
Copper-induced reduction in myocardial fibrosis is associated with increased matrix metalloproteins in a rat model of cardiac hypertrophy
Yinjie
Liu
,
Ying
Xiao
,
Jiaming
Liu
,
Li
Feng
and
Y. James
Kang
 *
*
Regenerative Medicine Research Center, Sichuan University West China Hospital, Chengdu, Sichuan 610041, China. E-mail: yjkang01@louisville.edu; Fax: +86 028-8516-4088; Tel: +86 028-8516-4037
First published on 12th December 2017
Abstract
Trientine (TETA), a copper (Cu) chelator, is capable of replenishing Cu in the heart, and Cu repletion reduces cardiac fibrosis in a rodent model of cardiac hypertrophy. This study was undertaken to explore possible mechanisms by which Cu repletion diminishes cardiac fibrosis. Adult male Sprague-Dawley rats were subjected to ascending aortic constriction to induce cardiac hypertrophy. Four months after the operation, cardiac hypertrophy along with fibrosis was fully developed. TETA treatment was then followed at a dose of 21.9 mg kg−1, twice a day, administered orally for six weeks. At the end of the treatment, the hearts were harvested and all of the tissue samples were subjected to qRT-PCR, western blot, Sirius red staining, hydroxyproline assay, and immunostaining analyses. TETA treatment significantly increased the content of Cu in the hypertrophied myocardium, decreased type III collagen deposition and reduced cardiac fibrosis. On the other hand, this treatment did not alter the increase in fibroblasts induced by pressure overload, but significantly increased matrix metalloproteinase-2 (MMP-2), which is the enzyme mainly responsible for degradation of collagens in the heart. In addition, the mRNA and protein levels of tissue inhibitors of matrix metalloproteinase-1 and -2 (TIMP-1 and TIMP-2) were both remarkably increased in the hypertrophic myocardium, and normalized after TETA treatment. The data thus demonstrated that the reduction in cardiac fibrosis by TETA-induced Cu repletion is associated at least in part with an enhanced MMP-2 activity, leading to collagen degradation.
1. Introduction
Prolonged pressure overload decreases cardiac copper (Cu) concentrations and promotes cardiac fibrosis,1,2 leading to heart failure.3,4 Previous studies have demonstrated that dietary Cu supplementation replenishes cardiac Cu and reduces cardiac fibrosis, consequently preventing the pathogenesis of heart failure even in the presence of sustained pressure overload.2,5 However, the molecular mechanism by which Cu repletion reduces cardiac fibrosis remains elusive.Cardiac fibrosis results from an imbalance of collagen homeostasis.6–8 Upon pressure overload, cardiomyocytes become hypertrophic and fibroblasts are activated to produce collagens, especially collagens I and III.9,10 These fibers around hypertrophied cardiomyocytes limit the energy supply, augment the hypoxic state and cause myocardial cell death, finally leading to decompensated heart failure.10,11 Hence, normalizing cardiac collagen homeostasis would be an effective approach to reduce cardiac fibrosis.
Disturbed collagen homeostasis is related to both the production and the degradation of collagens.12,13 The degradation of collagens is affected by the changes in matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs).14,15 Previous studies have shown that cardiac fibrosis was fully developed at 8 weeks after ascending aortic constriction,16 and the level of MMP-2 increased at 3 weeks and returned to the control level at 8 weeks, but the level of MMP-9 was increased at 8 weeks after surgery.17 Although both MMP-2 and MMP-9 are gelatinases and are capable of degrading collagens in mammalian systems,18,19 the major enzyme responsible for degradation of collagens in the heart is MMP-2.20 The activation of these enzymes is often accompanied by an increase in TIMPs for counteraction with their activity of collagen degradation. Interestingly, it was observed that the expression of TIMP-1 and TIMP-2 was increased at 8 weeks after pressure overload surgery.17 Therefore, the down-regulated MMP-2 activity and the up-regulated TIMPs together suppress collagen degradation and promote cardiac fibrosis in the heart.
Trientine (TETA), a Cu chelator, is not only capable of reducing serum Cu levels and increasing the excretion of Cu from urine,21 but also capable of replenishing Cu in the heart.22 TETA thus provides a tool to replete Cu concentrations in the hypertrophic myocardium. Thus, we undertook this study to test the hypothesis that Cu repletion by TETA treatment enhances collagen degradation, leading to reduction in cardiac fibrosis. We found that enhanced collagen III degradation due to an augmentation in MMP-2 activity and a reduction in TIMP-1 and -2 would crucially contribute to Cu repletion-induced reduction in myocardial fibrosis in a rat model of cardiac hypertrophy.
2. Materials and methods
2.1 Animal model and TETA treatment
Male Sprague-Dawley rats (6–8 weeks old, on average 220 g) were obtained from Chengdu Da-Shuo Experimental Animal Breeding and Research Center, an accredited rodent animal center in Sichuan Province, China. The rats were accustomed to the conditions for at least 1 week in the Animal Care and Use Center. The animals were handled in strict accordance with good animal practice approved by the Laboratory Animal Management Committee of Sichuan Province (LAMC).All rats were randomly divided into sham operated control (n = 10) and ascending aorta constriction (AAC) groups (n = 25). As mentioned previously,16 the rats were subjected to AAC to induce pathological cardiac hypertrophy. Briefly, the aortic arch of the rat was exposed, a prepared piece of a 6-0 silk suture was used to decrease the aortic diameter by one half and then the chest cavity and all layers of muscle and skin were closed with 5-0 nylon sutures. The sham-operated controls underwent the same surgical procedure without aortic banding. To reduce pain after surgery, the analgesic dezocine (0.8 mg kg−1) was given intramuscularly and once daily for two days. Four months after the operation, cardiac hypertrophy along with fibrosis was determined by histopathological examination in three rats randomly selected from the AAC group. At the same time, the AAC group was divided into a normal saline-treated group (AAC-NS, n = 11) and a TETA-treated group (AAC-TETA, n = 11).
