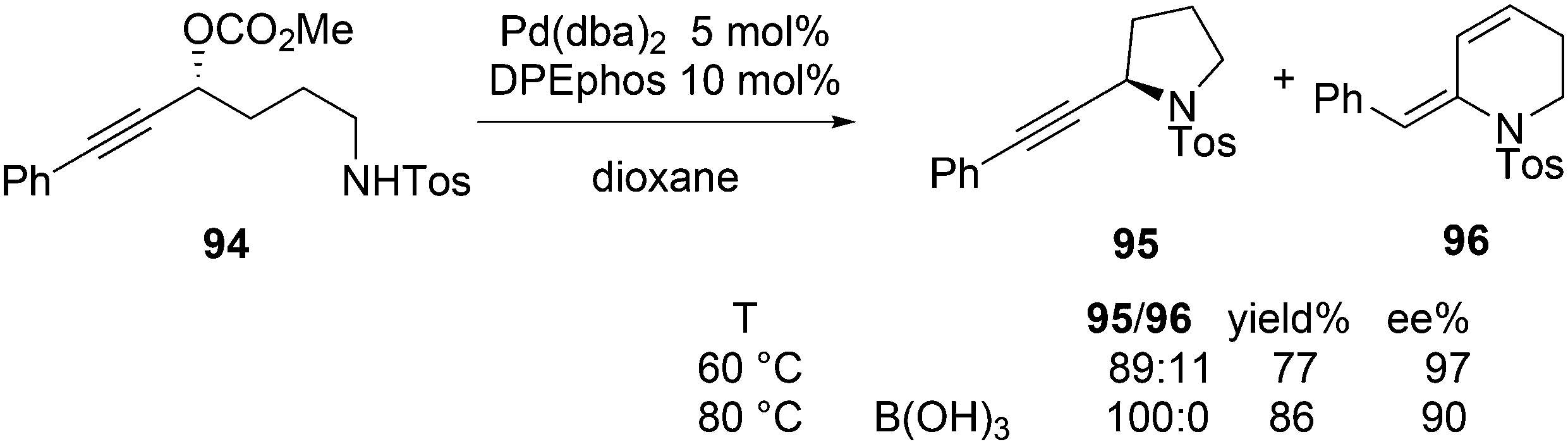 | ||
| Scheme 143 Intramolecular allylic amination. | ||
Kamer and co-workers used chiral DPEphos derivatives 29a–d (Scheme 8) in the allylic alkylation of l,3-diphenyl-2-propenyl acetate and cyclohex-2-enyl acetate with dimethyl malonate and obtained a modest 60% ee for the latter substrate.87
Jay and Barker developed a one-pot procedure for the conversion of allylic isocyanates to allylureas (Scheme 144).452 Wide bite angle ligands such as DPEphos and Xantphos were needed to obtain good conversion to the isocyanates. The amine was added in a second step.
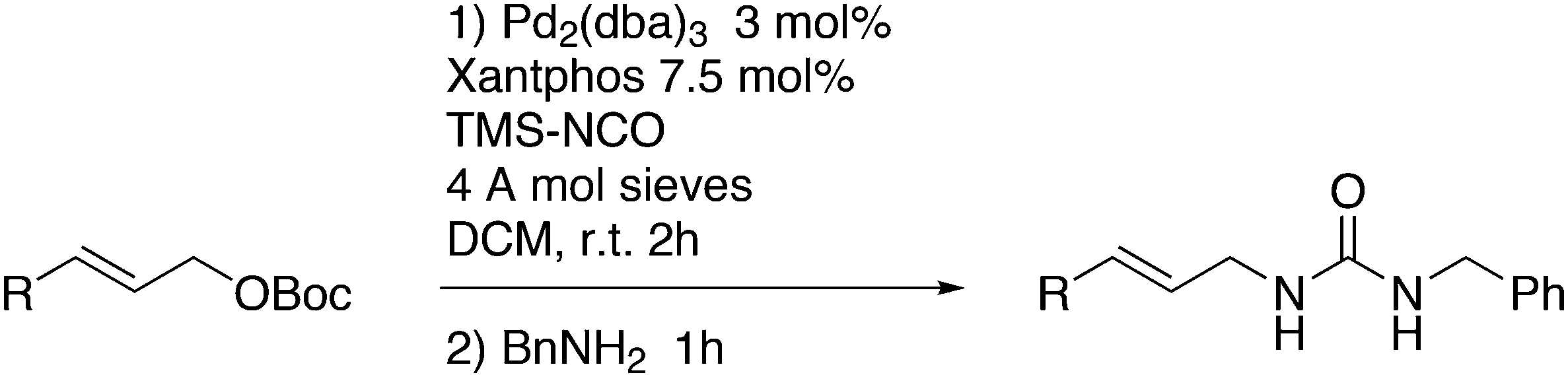 | ||
| Scheme 144 Synthesis of allylureas. | ||
A peculiar allylic palladium reaction is the cross-coupling of allyl acetates and aryl bromides (and pseudohalides), in which the allylic acetates are in situ converted to allylindium compounds with indium metal (Scheme 145).453 After Xantphos (97% GC yield), the next best yield was from dppf (74%), while all of the other ligands gave <30%.
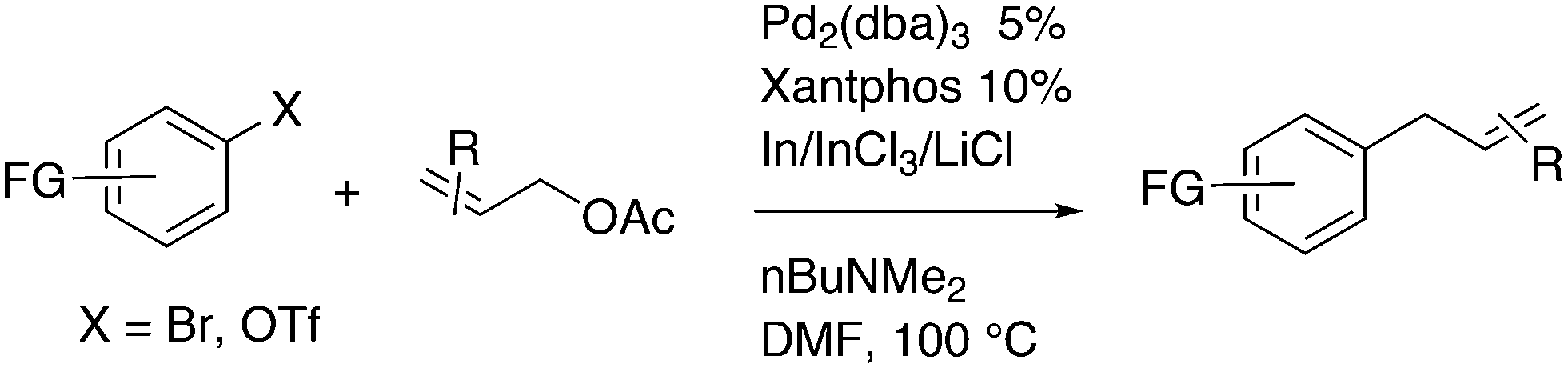 | ||
| Scheme 145 Allyl–aryl cross-coupling. | ||
Kleij and co-workers reported a practical methodology for the stereoselective synthesis of tri- and tetrasubstituted allylic amines based on Pd-catalysed conversion of allyl surrogates readily obtained from cyclic vinyl carbonates (Scheme 146).454 PPh3 showed a strong solvent effect on the selectivity, with DMF giving the highest Z content. The Z-isomer can be obtained exclusively; the favoured ligand for high selectivity is DPEphos (99%), while Xantphos and dppf also gave good conversion but lower selectivity in DMF (75%). Detailed DFT calculations on the DPEphos catalyst explained the preference for the Z-isomer, but not the ligand effect per se.
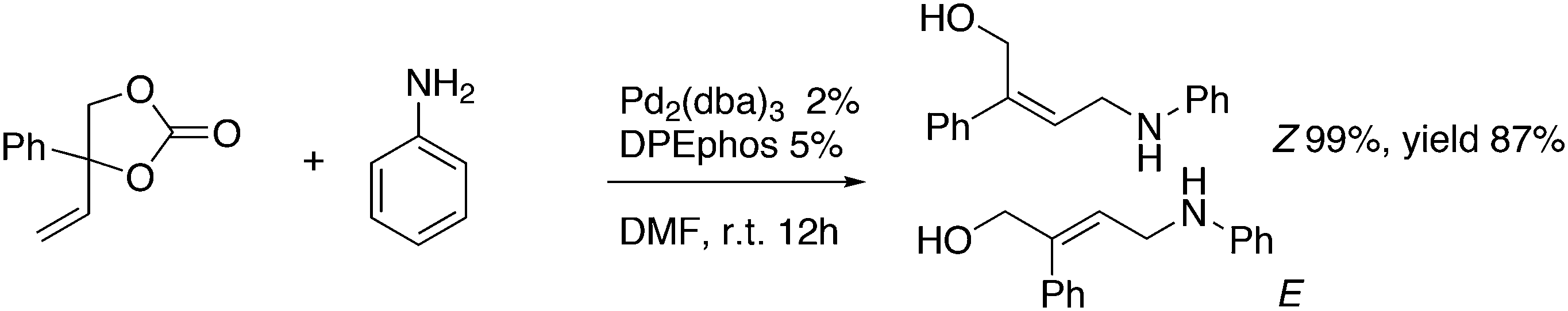 | ||
| Scheme 146 Conversion of cyclic carbonates. | ||
Propargylic compounds were selectively converted into multifunctional compounds according to Scheme 147.455 Oxidative addition to Pd(0), coupling with the 1,3-dicarbonyl component, rearrangement into an allyl intermediate, and finally an N-nucleophile was added to the allylpalladium. This process enables the installation of an all-carbon quaternary center and new C–C and C–N bonds in a single operation in moderate to high yields. The preferred ligand is Xantphos, for which a broad scope was demonstrated.
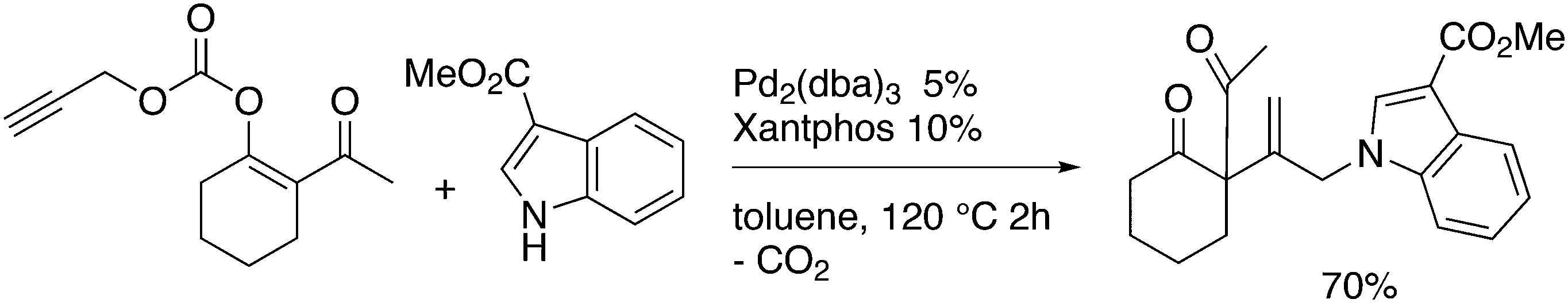 | ||
| Scheme 147 Conversion of propargylic enol carbonates. | ||
Luo and co-workers described a synergistic chiral primary amine/achiral palladium catalysed enantioselective terminal addition to allenes with α-branched β-ketocarbonyls and aldehydes (Scheme 148).456 The reactions afford allylic adducts bearing acyclic all-carbon quaternary centers with high regio- and enantioselectivity. A wide range of allenes, including those with aromatic groups or 1,1′-disubstituted, could be employed, thus expanding the scope of typical asymmetric allylic alkylation reactions. As the phosphine component, Xantphos and DPEphos gave the best results while triphenylphosphine and dppe gave no conversion, and dppf only traces. The chiral amine forms an enamine intermediate that attacks the π-allylpalladium intermediate formed from (bisphosphine)PdH+ and allene.
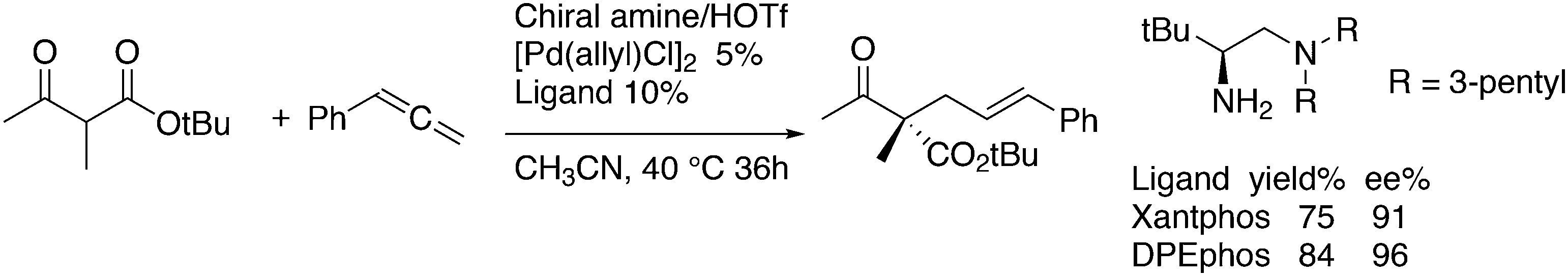 | ||
| Scheme 148 Enantioselective addition to allenes. | ||
More recently, Breit and co-workers applied various ligands in the rhodium catalysed addition of sulfonylhydrazines to allenes to give regioselectively branched allylic sulfones (with the loss of hydrogen and nitrogen).457 With the use of Rh(I)/DPEphos/benzoic acid as the catalyst system, branched allylic sulfones can be obtained in excellent yields and regioselectivities, Scheme 149. Previously, Yamamoto reported that dppf/acetic acid gave high selectivities for the linear products.458 The mechanism involves the oxidative addition of hydrazine to Rh(I), loss of N2 and H2, insertion of allene into Rh–H, and finally elimination of the product.
 | ||
| Scheme 149 Addition of sulfonylhydrazines to allenes. | ||
Earlier, Breit et al. reported a similar result for the addition of sulfonylhydrazines to terminal alkynes and, in this instance, branched allyl sulfones were also formed.459 As in Scheme 141 above, the reaction with alkynes requires more transfers of H-atoms. The preferred catalyst system is the same, Rh/DPEphos/benzoic acid. Addition of benzoic acid to terminal alkynes to give branched allyl benzoates can also be accomplished with a Rh/DPEphos catalyst.460 This reaction sequence starts with the Rh catalysed protonation of the terminal carbon atom.461
Rhodium complexes of Xantphos and DPEphos have been used as catalysts for allylic coupling. Regarding (allyl)RhCl2(bisphosphine), it was noted that both catalysis and complexes showed a broad variation.314 Small bite angle ligands gave η1 allyl and cinnamyl complexes while DPEphos and Xantphos gave η3 complexes. The η1 allyl group binds with the methylene carbon atom to Rh. Via SN2′ attack, the former gave mainly branched products, while the latter produced mainly linear products, as in the case of Pd as the metal. The mechanism is clearly different from that of the reactions above reported by Breit and Yamamoto.
Since Pd-Xantphos is a good combination for carbonylative coupling and allylic substitution reactions, a combination of the two reactions seems worth trying. Indeed, Huang and co-workers found that allylamines can be carbonylated to unsaturated amides (Scheme 150) with Xantphos as by far the best catalyst at 10 bar of CO.462
 | ||
| Scheme 150 Carbonylation of allylamines. | ||
Beller et al. subsequently used allylalcohols as the starting material in the presence of amines and also found Xantphos to outperform various ligands such as dppf, BINAP and DPEphos for the reaction shown in Scheme 151.463 Small amounts of HCl and water improved the result (90%). Whereas in most cases the combination of PdCl2 with Xantphos gave the best results, sterically hindered substrates performed better in the presence of simple triphenylphosphine, and primary anilines gave the best results using cataCXium®PCy.
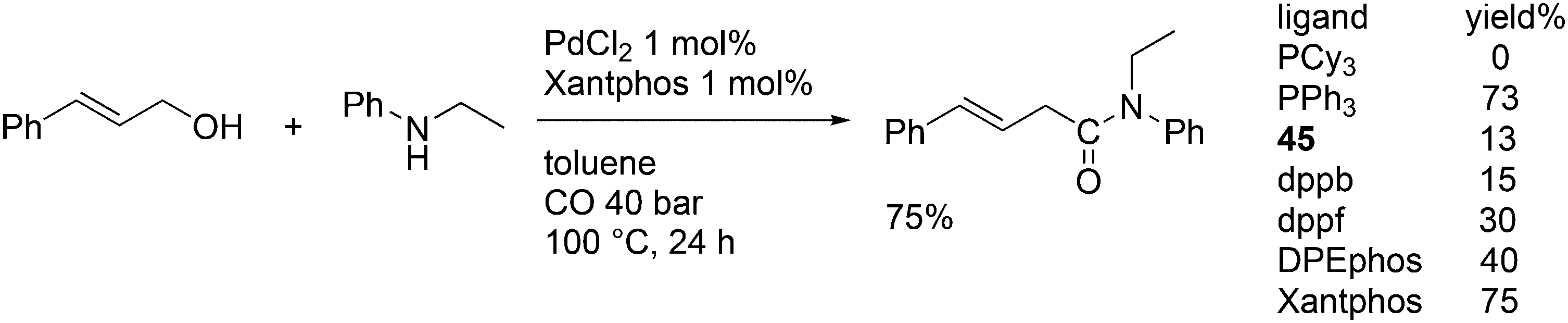 | ||
| Scheme 151 Conversion of allylic alcohols to amides. | ||
In a ligand screening for the reaction shown in Scheme 152, Zhu and Rawal464 found that bidentates performed better than many monodentates (<20%) in the order DPEphos (90%) > Xantphos > dppf. This was the first general method for the regioselective C3-benzylation of 3-substituted indoles.
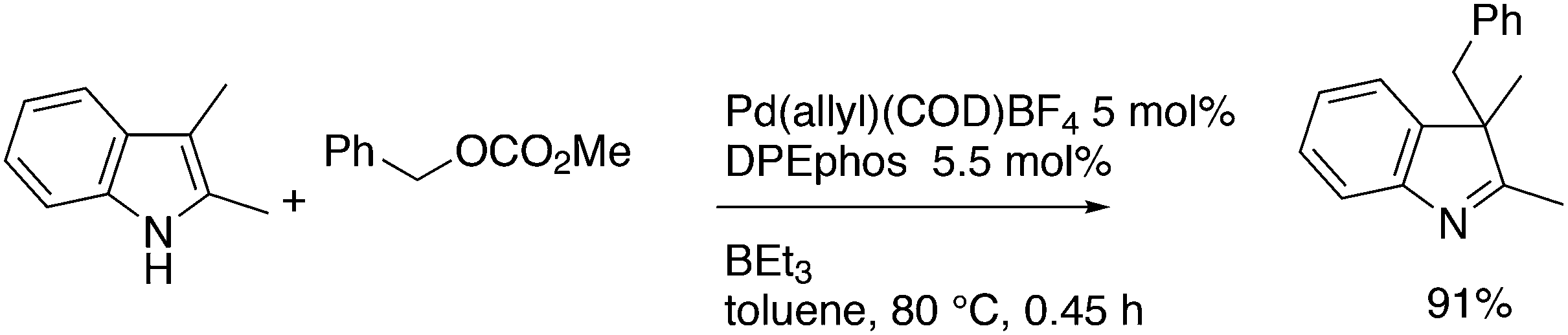 | ||
| Scheme 152 C-Benzylation of substituted indoles. | ||
Platinum/Xantphos-like ligands play a dominant role in a few allylation reactions. The first one is an allylation of amines with allylalcohols as the allyl donor. This is very neat as the alcohol does not have to be converted to a leaving group and water is produced in the coupling reaction instead of salts, Scheme 153.465 A wide range of ligands were tested and DPEphos (91%) and Xantphos (86%) stood out, while the usual competitors, dppf (29%) and BINAP (4%), were behind. A broad amine scope was established. The DPEphos ligand was essential for obtaining high catalyst activity and high monoallylation selectivity of primary amines, allowing the formation of a variety of monoallylation products in good to excellent yield. Substituted allylalcohols may suffer from a regioselectivity issue. Le Floch and co-workers showed that the bite angle was responsible for these results.466 The DFT study showed that the mechanism of the platinum-catalysed allylation of amines with allyl alcohols differs from the palladium-catalysed process, since it involves an associative ligand-exchange step under the formation of a tetracoordinate 18-electron complex. DPEphos and Xantphos destabilized inactive PtH sinks of the catalysts, according to their calculations. This reaction was studied before for palladium and monodentates.467
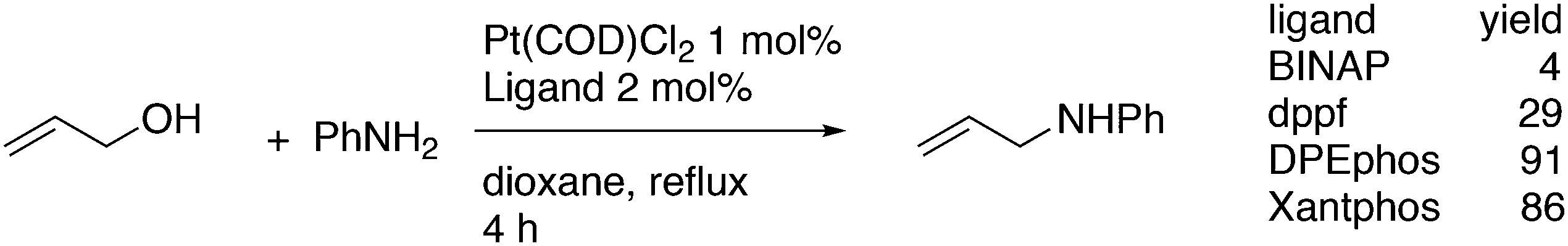 | ||
| Scheme 153 Allylation of amines with allylic alcohols. | ||
An extension of the ligand range by Ohshima et al. did not lead to more effective ligands, but interestingly, they found a very low reactivity when using the methylene-linked variant of DPEphos (bite angle 111°), which strongly suggests the importance of the linker oxygen in DPEphos and Xantphos.468 No indications were found, experimentally or theoretically, for the coordination of oxygen to Pt and it was proven that the effect is not electronic either, since p-MeO groups slowed the reaction. The authors tentatively proposed that the effects may be due to an increase in the leaving ability of the hydroxy group through hydrogen bonding.
The transition metal catalysed nucleophilic allylic substitution of allylic alcohols via π-allylic species was reviewed by Bruneau and co-workers.469
Co/Xantphos was recently reported as a catalyst for C(sp3)–H activation, followed by CO2 capturing (Scheme 154).470 Formation of a carboxylic acid in this way is thermodynamically uphill and in this instance, the loss is compensated by the stoichiometric consumption of AlMe3 and formation of a dimethylaluminum carboxylate. In conventional allylic activation using high valent Pd or Rh salts, a proton is abstracted (often by acetate) and high valent π-allyl complexes are formed that undergo the common nucleophilic substitution. In the present scheme, a low valent Co complex is formed and methane is formed. So far, Xantphos is the only ligand that has given moderate to high yields of the linear acids.
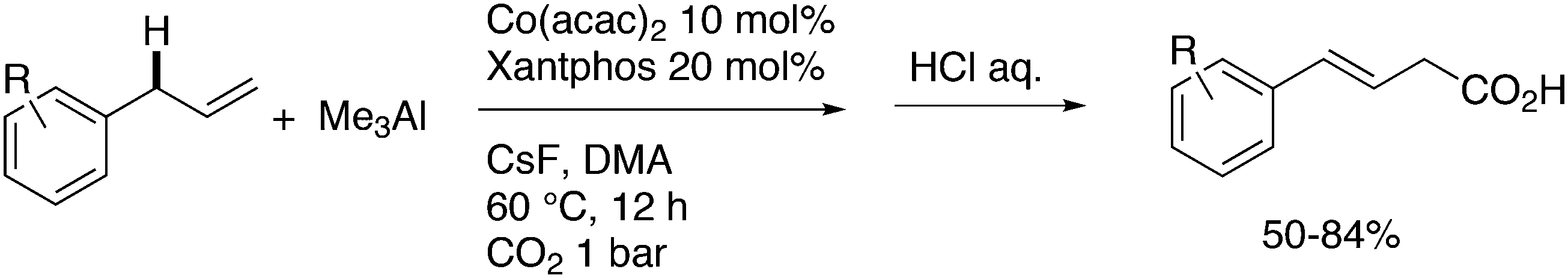 | ||
| Scheme 154 C–H activation and CO2 coupling with allylarenes. | ||
3.7. Various reactions
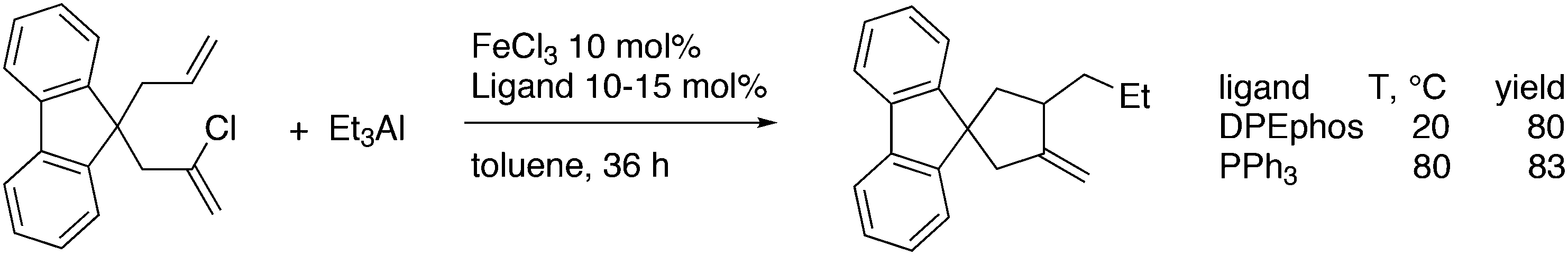 | ||
| Scheme 155 Fe catalysed intramolecular coupling of a vinyl chloride and alkene. | ||
Nakamura and co-workers reported an alkyl alkyl cross coupling reaction using Fe, in which Xantphos afforded the best results among the ligands investigated.475Scheme 156 shows a typical result. The nitrile group is not affected by the Grignard reagent, since it is less reactive when the borate is formed. When BH3 was used instead of trialkylboron, an efficient cross-coupling of the resulting alkylboron and the alkyl halide was obtained.
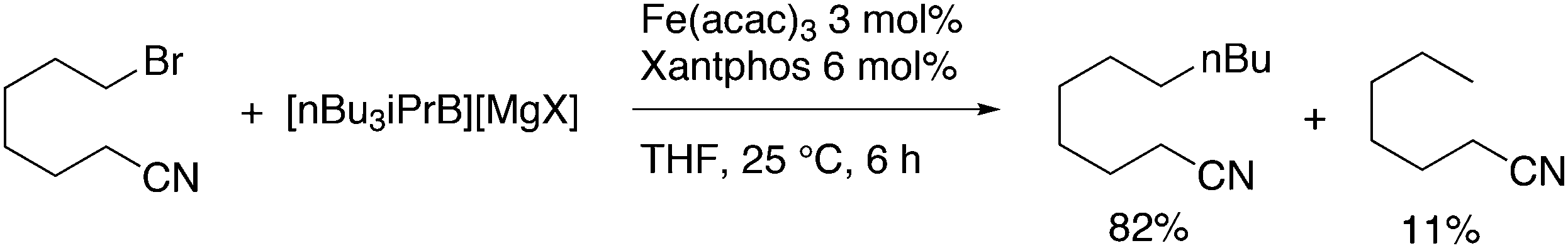 | ||
| Scheme 156 Fe catalysed alkyl alkyl cross-coupling. | ||
Xantphos is the only bisphosphine ligand shown to date to be effective in promoting iron-catalysed Kumada and Suzuki–Miyaura alkyl–alkyl cross-coupling reactions (with NHC ligands excelling for biaryl couplings),476 while benzene-1,2-diyl and ethane-1,2-diyl bisphosphines are selective ligands for alkyl halide arylmetal couplings. These findings were recently analysed by Neidig and co-workers in magnetic circular dichroism and DFT studies.477 They reported that coordination of Xantphos to FeCl2 results in a significantly reduced ligand field, relative to the other ligands. A large bite angle leads to reduced Fe–P Mayer bond orders,478 which could play a role in the unique ability of Xantphos to be an effective ligand in alkyl–alkyl cross-couplings.
