
Catalyst-free chemoselective conjugate addition and reduction of α,β-unsaturated carbonyl compounds via a controllable boration/protodeboronation cascade pathway†
Xi
Huang
,
Junjie
Hu
,
Mengying
Wu
,
Jiayi
Wang
,
Yanqing
Peng
and
Gonghua
Song
 *
*
Shanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology, Shanghai, 200237, PR China. E-mail: ghsong@ecust.edu.cn; Fax: +86-21-64252603
First published on 27th November 2017
Abstract
A novel, efficient transition-metal-free and controllable boration/protodeboronation strategy has been developed for the chemoselective conjugate addition and 1,4-reduction of α,β-unsaturated carbonyl compounds. Without any metal-catalyst or base, a series of β-boration products of α,β-unsaturated carbonyl compounds was easily obtained in moderate to excellent yields in a mixed solvent of ethanol and water. The presence of a catalytic amount of Cs2CO3 can effectively induce further protodeboronation reaction towards 1,4-reduction products at higher reaction temperature. Therefore, by slightly changing the reaction conditions, the boration or reduction products of α,β-unsaturated carbonyl compounds can be controllably obtained. Mechanistic studies revealed that Cs2CO3 played the key role in activating the protodeboronation step. This transition-metal-catalyst-free and product controllable method provides a useful and eco-friendly tool for the highly chemoselective preparation of the β-boration products and 1,4-reduction products of α,β-unsaturated carbonyl compounds.
Introduction
The development of easy, practical and environmentally friendly chemical reactions has become an important consideration in the current chemical research. In this context, transition-metal-free organic reactions have received much attention as they can not only prevent the formation of toxic metal residues in pharmaceutical products, but also considerably reduce the risk of environmental pollution.Organoboron compounds are of great importance in organic synthesis, not only for their special characteristics and biological activities,1 but also for their use as key reagents with broad utility in the field of organic synthesis.2 As to the introduction of a boronate group at the β-position of carbonyl, the catalytic conjugate addition of diboron reagents to α,β-unsaturated carbonyl compounds has attracted more and more attention in recent years.3 This conjugate borylation has been either catalyzed by different metal complexes, such as palladium,4 platinum,5 rhodium,6 copper,7 and nickel8 complexes, or carried out with N-heterocyclic carbenes.9 Although these methods provide useful tools for the introduction of boron into an organic framework, they also exhibit some drawbacks such as high catalyst loading, complex and expensive ligands, harsh conditions, long reaction times and narrow substrate scope. The development of a simple, effective and catalyst-free method for the β-boration of α,β-unsaturated carbonyl compounds has remained an elusive goal. On the other hand, more recently, Song and co-workers reported a chemoselective 1,4-reduction of α,β-unsaturated ketones through an interesting transformation of the C–B bond of an alkylboronic ester into a C–H bond (Scheme 1a).10 Immediately following this work, analogous Cu-catalyzed chemoselective borylation and reduction of α,β-unsaturated carbonyl compounds were reported (Scheme 1b).11 However, Cu catalysts for both borylation and reduction reactions, over 2 equiv. of base for reduction, and long reaction times were necessary to achieve moderate to good yields of β-boration and 1,4-reduction products.
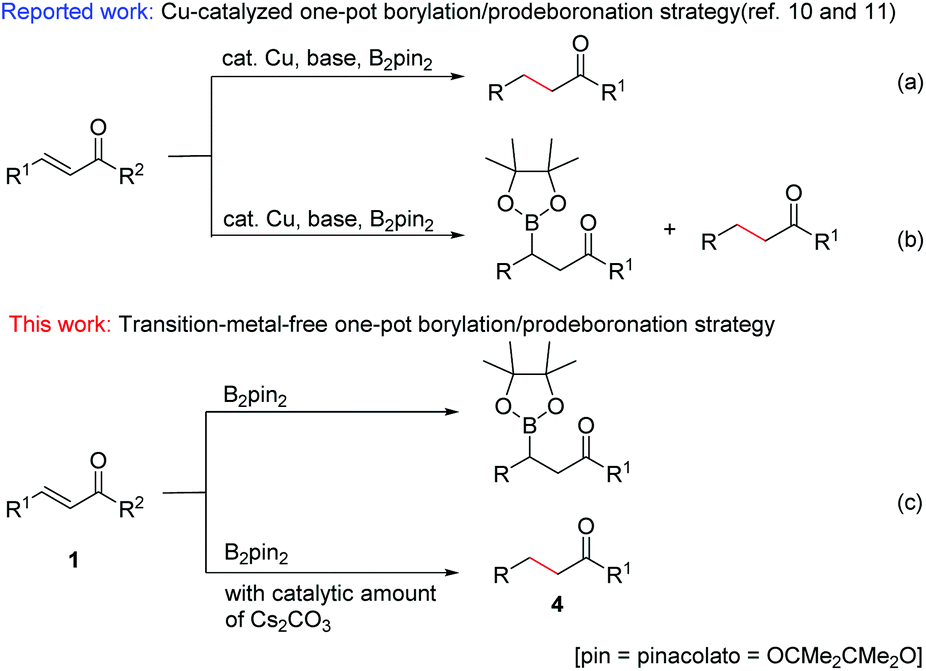 | ||
| Scheme 1 Boration/protodeboronation of α,β-unsaturated compounds. | ||
Based on the understanding of the boration/protodeboronation pathway, herein we report for the first time a transition-metal-free chemoselective conjugate addition and reduction of α,β-unsaturated carbonyl compounds via a controllable boration/protodeboronation strategy (Scheme 1c). With appropriate adjustment of reaction conditions, various β-boration products or reduction products of α,β-unsaturated carbonyl compounds such as chalcones and cinnamic esters can be obtained controllably in moderate to excellent yields. This novel, simple and green strategy provides a useful tool for the highly chemoselective β-boration and 1,4-reduction of α,β-unsaturated carbonyl compounds with wide substrate compatibility.
