DOI:
10.1039/C7FO01210A
(Paper)
Food Funct., 2018,
9, 124-133
Preventive effects of taurine against D-galactose-induced cognitive dysfunction and brain damage
Received
5th August 2017
, Accepted 5th October 2017
First published on 5th October 2017
Abstract
Oxidative stress arising from life processes or environmental influences and its resultant cellular dysfunctions are major causes of neurodegenerative disorders. The objectives of this study were to investigate whether taurine (Tau) can prevent D-galactose-induced cognitive dysfunction and brain oxidative damage. Mice given with Tau supplementation (100 and 400 mg per kg BW per day) spent shorter (p < 0.05) time in searching target in D-galactose (100 mg per kg BW per day) treated mice in a water maze reference memory experiment. Moreover, Tau supplementation extended (p < 0.05) the searching period around the target quadrant in the probe test of the water maze, and neuronal degeneration and nucleus shrinkage in the hippocampus dentate gyrus area of D-galactose treated mice were observed to be attenuated. Tau also downregulated (p < 0.05) expression of the glial fibrillary acidic protein (Gfap) and of the cluster of differentiation marker Cd11b; meanwhile, it strengthened (p < 0.05) antioxidant capacity and lowered (p < 0.05) the accumulation of advanced glycation end-products (AGEs) in the brain. Therefore, Tau could be effective to ameliorate oxidative damage and inflammation in the brain, and apoptosis of brain cells, which further lessen the cognitive dysfunction.
Introduction
According to a statistical report from the United Nations (2015),1 there were 205 million older persons (over 60-years old) worldwide, of which China, India, and the United States of America together accounted for more than 100 million. In 50 years, the total population of older persons will triple (approximately 606 million). The number of countries with more than 10 million older persons in 2000 had increased to 12 compared to that in 1950. A similar or even higher estimate in an aged population was also conjectured in Taiwan. It has been reported that in the aged society the occurrences of dementia and disability dramatically increase. Daily routine duties are always restricted by cognitive damage. It has been believed that the oxidative stress produced from the environment and its resultant cellular dysfunctions are the major causes of neurodegenerative disorders.2,3 However, until now there has not been an effective therapy for neurodegenerative disorders. Hence, food supplements that can reverse or ameliorate cognitive impairments are urgently needed, especially with the increase in the aged population. Taurine (Tau) is a β-amino acid found in high concentrations in many mammalian tissues, such as the brain, liver, retina, myocardium, skeletal muscle, platelets, and leukocytes. Although Tau can be synthesized from methionine and cysteine, the rate of its biosynthesis is limited in mammals. On account of limited biosynthetic capacity in the liver (cats)4 and bile acid conjugation in mammals more generally,5,6 Tau supplementation through the diet is essential, and is met predominantly through eggs, meat, and seafood. Tau also acts as an antioxidant and plays a role in detoxification, membrane stabilization and osmoregulation, neuromodulation, and brain and retinal development in mammals. However, it is still unclear how Tau improves learning ability and memory, or what the protective mechanism is. D-Galactose is normally metabolized by D-galactokinase and galactose-1-phosphate uridyltransferase in animals.7 When a dose of D-galactose is beyond the capacities of these two enzymes, galactitol is formed in tissues as an alternative result, thus leading to osmotic stress and reactive oxygen species (ROS) generation via aldose/D-xylose reductase.7 Hence, an overconsumption of D-galactose accelerates the production of advanced glycation end products (AGEs) which accumulate in brain cells, thus initiating oxidative stress, reducing the levels of antioxidant enzymes, and causing a decline in neurogenesis, as well as promoting neuro-inflammation and neuronal apoptosis. Hence, a chronic exposure to D-galactose usually causes neurobehavioral changes and cognitive impairment in mice.2,8 Although it has been reported that Tau supplementation can decrease oxidative stress, cell apoptosis, and vacuolar changes in the brain,9 unfortunately observations on the direct (behavioral) and molecular changes in the brain of D-galactose-treated rats have so far been unavailable.
Based on the well-known membrane stabilization and osmoregulation, antioxidant, and anti-inflammatory effects of Tau, the objective of this study was to investigate if Tau could improve cognitive dysfunction and oxidative damage in the brains of D-galactose-treated mice. Hence, the major investigation of this study was divided into three parts: (1) learning and memory behavioral tests (Morris water maze: reference memory and probe tasks); (2) histological analysis of the hippocampus; and (3) possible molecular protective mechanism, i.e. antioxidant capacity, inflammatory responses, apoptosis, and activation of glial cells.
Materials and methods
Animal and diets
The animal use and protocol was reviewed and approved by the National Taiwan University Animal Care Committee (IACUC no.: 101-099). Forty-five male mice (imprinting control region, ICR) of 12-week age were purchased from the BioLASCO Taiwan Co., Ltd. (Taipei, Taiwan). Three mice with an ear tag (no. 1, 2, or 3) were housed in each cage in an animal room at 22 ± 2 °C with a 12/12 h light–dark cycle. Chow diets (Laboratory Rodent Diet 5001, PMI® Nutrition International/Purina Mills LLC, USA) and water were provided ad libitum for one week of acclimation. After that, mice were randomly divided into five groups (9 mice per group): (1) control (CON): 0.3 mL saline and 0.2 mL distilled water (oral gavage); (2) DG: 100 mg D-galactose per kg BW in 0.3 mL saline and 0.2 mL distilled water (oral gavage); (3) DG_LT: 100 mg D-galactose per kg BW in 0.3 mL saline and 100 mg taurine (Tau) per kg BW in 0.2 mL distilled water (oral gavage); (4) DG_HT: 100 mg D-galactose per kg BW in 0.3 mL saline and 400 mg Tau per kg BW in 0.2 mL distilled water (oral gavage); (5) DG_AG: 100 mg D-galactose per kg BW in 0.3 mL saline and 50 mg aminoguanidine hydrochloride per kg BW in 0.2 mL distilled water (oral gavage) for 9 weeks. It has been reported that a dose of 43 mg Tau per kg BW via an i.p. injection before or after ozone (0.7–0.8 ppm) exposure for 4 h can ameliorate the long-term memory of mature rats (540-day old) and lipid peroxidation in the hippocampus of old (900-day old) and in the striatum of young (47-day old) and old rats.10 With adjustment for body surface area,11 the i.p. dose for mice is 71.7 mg taurine per kg BW. Hence, oral-gavage doses of 100 and 400 mg taurine per kg BW were chosen in this study. Because aminoguanidine hydrochloride (AG) can ameliorate the formation of advanced glycation end-products (AGEs) in D-galactose-treated mice through interacting with 3-deoxyglucosone,12,13 the DG_AG group was used as a positive control group. D-Galactose was purchased from Bio-serv Co., Flemington, NJ, USA. Taurine was kindly supplied by Forever Chemical Co. Ltd (Tauyan City, Taiwan) with a purity of 99.6%. Saline and D-galactose were provided daily by a subcutaneous injection on the back. The body weight of each mouse was measured at the beginning and end of each experiment. Average daily feed and water intakes were expressed as obtaining daily feed and water intakes on per mouse daily basis.
Behavioral tests
The Morris water maze evaluation was applied as the behavioral test.2 In the 56th day of the experiment (end of the 8th week), all the mice per group were trained three times for searching for the visible platform during a 60 s period. The apparatus for the Morris water maze consisted of a circular water tank (100 cm in diameter; 80 cm in height), a stainless steel platform (4.3 cm in diameter; 16 cm in height), high-contrast spatial cues in the interior of the pool, and a computerized tracking system with a recording video camera and software (TM-02 Animal Video Behavior System, Diagnostic & Research Instruments Co., Ltd, Taipei, Taiwan). The tank was filled with water (23 ± 1 °C) to a depth of 17 cm. The water was rendered opaque by adding skimmed milk and a blue color reagent (Food color, Blue, Ever Style Foodstuff Industrial Co., Ltd, Taipei, Taiwan). The water maze was divided into four equal quadrants (I: right; II: target; III: left; IV: opposite). For the reference memory task, the platform was placed at the midpoint of the II quadrant. On the first day (57th day), each mouse was placed in the III quadrant to locate the platform. The mice were allowed to swim for 60 s to find the platform and then rest on it for 15 s. If mice did not reach the platform within 60 s, they were gently guided to the platform and also allowed to rest for 15 s. The escape latency, the period needed to reach the platform, was recorded and the swimming speed per mouse was also calculated. On the successive days (58th∼60th day), all mice were individually placed in the orders of the IV, I, and II quadrant to locate the platform, respectively. On the 61st day of experiment, the platform was removed from the water tank, and meanwhile, the probe trial, which evaluates the ability of remembering the place of platform, was carried out. In the probe trail, the mice were allowed to swim freely from the III quadrant for 60 s, as well as the total spent period and numbers of crossing over the target quadrant (II quadrant) within the given time were recorded.