Based on the dosages of TETA used for the treatment of Wilson’ s disease in children, varying from 500 to 750 mg per day (equivalent to ∼12.5–18.8 mg kg−1 per day in 40 kg children),23,24 TETA at a dose of 21.9 mg kg−1 per day (equivalent to ∼ 6 mg kg−1 day in 40 kg children), around one half of the recommended minimum dosage, was administered orally for six weeks in the AAC-TETA group, while the surgery-matched rats received the same volume (5 ml kg−1) of normal saline twice a day.
2.2 Tissue preparation
Animal harvest was performed by euthanasia via intravenous injection of 10% potassium chloride (2 ml kg−1) under anesthesia (10% chloral hydrate 35 mg kg−1) at the end of the treatment. After being perfused with 30 ml of cold PBS containing 0.1 ml of 1% heparin, the heart was divided into three parts: one part was stored at 4 °C for Cu concentration detection, hydroxyproline assay and quantitative reverse-transcription polymerase chain reaction (qRT-PCR); another part was cut into several pieces and preserved in liquid nitrogen for western blot; and the remaining part was embedded in OCT gel and then frozen in liquid nitrogen to prepare frozen sections for Sirius red staining, and immunohistochemical and immunofluorescence analyses.2.3 Cu concentrations in heart tissue
Heart tissue samples stored at −80 °C were lyophilized under vacuum and weighed. The samples were then digested with concentrated nitric acid at 60 °C overnight. Ultrapure water was added to dilute HNO3 to a final concentration of 2%. Cu concentrations were determined by graphite furnace atomic absorption spectrometry (ICE3500, Thermo). The amount of Cu was corrected for the dry tissue weight.2.4 Measurement of the collagen volume fraction (CVF)
The collagens were visualized using Sirius red, while other non-collagen proteins were shown using Fast Green. Sections stained with Sirius red were examined under a microscope (Nikon, 80i) with a 10× ocular lens and a 4× objective lens and photographed using a SPOT FLEX. The CVF was measured using Image Pro-Plus 6.0 (IPP 6.0) and calculated as the proportion of Sirius red positive areas in the total section areas.2.5 Measurement of the average size of cardiomyocytes
All the sections were fixed and incubated with 20 μg ml−1 FITC-conjugated lectin collected from Triticum vulgaris (WGA, Sigma, L4895) to label the cell membrane at 37 °C for 1 hour and then photographed under a confocal microscope. Ten visual fields of the left ventricle of each sample were collected and the average size of about 300 cells in each heart was determined using IPP 6.0 software.2.6 Immunohistochemistry and immunofluorescence analysis
Briefly, the slides were blocked with 3% hydrogen peroxide for 20 min and followed by incubation with 1% bovine serum albumin at 37 °C for 1 h under dark conditions. After being washed with PBS 3 times, 5 min each time, the slides were further incubated with the primary antibodies rabbit anti-rat type I collagen and type III collagen polyclonal antibodies (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200, Abcam) overnight in a refrigerator at 4 °C. Then the slides were incubated with HRP labeled secondary antibody mouse anti-rabbit antibody (CST) at 37 °C for 1 h. Finally, freshly mixed diaminobenzidine (DAB, CST) was added according to the manufacturer's instruction, whereas the nuclei were counterstained with hematoxylin.
:200, Abcam) overnight in a refrigerator at 4 °C. Then the slides were incubated with HRP labeled secondary antibody mouse anti-rabbit antibody (CST) at 37 °C for 1 h. Finally, freshly mixed diaminobenzidine (DAB, CST) was added according to the manufacturer's instruction, whereas the nuclei were counterstained with hematoxylin.
The staining on the tissue sections was detected using a light microscope (Nikon, 80i) and the images were digitized (SPOT FLEX). Under 200-fold magnification, three visual fields were randomly observed from each slide, defining the average optical density with a positive expression (integrated optical density/area) for semi-quantitative analysis.
For immunofluorescence analysis, the slides were blocked with 1% BSA as described above and then incubated with the first antibody, mouse anti-human vimentin monoclonal antibody (1:400, Abcam), at 37 °C for 2 h and then overnight in a refrigerator at 4 °C. After being washed with PBS, the slides were incubated with Alexa Fluor 568 goat anti-rabbit IgG (1:1000, Thermo Fisher). And then the nuclei were stained by incubation with DAPI (Sigma) for confocal observation (Nikon, A1, ECLIPSE Ti).
2.7 Western blot analysis
Tissues were extracted in RIPA buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP-40, and 0.1% SDS) to collect total proteins. Then 1% EDTA-free protease inhibitor cocktail (Roche Diagnostics, Germany) was added to protect proteins from degradation. Further, the protein concentrations of collected supernatants were determined using a BCA protein assay kit (Thermo Fisher, USA) and then mixed with 5× loading buffer and denatured in boiling water.Equal amounts of protein (30 μg) from each sample were separated by 10% SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Bio-Rad Laboratories, USA). Membranes were blocked in 5% nonfat dry milk in Tris–HCl buffer solution (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, and 0.1% Tween-20) for 1 h at room temperature, and then incubated with primary antibodies, MMP-2 (1:1000, Abcam), MMP-9 (1:1000, Abcam), TIMP-1 (1:1000, Abcam), TIMP-2 (1:1000, Abcam) and mouse anti-GAPDH monoclonal antibodies (Zhongshan) overnight at 4 °C. Appropriate secondary antibodies were further used to incubate the membranes. Finally, the protein bands were observed by adding a chemiluminescent horseradish peroxidase substrate (Millipore Corporation, Germany) and analyzed using vilber fusion software (Bio-Rad Laboratories, USA).
