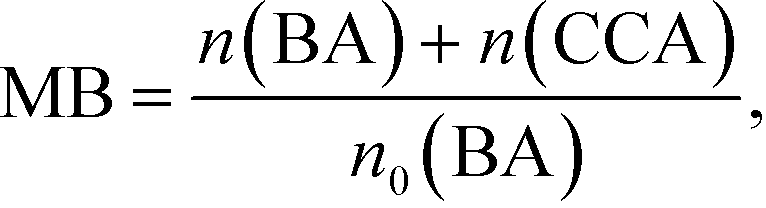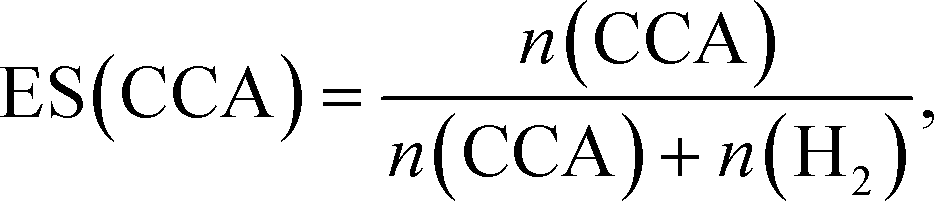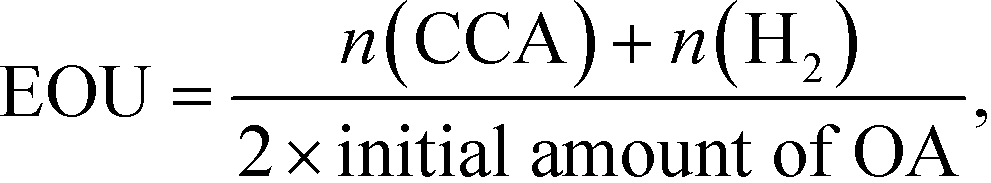Ring hydrogenation of aromatic compounds in aqueous suspensions of an Rh-loaded TiO2 photocatalyst without use of H2 gas†
Received
20th September 2017
, Accepted 13th November 2017
First published on 13th November 2017
Abstract
There are various possibilities of co-catalyst-assisted photocatalytic reduction (CPR) over a titanium(IV) oxide (TiO2) photocatalyst, especially H2-free and chemoselective CPR. We examined the photoinduced ring hydrogenation of aromatics having a carboxyl group over metal-loaded TiO2 under H2-free conditions and found that the aromatics were almost quantitatively hydrogenated to the corresponding cyclohexanes having a carboxyl group when rhodium, water and oxalic acid were used as a metal co-catalyst, solvent and hole scavenger, respectively. The effects of different metal co-catalysts, solvents and hole scavengers on the ring hydrogenation were also examined. Based on the results obtained under various conditions, the light dependency and adsorption behavior of the aromatics and hole scavengers, the functions of TiO2 and the co-catalyst, and the reaction process are discussed.
1. Introduction
Since a photocatalytic reaction satisfies almost all of the 12 proposed requirements for green chemistry,1 photocatalytic conversion of various organic compounds has recently been studied by many researchers.2 Recently, the number of reports on reductive conversion of organic compounds by using photogenerated electrons has been increasing.3,4 However, most of the reports deal with reduction (hydrogenation) of nitrobenzenes to aminobenzenes.5–15 Our research group has explored new photocatalytic reduction systems under hydrogen (H2)-free conditions with focus on a) selective and chemoselective photocatalytic reduction and b) co-catalyst-assisted photocatalytic reduction (CPR). As a typical case of a), 3-nitrostyrene was chemoselectively reduced to 3-aminostyrene without hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond to 3-ethylaniline in a suspension of a TiO2 photocatalyst.16 As a case of a), we also found that p-chlorobenzaldehyde was chemoselectively converted to p-chlorobenzyl alcohol in an alcoholic suspension of TiO2 in which the chloro group was preserved.17 As a case of b), benzonitrile and styrene were successfully hydrogenated to benzylamine and ethylbenzene, respectively, in alcohol suspensions of a palladium-loaded TiO2 photocatalyst, although the reduction potentials of benzonitrile and styrene are believed to be much higher than the potential of the conduction band of TiO2.18,19 As a case that satisfies both a) and b), i.e., chemoselective CPR (CCPR), we reported the chemoselective reduction of (2,3-epoxypropyl)benzene to allylbenzene (silver co-catalyst)20 and the semihydrogenation of alkynes to alkenes (copper co-catalyst).21 We think that various possibilities can happen in CCPR because many metal co-catalysts can be applied to photocatalytic reactions.
C double bond to 3-ethylaniline in a suspension of a TiO2 photocatalyst.16 As a case of a), we also found that p-chlorobenzaldehyde was chemoselectively converted to p-chlorobenzyl alcohol in an alcoholic suspension of TiO2 in which the chloro group was preserved.17 As a case of b), benzonitrile and styrene were successfully hydrogenated to benzylamine and ethylbenzene, respectively, in alcohol suspensions of a palladium-loaded TiO2 photocatalyst, although the reduction potentials of benzonitrile and styrene are believed to be much higher than the potential of the conduction band of TiO2.18,19 As a case that satisfies both a) and b), i.e., chemoselective CPR (CCPR), we reported the chemoselective reduction of (2,3-epoxypropyl)benzene to allylbenzene (silver co-catalyst)20 and the semihydrogenation of alkynes to alkenes (copper co-catalyst).21 We think that various possibilities can happen in CCPR because many metal co-catalysts can be applied to photocatalytic reactions.
Ring hydrogenation of aromatics is an important method to produce compounds consisting of a cyclohexane structure. For example, cyclohexanecarboxylic acid (CCA), which is an important intermediate of pharmaceutical and related compounds, is generally produced by ring hydrogenation of benzoic acid (BA).22 In the case of substituted aromatics, hydrogenation (reduction) of the functional groups also occurs as a side reaction and produces aromatic compounds of which the functional group is reduced. In the case of BA, benzaldehyde and benzyl alcohol were formed by hydrogenation of the carboxyl group of BA.23 Ring hydrogenation of BA was achieved over metal catalysts supported on suitable supports such as rhodium (Rh), ruthenium and NiZrB.24–27 However, these catalyst systems require the addition of an excess amount of H2 gas as a hydrogen source in a closed reactor. Since special care should be given during a reaction using H2, a safer catalytic reaction system driving selective ring hydrogenation without the use of H2 is desired. As described above, there are various possibilities of CCPR, and H2-free CCPR can be applied to ring hydrogenation of aromatics having a carboxyl group to corresponding alicyclic compounds.