Collection of sera and brains of experimental mice, as well as determination of serum biochemical values
After the behavioral tests (9th week), mice were sacrificed by CO2 asphyxiation. Blood samples were also collected by an intracardiac puncture after overnight fasting. Sera were separated from blood samples by centrifugation at 3000g for 10 min and then stored at −80 °C for further analyses. The brain tissues of each mouse were removed and weighed individually. Three brains from different mice per group (no. 1 mouse in each cage per group) were picked for the histological analysis, and the others were stored at −80 °C for further analyses. The serum biochemical parameters, i.e. triglyceride (TG), cholesterol (T-Cho), aspartate aminotransferase (AST), and alanine aminotransferase (ALT), were determined by using commercial enzymatic kits and a SPOTCHEM™ EZ SP-4430 automated analyzer (ARKRAY, Inc., Kyoto, Japan).
Histological analysis
Based on a previous study,3 hematoxylin and eosin (H&E) staining was used to show the morphologies in the dentate gyrus area in the mouse hippocampus. After the brains of three randomly picked mice (n = 3 per group) had been harvested, the hippocampus and cerebral cortex of the brain tissues located in the 6 mm width area from the interaural line toward the direction of the olfactory bulb were cut out using rodent brain matrices (Sunpoint Co., Taoyuan City, Taiwan). They were immediately fixed in a 10% neutral-buffered formalin solution for 24 h and then dehydrated in a graded alcohol series (30, 50, 70, 95, and 99.5%). After dehydration, the brain tissues were cleaned in xylene, and embedded in paraffin. The blocks were later sliced to 5 μm thick slices using a microtome, and de-waxed in xylene for 20 min and rehydrated in graded alcohol. The slices were stained with hematoxylin and eosin. Photomicrographs under H&E staining were obtained using a Zeiss Axioskop microscope AxioCam ERc 5s camera system with AxioVision Release 4.8.2 (06-2010) software (Carl Zeiss Microscopy, LLC, Thornwood, NY, USA).
Preparation of brain homogenates
Except for the three brains selected for histological analyses, the right sides of the brains (n = 6 per group) were homogenized for further analyses. The brain homogenate (10%, w/v) was made in phosphate buffered saline (PBS, pH 7.0, containing 0.25 M sucrose), and the supernatant was collected by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 min. The protein content in the supernatant was measured using a Bio-Rad protein assay kit (Cat#: 500-0006, Bio-Rad).
000g for 30 min. The protein content in the supernatant was measured using a Bio-Rad protein assay kit (Cat#: 500-0006, Bio-Rad).
Measurements of lipid peroxidation and antioxidative capacities in sera and brains
Thiobarbituric acid reactive substances (TBARS) and reduced glutathione (GSH) values in the sera and brains were analyzed according to the methods described by Chou et al. (2014).8 TBARS and reduced GSH values were calculated by taking the extinction coefficients of malondialdehyde (MDA) to be 1.56 × 105 M−1 cm−1 at 535 nm and TNB to be 1.36 × 104 M−1 cm−1 at 412 nm. The activities of superoxide dismutase (SOD) and catalase (CAT) in the brains were analyzed according to the methods described by Lin et al. (2017).14 Brain SOD was measured by the inhibitory effect of SOD on pyrogallol autoxidation. The SOD activities were calculated based on the inhibition of the standard curve. The calculation was shown as: Inhibition (%) = 1 − [(Δ420 nm of sample or standard/min)/(Δ420 nm of calibration/min)]. The unit of SOD activity was unit per mg protein. Regarding the measurement of CAT activity, a clear supernatant was collected from 90 μL of 10% brain filtrate mixed with 10 μL of 10% Triton X-100 after a centrifugation at 9000g for 10 min at 4 °C. A mixture was then made of 10 μL of clear supernatant and 990 μL of PBS (pH 7.0). A 500 μL mixture with 250 μL of H2O2 (30 mM) was made and then the difference in absorbance between 0 and 60 s (Δ240 nm) was recorded at 240 nm. The CAT activity was taken using the extinction coefficient (39.4 M−1 cm−1) of H2O2, and the specific activity of CAT was expressed in terms of unit per mg protein. Glutathione peroxidase (GPx) activity in the brain was assayed following the commercial manufacturer's directions (Randox Laboratories Ltd, Antrim, UK). Briefly, GPx catalyzes the oxidation of reduced GSH by cumene hydroperoxide. The oxidized GSH is converted to the reduced form in the presence of glutathione reductase and nicotinamide adenine dinucleotide phosphate (NADPH). In the reaction NADPH is oxidized to NADP+ simultaneously. The decrease in absorbance between 1 and 2 min readings at 340 nm (Δ340 nm) was then measured. Brain GPx activity was calculated by the following equation: GPx activity (unit per mg protein) = 8412 × (Δ340 nm)/protein concentration of brain homogenate.
Western blotting analysis of advanced glycation end-products (AGEs)
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate proteins according to molecular weight. After the protein separation, the proteins on the gel were transferred to a polyvinylidene difluoride (PVDF) membrane and the antigen–antibody reaction was performed to detect specific protein expression. The protocol is described briefly below. Brain homogenate and 5× protein dye were mixed in a ratio of 4:1 (v/v). Then the mixture was heated at 95 °C for 10 min. After cooling on ice for 10 min, the mixture was loaded onto SDS-PAGE comprising 8% running gel and 5% stacking gel. 80 volts were used to separate the proteins in the brain homogenates. After the electrophoresis, the proteins on the gel were transferred to a PVDF membrane (Immobilon®-P Transfer Membrane, Millipore Co., Billerica, MA, USA) by a semi-dry blotter (Trans-blot® SD cell, Bio-Rad Laboratories, Inc., New York City, NY, USA). After the transfer, the membrane was blocked by placing in 2.5% bovine serum albumin (Bio-Rad Laboratories Inc., Hercules, CA, USA) for 30 min. The specific primary antibody [β-actin (Cat. no.: MAB1501, Merck Millipore Co., Darmstadt, Germany) or polyclonal AGEs (Cat. no.: bs-1158R, Bioss Inc., Woburn, MA, USA)] was added into the PVDF membrane, which was then kept at 4 °C overnight. Washing buffer was then used to wash the membrane 4 times, for 5 min each time. After washing, a secondary antibody [Goat anti-mouse IgG (H + L) secondary antibody, horseradish peroxidase (HRP) conjugate, Thermo Fisher Scientific Taiwan Co., Ltd, Taipei, Taiwan] was added to the membrane, which was then kept at room temperature for 2 h. The membrane was then washed as previously described. Because the anti-AGE antibody was suitable for the detection of different AGE proteins in tissues, as indicated by the protocol, several protein bands for AGE were revealed.15 Protein bands were visualized with an enhanced chemiluminescence (ECL) kit (Immobilon™ Western, Millipore Co., Billerica, MA, USA) under a MultiGel-21 (TOP BIO Co., New Taipei City, Taiwan). Image J. software (National Institutes of Health, Bethesda, MD, USA) was used to quantify the optical density of protein bands, with the value of the β-actin band as a reference.