Results and discussion
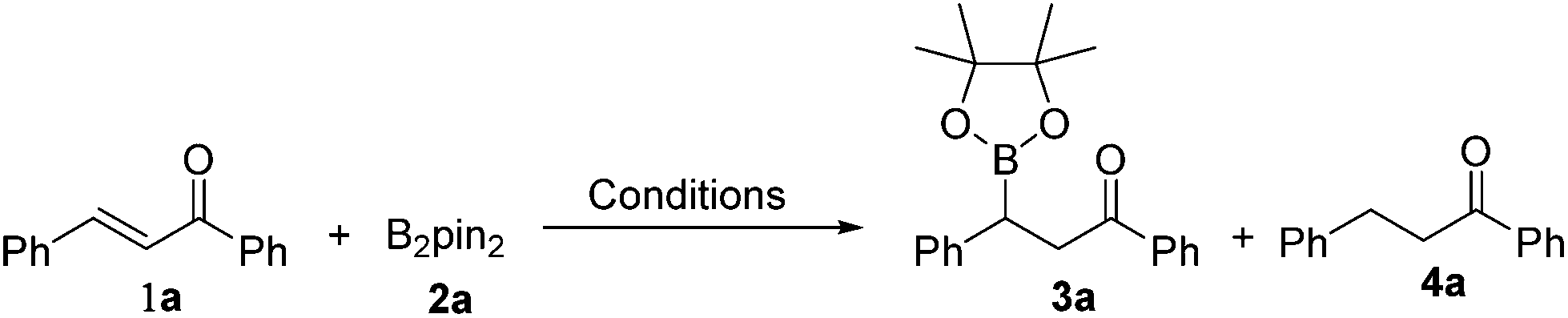
By employing the transition-metal-free chemoselective β-boration and reduction of chalcone with bis(pinacolato)diboron as the model reaction, a range of reaction conditions was investigated as shown in Table 1. Compared to Et3N, other bases were found to be less effective for the transformation to the desired β-boration product 3a (entries 1–4). When the reaction was conducted in a mixed solvent of ethanol and water (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), a significant increase in the yield of 3a was observed (entry 5). An excellent yield of 3a was obtained by prolonging the reaction time to 30 minutes (entry 6). Interestingly, in the absence of any base, the borylation procedure also proceeded smoothly with a longer reaction time (entry 7). These surprising results implied that the β-boration could be performed under both transition-metal-free and base-free conditions. Also, it was brought to our attention that when a catalytic amount of Cs2CO3 was used, the reaction preferred to give reduced product 4a rather than the β-boration product 3a (entry 1). The ratio of the reduction product 4a to the β-boration product 3a increased remarkably as the reaction temperature was raised to 100 °C with the assistance of microwave irradiation (entry 8). Eventually, the reaction afforded the reduced product 4a almost exclusively when it was performed at 110 °C with (entry 9), or without the assistance of microwave irradiation (entry 10). These results indicated that the presence of Cs2CO3 and high temperature were in favour of the deborylation of β-boration products derived from α,β-unsaturated carbonyl compounds.
:1), a significant increase in the yield of 3a was observed (entry 5). An excellent yield of 3a was obtained by prolonging the reaction time to 30 minutes (entry 6). Interestingly, in the absence of any base, the borylation procedure also proceeded smoothly with a longer reaction time (entry 7). These surprising results implied that the β-boration could be performed under both transition-metal-free and base-free conditions. Also, it was brought to our attention that when a catalytic amount of Cs2CO3 was used, the reaction preferred to give reduced product 4a rather than the β-boration product 3a (entry 1). The ratio of the reduction product 4a to the β-boration product 3a increased remarkably as the reaction temperature was raised to 100 °C with the assistance of microwave irradiation (entry 8). Eventually, the reaction afforded the reduced product 4a almost exclusively when it was performed at 110 °C with (entry 9), or without the assistance of microwave irradiation (entry 10). These results indicated that the presence of Cs2CO3 and high temperature were in favour of the deborylation of β-boration products derived from α,β-unsaturated carbonyl compounds.
|
|
|||
|---|---|---|---|
| Entry | Conditions | Yield of 3a [%] | Yield of 4a [%] |
|
a Reaction conditions: 0.5 mmol of chalcone (1a), 1.5 equiv. of bis(pinacolato)diboron (2a), base, 1.5 mL of ethanol in a sealed tube heated at 70 °C for 20 min. GC yield (dodecane as an internal standard).
b The reaction was conducted in a mixed solvent (ethanol:water = 6:1).
c The reaction was performed at 100 °C with the assistance of microwave irradiation at a maximum power of 300 W.
d The reaction was performed at 110 °C with the assistance of microwave irradiation at a maximum power of 300 W.