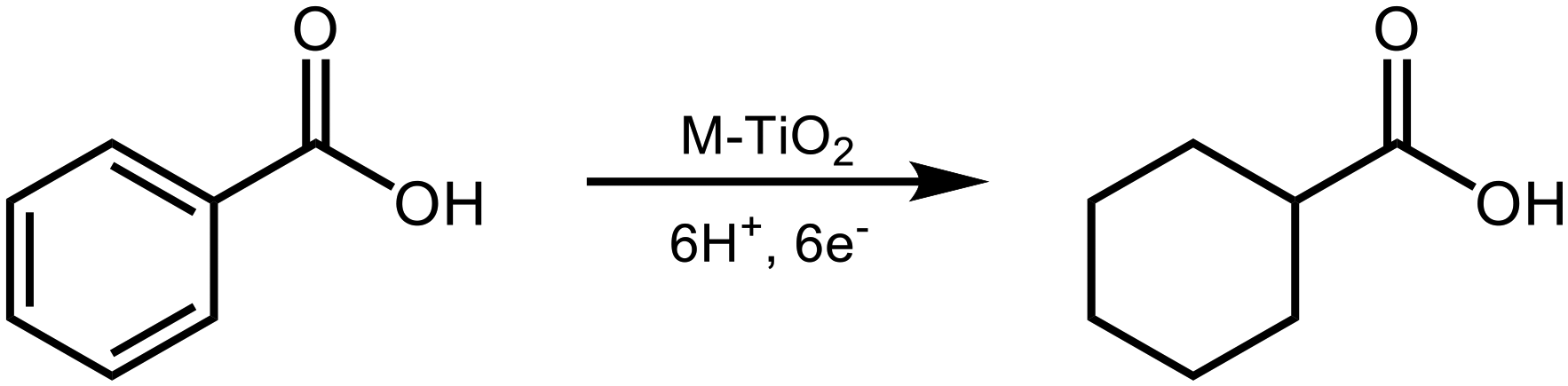
In this study, we chose BA as a model compound of aromatics having a carboxyl group and examined the photoinduced H2-free ring hydrogenation of BA over metal-loaded TiO2, in the presence of a hole scavenger that works as an electron and hydrogen source (Scheme 1).
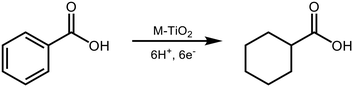 |
| | Scheme 1 Photoinduced ring hydrogenation of benzoic acid in an aqueous suspension of metal (M)-loaded TiO2 under H2-free conditions. | |
2. Experimental
2.1. Preparation of metal-loaded TiO2
Various metals as co-catalysts (Au, Ag, Cu, Pt and Rh) were loaded onto TiO2 (ST-01, Ishihara) by using the photodeposition method. TiO2 powder was suspended in 10 cm3 of an aqueous methanol solution (10 vol%) containing a metal source in a test tube. HAuCl4, AgNO3, CuCl2, PdCl2, H2PtCl6 and RhCl3 were used as metal sources. Each test tube was sealed with a rubber septum under argon (Ar) and then photoirradiated for 90 min at λ > 300 nm using a 400 W high-pressure mercury lamp (Eikosha, Osaka) with magnetic stirring in a water bath continuously kept at 298 K. The resulting powder was washed repeatedly with distilled water and dried for 2 h in vacuo. It was confirmed by atomic absorption spectrometry that the metals were almost quantitatively loaded onto TiO2.
2.2. Photocatalytic reaction
In a typical run, metal-loaded TiO2 (50 mg) was suspended in 5 cm3 of water containing BA (ca. 50 μmol) and OA (ca. 300 μmol) as a hole scavenger in a test tube, which was sealed with a rubber septum and then photoirradiated under Ar at 298 K with a high-pressure mercury lamp. The amounts of BA and CCA were determined using an FID-type gas chromatograph (GC-2025, Shimadzu) equipped with a DB-1 column. Chlorobenzene was used as an internal standard sample. Chlorobenzene (7 mm3) and diethyl ether (2 cm3) were added to the reaction solution (3 cm3). After the mixture had been stirred for 7 min, BA and CCA in the diethyl ether phase were analyzed. The amounts of BA and CCA were determined from the ratios of their peak areas to the peak area of chlorobenzene. The amount of H2 as the reduction product of protons (H+) and the amount of CO2 as the oxidation product of OA were determined using a TCD-type gas chromatograph (GC-8A, Shimadzu) equipped with an MS-5A column and a Porapak QS column. A multi-wavelength irradiation monochromator (MM-3, Bunkoukeiki Co., Ltd) was used to obtain the apparent quantum efficiency (AQE), and the light intensity was determined by using a spectroradiometer (USR-45D, Ushio Inc.).
2.3. Adsorption experiment
Rh-Loaded TiO2 (200 mg) was suspended in 5 cm3 of water containing BA (ca. 50 μmol) and OA (ca. 300 μmol), methanol containing BA or acetonitrile containing BA (ca. 50 μmol) and OA (ca. 300 μmol) in a test tube. The test tube was sealed with a rubber septum under Ar and the mixture was stirred in a water bath at 293 K for 20 hours in the dark. The amount of BA was determined in the same way as that for the photocatalytic reaction (section 2.2). When methanol and acetonitrile were used as solvents, no diethyl ether was added. The amount of OA in the liquid phase was determined using a Jasco PU-2800 Plus ion chromatograph equipped with an IC NI-424 column (Shodex, Japan). Before analysis using the ion chromatograph, the pH value of the solution containing OA was adjusted to 10 with an aqueous solution of sodium hydroxide.
2.4. Catalytic reaction under H2 conditions
Rh-Loaded TiO2 (50 mg) was placed in a test tube. The test tube was sealed with a rubber septum under H2. Into the sealed test tube, 5 cm3 of water containing BA (ca. 50 μmol) and OA (ca. 300 μmol), water containing BA (ca. 50 μmol) and methanol (ca. 300 μmol) or methanol containing BA (ca. 50 μmol) was injected with another needle being inserted into the rubber septum to keep the internal pressure at 1 atm. After injection of the reaction solution, the mixture was stirred in a water bath at 293 K in the dark. The amount of BA was determined in the same way as that for the photocatalytic reaction (section 2.2.).
3. Results and discussion
3.1. Effects of different metal co-catalysts
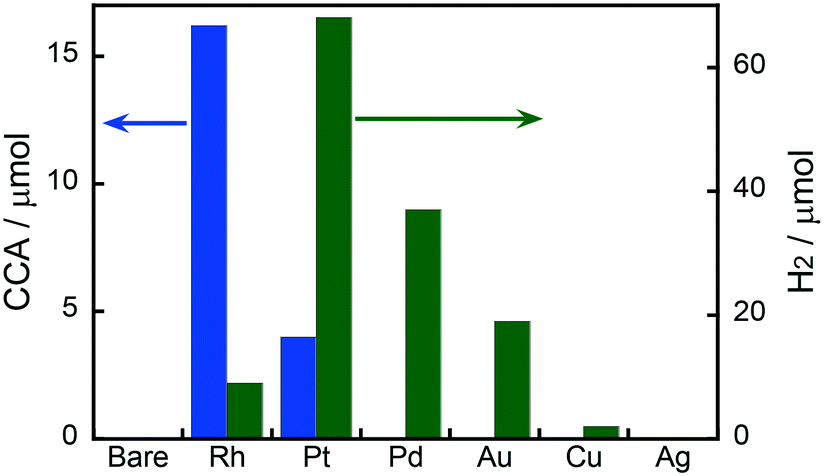
Fig. 1 shows the effects of different metal co-catalysts (1.0 wt%) on the yields of CCA produced in the photoinduced ring hydrogenation of BA in aqueous suspensions of metal-loaded TiO2 under irradiation with UV light for 15 min. No CCA was formed when bare TiO2 was used as the photocatalyst. The color of bare TiO2 became blue just after photoirradiation, indicating that some of the Ti4+ ions in TiO2 were reduced to Ti3+ by photogenerated electrons. When Au-, Ag-, Cu- and Pd-loaded TiO2 samples were used, no CCA was formed, indicating that bare TiO2 and these metals were inactive for ring hydrogenation of BA. In contrast to these metal-loaded TiO2 samples, when Pt and Rh were loaded onto TiO2, these photocatalysts, i.e., Pt–TiO2 and Rh–TiO2, hydrogenated BA, resulting in the production of CCA. Rh–TiO2 showed a much higher CCA yield than the other co-catalysts loaded onto TiO2 samples. In the case of Au, Cu, Pd, Pt and Rh, H2 was formed as the product of reduction of H+ by photogenerated electrons (2H+ + 2e− → H2), although the amount of H2 was dependent on the type of co-catalyst. Formation of CCA and/or H2 means that two types of reactions (BA hydrogenation and H2 formation) compete over metal-loaded TiO2 photocatalysts.