Messenger ribonucleic acid (mRNA) expression analyses
Except for the three brains selected for histological analyses, the left sides of the brains (n = 6 per group) was stored in RNAlater RNA stabilization reagent (Qiagen Taiwan Co., Taipei, Taiwan) at 4 °C overnight and then kept at −80 °C. The mRNA in brain tissues was extracted by using a commercial kit (Trisure™, Bioline, London, UK), and the complementary deoxyribonucleic acid (cDNA) was synthesized from mRNA by a reverse transcription reaction according to the manufacturer's protocol provided by GoScript™ Reverse Transcriptase (Promega Co., Madison, Fitchburg, WI, USA). According to the principle of quantitative real-time polymerase chain reaction (PCR), all the primers were designed for the specific gene sequences in order to amplify the specific cDNA fragments as: glyceraldehyde 3-phosphate dehydrogenase (Gapdh, GenBank no.: NM_001289726.1) F: 5′-AACCTGCCAAGTATGATGA-3′; R: 5′-GGAGTTGCTGTTGAAGTC-3′; receptor for advanced glycation end-products (Rage, GenBank no.: NM_007425.3) F: 5′-CAGCATCAGGGTCACAGAAAC-3′; R: 5′-CGTTTTCGCCACAGGATAGC-3′; nicotinamide adenine dinucleotide phosphate-oxidase 2 (Nox2, GenBank no.: FJ168469.1) F: 5′-AGCTATGAGGTGGTGATGTTAGTGG-3′; R: 5′-CACAATATTTGTACCAGACAGACTTGAG-3′; nuclear factor kappa B (NFκB, GenBank no.: NM_008689.2) F: 5′-TTCACCATGGCAGACGATGA-3′; R: 5′-TTGAAGGTATGGGCCATCTGT-3′; interleukin 1 beta (Il-1β, GenBank no.: NM_008361.3) F: 5′-GCCACCTTTTGACAGTGATG-3′; R: 5′-AGTGATACTGCCTGCCTGAA-3′; interleukin 6 (Il-6, GenBank no.: NM_031168.1) F: 5′-CGGCCTTCCCTACTTCACAA-3′; R: 5′-GCCATTGCACAACTCTTTTCTC-3′; tumor necrosis factor alpha (Tnfα, GenBank no.: NM_013693.3) F: 5′-CCCCACTCTGACCCCTTTAC-3′; R: 5′-ACTGTCCCAGCATCTTGTGT-3′; cyclooxygenase 2 (Cox2, GenBank no.: NM_011198.3) F: 5′-TGGGTTCACCCGAGGACTG-3′; R: 5′-GGGGATACACCTCTCCACCAA-3′; B-cell lymphoma 2 (Bcl2, GenBank no.: NM_009741.4) F: 5′-TGGGATGCCTTTGTGGAACT-3′; R: 5′-TTGTTTGGGGCAGGTTTGTC-3′; B-cell lymphoma-extra large (BclxL, GenBank no.: NM_009743.4) F: 5′-TGACCACCTAGAGCCTTGGA-3′; R: 5′-AAGAGTGAGCCCAGCAGAAC-3′; BCL2-associated X protein (Bax, GenBank no.: NM_007527.3) F: 5′-TTTGCTACAGGGTTTCATCCA-3′; R: 5′-CCCAGTTGAAGTTGCCATCAG-3′; caspase 3 (Casp3, GenBank no.: NM_009810.3) F: 5′-GGAGCTTGGAACGCTAAGAAA-3′; R: 5′-TGTCCAGATAGATCCCAGAGT-3′; glial fibrillary acidic protein (Gfap, GenBank no.: NM_010277.3) F: 5′-GGCGAAGAAAACCGCATCA-3′; R: 5′-CCTCACCATCCCGCATCTC-3′; cluster of differentiation molecule 11B (Cd11b, GenBank no.: NM_008401.2) F: 5′-AATTGAGGGCACGCAGACA-3′; R: 5′-CAGTCAAAGCTCCCCACAGA-3′. The sizes of the reaction products are as follows: Gapdh, 119 bp; Rage, 150 bp; Nox2, 88 bp; NFκB, 135 bp; Il-1β, 165 bp; Il-6, 149 bp; Tnfα, 157 bp; Cox2, 137 bp; Bcl2, 162 bp; BclxL, 165 bp; Bax, 248 bp; Casp3, 131 bp; Gfap, 145 bp; Cd11b, 120 bp. In carrying out quantitative real time PCR, 2 μL of cDNA (total 50 ng per reaction), 5 μL of SYBR Green (Fast SYBR® Green Master Mix (2×), Applied Biosystems, Molecular Probes Inc., Eugene, OR, USA), 2.4 μL of RNase free water, and 0.6 μL of the target primer (forward and reverse primer) were mixed together and the fluorescence signal was detected by using a StepOne Real time PCR system (Applied Biosystems, Foster, CA, USA). Gapdh was used as an internal control, and the values of all the genes in the DG, DG_LT, DG_HT, and DG_AG mice were expressed relative to the average value of CON mice, which was set at 1.0.
Statistical analysis
The experiment was conducted using a completely randomized design. When a significant difference (p < 0.05) among groups was detected by the one-way analysis of variance (ANOVA), differences among treatments were tested using the Least Significant Difference (LSD) test. All statistical analyses of the data were performed using Statistical Analysis Software (SAS Institute Inc., Cary, NC, USA, 2002).
Results
Effects of taurine (Tau) on growth performance and serum biochemical values
There was not (p > 0.05) a difference in initial body weight among the groups, but a high dose of Tau supplementation (DG_HT group) resulted in a lower (p < 0.05) final body weight. During the experimental period, the calculated daily food and water intakes among the groups did not (p < 0.05) alter, and neither did the brain weight (Table 1). Regarding the serum biochemical values, D-galactose with Tau treatments did not (p > 0.05) influence T-Cho, AST, and ALT values, but it lowered (p < 0.05) TG values, especially in the DG_HT group compared to other groups (Table 1).
Table 1 Growth performance, serum biochemical parameters, serum and brain TBARS, and serum and brain levels of reduced GSH in experimental mice
| |
CONb |
DGb |
DG_LTb |
DG_HTb |
DG_AGb |
|
Values are given as mean ± SEM (n = 9, except food intake and water intake, n = 3, as well as liver TBARS and reduced GSH levels, n = 6). Mean values within each test parameter without letters in common (a–c) indicate a significant difference (p < 0.05).
CON: 0.3 ml saline and 0.2 ml distilled water; DG: 100 mg D-galactose per kg BW in 0.3 ml saline and distilled water; DG_LT: 100 mg D-galactose per kg BW and 100 mg taurine per kg BW; DG_HT: 100 mg D-galactose per kg BW and 400 mg taurine per kg BW; DG_AG: 100 mg D-galactose per kg BW and 50 mg aminoguanidine per kg BW.