2.8 Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the myocardium in liquid nitrogen, and then dissolved in Trizol reagent (Invitrogen, USA) according to the manufacturer's instructions. The integrity of total RNA was detected by Sepharose gio-gel, and the concentration was quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher, USA). RNA was reverse transcribed to complementary DNA (cDNA) using a PrimeScript® RT reagent kit (TaKaRa, Japan) in an MJ Mini Personal Thermal Cycler (Bio-Rad, USA). The amount of cDNA corresponding to 50 ng of RNA was amplified using a SYBR Green PCR kit (Bio-Rad Laboratories) with the primers for MMP-2, MMP-9, TIMP-1, TIMP-2 and ribosomal protein S18 (RPS18). The primer sequences (Table 1) were designed and synthesized by Invitrogen.| Gene | Primer |
|---|---|
| MMP-2 | Forward primer: GGGTGGTGGTCACAGCTATT |
| Reverse primer: CCCAGCCAGTCCGATTTGAT | |
| MMP-9 | Forward primer: AGGACGGTCGGTATTGGAAG |
| Reverse primer: GTGCGGGCAATAAGAAAGGG | |
| TIMP-1 | Forward primer: ACAGCTTTCTGCAACTCGGA |
| Reverse primer: CGTCGAATCCTTTGAGCATC | |
| TIMP-2 | Forward primer: TAATTGCAGGGAAGGCGGAA |
| Reverse primer: CGTGTCCCAGGGCACAATAA | |
| RPS18 | Forward primer: CCTTCGCTATCACTGCCATT |
| Reverse primer: TGGCCAGAACCTGGCTATAC | |
2.9 Statistical analysis
Data are presented as means ± SEM. SPSS 22.0 and Prism 6.0 were applied to perform statistical processing. An independent-sample t-test was applied when normality (and homogeneity of variance) assumptions were satisfied; otherwise, a nonparametric test would be used. Comparisons were done between the Sham and AAC groups, Sham-NS and AAC-NS groups, and AAC-NS and AAC-TETA groups. For all analyses, P values <0.05 were considered significant.3. Results
3.1 Cu repletion in the hypertrophied myocardium after TETA treatment
Cardiac Cu concentrations were measured by atomic absorption spectrometry to confirm the efficacy of TETA in replenishing cardiac Cu levels. As shown in Fig. 1, the cardiac Cu concentrations were decreased in the hypertrophic myocardium compared to those of the sham-operated controls, and TETA treatment significantly increased the Cu concentrations in the hypertrophic myocardium.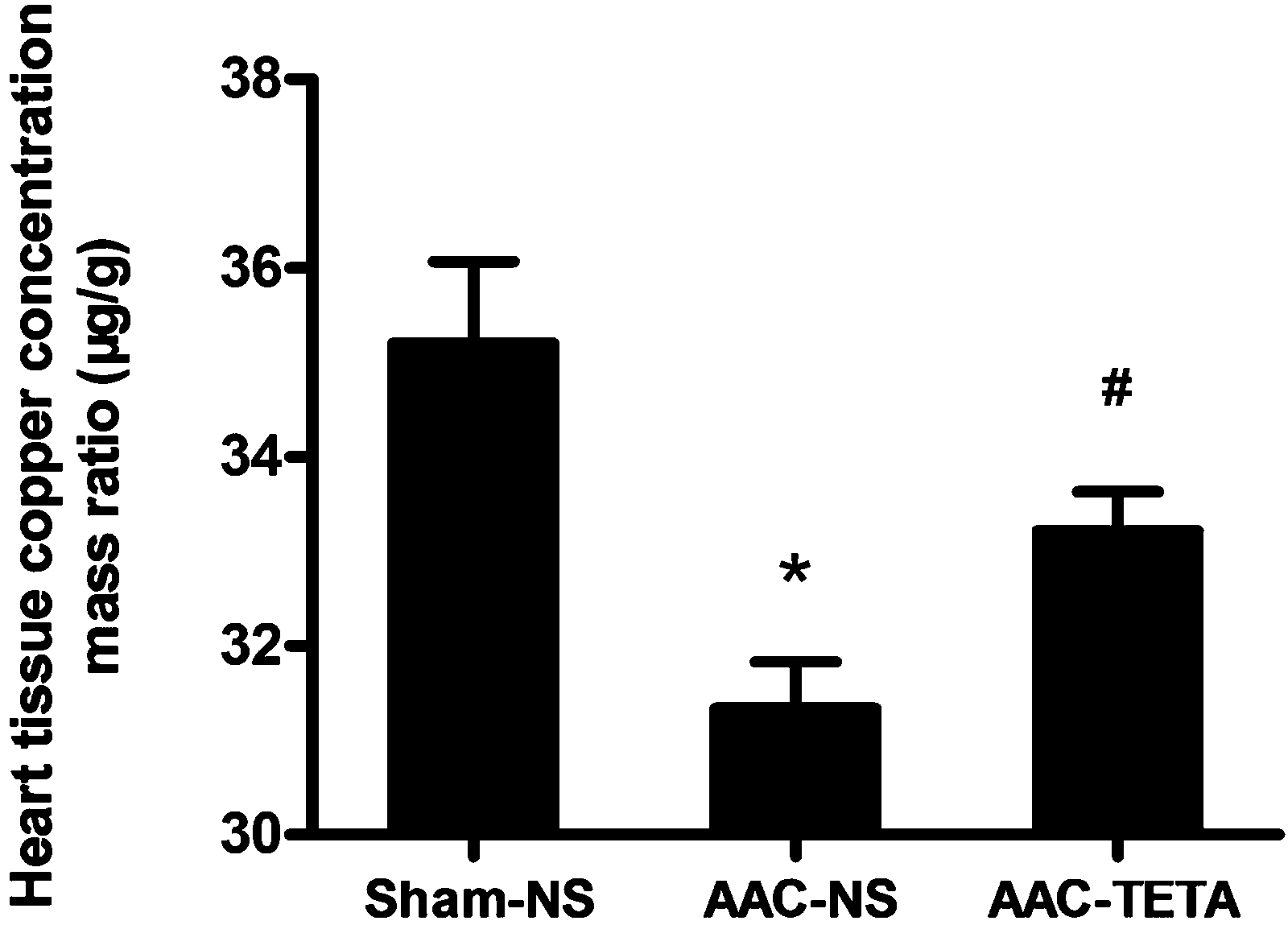 | ||
| Fig. 1 Cu concentrations in the hypertrophied myocardium and the effect of TETA treatment. The hypertrophied myocardium was induced by ascending aortic constriction (AAC) for four months and then TETA treatment was followed at a dose of 21.9 mg kg−1, twice a day, administered orally for six weeks. Cu concentrations were determined by graphite furnace atomic absorption spectrometry. Data are presented as means ± SEM and taken from n = 6–7. *P < 0.05 versus the Sham-NS group. #P < 0.05 versus the AAC-NS group. | ||