A complex of CoCl2 and Xantphos was used by Vogt et al. in the hydrovinylation of styrene, but compared to dppe and dppp, the conversion was low.479 Gosmini and co-workers reported that the formation of arylzinc reagents from the aryl halides could be accelerated by the intermediacy of low-valent Co complexes as catalysts.480 Following these findings, Yoshikai outlined in several publications how a fast cobalt-Xantphos-catalysed LiCl-mediated system was developed for the direct and expedient preparation of arylzinc reagents in THF from the corresponding aryl iodides, bromides, and chlorides.481,482 Thus, aryl halides were converted to arylzinc species in moderate to high yield (Scheme 157), which could be used in a one-pot procedure in various cross-coupling reactions (not shown here). Cobalt(II) chloride was reduced by zinc and the aryl halide was oxidatively added to Co(I) followed by aryl-halide exchange with ZnCl2. DPEphos also gave high conversions but dppe, dppf and PPh3 gave much lower yields. The presence of LiCl was essential for high conversions as explained by Liu et al.483 as well as for the insertion of Zn in the aryl halide bond as discovered by Knochel et al.484
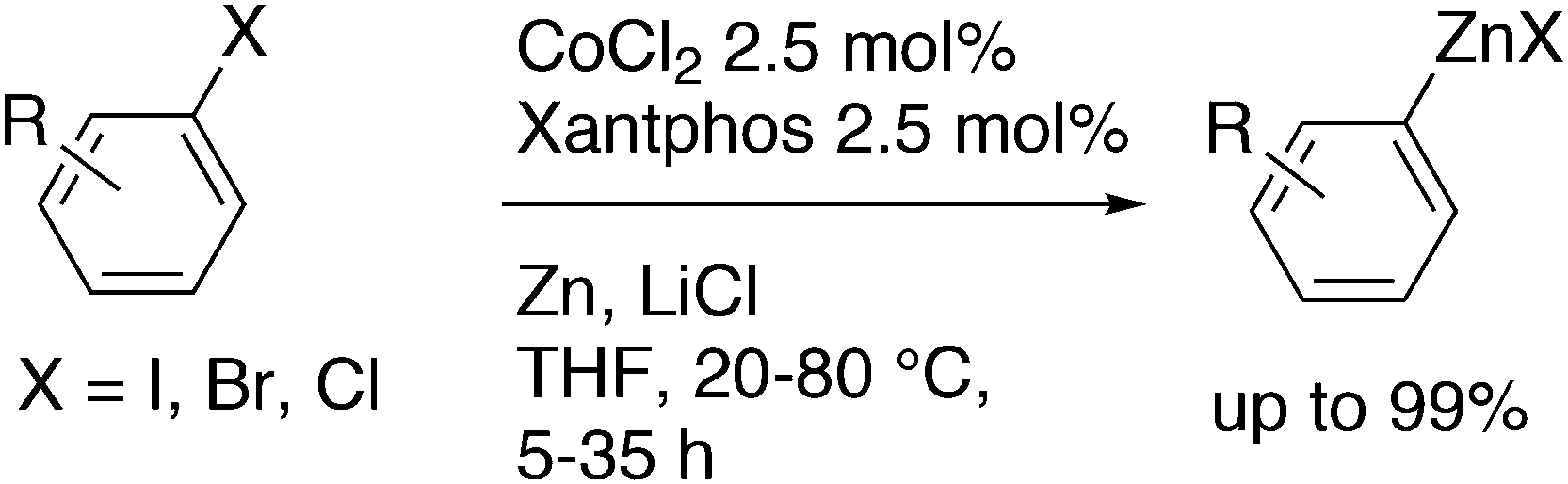 | ||
| Scheme 157 Co catalysed arylzinc formation. | ||
A bimetallic Co/Xantphos/Cr catalyst system converts aryl iodies and bromides to boron esters via the intermediacy of arylzinc compounds and stoichiometric amounts of LiCl and TMSCl (Scheme 158).485 In part, this resembles the previous system, but the role of CrCl3(THF)3 is curious, since the reaction does not take place in its absence.
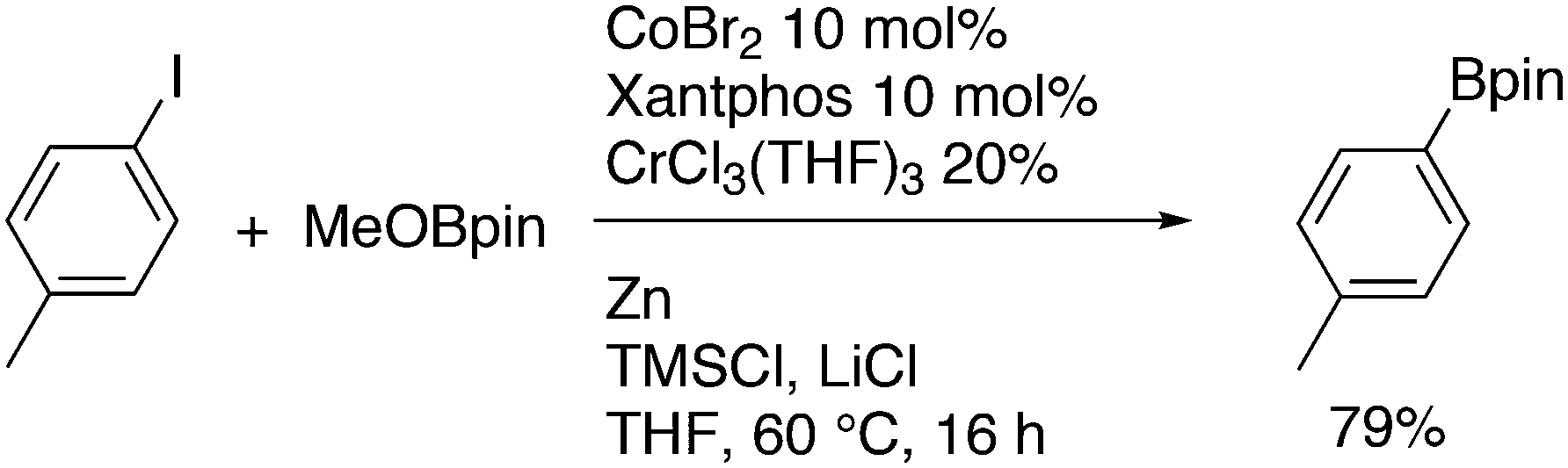 | ||
| Scheme 158 Borylation of tolyl iodide with a Co/Cr catalyst. | ||
In addition to the catalytic formation of hydrocarbylzinc compounds, Co/Xantphos can also be involved in alkenylation reactions of alkynes as is shown in the following example. Alkenylzinc is added highly selectively to alkynes, yielding 1,3-dienes with the use of CoF2 and Xantphos as the catalyst (Scheme 159). Xantphos carries tBu groups in this instance instead of phenyl groups, which are sterically encumbered and electron-rich ligands. Other ligands and halides gave lower yields.
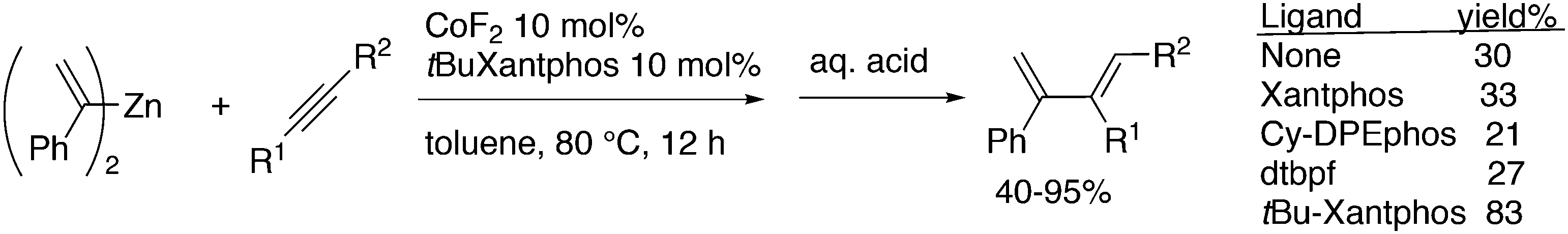 | ||
| Scheme 159 Vinyl addition to alkynes by CoF2 and Xantphos. | ||
(PPh3)3CoCl catalyses the cross-coupling of aryl iodides and bromides with lithium silylamides (Scheme 160).486 The yields were quantitative in many cases for aryl iodides when BINAP, dppf, or DPEphos were added to the PPh3 based catalyst. For cost reasons, DPEphos was used in the optimization and scope studies. The results support a closed-shell mechanism catalysed by a Co(I) species, but a Co(III) intermediate was not identified.
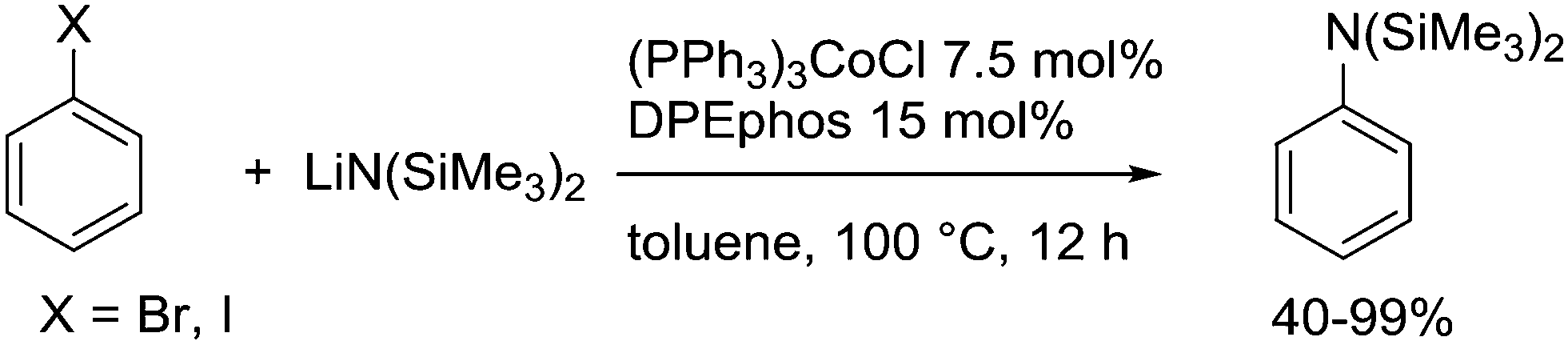 | ||
| Scheme 160 N–C bond formation with Co/DPEphos. | ||
A catalyst derived from Co(acac)2 and Xantphos catalysed the hydrosilylation of styrenes with phenylsilane to almost quantitatively give the branched product. Diphenylsilane gave mixtures, but when dppf was used, the linear product was obtained (Scheme 161).487 A very broad scope was reported. The mechanism was studied by deuteration experiments, NMR, and mass spectroscopy. A (Xantphos)CoH species was observed in situ and it was thus concluded that a Chalk–Harrod mechanism took place. More bulky silanes gave more branched product for Xantphos also.
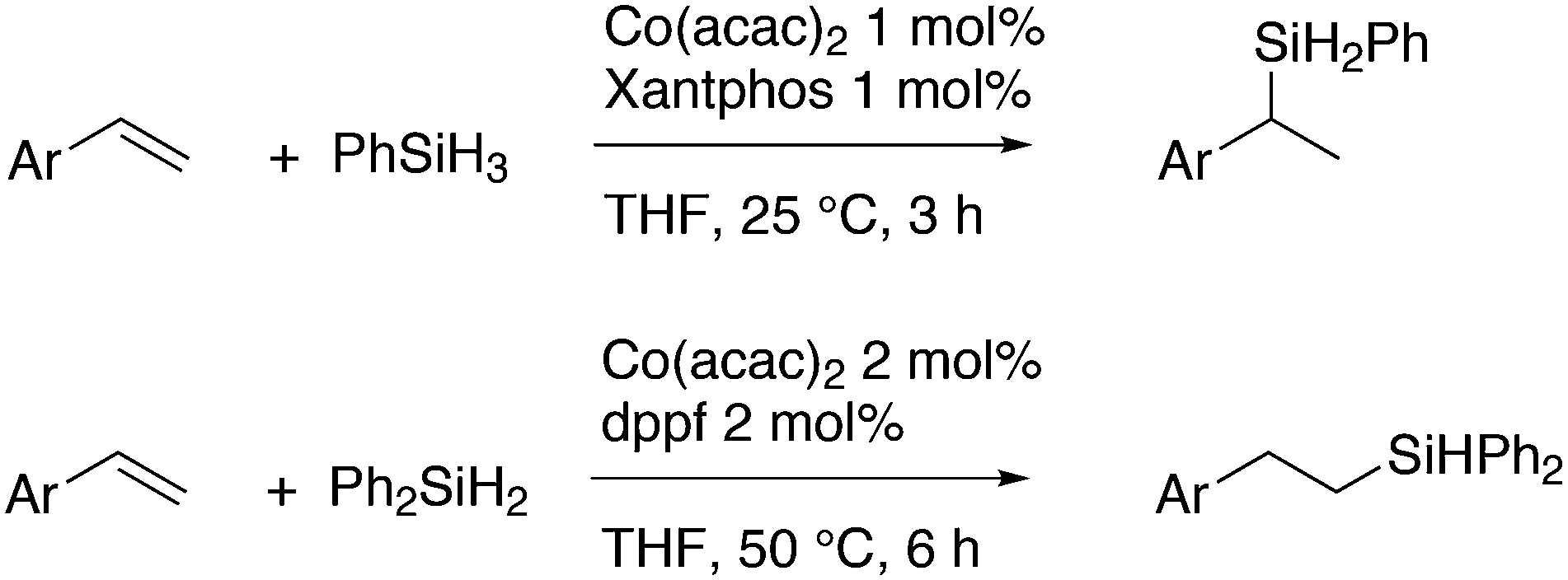 | ||
| Scheme 161 Ligand effects in the Co catalysed hydrosilylation of styrenes. | ||
A nickel bisphosphole-Xantphos complex 97 and several analogous complexes were synthesized by Le Floch et al. and the structures were investigated.488 Complex 97 shows a high-spin low-spin equilibrium at ordinary temperatures. In the presence of MAO, the complex gave a fast and selective ethene dimerization catalyst with TOF for 1-butene up to 43000 mol mol−1 h−1 (20 °C, 30 bar), three times faster than the Xantphos based catalyst.
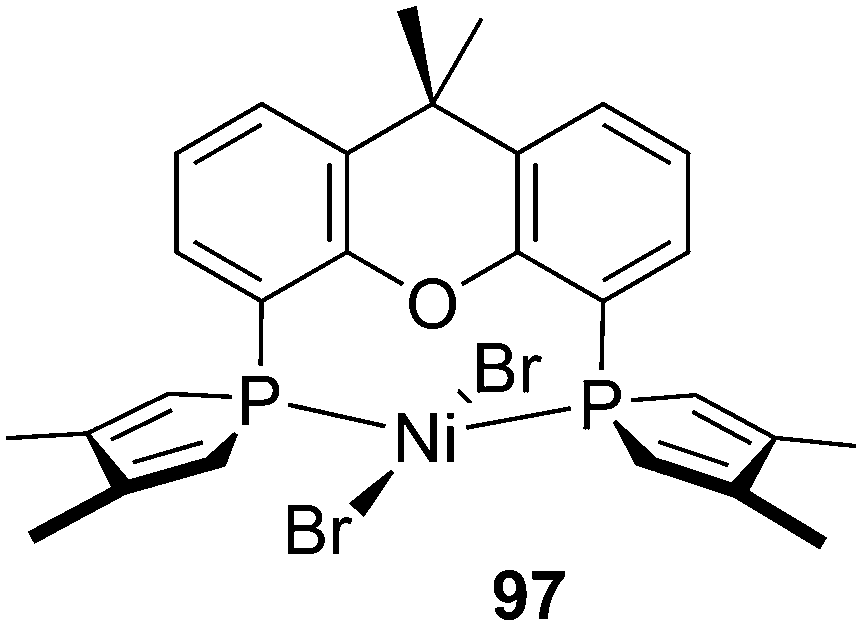
A decarboxylative coupling reaction with an alkynyl carboxylic acid and aryl iodides in the presence of a nickel catalyst was developed by Lee and co-workers (Scheme 162).489 When the reaction was conducted with NiCl2, Xantphos, Mn as reductant, and Cs2CO3 as the base, the desired diaryl alkynes were formed in moderate to good yields. Furthermore, this method does not produce the diyne, which is formed in the homocoupling of alkynyl carboxylic acids.
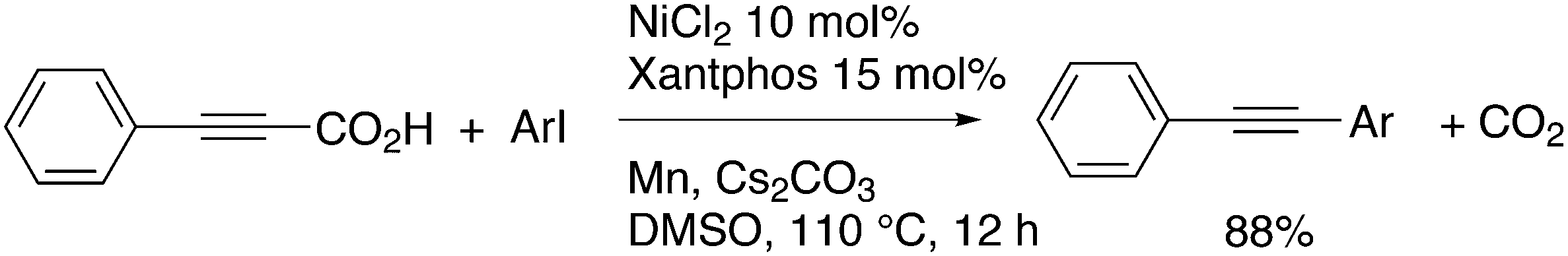 | ||
| Scheme 162 Decarboxylative coupling. | ||
A nickel(0)-catalysed method for the synthesis of quinazolinediones from isatoic anhydrides and isocyanates was described by Beutner et al.490 High-throughput ligand screening revealed that Xantphos was the optimal ligand for this transformation (Scheme 163). Subsequent optimization studies, supported by kinetic analysis, significantly expanded the reaction scope. Interestingly, the example shown for o-tolyl isocyanate gave a much higher yield than phenyl isocyanate. The reaction exhibits a case of substrate inhibition kinetics with respect to the isocyanate.
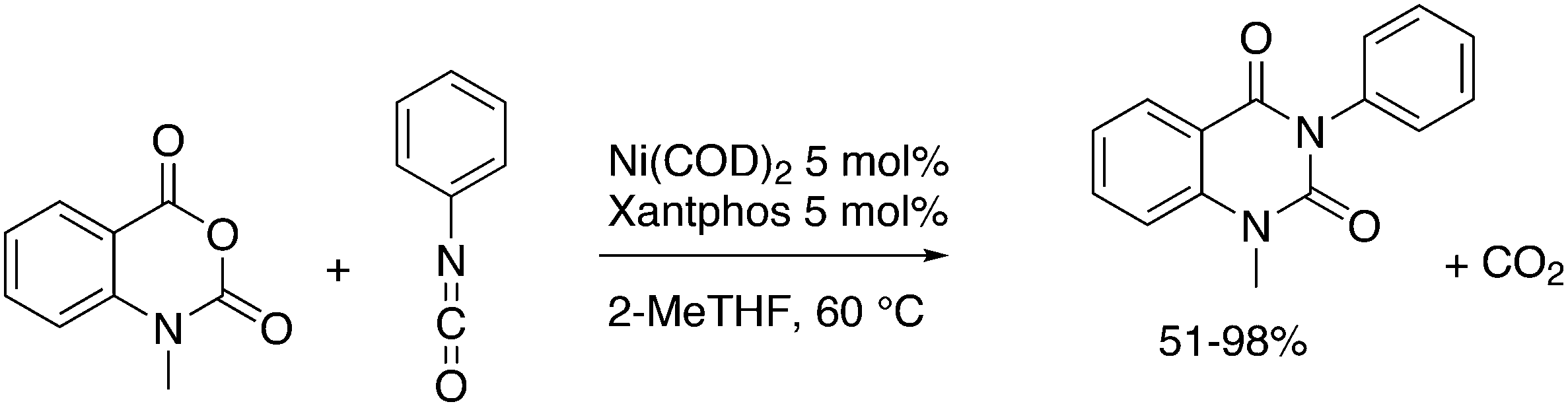 | ||
| Scheme 163 Reaction of carboxylic anhydrides with aryl isocyanates. | ||
A peculiar room temperature cyanation of aryl halides was reported recently by Beller and co-workers for nickel and Xantphos as the ligand and Zn(CN)2 as the cyanide (CN−) source.491 The method involves the use of arylnickel chloride Xantphos as the catalyst and yields of 60–95% were obtained.
 | ||
| Scheme 164 Allylic borylation catalysed by Cu(I) complexes. | ||
Copper(I)-catalysed borylative cyclization reactions of alkenyl halides and pseudo halides (Scheme 165) were discovered by Ito et al.494 A range of ligands were studied and Xantphos gave excellent results (<1% of the alkene byproduct), with dppf being second best. Chlorides and bromides gave the highest yields.
 | ||
| Scheme 165 Borylative cyclization of alkenyl halides. | ||
Yao Fu and co-workers discovered an interesting intermolecular alkylation/borylation with control of the regioselectivity for 98 or 99.495 Cy-Xantphos afforded higher yields than Xantphos and a curious control of the regioselectivity (Scheme 166); Xantphos gave mainly primary boronates, Cy-Xantphos secondary boronates. However, when propyl iodide was used with the latter ligand, the main product was the primary one, 98. The authors note that so far, little use has been made of substituents at phosphorus other than Ph, and it is seen that it has mayor effects on rate and selectivity in these examples. The two ligands that were used in these reactions are also suitable for regiodivergent copper-catalysed hydroboration and deuteroboration reactions in the presence of (deuterio) methanol.
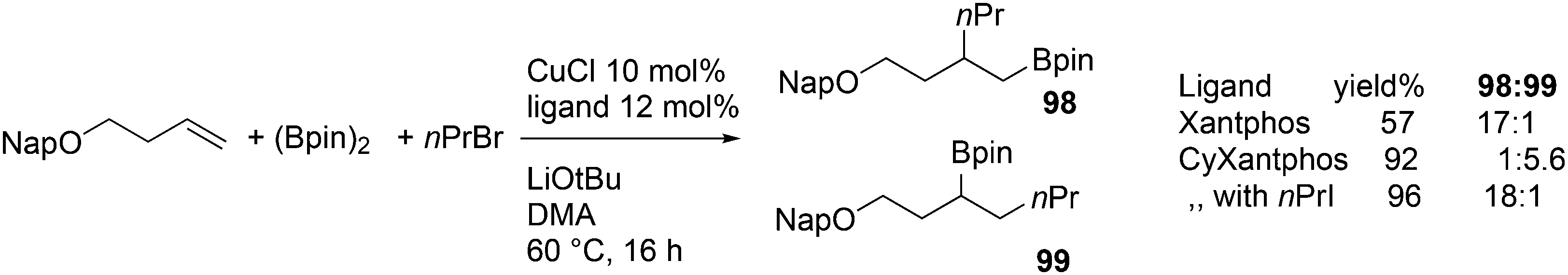 | ||
| Scheme 166 Alkylation/borylation of alkenes with Cy-Xantphos. | ||
Many metal complexes catalyse the silylation reaction of alcohols with silanes, and iridium complexes with weakly coordinating anions are among the fastest catalysts.496 Copper alkoxides in the presence of phosphines show activity for this reaction and the rate depends strongly on the ligand applied. A convenient system was reported by Ito et al. (Scheme 167) based on phosphine complexes of Cu(OtBu).497 They noted that Xantphos gave much faster catalysts than other bidentate phosphines, which they ascribed in part to the inhibition of the agglomeration of the intermediate Cu hydrides. In a competition experiment with primary and secondary alcohols, the former were favoured for HSiMe2Ph as the silane, which was more pronounced when the more bulky DTBM-Xantphos was used.
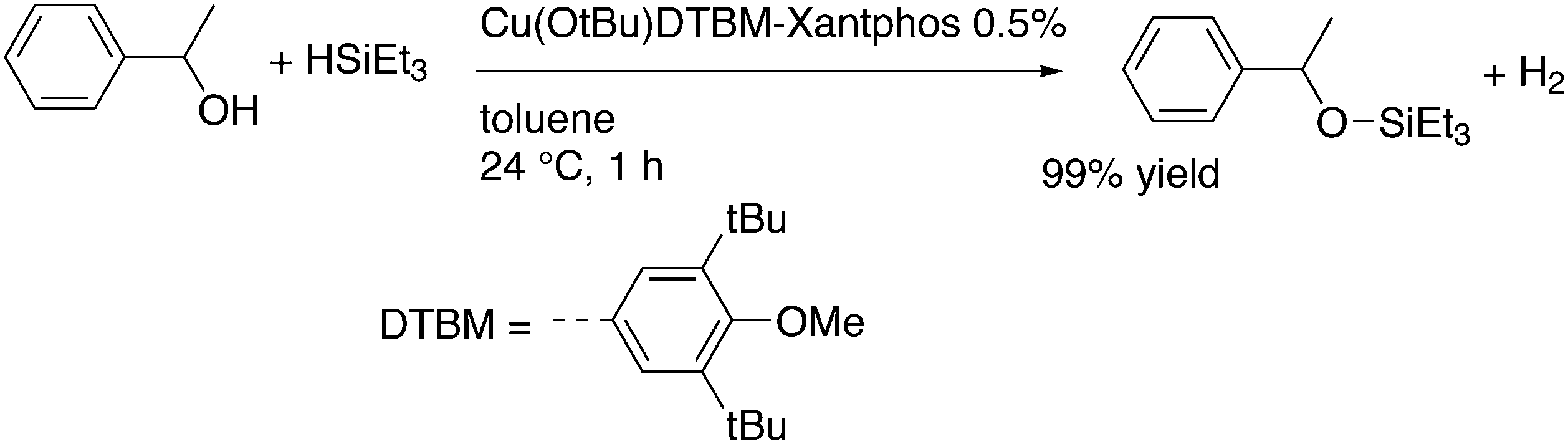 | ||
| Scheme 167 Cu catalysed dehydrogenative coupling of silanes and alcohols. | ||
The hydrogen generated in the silylation of alcohols can be utilised in certain instances for hydrogenation of unsaturates as was shown by Yun et al. in the reaction of alcohols with polymethylhydrosiloxane (PMHS), a cheap source for Si–H groups.498 The complexes were generated in situ from Cu(OAc)2, ligand and PMHS by an easy and practical method. Thus, α,β-unsaturated nitriles were efficiently hydrogenated (Scheme 168) with, in the order of reactivity, Xantphos, BINAP, and DPEphos as the ligands. Dppp was two orders of magnitude slower, while dppb gave an inactive system.
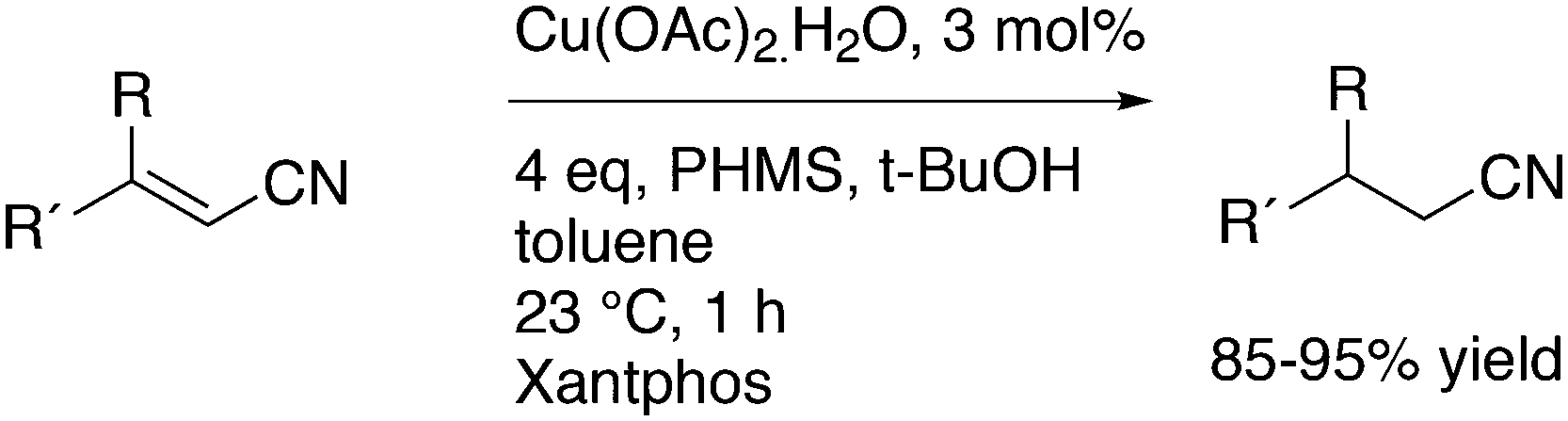 | ||
| Scheme 168 Alkene hydrogenation with Cu/Xantphos/PMHS. | ||
A similar catalyst system was used for the semihydrogenation of alkynes (Scheme 169).499 Since Xantphos gave 10% of the alkane as by-product, a further ligand scouting was conducted and the 4-CF3C6H4-Xantphos analogue came out as a selective and fast ligand to the cis alkene product. tBu2P-Xantphos gave very low conversions and in this case, electron-poor phosphines were the preferred ligands. A variety of alkynes was successfully hydrogenated to the cis alkenes.
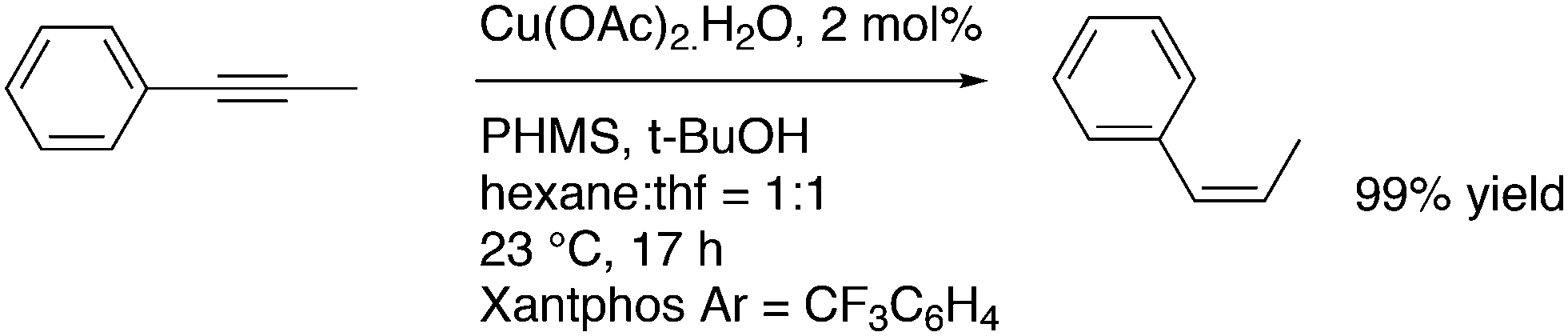 | ||
| Scheme 169 Semihydrogenation of alkynes. | ||
Many metal based catalysts have been developed in the last two decades, which can insert the carbenes generated from diazo compounds into N–H bonds.500 Ramakrishna and Sivasankar studied three complexes of Cu perchlorate with dppf, Xantphos and a dibenzodiazepine based bisphosphine (Scheme 170) as ligands, and the latter, in most cases, gave the highest conversion.