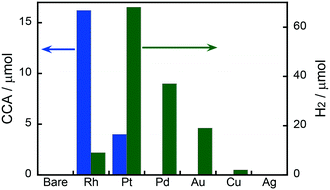 |
| | Fig. 1 Effects of different metal co-catalysts (1.0 wt%) on the yields of CCA and H2 produced in photoinduced ring hydrogenation of BA in aqueous suspensions of metal-loaded TiO2 for 15 min under irradiation with UV light from a high-pressure mercury lamp. | |
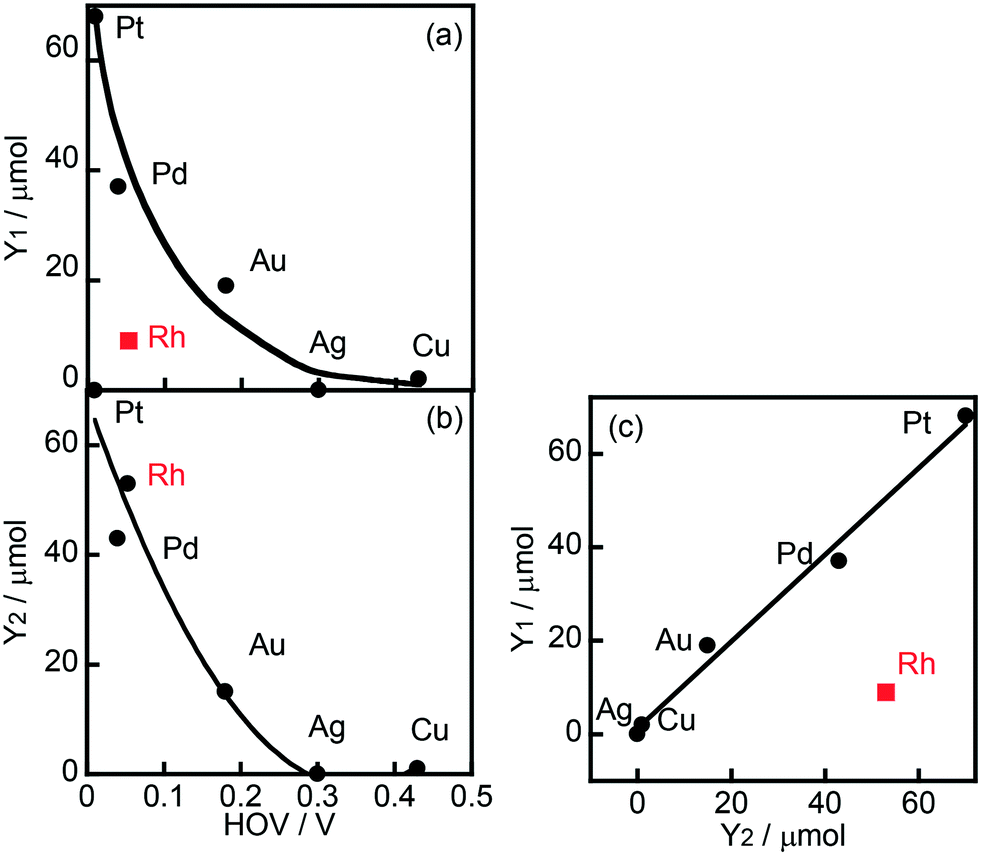
Fig. 2(a) shows the correlation between the yield of H2 produced in the photocatalytic ring hydrogenation of BA (Y1) and the hydrogen overvoltage (HOV) of metal electrodes. The use of an Rh co-catalyst resulted in a very low yield of H2 that was very different from the value predicted from HOV. For comparison, a photocatalytic reaction in the absence of BA was also examined and only reduction of H+ by e− occurred under this condition. The yield of H2 produced in the photocatalytic reaction in the absence of BA (Y2) was plotted against HOV (Fig. 2(b)), showing that the yield gradually decreased with the increase in HOV including Rh. The change in H2 yield without exception means that reduction of H+ by e− occurred on the metal co-catalyst and the reaction rate is determined by the HOV of the co-catalyst. Fig. 2(c) shows the correlation between the H2 yields in the presence and absence of BA. A linear correlation with a slope close to unity was observed for all of the co-catalysts except Rh. This correlation means that H2 formation, i.e., reduction of H+ by e−, occurs regardless of whether BA is present or not. In other words, these co-catalysts have no or little interaction with BA. On the other hand, H2 evolution over Rh greatly decreased when BA was present in the reaction system, suggesting that Rh interacts with BA and provides a new route with low activation energy for ring hydrogenation of BA to CCA.
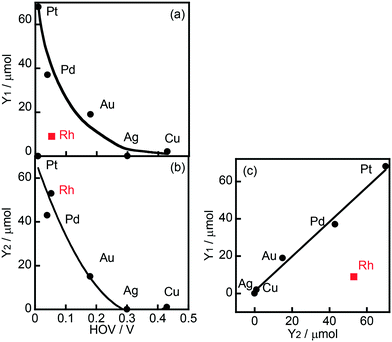 |
| | Fig. 2 (a) Correlation between hydrogen overvoltage (HOV) of metal electrodes and yield of H2 produced in the photocatalytic ring hydrogenation of BA in the presence of OA (Y1, Fig. 1), (b) correlation between HOV and yield of H2 produced under BA-free conditions (Y2), and (c) correlation between Y1 and Y2. | |
3.2. Photoinduced ring hydrogenation of BA in an aqueous suspension of Rh–TiO2
The effect of the amount of Rh loading on CCA yield was examined, and the results of photoirradiation for 15 min are shown in Fig. S1.† The highest CCA yield was obtained at 1.0 wt%, indicating that the photocatalytic performance was affected positively and negatively by the Rh particles loaded on TiO2. The number of reduction sites for ring hydrogenation increased and the light-shading effect also increased with the increase in Rh loading. Fig. S2† shows the TEM and HAADF STEM-EDX images, revealing that Rh nanoparticles were loaded onto TiO2.