e The reaction was conducted in a pressure-tight tube at 110 °C under conventional heating.
|
|||
| 1 | Cs2CO3 (0.1 eq.), 20 min | 6 | 8 |
| 2 | K2CO3 (0.1 eq.), 20 min | 15 | 4 |
| 3 | DIPEA (0.1 eq.), 20 min | 19 | <1 |
| 4 | Et3N (0.1 eq.), 20 min | 37 | <1 |
| 5b | Et3N (0.1 eq.), 20 min | 85 | <1 |
| 6b | Et3N (0.1 eq.), 30 min | 98 | <1 |
| 7b | No base, 2 h | 96 | <1 |
| 8c | Cs2CO3 (0.1 eq.), 20 min | 16 | 82 |
| 9d | Cs2CO3 (0.1 eq.), 20 min | <1 | 96 |
| 10e | Cs2CO3 (0.1 eq.), 3 h | <1 | 93 |
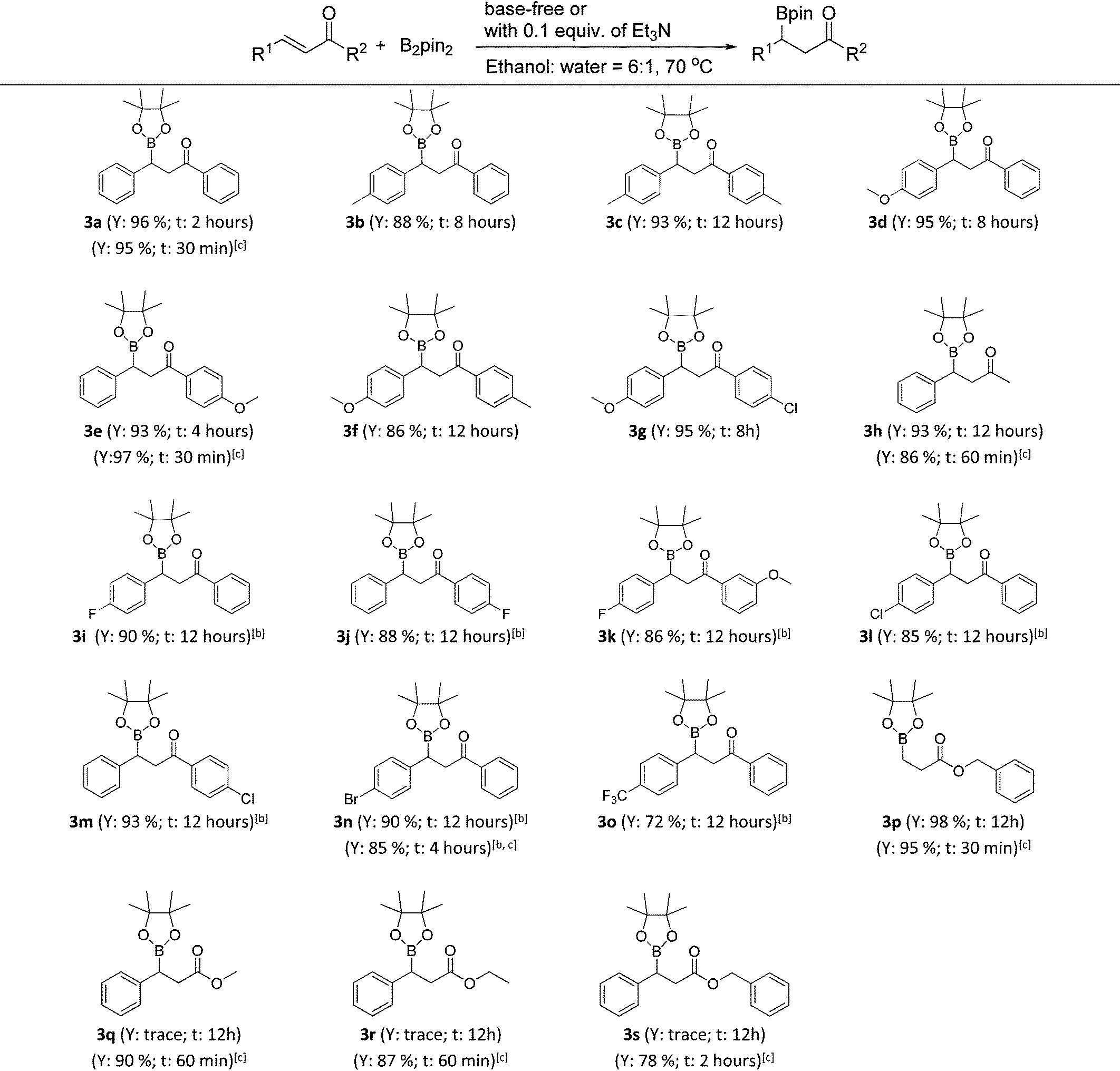
With the optimized conditions in hand, the catalyst- and base-free β-boration of a range of α,β-unsaturated carbonyl compounds was investigated (Table 2). To our delight, the reactions of various chalcones with electron-donating substituents (3a–3g) and the enone with a methyl group (3h) gave the corresponding β-boration products in good to excellent yields. For chalcones bearing electron-withdrawing groups (3i–3o), satisfactory yields were obtained by simply changing the reaction medium to a mixed solvent of ethanol and cyclohexane (6:1). Also, the corresponding β-boration product of benzyl acrylate (3p) was achieved in excellent yields. When cinnamates were applied to the reaction under identical conditions, only trace amounts of products were obtained according to GC analysis (3q–3s). Fortunately, it was found that the addition of 0.1 equiv. of Et3N could make the β-boration of cinnamates performable (Table 2, footnote c). Further investigation demonstrated that the presence of a catalytic amount of Et3N could accelerate the β-boration reaction rates of various enones (Table 2, 3a, 3e, 3h, 3n and 3p, footnote c). Therefore, the present work provides an excellent approach for β-boration with high substituent compatibility under catalyst- and base-free conditions and it is more eco-friendly than previous reports.