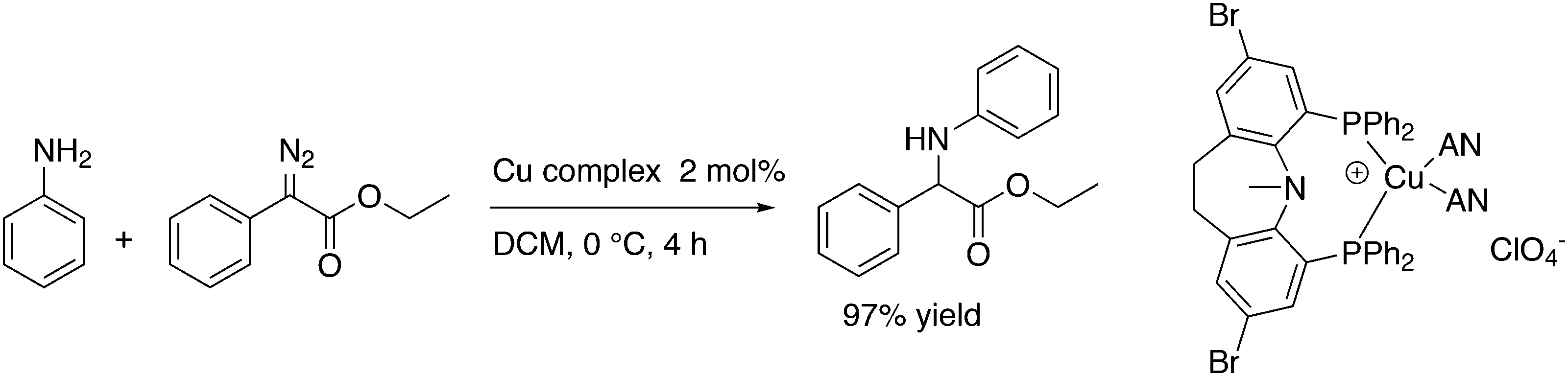 | ||
| Scheme 170 Carbene insertion into NH bonds by Cu(I) bisphosphine catalysts. | ||
Sawamura et al. continued their studies on Cu(I)–Xantphos systems with the intramolecular hydroalkoxylation of unactivated terminal alkenes, giving five- and six-membered ring ethers (Scheme 171).501 This system is applicable to both primary and secondary alcohols. A reaction pathway involving the addition of the Cu–O bond across the C–C double bond was proposed. Mesitylcopper is used as the precursor, which rapidly reacts to give the alkoxide when the substrate alkenylalcohol is added. Several chiral bisphosphines were explored and ees up to 70% were obtained.
 | ||
| Scheme 171 Cu/Xantphos catalysed addition of alcohols to alkenes. | ||
The subtle role of Xantphos in Cu catalysed reactions may well lie in the less ready formation of dimeric or oligomeric species, as cited above. In addition, the Xantphos complexes are often three coordinate, which may facilitate the coordination of a susbstrate, while coordination of a second ligand, for instance, is hindered. Also, as cited above, and found in other studies,502 the wide bite angle weakens the Cu–P bond and this will in turn influence the interaction of Cu with substrates.
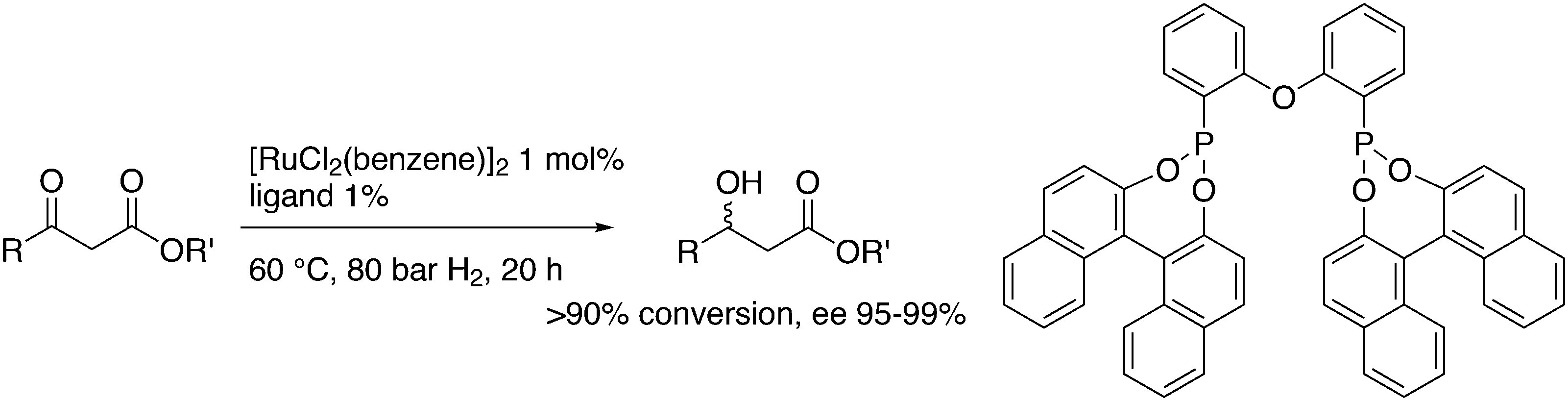 | ||
| Scheme 172 Asymmetric hydrogenation of β-keto esters with a DPEphos derived ligand. | ||
Ruthenium Xantphos catalysts have been used in many hydrogen borrowing reactions with as the most notable example being the hydrogenolysis of C–O bonds of model compounds for biomass.504 Nichols et al. found in a ligand screening of the reaction shown in Scheme 173 a unique result for Xantphos, which showed quantitative conversion of 2-phenoxy-1-phenylethan-1-ol to acetophenome and phenol, while other ligands showed no or little conversion (e.g. dppp 0%, dppf 6%). A lignin related polymer could be depolymerized to 4-hydroxyacetophenone with the same catalyst at slightly higher temperature. A hydrogen borrowing mechanism was proposed.
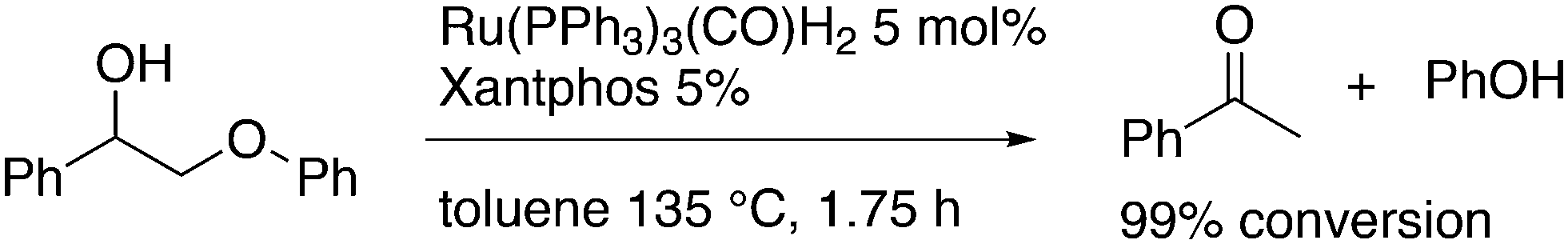 | ||
| Scheme 173 C–O bond hydrogenolysis. | ||
DFT calculations by Paton and co-workers on this reaction indicated that the most likely mechanism involves dehydrogenation of the alcohol as the first step.505 Then, oxidative addition occurs breaking the C–O bond giving Ru(II) enolate-phenolate, the most stable intermediate encountered in the cycle. This intermediate complex reacts with hydrogen to yield the products in two steps, phenol being released first. The exceptional favourable behaviour of Xantphos with respect to other ligands was not explicitly explained.
More recently, Kamer and co-workers studied the electronic and bite angle effect of Xantphos modifications on the rate and selectivity of the reaction of Scheme 174.506 An enhanced conversion was observed with increasing σ-donor strength of the ligands (Scheme 174); the ligands included substituted aryl Xantphos ligands (4-MeO, 4-Me, 4-F, and 4-CF3). Kinetic and thermodynamic data suggest that the oxidative addition of the dehydrogenated model compound to the bisphosphine Ru(0) complex is rate limiting. The bite angle variation (102–121°) showed that among the series 1, 2, 4, 6, 7, and 10, the initial ligand Xantphos 1 gave the fastest catalyst.
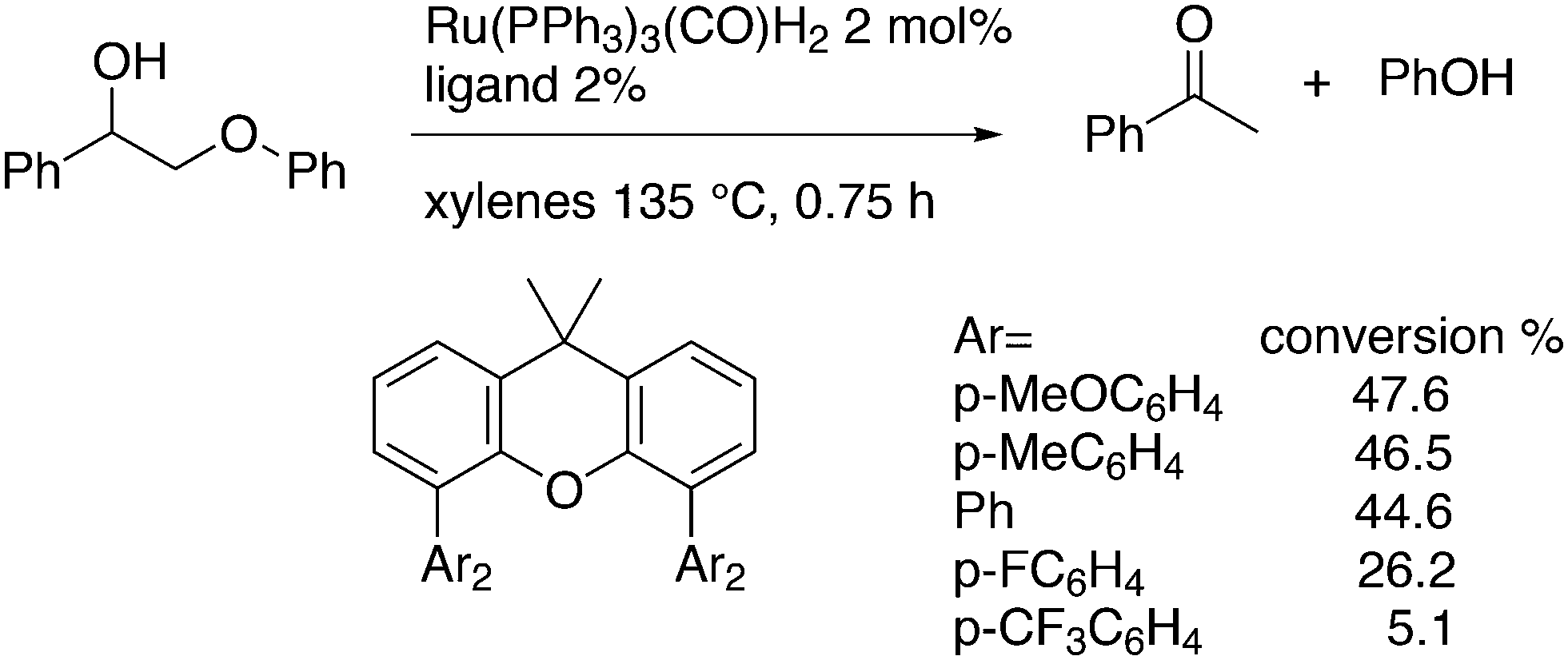 | ||
| Scheme 174 Electronic ligand effect on conversion of lignin model. | ||
Carboxylic acids can be added to alkenes by cationic Ru(II) bisphosphine complexes with modest rates as shown in Scheme 175.507 Xantphos affords a catalyst that is about one order of magnitude faster than dppp.
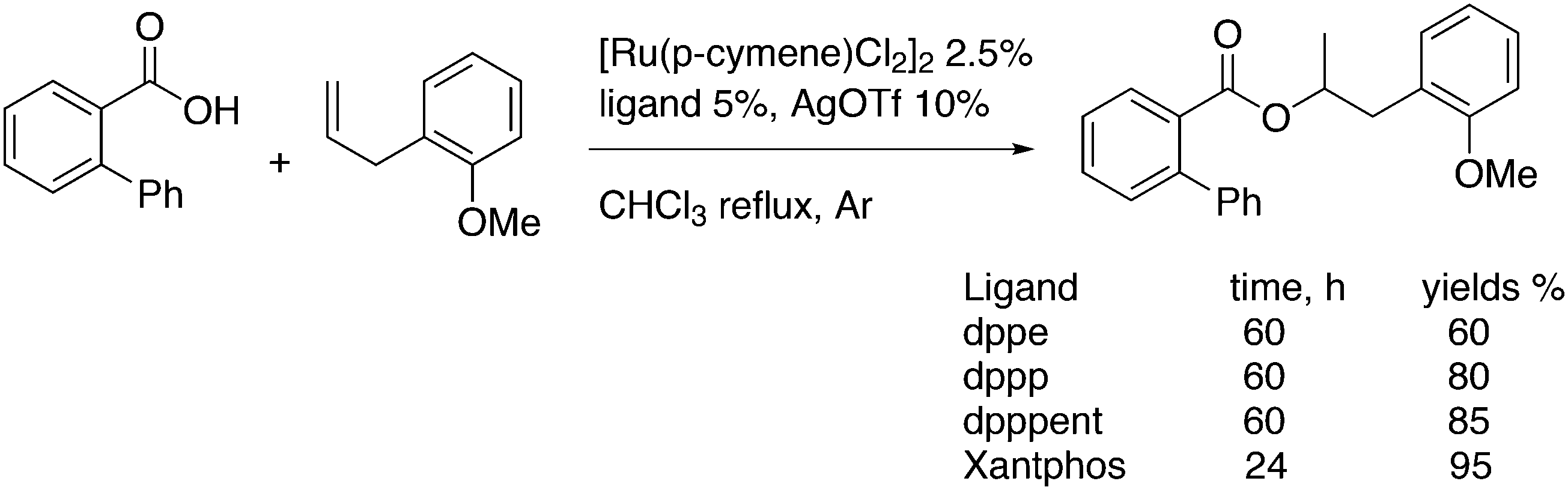 | ||
| Scheme 175 Addition of carboxylic acids to alkenes. | ||
An interesting hydrogen borrowing reaction is shown in Scheme 176.508 Some 2,5-disubstituted furans were prepared using a ruthenium-based catalyst with Xantphos at 1 mol% loading. When the reaction was carried out in the presence of benzylamines, benzylpyrroles were formed in high yield.509
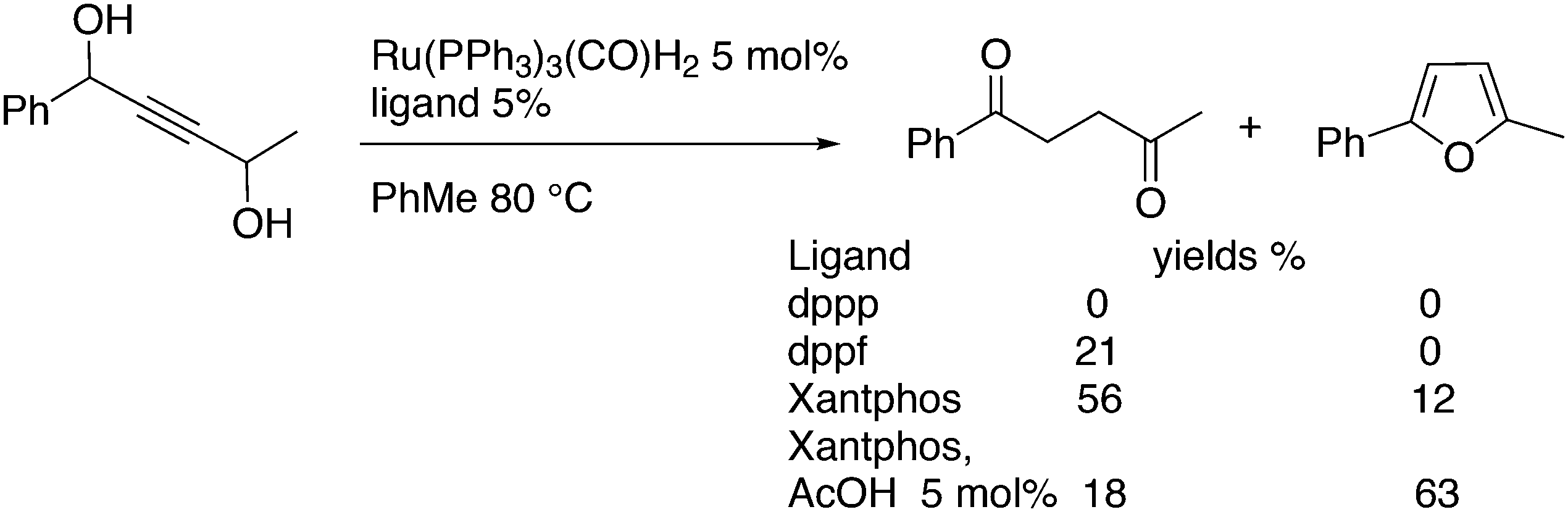 | ||
| Scheme 176 Conversion of alkyne diols to furans. | ||
Prior to this, Williams and co-workers reported C–C bond formation between an alcohol and an acidic C–H bond via hydrogen borrowing (Scheme 177).510 Xantphos (100%), iPr-dppf (91%) and dppf (56%) were good ligands and surprisingly, DPEphos gave very low conversion (8%). The authors noted that Xantphos had a remarkable ligand acceleration effect and proved crucial to the high reactivity of the ruthenium catalyst. Good conversions were also obtained for the alkylation of several other activated methylene compounds.
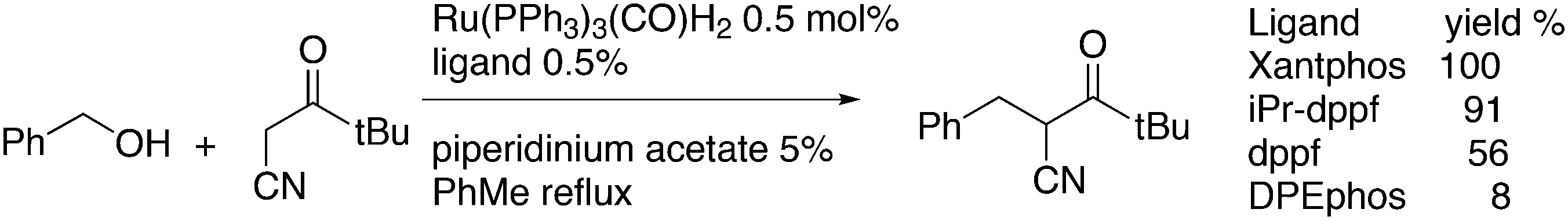 | ||
| Scheme 177 C–C bond formation. | ||
Marimuthu and Friedrich obtained fast hydrogenation of acetophenone with isopropylalcohol with Ru and Xantphos-based ligands and TOF up to 3000 mol mol−1 h−1.511 The preferred Ru precursor is [Ru(p-cymene)Cl2]2 at 80 °C and KOtBu as the base. Other Xantphos like ligands also gave high rates. Dutta and co-workers compared Xantphos and DPEphos Ru complexes, Ru(ligand)(CO)2Cl2, in the hydrogen transfer reaction in the presence of NaOH and they found only slight differences in the activity of the two ligands.512 They reported that transfer to benzaldehyde was more than a factor of 10 faster than transfer to acetophenone with these catalysts. While DPEphos is not a chiral ligand, together with a chiral diamine it was used in a very effective asymmetric hydrogenation reaction (Scheme 178).513
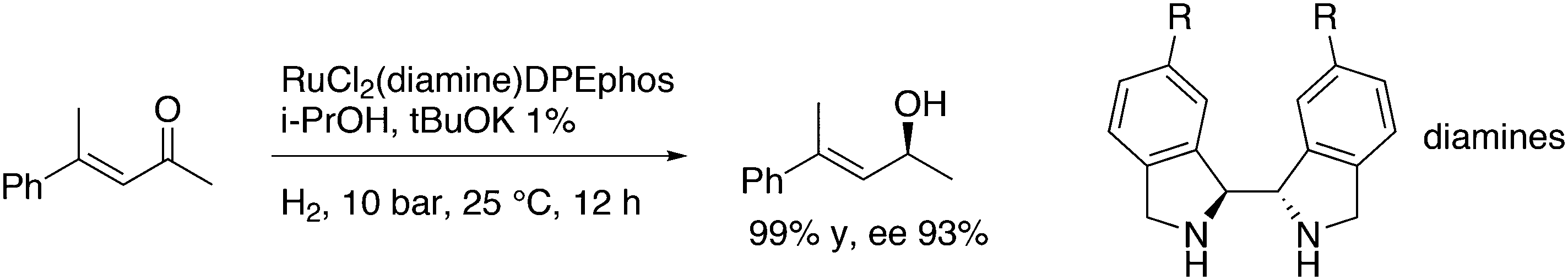 | ||
| Scheme 178 DPEphos as a co-ligand in asymmetric hydrogen transfer. | ||
Amination of alcohols represents the typical example of the “hydrogen borrowing” methodology,514 also known as the “hydrogen auto-transfer” reaction,515 as shown in Scheme 179. The catalyst dehydrogenates the alcohol to ketone, the reaction of ketone and amine takes place and the hydrogen released earlier is reincorporated, giving the amine. The ruthenium catalysts require relatively high temperatures (e.g. 150 °C), perhaps for the dehydrogenation step, which is thermodynamically prohibited by lower temperatures and thus a direct transfer of hydrogen from alcohol to imine might lead to milder conditions. Selectivity to primary, secondary or tertiary amines is an issue in this chemistry.
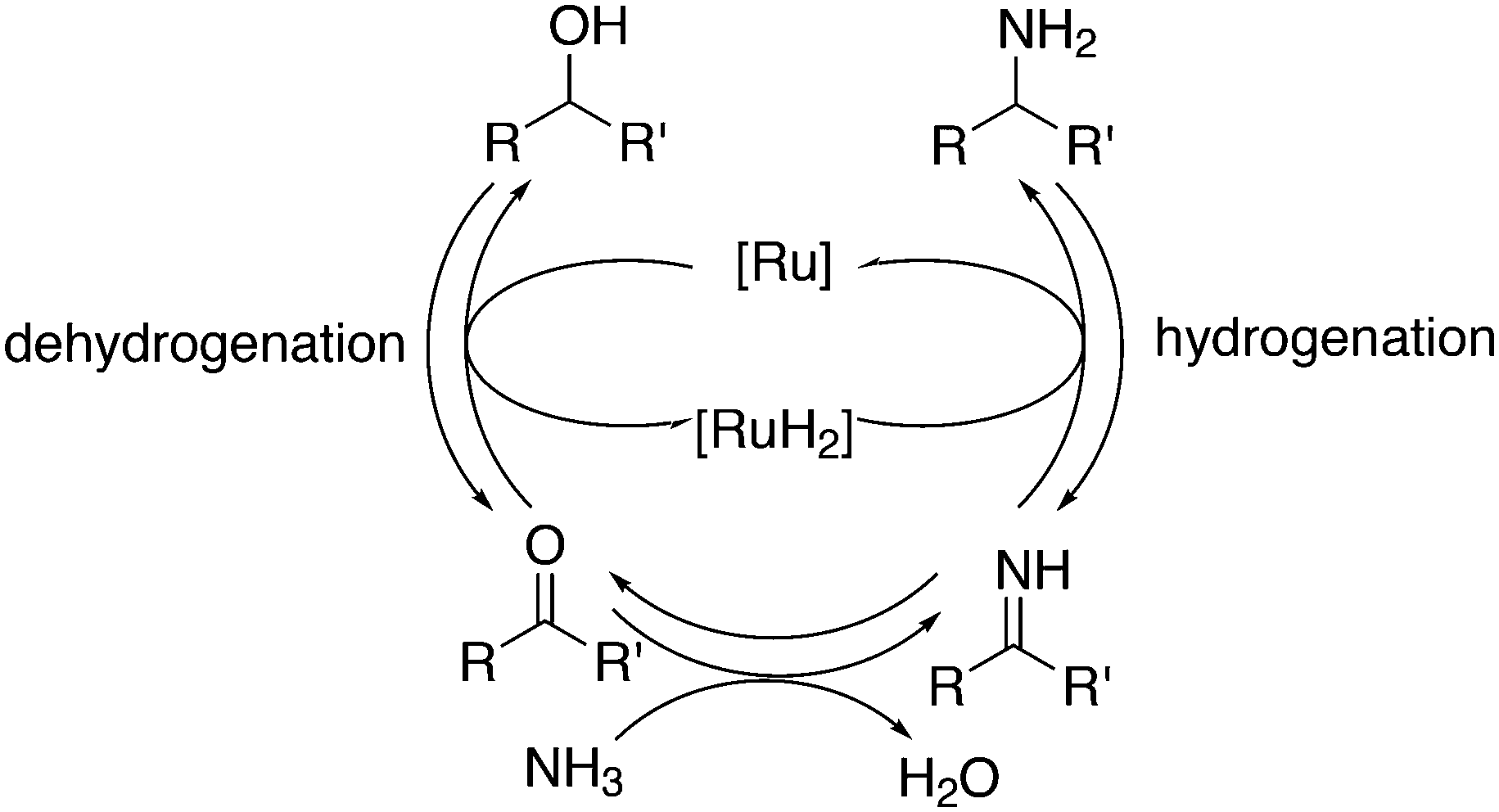 | ||
| Scheme 179 Borrowing hydrogen mechanism in alcohol amination. | ||
The first examples of such reactions date back to 1981,516 but since 2007, faster and more selective systems based on pincer ligands, including DPEphos and Xantphos were reported.517–519 When Ru/Xantphos, reported above by Williams et al., was first used in alcohol amination, the amine yield was modest and mainly ester was obtained at 100% conversion.520 In this work, dppf gave excellent results for amines. In later studies, they also added DPEphos to the range of preferred ligands.521 Beller and co-workers and Deutsch and co-workers did a screening of “common” ligands and although Xantphos performed the best (Scheme 180), DPEphos and dppb gave results that were pretty close to those of the more expensive Xantphos.522,523 In view of the good results for dppb, one may tentatively conclude that the pincer behaviour of DPEphos is not critical in this reaction. Recent applications of these ligand systems were reported by Kann et al.524
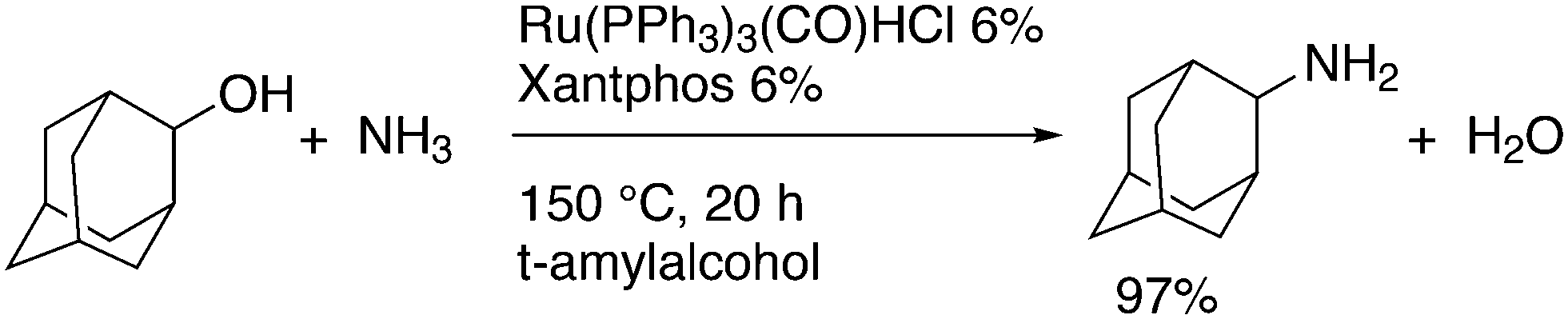 | ||
| Scheme 180 Amination of secondary alcohols with Ru/Xantphos. | ||
Vogt et al. reported on the catalytic activity and fluxional behavior of complexes based on RuHCl(CO)(PPh3)3 and Xantphos-type ligands in the reaction of cyclohexanol and ammonia (Scheme 181).525 Sixantphos gave the highest TOF. In a mechanistic study, Vogt et al. found that the addition of ketone accelerated the reaction.526 The authors pointed out that the “hydrogen shuttling” processes tend to be intrinsically hampered by a low steady-state concentration of intermediates. When the presumed catalyst (Xantphos)Ru(CO)H2 was used as the precursor no activity was observed, which makes sense according to the authors, because the sequence starts with the dehydrogenation of cyclohexanol. When 10% of cyclohexanone was added, the catalytic activity was restored.
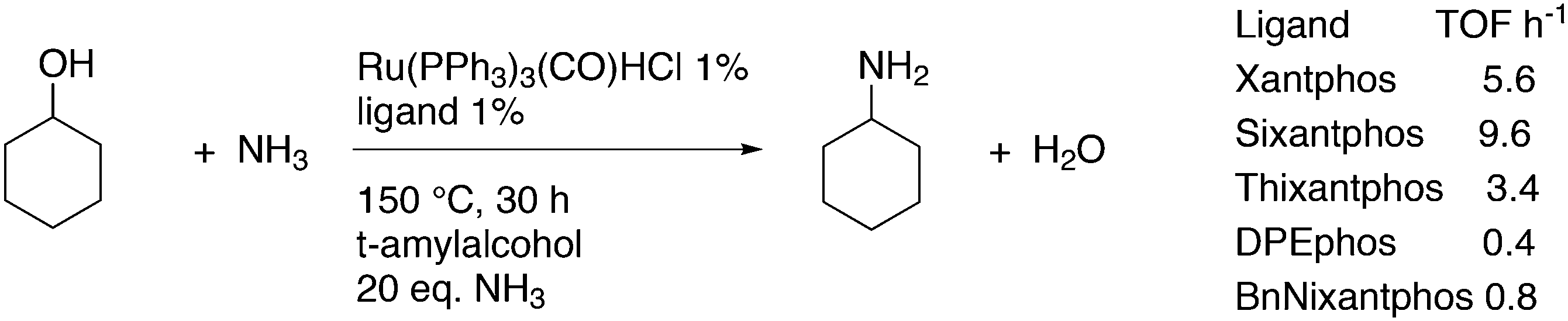 | ||
| Scheme 181 Xantphos ligands in the amination of cyclohexanol. | ||
In the mechanistic schemes, pincer complexes are often presented for DPEphos and Xantphos since they were also observed in the NMR analyses of the reaction mixtures. However, the presence of the O-atom is not a prerequisite because dppb and triphos527 also give active systems. The intermediates in the latter catalyst are cationic species rather than neutral species as found for Xantphos.