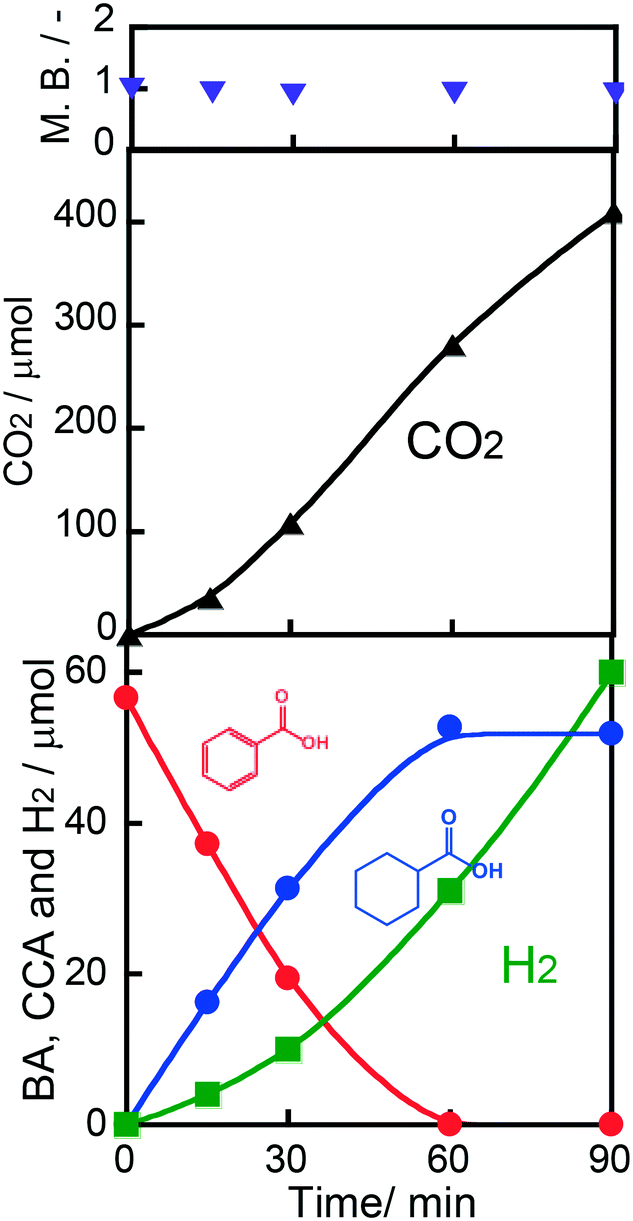
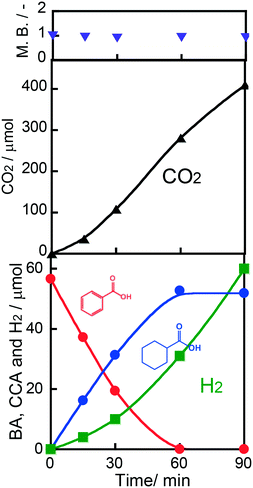
Fig. 3 shows time courses of BA remaining, CCA formed, and H2 and CO2 evolved in an aqueous suspension of 1.0 wt% Rh–TiO2 under irradiation with UV light. Just after irradiation, the amount of BA monotonously decreased, while CCA was formed. After 60 min, BA was almost completely consumed and CCA was obtained in a high yield (93%). To evaluate the selectivity to CCA and intermediates in the hydrogenation of BA, an indicator, i.e., material balance (MB), was calculated using eqn (1):
| | 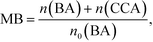 | (1) |
where
n(BA) and
n(CCA) are the amounts of BA and CCA during the photocatalytic reaction, respectively, and
n0(BA) is the amount of BA before the photocatalytic reaction. During the reaction, the value of MB was
ca. 0.9. However, no other product such as benzaldehyde, benzyl alcohol or toluene was detected in the reaction mixture. A small amount of a water-soluble by-product may be produced. As also shown in
Fig. 3, H
2 evolution was predominant under excessive photoirradiation after consumption of BA, indicating that active hydrogen species (AHS) over Rh were continuously formed by proton reduction over the Rh co-catalyst. We noted that the amount of CCA did not decrease after complete consumption of BA. Since an excess of OA used as a hole scavenger effectively consumed positive holes, the possibility of CCA re-oxidation and over-hydrogenation was eliminated.
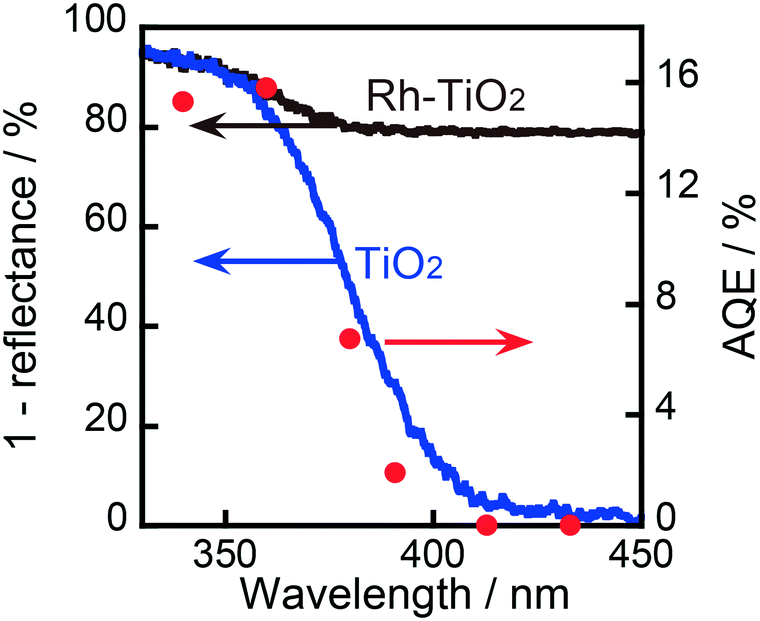
An action spectrum is a strong tool for determining whether a photoinduced process is the rate-determining step in the reaction observed. To obtain an action spectrum in this reaction system, ring hydrogenation of BA in aqueous suspensions of 1.0 wt% Rh–TiO2 was carried out at 298 K under irradiation with monochromated light from a Xe lamp. The apparent quantum efficiency (AQE) at each centered wavelength of light was calculated from the ratio of sextuple the amount of CCA formed and the amount of photons irradiated using eqn (2):
| |  | (2) |
 |
| | Fig. 3 Time courses of BA remaining, CCA formed, and H2 and CO2 evolved in an aqueous suspension of 1.0 wt% Rh–TiO2 under irradiation with UV light from a high-pressure mercury lamp. | |
As shown in Fig. 4, the AQE in CCA formation was in agreement with the absorption spectrum of TiO2. Therefore, it can be concluded that the ring hydrogenation of BA in an aqueous suspension was induced by the photoabsorption of TiO2. As also shown in Fig. 4, the AQE reached 16% at 360 nm, indicating that the photoinduced ring hydrogenation of BA to CCA occurred with high efficiency of photon utilization. The action spectrum of the evolution of H2 (by-product in this case) is shown in Fig. S3† and the value of AQE was determined to be 2% at 360 nm (eqn (S1)†). For comparison, H2 formation from an aqueous solution of OA in the absence of BA was examined and the action spectrum of the formation of H2 (main product, (COOH)2 → H2 + 2CO2) is also shown in Fig. S3.† The value of AQE was determined to be 10% at 360 nm which is smaller than that of AQE in CCA formation (16%, Fig. 4). These results support the idea that Rh interacts with BA and provides a new route with low activation energy for ring hydrogenation of BA to CCA.
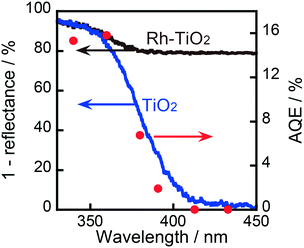 |
| | Fig. 4 Absorption spectrum (left axis) and action spectrum of TiO2 and 1.0 wt% Rh–TiO2 in the ring hydrogenation of BA (right axis). | |
3.3. Effects of reaction conditions on the ring hydrogenation of BA
The effects of various reaction conditions on the ring hydrogenation of BA to CCA in aqueous suspensions of Rh–TiO2 at 298 K are shown in Table 1. First, in order to examine the durability of the Rh–TiO2 photocatalyst in this reaction system, Rh–TiO2 was used repeatedly. After reaction for 60 min (entry 1), Rh–TiO2 particles were recovered by simple filtration from the reaction mixture and were re-used. Entries 2 and 3 show that the Rh–TiO2 photocatalysts were reusable at least twice without notable loss of activity.