|
a Reaction conditions: 0.5 mmol of α,β-unsaturated carbonyl compounds, 1.5 equiv. of bis(pinacolato)diboron (2a), 1.5 mL of mixed solvent of ethanol and water (6:1) in a sealed tube heated at 70 °C (General procedure B). Y = isolated yield; t = reaction time.
b A mixed solvent of ethanol and cyclohexane (6:1) was used as the reaction solvent.
c With the addition of 0.1 equiv. of Et3N (General procedure C).
|
|---|
|
Next, the controllable chemoselective 1,4-reduction was applied to various α,β-unsaturated carbonyl compounds (Table 3). To our delight, all substituted chalcones and cinnamates were compatible with this transformation and the corresponding reduction products were obtained in good to excellent yields with properly chosen reaction conditions (4a–4w). In general, a little stronger reaction conditions were needed for chalcones with an electron-donating substituent (4k–4p) and those with multiple or large steric hindrance substituents (4q–4t) as well as for cinnamates (4u–4w). In addition, the strategy was also applicable for α,β-unsaturated enones with a heterocycle (4x) or an alkyl group (4y). Notably, when 1,5-diphenylpenta-2,4-dien-1-one (1z) was tried, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond at the α,β-position of carbonyl was chemoselectively reduced while the CC bond at the γ,δ-position remained untouched. It was interesting that when benzyl acrylate was adopted, the β-boration intermediate of benzyl acrylate (3p) was isolated in a yield of 68%, while only a trace amount of the reduction product, namely benzyl propionate, was detected even in the presence of 0.3 equiv. of Cs2CO3 and with heating up to 150 °C. This phenomenon was consistent with the previous report that the presence of an aromatic ring at the β-position of α,β-unsaturated carbonyl compounds was necessary for the protodeboronation of the β-boration intermediates.11b
C double bond at the α,β-position of carbonyl was chemoselectively reduced while the CC bond at the γ,δ-position remained untouched. It was interesting that when benzyl acrylate was adopted, the β-boration intermediate of benzyl acrylate (3p) was isolated in a yield of 68%, while only a trace amount of the reduction product, namely benzyl propionate, was detected even in the presence of 0.3 equiv. of Cs2CO3 and with heating up to 150 °C. This phenomenon was consistent with the previous report that the presence of an aromatic ring at the β-position of α,β-unsaturated carbonyl compounds was necessary for the protodeboronation of the β-boration intermediates.11b