The rich chemistry of Ru(Xantphos/DPEphos) was explored by the groups of Williams and Whittlesy, but here we will mention only a few catalytic examples. Oxime ethers can be converted to nitriles by Ru(PPh3)3(CO)H2 as the catalyst precursor and ligands are added as shown in Scheme 182.528 The reaction shows an unexpected ligand effect, but it should be borne in mind that there is always PPh3 present in addition to the ligand examined. The reaction was optimized for Xantphos and yields up to 93% were obtained in this clean preparation of nitriles.
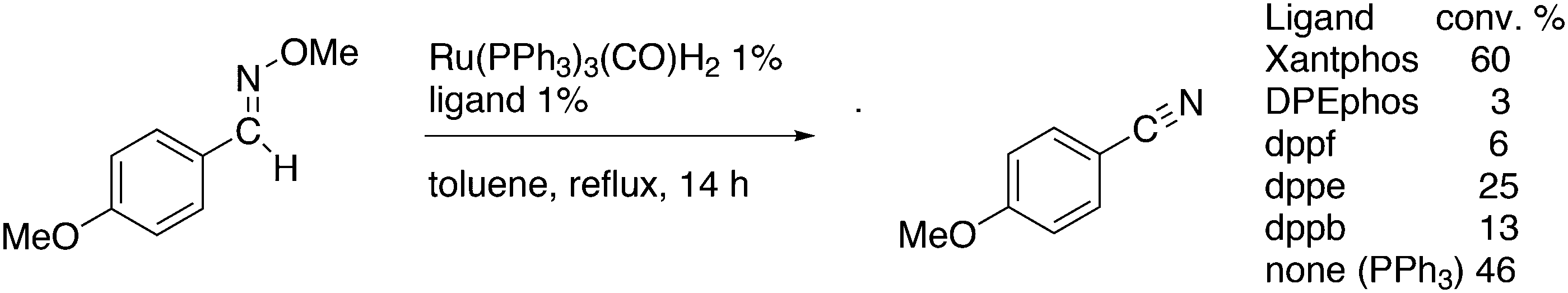 | ||
| Scheme 182 Conversion of oxime ethers into nitriles. | ||
Only the wide bite angle ligands BINAP, Xantphos and DPEphos led to the dehydrogenative coupling of benzylalcohol and the aniline amide shown in Scheme 183.529 Xantphos gave full conversion to 91% of the saturated product and 9% of the unsaturated one. Crotonitrile was added as the oxidant (hydrogen acceptor) and ammonium chloride was found to inhibit the dehydrogenation step to the unsaturated byproduct, but it did not affect the dehydrogenation of alcohol to aldehyde.
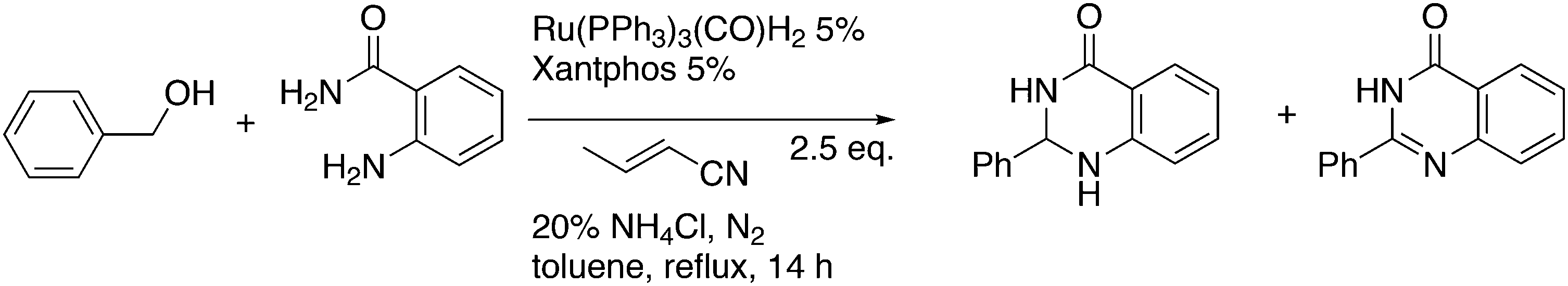 | ||
| Scheme 183 Oxidative coupling of alcohol and aniline amide. | ||
A similar reaction occurs with 1,2-diaminobenzenes with either benzylacohol or benzaldehyde and crotonitrile as the hydrogen acceptor (Scheme 184).530 Piperidinium acetate was added as an organocatalyst, which more than doubled the rate.
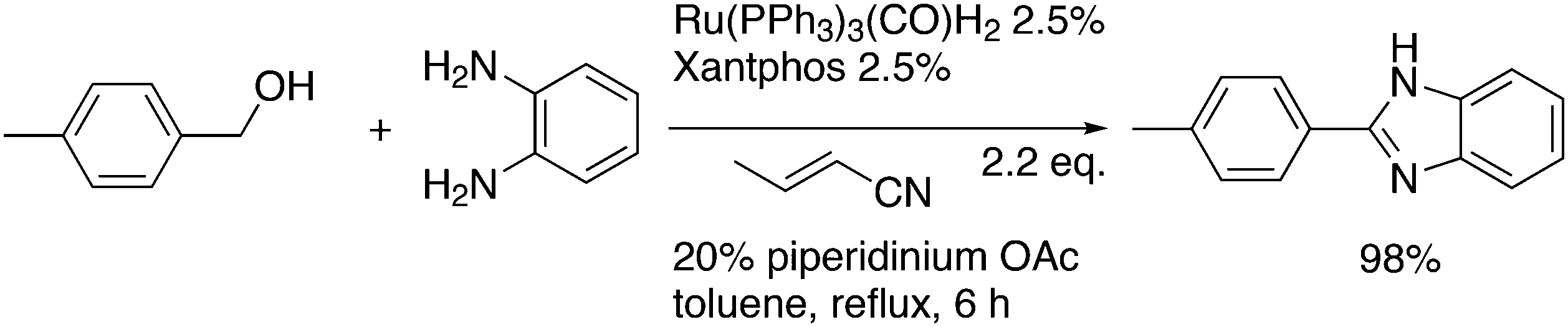 | ||
| Scheme 184 Formation of benzimidazoles. | ||
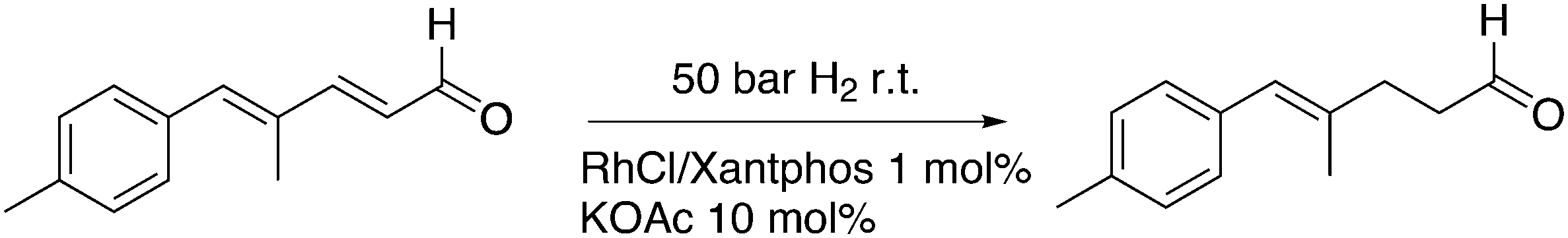 | ||
| Scheme 185 Selective hydrogenation of dienals. | ||
Weller, Willis and co-workers used a cationic alkyne adduct 100 (Scheme 186) that could be converted to a cationic Rh(III) dihydride 101, which reacted with thioethers with cleavage of the C–S bond to give 102.534 The reaction needs assistance of an ortho donor group that acts as the tug-in group of the substrate.
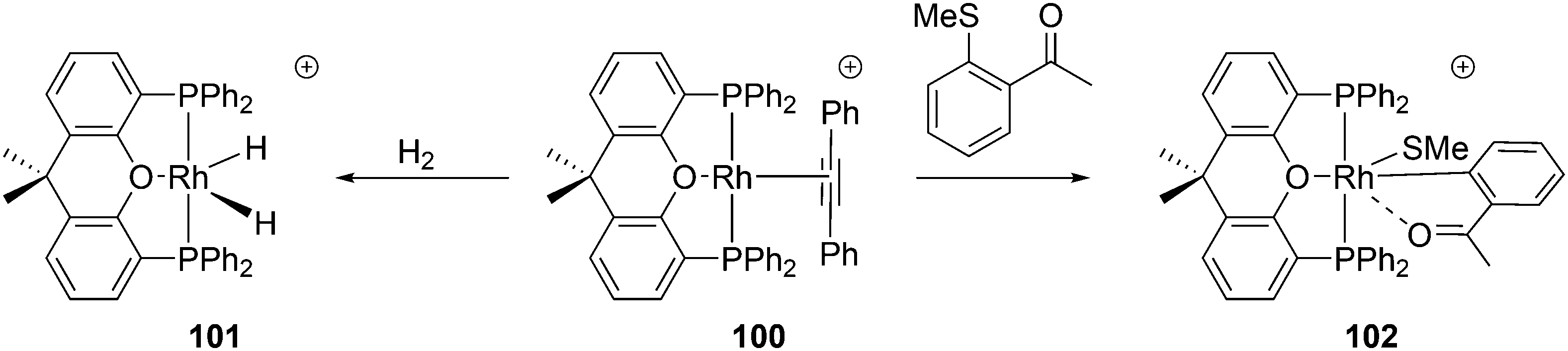 | ||
| Scheme 186 Reactions of Rh(Xantphos)(alkyne) (the BArF4− ligand is omitted throughout). | ||
The complexes are active in several reactions such as the trimerization of alkynes and the insertion of alkynes in the C–S bond (Scheme 187).
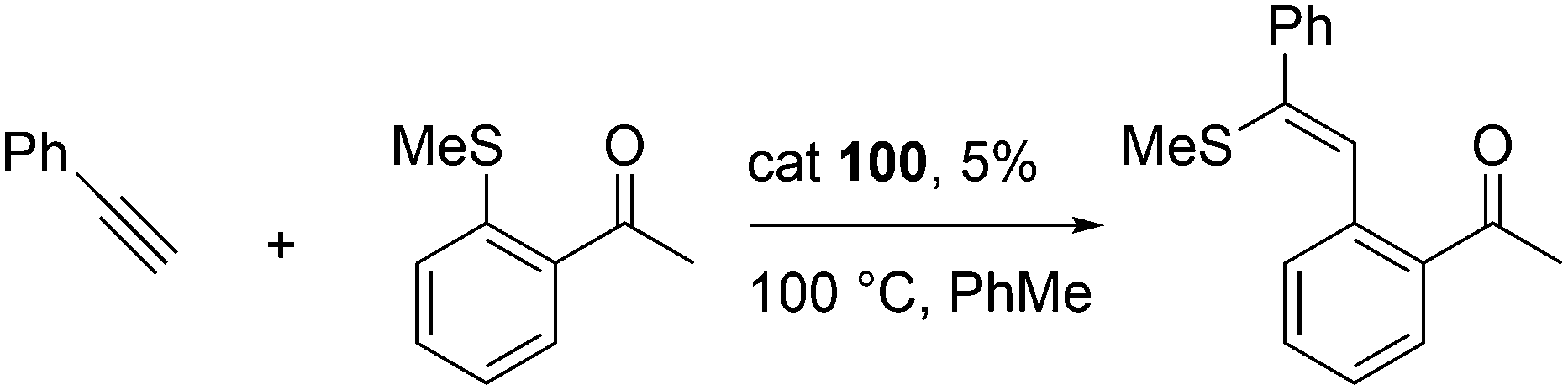 | ||
| Scheme 187 Reaction of alkyne and methyl aryl thioether with adjacent donor group. | ||
Catalyst 101 reacts with boranes to give adducts, which are catalyst precursors in the dehydrogenative polymerization of H3B·MeNH2.535 In the presence of the aminoboranes, P–C activation takes place and Xantphos based phosphido dimers form, which are the actual catalysts. Dehydrogenation of H3B·NMe3 leads to homocoupled B–B products.536
Complex 103 reacts with aminoborane to form an adduct, which undergoes insertion of alkenes in a B–H bond (Scheme 188).537 The resulting complex 104 serves as the catalyst for borane addition to the same alkenes.
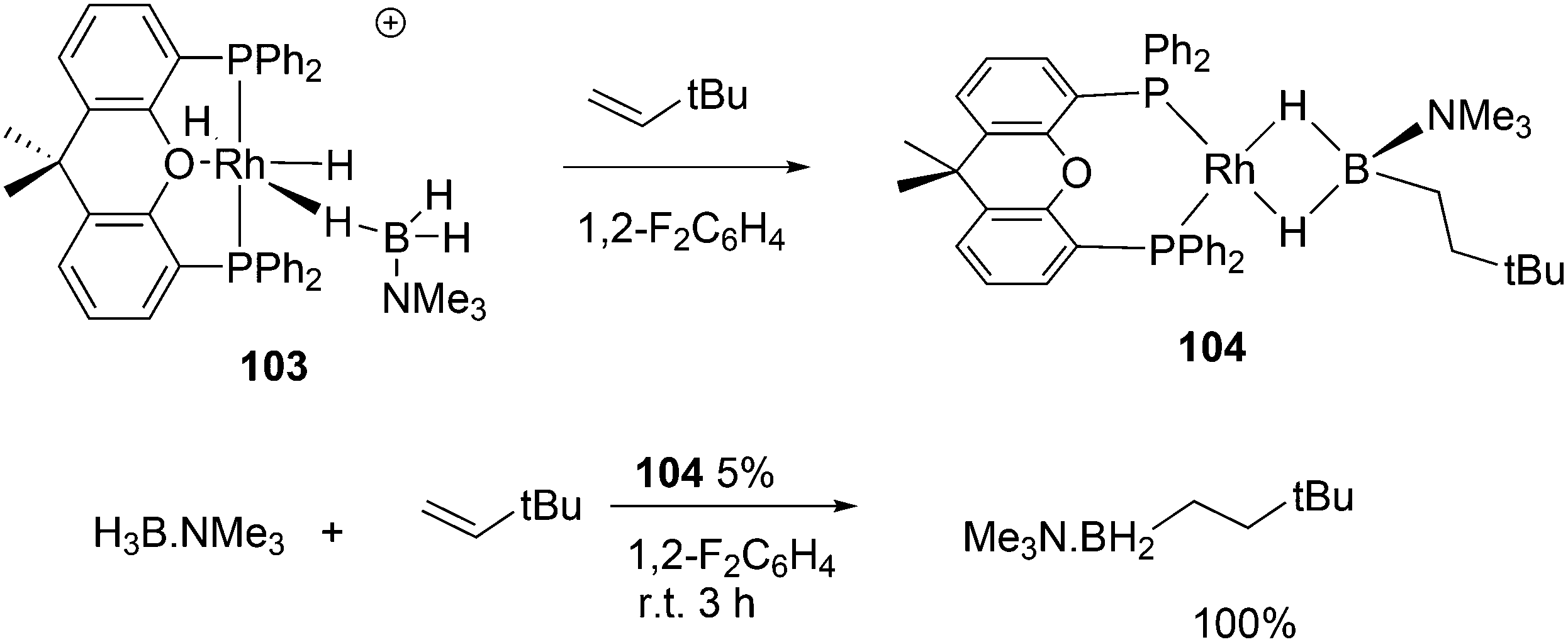 | ||
| Scheme 188 Alkene hydroboration catalyst. | ||
Arylnitriles can be converted to boronic esters with 2,2′-bi(dioxaborinane) and RhCl(Xantphos) as the catalyst, as shown in Scheme 189.538 While the behaviour of the nitrile seems like that of a pseudohalide, stoichiometric reaction with (Bpin)2 by Esteruelas and co-workers on iPrXantphos showed that the mechanism is more interesting.539 The isolated intermediates showed that after formation of a Rh–B bond, the nitrile inserts into this bond and this intermediate rearranges to the phenylrhodium and a boryl cyanide. Phenylrhodium reacts with (Bpin)2 to give phenylboronate.
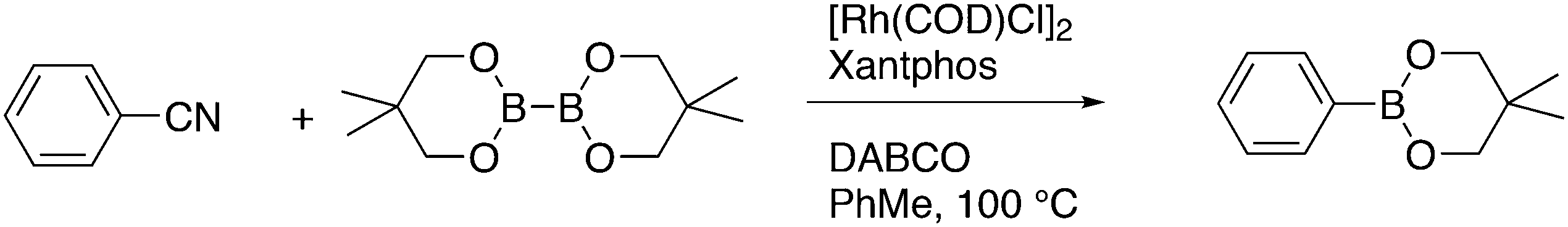 | ||
| Scheme 189 Conversion of aryl nitriles to aryl boronates. | ||
The square-planar rhodium hydride of iPrXantphos can be converted to the cationic complex shown in Scheme 190, which is a fast catalyst for the monoalcoholysis of H2SiPh2.540
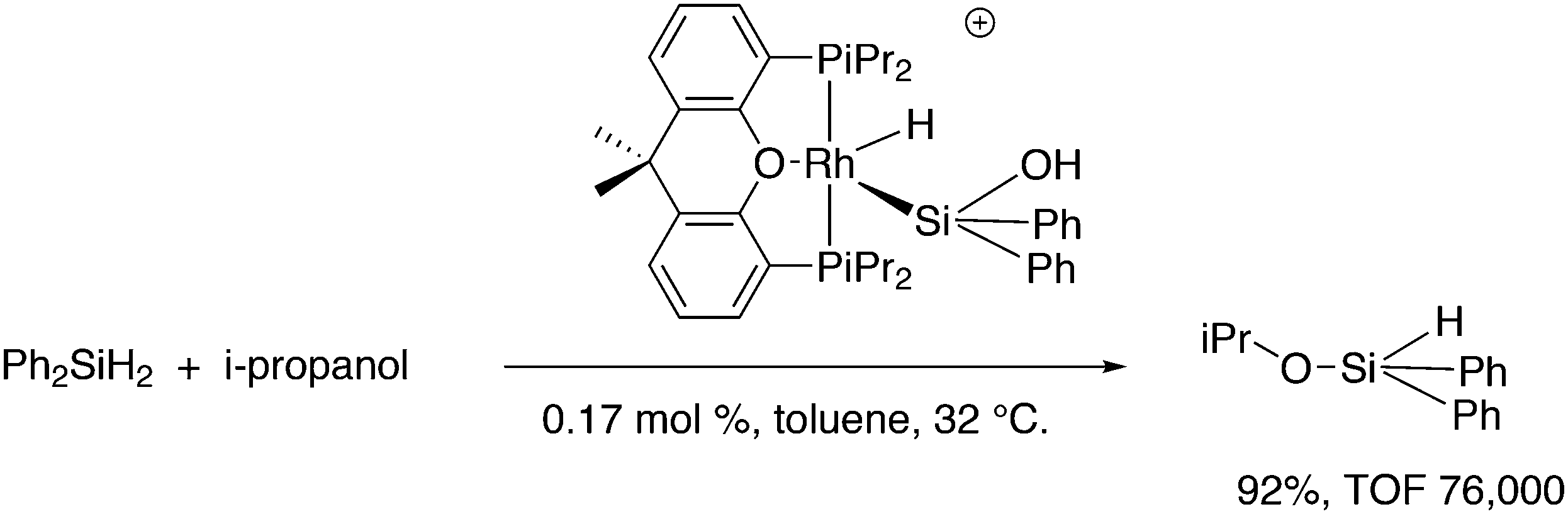 | ||
| Scheme 190 Alcoholysis of silanes. | ||
Decarbonylation of alkynylketones and alkynyl-1,2-diketones with the use of RhCl-Xantphos was reported by Dong and co-workers.541,542 In the first publication concerning alkynyl ketones, the authors conducted DFT calculations, which proved that rhodium inserts into the carbonyl–alkynyl bond. The rate for this reaction did not differ much for dppp or Xantphos, but the reductive elimination for the dppp complex was much slower than that of the Xantphos intermediate. In the second publication an interesting ligand effect was reported as shown in Scheme 191; dppe gave monodecarbonylation from alkynyl α-diketones, while Xantphos gave double decarbonylation. There are no calculations available yet to explain this behaviour.
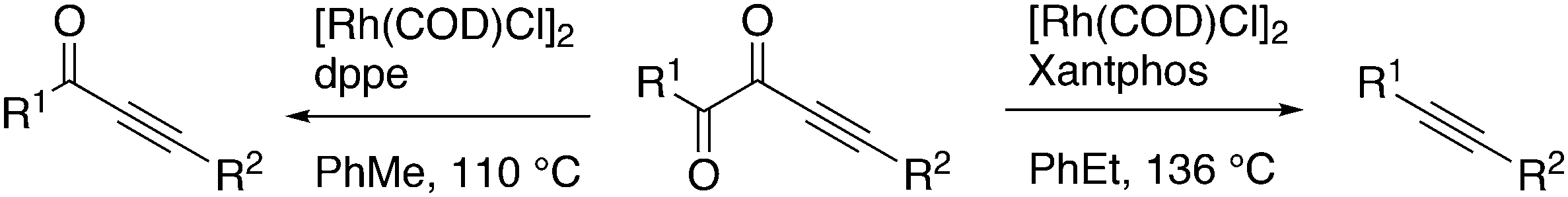 | ||
| Scheme 191 Rh-Diphos catalysed decarbonylation. | ||
In their search for pincer ligands useful for alkane hydrogen transfer reactions, Krogh-Jespersen, Goldberg and co-workers studied rhodium hydrides of tBuXantphos.543 The square planar chloride complex undergoes oxidative addition to the chlorodihydride rhodium complex, which affords rhodium(I) hydride upon treatment with strong base (Scheme 192) or the cationinic dihydride when treated with a chloride abstractor. Interestingly the monohydride does not undergo oxidative addition of H2, in spite of its presumed high electron density. It undergoes insertion of ethene to form the unstable ethylrhodium complex. With hexene-1, no insertion products were observed, but it does isomerize to hexene-2, showing that labile alkyl intermediates are accessible.
 | ||
| Scheme 192 Synthesis of tBu-Xantphos Rh–H complexes. | ||
Related iridium chemistry has also been explored by Weller and co-workers.544 Thus Ir(κ2-Xantphos)(COD)BArF4 reacts with H2 to give cationic dihydrides Ir(κ3-Xantphos)H2(acetone)BArF4 as shown above for Rh.
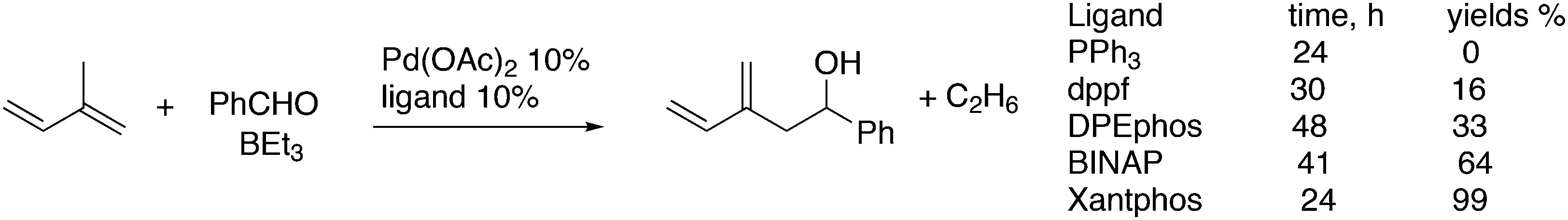 | ||
| Scheme 193 Allylation of benzaldehyde with dienes. | ||
Sauthier and co-workers reported a combined alkoxycarbonylation and allylation reaction catalysed by the same Pd catalyst, preferably ligated by Xantphos, Scheme 194.546 Prolonged reaction times gave 80% double allylation.
 | ||
| Scheme 194 Combined methoxycarbonylation and allylation. | ||
Another example of a combined radical and organometallic-catalysed reaction involves the functionalization of benzylic C–H bonds with a carboxylic ester group.547 Thus, an excess of toluene was reacted with an oxidant (tBuOOtBu), CO, ethanol, and a Pd/Xantphos complex as the carbonylation catalyst. Yields based on peroxide were 80% and several hundreds of turnovers were achieved at 120 °C and 10 bar CO. The authors proposed that the sequence starts with the abstraction of a hydrogen atom and that two radicals react with Pd(0) (Scheme 195). Incorporation of tBuO in the product is low and the reaction is much faster for EtOH. It might well be that CO insertion occurs in the Pd–benzyl bond; rate differences for alcohols and Pd-acyl complexes are well established.548
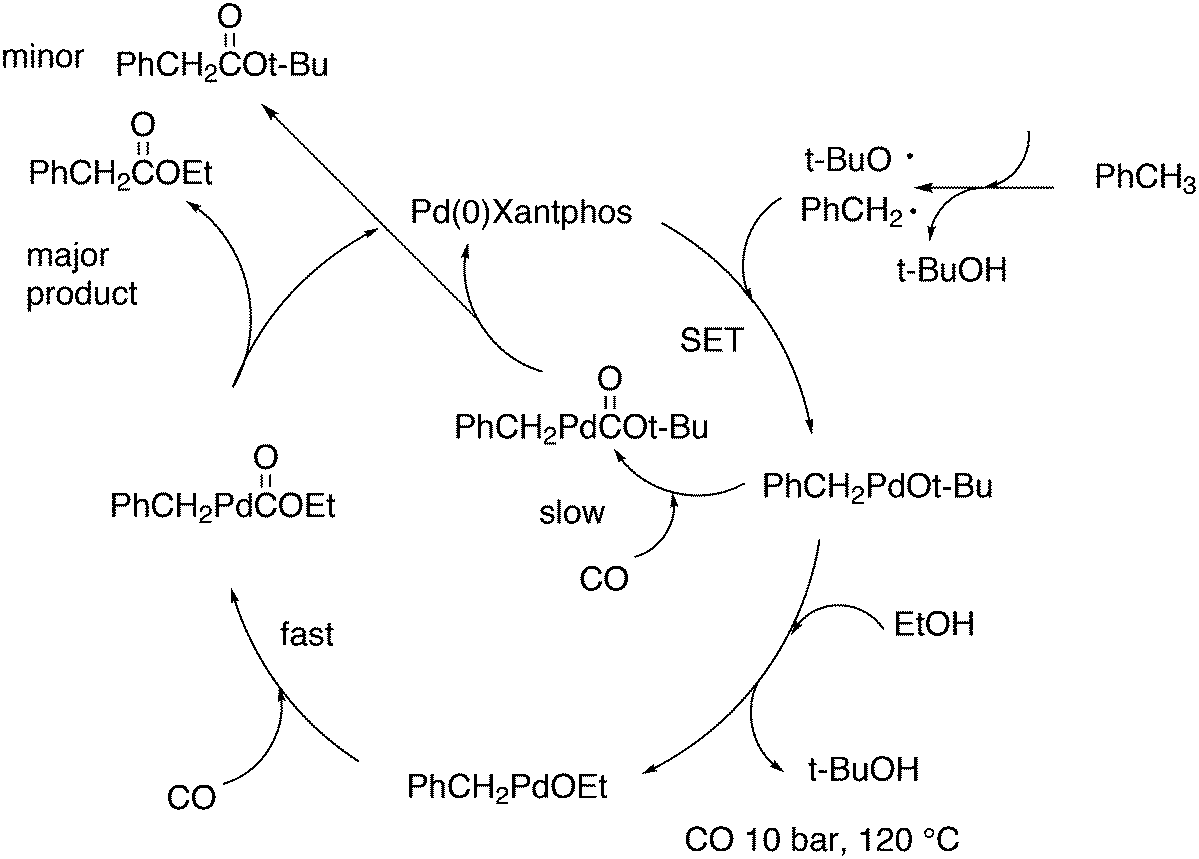 | ||
| Scheme 195 Radical initiated alkoxycarbonylation of toluene (Xantphos omitted on Pd(II)). | ||
Skrydstrup and co-workers developed a protocol for the rapid introduction of a 11C label as an acetyl group in proteins for PET studies.549 The acetyl group was generated on a methylpalladium precursor with 11CO (Scheme 196). Since t1/2 is only 20.4 min, the reactions used must be very fast and efficient. A range of mono- and bidentate phosphines was tested for the stoichiometric transfer of the acetyl 12C group to a pendant lysine residue in a protein molecule. Xantphos and dppf gave satisfactory results and XantphosPdMeCl, although not very stable, was used in the final experiment with 11CO. Trapping efficiencies were up to 96% and radiochemical yields up to 21%. Water had to be rigorously excluded as the acetylpalladium complex rapidly reacts with water to give acetic acid.546
 | ||
| Scheme 196 Labeling of a lysine residue with 11C acetylation. | ||
Jiao, Huang, et al. studied the palladium-catalysed hydroaminocarbonylation of alkenes with amines (Scheme 197).550 All reactions were carried out in the presence of chloride and the addition of a weak acid in stoichiometric amounts with respect to amine was necessary. The most effective acid was NH2OH·HCl, and stronger acids such as ptsa gave lower conversions. It was suggested that (ligand)PdHCl is a catalyst intermediate undergoing alkene insertion, which would react with amine to give inactive Pd(0); protonation of the amine substrate avoids this. Mixtures of linear and branched products were obtained and both aromatic and aliphatic alkenes could be converted.