Table 1 Effect of various reaction conditions on the hydrogenation of BA to CCA in aqueous suspensions of Rh–TiO2 at 298 K
| Entry |
Substrate |
Hole scavenger |
Solvent |
Photocatalyst |
UV light |
Time/min |
Atmosphere |
Yielda/μmol |
H2/μmol |
| Type |
Amount/μmol |
|
Yield of corresponding alicyclic compound.
Second use.
Third use.
Not detected.
Phthalic acid.
Amount of H2: 100 μmol.
|
| 1 |
BA |
OA |
300 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
53 |
31 |
| 2b |
BA |
OA |
300 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
52 |
34 |
| 3c |
BA |
OA |
300 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
53 |
32 |
| 4 |
BA |
OA |
300 |
H2O |
Rh–TiO2 |
On |
15 |
Ar |
16 |
4 |
| 5 |
BA |
OA |
250 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
48 |
42 |
| 6 |
BA |
OA |
200 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
47 |
36 |
| 7 |
BA |
OA |
150 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
32 |
33 |
| 8 |
BA |
OA |
50 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
10 |
15 |
| 9 |
BA |
OA |
300 |
H2O |
Rh–TiO2 |
(Dark) |
15 |
Ar |
n. d.d |
n. d. |
| 10 |
BA |
OA |
300 |
H2O |
— |
On |
15 |
Ar |
n. d. |
n. d. |
| 11 |
BA |
OA |
300 |
H2O |
— |
(Dark) |
15 |
Ar |
n. d. |
n. d. |
| 12 |
PAe |
OA |
300 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
49 |
37 |
| 13 |
Benzene |
OA |
300 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
n. d. |
42 |
| 14 |
Toluene |
OA |
300 |
H2O |
Rh–TiO2 |
On |
60 |
Ar |
n. d. |
47 |
| 15 |
BA |
Methanol |
300 |
H2O |
Rh–TiO2 |
On |
15 |
Ar |
1 |
7 |
| 16 |
BA |
Methanol |
— |
Methanol |
Rh–TiO2 |
On |
15 |
Ar |
n. d. |
62 |
| 17 |
BA |
Ethanol |
— |
Ethanol |
Rh–TiO2 |
On |
15 |
Ar |
n. d. |
85 |
| 18 |
BA |
2-Propanol |
— |
2-Propanol |
Rh–TiO2 |
On |
15 |
Ar |
n. d. |
41 |
| 19 |
BA |
OA |
300 |
Acetonitrile |
Rh–TiO2 |
On |
15 |
Ar |
n. d. |
22 |
| 20 |
BA |
OA |
300 |
H2O |
Rh–TiO2 |
(Dark) |
15 |
H2 |
18 |
— |
| 21 |
BA |
OA |
300 |
H2O |
Rh–TiO2 |
(Dark) |
15 |
H2/Arf |
2 |
— |
| 22 |
BA |
OA |
300 |
H2O |
— |
(Dark) |
15 |
H2 |
n. d. |
— |
| 23 |
BA |
Methanol |
300 |
H2O |
Rh–TiO2 |
(Dark) |
15 |
H2 |
10 |
— |
| 24 |
BA |
Methanol |
300 |
Methanol |
Rh–TiO2 |
(Dark) |
15 |
H2 |
2 |
— |
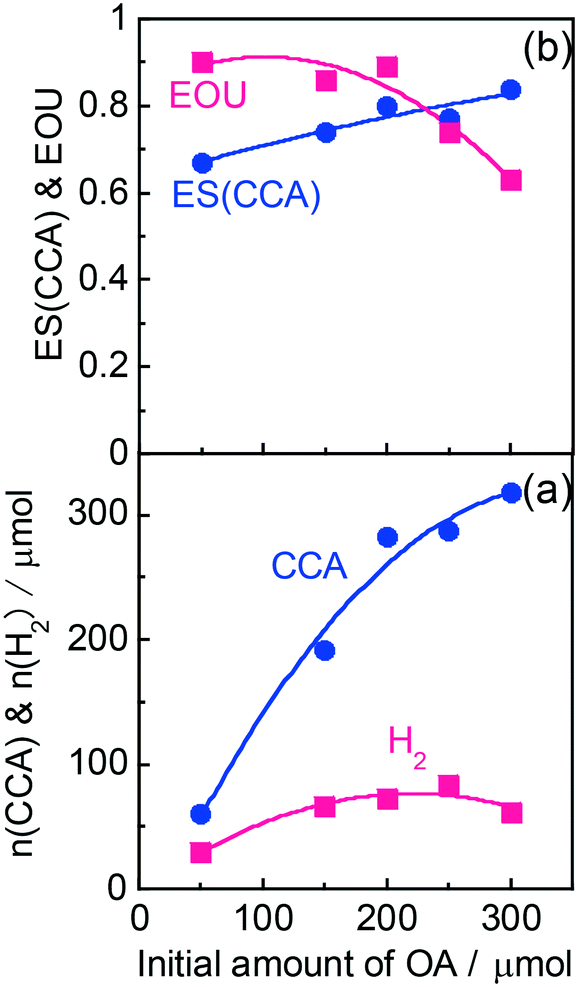
According to Scheme 1, the conversion of one molecule of BA to CCA requires six electrons, which corresponds to three molecules of OA (3(COOH)2 → 6CO2 + 6H+ + 6e−). Therefore, at least 150 μmol of OA is necessary for hydrogenation of 50 μmol of BA. The effects of different amounts of OA were also examined, and the results are shown in entries 5–8 in Table 1. High yields of CCA were obtained when the amount of OA was reduced to 250 and 200 μmol (entries 5 and 6). Under stoichiometric conditions (150 μmol), the yield of CCA decreased because a competitive reaction (H2 evolution) consuming photogenerated electrons simultaneously occurred as shown in Fig. 3 (entry 7). When the reaction was carried out with 50 μmol of OA, the CCA yield further decreased due to the shortage of the hole scavenger for this reaction. The effect of the amount of OA on this reaction is further discussed on the basis of utilization of photogenerated electrons. The amount of electrons used for the ring hydrogenation of BA (production of CCA, n(CCA)) increased with the increase in the amount of OA (Fig. 5(a)). Here, a new indicator, i.e., electron selectivity for CCA formation (ES(CCA)), was calculated according to eqn (3):
| | 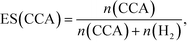 | (3) |
where
n(H
2) is the amount of electrons used for H
2 evolution (
Fig. 5(b)). The value of ES(CCA) increased with the increase in the amount of OA. However, the efficiency of OA utilization (EOU) determined using
eqn (4),
| | 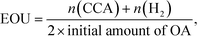 | (4) |
decreased, probably because unreacted OA remained after 60 min of reaction (also shown in
Fig. 5(b)). From the results of ES(CCA) and EOU, it can be concluded that the appropriate amount of OA is about 200 μmol, satisfying both high ES(CCA) (0.80) and EOU (0.89).