| a Reaction conditions: 0.5 mmol of α,β-unsaturated carbonyl compounds, 1.5 equiv. of bis(pinacolato)diboron (2a), 0.1 equiv. of Cs2CO3, 1.5 mL of ethanol in a sealed microwave tube at 110 °C with the assistance of microwave irradiation at a maximum power of 300 W (General procedure D). Y = isolated yield; t = reaction time, the heating up time to 110 °C was not included. b 0.2 equiv. of Cs2CO3 were used. c 0.3 equiv. of Cs2CO3 were used. d 2 equiv. of bis(pinacolato)diboron (2a) were used. e The reaction was performed at 130 °C with the assistance of microwave irradiation. f The reaction was performed at 150 °C with the assistance of microwave irradiation. g TBA (tert-butyl alcohol) was used as the reaction solvent in the case of transesterification. |
|---|
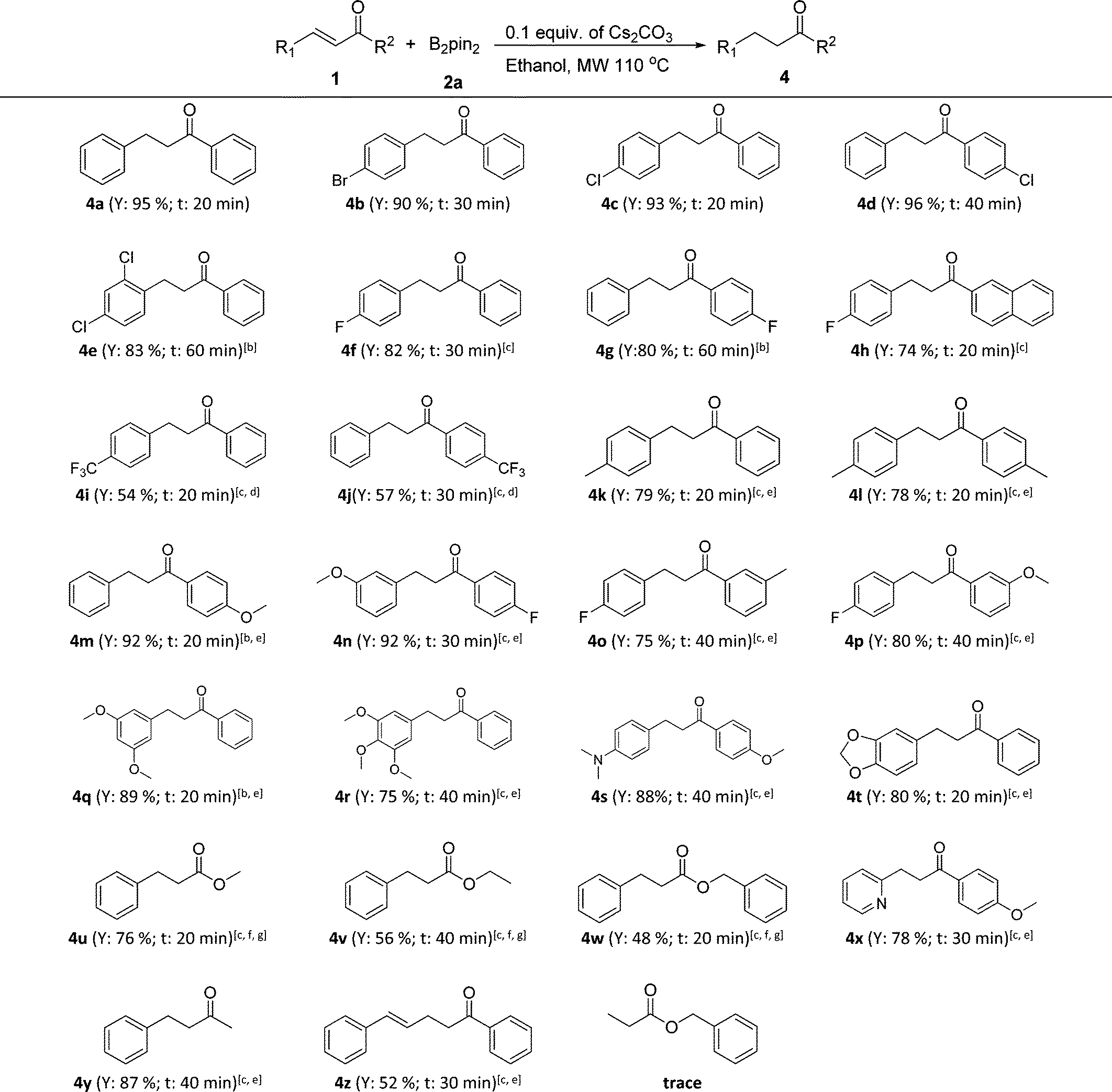
|
To preclude the catalytic action of trace metal impurities from the glassware, stirring bar, and Cs2CO3, the concentration of common transition metals such as Fe, Cu, Ni, Co, Zn, Cr, Mn, Rh, Pd and Pt in both boration and reduction reaction mixtures was measured by ICP-AES. It was found that the contents of Fe and Zn were <5 mg kg−1 and those of all other metals were <2 mg kg−1 (Section 5 in the ESI†). The ICP-AES test result confirms that this controllable boration/protodeboronation process is practically performable under transition-metal-free conditions.
To gain insight into the hydrogen source of the β-boration and reduction products in this boration/protodeboronation strategy, a few isotope-labeled experiments were conducted (Scheme 2). Firstly, the 1,4-reduction of chalcone (1a) was performed in CD3OD (Scheme 2a). The result revealed that the deuteration ratio on the β-position and α-position of the reduction product was 94% and 185%, respectively (determined by 1H NMR and HRMS), which clearly demonstrated that the solvent CD3OD also served as the hydrogen source in this boration/protodeboronation strategy. In the following trials, the β-boration reaction of chalcone (1a) was carried out under the standard conditions except for the substitution of alcohol or water in the mixed solvent with CD3OD (Scheme 2b) or D2O (Scheme 2c), respectively. Interestingly, the little bit of the deuterated β-boration product (Scheme 2b) or the absence of the deuterated β-boration product (Scheme 2c) (determined by 1H NMR) jointly indicated that the hydrogen was mainly from the non-deuterated methanol or water in the mixed solvent. The β-boration reaction of chalcone (1a) was then performed in a mixed solvent of CH3OH and CD3OD (1:1) (Scheme 2d). The ratio of hydrogen to deuterium on the α-position of the β-boration product was 17:3. The isotope-labeled experiments revealed that both water and methanol could provide a hydrogen source for the transformation while a proton was relatively more competitive than its deuterated counterpart. This phenomena is consistent with the isotope effect.12 Therefore, it is the alcohol and/or water in the mixed solvent that supplies the hydrogen source for both β-boration and reduction reactions.