|
|
Performance
|
| Initial body weight (g)a |
39.79 + 0.31a |
39.53 + 0.78a |
39.30 + 0.40a |
39.01 + 0.56a |
39.64 + 0.73a |
| Final body weight (g)a |
41.90 + 0.58a |
40.78 + 0.55a |
41.23 + 0.63a |
37.04 + 1.47b |
42.42 + 1.07a |
| Food intake (g per mouse per day)a |
6.33 + 0.11a |
6.77 + 0.35a |
6.92 + 0.68a |
6.08 + 0.35a |
6.26 + 0.41a |
| Water intake (ml per mouse per day)a |
6.81 + 0.43a |
7.03 + 0.30a |
6.75 + 0.39a |
6.17 + 0.42a |
6.58 + 0.83a |
| Brain (g)a |
0.55 ± 0.01a |
0.55 ± 0.01a |
0.55 ± 0.01a |
0.53 ± 0.01a |
0.54 ± 0.01a |
|
Serum biochemical value
|
| TG (mg dL−1)a |
111.00 ± 5.25ab |
112.44 ± 7.74ab |
98.78 ± 6.11b |
68.56 ± 5.89c |
123.11 ± 5.05a |
| T-Cho (mg dL−1)a |
145.78 ± 6.39a |
149.44 ± 7.99a |
141.78 ± 1.79a |
125.22 ± 4.50a |
142.44 ± 6.48a |
| AST (IU L−1)a |
54.33 ± 2.81a |
57.67 ± 6.44a |
52.56 ± 2.52a |
67.22 ± 6.80a |
51.11 ± 1.96a |
| ALT (IU L−1)a |
44.22 ± 5.67a |
44.78 ± 3.38a |
37.78 ± 2.61a |
52.89 ± 6.88a |
41.44 ± 8.04a |
|
Serum TBARS and reduced GSH levels
|
| TBARS (nmol MDA eq. per mL serum)a |
52.42 ± 1.76b |
62.21 ± 3.64a |
56.18 ± 1.20ab |
35.96 ± 4.76c |
53.59 ± 2.83ab |
| Reduced GSH (nmol mL−1 serum)a |
238.58 ± 11.89a |
203.98 ± 6.88b |
248.21 ± 9.17a |
246.23 ± 13.30a |
256.01 ± 14.83a |
|
Brain TBARS and reduced GSH levels
|
| TBARS (nmol MDA eq. per mg protein)a |
1.11 ± 0.20b |
2.75 ± 0.43a |
1.81 ± 0.38ab |
1.26 ± 0.20b |
1.79 ± 0.47ab |
| Reduced GSH (nmol mg−1 protein)a |
231.58 ± 11.65a |
201.71 ± 2.53b |
220.73 ± 6.87ab |
242.03 ± 6.05a |
241.33 ± 9.27a |
Effects of taurine on learning and memory abilities
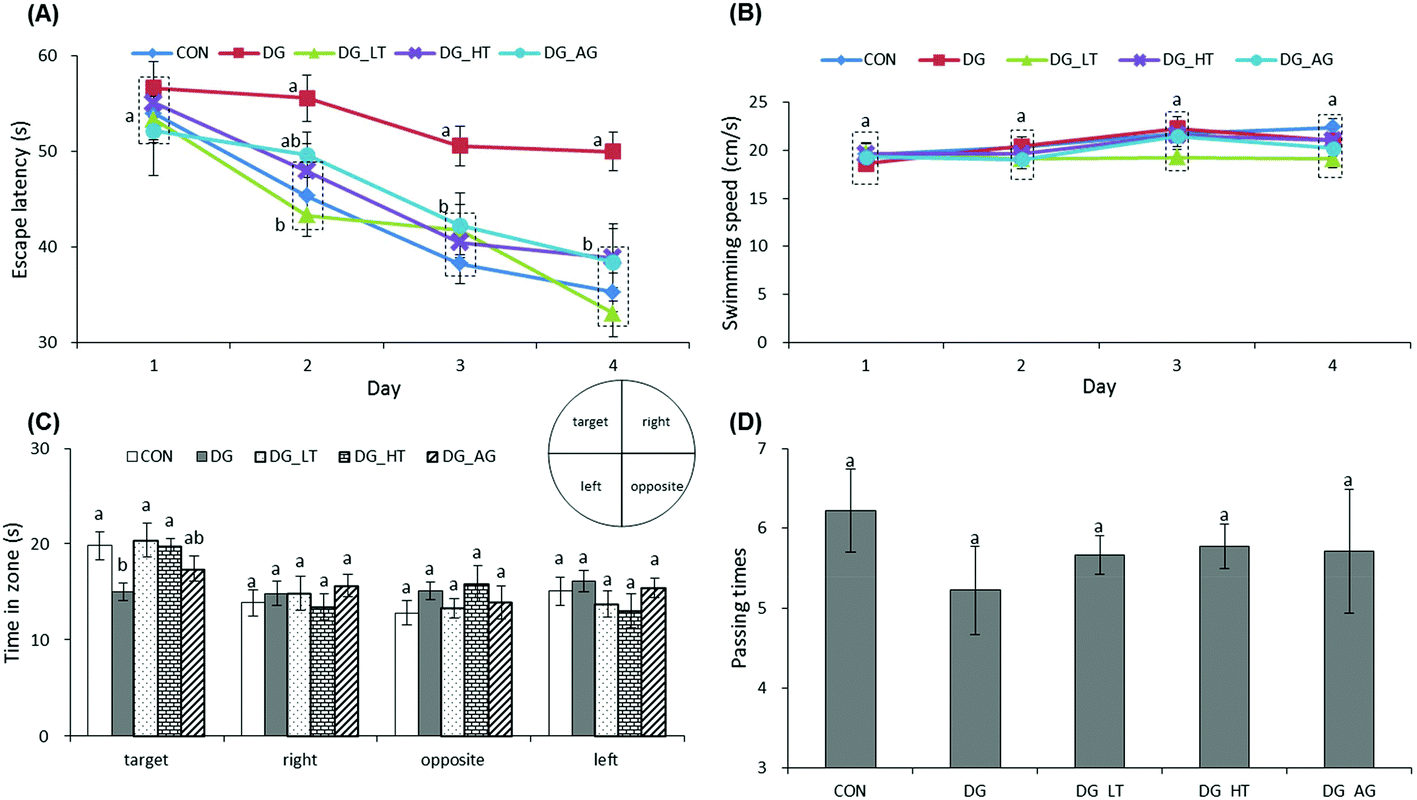
The escape latency and swimming speed of mice in reference memory tests are illustrated in Fig. 1A and B. On the first day (57th day) of the Morris water maze evaluation, no (p > 0.05) differences in escape latency were observed among all the groups. After the second day (58th day) of the Morris water maze evaluation, CON and Tau supplemented groups (DG_LT and DG_HT) had significantly shortened (p < 0.05) escape latencies compared to the DG group when the effect (p < 0.05) of aminoguanidine was observed after the 3rd-day test. The swimming speeds during the 4-day test period were also not (p > 0.05) different among groups. In the probe trial, although there were no (p > 0.05) differences in the period spent in the right, opposite, or left quadrant among groups (Fig. 1C), shorter (p < 0.05) periods spent in the target quadrant were recorded for the DG group compared to the CON group, whereas Tau supplementation increased (p < 0.05) that period. In addition, among the groups, only a small tendency toward fewer numbers crossing over the target quadrant in the DG group was observed (Fig. 1D).
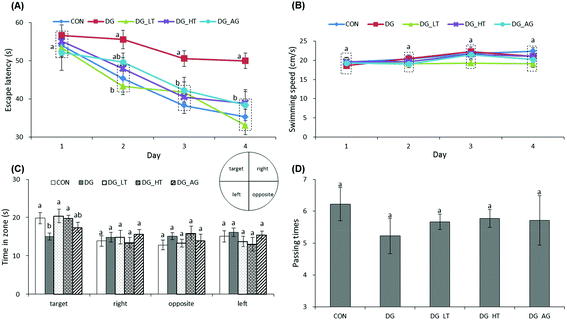 |