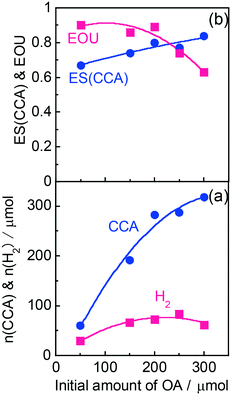 |
| | Fig. 5 Effect of the amount of OA on (a) the amount of electrons used for production of CCA and H2 (n(CCA) and n(H2), respectively) and (b) the electron selectivity for CCA (ES(CCA)) and efficiency of OA utilization (EOU). | |
Entry 4 shows the result after 15 min of photoirradiation. Entries 9–11 show the results of three blank reactions: 1) in the presence of Rh–TiO2 in the dark (thermocatalytic reaction over Rh–TiO2, entry 9), 2) in the absence of Rh–TiO2 with light irradiation (photochemical reaction, entry 10), and 3) in the absence of Rh–TiO2 in the dark (non-catalytic thermal reaction between BA and OA, entry 11). These results indicate that the Rh–TiO2 photocatalyst and photoirradiation are indispensable for the ring hydrogenation of BA.
The photoinduced ring hydrogenation of phthalic acid to 1,2-cyclohexanedicarboxylic acid was achieved over Rh–TiO2 (entry 12). However, benzene and toluene were not converted to the corresponding alicyclic compounds and only H2 was evolved (entries 13 and 14), indicating the high chemoselectivity of the Rh–TiO2 photocatalyst for ring hydrogenation of carboxylic acids.
Effects of the hole scavenger and solvent were also examined as shown in entries 15–19. When methanol was used as a hole scavenger instead of OA in an aqueous suspension of Rh–TiO2, the reaction rate for the ring hydrogenation of BA was very low (entry 15), probably due to the small amount of adsorption on the surface of TiO2. In other words, OA is an excellent hole scavenger for this reaction. The adsorption behavior of OA and BA will be discussed in the next section. No hydrogenation occurred in the photoirradiation of BA in an acetonitrile suspension of Rh–TiO2 in the presence of OA, although H2 was evolved (entry 19). This result means that the reaction medium is also an important factor to drive the ring hydrogenation of BA and that an aqueous medium and OA as the hole scavenger are an indispensable combination for efficient hydrogenation of BA. When alcohols are used as solvents, they act as hole scavengers in addition to the reaction medium. In previous studies, we found that alcohols worked well as solvents and hole scavengers for photocatalytic hydrogenation of unsaturated carbon–carbon bonds.24,26 However, in the case of ring hydrogenation in alcohols in the absence of OA, only H2 was produced with no CCA being formed (entries 16–18). These results support the importance of the combination of an aqueous medium and OA as the hole scavenger for the photocatalytic ring hydrogenation of BA.
3.4. Adsorption experiments
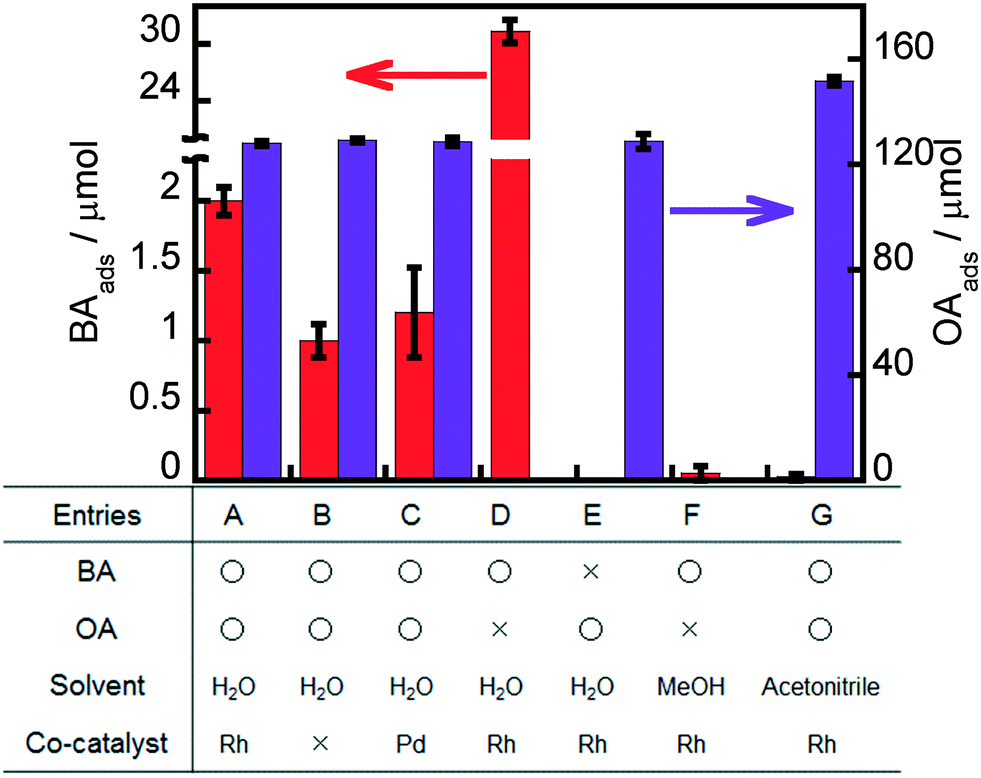
Some adsorption experiments were carried out to understand the effects of the metal co-catalyst, OA and solvent on BA adsorption to Rh–TiO2, and the results are shown in Fig. 6. A large amount of OA molecules was adsorbed on Rh–TiO2 in water regardless of the presence or absence of BA (entries A and E). A large concentration of OA molecules on the surface of Rh–TiO2 greatly contributes to hole scavenging under light irradiation, resulting in a high reaction rate of the ring hydrogenation of BA. On the other hand, the adsorption behavior of BA in water was affected by the presence of OA, and the amount of BA greatly decreased in the presence of OA (entries A and D). These results indicate that the adsorption of BA and OA on Rh–TiO2 was competitive and that OA adsorption was predominant under the conditions of a photocatalytic reaction. However, 2.0 μmol of BA was adsorbed on Rh–TiO2 even in the presence of OA. The balanced adsorption of OA and BA is one of the reasons for the effective ring hydrogenation of BA. The amounts of BA adsorbed on Rh–TiO2 in methanol and acetonitrile were below the detection limit (entries F and G). Therefore, the negligible adsorption of BA to Rh–TiO2 in methanol and acetonitrile can be attributed to predominant H2 formation without ring hydrogenation of BA in alcohol suspensions (entries 16–19 in Table 1). In order to discuss more the effect of Rh, the results for the adsorption of BA and OA on bare TiO2 and Pd-loaded TiO2 were also added as shown in entries B and C, indicating that the amount of BA adsorbed on Rh–TiO2 was larger than those of BA adsorbed on bare TiO2 and Pd–TiO2. The difference between the amounts of BA adsorbed on metal-loaded TiO2 and bare TiO2 can be regarded as the amount of BA adsorbed on the metal particles. Entries B and C show that only a small amount of BA was adsorbed on Pd particles. On the other hand, the amount of BA adsorbed on Rh particles was much larger (entry A). The difference in the adsorption properties of Rh and Pd to BA is one of the important reasons for the high efficiency of the Rh–TiO2 photocatalyst in the ring hydrogenation of BA.