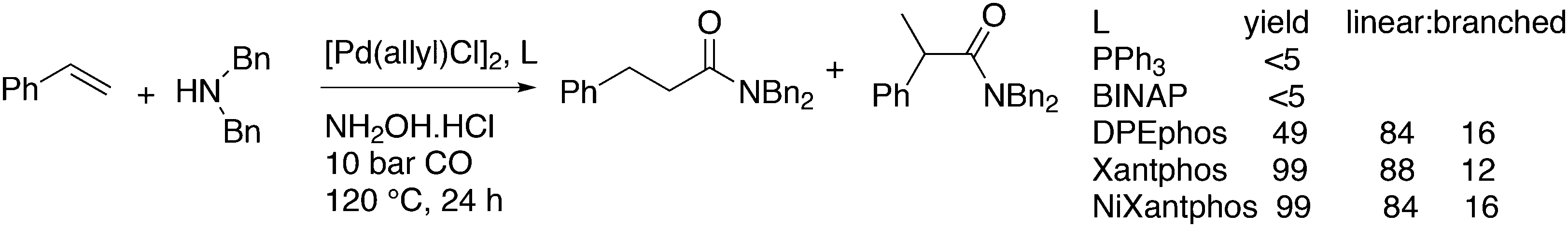 | ||
| Scheme 197 Hydroaminocarbonylation of alkenes. | ||
1,2-Allenes were successfully alkoxycarbonylated with aliphatic alcohols by Liu, Beller, et al. using the procedure popularized by Drent, i.e. the catalyst is made in situ from Pd(OAc)2, ptsa and ligand.551 Two interesting ligand systems were found: 2-PyPPh2, whic gave selectively α-β-unsaturated branched esters, and Xantphos, which gave the linear product, a β-γ-unsaturated ester (Scheme 198). DPEphos and dppf, the common look-alikes of Xantphos, gave only 20 and 2% yield respectively. DPEphos gave mainly butyl R-allyl ether as the product. The authors proposed for both reactions that the cycle starts with a Pd–H intermediate, as in most non-oxidative carbonylation reactions with Pd catalysts. Note that 2-PyPPh2 is also the preferred ligand for propyne methoxycarbonylation to methyl methacrylate, as reported by Drent and Budzelaar, which must pass through the same intermediate.552
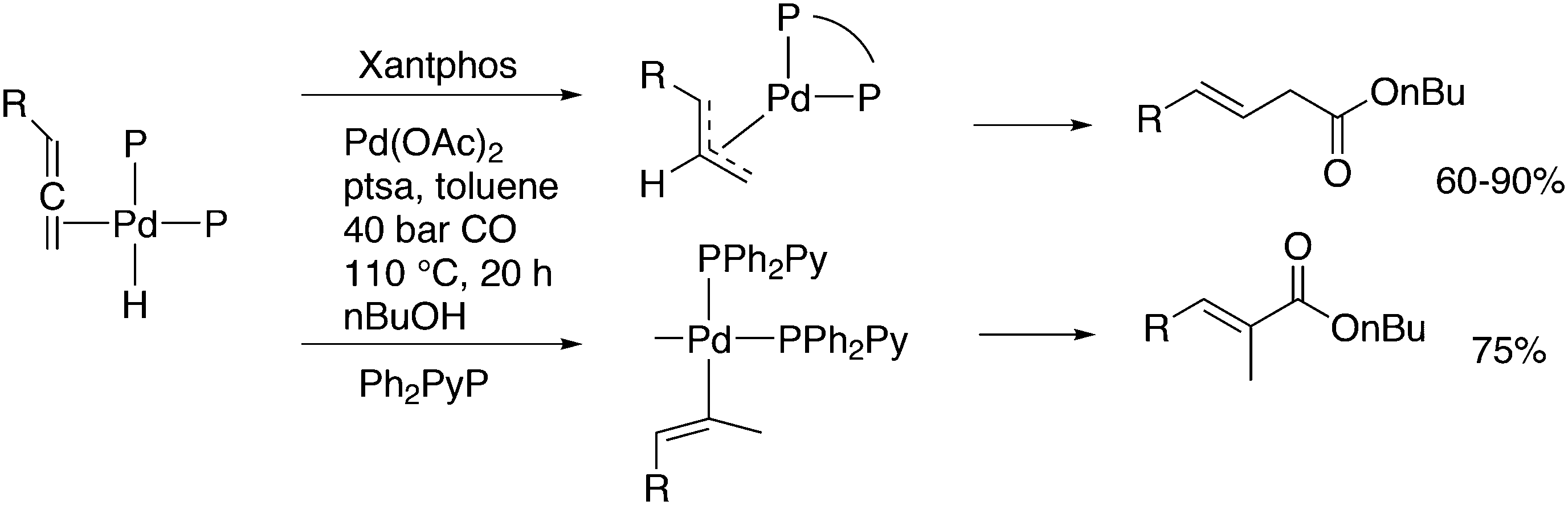 | ||
| Scheme 198 Alkoxycarbonylation of allenes. | ||
Manoury and co-workers studied the intramolecular alkoxycarbonylation with a PdCl2/SnCl2 catalyst modified with a broad spectrum of phosphine ligands (Scheme 199).553 The interesting results have not yet been fully explained. The monodentate phosphines gave mainly the six-membered lactone 105, while the bidentate phosphines gave >75% of the five-membered ring 106, both as a mixture of diastereoisomers. Only the two extreme results are shown, viz. PCy3 gave the highest selectivity to 105. For the bidentates, the rate increased with the bite angle and for Xantphos, the selectivity peaked at 99%. This is an extraordinary result, since the formation of 106 requires a branched alkyl species, while in all carbonylation reactions, be it for Pd or Rh, Xantphos-like ligands have a preference for linear alkyl formation. For intermolecular hydroxycarbonylation, the “normal” behaviour was reported, i.e. Xantphos prefers linear products, and monophoshines produce more branched products.554 The addition of SnCl2 to a PdCl2 catalyst strongly influences the regioselectivity in intermolecular alkoxycarbonylation with PPh3 as the ligand; it reverses the selectivity from mainly branched to mainly linear.555 We assume that the initial alkene insertion does not determine the final product and that all insertions are reversible. DFT calculations have shown that the attack by alcohol on the metal–acyl bond involves coordination of the alcohol to the metal centre and the reaction resembles a reductive elimination.556 In the intermolecular reaction, tertiary alcohols react extremely slowly. On this latter part of the reaction, monodentates and bidentates may exert different influences.
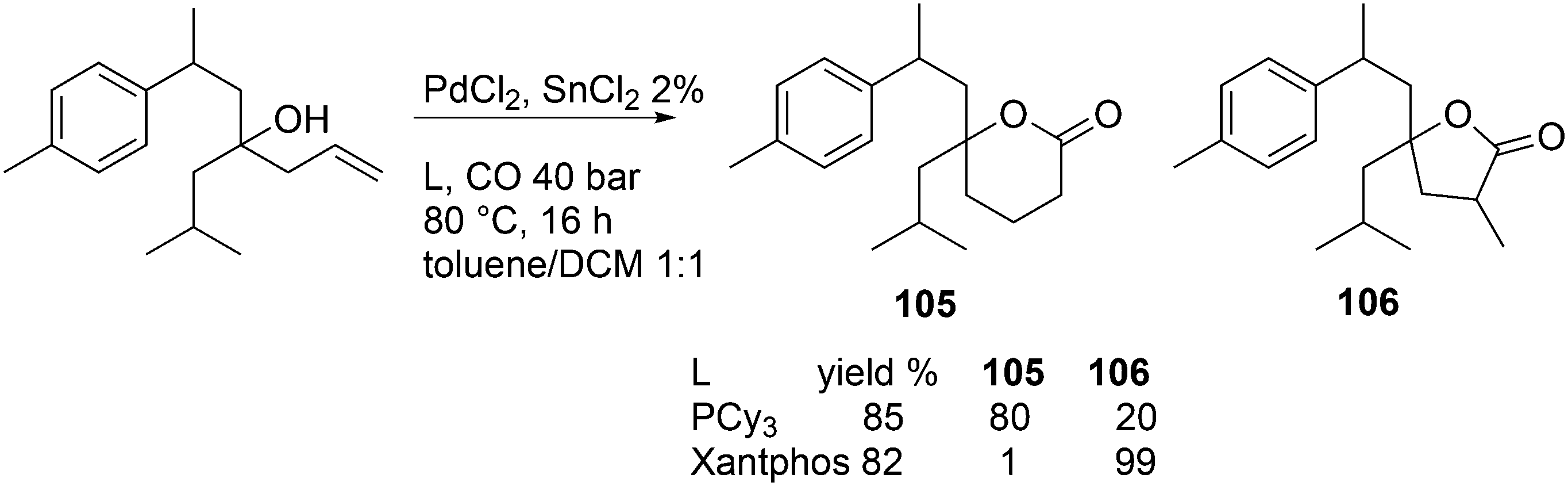 | ||
| Scheme 199 Intramolecular alkoxycarbonylation. | ||
Sun and co-workers developed a multistep one-pot reaction for the formation of benzoxazoles, benzothiazoles, quinazolinones, etc. from nitroaromatics, which uses Pd catalysts, Fe(CO)5 as the reductant, and MW heating (Scheme 200).557 The reaction works fine without a ligand on Pd for the five-membered derivatives. Optimization of the reaction yielding quinazolinones showed that bidentate phosphine ligands enhanced the yields (none 20%; dppp 42%; Xantphos 87%) under identical conditions. The Pd catalysed reduction of nitroaromatics is well known,558 also with Fe carbonyls,559 but unfortunately, phosphine ligands were found to be short-lived and oxidized by the nitro substrates.560
 | ||
| Scheme 200 One-pot formation of benzoxazoles and quinazolinones. | ||
Decarbonylation of carboxylic acids to alkenes was reported by Gooßen and Rodríguez, Scheme 201.561 See also 3.3.1.2 in which the intermediate Pd–aryl obtained from an acid anhydride was used in a cross-coupling reaction.221 PdBr2 instead of PdCl2 gave 100% yield of the completely isomerized product, and less polar solvents were less effective. Pivalic acid can be recycled.
 | ||
| Scheme 201 Conversion of carboxylic acids to alkenes. | ||
Additions of amines to 1,2-dienes can be catalysed by Pt complexes as shown by Toups and Widenhoefer; this had been known as a stoichiometric reaction for years.562 A range of ligands were studied and typical results are shown in Scheme 202 (only the E product is shown). The authors proposed a Wacker type addition of amine to the Pt-coordinated allene, followed by protonation of the Pt–C bond, which we would disfavour on cationic Pd and Pt systems.
 | ||
| Scheme 202 Addition amines to allenes with a Pt catalyst. | ||
Seayad et al. studied the Pd-catalysed alkylation of amines by primary and secondary alcohols (Scheme 203).563 Palladium chloride was used as the catalyst for the reaction of aniline and benzyl alcohol and a wide range of ligands was screened. High yields were reached with Xantphos, tBuXantphos, and tBu-dppf, but not for dppf, BINAP, DPEphos, CyDPEphos, etc. Surprisingly, dppe also gave good results; this ligand does not resemble the electron donating and wide bite angle ligands that gave high activity. Throughout the catalytic cycle, Pd remained divalent and perhaps both the donor character of the tBu substituted ligands and the small bite angle of dppe played a favourable role in this.
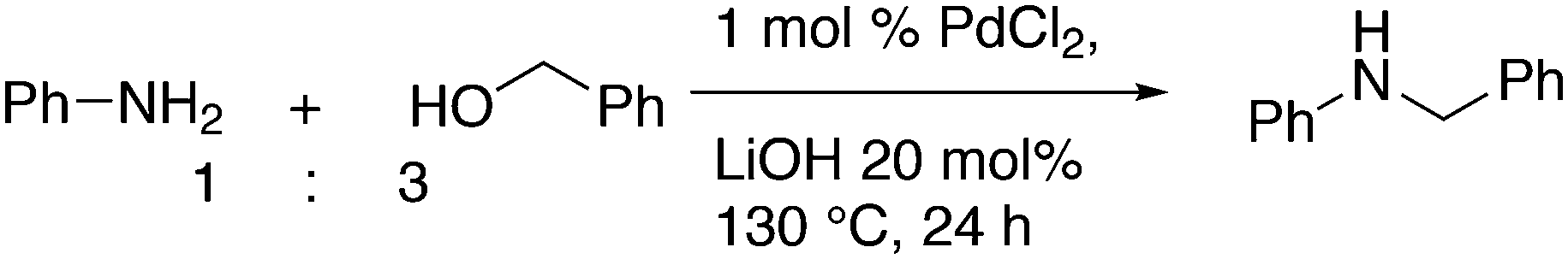 | ||
| Scheme 203 Alkylation of aniline with benzyl alcohol. | ||
Liu et al. described a new method for the synthesis of chromeno[2,3-b]indoles, which are desired as both natural products and drug molecules.564 Their synthesis requires many steps and harsh temperatures. The new synthesis involves the Pd catalysed reaction of salicylaldehyde and 2-bromoindole for the unsubstituted compound (Scheme 204). It was found that the reaction can be catalysed by Pd2(dba)3 and phosphine ligands. Xantphos gave the highest yield (90%), followed by dppf (80%) and dppb (52%). Although this ligand effect suggests a cross-coupling mechanism and a rate limiting reductive elimination, with control reactions the authors found no indications for a cross-coupling. Instead, they proposed a nucleophilic attack by the phenolate as the first step, with the formation of a Pd–C-3 bond that undergoes insertion of the aldehyde. A broad scope of the reaction was reported.
 | ||
| Scheme 204 A new synthesis of chromeno[2,3-b]indoles. | ||
In their work on selective 15N isotope substituted nucleotides, Caner and Vilarrasa developed a protocol for sequential C–O and C–N bond forming reactions and out of 5 ligand systems, Xantphos gave the best results (Scheme 205).565 The isotope in the 1-position was accomplished via a series of ring-opening/closure reactions566 and the final C–N coupling also gave an opportunity to introduce15N.
 | ||
| Scheme 205 Subsequent C–O and C–N bond formation (Rf = tri-O-protected β-ribofuranosyl). | ||
Double borylation of 2,2′-di(bromophenyl) ether can be accomplished by iPr2NBH2 and a palladium phosphine catalyst (Scheme 206).567 When unbridged aryl halides are used, only monoarylation of boron takes place. In the aqueous workup, the amine group is replaced by hydroxyl. CyJohnphos as the ligand also gave high yield, provided that KI was added. A broad scope was reported for the reaction. The boron products can be converted with a variety of 1,2-dihaloarenes to cyclic compounds and Pd/CyJohnphos as the catalyst.
 | ||
| Scheme 206 Double arylation of borane. | ||
Nechaev et al. compared many phosphine ligands for the borylation of aryl halides and pseudohalides with B2(pin)2.568 For aryl bromides, DPEphos gave the highest yield, but for chlorobenzene it gave zero conversion, while Xphos gave 99% yield. Scheme 207 presents a few typical results.
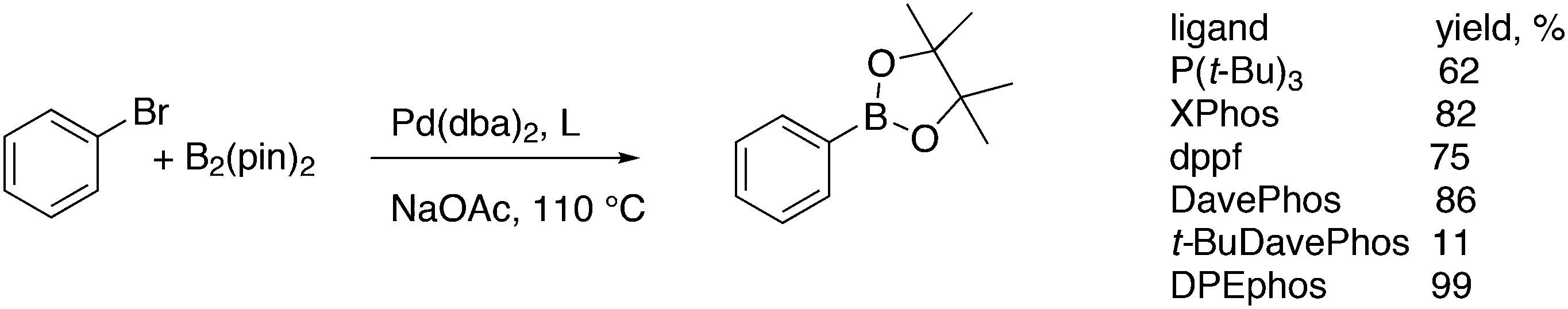 | ||
| Scheme 207 Borylation of bromobenzene. | ||
Palladium/bisphosphine catalysts can be used for hydrogen transfer of formic acid to enones in which the C–C double bond is hydrogenated as shown by Liu, Zeng, et al.569 Several bidentates were explored, but the first screening showed Xantphos as the best ligand in toluene and the conversion could be further increased to 99% in protic solvents (Scheme 208). It is interesting that in such a heterolytic hydrogenation system the double carbon bond is hydrogenated rather than the polar CO bond (commercially Pd is used as a heterogeneous catalyst for this conversion). The authors propose a mechanism in which both hydrogen atoms pass through Pd–H rather than an enolate mechanism and a proton. The scope was extended to 25 more related substrates with the use of water and Xantphos.
 | ||
| Scheme 208 Hydrogen transfer hydrogenation of enones. | ||
Hu and Snyder recently developed a total synthesis of presilphiperfolan-8-ol, a natural product isolated from plants (Eriophyllum staechadifolium and Flourensia heterolepis occurring in California and the Americas, respectively), which had not been synthesized before.570 The key feature is the strained trans nature of the [3,3,0]-bicyclooctane fragment (Scheme 209). The intriguing step is the tandem formation of the two rings with a Pd catalyst. Several ligands gave moderate to good results, and with the use of DPEphos, 75% was reported. The reaction cannot stop after the first insertion step as a Heck–Mizoroki reaction, since there is no β-hydrogen and thus the next alkene is inserted. The trans configuration is obtained in the next step (not shown), via epoxidation of the internal double bond while the methylene fragment is converted to a ketone.
 | ||
| Scheme 209 Key step in the synthesis of (−)-presilphiperfolan-8-ol from R-pulegone. | ||
000 were achieved. The rates increase in the order dppf < DPEphos < homoxantphos < Sixantphos. Kinetic behaviour and rates are very similar to those of the well known Rh catalysts, as developed by Wilkinson and Osborn. The study is supported by DFT calculations, which showed that in the hydride migration step the P–Re–P angle was widened by 6°, favoring the presence of the large-bite-angle bisphosphines in this elementary step. The authors comment further that the large-bite-angle bisphosphines also support Re(I)/Re(III) redox changes, favoring the generation of five-coordinate Re(I) species by ligand dissociation and seven-coordinate Re(III) intermediates required in oxidative addition processes. These two elements explain the large-bite-angle influence of the bisphosphines on the catalytic reaction.
 | ||
| Scheme 210 Rhenium hydride complexes. | ||
The Re complexes with wide bite angle bisphosphines were also found to be good homogeneous catalysts for the hydrogenation of nitriles at somewhat higher temperatures than those used for alkene hydrogenation (120–140 °C).573 Sixantphos afforded the fastest catalyst with TOF up to 250 h−1 at 140 °C, 50 bar H2, and Et3SiH added as a stabilizer. Benzonitrile gave dibenzylamine as the main product in around 80% yield, which is not uncommon for nitrile hydrogenation. Cyclohexylamine gave as much as 95% dicyclohexylamine.
He et al. reported on a three-component reaction of propargylic alcohols, CO2, and β-aminoalcohols.574 Several products can be envisaged for this reaction, but with the use of Ag as the catalyst ligated by phosphine ligands, the reaction takes the course shown in Scheme 211. Several phosphines gave some product (<20%, dppf, dppp, dpp-benzene, tri-2-furylphosphine), but since Xantphos performed the best, the reaction was optimized for this ligand. A wide range of 2-oxazolidinones could be prepared in this way.
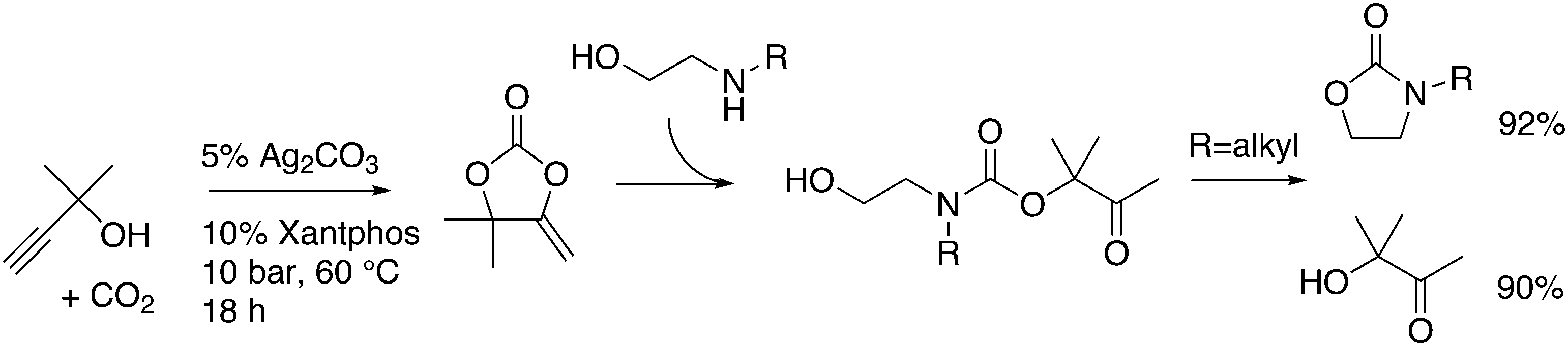 | ||
| Scheme 211 Ag/phosphine catalysed reaction of propargyl alcohol, CO2 and aminoalcohol. | ||
Sun and co-workers recently reported an interesting complementary behaviour of Au and Ag catalysts in the insertion of vinylcarbene into O–H bonds starting from vinyldiazoacetates.575 The substrates are 2-pyridones, but insertion takes place selectively in the O–H bond of the phenolic hydroxylpyridine isomer (Scheme 212). Several metal salts catalysed the reaction in moderate yields (Cu, Rh, NHC-Au) but cationic Au complexes Xantphos(AuNTf2)2 gave high yields and >95% selectivity for 107. When AgSbF6 was used and no ligands added, 108 was obtained in 83% yield and 90% selectivity. N–H insertion products were not observed at all and no catalytic isomerisation was detected. The vinologous reactivity for O–H insertions in methanol and benzylalcohol was observed before by Hansen and Davies, also for AgOTf.576
 | ||
| Scheme 212 Vinylcarbene insertion into 2-pyridone. | ||
As we have seen above, Au(I) forms bimetallic complexes with Xantphos, i.e. each P-donor coordinates to one Au cation. Paradies et al. investigated the effect of two nearby Au cations on the intermolecular addition of sulfonamides to alkenes (Scheme 213).577 The AuCl complexes were in situ dechlorinated by Ag salts, triflates and the like. Yields for the hydrosulfonamidation ranged from 50 to 90% and the best results stemmed from the dicationic catalysts. A strong effect of anions and solvents was noted. The authors proposed that there is no particular bimetallic effect caused by the proximaty of the two cations, but an increased electrophilicity of the dicationic species explains the higher reactivity.
 | ||
| Scheme 213 Monometallic and bimetallic Au catalysts in the hydrosulfonamidation of alkenes. | ||
Above, we referred to the dehydrogenative coupling of silanes and alcohols, a reaction catalysed by many cationic transition metal complexes.495 Having discovered the favourable action of Xantphos for Cu catalysts, Sawamura and co-workers also studied Au(I) complexes for this reaction (Scheme 214).578 Gold chloride was used for the reaction and cationic complexes were not tested. Xantphos gave the only practical catalyst, although the Cu complex is still faster. It was proposed that the unique formation of three-coordinate complexes by Xantphos was important and that Cl− can most likely be easily replaced in this complex. Primary, secondary, and tertiary alcohols could be transformed with high efficiency.
 | ||
| Scheme 214 Dehydrogenative silylation of alcohols. | ||
Jean, Mezailles, et al. studied this reaction by DFT and developed a catalyst based on phosphole ligand 92Scheme 142, omitting the Ph groups in the calculations.448,579 A dimeric Au2H+ was found to be a likely intermediate as it was the only observed species in the catalytic system. Complete conversion was reached in 24 hours at 50 °C, substrate ratio 1:1, 1% catalyst, and it seems that the cationic complexes are not significantly faster than the chloride systems.
3.8. Concluding remarks and outlook
In the past few decades, the formation of C–C and C–X bonds with the aid of metal catalysts has undergone tremendous growth in the number of reaction types and catalysts, which still continues at an increasing pace. Ligands form an important constituent of the catalyst and a variety of privileged ligand groups have been developed. In this review we have highlighted the catalysis of P-ligands with the generic group 2,2′-bis(PR2)diphenyl ether. First, the syntheses were reviewed with attention to accessibility and potential modifications. Subsequently, we showcased a number of reactions in which the Xantphos ligands gave exceptional results that were mostly positive, but sometimes negative. This review is not comprehensive since that would result in close to 3000 references to date; the present selection contains about 20% of these.Most Xantphos ligands can be made via a straightforward method in few steps and the availability of the backbone is crucial. Diphenyl ether and ditolyl ether are on the market on a multi-ton scale and can be used for the synthesis of DPEphos, Sixantphos, and Phosxantphos via lithiation chemistry, and the backbone of Thixantphos via Friedel–Crafts chemistry, followed by lithiation, which is probably the cheapest route. Several of these have been made on the multi-kg scale and can now be ordered on a large scale. Several authors have noted in their work the often similar behaviour in catalysis of dppf and DPEphos, remarking that cost-wise the latter is much more attractive. Several water-soluble ligands have been reported and Sulfoxantphos 14 is most often used. Nixantphos is made from phenoxazine via lithiation after protecting the N–H group; phenoxazine can be made from o-aminophenol with iodide and is available on the multi-kg scale. Nixantphos has often been the choice for immobilization on solids or polymeric materials. Chiral ligands have scarcely been explored as was pointed out in 2.2.5. So far, the asymmetric reaction studied most has been AAA, for which a plethora of other ligands is available. Since wide bite angle ligands are not particualarly favoured for hydrogenation in square-planar complexes, there is little demand for chiral variants. Perhaps useful asymmetric applications can be found in the less known reactions collected in this review (3.7).
Rhodium/bisphosphine catalysed hydroformylation (3.1.1) has been the subject of many studies and a lot of insight has been generated. In most cases, a bis-equatorially coordinated bisphosphine rhodium intermediate favours the insertion of 1-alkenes into the 1-alkylrhodium species. Most authors have assigned this to non-bonding interactions between substrate and ligand. Steric variations in the ligand groups discussed here are unfortunately scarce, but this might provide further insight. Whether alkene coordination or migratory insertion is the rate limiting step has not yet been established; elaborate isotope studies may give an answer to this, but the one example that was studied (Xantphos) did not lead to a definite answer. The coordination mode per se does not determine the preference for the linear product, as there are several exceptions to the rule; e.g. Xantphos coordinates in the pentagonal resting state predominantly in the axial–equatorial mode. Since both rate and linearity increase when electron-withdrawing ligands are used, alkylphosphine derivatives are not of interest, unlike new mechanisms that become operative, such as carbene mechanisms.580 A modest number of EWG-containing aryl groups and phosphonite groups have been investigated.
In palladium catalysed methoxycarbonylation/oligomerisation/polymerisation of CO and alkenes, the Xantphos group played a role in the rationalization of the ligand effect on the product distribution. The ligands gave oligomerisation, similar to PPh3, or were not active if stable trans intermediates could be formed. There is an important steric effect for the production of ester, bulky ligands with intermediate angles providing the best catalysts for ester formation (BDtBPX, the Lucite or Alpha ligand). In view of the increasing appreciation for remote steric effects of ligands in catalysis, this is also an option for the present ligand group. Since most ligand effects of Xantphos are explained as steric, this modification could lead to major advances. Bifunctional ligands are also gaining ground, among which we find 2-pyridyl substituted phosphines that aid proton transfer in Pd catalysed carbonylation reactions.581 Beller has shown that this works very well in xylene and ferrocene based ligands, but Xantphos, containing the same substitution, was inactive in the methoxycarbonylation of alkenes.582
Wide bite angle ligands were introduced in Ni, Pd catalysed hydrocyanation and cross-coupling as it was expected that reductive elimination would be enhanced by ligands preferring angles in tetrahedral species. This turned out to be the case, but not for the initially thought electronic effects, but simply for steric reasons, since DFT calculations show that the electronic bite angle effect plays a minor role. Thus, the ferrocene ligands with more sterically bulky phosphine groups show the same behaviour. Since sterics also dominate for these reactions, remote steric effects may improve the catalysts. For aryl chlorides, Xantphos ligands were not suited until deprotonated Nixantphos turned out to be an efficient catalyst, as was discovered by the Walsh group (3.3.1.13). More electron-rich Xantphos might also offer a possibility to convert aryl chlorides.
As stated in 3.3.2 (C–C) and 3.4.2 (C–N), in carbonylative cross-coupling reactions Xantphos often stands out but detailed explanations have not been given. Perhaps five-coordinate intermediates are more easily accessible in complexes with flexible ligands.
In C–N (3.4.1) bond formation, Xantphos was shown to be particularly useful for weak N-nucleophiles. This may be due to the low concentration of the critical intermediate arylPd(N-nucleophile) and thus the ligand most facilitating the reductive elimination may lead to the most effective catalyst. Addition of amines to dienes proceeds well with Xantphos, most likely in view of the right balance between the stability and reactivity of the allylic intermediates.
For C–P bond formation (3.5.1), the R2PH precursors form stable metal complexes and in general, they are not suitable for cross-coupling with transition metal catalysts. Instead, pentavalent phosphorus compounds R2P(O)H were used in both coupling reactions with halides and addition reactions to alkynes and alkenes. Hypophosphorous acid H2P(O)OH and its esters are very versatile starting materials and Xantphos stands out as the ligand. This reaction is of industrial interest for the production of flame retarding materials.
Reactions involving allylic fragments often take advantage of more bulky bidentates or wide bite angle bidentates, as in the “Trost” ligands (3.6). Chiral variants of the Xantphos ligands might be desirable in some instances.