| | Fig. 1 Effect of taurine on learning and memory impairments of D-galactose-treated mice. Data are given as mean ± SEM (n = 9). (A) Mean of escape latency in the hidden platform test. Mice were trained with 4 trials per day for 4 days. (B) Mean of swimming speed during 4 training days. (C) In the probe trial on the fifth day. Comparison of the period spent in quadrants on day 5 (the hidden platform was previously placed in the target quadrant). (D) Comparison of numbers crossing over the target quadrant on day 5. Data points within the same testing day, data bars within the same quadrant, or data bars without letters in common (a–b) indicate a significant difference (p < 0.05). | |
Effects of taurine on histological observations of dentate gyrus and advanced glycation end product (AGE) levels in brains
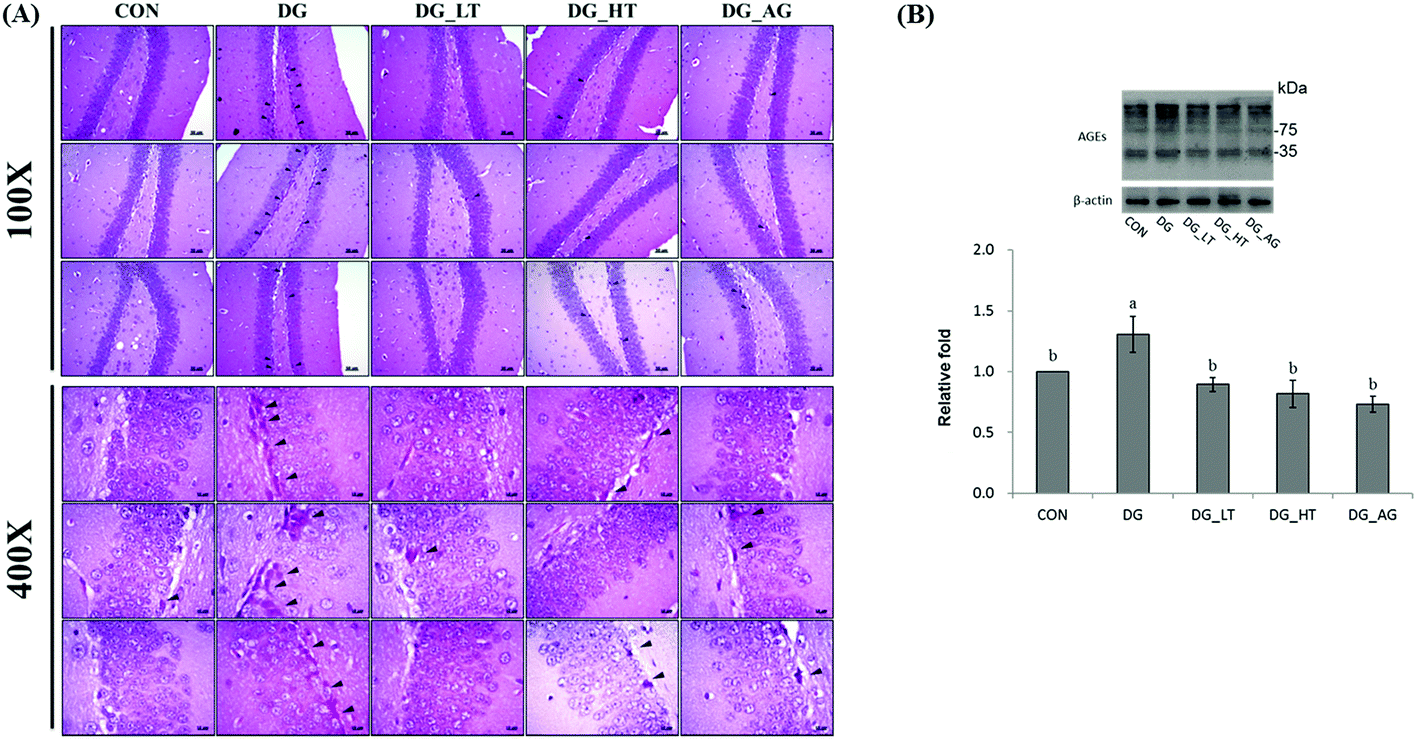
Well-organized dentate gyrus in the hippocampus with intact brain plates were observed in the CON group, but intensely contracted basophilic neuron bodies (dark arrow heads) occurred in the dentate gyrus of mice treated with D-galactose (Fig. 2A). Meanwhile, Tau or aminoguanidine supplementation clearly reduced these observations in D-galactose-treated mice. Because the anti-AGE antibody was suitable for the detection of different AGE products in tissues, as the protocol indicated, several protein bands for AGE in brains were displayed and quantified together. The level of AGE proteins in the right side of the brains of D-galactose-treated mice without Tau or aminoguanidine (DG group) was obviously higher (p < 0.05) than for the CON group, whereas Tau or aminoguanidine co-treatment lowered the AGE protein expression, even when this was not (p > 0.05) different from that of the CON group (Fig. 2B).
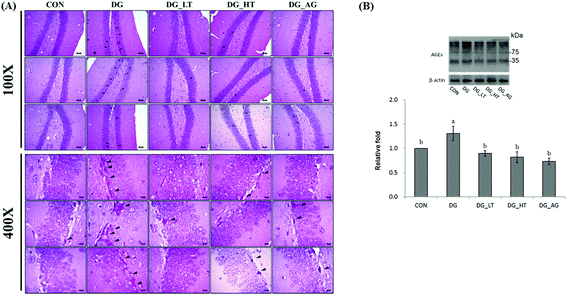 |
| | Fig. 2 (A) Morphological features of the dentate-gyrus area in mouse hippocampus from each group were evaluated by hematoxylin and eosin (HE) staining. The presence of dark neurons is characterized by contracted and intensely basophilic neuron necrosis bodies (black arrowheads) (magnification: 10 × 10 and 10 × 40); (B) protein levels of advanced glycation end products (AGEs) in brains of the experimental mice. Data are given as mean ± SEM (n = 6). Data bars without letters in common (a–b) indicate a significant difference (p < 0.05). | |
Effects of taurine on serum and brain antioxidant capacities
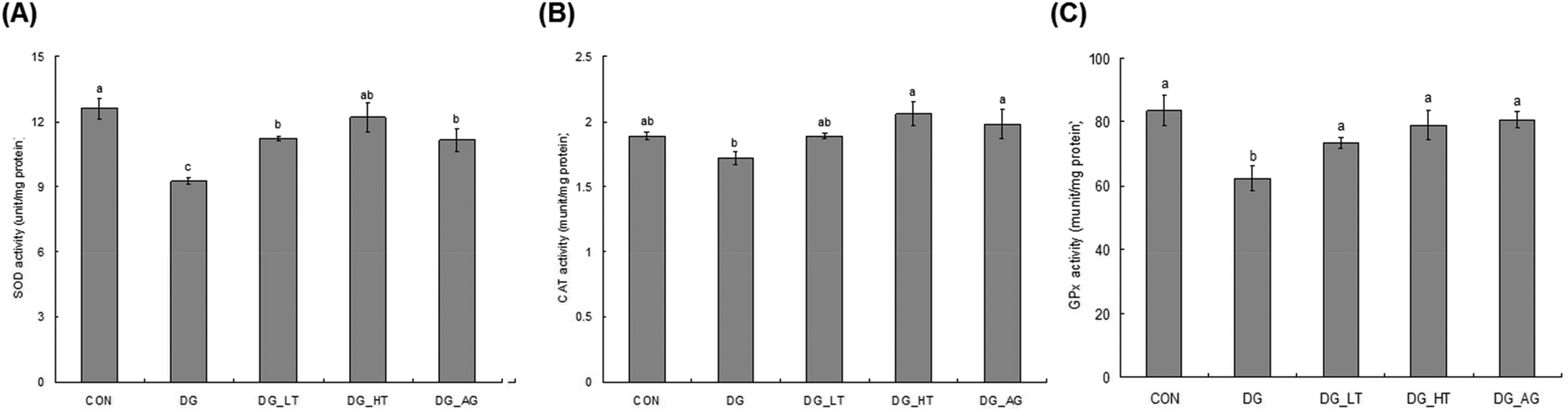
D-Galactose treatment resulted in a significant lowering of reduced GSH levels and an increase of TBARS levels in sera and brains compared to those of mice without D-galactose (CON group), respectively (p < 0.05) (Table 1). Meanwhile, Tau cotreatment, especially a high dosage of Tau, apparently lowed (p < 0.05) TBARS levels and increased (p < 0.05) reduced GSH levels in sera and brains. In addition, activities of brain antioxidant enzymes (SOD, CAT, and GPx) were reduced (p < 0.05) in D-galactose-treated mice; however, these activities were not diminished (p < 0.05) when there was co-treatment with Tau or aminoguanidine (Fig. 3).