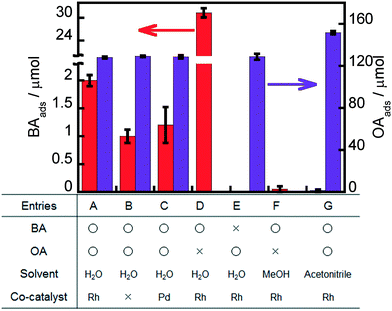 |
| | Fig. 6 Amounts of BA and/or OA adsorbed onto Rh–TiO2, Pd–TiO2 or TiO2 for 20 h in the dark. | |
3.5. Dark reactions under H2
Since H2 is formed as a by-product in the photocatalytic ring hydrogenation of BA in the presence of a hole scavenger, some dark reactions (thermal reactions) under H2 (1 atm) were examined at 298 K to understand this reaction (entries 20 and 22–24). In the dark, CCA was formed in an aqueous suspension of Rh–TiO2 (entry 20) but was not formed in the absence of Rh–TiO2 (entry 22), indicating that supported Rh particles were active for ring hydrogenation in the presence of H2. This process consists of 1) the formation of AHS on the surface of Rh particles by dissociative adsorption of H2 and 2) the insertion of AHS into the aromatic ring of BA. A thermal catalytic reaction under H2 also occurred in an aqueous suspension of Rh–TiO2 in the presence of methanol (entry 23). We noted that the reaction rate of the thermal reaction in the methanol suspension of Rh–TiO2 under H2 was very low (entry 24) compared with the reaction rate in aqueous media (entries 20 and 23). The adsorption experiment described in the previous section clearly showed that BA was hardly adsorbed on Rh–TiO2 in methanol. Therefore, the result of entry 24 can be explained by this adsorption effect. When the amount of H2 was reduced from 1 atm (corresponding to 1300 μmol) to 100 μmol, the dark reaction rate drastically decreased (entry 21). This result indicates that a large amount of H2 (high partial pressure of H2) is necessary for an efficient dark reaction.
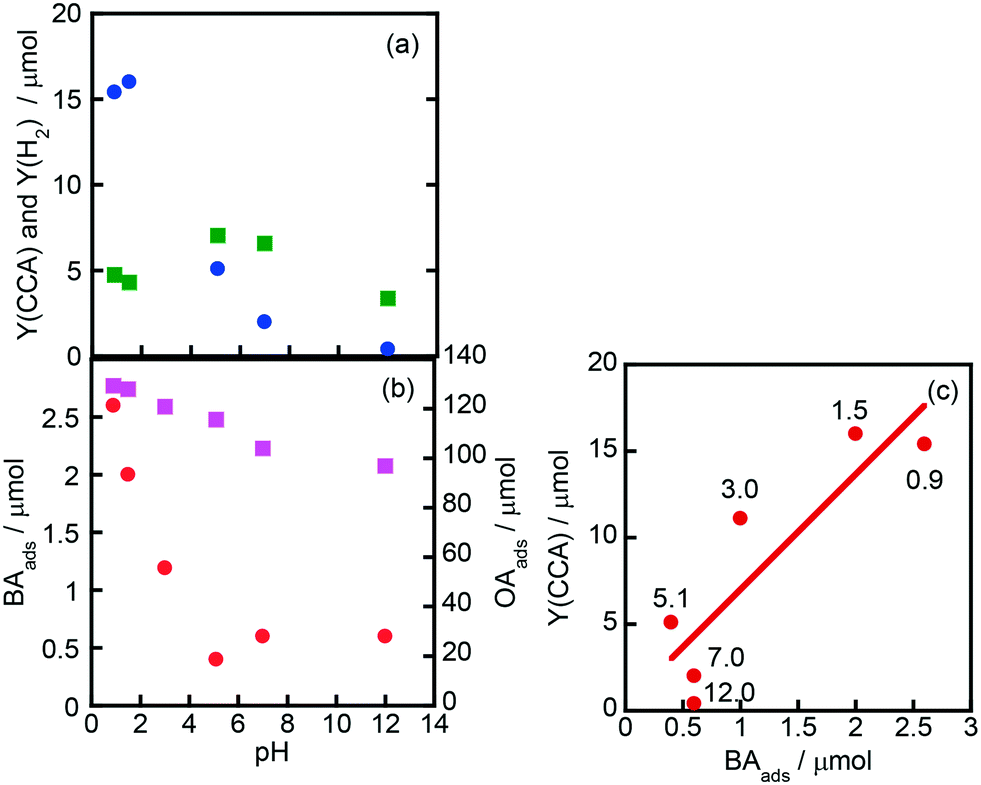
3.6. Effect of pH
Fig. 7(a) shows the effects of initial pH of the aqueous suspension of Rh–TiO2 on CCA yield (Y(CCA)) and H2 yield (Y(H2)), indicating that acidic conditions are appropriate for the ring hydrogenation of BA. On the other hand, H2 evolution was less affected by the pH of the suspension. To understand the effect of pH on Y(CCA), adsorption experiments were carried out under various pH conditions (Fig. 7(b)). The amount of OA adsorbed gradually decreased with the increase in pH under acidic conditions; however, the amount was still large under basic conditions. The amount of BA adsorbed was much smaller than that of OA as shown in Fig. 6, and the amount of BA adsorbed decreased with the increase in pH of the suspension. We noticed that the behavior of BA adsorption against pH was similar to that of Y(CCA), and a relatively clear correlation was observed between BA adsorption and Y(CCA), especially under acidic conditions (Fig. 7(c)). This plot indicates that the reaction rate of BA hydrogenation was dictated by the adsorption of BA, at least under acidic conditions.
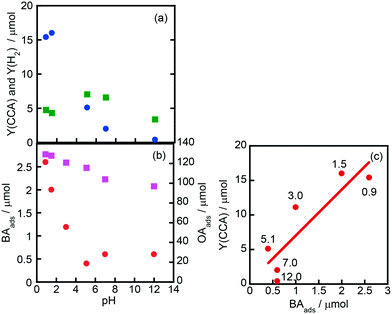 |
| | Fig. 7 Effects of initial pH of the aqueous suspension of Rh–TiO2 on (a) CCA yield (Y(CCA), circles) and H2 yield (Y(H2), squares) and (b) the amounts of BA (BAads, circles) and OA (OAads, squares) adsorbed and (c) the correlation between Y(CCA) and BAads. The reaction conditions except pH were the same as those in the experiment for which the results are shown in Fig. 1. The pH of the reaction solution was adjusted with an aqueous solution of sulfuric acid and sodium hydroxide. The values shown in (c) are pH values of the suspension. | |
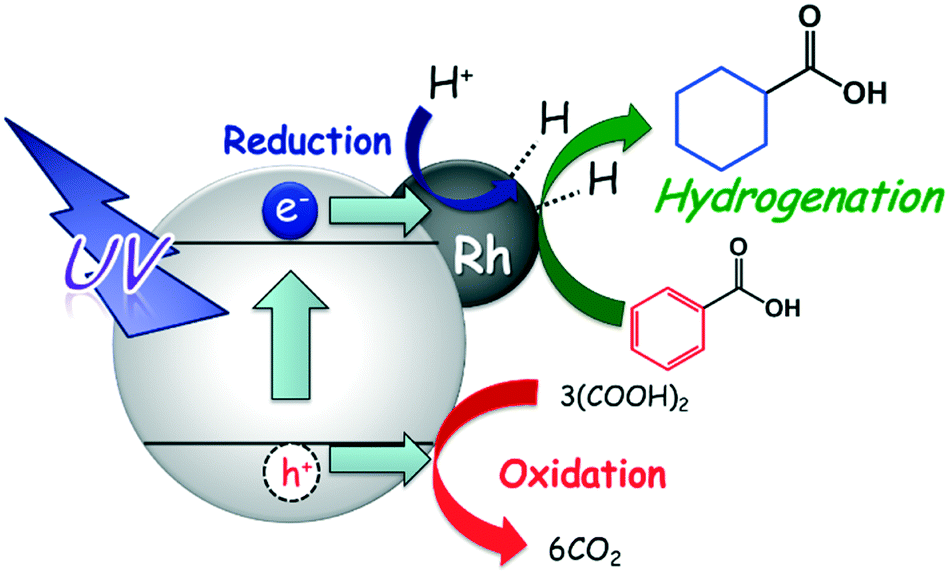
3.7. Expected reaction process
The expected reaction process of the photoinduced ring hydrogenation of BA to CCA over Rh–TiO2 is shown in Fig. 8: 1) by irradiation with UV light, photogenerated electrons (e−) and positive holes (h+) are formed in the conduction and valence bands of TiO2, and OA is oxidized by h+, 2) H+ is reduced by e−, resulting in the formation of AHS over Rh particles and 3) the aromatic ring of BA is successively hydrogenated by AHS over Rh particles, resulting in the formation of CCA. Since CCA was formed in the presence of H2 in the dark at 298 K, AHS would be essentially the same as that formed in the photocatalytic process. Self-coupling of AHS resulted in H2 evolution. The rate of H2 production increased after consumption of BA, which means that the active species formed on Rh are active for the ring hydrogenation of BA.