Section 3.7 shows a great variety of reactions using a range of transition metals. For these less explored reactions, an approach by ligand design does not seem feasible, especially not since sometimes only one ligand gives the desired or unexpected result. Indeed, most reactions have been found by trial and error, but it is generally recognized that de Xantphos ligands may bring a surprise. Only in very few cases have these studies found follow-up with mechanistic investigations supported by DFT calculations, except for the classic reactions hydroformylation and reductive elimination. One notable example among the new reactions was mentioned above (3.7.6): the hydrogenation of alkenes by novel Re complexes with wide bite angle bisphosphine ligands, for which a detailed study was completed and catalysts were developed that compete with the classic hydrogenation catalysts of Rh and group 10 metals.
The presented selection of examples is illustrative of the unique performance of wide bite angle phosphorus donor ligands, of which Xantphos is one of the most prominent examples in many catalytic reactions. We anticipate that new applications of this class of ligands will continue to appear, not only in catalysis, but also in photochemistry, energy and material science applications.
Abbreviations
| AAAA | Asymmetric allylic alkylation |
| BINAP | 2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene |
| BISBI | 2,2′-Bis(diphenylphosphinomethyl)-1,1′-binaphthalene |
| β n | Calculated natural bite angle |
| BpinH | 4,4,5,5-Tetramethyl-1,3,2-dioxaborolane |
| BrettPhos | 2-(Dicyclohexylphosphino)3,6-dimethoxy-2′,4′,6′-triisopropyl-1,1′-biphenyl |
| CPME | Cyclopentylmethyl ether |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DABSO | 1,4-Diazabicyclo[2.2.2]octane bis(sulphur dioxide) |
| dba | Dibenzylideneacetone |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DBFphos | 4,6-Bis(diphenylphosphino)dibenzofuran |
| DCCP | Deprotonative cross-coupling process |
| DFT | Density functional theory |
| DIPAMP | 1,2-Bis(phenyl,o-anisylphosphino)ethane |
| DIOP | Bis(diphenylphosphino) |
| DME | 1,2-Dimethoxyethane |
| DMF | Dimethylformamide |
| DPEphos | 2,2′-Bis(diphenylphosphino)diphenyl ether |
| dppb | 1,4-Bis(diphenylphosphino)butane |
| dppe | 1,2-Bis(diphenylphosphino)ethane |
| dppf | 1,1′-Bis(diphenylphosphino)ferrocene |
| dppm | Bis(diphenylphosphino)methane |
| dppp | 1,3-Bis(diphenylphosphino)propane |
| Duphos | 1,2-Bis(2,5-dimethylphospholan-1-yl)benzene |
| EH | Extended Hückel |
| EWG | Electron withdrawing group |
| Josiphos | (R)-1-[(SP)-2-(Diphenylphosphino)ferrocenyl]ethyldicyclohexylphosphine |
| KOtBu | Potassium-tert-butoxide |
| LiHMDS | Lithium hexamethyldisilazide |
| MAO | Methylaluminoxane |
| S-MonoPhos | (S)-(+)-(3,5-Dioxa-4-phosphacyclohepta[2,1-a;3,4-a']dinaphthalen-4-yl)dimethylamine |
| NHC | N-heterocyclic-carbene |
| Nixantphos | 4,6-Bis(diphenylphosphino)-10H-phenoxazine |
| Phanephos | 4,12-Bis(diphenylphosphino)-[2.2]-paracyclophane |
| POP group | 10H-Phenoxaphosphinyl |
| POP ligand | 2,2′-Bis(diphenylphosphinoethyl) ether |
| QM/MM | Quantum mechanical/molecular mechanics |
| RuPhos | 2-Dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl |
| SET | Single electron transfer |
| SILP | Supported ionic liquid-phase |
| SPANphos | 4,4,4′,4′,6,6′-Hexamethyl-2,2′-spirobi[chromane]-8,8′-diylbis(diphenylphosphane) |
| SPhos | 2-Dicyclohexylphosphino-2′,6′-dimethoxybiphenyl |
| Sulfoxantphos | 4,5-Bis(diphenylphosphino)-9,9-dimethyl-2,7-disulfoxanthene disodium salt |
| THF | tetrahydrofuran |
| TMEDA | N,N,N′,N′-1,2-Ethylenediamine |
| Xantphos | 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene |
| XPhos | 2-Dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl |
Conflicts of interest
There are no conflicts to declare.References
-
P. W. N. M. van Leeuwen and J. C. Chadwick, Homogeneous Catalysts, Wiley VCH, Weinheim, Germany, 2011, ch. 1 Search PubMed
.
- W. Reppe and W. J. Schweckendiek, Justus Liebigs Ann. Chem., 1948, 560, 104 CrossRef CAS
- One synthetic route to (PPh3)2NiBr2 involved the reaction of nickel carbonyl with bromobenzene giving diphenyl, a C–C coupling reaction: W. J. Schweckendiek, DE patent 817909 (BASF, Ludwigshafen), 1951 (Submitted in France 1946)
- J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc. A, 1966, 1711 RSC
- P. S. Hallman, D. Evans, J. A. Osborn and G. Wilkinson, Chem. Commun., 1967, 305 RSC
- J. F. Young, J. A. Osborn, F. H. Jardine and G. Wilkinson, Chem. Commun., 1965, 131 RSC
-
L. H. Slaugh, BE Pat., 606408, 619344, and 621662, 1961–1963 (to Shell Internationale Research Maatschappij B. V. Neth.); Chem. Abstr., 1963, 59, 461609 and 1963, 59, 414728 Search PubMed
- K. Issleib and D. W. Mueller, Chem. Ber., 1959, 92, 3175 CrossRef CAS
- J. Chatt and W. Hart, J. Chem. Soc., 1960, 1378 RSC
- D. L. Thorn and R. Hoffmann, J. Am. Chem. Soc., 1978, 100, 2079 CrossRef CAS
- M. Iwamoto and S. Yuguchi, J. Org. Chem., 1966, 31, 4290 CrossRef CAS
- W. S. Knowles, M. J. Sabacky and B. D. Vineyard, Adv. Chem. Ser., 1974, 132, 274 CrossRef CAS
- H. B. Kagan and T.-P. Dang, J. Am. Chem. Soc., 1972, 94, 6429 CrossRef CAS
- M. van den Berg, A. J. Minnaard, E. P. Schudde, J. van Esch, A. H. M. de Vries, J. G. de Vries and B. L. Feringa, J. Am. Chem. Soc., 2000, 122, 11539 CrossRef CAS
-
P. W. N. M. van Leeuwen, P. A. M. Grotenhuis and B. L. Goodall, (to Shell Internationale Research Maatschappij B. V. Neth.). Eur. Pat. Appl., 309056, 1989, Priority: GB 87-22460
-
T. J. Devon, G. W. Phillips, T. A. Puckette, J. L. Stavinoha and J. J. Vanderbilt, (to Texas Eastman) US Pat., 4694109, 1987; Chem. Abstr., 1988, 108, 7890 Search PubMed
- M. Kranenburg, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 3081 CrossRef CAS
-
P. C. J. Kamer, M. Kranenburg and P. W. N. M. van Leeuwen, Book of Abstracts, XVlth International Conference Organometallic Chemistry, University of Sussex, The Royal Society of Chemistry - Dalton Division, abstract no. OC. 18, 10-15 July 1994 Search PubMed
- S. Hillebrand, J. Bruckmann, C. Krueger and M. W. Haenel, Tetrahedron Lett., 1995, 36, 75 CrossRef CAS
-
P. C. J. Kamer, M. Kranenburg, P. W. N. M. van Leeuwen and J. G. de Vries, BE patent appl., 9400470, May 6, 1994; PCT Int. Appl., WO 9530680, 1995; USP5817848, 1998, to DSM N.V. Heerlen, the Netherlands Search PubMed
- M. W. Haenel, D. Jakubik, E. Rothenberger and G. Schroth, Chem. Ber., 1991, 124, 1705 CrossRef CAS
- M. W. Haenel, H. Fieseler, D. Jakubik, B. Gabor, R. Goddard and C. Krueger, Tetrahedron Lett., 1995, 34, 2107 CrossRef
- E. M. Vogl, J. Bruckmann, C. Krueger and M. W. Haenel, J. Organomet. Chem., 1996, 520, 249 CrossRef CAS
- E. M. Vogl, J. Bruckmann, M. Kessler, C. Krueger and M. W. Haenel, Chem. Ber./Recl, 1997, 130, 1315 CrossRef CAS
- J. I. van der Vlugt, R. van Duren, G. D. Batema, R. den Heeten, A. Meetsma, J. Fraanje, K. Goubitz, P. C. J. Kamer, P. W. N. M. van Leeuwen and D. Vogt, Organometallics, 2005, 24, 5377 CrossRef CAS
- Strictly speaking the backbones in which X is not C are not xanthenes, but phenoxazine for X = N, phenoxathiine for X = S, phenoxaphosphinine for X = P, etc. To underscore the relationship we maintained xantphos in most names.
- L. A. van der Veen, P. H. Keeven, G. C. Schoemaker, J. N. H. Reek, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, Organometallics, 2000, 19, 872 CrossRef CAS
- Commercially available phosphines are Xantphos, (including Pd and Cu complexes, and Buchwald's metalated phenylaniline precursor, CAS 1445085-97-1), the cyclohexyl and t-butyl homologs of Xantphos, Nixantphos, DPEphos and its cyclohexyl, o-tolyl and t-butyl analogs, and from a small number of suppliers Thixantphos, Sixantphos, and chiral MePhXantphos.
- M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801 CrossRef CAS PubMed
-
W. Ahlers, M. Roeper, P. Hofmann, D. C. Warth and R. Paciello, PCT Int. Appl., WO2001058589, 2001 (to BASF, Ludwigshafen)
- O. Grossman and D. Gelman, Org. Lett., 2006, 8, 1189 CrossRef CAS PubMed
- L. Kaganovsky, K. Rueck-Braun and D. Gelman, J. Organomet. Chem., 2010, 695, 260 CrossRef CAS
- M. A. Zuideveld, B. H. G. Swennenhuis, D. K. M. Boele, Y. Guari, G. P. F. van Strijdonck, J. N. H. Reek, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz, A. L. Spek and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 2002, 2308 RSC
- J. Yin and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 6043 CrossRef CAS PubMed
- P. Dierkes and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans.: Inorg. Chem., 1999, 1519 RSC
- Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890 RSC
- P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741 CrossRef CAS PubMed
- M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099 RSC
- G. Cavinato and L. Toniolo, Molecules, 2014, 19, 15116 CrossRef PubMed
- P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895 CrossRef CAS PubMed
- J. S. Nowick, P. Ballester, F. Ebmeyer and J. Rebek, J. Am. Chem. Soc., 1990, 112, 8902 CrossRef CAS
- J. Clayden, A. Lund, L. Vallverdu and M. Helliwell, Nature, 2004, 431(7011), 966 CrossRef CAS PubMed
- A. F. Holleman, Org. Synth., 1941, 1, 552 Search PubMed
- Z. Freixa, M. S. Beentjes, G. D. Batema, C. B. Dieleman, G. P. F. van Strijdonck, J. N. H. Reek, P. C. J. Kamer, J. Fraanje, K. Goubitz and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2003, 42, 1284 CrossRef CAS PubMed
-
J. P. Wolfe, e-EROS Encyclopedia of Reagents for Organic Synthesis, Wiley, Chichester, UK, 2011, pp. 1–13 Search PubMed
- Y. M. Al-Hiari, S. J. Bennett, B. Cox, R. J. Davies, A. I. Khalaf, R. D. Waigh and A. J. Worsley, J. Heterocycl. Chem., 2005, 42, 647 CrossRef CAS
- M. Feigel, Liebigs Ann. Chem., 1989, 459 CrossRef CAS
- Y. Terao, T. Satoh, M. Miura and M. Nomura, Bull. Chem. Soc. Jap., 1999, 72, 2345 CrossRef CAS
- L. A. van der Veen, M. D. K. Boele, F. R. Bregman, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, J. Fraanje, H. Schenk and C. Bo, J. Am. Chem. Soc., 1998, 120, 11616 CrossRef CAS
- J. W. Raebiger, A. Miedaner, C. J. Curtis, S. M. Miller, O. P. Anderson and D. L. DuBois, J. Am. Chem. Soc., 2004, 126, 5502 CrossRef CAS PubMed
- M. Schroeder Goedheijt, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Mol. Catal. A: Chem., 1998, 134, 243 CrossRef
- W. P. Mul, K. Ramkisoensing, P. C. J. Kamer, J. N. H. Reek, A. J. van der Linden, A. Marson and P. W. N. M. van Leeuwen, Adv. Synth. Catal., 2002, 344, 293 CrossRef CAS
- R. P. J. Bronger, S. M. Silva, P. C. J. Kamer and P. W. N. M. van Leeuwen, Dalton Trans., 2004, 1590 RSC
- S. M. Silva, R. P. J. Bronger, Z. Freixa, J. Dupont and P. W. N. M. van Leeuwen, New J. Chem., 2003, 27, 1294 RSC
- T. Hamerla, A. Rost, Y. Kasaka and R. Schomaecker, ChemCatChem, 2013, 5, 1854 CrossRef CAS
- T. Pogrzeba, D. Müller, T. Hamerla, E. Esche, N. Paul, G. Wozny and R. Schomäcker, Ind. Eng. Chem. Res., 2015, 54, 11953 CrossRef CAS
- A. Riisager, P. Wasserscheid, R. van Hal and R. Fehrmann, J. Catal., 2003, 219, 452 CrossRef CAS
- T. Gaide, J. M. Dreimann, A. Behr and A. J. Vorholt, Angew. Chem., Int. Ed., 2016, 55, 2924 CrossRef CAS PubMed
- M. Schreuder Goedheijt, B. E. Hanson, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2000, 122, 1650 CrossRef
- A. Buhling, P. C. J. Kamer, P. W. N. M. van Leeuwen, J. W. Elgersma, K. Goubitz and J. Fraanje, Organometallics, 1997, 16, 3027 CrossRef CAS
- M. Mokhadinyana, S. L. Desset, D. B. G. Williams and D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2012, 51, 1648 CrossRef CAS PubMed
- D. J. Adams, D. J. Cole-Hamilton, D. A. J. Harding, E. G. Hope, P. Pogorzelec and A. M. Stuart, Tetrahedron, 2004, 60, 4079 CrossRef CAS
- A. J. Sandee, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 8468 CrossRef CAS PubMed
- F. Marras, J. Wang, M.-A. Coppens and J. N. H. Reek, Chem. Commun., 2010, 46, 6587 RSC
- N. J. Meehan, A. J. Sandee, J. N. H. Reek, P. C. J. Kamer, P. W. N. M. van Leeuwen and M. Poliakoff, Chem. Commun., 2000, 1497 RSC
- T. E. Kunene, P. B. Webb and D. J. Cole-Hamilton, Green Chem., 2011, 13, 1476 RSC
- A. M. Kluwer, C. Simons, Q. Knijnenburg, J. I. van der Vlugt, B. de Bruin and J. N. H. Reek, Dalton Trans., 2013, 42, 3609 RSC
- D. de Groot, B. F. M. de Waal, J. N. H. Reek, A. P. H. J. Schenning, P. C. J. Kamer, E. W. Meijer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 8453 CrossRef CAS PubMed
- R. P. J. Bronger, J. P. Bermon, J. N. H. Reek, P. C. J. Kamer, P. W. N. M. van Leeuwen, D. N. Carter, P. Licence and M. Poliakoff, J. Mol. Catal. A: Chem., 2004, 224, 145 CrossRef CAS
- R. Chen, R. P. J. Bronger, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, J. Am. Chem. Soc., 2004, 126, 14557 CrossRef CAS PubMed
- J. N. H. Reek, S. Arévalo, R. van Heerbeek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Adv. Catal., 2006, 49, 71 CAS
- S. Ricken, P. W. Osinski, P. Eilbracht and R. Haag, J. Mol. Catal. A: Chem., 2006, 257, 78 CrossRef CAS
- G. E. Oosterom, S. Steffens, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Top. Catal., 2002, 19, 61 CrossRef CAS
- M. Dieguez, O. Pamies, Y. Mata, E. Teuma, M. Gomez, F. Ribaudo and P. W. N. M. van Leeuwen, Adv. Synth. Catal., 2008, 350, 2583 CrossRef CAS
- S. Shylesh, D. Hanna, S. Werner and A. T. Bell, ACS Catal., 2012, 2, 487 CrossRef CAS
- R. P. J. Bronger, S. M. Silva, P. C. J. Kamer and P. W. N. M. van Leeuwen, Chem. Commun., 2002, 3044 RSC
- J. J. Carbó, F. Maseras, C. Bo and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 7630 CrossRef
- W. Goertz, P. C. J. Kamer, P. W. N. M. van Leeuwen and D. Vogt, Chem. – Eur. J., 2001, 7, 1614 CrossRef CAS
- R. van Duren, L. L. J. M. Cornelissen, J. I. van der Vlugt, J. P. J. Huijbers, A. M. Mills, A. L. Spek, C. Müller and D. Vogt, Helv. Chim. Acta, 2006, 89, 1547 CrossRef CAS
- M. T. Reetz and X. Li, J. Am. Chem. Soc., 2006, 128, 1044 CrossRef CAS PubMed
- C. B. Dieleman, P. C. J. Kamer, J. N. H. Reek and P. W. N. M. van Leeuwen, Helv. Chim. Acta, 2001, 84, 3269 CrossRef CAS
- Y. Hamada, F. Matsuura, M. Oku, K. Hatano and T. Shioiri, Tetrahedron Lett., 1997, 38, 8961 CrossRef CAS
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed
- J. Holz, K. Rumpel, A. Spannenberg, R. Paciello, H. Jiao and A. Börner, ACS Catal., 2017, 7, DOI:10.1021/acscatal7b02434
- P. Dierkes, S. Ramdeehul, L. Barloy, A. De Cian, J. Fischer, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. A. Osborn, Angew. Chem., Int. Ed., 1998, 37, 3116 CrossRef CAS
- M. J. Burk, J. Am. Chem. Soc., 1991, 113, 8518 CrossRef CAS
- S. Ramdeehul, P. Dierkes, R. Aguado, P. C. J. Kamer, P. W. N.
M. van Leeuwen and J. A. Osborn, Angew. Chem., Int. Ed., 1998, 37, 3118 CrossRef CAS
- M. J. Burk and J. E. Feaster, J. Am. Chem. Soc., 1992, 114, 6266 CrossRef CAS
- C. F. Czauderna, A. G. Jarvis, F. J. L. Heutz, D. B. Cordes, A. M. Z. Slawin, J. I. van der Vlugt and P. C. J. Kamer, Organometallics, 2015, 34, 1608 CrossRef CAS
- H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119 CrossRef PubMed
- S. H. Chikkali, R. Bellini, B. de Bruin, J. I. van der Vlugt and J. N. H. Reek, J. Am. Chem. Soc., 2012, 134, 6607 CrossRef CAS PubMed
- L. A. van der Veen, P. K. Keeven, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 2000, 2105 RSC
- A. Gual, C. Godard, S. Castillon and C. Claver, Tetrahedron: Asymmetry, 2010, 21, 1135 CrossRef CAS
- D. A. C. Molina, C. P. Casey, I. Müller, K. Nozaki and C. Jäkel, Organometallics, 2010, 29, 3362 CrossRef
- S. H. Chikkali, J. I. van der Vlugt and J. N. H. Reek, Coord. Chem. Rev., 2014, 262, 1 CrossRef CAS
-
Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer Academic Publishers, Dordrecht, Netherlands, 2000 Search PubMed
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS PubMed
- S. K. Sharma and R. V. Jasra, Catal. Today, 2015, 247, 70 CrossRef CAS
-
L. H. Slaugh and R. D. Mullineaux, US Pat., 3239569 and 3239570, 1966 (to Shell Devco); Chem. Abstr., 1964, 64, 15745 and 19420 Search PubMed
-
J. L. Van Winkle, S. Lorenzo, R. C. Morris and R. F. Mason, US Pat. (to Shell Devco), 3420898, 1969; Chem. Abstr., 1967, 66, 65101 Search PubMed
- J. F. Young, J. A. Osborn, F. A. Jardine and G. Wilkinson, Chem. Commun., 1965, 131 RSC
- J. M. Brown and A. G. Kent, J. Chem. Soc., Perkin Trans. 2, 1987, 1597 RSC
- J. D. Unruh and J. R. Christenson, J. Mol. Catal., 1982, 14, 19 CrossRef CAS
- G. Consiglio, C. Botteghi, C. Salomon and P. Pino, Angew. Chem., Int. Ed. Engl., 1973, 85, 665 Search PubMed
- C. Crocker, R. J. Errington, R. Markham, C. J. Moulton, K. J. Odell and B. L. Shaw, J. Am. Chem. Soc., 1980, 102, 4373 CrossRef CAS
- T. A. Puckette, Top. Catal., 2012, 55, 421 CrossRef CAS
- C. P. Casey, G. T. Whiteker, M. G. Melville, L. M. Petrovich, J. A. Gavney Jr. and D. R. Powell, J. Am. Chem. Soc., 1992, 114, 5535 CrossRef CAS
-
E. Billig, A. G. Abatjoglou and D. R. Bryant, US Pat. 469498, U.S. Pat. 4668651, U.S. Pat. 4748261, 1987 (to Union Carbide Corp.); Chem. Abstr., 1987, 107, 7392r
- A. van Rooy, P. C. J. Kamer, P. W. N. M. van Leeuwen, K. Goubitz, J. Fraanje, N. Veldman and A. L. Spek, Organometallics, 1996, 15, 835 CrossRef CAS
-
P. W. N. M. van Leeuwen and Z. Freixa, in Modern Carbonylation Methods, ed. L. Kollar, Wiley VCH, Weinheim, 2008, p. 1 Search PubMed
- Z. Freixa and P. W. N. M. van Leeuwen, Dalton Trans., 2003, 1890 RSC
- E. Zuidema, L. Escorihuela, T. Eichelsheim, J. J. Carbó, C. Bo, P. C. J. Kamer and P. W. N. M. van Leeuwen, Chem. – Eur. J., 2008, 14, 1843 CrossRef CAS PubMed
- L. A. van der Veen, P. H. Keeven, G. C. Schoemaker, J. N. H. Reek, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, Organometallics, 2000, 19, 872 CrossRef CAS
-
(a) P. C. J. Kamer, A. van Rooy, G. C. Schoemaker and P. W. N. M. van Leeuwen, Coord. Chem. Rev., 2004, 248, 2409 CrossRef CAS
- L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1999, 18, 4765 CrossRef CAS
- M. Miyazawa, S. Momose and K. Yamamoto, Synlett, 1990, 711 CrossRef CAS
- S. C. van der Slot, P. C. J. Kamer, P. W. N. M. van Leeuwen, J. Fraanje, K. Goubitz, M. Lutz and A. L. Spek, Organometallics, 2000, 19, 2504 CrossRef CAS
- J. Mormul, M. Mulzer, T. Rosendahl, F. Rominger, M. Limbach and P. Hofmann, Organometallics, 2015, 34, 4102 CrossRef CAS
- B. Hamers, E. Kosciusko-Morizet, C. Mueller and D. Vogt, ChemCatChem, 2009, 1, 103 CrossRef CAS
- O. Diebolt, H. Tricas, Z. Freixa and P. W. N. M. van Leeuwen, ACS Catal., 2013, 3, 128 CrossRef CAS
- S. C. van der Slot, J. Duran, J. Luten, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 2002, 21, 3873 CrossRef CAS
- Y. Yan, X. Zhang and X. Zhang, J. Am. Chem. Soc., 2006, 128, 16058 CrossRef CAS PubMed
- E. Zuidema, P. E. Goudriaan, B. H. G. Swennenhuis, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Lutz and A. L. Spek, Organometallics, 2010, 29, 1210 CrossRef CAS
- Y. Ohtsuka, O. Kobayashi and T. Yamakawa, J. Fluorine Chem., 2014, 161, 34 CrossRef CAS
- S. Simaan and I. Marek, J. Am. Chem. Soc., 2010, 132, 4066 CrossRef CAS PubMed
-
J. J. Carbó, F. Maseras and C. Bo, Rhodium diphosphine hydroformylation in Catalysis by Metal Complexes, ed. B. R. James and P. W. N. M. van Leeuwen, 2002, (Computational Modeling of Homogeneous Catalysis), vol. 25, p. 161 Search PubMed
- E. Zuidema, E. Daura-Oller, J. J. Carbo, C. Bo and P. W. N. M. van Leeuwen, Organometallics, 2007, 26, 2234 CrossRef CAS
- C. R. Landis and J. Uddin, J. Chem. Soc., Dalton Trans., 2002, 729 RSC
- M. Kumar, R. V. Chaudhari, B. Subramaniam and T. A. Jackson, Organometallics, 2015, 34, 1062 CrossRef CAS
- O. Saidi, S. Liu and J. Xiao, J. Mol. Catal. A: Chem., 2009, 305, 130 CrossRef CAS
- T. Kégl, RSC Adv., 2015, 5, 4304 RSC
- P. D. Achord, P. Kiprof and B. Barker, J. Mol. Struct.: THEOCHEM, 2008, 849, 103 CrossRef CAS
- M. A. Carvajal, S. Kozuch and S. Shaik, Organometallics, 2009, 28, 3656 CrossRef CAS
- M. Vilches-Herrera, L. Domke and A. Boerner, ACS Catal., 2014, 4, 1706 CrossRef CAS
- V. Goldbach, P. Roesle and S. Mecking, ACS Catal., 2015, 5, 5951 CrossRef CAS
- R. P. J. Bronger, J. P. Bermon, J. Herwig, P. C. J. Kamer and P. W. N. M. van Leeuwen, Adv. Synth. Catal., 2004, 346, 789 CrossRef CAS
-
H. Bohnen and J. Herwig, World Patent WO02/068369 (to Celanese Chemicals Europe GmbH) 2002; Chem. Abstr., 2002, 137, 187386 Search PubMed
- L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 336 CrossRef CAS
- B. P. Bondzic, J. Mol. Catal. A: Chem., 2015, 408, 310 CrossRef CAS
- D. Konya, K. Q. Almeida Lenero and E. Drent, Organometallics, 2006, 25, 3166 CrossRef CAS
- O. Diebolt, C. Mueller and D. Vogt, Catal. Sci. Technol., 2012, 2, 773 CAS
- O. Diebolt, C. Cruzeuil, C. Müller and D. Vogt, Adv. Synth. Catal., 2012, 354, 670 CrossRef CAS
- K. Takahashi, M. Yamashita, T. Ichihara, K. Nakano and K. Nozaki, Angew. Chem., Int. Ed., 2010, 49, 4488 CrossRef CAS PubMed
- K. Takahashi, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 18746 CrossRef CAS PubMed
- Y. Yuki, K. Takahashi, Y. Tanaka and K. Nozaki, J. Am. Chem. Soc., 2013, 135, 17393 CrossRef CAS PubMed
- A. Moballigh, A. Seayad, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2003, 125, 10311–10318 CrossRef PubMed
- M. Ahmed, R. P. J. Bronger, R. Jackstell, P. C. J. Kamer, P. W. N. M. van Leeuwen and M. Beller, Chem. – Eur. J., 2006, 12, 8979 CrossRef CAS PubMed
- K. C. B. Oliveira, A. G. Santos and E. N. Dos Santos, Appl. Catal., A, 2012, 445–446, 204 CrossRef CAS
- B. Hamers, P. S. Bäuerlein, C. Müller and D. Vogt, Adv. Synth. Catal., 2008, 350, 332 CrossRef CAS
- S. Raoufmoghaddam, E. Drent and E. Bouwman, ChemSusChem, 2013, 6, 1759 CrossRef CAS PubMed
- P. Köhling, A. M. Schmidt and P. Eilbracht, Org. Lett., 2003, 5, 3213 CrossRef PubMed
- A. M. Schmidt and P. Eilbracht, J. Org. Chem., 2005, 70, 5528 CrossRef CAS PubMed
- G. Makado, T. Morimoto, Y. Sugimoto, K. Tsutsumi, N. Kagawa and K. Kakiuchi, Adv. Synth. Catal., 2010, 352, 299 CrossRef CAS
- M. Uhlemann, S. Doerfelt and A. Boerner, Tetrahedron Lett., 2013, 54, 2209 CrossRef CAS
- J. H. Kim, T. Song and Y. K. Chung, Org. Lett., 2017, 19, 1248 CrossRef CAS PubMed
- S. K. Murphy, J.-W. Park, F. A. Cruz and V. M. Dong, Science, 2015, 347(6217), 56 CrossRef CAS PubMed
- P. Meessen, D. Vogt and W. Keim, J. Organomet. Chem., 1998, 551, 165 CrossRef CAS
- L. A. van der Veen, P. K. Keeven, P. C. J. Kamer and P. W. N. M. van Leeuwen, Chem. Commun., 2000, 333 RSC