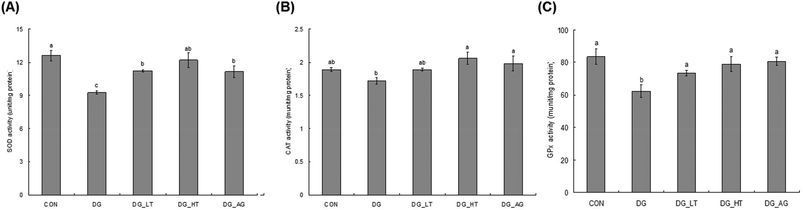 |
| | Fig. 3 Brain antioxidant enzymatic activities: (A) superoxide dismutase (SOD), (B) catalase (CAT), and (C) glutathione peroxidase (GPx) of experimental mice. * Data are given as mean ± SEM (n = 6). Data bars without letters in common (a–b) indicate a significant difference (p < 0.05). | |
Effect of taurine on brain inflammation and apoptosis
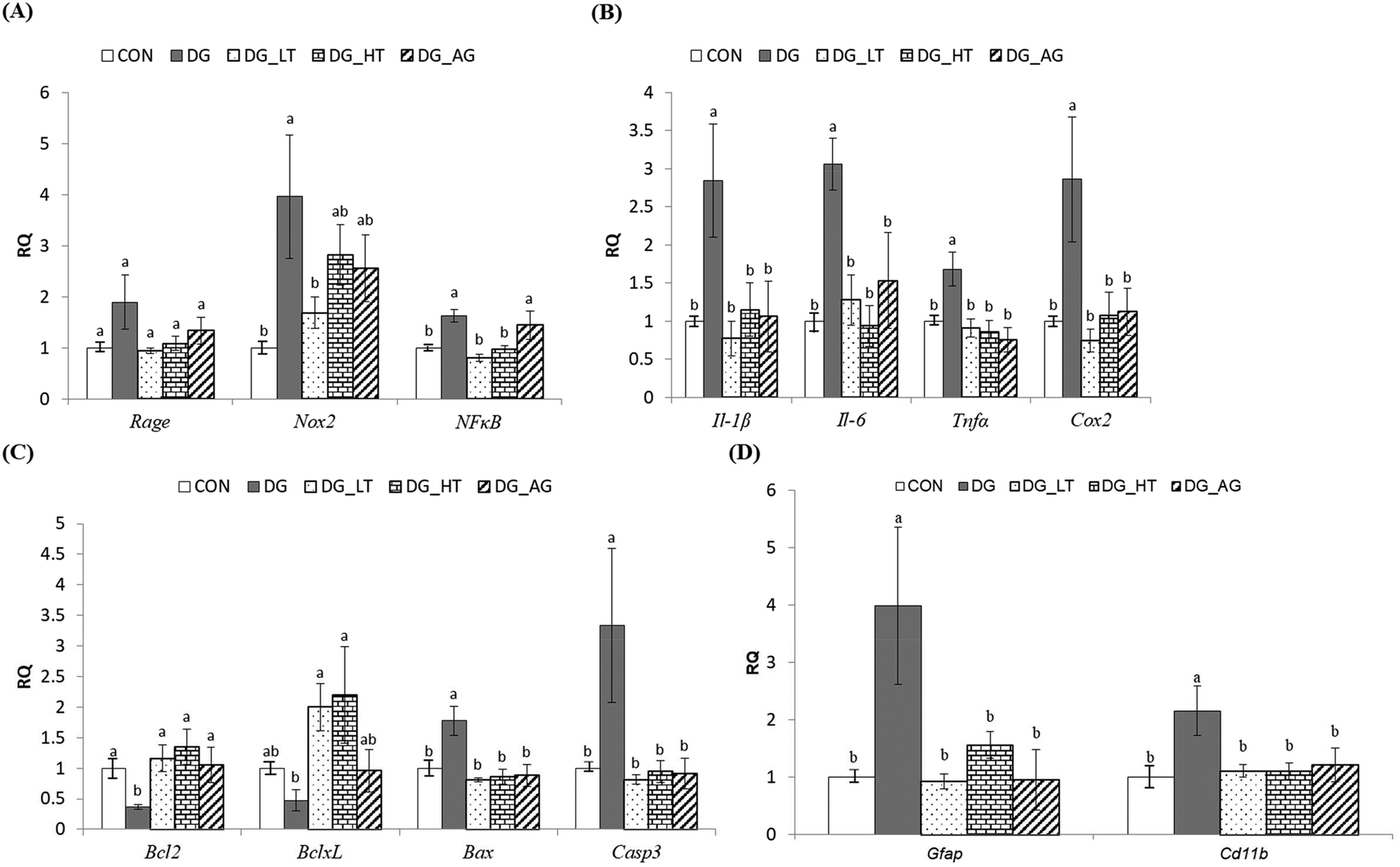
In order to clarify the protective mechanism of Tau on D-galactose-induced oxidative damage in the brain, the mRNA expression of genes involved in inflammation, apoptosis, and the activation of glial cells in brains was investigated (Fig. 4). Although there was only a tendency toward a high Rage expression in brains of the DG group compared to the other groups, the upregulated (p < 0.05) expression of Nox2 and NFκB in brain cells by D-galactose treatment was markedly diminished by Tau co-treatment (Nox2: DG_LT group, p < 0.05; NFκB: both DG_LT and DG_HT groups, p < 0.05) (Fig. 4A). Regarding pro-inflammatory cytokines, Il-1β, Il-6, Tnfα and Cox2 were also dramatically upregulated (p < 0.05) by D-galactose treatment, and Tau or aminoguanidine reversed (p < 0.05) their expression, to levels that were similar (p > 0.05) to those of the CON group (Fig. 4B). In the regulation of cellular apoptosis (Fig. 4C), the DG group showed increased (p < 0.05) gene expression of pro-apoptotic genes, i.e. Bax and Casp3, but decreased (p < 0.05) expression of anti-apoptotic genes, i.e. Bcl2 and BclxL. Tau or aminoguanidine co-treatment normalized (p < 0.05) the expression of all these genes. Furthermore, upregulated (p < 0.05) gene expression of markers for glial cell activation, i.e. Gfap and Cd11b, was only detected in the DG groups, as compared with the CON groups, while Tau or aminoguanidine co-treatment resulted in similar (p > 0.05) expression to that in the CON groups.
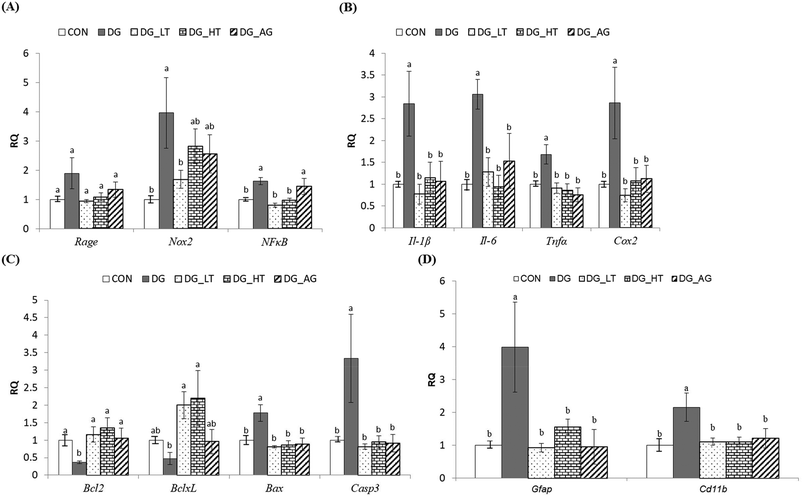 |
| | Fig. 4 The expression of genes involved in inflammation, apoptosis, and activation of the glial cells of brains of the experimental mice. Total RNA was isolated and analyzed by qPCR for the expression of inflammation-related genes. (A) Rage, Nox2, and NFκB, (B) Il-1β, Il-6, and Tnfα, and Cox2, (C) Bcl2, BclxL, Bax, and Casp3, and (D) Gfap and Cd11b. The values for mRNA were normalized to the value for GAPDH. The data are given as mean ± SEM (n = 6). For each gene, data bars without letters in common (a–b) indicate a significant difference (p < 0.05). | |
Discussion
A D-galactose-induced age-related disease model has been frequently used for brain aging and anti-aging pharmacological studies.2 Aminoguanidine hydrochloride can ameliorate the formation of AGEs in D-galactose-treated mice through interacting with 3-deoxyglucosone;12,13 hence it is used as a positive control group in this study. As a previous study reported, a high-fat diet or genetic obesity causes a depletion of blood Tau levels and promotes further obesity, which forms a vicious cycle.16 That is, Tau supplementation can interrupt this vicious cycle and increase the basal metabolic rate, thus reducing body weight, and parametrical white adipose tissue, body fat, and adipocyte size (%). In addition, decreased serum and liver TG values in rats fed a high-fat diet supplemented with Tau is highly related to upregulation of expression of peroxisome proliferator-activated receptor-alpha and uncoupling protein 2 genes, thus increasing energy expenditure.5 Hence, based on the current results, although serum TG levels were reduced by taurine supplementation, D-galactose induction, and Tau and AG supplementations did not change the physiological status during the experimental period (Table 1).