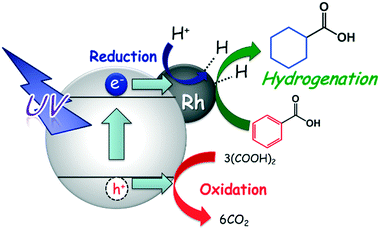 |
| | Fig. 8 Expected reaction process of the photocatalytic hydrogenation of BA to CCA over Rh–TiO2. | |
4. Conclusions
We examined the photoinduced ring hydrogenation of BA in an aqueous suspension of metal (M)-loaded TiO2 in the presence of OA under H2-free conditions. Among the metal-loaded TiO2 samples examined in this study, Rh–TiO2 showed distinctive photocatalytic activity for the largest production of CCA, and the apparent quantum efficiency reached 16% at 360 nm. A large amount of OA molecules was adsorbed on Rh–TiO2 in water, contributing to efficient hole scavenging under light irradiation, and the reaction rate for CCA formation was mainly determined by the amount of BA adsorbed. The ring hydrogenation of BA to CCA under the present conditions consisted of two processes: 1) the photocatalytic production of AHS and 2) the thermocatalytic hydrogenation of BA over Rh particles.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Numbers 17H03462, 15J11412, 16K18292 and 17H04967. This work was also supported by the MEXT-Supported Program for the Strategic Research Foundation at Private Universities 2014–2018, subsidy from MEXT and Kindai University. K. N. is grateful to the Japan Society for the Promotion of Science (JSPS) for a Research Fellowship for young scientists. A. T. is grateful for financial support from the Faculty of Science and Engineering, Kindai University.
Notes and references
-
P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed
 .
.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS .
- G. Palmisano, V. Augugliaro, M. Pagliarob and L. Palmisano, Chem. Commun., 2007, 3425–3437 RSC .
- G. Palmisano, E. Garcia-Lopez, G. Marci, V. Loddo, S. Yurdakal, V. Augugliaro and L. Palmisano, Chem. Commun., 2010, 46, 7074–7089 RSC .
- F. Mahdavi, T. C. Bruton and Y. Li, J. Org. Chem., 1993, 58, 744–746 CrossRef CAS .
- V. Brezova, A. Blažkova, I. Šurina and B. Havlinova, J. Photochem. Photobiol., A, 1997, 107, 233–237 CrossRef CAS .
- J. L. Ferry and W. H. Glaze, Langmuir, 1998, 14, 3551–3555 CrossRef CAS .
- O. V. Makarova, T. Rajh, M. C. Thurnauer, A. Martin, P. A. Kemme and D. Cropek, Environ. Sci. Technol., 2000, 34, 4797–4803 CrossRef CAS .
- V. Brezova, P. Tarabek, D. Dvoranova, A. Staško and S. Biskupič, J. Photochem. Photobiol., A, 2003, 155, 179–198 CrossRef CAS .
- H. Tada, T. Ishida, A. Takao and S. Ito, Langmuir, 2004, 20, 7898–7900 CrossRef CAS PubMed .
- H. Tada, T. Ishida, A. Takao, S. Ito, S. Mukhopadhyay, T. Akita, K. Tanaka and H. Kobayashi, ChemPhysChem, 2005, 6, 1537–1543 CrossRef CAS PubMed .
- S. O. Flores, O. R.-Bernij, M. A. Valenzuela, I. Cordova, R. Gomez and R. Gutierrez, Top. Catal., 2007, 44, 507–511 CrossRef CAS .
- H. Kominami, S. Iwasaki, T. Maeda, K. Imamura, K. Hashimoto, Y. Kera and B. Ohtani, Chem. Lett., 2009, 15, 410–411 CrossRef .
- K. Imamura, S. Iwasaki, T. Maeda, K. Hashimoto, B. Ohtani and H. Kominami, Phys. Chem. Chem. Phys., 2011, 13, 5114–5119 RSC .
- K. Imamura, T. Yoshikawa, K. Hashimoto and H. Kominami, Appl. Catal., B, 2013, 134–135, 193–197 CrossRef CAS .
- K. Imamura, K. Hashimoto and H. Kominami, Chem. Commun., 2012, 48, 4356–4358 RSC .
- M. Fukui, A. Tanaka, K. Hashimoto and H. Kominami, Chem. Lett., 2016, 45, 985–987 CrossRef CAS .
- K. Imamura, T. Yoshikawa, K. Nakanishi, K. Hashimoto and H. Kominami, Chem. Commun., 2013, 49, 10911–10913 RSC .
- K. Imamura, Y. Okubo, T. Ito, A. Tanaka, K. Hashimoto and H. Kominami, RSC Adv., 2014, 4, 19883–19886 RSC .
- H. Kominami, S. Yamamoto, K. Imamura, A. Tanaka and K. Hashimoto, Chem. Commun., 2013, 49, 4558–4560 RSC .
- H. Kominami, M. Higa, T. Nojima, T. Ito, K. Nakanishi, K. Hashimoto and K. Imamura, ChemCatChem, 2016, 8, 2019–2022 CrossRef CAS .
- B. S. Moore, H. Cho, R. Casati, E. Kennedy, K. A. Reynolds, U. Mocek, J. M. Beale and H. G. Floss, J. Am. Chem. Soc., 1993, 115, 5254–5266 CrossRef CAS .
- N. Perret, X. Wang, J. J. Delgado, G. Blanco, X. Chen, C. M. Olmos, S. Barnal and M. A. Keane, J. Catal., 2014, 317, 114–125 CrossRef CAS .
- T. Harada, S. Ikeda, Y. H. Ng, T. Sakata, H. Mori, T. Torimoto and M. Matsumura, Adv. Funct. Mater., 2008, 18, 2190–2196 CrossRef CAS .
- Z. Jiang, G. Lan, X. Liu, H. Tang and Y. Li, Catal. Sci. Technol., 2016, 6, 7259–7266 CAS .
- X. Wen, Y. Cao, X. Qiao, L. Niu, L. Huo and G. Bai, Catal. Sci. Technol., 2015, 5, 3281–3287 CAS .
- H. Wang and F. Zhao, Int. J. Mol. Sci., 2007, 8, 628–634 CrossRef CAS .
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1. See DOI: 10.1039/c7cy01929g |
| ‡ Present address: Department of Chemistry and Biotechnology, Faculty of Science and Technology, Kochi University, 2-5-1, Akebono, Kochi 780-8520, Japan. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Ryosuke
Yagi
a,
Ryosuke
Yagi