3.2 Changes in cardiac function after TETA treatment
Alterations in cardiac function were measured by echocardiography. After the AAC surgery (AAC-NS), the ejection fraction (EF) and fractional shortening (FS) were both increased in comparison with those of the sham operated controls (Sham-NS). This increase continued for 3 weeks after the surgery, and then the EF and FS decreased and reached the levels which were the same as those of the sham controls at the end of the experiment. However, no decrease in both EF and FS was observed in the AAC-TETA group was not observed; the high EF and FS sustained until the end of the experiment (Fig. 2A and B).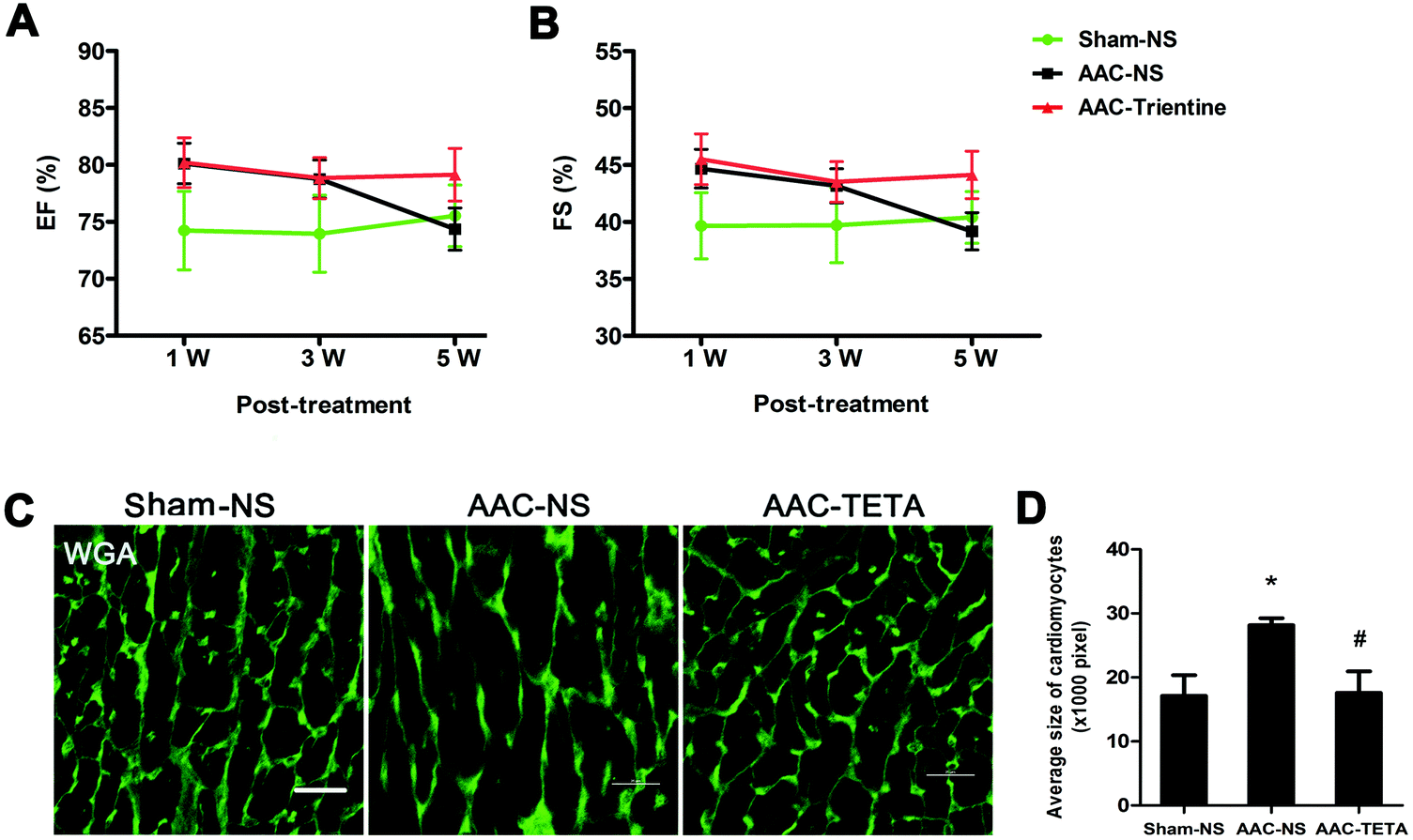 | ||
| Fig. 2 Changes in cardiac function and the morphology of cardiomyocytes before and after TETA treatment. Changes in cardiac function were determined by echocardiography. (A) Ejection fraction (EF) and (B) fractional shortening (FS) at different time points. (C) Cell membranes labeled with FITC-conjugated WGA (green). (D) Comparison of the average sizes of cardiomyocytes in the Sham-NS, AAC-NS and AAC-TETA groups. Data are presented as means ± SEM and taken from n = 8–10. *P < 0.05 versus the Sham-NS group. #P < 0.05 versus the AAC-NS group. | ||
3.3 Changes in the size of cardiomyocytes after TETA treatment
The cell membranes of cardiomyocytes were stained with FITC-conjugated WGA to determine the size of cardiomyocytes (Fig. 2C). The average size of cardiomyocytes in the AAC-NS group was significantly increased in comparison with that of the Sham-NS group. TETA treatment reversed the enlarged size of cardiomyocytes, making the size comparable with that of the Sham-NS group (Fig. 2C and D).3.4 Changes in cardiac collagens after TETA treatment
Myocardial tissue sections were stained with Sirius red to define the total collagens in the myocardium. As shown in Fig. 3A, four months after AAC surgery, collagens stained red were markedly increased in the endocardium of the hypertrophied hearts compared with the sham operated control heart tissues. Six weeks after TETA treatment, the excessive collagen deposition was significantly diminished (Fig. 3D and E). This reduction in the collagen content was further confirmed by the measurement of the hydroxyproline content (Fig. 3C). Immunohistochemical staining further revealed a remarkable increase in type III collagen in the hypertrophied myocardium and this increase was significantly decreased by about 30% after TETA treatment (Fig. 3H and I). On the other hand, type I collagen did not change in the fibrotic heart before or after TETA treatment (Fig. 3F and G). Fibroblasts, which were determined by anti-vimentin immunofluorescence staining, were significantly increased in the fibrotic heart compared to the sham control myocardium, but this increase did not change after TETA treatment (Fig. 3J and K).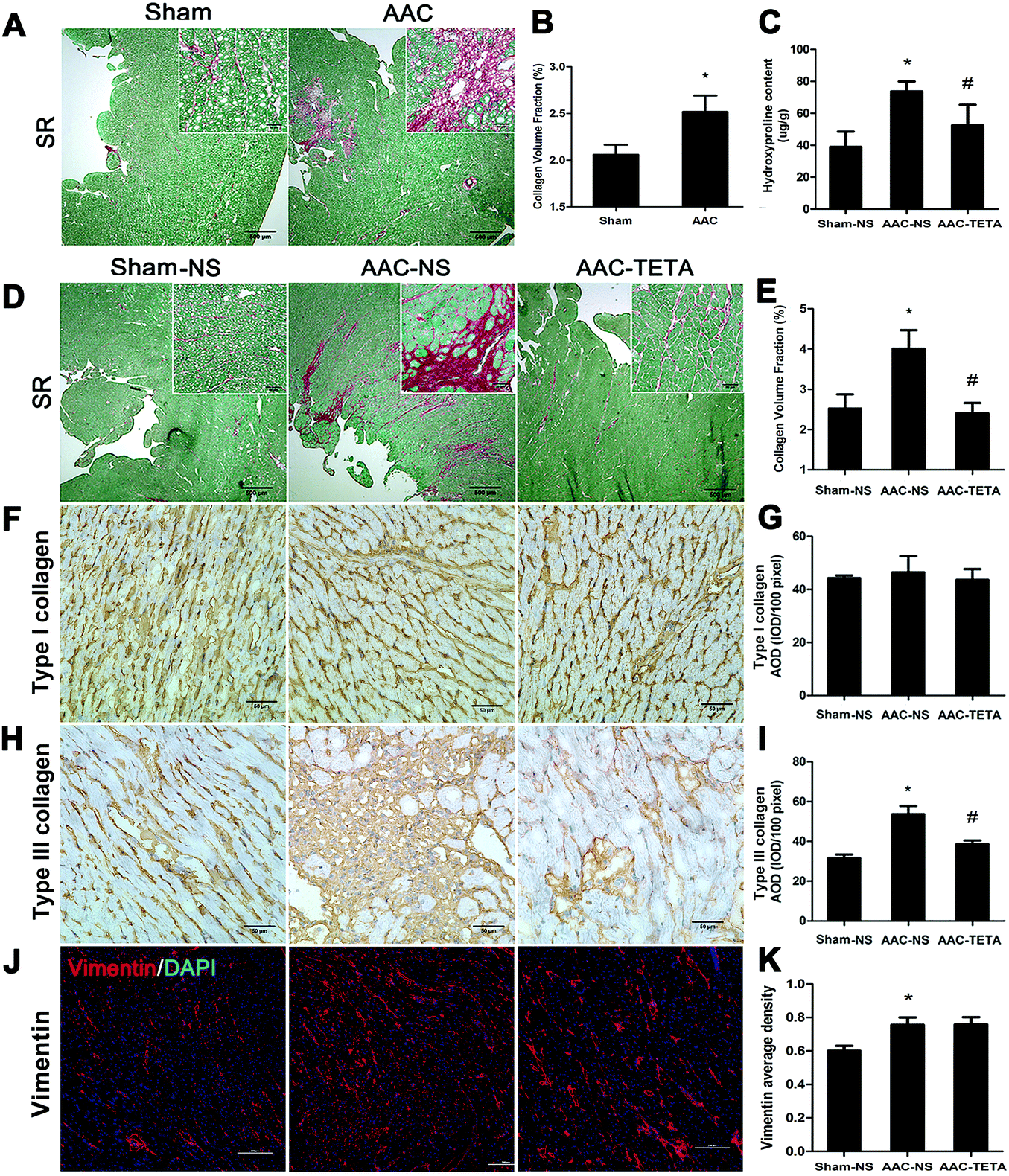 | ||