- S. C. Tang and L. Kim, J. Mol. Catal., 1982, 14, 231 CrossRef CAS
- P. W. N. M. van Leeuwen, C. F. Roobeek, R. L. Wife and J. H. G. Frijns, J. Chem. Soc., Chem. Commun., 1986, 31 RSC
- C. Botteghi, S. Paganelli, U. Matteoli, A. Scrivanti, R. Ciorciaro and L. M. Venanzi, Helv. Chim. Acta, 1990, 73, 284 CrossRef CAS
- Y. Kawabata, T. Hayashi and I. Ogata, J. Chem. Soc., Chem. Commun., 1979, 462 RSC
- T. Hayashi, Y. Kawabata, T. Isoyama and I. Ogata, Bull. Chem. Soc. Jpn., 1981, 54, 3438 CrossRef CAS
- A. Bedekovits, L. Kollár and T. Kégl, Inorg. Chim. Acta, 2010, 363, 2029 CrossRef CAS
- R. P. Dias and W. R. Rocha, Organometallics, 2011, 30, 4257 CrossRef CAS
- T. Papp, L. Kollár and T. Kégl, Organometallics, 2013, 32, 3640 CrossRef CAS
- L. A. van der Veen, P. K. Keeven, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 2000, 2105 RSC
- R. van Duren, J. I. van der Vlugt, H. Kooijman, A. L. Spek and D. Vogt, Dalton Trans., 2007, 1053 RSC
- C. M. Crudden and H. Alper, J. Org. Chem., 1994, 59, 3091 CrossRef CAS
- P. P. Deutsch and R. Eisenberg, Organometallics, 1990, 9, 709 CrossRef CAS
- D. J. Fox, S. B. Duckett, C. Flaschenriem, W. W. Brennessel, J. Schneider, A. Gunay and R. Eisenberg, Inorg. Chem., 2006, 45, 7197 CrossRef CAS PubMed
- I. Piras, R. Jennerjahn, R. Jackstell, A. Spannenberg, R. Franke and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 280 CrossRef CAS PubMed
- J. Ternel, J.-L. Couturier, J.-L. Dubois and J.-F. Carpentier, ChemCatChem, 2015, 7, 513 CrossRef CAS
- A. Behr, A. Kämper, R. Kuhlmann, A. J. Vorholt and R. Franke, Catal. Sci. Technol., 2016, 6, 208 Search PubMed
- S. Kusumoto, T. Tatsuki and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 8458 CrossRef CAS PubMed
- K. Takahashi, M. Yamashita, Y. Tanaka and K. Nozaki, Angew. Chem., Int. Ed., 2012, 51, 4383 CrossRef CAS PubMed
-
E. Drent, Eur. Pat. Appl., EP220767, 1987 (to Shell); Chem. Abstr., 1987, 107, 439199 Search PubMed
- E. Drent and P. H. M. Budzelaar, J. Organomet. Chem., 2000, 593, 211 CrossRef
- R. Jennerjahn, I. Piras, R. Jackstell, R. Franke, K.-D. Wiese and M. Beller, Chem. – Eur. J., 2009, 15, 6383 CrossRef CAS PubMed
- Y. Katafuchi, T. Fujihara, T. Iwai, J. Terao and Y. Tsuji, Adv. Synth. Catal., 2011, 353, 475 CrossRef CAS
- Z. Freixa, P. C. J. Kamer, M. Lutz, A. L. Spek and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2005, 44, 4385 CrossRef CAS PubMed
-
S. Gaemers and J. G. Sunley, PCT Int. Appl., WO2004101488, 2004 (to BP Chemicals Limited, UK); Chem. Abstr., 2004, 142, 1015990
- G. L. Williams, C. M. Parks, C. R. Smith, H. Adams, A. Haynes, A. J. H. M. Meijer, G. J. Sunley and S. Gaemers, Organometallics, 2011, 30, 6166 CrossRef CAS
- L. D. Dingwall, A. F. Lee, J. M. Lynam, K. Wilson, L. Olivi, J. M. S. Deeley, S. Gaemers and G. J. Sunley, ACS Catal., 2012, 2, 1368 CrossRef CAS
- E. Amadio, Z. Freixa, P. W. N. M. van Leeuwen and L. Toniolo, Catal. Sci. Technol., 2015, 5, 2856 CAS
- T. Gaide, A. Behr, M. Terhorst, A. Arns, F. Benski and A. J. Vorholt, Chem. Ing. Tech., 2016, 88, 158 CrossRef CAS
- K. A. Ostrowski, D. Vogelsang, T. Seidensticker and A. J. Vorholt, Chem. – Eur. J., 2016, 22, 1840 CrossRef CAS PubMed
- C. A. Tolman, R. J. McKinney, W. C. Seidel, J. D. Druliner and W. R. Stevens, Adv. Catal., 1985, 33, 1 CAS
-
P. W. N. M. van Leeuwen in Homogeneous Catalysis: Understanding of the Art, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2004, p. 229 Search PubMed
- M. Kranenburg, P. C. J. Kamer, P. W. N. M. van Leeuwen, D. Vogt and W. Keim, J. Chem. Soc., Chem. Commun., 1995, 2177 RSC
- W. Goertz, W. Keim, D. Vogt, U. Englert, M. D. K. Boele, L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1998, 2981 RSC
- W. Goertz, P. C. J. Kamer, P. W. N. M. van Leeuwen and D. Vogt, Chem. Commun., 1997, 1521 RSC
- W. Goertz, P. C. J. Kamer, P. W. N. M. van Leeuwen and D. Vogt, Chem. – Eur. J., 2001, 7, 1614 CrossRef CAS
- J. I. van der Vlugt, A. C. Hewat, S. Neto, R. Sablong, A. M. Mills, M. Lutz, A. L. Spek, C. Mueller and D. Vogt, Adv. Synth. Catal., 2004, 346, 993 CrossRef CAS
- L. Bini, C. Mueller, J. Wilting, L. Von Chrzanowski, A. L. Spek and D. Vogt, J. Am. Chem. Soc., 2007, 129, 12622 CrossRef CAS PubMed
- M. E. Tauchert, D. C. M. Warth, S. M. Braun, I. Gruber, A. Ziesak, F. Rominger and P. Hofmann, Organometallics, 2011, 30, 2790 CrossRef CAS
- P. Elmes and W. R. Jackson, J. Am. Chem. Soc., 1979, 101, 6128 CrossRef CAS
- M. Hodgson, D. Parker, R. J. Taylor and G. Ferguson, Organometallics, 1988, 7, 1761 CrossRef CAS
- J. E. Marcone and K. G. Moloy, J. Am. Chem. Soc., 1998, 120, 8527 CrossRef CAS
- S. Otsuka, J. Organomet. Chem., 1980, 200, 191 CrossRef CAS
- A. C. Balazs, K. H. Johnson and G. M. Whitesides, Inorg. Chem., 1982, 21, 2162 CrossRef CAS
- T. Gathy, O. Riant, D. Peeters and T. Leyssens, J. Organomet. Chem., 2011, 696, 3425 CrossRef CAS
- J. W. Raebiger, A. Miedaner, C. J. Curtis, S. M. Miller, O. P. Anderson and D. L. DuBois, J. Am. Chem. Soc., 2004, 126, 5502 CrossRef CAS PubMed
- A. D. Wilson, A. J. M. Miller, D. L. DuBois, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 2010, 49, 3918 CrossRef CAS PubMed
- K. Almeida Lenero, M. Kranenburg, Y. Guari, P. C. J. Kamer, P. W. N. M. van Leeuwen, S. Sabo-Etienne and B. Chaudret, Inorg. Chem., 2003, 42, 2859 CrossRef PubMed
- T. Papp, L. Kollár and T. Kégl, J. Quantum Chem., 2014, 528072 Search PubMed
- E. Zuidema, P. W. N. M. van Leeuwen and C. Bo, Organometallics, 2005, 24, 3703 CrossRef CAS
- P. Anstaett and F. Schoenebeck, Chem. – Eur. J., 2011, 17, 12340 CrossRef CAS PubMed
- W.-J. van Zeist and F. M. Bickelhaupt, Dalton Trans., 2011, 40, 3028 RSC
- W. J. van Zeist, R. Visser and F. M. Bickelhaupt, Chem. - Eur. J., 2009, 15, 6112 CrossRef CAS PubMed
- I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953 RSC
- J. A. Gillespie, D. L. Dodds and P. C. J. Kamer, Dalton Trans., 2010, 39, 2751 RSC
- C. M. Donahue, S. P. McCollom, C. M. Forrest, A. V. Blake, B. J. Bellott, J. M. Keith and S. R. Daly, Inorg. Chem., 2015, 54, 5646 CrossRef CAS PubMed
- D. L. Lichtenberger and M. E. Jatcko, J. Coord. Chem., 1994, 32, 79 CrossRef CAS
-
E. E. Brunel and C. Kenneth, US Pat., 5847191A (to DuPont), 1998; Chem. Abstr., 1998, 130, 26464 Search PubMed
- M. Kranenburg, P. C. J. Kamer and P. W. N. M. van Leeuwen, Eur. J. Inorg. Chem., 1998, 155 CrossRef CAS
- J. P. Sadighi, M. C. Harris and S. L. Buchwald, Tetrahedron Lett., 1998, 39, 5327 CrossRef CAS
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1998, 120, 3694 CrossRef CAS
-
(a) Y. Guari, D. S. van Es, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Tetrahedron Lett., 1999, 40, 3789 CrossRef CAS
-
(a) J. F. Hartwig, M. Kawatsura, S. I. Hauck, K. H. Shaughnessy and L. M. Alcazar-Roman, J. Org. Chem., 1999, 64, 5575 CrossRef CAS PubMed
- Y. Ji, R. E. Plata, C. S. Regens, M. Hay, M. Schmidt, T. Razler, Y. Qiu, P. Geng, Y. Hsiao, T. Rosner, M. D. Eastgate and D. G. Blackmond, J. Am. Chem. Soc., 2015, 137, 13272 CrossRef CAS PubMed
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314 CrossRef CAS PubMed
- M. S. Stephan, A. J. J. M. Teunissen, G. K. M. Verzijl and J. G. de Vries, Angew. Chem., Int. Ed., 1998, 37, 662 CrossRef CAS
- K. H. Vardhan Reddy, J.-D. Brion, S. Messaoudi and M. Alami, J. Org. Chem., 2016, 81, 424 CrossRef CAS PubMed
- D. Mitchell, D. M. Coppert, H. A. Moynihan, K. T. Lorenz, M. Kissane, O. A. McNamara and A. R. Maguire, Org. Process Res. Dev., 2011, 15, 981 CrossRef CAS
- M. S. Joshi and F. C. Pigge, ACS Catal., 2016, 6, 4465 CrossRef CAS
- M. Liu, M. Ye, Y. Xue, G. Yin, D. Wang and J. Huang, Tetrahedron Lett., 2016, 57, 3137 CrossRef CAS
- M. Beaupérin, E. Fayad, R. Amardeil, H. Cattey, P. Richard, S. Brandès, P. Meunier and J.-C. Hierso, Organometallics, 2008, 27, 1506 CrossRef
- J. Huang, J. Chan, Y. Chen, C. J. Borths, K. D. Baucom, R. D. Larsen and M. M. Faul, J. Am. Chem. Soc., 2010, 132, 3674 CrossRef CAS PubMed
- A. López-Pérez, J. Adrio and J. C. Carretero, Org. Lett., 2009, 11, 5514 CrossRef PubMed
- J.-C. Shi and E.-I. Negishi, J. Organomet. Chem., 2003, 687, 518 CrossRef CAS
- E.-I. Negishi, J.-C. Shi and X. Zeng, Tetrahedron, 2005, 61, 9886 CrossRef CAS
- F. Liron, C. Fosse, A. Pernolet and E. Roulland, J. Org. Chem., 2007, 72, 2220 CrossRef CAS PubMed
- Z. Huang, M. Qian, D. J. Babinski and E.-I. Negishi, Organometallics, 2005, 24, 475 CrossRef CAS
- R. Martin and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3844 CrossRef CAS PubMed
- L. Wu and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 15824 CrossRef CAS PubMed
- M. Fukushima, D. Takushima and M. Kimura, J. Am. Chem. Soc., 2010, 132, 16346 CrossRef CAS PubMed
- T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed. Engl., 1997, 36, 1740 CrossRef CAS
- R. Martín and S. L. Buchwald, Org. Lett., 2008, 10, 4561 CrossRef PubMed
- R. Martín and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 7236 CrossRef PubMed
- M. Carril, R. SanMartin, F. Churruca, I. Tellitu and E. Domínguez, Org. Lett., 2005, 7, 4787 CrossRef CAS PubMed
- L. Ackermann and V. P. Mehta, Chem. – Eur. J., 2012, 18, 10230 CrossRef CAS PubMed
- L. Wu and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 15824 CrossRef CAS PubMed
- Á. Gutiérrez-Bonet, F. Juliá-Hernández, B. de Luis and R. Martin, J. Am. Chem. Soc., 2016, 138, 6384 CrossRef PubMed
- M. Beaudoin Bertrand and J. P. Wolfe, Org. Lett., 2007, 9, 3073 CrossRef PubMed
- A. A. Ruch, S. Handa, F. Kong, V. N. Nesterov, D. R. Pahls, T. R. Cundari and L. M. Slaughter, Org. Biomol. Chem., 2016, 14, 8123 CAS
- G. Yang, X. Jiang, Y. Liu, N. Li, G. Yin and C. Yu, Asian J. Org. Chem., 2016, 5, 882 CrossRef CAS
- C. Alonso, E. Martinez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed
- V. V. Grushin and W. J. Marshall, J. Am. Chem. Soc., 2006, 128, 12644 CrossRef CAS PubMed
- E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679 CrossRef CAS PubMed
- P. Anstaett and F. Schoenebeck, Chem. – Eur. J., 2011, 17, 12340 CrossRef CAS PubMed
- V. I. Bakhmutov, F. Bozoglian, K. Gómez, G. González, V. V. Grushin, S. A. Macgregor, E. Martin, F. M. Miloserdov, M. A. Novikov, J. A. Panetier and L. V. Romashov, Organometallics, 2012, 31, 1315 CrossRef CAS
- J. Jover, F. M. Miloserdov, J. Benet-Buchholz, V. V. Grushin and F. Maseras, Organometallics, 2014, 33, 6531 CrossRef CAS
- M. C. Nielsen, K. J. Bonney and F. Schoenebeck, Angew. Chem., Int. Ed., 2014, 53, 5903 CrossRef CAS PubMed
-
P. W. N. M. van Leeuwen, Homogeneous catalysis; Understanding the Art. Dordrecht, Springer, 2004 Search PubMed
- Z. Feng, Q.-Q. Min and X. Zhang, Org. Lett., 2016, 18, 44 CrossRef CAS PubMed
- Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270 CrossRef CAS PubMed
- X.-F. Xia, S.-L. Zhu, Y. Li and H. Wang, RSC Adv., 2016, 6, 51703 RSC
- S. Yang, Q. Xu and M. Shi, Chem. – Eur. J., 2016, 22, 10387 CrossRef CAS PubMed
- M. Sundermeier, A. Zapf, S. Mutyala, W. Baumann, J. Sans, S. Weiss and M. Beller, Chem. - Eur. J., 2003, 9, 1828 CrossRef CAS PubMed
- M. R. Pitts, P. McCormack and J. Whittall, Tetrahedron, 2006, 62, 4705 CrossRef CAS
- D. C. Koester, M. Kobayashi, D. B. Werz and Y. Nakao, J. Am. Chem. Soc., 2012, 134, 6544 CrossRef CAS PubMed
- K. Fukumoto, A. A. Dahy, T. Oya, K. Hayasaka, M. Itazaki, N. Koga and H. Nakazawa, Organometallics, 2012, 31, 787 CrossRef CAS
- Y. Miyazaki, N. Ohta, K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 3732 CrossRef CAS PubMed
- Y. Xie, J. Hu, Y. Wang, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 20613 CrossRef CAS PubMed
- Y. Xie, J. Hu, P. Xie, B. Qian and H. Huang, J. Am. Chem. Soc., 2013, 135, 18327 CrossRef CAS PubMed
- X. Qi, S. Liu and Y. Lan, Organometallics, 2016, 35, 1582 CrossRef CAS
- J. Hu, Y. Xie and H. Huang, Angew. Chem., Int. Ed., 2014, 53, 7272 CrossRef CAS PubMed
- M. B. Hay and J. P. Wolfe, Angew. Chem., Int. Ed., 2007, 46, 6492 CrossRef CAS PubMed
- D. Jiang, J. Peng and Y. Chen, Tetrahedron, 2008, 64, 1641 CrossRef CAS
- D. Jiang, J. Peng and Y. Chen, Org. Lett., 2008, 10, 1695 CrossRef CAS PubMed
- N. Sharma, Z. Li, U. K. Sharma and E. V. Van der Eycken, Org. Lett., 2014, 16, 3884 CrossRef CAS PubMed
- T. Fujita, K. Sugiyama, S. Sanada, T. Ichitsuka and J. Ichikawa, Org. Lett., 2016, 18, 248 CrossRef CAS PubMed
- L. Zhang, J. Panteleev and M. Lautens, J. Org. Chem., 2014, 79, 12159 CrossRef CAS PubMed
- D. R. White and J. P. Wolfe, Org. Lett., 2015, 17, 2378 CrossRef CAS PubMed
- J. Liu, X. Xu, J. Li, B. Liu, H. Jianga and B. Yin, Chem. Commun., 2016, 52, 9550 RSC
- F. Gao, B. S. Kim and P. J. Walsh, Chem. Commun., 2014, 50, 10661 RSC
- J. Zhang, A. Bellomo, N. Trongsiriwat, T. Jia, P. J. Carroll, S. D. Dreher, M. T. Tudge, H. Yin, J. R. Robinson, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 6276 CrossRef CAS PubMed
- S. A. Baker Dockrey, A. K. Makepeace and J. R. Schmink, Org. Lett., 2014, 16, 4730 CrossRef CAS PubMed
- D. Wang, H.-G. Chen, X.-C. Tian, X.-X. Liang, F.-Z. Chen and F. Gao, RSC Adv., 2015, 5, 107119 RSC
- B.-S. Kim, J. Jiménez, F. Gao and P. J. Walsh, Org. Lett., 2015, 17, 5788 CrossRef CAS PubMed
- A. R. Rivero, B.-S. Kim and P. J. Walsh, Org. Lett., 2016, 18, 1590 CrossRef CAS PubMed
- X. Yang, B.-S. Kim, M. Li and P. J. Walsh, Org. Lett., 2016, 18, 2371 CrossRef CAS PubMed
- M. Li, B. Yucel, J. Adrio, A. Bellomo and P. J. Walsh, Chem. Sci., 2014, 5, 2383 RSC
- M. Li, M. Gonzalez-Esguevillas, S. Berritt, X. Yang, A. Bellomo and P. J. Walsh, Angew. Chem., Int. Ed., 2016, 55, 2825 CrossRef CAS PubMed
- C. Mollar, M. Besora, F. Maseras, G. Asensio and M. Medio-Simon, Chem. – Eur. J., 2010, 16, 13390 CrossRef CAS PubMed
- A. A. C. Braga, G. Ujaque and F. Maseras, Organometallics, 2006, 25, 3647 CrossRef CAS
- M. García-Melchor, A. A. C. Braga, A. Lledos, G. Ujaque and F. Maseras, Acc. Chem. Res., 2013, 46, 2626 CrossRef PubMed
- X. Qi, S. Liu and Y. Lan, Organometallics, 2016, 35, 1582 CrossRef CAS
- L.-Z. Yu, Z.-Z. Zhu, X.-B. Hu, X.-Y. Tang and M. Shi, Chem. Commun., 2016, 52, 6581 RSC
- J. Albaneze-Walker, C. Bazaral, T. Leavey, P. G. Dormer and J. A. Murry, Org. Lett., 2004, 6, 2097 CrossRef CAS PubMed
- X. Qi, C.-L. Li, L.-B. Jiang, W.-Q. Zhang and X.-F. Wu, Catal. Sci. Technol., 2016, 6, 3099 CAS
- J. T. Joseph, A. M. Sajith, R. C. Ningegowda and S. Shashikanth, Adv. Synth. Catal., 2017, 359, 419 CrossRef CAS
- H. Wang, J. Cai, H. Huang and G.-J. Deng, Org. Lett., 2014, 16, 5324 CrossRef CAS PubMed
- Y. Huang and H. Alper, J. Org. Chem., 1991, 56, 4534 CrossRef CAS
- X.-F. Wu, B. Sundararaju, P. Anbarasan, H. Neumann, P. H. Dixneuf and M. Beller, Chem. – Eur. J., 2011, 17, 8014 CrossRef CAS PubMed
- T. L. Andersen, M. W. Frederiksen, K. Domino and T. Skrydstrup, Angew. Chem., Int. Ed., 2016, 55, 10396 CrossRef CAS PubMed
- H.-Y. Zhao, Z. Feng, Z. Luo and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 10401 CrossRef CAS PubMed
- Q. Wang, Y.-T. He, J.-H. Zhao, Y.-F. Qiu, L. Zheng, J.-Y. Hu, Y.-C. Yang, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2016, 18, 2664 CrossRef CAS PubMed
- A. Takemiya and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 14800 CrossRef CAS PubMed
- Y. Hirata, M. Tanaka, A. Yada, Y. Nakao and T. Hiyama, Tetrahedron, 2009, 65, 5037 CrossRef CAS
- D. D. Dawson and E. R. Jarvo, Org. Process Res. Dev., 2015, 19, 1356 CrossRef CAS PubMed
- P. Kumar, S. Prescher and J. Louie, Angew. Chem., Int. Ed., 2011, 50, 10694 CrossRef CAS PubMed
- N. D. Staudaher, R. M. Stolley and J. Louie, Chem. Commun., 2014, 50, 15577 RSC
- J. Jover, F. M. Miloserdov, J. Benet-Buchholz, V. V. Grushin and F. Maseras, Organometallics, 2014, 33, 6531 CrossRef CAS
- X. Cao, S.-C. Sha, M. Li, B.-S. Kim, C. Morgan, R. Huang, X. Yang and P. J. Walsh, Chem. Sci., 2016, 7, 611 RSC
- R. Motoki, M. Kanai and M. Shibasaki, Org. Lett., 2007, 9, 2997 CrossRef CAS PubMed
- M. Sawamura, H. Hamashima and Y. Ito, J. Am. Chem. Soc., 1992, 114, 8295 CrossRef CAS
- Y. Asano, H. Ito, K. Hara and M. Sawamura, Organometallics, 2008, 27, 5984 CrossRef CAS
- R. D. Stephens and C. E. Castro, J. Org. Chem., 1963, 28, 3313 CrossRef CAS
- Z. Li, R. Li, L. Jiang and Z. Li, Molecules, 2015, 20, 15023 CrossRef CAS PubMed
- J. Aziz, T. Baladi and S. Piguel, J. Org. Chem., 2016, 81, 4122 CrossRef CAS PubMed
- G. L. Moxham, H. Randell-Sly, S. K. Brayshaw, A. S. Weller and M. C. Willis, Chem. – Eur. J., 2008, 14, 8383 CrossRef CAS PubMed
- R. J. Pawley, G. L. Moxham, R. Dallanegra, A. B. Chaplin, S. K. Brayshaw, A. S. Weller and M. C. Willis, Organometallics, 2010, 29, 1717 CrossRef CAS
- A. B. Pritzius and B. Breit, Angew. Chem., Int. Ed., 2015, 54, 15818 CrossRef CAS PubMed
- R. J. van Haaren, E. Zuidema, J. Fraanje, K. Goubitz, P. C. J. Kamer, P. W. N. M. van Leeuwen and G. P. F. van Strijdonck, C. R. Chim., 2002, 5, 431 CrossRef CAS
- T. M. Beck and B. Breit, Org. Lett., 2016, 18, 124 CrossRef CAS PubMed
- F. A. Cruz, Z. Chen, S. I. Kurtoic and V. M. Dong, Chem. Commun., 2016, 52, 5836 RSC
- R. S. Manan and P. Zhao, Nat. Commun., 2016, 7, 11506, DOI:10.1038/ncomms11506
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed
- J. P. Wolfe and S. L. Buchwald, J. Org. Chem., 2000, 65, 1144 CrossRef CAS PubMed
- S. Shekhar, P. Ryberg, J. F. Hartwig, J. S. Mathew, D. G. Blackmond, E. R. Strieter and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 3584 CrossRef CAS PubMed
- S. Tasler, J. Mies and M. Lang, Adv. Synth. Catal., 2007, 349, 2286 CrossRef CAS
- J. H. Ryu, J. A. Lee, S. Kim, Y. A. Shin, J. Yang, H. Y. Han, H. J. Son, Y. H. Kim, J. H. Sa, J.-S. Kim, J. Lee, J. Lee and H. Park, J. Med. Chem., 2016, 59, 10176 CrossRef CAS PubMed
- S. Birksø Larsen, B. Bang-Andersen, T. Nørskov Johansen and M. Jørgensen, Tetrahedron, 2008, 64, 2938 CrossRef
- A. Kuwahara, K. Nakano and K. Nozaki, J. Org. Chem., 2005, 70, 413 CrossRef CAS PubMed
- C. J. Chapman, A. Matsuno, C. G. Frost and M. C. Willis, Chem. Commun., 2007, 3903 RSC
- F. N. Ngassa, K. A. DeKorver, T. S. Melistas, E. A.-H. Yeh and M. K. Lakshman, Org. Lett., 2006, 8, 4614 CrossRef PubMed
- T. Takamura-Enya, S. Enomoto and K. Wakabayashi, J. Org. Chem., 2006, 71, 5599 CrossRef CAS PubMed
- A. Chenna, R. C. Gupta, R. R. Bonala, F. Johnson and B. Hang, Nucleosides, Nucleotides Nucleic Acids, 2008, 27, 979 CAS
- P. F. Thomson, P. Lagisetty, J. Balzarini, E. De Clercq and M. K. Lakshman, Adv. Synth. Catal., 2010, 352, 1728 CrossRef CAS PubMed
- P. Lagisetty, L. M. Russon and M. K. Lakshman, Angew. Chem., Int. Ed., 2006, 45, 3660 CrossRef CAS PubMed
- K. W. Anderson, M. Mendez-Perez, J. Priego and S. L. Buchwald, J. Org. Chem., 2003, 68, 9563 CrossRef CAS PubMed
- J. D. Hicks, A. M. Hyde, A. Martinez Cuezva and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 16720 CrossRef CAS PubMed
- R. E. Tundel, K. W. Anderson and S. L. Buchwald, J. Org. Chem., 2006, 71, 430 CrossRef CAS PubMed
- L. M. Klingensmith, E. R. Strieter, T. E. Barder and S. L. Buchwald, Organometallics, 2006, 25, 82 CrossRef CAS
- J. Yin and S. L. Buchwald, Org. Lett., 2000, 2, 1102 CrossRef
- S. Piguel and M. Legraverend, J. Org. Chem., 2007, 72, 7026 CrossRef CAS PubMed
- B. Li, L. Samp, J. Sagal, C. M. Hayward, C. Yang and Z. Zhang, J. Org. Chem., 2013, 78, 1273 CrossRef CAS PubMed
- W. Yang, H. Ma, Q. Yang, J. Wang, Y. Liu, Q. Yang, J. Wu, C. Song and J. Chang, Org. Biomol. Chem., 2017, 15, 379 CAS
- K. Fujita, M. Yamashita, F. Puschmann, M. Martinez Alvarez-Falcon, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9044 CrossRef CAS PubMed
- G. A. Artamkina, A. G. Sergeev and I. P. Beletskaya, Russ. J. Org. Chem., 2002, 38, 538 CrossRef CAS
- G. A. Artamkina, A. G. Sergeev and I. P. Beletskaya, Tetrahedron Lett., 2001, 42, 4381 CrossRef CAS
- A. G. Sergeev, G. A. Artamkina and I. P. Beletskaya, Russ. J. Org. Chem., 2003, 39, 1741 CrossRef CAS
- S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 10253 CrossRef
- J. Barluenga, C. Valdés, G. Beltrán, M. Escribano and F. Aznar, Angew. Chem., Int. Ed., 2006, 45, 6893 CrossRef CAS PubMed
- T. D. Montgomery and V. H. Rawal, Org. Lett., 2016, 18, 740 CrossRef CAS PubMed
- A. M. Johns, N. Sakai, A. Ridder and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9306 CrossRef CAS PubMed
- M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 9546 CrossRef CAS
- A. M. Johns, M. Utsunomiya, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 1828 CrossRef CAS PubMed
- R. J. van Haaren, H. Oevering, B. B. Coussens, G. P. F. van Strijdonck, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Eur. J. Inorg. Chem., 1999, 1237 CrossRef CAS
- A. M. Johns, Z. Liu and J. F. Hartwig, Angew. Chem., Int. Ed., 2007, 46, 7259 CrossRef CAS PubMed
- A. Perrier, M. Ferreira, J. N. H. Reek and J. I. van der Vlugt, Catal. Sci. Technol., 2013, 3, 1375 CAS
- N. Sakai, A. Ridder and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 8134 CrossRef CAS PubMed
- J. D. Neukom, N. S. Perch and J. P. Wolfe, Organometallics, 2011, 30, 1269 CrossRef CAS
- J. A. Fritz, J. S. Nakhla and J. P. Wolfe, Org. Lett., 2006, 8, 2531 CrossRef CAS PubMed
- B. P. Zavesky, N. R. Babij, J. A. Fritz and J. P. Wolfe, Org. Lett., 2013, 15, 5420 CrossRef CAS PubMed
- Y. Zhang, K. A. DeKorver, A. G. Lohse, Y.-S. Zhang, J. Huang and R. P. Hsung, Org. Lett., 2009, 11, 899 CrossRef CAS PubMed
- J. Peng, D. Jiang, W. Lina and Y. Chen, Org. Biomol. Chem., 2007, 5, 1391 CAS
- R. S. Pathare, S. Sharma, S. Elagandhula, V. Saini, D. M. Sawant, M. Yadav, A. Sharon, S. Khan and R. T. Pardasani, Eur. J. Org. Chem., 2016, 5579 CrossRef CAS
- J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460 CrossRef CAS PubMed
- J. R. Martinelli, D. M. M. Freckmann and S. L. Buchwald, Org. Lett., 2006, 8, 4843 CrossRef CAS PubMed
- J. R. Martinelli, D. A. Watson, D. M. M. Freckmann, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2008, 73, 7102 CrossRef CAS PubMed
- S. D. Friis, T. Skrydstrup and S. L. Buchwald, Org. Lett., 2014, 16, 4296 CrossRef CAS PubMed
- E. R. Murphy, J. R. Martinelli, N. Zaborenko, S. L. Buchwald and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 1734 CrossRef CAS PubMed
- A. Deagostino, P. Larini, E. G. Occhiato, L. Pizzuto, C. Prandi and P. Venturello, J. Org. Chem., 2008, 73, 1941 CrossRef CAS PubMed
- S. V. F. Hansen, Z. E. Wilson, T. Ulven and S. V. Ley, React. Chem. Eng., 2016, 1, 280 CAS
- T. L. Andersen, S. D. Friis, H. Audrain, P. Nordeman, G. Antoni and T. Skrydstrup, J. Am. Chem. Soc., 2015, 137, 1548 CrossRef CAS PubMed