As is well known, the hippocampus is a specific region for short- and long-term spatial memories and plays an important role in the acquisition, modification, and amalgamation of messages. Through the use of the Morris water maze, a learning behavioral dysfunction (longer escape latency and swimming distance) of mice treated with D-galactose was observed.2 In general, swimming paths in the Morris water maze distinguish marginal, random, taxis, and straight strategies.2 No matter which methods are selected, the escape latency and swimming path distance should be examined for the evaluation of spatial learning and memory abilities. The escape latency (the period needed to reach the platform) and period spent of crossing over the target quadrant indicates how quickly the animal can learn and memorize the location of the platform. Tau can act as an antioxidant and plays a role in the protection of the liver from chronic alcohol damage17 and the mitigation of oxidative insult induced in rat blood by potassium bromate.18 The functional behaviors of different aspects of learning and memory have divergent neuroanatomical locations within the lateral amygdala, dorsal striatum, and hippocampus. The dentate gyrus is one of the few regions of the adult brain where neurogenesis takes place and its granule cells are critically involved in spatial behavior. AGE production is one of the mechanisms of D-galactose-induced aging.12,19 It has also been demonstrated that AGE production due to ROS induction in the brain upregulates amyloid precursor protein in the pathophysiological processes of aging and degeneration.20 Tau is abundant in neurons and plays an important role in the assembly and stabilization of microtubules, and maintains the cytoskeletal structure. Because Tau is able to cross the blood–brain barrier, its level in the brain is increased when Tau is administered therapeutically.21 Tau promotes axonal outgrowth, and it is also necessary for maintaining axonal morphology and transport. Recently, it was also observed that Tau supplementation rescues cognitive deficits in APP/PS1 of Alzheimer's mice up to the level of age-matched wild-type mice in Y-maze and passive avoidance tests, and meanwhile, also slightly decreases the insoluble fraction of β-amyloid in the cortex.22 Moreover, our results showed that Tau supplementation can improve learning and memory abilities in D-galactose-treated mice, which is largely due to the presence of fewer morphological changes of the dentate gyrus and less AGE accumulation in the brain (Fig. 1 & 2).
Oxidative loads in the brain are involved in age-related impairments, i.e. learning and memory deficits.23 When the dose of D-galactose intake is beyond the capacities of D-galactokinase and galactose-1-phosphate uridyltransferase, the generation of lipid peroxide, oxidative stress, and AGEs, as well as changes in learning and memory capacities, are observed in rodents.2,7,8D-Galactose treatment results in increases in brain MDA and protein carbonyl levels, but decreases in reduced GSH levels, as well as in SOD, CAT, and GPx activities.2 Tau leads to an enhancement of brain antioxidant capacities; thus the decreases in reduced GSH levels and antioxidant enzymatic activities, i.e. GPx, CAT, and SOD, as well as the increased TBARS values in brain tissues of a diabetic rat model [intracerebroventricular injection of streptozotocin (STZ), 3 mg per kg BW,] are reversed by pretreatment with Tau (50 mg per kg BW) to the values observed in sham-operated rats (without the injection of STZ).24 Moreover, a previous study indicated that the interaction of AGEs with their receptor [receptor for AGEs (RAGE)] causes oxidative stress and inflammatory reactions, even leading to cell death.25 Our study also demonstrated that an aggregation of AGEs in brain tissues of D-galactose-treated mice is decreased (p < 0.05) by Tau co-treatment (Fig. 2), which also corresponds to the higher brain antioxidant capacities in Tau co-treated groups (Table 1 and Fig. 3).
The oxidative stress not only results in neuronal degeneration, but also impairs normal function of astrocytes (one of the most abundant glial cell types). Impaired astrocytes may decrease their antioxidant capacities and produce NO via an up-regulation of iNOS expression, which inhibits neuronal respiration, resulting in a release of glutamate. Cytokines play an important role in modulating the inflammatory response and inducing proliferation of the central nervous system (astrocytes and microglia). AGEs lead to oxidative stress and the activation of the transcription factor NFκB pathways through RAGE.26 Regarding the mechanism, inflammation-related molecules and factors (cytokines) are synthesized and mediated through an NFκB-dependent signaling pathway. NFκB acts as a key player in physiological processes, such as synaptic plasticity, which are highly related to learning and memory in the hippocampus (granule cells and pyramidal neurons of CA1 and CA3). Therefore, NFκB not only affects a variety of pro-inflammatory cytokines (Il-1, Il-6, and TNFα) but also increases the expression of pro-inflammatory enzymes (Cox2), thus causing secondary neurotoxicity-exacerbated inflammation-induced neurodegeneration.27 Moreover, chronic D-galactose administration has been reported to cause aging syndromes, such as age-related cognitive decline in mice, along with increased inflammation and reduced hippocampal neurogenesis.19 In the prefrontal cortex of D-galactose-treated mice, microglial cells and astrocytes were activated, and then the expression of Cd11b and Gfap w increased, further upregulating expression of iNOS, Cox2, Il-1β, Il-6, and Tnfα.26 Aminoguanidine can prevent the reactive gliosis induced by Aβ25–35 injection, a fragment of the Alzheimer's amyloid β-peptide, through the cell immunoreactivity for Gfap and Cd11b, as well as through decreased Il-1β and Tnfα expression and diminished production of NO.28 In addition, aminoguanidine can also inhibit Casp3 and delay the final stages of apoptosis leading to neuronal death. Under some cell-damage conditions, only 200 μm to 1 mm Tau could protect the hippocampus via the activation of glycine receptors.29 In the brain, Tau administration interacted directly with the N-methyl-D-aspartic acid receptor (NMDA receptor), and then reduced cell damage and cytokine expressions after brain injury.21 It is believed that the neuroprotective effect of Tau is mediated via its anti-inflammatory properties in the hindrance of TLR-4/MyD88 signaling (NFκB, Il-1β, and Tnfα).30 In addition, the neuron injury mediated by apoptosis is closely correlated with dementia and motor neuron disability. Apoptosis has been associated with various pathological conditions and is frequently used as an index of brain injuries. The anti-apoptotic proteins (Bcl2 and BclxL) and Bax, acting as the upstream, in mitochondria regulated the release of pro-apoptotic proteins (cytochrome c) into the cytosol, which have an important influence on apoptosis.3Bax and Casp3 expressions were increased, and Bcl2 expression was decreased in the hippocampus of D-galactose treated rats.31 Based on our results, it is clear that the pro-apoptotic status of brain cells in D-galactose treated mice is ameliorated via Tau supplementation, and, meanwhile, the anti-inflammatory response and its inactivation of glial cells are also revealed. In the morphological observations of the dentate gyrus in mice, the contracted and intensely basophilic neuron necrosis could be highly related to those regulations of molecular mechanisms (Fig. 2A and 4).
Conclusion
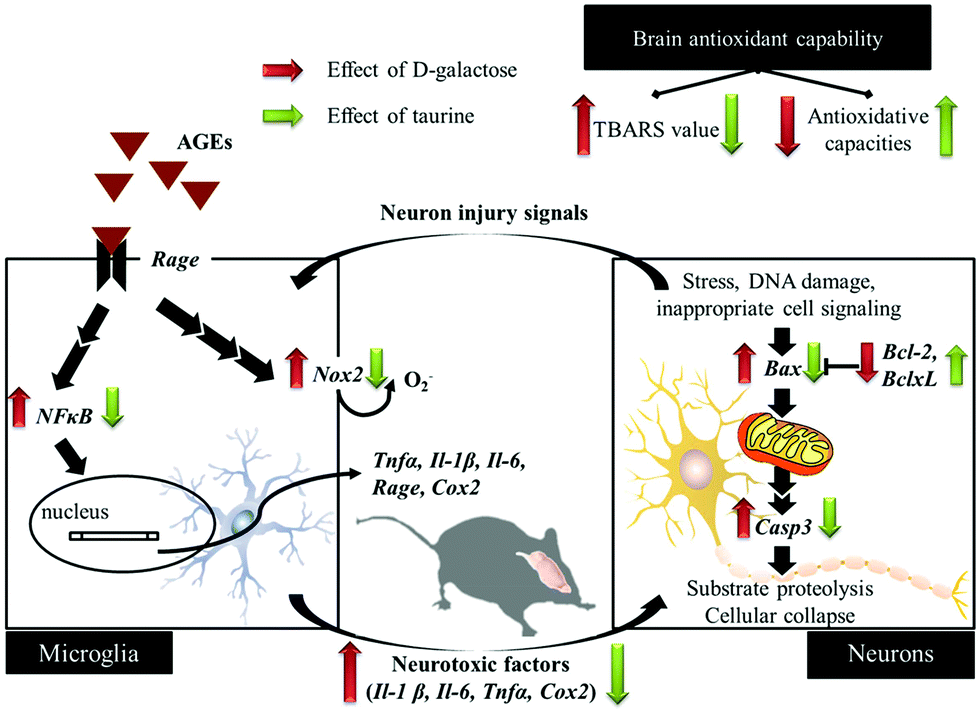
D-Galactose induction for 9 weeks indeed induced age-related syndromes (learning and memory defects), increased oxidative stress, and upregulated inflammation and apoptosis in brain cells and glial-cell activation (Fig. 5). According to the results in this study, Tau supplementation improved learning and memory impairment of D-galactose-treated mice, which is highly related to significantly increased brain antioxidant capacities and decreased inflammatory responses, apoptosis, and glial-cell activation. Meanwhile, those protective effects are as similar as aminoguanidine's. Thus, Tau can be suggested as a daily dietary supplement to help a prevention against the onset of oxidative-stress-induced cognitive dysfunction.