| Fig. 3 Changes in cardiac collagens and fibroblasts in the hypertrophied myocardium. (A) Total collagen deposition determined by Sirius red staining at 4 months post AAC surgery before TETA treatment. Red, total collagens; green, non-collagenous proteins. (B) Quantitative analysis of the collagen volume fraction (CVF) in the AAC and Sham groups. Data are presented as means ± SEM and taken from n = 3. *P < 0.05 versus the Sham group. (C) Hydroxyproline content in the Sham-NS, AAC-NS and AAC-TETA groups after TETA treatment. (D) Changes in total collagens in the hypertrophied myocardium at the end of 6 weeks post TETA treatment. Scale bar, 500 μm in the visual field; and scale bar, 50 μm in the smaller visual field at the top right corner of each section. (E) Quantitative analysis of the CVF in the Sham-NS, AAC-NS and AAC-TETA groups. (F and H) Type I and type III collagen concentrations and localizations determined by immunohistochemistry. Bar = 500 μm. Brown, type I or type III collagen; blue, nuclei stained with hematoxylin. (G and I) Average optical density (AOD) values of type I and type III collagen staining among the three groups. (J) Fibroblasts shown by anti-vimentin immunofluorescence staining (red) and counterstained with DAPI to show nuclei (blue). Bar = 200 μm. (K) Average optical density (AOD) values of vimentin staining among the three groups. Data are means ± SEM and taken from n = 5–8. *P < 0.05 versus the Sham-NS group. #P < 0.05 versus the AAC-NS group. | ||
3.5 Changes in MMP-2, MMP-9, TIMP-1 and TIMP-2 after TETA treatment
The changes in MMP-2 and MMP-9, gelatinases that are involved in collagen homeostasis by cleaving collagens, were defined by their protein levels. Western blot analyses revealed that MMP-9 protein levels did not change in the fibrotic heart before or after TETA treatment (Fig. 4A and B), although its mRNA levels were increased in the hypertrophied myocardium (Fig. 4C). In contrast, MMP-2 protein levels were significantly reduced (by ∼25%) in the hypertrophied myocardium in comparison with the sham operated controls, and after TETA treatment the MMP-2 protein levels were not only recovered, but also further increased (Fig. 4A and D), although its mRNA levels of MMP-2 did not change (Fig. 4E). | ||