- K. C. Kong and C. H. Cheng, J. Am. Chem. Soc., 1991, 113, 6313 CrossRef CAS
- M. Stefko, R. Pohl and M. Hocek, Tetrahedron, 2009, 65, 4471 CrossRef CAS
- L.-J. Gu, Y.-S. Wang, H.-T. Zhang, H.-J. Tang, G.-P. Li and M.-L. Yuan, ChemCatChem, 2016, 8, 2206 CrossRef CAS
- F. M. Miloserdov, C. L. McMullin, M. Martínez Belmonte, J. Benet-Buchholz, V. I. Bakhmutov, S. A. Macgregor and V. V. Grushin, Organometallics, 2014, 33, 736 CrossRef CAS
- H. Li, X. Fang, R. Jackstell, H. Neumann and M. Beller, Chem. Commun., 2016, 52, 7142 RSC
- H. Ohmiya, T. Moriya and M. Sawamura, Org. Lett., 2009, 11, 2145 CrossRef CAS PubMed
- K. T. J. Loones, B. U. W. Maes and R. A. Dommisse, Tetrahedron, 2007, 63, 8954 CrossRef CAS
- R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, Angew. Chem., Int. Ed., 2012, 51, 3953 CrossRef CAS PubMed
- M. Mailig, R. P. Rucker and G. Lalic, Chem. Commun., 2015, 51, 11048 RSC
- A. L. Reznichenko and K. C. Hultszch, Org. React., 2015, 88, 1 Search PubMed
- M. Utsunomiya, R. Kuwano, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 5608 CrossRef CAS PubMed
- A. Couce-Rios, A. Lledós and G. Ujaque, Chem. – Eur. J., 2016, 22, 9311 CrossRef CAS PubMed
- O. Eisenstein and R. Hoffman, J. Am. Chem. Soc., 1981, 103, 4308 CrossRef CAS
- L.-B. Han and M. Tanaka, J. Am. Chem. Soc., 1996, 118, 1571 CrossRef CAS
- L.-B. Han, N. Choi and M. Tanaka, Organometallics, 1996, 15, 3259 CrossRef CAS
- A. Allen Jr., L. Ma and W. Lin, Tetrahedron Lett., 2002, 43, 3707 CrossRef
- S. Deprele and J.-L. Montchamp, J. Am. Chem. Soc., 2002, 124, 9386 CrossRef CAS PubMed
- P. Ribiere, K. Bravo-Altamirano, M. I. Antczak, J. D. Hawkins and J.-L. Montchamp, J. Org. Chem., 2005, 70, 4064 CrossRef CAS PubMed
- S. Deprele and J.-L. Montchamp, Org. Lett., 2004, 6, 3805 CrossRef CAS PubMed
- K. Bravo-Altamirano and J.-L. Montchamp, Tetrahedron Lett., 2007, 48, 5755 CrossRef CAS PubMed
- Y. Belabassi, K. Bravo-Altamirano and J.-L. Montchamp, J. Organomet. Chem., 2011, 696, 106 CrossRef CAS
- K. Bravo-Altamirano, L. Coudray, E. L. Deal and J.-L. Montchamp, Org. Biomol. Chem., 2010, 8, 5541 CAS
- K. Bravo-Altamirano and J.-L. Montchamp, Org. Lett., 2006, 8, 4169 CrossRef CAS PubMed
- L. Coudray and J.-L. Montchamp, Eur. J. Org. Chem., 2008, 4101 CrossRef CAS PubMed
- K. Bravo-Altamirano, I. Abrunhosa-Thomas and J.-L. Montchamp, J. Org. Chem., 2008, 73, 2292 CrossRef CAS PubMed
- L. Coudray, K. Bravo-Altamirano and J.-L. Montchamp, Org. Lett., 2008, 10, 1123 CrossRef CAS PubMed
- A. Fers-Lidou, O. Berger and J.-L. Montchamp, Molecules, 2016, 21, 1295 CrossRef PubMed
-
M. Hill, W. Krause and M. Sicken, PCT Int. Appl., WO2010051891, 2010 (to Clariant International Ltd, Switz.); Chem. Abstr., 2010, 152, 569243 (one example)
- M. Kalek and J. Stawinski, Tetrahedron, 2009, 65, 10406 CrossRef CAS
- G. Laven, M. Kalek, M. Jezowska and J. Stawinski, New J. Chem., 2010, 34, 967 RSC
- L. Botez, G. B. de Jong, J. C. Slootweg and B.-J. Deelman, Eur. J. Org. Chem., 2017, 434 CrossRef CAS
- A. J. Bloomfield and S. B. Herzon, Org. Lett., 2012, 14, 4370 CrossRef CAS PubMed
- M. Kosugi, T. Shimizu and T. Migita, Chem. Lett., 1978, 13 CrossRef CAS
- G. Mann, D. Baranano, J. F. Hartwig, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120, 9205 CrossRef CAS
- U. Schlopfer and A. Schlapbach, Tetrahedron, 2001, 57, 3069 CrossRef
- M. Murata and S. L. Buchwald, Tetrahedron, 2004, 60, 7397 CrossRef CAS
- G.-Y. Gao, A. J. Colvin, Y. Chen and X. P. Zhang, J. Org. Chem., 2004, 69, 8886 CrossRef CAS PubMed
- M. A. Fernandez-Rodriguez, Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 2180 CrossRef CAS PubMed
- C. Mispelaere-Canivet, J.-F. Spindler, S. Perrioa and P. Beslin, Tetrahedron, 2005, 61, 5253 CrossRef CAS
- T. Itoh and T. Mase, Org. Lett., 2004, 6, 4587 CrossRef CAS PubMed
- R. Ahmed Atto Al-Shuaeeb, S. Kolodych, O. Koniev, S. Delacroix, S. Erb, S. Nicolay, J.-C. Cintrat, J.-D. Brion, S. Cianférani, M. Alami, A. Wagner and S. Messaoudi, Chem. – Eur. J., 2016, 22, 11365 CrossRef PubMed
- B. Chekal, D. Damon, D. LaFrance, K. Leeman, C. Mojica, A. Palm, M. St. Pierre, J. Sieser, K. Sutherland, R. Vaidyanathan, J. Van Alsten, B. Vanderplas, C. Wager, G. Weisenburger, G. Withbroe and S. Yu, Org. Process Res. Dev., 2015, 19, 1944 CrossRef CAS
- S. A. Baker Dockrey, A. K. Makepeace and J. R. Schmink, Org. Lett., 2014, 16, 4730 CrossRef CAS PubMed
- B. Yucel and P. J. Walsh, Adv. Synth. Catal., 2014, 356, 3659 CrossRef CAS PubMed
- N. Abidi and J. R. Schmink, J. Org. Chem., 2015, 80, 4123 CrossRef CAS PubMed
- J. R. Schmink, S. A. Baker Dockrey, T. Zhang, N. Chebet, A. van Venrooy, M. Sexton, S. I. Lew, S. Chou and A. Okazaki, Org. Lett., 2016, 18, 6360 CrossRef CAS PubMed
- J. Mao, T. Jia, G. Frensch and P. J. Walsh, Org. Lett., 2014, 16, 5304 CrossRef CAS PubMed
- K. M. Wager and M. H. Daniels, Org. Lett., 2011, 13, 4053 CrossRef PubMed
- J. E. Gómez, W. Guo and A. W. Kleij, Org. Lett., 2016, 18, 6042 CrossRef PubMed
- S. Cacchi, G. Fabrizi, A. Goggiamani, L. M. Parisi and R. Bernini, J. Org. Chem., 2004, 69, 5608 CrossRef CAS PubMed
- E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 12679 CrossRef CAS PubMed
- G. Maitro, S. Vogel, G. Prestat, D. Madec and G. Poli, Org. Lett., 2006, 8, 5951 CrossRef CAS PubMed
- F. Colobert, R. Ballesteros-Garrido, F. R. Leroux, R. Ballesteros and B. Abarca, Tetrahedron Lett., 2007, 48, 6896 CrossRef CAS
- T. Jia, A. Bellomo, S. Montel, M. Zhang, K. EL Baina, B. Zheng and P. J. Walsh, Angew. Chem., Int. Ed., 2014, 53, 260 CrossRef CAS PubMed
- T. Jia, M. Zhang, I. K. Sagamanova, C. Y. Wang and P. J. Walsh, Org. Lett., 2015, 17, 1168 CrossRef CAS PubMed
- C. Lai and B. J. Backes, Tetrahedron Lett., 2007, 48, 3033 CrossRef CAS
-
B. F. M. Kimmich, H. E. Toomey, X. Yao, G. P. Torrence, J. C. van der Waal and M. J. Doyle, US2010/0286440, 2010 (to Celanese)
- D. Fujino, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2014, 136, 6255 CrossRef CAS PubMed
- M. F. Cain, R. P. Hughes, D. S. Glueck, J. A. Golen, C. E. Moore and A. L. Rheingold, Inorg. Chem., 2010, 49, 7650 CrossRef CAS PubMed
- J.-E. Lee, J. Kwon and J. Yun, Chem. Commun., 2008, 733 RSC
- H.-Y. Jung, X. Feng, H. Kim and J. Yun, Tetrahedron, 2012, 68, 3444 CrossRef CAS
- W. Yuan, L. Song and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 3140 CrossRef CAS PubMed
- K. Semba, M. Shinomiya, T. Fujihara, J. Terao and Y. Tsuji, Chem. – Eur. J., 2013, 19, 7125 CrossRef CAS PubMed
- W. J. Jang, W. L. Lee, J. H. Moon, J. Y. Lee and J. Yun, Org. Lett., 2016, 18, 1390 CrossRef CAS PubMed
- H. Ito, Y. Sasaki and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 15774 CrossRef CAS PubMed
- L. Mao, K. J. Szabó and T. B. Marder, Org. Lett., 2017, 19, 1204 CrossRef CAS PubMed
- H. Ito, T. Miya and M. Sawamura, Tetrahedron, 2012, 68, 3423 CrossRef CAS
- H. Ito, Y. Kosaka, K. Nonoyama, Y. Sasaki and M. Sawamura, Angew. Chem., Int. Ed., 2008, 47, 7424 CrossRef CAS PubMed
- W. Su, T.-J. Gong, X. Lu, M.-Y. Xu, C.-G. Yu, Z.-Y. Xu, H.-Z. Yu, B. Xiao and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 12957 CrossRef CAS PubMed
- H. C. Johnson, C. L. McMullin, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2013, 52, 9776 CrossRef CAS PubMed
- H. C. Johnson, E. M. Leitao, G. R. Whittell, I. Manners, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2014, 136, 9078 CrossRef CAS PubMed
- H. C. Johnson, R. Torry-Harris, L. Ortega, R. Theron, J. S. McIndoe and A. S. Weller, Catal. Sci. Technol., 2014, 4, 3486 CAS
- M. Tobisu, H. Kinuta, Y. Kita, E. Rémond and N. Chatani, J. Am. Chem. Soc., 2012, 134, 115 CrossRef CAS PubMed
- M. A. Esteruelas, M. Oliván and A. Vélez, J. Am. Chem. Soc., 2015, 137, 12321 CrossRef CAS PubMed
- M. A. Esteruelas, M. Oliván and A. Vélez, Organometallics, 2015, 34, 1911 CrossRef CAS
- R. J. Pawley, M. A. Huertos, G. C. Lloyd-Jones, A. S. Weller and M. C. Willis, Organometallics, 2012, 31, 5650 CrossRef CAS
- M. C. Haibach, D. Y. Wang, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Chem. Sci., 2013, 4, 3683 RSC
- A. Lumbroso, P. Koschker, N. R. Vautravers and B. Breit, J. Am. Chem. Soc., 2011, 133, 2386 CrossRef CAS PubMed
- B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed
- R. J. van Haaren, K. Goubitz, J. Fraanje, G. P. F. van Strijdonck, H. Oevering, B. Coussens, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Inorg. Chem., 2001, 40, 3363 CrossRef CAS PubMed
- S. C. Milheiro and J. W. Faller, J. Organomet. Chem., 2011, 696, 879 CrossRef CAS
- G. Mora, B. Deschamps, S. van Zutphen, X. F. Le Goff, L. Ricard and P. Le Floch, Organometallics, 2007, 26, 1846 CrossRef CAS
- D. S. B. Daniels, A. S. Jones, A. L. Thompson, R. S. Paton and E. A. Anderson, Angew. Chem., Int. Ed., 2014, 53, 1915 CrossRef CAS PubMed
- L. P. Jay and T. J. Barker, Eur. J. Org. Chem., 2016, 1829 CrossRef CAS
- D. Seomoon and P. H. Lee, J. Org. Chem., 2008, 73, 1165 CrossRef CAS PubMed
- W. Guo, L. Martínez-Rodríguez, R. Kuniyil, E. Martin, E. C. Escudero-Adán, F. Maseras and A. W. Kleij, J. Am. Chem. Soc., 2016, 138, 11970 CrossRef CAS PubMed
- M. Kenny, D. J. Kitson and V. Franckevičius, J. Org. Chem., 2016, 81, 5162 CrossRef CAS PubMed
- H. Zhou, Y. Wang, L. Zhang, M. Cai and S. Luo, J. Am. Chem. Soc., 2017, 139, 3631 CrossRef CAS PubMed
- V. Khakyzadeh, Y.-H. Wang and B. Breit, Chem. Commun., 2017, 53, 4966 RSC
- S. Kamijo, M. Al-Masum and Y. Yamamoto, Tetrahedron Lett., 1998, 39, 691 CrossRef CAS
- K. Xu, V. Khakyzadeh, T. Bury and B. Breit, J. Am. Chem. Soc., 2014, 136, 16124 CrossRef CAS PubMed
- A. Lumbroso, P. Koschker, N. R. Vautravers and B. Breit, J. Am. Chem. Soc., 2011, 133, 2386 CrossRef CAS PubMed
- U. Gellrich, A. Meißner, A. Steffani, M. Kähny, H.-J. Drexler, D. Heller, D. A. Plattner and B. Breit, J. Am. Chem. Soc., 2014, 136, 1097 CrossRef CAS PubMed
- H. Yu, G. Zhang, Z.-J. Liu and H. Huang, RSC Adv., 2014, 4, 64235 RSC
- H. Li, H. Neumann and M. Beller, Chem. – Eur. J., 2016, 22, 10050 CrossRef CAS PubMed
- Y. Zhu and V. H. Rawal, J. Am. Chem. Soc., 2012, 134, 111 CrossRef CAS PubMed
- M. Utsunomiya, Y. Miyamoto, J. Ipposhi, T. Ohshima and K. Mashima, Org. Lett., 2007, 9, 3371 CrossRef CAS PubMed
- G. Mora, O. Piechaczyk, R. Houdard, N. Mezailles, X.-F. Le Goff and P. le Floch, Chem. – Eur. J., 2008, 14, 10047 CrossRef CAS PubMed
- O. Piechaczyk, C. Thoumazet, Y. Jean and P. Le Floch, J. Am. Chem. Soc., 2006, 128, 14306 CrossRef CAS PubMed
- T. Ohshima, Y. Miyamoto, J. Ipposhi, Y. Nakahara, M. Utsunomiya and K. Mashima, J. Am. Chem. Soc., 2009, 131, 14317 CrossRef CAS PubMed
- B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467 RSC
- K. Michigami, T. Mita and Y. Sato, J. Am. Chem. Soc., 2017, 139, 6094 CrossRef CAS PubMed
- T. Tamura and J. K. Kochi, J. Am. Chem. Soc., 1971, 93, 1487 CrossRef
- Ref. 1, pp. 279–282.
- A. Fuerstner, ACS Cent. Sci., 2016, 2, 778 CrossRef CAS PubMed
- D. Necas, M. Kotora and I. Cisarova, Eur. J. Org. Chem., 2004, 1280 CrossRef CAS
- T. Hatakeyama, T. Hashimoto, K. K. A. D. S. Kathriarachchi, T. Zenmyo, H. Seike and M. Nakamura, Angew. Chem., Int. Ed., 2012, 51, 8834 CrossRef CAS PubMed
- T. Hatakeyama, S. Hashimoto, K. Ishizuka and M. Nakamura, J. Am. Chem. Soc., 2009, 131, 11949 CrossRef CAS PubMed
- J. L. Kneebone, V. E. Fleischauer, S. L. Daifuku, A. A. Shaps, J. M. Bailey, T. E. Iannuzzi and M. L. Neidig, Inorg. Chem., 2016, 55, 272 CrossRef CAS PubMed
- A. J. Bridgeman, G. Cavigliasso, L. R. Ireland and J. Rothery, J. Chem. Soc., Dalton Trans., 2001, 2095 RSC
- M. M. P. Grutters, C. Mueller and D. Vogt, J. Am. Chem. Soc., 2006, 128, 7414 CrossRef CAS PubMed
- H. Fillon, C. Gosmini and J. Périchon, J. Am. Chem. Soc., 2003, 125, 3867 CrossRef CAS PubMed
- M.-Y. Jin and N. Yoshikai, J. Org. Chem., 2011, 76, 1972 CrossRef CAS PubMed
- K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed
- C.-Y. Liu, X. Wang, T. Furuyama, S. Yasuike, A. Muranaka, K. Morokuma and M. Uchiyama, Chem. – Eur. J., 2010, 16, 1780 CrossRef CAS PubMed
- A. Krasovskiy, V. Malakhov, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 6040 CrossRef CAS PubMed
- K. Komeyama, S. Kiguchi and K. Takaki, Chem. Commun., 2016, 52, 7009 RSC
- M. R. Brennan, D. Kim and A. R. Fout, Chem. Sci., 2014, 5, 4831 RSC
- C. Wang, W. J. Teo and S. Ge, ACS Catal., 2017, 7, 855 CrossRef CAS
- G. Mora, S. van Zutphen, C. Klemps, L. Ricard, Y. Jean and P. Le Floch, Inorg. Chem., 2007, 46, 10365 CrossRef CAS PubMed
- Y. Son, H.-S. Kim, J.-H. Lee, J. Jang, C.-F. Lee and S. Lee, Tetrahedron Lett., 2017, 58, 1413 CrossRef CAS
- G. L. Beutner, Y. Hsiao, T. Razler, E. M. Simmons and W. Wertjes, Org. Lett., 2017, 19, 1052 CrossRef CAS PubMed
- D. D. Beattie, T. Schareina and M. Beller, Org. Biomol. Chem., 2017, 15, 4291 CAS
- H. Ito, T. Miya and M. Sawamura, Tetrahedron, 2012, 68, 3423 CrossRef CAS
- H. Ito, C. Kawakami and M. Sawamura, J. Am. Chem. Soc., 2005, 127, 16034 CrossRef CAS PubMed
- K. Kubota, E. Yamamoto and H. Ito, J. Am. Chem. Soc., 2013, 135, 2635 CrossRef CAS PubMed
- W. Su, T.-J. Gong, X. Lu, M.-Y. Xu, C.-G. Yu, Z.-Y. Xu, H.-Z. Yu, B. Xiao and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 12957 CrossRef CAS PubMed
- X. L. Luo and R. H. Crabtree, J. Am. Chem. Soc., 1989, 111, 2527 CrossRef CAS
- H. Ito, A. Watanabe and M. Sawamura, Org. Lett., 2005, 7, 1869 CrossRef CAS PubMed
- D. Kim, B.-M. Park and J. Yun, Chem. Commun., 2005, 1755 RSC
- K. Semba, T. Fujihara, T. Xu, J. Terao and Y. Tsuji, Adv. Synth. Catal., 2012, 354, 1542 CrossRef CAS
- K. Ramakrishna and C. Sivasankar, J. Organomet. Chem., 2016, 805, 122 CrossRef CAS
- H. Murayama, K. Nagao, H. Ohmiya and M. Sawamura, Org. Lett., 2015, 17, 2039 CrossRef CAS PubMed
- S. Fischer, D. Hollmann, S. Tschierlei, M. Karnahl, N. Rockstroh, E. Barsch, P. Schwarzbach, S.-P. Luo, H. Junge, M. Beller, S. Lochbrunner, R. Ludwig and A. Brückner, ACS Catal., 2014, 4, 1845 CrossRef CAS
- M. T. Reetz and X. Lia, Adv. Synth. Catal., 2006, 348, 1157 CrossRef CAS
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554 CrossRef CAS PubMed
- S. C. Chmely, S. Kim, P. N. Ciesielski, G. Jiménez-Osés, R. S. Paton and G. T. Beckham, ACS Catal., 2013, 3, 963 CrossRef CAS
- L. Shaw, D. M. U. K. Somisara, R. C. How, N. J. Westwood, P. C. A. Bruijnincx, B. M. Weckhuysen and P. C. J. Kamer, Catal. Sci. Technol., 2017, 7, 619 CAS
- Y. Oe, T. Ohta and Y. Ito, Tetrahedron Lett., 2010, 51, 2806 CrossRef CAS
- S. J. Pridmore, P. A. Slatford and J. M. J. Williams, Tetrahedron Lett., 2007, 48, 5111 CrossRef CAS
- S. J. Pridmore, P. A. Slatford, A. Daniel, M. K. Whittlesey and J. M. J. Williams, Tetrahedron Lett., 2007, 48, 5115 CrossRef CAS
- P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, Tetrahedron Lett., 2006, 47, 6787 CrossRef CAS
- T. Marimuthu and H. B. Friedrich, ChemCatChem, 2012, 4, 2090 CrossRef CAS
- B. Deb, B. J. Borah, B. J. Sarmah, B. Das and D. K. Dutta, Inorg. Chem. Commun., 2009, 12, 868 CrossRef CAS
- X. Chen, H. Zhou, K. Zhang, J. Li and H. Huang, Org. Lett., 2014, 16, 3912 CrossRef CAS PubMed
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555 CrossRef CAS
- G. Guillena, D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 235 CrossRef PubMed
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc., Chem. Commun., 1981, 611 RSC
- O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Chem. Commun., 2010, 46, 1541 RSC
- C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661 CrossRef CAS PubMed
- D. Pingen, C. Mueller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130 CrossRef CAS PubMed
- M. H. S. A. Hamid and J. M. J. Williams, Chem. Commun., 2007, 725 RSC
- M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766 CrossRef CAS PubMed
- S. Imm, S. Bähn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599 CrossRef CAS PubMed
- W. Baumann, A. Spannenberg, J. Pfeffer, T. Haas, A. Koeckritz, A. Martin and J. Deutsch, Chem. – Eur. J., 2013, 19, 17702 CrossRef CAS PubMed
- A. S. Stalsmeden, J. L. Belmonte Vazquez, K. van Weerdenburg, R. Rae, P.-O. Norrby and N. Kann, ACS Sustainable Chem. Eng., 2016, 4, 5730 CrossRef
- D. Pingen, T. Lebl, M. Lutz, G. S. Nichol, P. C. J. Kamer and D. Vogt, Organometallics, 2014, 33, 2798 CrossRef CAS
- D. Pingen, M. Lutz and D. Vogt, Organometallics, 2014, 33, 1623 CrossRef CAS
- E. J. Derrah, M. Hanauer, P. N. Plessow, M. Schelwies, M. K. da Silva and T. Schaub, Organometallics, 2015, 34, 1872 CrossRef CAS
- N. Anand, N. A. Owston, A. J. Parker, P. A. Slatforda and J. M. J. Williams, Tetrahedron Lett., 2007, 48, 7761 CrossRef CAS
- A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Biomol. Chem., 2012, 10, 240 CAS
- A. J. Blacker, M. M. Farah, M. I. Hall, S. P. Marsden, O. Saidi and J. M. J. Williams, Org. Lett., 2009, 11, 2039 CrossRef CAS PubMed
-
C. Saudan and L. Saudan, US Pat., 8871982, 2014 (to Firmenich SA, Switz.); Chem. Abstr., 2016:2024370 Search PubMed
- E. B. Walczuk, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2003, 42, 4665 CrossRef CAS PubMed
- C. J. Scheuermann and C. Jaekel, Adv. Synth. Catal., 2008, 350, 2708 CrossRef
- P. Ren, S. D. Pike, I. Pernik, A. S. Weller and M. C. Willis, Organometallics, 2015, 34, 711 CrossRef CAS
- H. C. Johnson and A. S. Weller, Angew. Chem., Int. Ed., 2015, 54, 10173 CrossRef CAS PubMed
- H. C. Johnson, C. L. McMullin, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2013, 52, 9776 CrossRef CAS PubMed
- H. C. Johnson, R. Torry-Harris, L. Ortega, R. Theron, J. S. McIndoe and A. S. Weller, Catal. Sci. Technol., 2014, 4, 3486 CAS
- M. Tobisu, H. Kinuta, Y. Kita, E. Rémond and N. Chatani, J. Am. Chem. Soc., 2012, 134, 115 CrossRef CAS PubMed
- M. A. Esteruelas, M. Oliván and A. Vélez, J. Am. Chem. Soc., 2015, 137, 12321 CrossRef CAS PubMed
- M. A. Esteruelas, M. Oliván and A. Vélez, Inorg. Chem., 2013, 52, 12108 CrossRef CAS PubMed
- A. Dermenci, R. E. Whittaker, Y. Gao, F. A. Cruz, Z.-X. Yu and G. Dong, Chem. Sci., 2015, 6, 3201 RSC
- R. E. Whittaker and G. Dong, Org. Lett., 2015, 17, 5504 CrossRef CAS PubMed
- M. C. Haibach, D. Y. Wang, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Chem. Sci., 2013, 4, 3683 RSC
- A. J. Pontiggia, A. B. Chaplin and A. S. Weller, J. Organomet. Chem., 2011, 696, 2870 CrossRef CAS
- M. Fukushima, D. Takushima and M. Kimura, J. Am. Chem. Soc., 2010, 132, 16346 CrossRef CAS PubMed
- B. Wahl, S. Giboulot, A. Mortreux, Y. Castanet, M. Sauthier, F. Liron and G. Poli, Adv. Synth. Catal., 2012, 354, 1077 CrossRef CAS
- P. Xie, Y. Xie, B. Qian, H. Zhou, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 9902 CrossRef CAS PubMed
- P. W. N. M. van Leeuwen, M. A. Zuideveld, B. H. G. Swennenhuis, Z. Freixa, P. C. J. Kamer, K. Goubitz, J. Fraanje, M. Lutz and A. L. Spek, J. Am. Chem. Soc., 2003, 125, 5523 CrossRef CAS PubMed
- T. L. Andersen, P. Nordeman, H. F. Christoffersen, H. Audrain, G. Antoni and T. Skrydstrup, Angew. Chem., Int. Ed., 2017, 56, 4549 CrossRef CAS PubMed
- G. Zhang, X. Ji, H. Yu, L. Yang, P. Jiao and H. Huang, Tetrahedron Lett., 2016, 57, 383 CrossRef CAS
- J. Liu, Q. Liu, R. Franke, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2015, 137, 8556 CrossRef CAS PubMed
- E. Drent, P. Arnoldy and P. H. M. Budzelaar, J. Organomet. Chem., 1994, 475, 57 CrossRef CAS
- J. El Karroumia, A. El Haiba, E. Manoury, A. Benharref, J. C. Darana, M. Gouygoua and M. Urrutigoïtya, J. Mol. Catal. A: Chem., 2015, 401, 18 CrossRef
- I. del Rio, N. Ruiz, C. Claver, L. A. van der Veen and P. W. N. M. van Leeuwen, J. Mol. Catal. A: Chem., 2000, 161, 39 CrossRef CAS
- Y. G. Noskov and E. S. Petrov, Kinet. Catal., 1993, 34, 902 Search PubMed
- E. Zuidema, C. Bo and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2007, 129, 3989 CrossRef CAS PubMed
- W.-H. Lin, W.-C. Wu, M. Selvarajua and C.-M. Sun, Org. Chem. Front., 2017, 4, 392 RSC
- F. Ragaini, Dalton Trans., 2009, 6251 RSC
- F. Ragaini, Organometallics, 1996, 15, 3572 CrossRef CAS
- P. Wehman, H. M. A. van Donge, A. Hagos, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Organomet. Chem., 1997, 535, 183 CrossRef CAS
- L. J. Gooßen and N. Rodríguez, Chem. Commun., 2004, 724 RSC
- K. L. Toups and R. A. Widenhoefer, Chem. Commun., 2010, 46, 1712 RSC
- T. T. Dang, B. Ramalingam, S. P. Shan and A. M. Seayad, ACS Catal., 2013, 3, 2536 CrossRef CAS
- J. Liu, N. Liu, Y. Yue, Y. Wang, K. Chen, J. Zhang, S. Zhao and K. Zhuo, Chem. – Asian J., 2017, 12, 401 CrossRef PubMed
- J. Caner and J. Vilarrasa, J. Org. Chem., 2010, 75, 4880 CrossRef CAS PubMed
- X. Ariza and J. Vilarrasa, J. Org. Chem., 2000, 65, 2827 CrossRef CAS PubMed
- T. Igarashi, M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2017, 56, 2069 CrossRef CAS PubMed
- P. B. Dzhevakov, M. A. Topchiy, D. A. Zharkova, O. S. Morozov, A. F. Asachenko and M. S. Nechaev, Adv. Synth. Catal., 2016, 358, 977 CrossRef CAS
- T. Liu, Y. Zeng, H. Zhang, T. Wei, X. Wua and N. Li, Tetrahedron Lett., 2016, 57, 4845 CrossRef CAS
- P. Hu and S. A. Snyder, J. Am. Chem. Soc., 2017, 139, 5007 CrossRef CAS PubMed
- B. Dudle, K. Rajesh, O. Blacque and H. Berke, J. Am. Chem. Soc., 2011, 133, 8168 CrossRef CAS PubMed
- B. Dudle, K. Rajesh, O. Blacque and H. Berke, Organometallics, 2011, 30, 2986 CrossRef CAS
- K. Rajesh, B. Dudle, O. Blacque and H. Berke, Adv. Synth. Catal., 2011, 353, 1479 CrossRef CAS
- Q.-W. Song, Z.-H. Zhou, M.-Y. Wang, K. Zhang, P. Liu, J.-Y. Xun and L.-N. He, ChemSusChem, 2016, 9, 2054 CrossRef CAS PubMed
- G. Xu, K. Liu, Z. Dai and J. Sun, Org. Biomol. Chem., 2017, 15, 2345 CAS
- J. H. Hansen and H. M. L. Davies, Chem. Sci., 2011, 2, 457 RSC
- J. M. Serrano-Becerra, A. F. G. Maier, S. González-Gallardo, E. Moos, C. Kaub, M. Gaffga, G. Niedner-Schatteburg, P. W. Roesky, F. Breher and J. Paradies, Eur. J. Org. Chem., 2014, 4515 CrossRef CAS
- H. Ito, K. Takagi, T. Miyahara and M. Sawamura, Org. Lett., 2005, 7, 3001 CrossRef CAS PubMed
- S. Labouille, A. Escalle-Lewis, Y. Jean, N. Mezailles and P. Le Floch, Chem. – Eur. J., 2011, 17, 2256 CrossRef CAS PubMed
- J. K. MacDougall, M. C. Simpson, M. J. Green and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1996, 1161 RSC
- L. Crawford, D. J. Cole-Hamilton and M. Bühl, Organometallics, 2015, 34, 438 CrossRef CAS
- K. Dong, R. Sang, X. Fang, R. Franke, A. Spannenberg, H. Neumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 5267 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2018 |