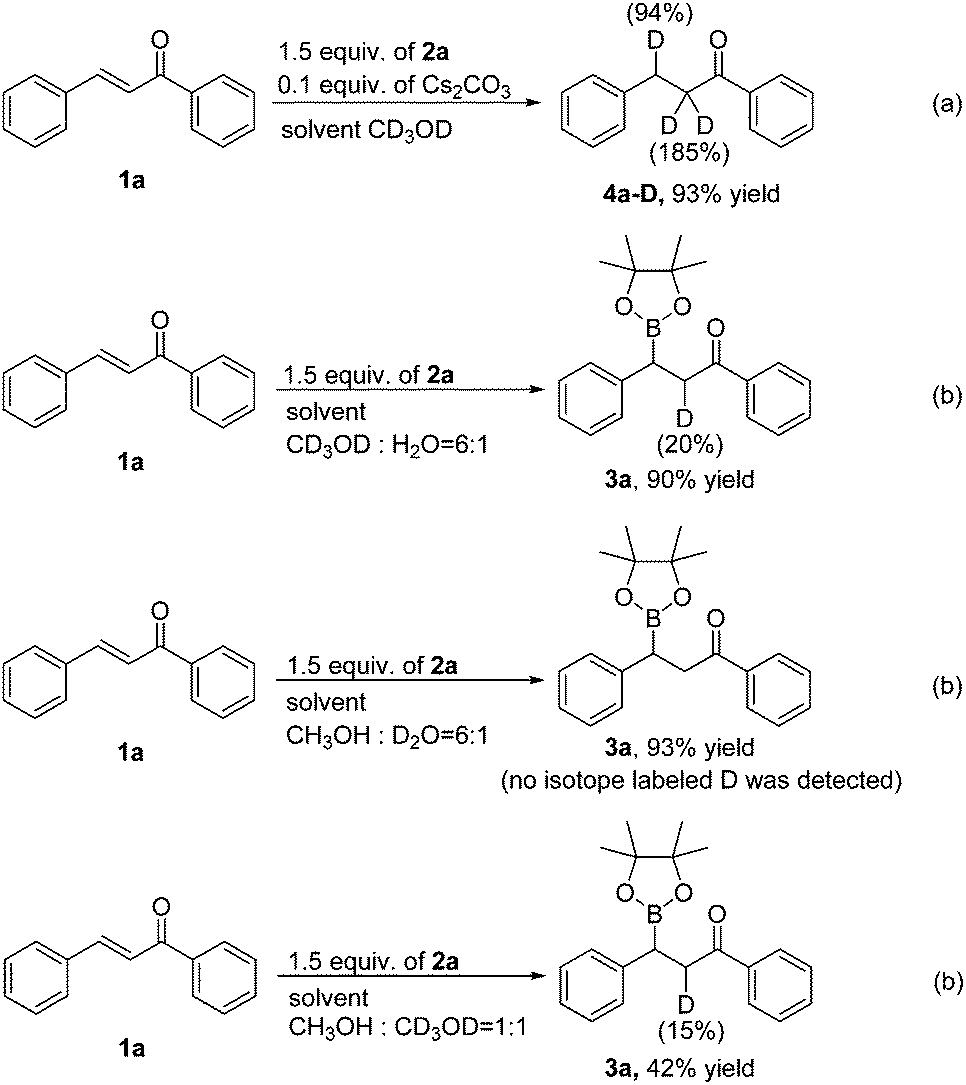 | ||
| Scheme 2 Hydrogen isotope labelling experiments. | ||
To identify the key factors affecting the whole boration/protodeboronation process, several control experiments were also conducted. It was found that no reaction would occur when B2pin2 was removed from the reaction system (Scheme 3a). Without the use of any base or catalyst, the β-boration product 3a was obtained in 38% yield in pure ethanol (Scheme 3b), and the β-boration product did not undergo any further transformation (Scheme 3c). These results indicated that this boration/protodeboronation cascade process is a base-controlled one under transition-metal-free conditions. This was further confirmed by the experimental results of the successful protodeboronation of 3a in the presence of Cs2CO3 (Scheme 3d). Thus, the boration or 1,4-reduction of an enone can be easily and effectively controlled by using or not using an appropriate base as an activating reagent. This product-controllable and transition-metal-free pathway is obviously superior to other protocols in terms of environmental compatibility.
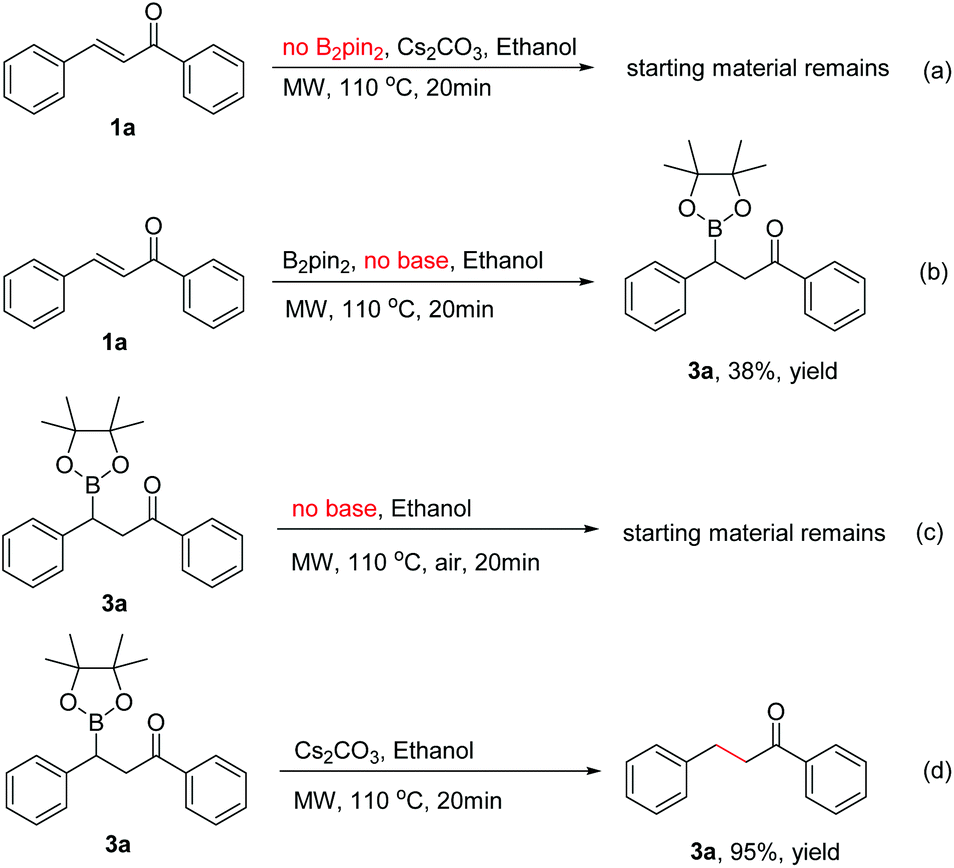 | ||
| Scheme 3 Control experiments. | ||
Based on the experimental results and the literature report,13 a plausible reaction mechanism for this boration/protodeboronation cascade reaction is proposed by using CD3OD as the hydrogen source (Scheme 4). The reaction is initiated by the formation of an anion CD3O− from deuterated methanol through a deprotonated process. The base served to promote the deprotonation of CD3OD thus accelerating the β-boration reaction, which was consistent with the results of experiments under catalyst-free conditions. As a Lewis acid, B2pin2 reacts with CD3O− to generate an anionic diborane species 5. The subsequent 1,4-addition of chalcone afforded