| Fig. 4 Protein and mRNA concentrations of MMPs and TIMPs. (A, B and D) Protein levels of MMP-9 and -2, using GAPDH as a loading control. (C and E) Relative mRNA levels (versus RPS18) of MMP-9 and -2. (F, G and I) Protein levels of TIMP-1 and -2. GAPDH served as a loading control. (H and J) Relative mRNA expression levels of TIMP-1 and -2. RPS18 served as a loading control. Data are means ± SEM and taken from n = 6–7. *P < 0.05 versus the Sham-NS group. #P < 0.05 versus the AAC-NS group. | ||
It was found that not only the protein levels of TIMP-1 (Fig. 4F and G) and TIMP-2 (Fig. 4F and I), but also their mRNA levels (Fig. 4H and J) were remarkably increased post AAC surgery, and Cu repletion completely eliminated this increase.
4. Discussion
Cu repletion facilitated remission of cardiac fibrosis and improved heart function in the presence of sustained pressure overload.2,5 TETA, a Cu chelator, was found to replenish Cu concentrations in the heart.22 In the present study, TETA at a safe dose of 21.9 mg kg−1, twice a day and administrated orally, significantly replenished cardiac Cu concentrations in the rat model of cardiac hypertrophy. This Cu repletion was also accompanied by reduction in cardiac fibrosis, as reported previously that dietary Cu supplementation was associated with reduction in myocardial fibrosis in a mouse model of pressure overload-induced cardiac hypertrophy.2 The present study revealed that this reduction in myocardial fibrosis was ascribed at least in part to the increase in MMP-2, leading to enhanced degradation of collagens, as evidenced by the reduction in type III collagens, the substrate of MMP-2.An imbalance of collagen homeostasis results in myocardial fibrosis.25 We assessed the changes in the content of collagens, as it was reported previously that the deposition of these collagens made a critical contribution to fibrosis.26,27 In our observation, the deposition of excessive type III collagens, not type I collagens, was significantly involved in the pathogenesis of myocardial fibrosis, as observed 4 months after AAC-induced pressure overload. This deposition was completely diminished after TETA-treatment.
How would the reduction in type III collagen take place? The imbalance between the production and the degradation of collagens causes improper collagen deposition in tissues. In the present study, it appeared that changes in MMP-2 would make an important contribution to the overt deposition of collagen III. In the hypertrophic myocardium, the protein level of MMP-2 was significantly decreased, and after Cu repletion, MMP-2 was not only recovered, but also further increased. The pattern of changes in MMP-2, decrease versus increase, was reversely paralleled with the changes in collagen type III deposition, increase versus decrease, suggesting that the capacity of collagen degradation under different conditions would be responsible for the fluctuation of collagen deposition.