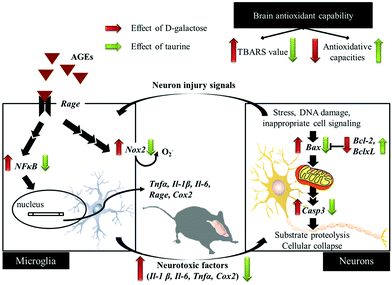 |
| | Fig. 5 Schematic diagram for the protective effects of taurine against D-galactose-induced oxidative damage in the brain. | |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the funding of this research by Ditmanson Medical Foundation Chia-Yi Christian Hospital Research Program (R105-010) and the Ministry of Science and Technology, Taiwan (Project: NSC 102-2313-B-002-039-MY3).
References
-
United Nations, World Population Ageing 1950-2050. Chapter II: Magnitude and speed of population ageing, 2015, http://www.un.org/esa/population/publications/worldageing19502050/pdf/80chapterii.pdf, [access date: August 3, 2017] Search PubMed.
- Y. Lei, J. Chen, W. Zhang, W. Fu, G. Wu, H. Wei, Q. Wang and J. Ruan, Food Chem., 2012, 135, 2702–2707 CrossRef CAS PubMed.
- W. Wu, X. Wang, Q. Xiang, X. Meng, Y. Peng, N. Du, Z. Liu, Q. Sun, C. Wang and X. Liu, Food Funct., 2014, 5, 158–166 CAS.
- S. E. Edgar, C. A. Kirk, Q. R. Rogers and J. G. Morris, J. Nutr., 1998, 128, 751–757 CAS.
- Y. Y. Chang, C. H. Chou, C. H. Chiu, K. T. Yang, Y. L. Lin, W. L. Weng and Y. C. Chen, J. Agric. Food Chem., 2011, 59, 450–457 CrossRef CAS PubMed.
- C. H. Chou, Y. Y. Chang, B. S. Tzang, C. L. Hsu, Y. L. Lin and Y. C. Chen, Food Chem., 2012, 135, 24–30 CrossRef CAS.
- K. Lai, L. J. Elsas and K. J. Wierenga, IUBMB Life, 2009, 61, 1063–1074 CrossRef CAS PubMed.
- C. H. Chou, S. Y. Wang, Y. T. Lin and Y. C. Chen, Int. J. Food Sci. Technol., 2014, 49, 1654–1662 CrossRef CAS.
- A. F. Aydın, J. Çoban, I. Doğan-Ekici, E. Betül-Kalaz, S. Doğru-Abbasoğlu and M. Uysal, Metab. Brain Dis., 2016, 31, 337–345 CrossRef PubMed.
- S. Rivas-Arancibia, C. Dorado-Martinez, G. Borgonio-Perez, M. Hiriart-Urdanivia, L. Verdugo-Diaz, A. Duran-Vazquez, L. Colin-Baranque and M. R. Avila-Costa, Environ. Res., 2000, 82, 7–17 CrossRef CAS PubMed.
- S. Reagan-Shaw, M. Nihal and N. Ahmad, FASEB J., 2008, 22, 659–661 CrossRef CAS PubMed.
- X. Song, M. Bao, D. Li and Y. M. Li, Mech. Ageing Dev., 1999, 108, 239–251 CrossRef CAS PubMed.
- P. J. Thornalley, Arch. Biochem. Biophys., 2003, 419, 31–40 CrossRef CAS PubMed.
- Y. L. Lin, S. Y. Tai, J. W. Chen, C. H. Chou, S. G. Fu and Y. C. Chen, Food Funct., 2017, 8, 1763–1774 CAS.
- L. Lu, W. H. Peng, W. Wang, L. J. Wang, Q. J. Chen and W. F. Shen, J. Zhejiang Univ., Sci., B, 2011, 12, 652–659 CrossRef CAS PubMed.
- N. Tsuboyama-Kasaoka, C. Shozawa, K. Sano, Y. Kamei, S. Kasaoka, Y. Hosokawa and O. Ezaki, Endocrinology, 2006, 147, 3276–3284 CrossRef CAS PubMed.
- Y. J. Fang, C. H. Chiu, Y. Y. Chang, C. H. Chou, H. W. Lin, M. F. Chen and Y. C. Chen, Food Res. Int., 2011, 44, 3105–3110 CrossRef CAS.
- M. K. Ahmad and R. Mahmood, Environ. Toxicol., 2016, 31, 304–313 CrossRef CAS PubMed.
- V. M. Doan, C. Chen, X. Lin, V. P. Nguyen, Z. Nong, W. Li, Q. Chen, J. Ming, Q. Xie and R. Huang, Food Funct., 2015, 6, 1712–1718 CAS.
- S. Chen, F. M. An, L. Yin, A. R. Liu, D. K. Yin, W. B. Yao and X. D. Gao, Neuroscience, 2013, 256, 137–146 CrossRef PubMed.
- J. Menzie, H. Prentice and J. Y. Wu, Brain Sci., 2013, 3, 877–907 CrossRef CAS PubMed.
- H. Y. Kim, H. V. Kim, J. H. Yoon, B. R. Kang, S. M. Cho, S. Lee, J. Y. Kim, J. W. Kim, Y. Cho, J. Woo and Y. Kim, Sci. Rep., 2014, 4, 7467–7474 CrossRef PubMed.
- B. Ye, H. Shen, J. Zhang, Y. G. Zhu, B. R. Ransom, X. C. Chen and Z. C. Ye, Glia, 2015, 63, 2208–2219 CrossRef PubMed.
- H. Javed, A. Khan, K. Vaibhav, M. Moshahid Khan, A. Ahmad, M. Ejaz Ahmad, A. Ahmad, R. Tabassum, F. Islam, M. M. Safhi and F. Islam, Neurol. Sci., 2013, 34, 2181–2192 CrossRef PubMed.
- I. Lubitz, J. Ricny, D. Atrakchi-Baranes, C. Shemesh, E. Kravitz, S. Liraz-Zaltsman, A. Maksin-Matveev, I. Cooper, A. Leibowitz, J. Uribarri, J. Schmeidler, W. Cai, Z. Kristofikova, D. Ripova, D. LeRoith and M. Schnaider-Beeri, Aging Cell, 2016, 15, 309–316 CrossRef CAS PubMed.
- J. Lu, D. M. Wu, Y. L. Zheng, B. Hu, Z. F. Zhang, Q. Ye, C. M. Liu, Q. Shan and Y. J. Wang, Cereb. Cortex, 2010, 20, 2540–2548 CrossRef PubMed.
- R. H. Shih, C. Y. Wang and C. M. Yang, Front. Mol. Neurosci., 2015, 8, 77–85 Search PubMed.
- A. Díaz, K. Rojas, B. Espinosa, R. Chávez, E. Zenteno, D. Limón and J. Guevara, Neuropeptides, 2014, 48, 153–159 CrossRef PubMed.
- F. Jia, M. Yue, D. Chandra, A. Keramidas, P. A. Goldstein, G. E. Homanics and N. L. Harrison, J. Neurosci., 2008, 28, 106–115 CrossRef CAS PubMed.
- C. Kim and Y. N. Cha, Amino Acids, 2014, 46, 89–100 CrossRef CAS PubMed.
- Z. Lan, J. Liu, L. Chen, Q. Fu, J. Luo, R. Qu, L. Kong and S. Ma, J. Ethnopharmacol., 2012, 141, 386–395 CrossRef CAS PubMed.
Footnotes |
| † Dom-Gene Tu, Yao-Ling Chang, and Chung-Hsi Chou contributed equally as co-first authors. |
| ‡ Yi-Chen Chen and Yuan-Yen Chang contributed equally as corresponding authors. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *df
*df