a borylated enolate 7, which was rapidly converted into boronic ester substituted ketone 9 in the presence of CD3OD. Finally, the protodeboronation of the key intermediate 3a in the presence of Cs2CO3 afforded the reduction product 4a along with compound 6. Compound 6 was detected by GC-MS in both boration and reduction reaction mixtures, which is in accordance with the proposed mechanism diagram. Considering that the Lewis acidity of boron could promote the complexation of CD3OD with the alkylborane, we assumed that an alkylboronic ester/CD3OD complex 10 was formed to activate the O–D bond homolysis producing compound 4a.14
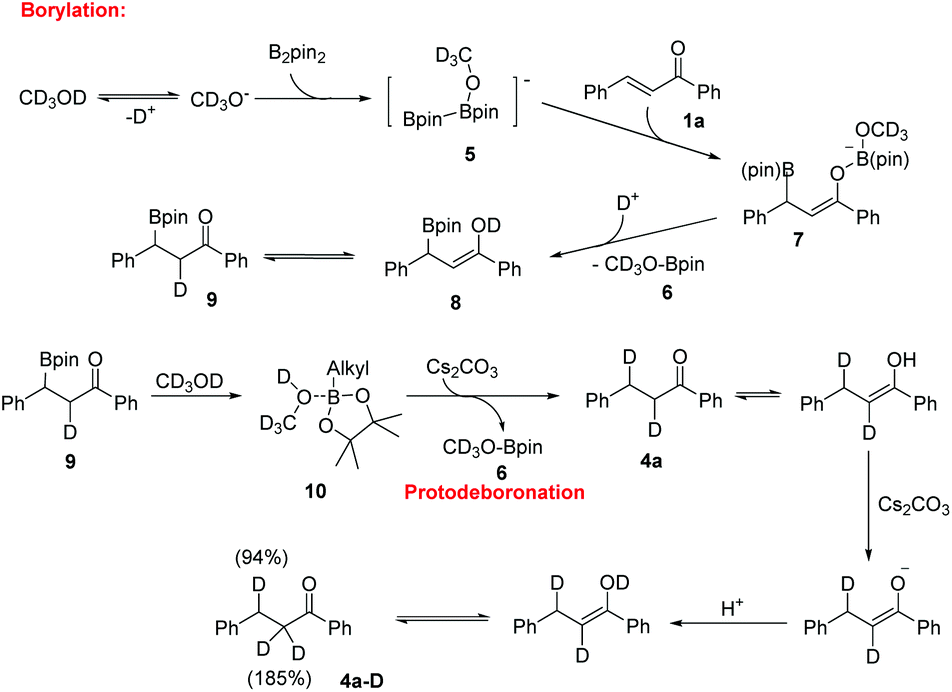 | ||
| Scheme 4 The plausible reaction mechanism. | ||
Conclusions
In conclusion, we firstly reported a highly efficient strategy for the controllable chemoselective conjugated addition and reduction of α,β-unsaturated carbonyl compounds with bis(pinacolato)diboron under transition-metal-free conditions. The boration/protodeboronation cascade process can be effectively controlled by an appropriate base. Moderate to excellent yields of β-boration products and reduction products could be achieved under mild conditions showing a broad substrate scope. This method provides an eco-friendly and product-controllable tool for the highly chemoselective preparation of the β-boration products and 1,4-reduction products of α,β-unsaturated carbonyl compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this study from the National Key Research and Development Plan (Grant No. 2017YFD0200504) and the National Natural Science Foundation of China (Grant No. 21572060) is gratefully acknowledged.Notes and references
- Y. Byun, J. Yan, A. S. A. Madhoun, J. Johnsamuel, W. Yang, R. F. Barth, S. Eriksson and W. Tjarks, J. Med. Chem., 2005, 48, 1188 CrossRef CAS PubMed.