The activity of MMPs is not only related to their protein levels, but also regulated by TIMPs. Interestingly, we observed here that TIMP-1 and -2 were significantly increased in the hypertrophic myocardium, and this increase was completely eliminated by Cu repletion through TETA treatment. The increase in TIMP-1 and -2, in combination with the decrease in MMP-2 protein levels in the hypertrophic myocardium, would allow an increase in collagen deposition as observed here. The reversal of this phenomenon was also true as evidenced by the observation that the increase in MMP-2 along with the decrease in TIMP-1 and -2 was accompanied by decreased collagen deposition.
It was interesting to observe that MMP-9 did not significantly change under different conditions although MMP-2 remarkably changed. Indeed, MMP-2 was shown to be capable of cleaving type III collagen.28
How did Cu repletion specifically modify the protein level of MMP-2? It was demonstrated that the MMP-2 expression is regulated by HIF-129 as it was identified that there is a typical HIF-1 binding site at the 5′ end of the MMP-2 gene sequence.30 We have previously demonstrated that Cu is required for HIF-1 transcriptional activity, although the expression of HIF-1 proteins is not affected by Cu.31,32 However, in the present study, we did not observe any change in MMP-2 mRNA levels in response to TETA treatment, suggesting that the up-regulation of the MMP-2 expression might not be a major contribution to the observed enhancement of MMP-2 activity. On the other hand, MMP-2 is zinc-dependent, and Cu is involved in zinc uptake.20,33 It has been shown that zinc concentrations in the serum and heart were increased upon Cu supplementation,33 along with upregulation of MMP-2.34
It appeared that MMP-2 played a major role in the degradation of collagens; however, other possibilities cannot be excluded. In our previous studies,8,35 we have observed that myocardial lysyl oxidase (LOX) changes significantly in response to changes in Cu concentrations. We observed that pressure overload increased LOX in the heart, and Cu supplementation normalized LOX activity.5 LOX is involved in the crosslinking of collagens and suppression of its activity would remit the crosslinking, assisting the degradation of collagens by MMPs.
It is important to note that the fibrolysis observed in the hypertrophied myocardium was associated with improvements in both the morphology of the cardiomyocytes and the overall cardiac function. The enlarged size of cardiomyocytes induced by pressure overload was significantly reversed by the TETA treatment, in agreement with earlier observations that Cu supplementation regressed cardiac hypertrophy.1,2 Cardiac contractility initially increased in response to pressure overload, reflecting a compensatory response. This increase was followed by a decrease due to decompensatory reaction upon sustained pressure overload. It was observed here that with TETA treatment the decline in cardiac contractility was prevented, indicating improved cardiac function under this circumstance.
In conclusion, we followed previous observations that TETA treatment replenished Cu concentrations in hypertrophic hearts and that Cu repletion reduced pressure overload-induced cardiac fibrosis to investigate possible mechanisms for Cu reduction of cardiac fibrosis. Our data indicate that enhanced collagen degradation due to increased MMP-2 and decreased TIMP-1 and -2 would primarily be responsible for the reduction in cardiac fibrosis upon Cu repletion. This fibrolysis response was associated with the reversal of cardiomyocyte hypertrophy and improvement in cardiac contractility.
Author contributions
All the authors participated in the design, analysis of the data, interpretation of the results, and review of the manuscript; Y. L., Y. X. and J. L. carried out the experiments; and Y. J. K., L. F. and Y. L. wrote the manuscript.Conflicts of interest
All the authors report no conflicts of interest.Acknowledgements
This work was supported by the National Science Foundation of China (grant number 81230004 to Y. J. Kang and grant number 81300109 to P. Han). The authors thank Suzhen Fan and Fulai Xue for technical support.References
- L. Zheng, P. Han, J. Liu, R. Li, W. Yin, T. Wang, W. Zhang and Y. J. Kang, Pharmacol. Ther., 2015, 148, 66–84 CrossRef CAS PubMed.
- Y. Jiang, C. Reynolds, C. Xiao, W. Feng, Z. Zhou, W. Rodriguez, S. C. Tyagi, J. W. Eaton, J. T. Saari and Y. J. Kang, J. Exp. Med., 2007, 204, 657–666 CrossRef CAS PubMed.
- S. Mahinrad, A. E. Vriend, J. W. Jukema, D. van Heemst, N. Sattar, G. J. Blauw, P. W. Macfarlane, E. N. Clark, A. J. M. de Craen and B. Sabayan, J. Alzheimer's Dis., 2017, 58, 275–283 CrossRef PubMed.
- L. Elsherif, R. V. Ortines, J. T. Saari and Y. J. Kang, Exp. Biol. Med., 2003, 228, 811–817 CrossRef CAS.
- W. M. Hughes, Jr., W. E. Rodriguez, D. Rosenberger, J. Chen, U. Sen, N. Tyagi, K. S. Moshal, T. Vacek, Y. J. Kang and S. C. Tyagi, Cardiovasc. Toxicol., 2008, 8, 137–144 CrossRef PubMed.
- N. L. Rosin, M. J. Sopel, A. Falkenham, T. D. Lee and J. F. Legare, Am. J. Physiol., 2015, 185, 631–642 CAS.
- K. Echegaray, I. Andreu, A. Lazkano, I. Villanueva, A. Saenz, M. R. Elizalde, T. Echeverria, B. Lopez, A. Garro, A. Gonzalez, E. Zubillaga, I. Solla, I. Sanz, J. Gonzalez, A. Elosegui-Artola, P. Roca-Cusachs, J. Diez, S. Ravassa and R. Querejeta, Rev. Esp. Cardiol., 2017, 70, 832–840 CrossRef.