- D. G. Hall, Boronic Acids, John Wiley & Sons, Weiheim, Germany, 2012 Search PubMed.
- E. Hartmann, D. J. Vyas and M. Oestreich, Chem. Commun., 2011, 47, 7917 RSC.
- (a) N. F. Pelz, A. R. Woodward, H. E. Burks, J. D. Sieber and J. P. Morken, J. Am. Chem. Soc., 2004, 126, 16328 CrossRef CAS PubMed; (b) A. Bonet, H. Gulyás, I. O. Koshevoy, F. Estevan, M. Sanaú, M. A. Úbeda and E. Fernández, Chem. – Eur. J., 2010, 16, 6382 CrossRef CAS PubMed.
- (a) Y. G. Lawson, M. J. G. Lesley, T. B. Marder, N. C. Norman and C. R. Rice, Chem. Commun., 1997, 21, 2051 RSC; (b) H. A. Ali, I. Goldberg and M. Srebnik, Organometallics, 2001, 20, 3962 CrossRef CAS; (c) N. J. Bell, A. J. Cox, N. R. Cameron, J. S. O. Evans, T. B. Marder, M. A. Duin, C. J. Elsevier, X. Baucherel, A. A. D. Tulloch and R. P. Tooze, Chem. Commun., 2004, 16, 1854 RSC.
- (a) G. W. Kabalka, B. C. Das and S. Das, Tetrahedron Lett., 2002, 43, 2323 CrossRef CAS; (b) J. B. Morgan, S. P. Miller and J. P. Morken, J. Am. Chem. Soc., 2003, 125, 8702 CrossRef CAS PubMed; (c) T. Shiomi, T. Adachi, K. Toribatake, L. Zhou and H. Nishiyama, Chem. Commun., 2009, 40, 5987 RSC.
- (a) H. Ito, H. Yamanaka, J. Tateiwa and A. Hosomi, Tetrahedron Lett., 2000, 41, 6821 CrossRef CAS; (b) K. Takahashi, T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2001, 625, 47 CrossRef CAS; (c) S. Mun, J.-E. Lee and J. Yun, Org. Lett., 2006, 8, 4887 CrossRef CAS PubMed; (d) J.-E. Lee and J. Yun, Angew. Chem., Int. Ed., 2008, 47, 145 CrossRef CAS PubMed; (e) J.-E. Lee, J. Kwon and J. Yun, Chem. Commun., 2008, 6, 733 RSC; (f) H. Chea, H.-S. Sim and J. Yun, Adv. Synth. Catal., 2009, 351, 855 CrossRef CAS; (g) M. Gao, S. B. Thorpe and W. L. Santos, Org. Lett., 2009, 11, 3478 CrossRef CAS PubMed; (h) H. Chea, H.-S. Sim and J. Yun, Bull. Korean Chem. Soc., 2010, 31, 551 CrossRef CAS; (i) R. Cano, D. J. Ramón and M. Yus, J. Org. Chem., 2010, 75, 3458 CrossRef CAS PubMed.
- (a) K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9, 5031 CrossRef CAS PubMed; (b) Y. Sumida, H. Yorimitsu and K. Oshima, J. Org. Chem., 2009, 74, 3196 CrossRef CAS PubMed; (c) V. Lillo, M. J. Geier, S. A. Westcottb and E. Fernández, Org. Biomol. Chem., 2009, 7, 4674 RSC.
- (a) K. Lee, A. R. Zhugralin and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7253 CrossRef CAS PubMed; (b) L. Wang, Z. Chen, M. Ma, W. Duan, C. Song and Y. Ma, Org. Biomol. Chem., 2015, 13, 10691 RSC; (c) Z. Niu, J. Chen, Z. Chen, M. Ma, C. Song and Y. Ma, J. Org. Chem., 2015, 80, 602 CrossRef CAS PubMed.
- W. Ding and Q. Song, Org. Chem. Front., 2016, 3, 14 RSC.
- (a) Q.-D. Wang, J.-M. Yang, D. Fang, J. Ren, B. Dong, B. Zhou and B.-B. Zeng, Tetrahedron Lett., 2016, 57, 2587 CrossRef CAS; (b) X.-F. Zhou, Y.-Y. Sun, Y.-D. Wu, J.-J. Dai, J. Xu and Y. Huang, Tetrahedron, 2016, 72, 5691 CrossRef CAS.
- (a) J. I. Ehrlich, C.-C. Hwang, P. F. Cook and J. S. Blanchard, J. Am. Chem. Soc., 1999, 121, 6966 CrossRef CAS; (b) A. Kohen and H.-H. Limbach, Isotope Effects in Chemistry and Biology, CRC Press, Taylor & Francis, Boca Raton, 2005 Search PubMed.
- H. Lu, Z. Geng, J. Li, D. Zou, Y. Wu and Y. Wu, Org. Lett., 2016, 18, 2774 CrossRef CAS PubMed.
- D. A. Spiegel, K. B. Wiberg, L. N. Schacherer, M. R. Medeiros and J. L. Wood, J. Am. Chem. Soc., 2005, 127, 12513 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02863f |
| This journal is © The Royal Society of Chemistry 2018 |