- Y. Xiao, X. Nie, P. Han, H. Fu and Y. J. Kang, Metallomics, 2016, 8, 973–980 RSC.
- C. Farquharson and S. P. Robins, J. Comp. Pathol., 1991, 104, 245–255 CrossRef CAS PubMed.
- J. B. Caulfield and T. K. Borg, Lab. Invest., 1979, 40, 364–372 CAS.
- X. M. Gao, H. Kiriazis, X. L. Moore, X. H. Feng, K. Sheppard, A. Dart and X. J. Du, Am. J. Physiol. Heart Circ. Physiol., 2005, 288, H2702–2707 CrossRef CAS PubMed.
- P. Kong, P. Christia and N. G. Frangogiannis, Cell. Mol. Life Sci., 2014, 71, 549–574 CrossRef CAS PubMed.
- A. H. Li, P. P. Liu, F. J. Villarreal and R. A. Garcia, Circ. Res., 2014, 114, 916–927 CrossRef CAS PubMed.
- T. Kinoshita, Y. Ishikawa, M. Arita, Y. Akishima-Fukasawa, K. Fujita, N. Inomata, T. Suzuki, A. Namiki, T. Mikami, T. Ikeda, J. Yamazaki, T. Ishii and Y. Akasaka, Cardiovasc. Pathol., 2014, 23, 92–100 CrossRef CAS PubMed.
- E. C. El Hajj, M. C. El Hajj, T. G. Voloshenyuk, A. J. Mouton, E. Khoutorova, P. E. Molina, N. W. Gilpin and J. D. Gardner, Alcohol.: Clin. Exp. Res., 2014, 38, 448–456 CrossRef CAS PubMed.
- J. Liu, P. Han, Y. Xiao, J. Liu and Y. J. Kang, Am. J. Physiol.: Heart Circ. Physiol., 2014, 307, H987–H995 CrossRef CAS PubMed.
- S. Givvimani, N. Tyagi, U. Sen, P. K. Mishra, N. Qipshidze, C. Munjal, J. C. Vacek, O. A. Abe and S. C. Tyagi, Arch. Physiol. Biochem., 2010, 116, 63–72 CrossRef CAS PubMed.
- B. D. Shogan, N. Belogortseva, P. M. Luong, A. Zaborin, S. Lax, C. Bethel, M. Ward, J. P. Muldoon, M. Singer, G. An, K. Umanskiy, V. Konda, B. Shakhsheer, J. Luo, R. Klabbers, L. E. Hancock, J. Gilbert, O. Zaborina and J. C. Alverdy, Sci. Transl. Med., 2015, 7, 286ra268 Search PubMed.
- R. T. Aimes and J. P. Quigley, J. Biol. Chem., 1995, 270, 5872–5876 CrossRef CAS PubMed.
- R. Visse and H. Nagase, Circ. Res., 2003, 92, 827–839 CrossRef CAS PubMed.
- J. Lu, Y. K. Chan, G. D. Gamble, S. D. Poppitt, A. A. Othman and G. J. Cooper, Drug Metab. Dispos., 2007, 35, 221–227 CrossRef CAS PubMed.
- L. Zhang, M. L. Ward, A. R. Phillips, S. Zhang, J. Kennedy, B. Barry, M. B. Cannell and G. J. Cooper, Cardiovasc. Diabetol., 2013, 12, 123 CrossRef PubMed.
- T. Dahlman, P. Hartvig, M. Lofholm, H. Nordlinder, L. Loof and K. Westermark, QJM., 1995, 88, 609–616 CAS.
- H. Y. Cho, R. A. Blum, T. Sunderland, G. J. Cooper and W. J. Jusko, J. Clin. Pharmacol., 2009, 49, 916–928 CrossRef CAS PubMed.
- K. T. Weber, J. S. Janicki, S. G. Shroff, R. Pick, R. M. Chen and R. I. Bashey, Circ. Res., 1988, 62, 757–765 CrossRef CAS PubMed.
- A. M. Ahmed, Folia Morphol., 2017, 76, 15–27 CrossRef CAS PubMed.
- K. T. Weber, J. Am. Coll. Cardiol., 1989, 13, 1637–1652 CrossRef CAS PubMed.
- B. Steffensen, U. M. Wallon and C. M. Overall, J. Biol. Chem., 1995, 270, 11555–11566 CrossRef CAS PubMed.
- S. W. Jing, Y. D. Wang, M. Kuroda, J. W. Su, G. G. Sun, Q. Liu, Y. J. Cheng and C. R. Yang, Acta Med. Okayama, 2012, 66, 399–407 CAS.
- P. S. Chen and Z. Feng, J. Gastroenterol. Hepatol., 2006, 21, A74 Search PubMed.
- Z. Zhang, L. Qiu, C. Lin, H. Yang, H. Fu, R. Li and Y. J. Kang, Metallomics, 2014, 6, 1889–1893 RSC.
- L. Qiu, X. Ding, Z. Zhang and Y. J. Kang, J. Pharmacol. Exp. Ther., 2012, 342, 561–567 CrossRef CAS PubMed.
- L. Elsherif, L. Wang, J. T. Saari and Y. J. Kang, J. Nutr., 2004, 134, 855–860 CAS.
- L. Elsherif, Y. Jiang, J. T. Saari and Y. J. Kang, Exp. Biol. Med., 2004, 229, 616–622 CrossRef CAS.
- Y. Xie, J. Chen, P. Han, P. Yang, J. Hou and Y. J. Kang, Exp. Biol. Med., 2012, 237, 853–859 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |