
Reactive oxygen species (ROS)-responsive nanomedicine for RNAi-based cancer therapy†
Yang
Li‡
a,
Hongzhen
Bai‡
 a,
Hebin
Wang
a,
Youqing
Shen
b,
Guping
Tang
*a and
Yuan
Ping
*c
a,
Hebin
Wang
a,
Youqing
Shen
b,
Guping
Tang
*a and
Yuan
Ping
*c
aInstitute of Chemical Biology and Pharmaceutical Chemistry, Department of Chemistry, Zhejiang University, Hangzhou 310028, P. R. China. E-mail: tangguping@zju.edu.cn
bCenter for Bionanoengineering and State Key Laboratory for Chemical Engineering, Department of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, P. R. China
cCollege of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310058, P. R. China. E-mail: pingyuan7@gmail.com; pingy@zju.edu.cn
First published on 15th November 2017
Abstract
Although much effort has been dedicated to the development of efficient siRNA delivery for cancer therapy, delivery nanomaterials that can particularly respond to reactive oxygen species (ROS), which are overproduced in the tissue and mitochondria of cancer cells, are still rare for the clinical translation of RNA interference (RNAi)-based therapy. To this end, we developed a ROS-responsive boronic vehicle with a lipid envelope for systemic vascular endothelial growth factor (VEGF) siRNA delivery so as to improve RNAi cancer therapy. We found that the efficiency of siRNA delivery largely relied on the ROS responsiveness of the carrier we have developed to mediate timely siRNA release, the PEG-functionalized lipid layer to shield the surface charge of polyplexes as well as the ability of the phenylboronic moiety to stabilize siRNA. The unique carrier nanostructure provides the efficient systemic transportation of siRNA to the tumor site for effective knockdown of the VEGF, which resulted in a significant antiangiogenesis effect and the effective inhibition of tumor growth in vivo. The current study defines a new systemic delivery strategy for siRNA by cooperatively integrating multifunctional lipid coatings with the ROS-responsive boronic polymer, which may potentially benefit RNAi-based therapy in the dawning era of precision nanomedicine for cancer therapy.
1. Introduction
Sequence-specific gene silencing mediated by RNA interference (RNAi) is a promising gene regulatory approach for cancer therapy.1 As the formation of new blood and lymph vessels (namely angiogenesis and lymphangiogenesis, respectively) is well known to facilitate tumor growth and metastasis, the inhibition of the vascular endothelial growth factor (VEGF) signaling pathway with small interference RNA (siRNA) becomes one of the most effective modalities for cancer treatments.2 To fulfill this goal, the efficient delivery of siRNA, which actually represents a major hurdle for successful clinical translation of RNAi-based therapies, is essential.3–6 Unlike DNA delivery for gene therapy, siRNA acts only in the cytosol and does not require the transportation of siRNA into the nucleus.7 This suggests that siRNA must be released into the cytosol so as to join the RNA-inducing silencing complex (RISC) for ablating target mRNA to exert its sequence-specific nuclease activity.8,9 Hence, efficient delivery of siRNA to intracellular compartments and timely release of siRNA into cytosols are of paramount importance to fully exert siRNA functions.10 To this end, tremendous efforts have been dedicated to the development of bioresponsive nanomaterials that can respond to intracellular stimuli, such as reactive oxygen species (ROS),11 redox,12 pH,13,14 enzymes,15 ATP,16,17etc., to improve the delivery efficiency of siRNA.18 Of particular interest are ROS-responsive materials, as the overproduction of ROS has been implicated in an array of pathological disorders, including cancer, and inflammatory, cardiovascular, and neurodegenerative diseases.19,20 Indeed, ROS-responsive nanomaterials represent a class of promising delivery vesicles for selectively transporting drug molecules to diseased sites by targeting oxidative microenvironments at different levels.20,21To date, several types of ROS-responsive nanomaterials, including those containing boronic ester,22–24 thioether,25,26 thioketal,27,28 selenium/tellurium,29,30 peroxalate ester31,32 and polyproline,33 have been explored for drug delivery applications. As an oxidation-responsive unit, phenylboronic ester/acid functional moieties have attracted considerable attention due to their greater sensitivity towards intracellular hydrogen peroxide (H2O2) (that is generated mostly in the mitochondria) over other ROS, and the merit that they are not associated with toxicity issues.21,34 The mechanism of ROS-mediated sensitivity depends on the oxidation-triggered hydrolysis of the phenylboronic acid/ester, which subsequently becomes a phenol to degrade the delivery vesicles for enhanced drug release.20,34 The recent findings also indicate that the exclusive interaction between the pendent phenylboronic acid (PBA) moieties and 3′-end ribose at both ends of the double-stranded siRNA and the hydrophobic interactions of PBA collaboratively contribute to the stability of polymer/siRNA complexes under simulated conditions.16 As siRNA is extremely vulnerable to degradation by plasma and tissue nucleases,35 this feature of PBA is critical to improve the siRNA serum stability.36 Therefore, we assume that taking advantage of the ROS-responsive characteristic of PBA with the desirable siRNA stabilization function would cooperatively contribute to the efficient delivery of siRNA in vitro and in vivo.
In this study, we adopted a biodegradable, ROS-responsive cationic polymer with pendent PBA moieties (termed PDP) that was developed by us previously as a siRNA delivery vehicle,37 in which the positive charge fully converted to neutral when PDP was exposed to endogenous H2O2 or other ROS. PDP could interact with siRNA by electrostatic interaction to form a PDP/siRNA complex, which was further coated with a layer of a lipid envelope which was able to effectively fuse with the cell membrane in an attempt to improve the efficiency of cellular uptake as well as to avoid endo/lysosomal trapping.37 The lipid envelope, which also contained non-fouling polyethylene glycol (PEG) and a cell-penetrating peptide (TAT), could greatly facilitate tumor targeting by the enhanced permeability and retention (EPR) effect and transmembrane delivery towards tumor cells. As shown in Scheme 1, the lipid bilayer consisted of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA), 1,2-distearoyl-sn-glycero-3-phosphoethanol-amine N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG) and DSPE-PEG-TAT in a certain proportion, the components of which were similar to the cell membrane phospholipid bilayer constitution.38,39 Therefore, the positive charge of PDP/siRNA became negative after the lipid coating to minimize the interaction with serum proteins, thereby facilitating the blood circulation.40 As one of the most potential cell-penetrating peptides, the cationic peptide TAT that is derived from human immunodeficiency virus type 1 (HIV-1)41,42 was introduced into the vehicle to improve the intracellular translocation process across the plasma membrane for enhanced therapeutic performance.43,44 In addition, the lipid-coated ROS-responsive cationic polymer also exhibited excellent blood compatibility and stability, making it promising for systemic siRNA delivery for tumor targeting and therapy. Collectively, the lipid envelope-coated, ROS-responsive boronic polymer represents an advanced vehicle for efficient siRNA delivery towards tumor tissues.
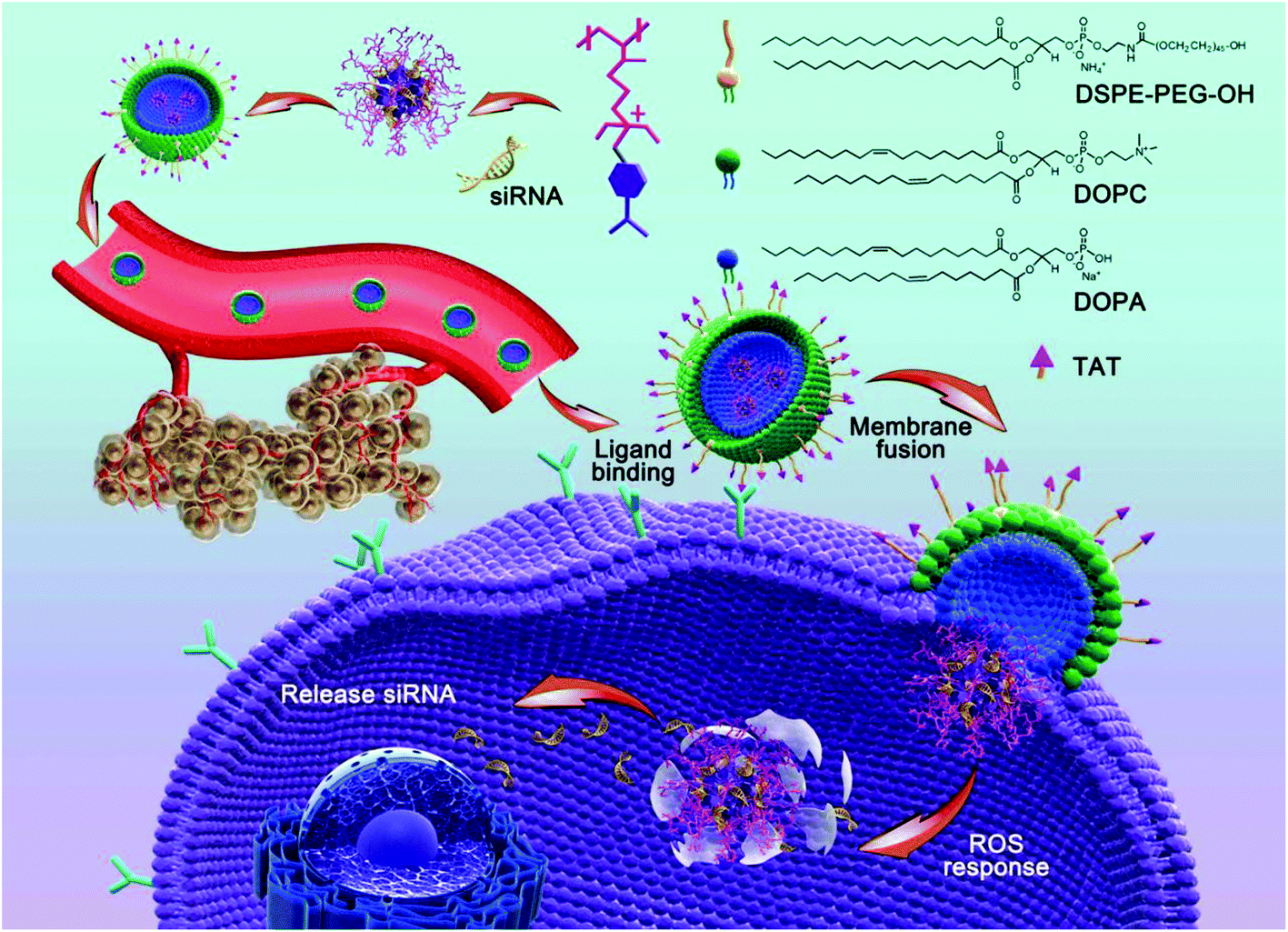 | ||
| Scheme 1 Schematic illustration of a lipid-coated ROS-responsive polymer for siRNA delivery. | ||
2. Materials and methods
Materials
Methoxypolyethylene glycols (mPEG, MW = 2000 Da), 1,10-carbonyldiimidazole (CDI), triethylamine (TEA, >99%), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St Louis, MO, USA). 1,2-Dioleoyl-sn-glycero-3-phosphate (DOPA), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-distearoyl-sn-glycero-3-phosphoethanol-amine N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol) 2000] (DSPE-PEG-MAL) were purchased from Laysan Bio. Inc. (USA). 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) was purchased from Beyotime Biotechnology. The TAT peptide was purchased from GL Biochem (Shanghai) Ltd. 5-Fu was purchased from J&K Chemical. The negative control siRNA (siNC), siRNA targeting green fluorescent protein (GFP-siRNA) and fluorescein-tagged negative-control siRNA (FAM-siRNA) were purchased from Biomics Biotechnologies Co, Ltd (Nantong, Jiangsu, China). VEGF siRNA (sense 5′-GGAGUACCCUGAUGAGAUCdTdT-3′; anti-sense 5′-CCUCAUGGGACUACUCUAGdTdT-3′) was prepared by GenePharma (Shanghai, China). Anti-VEGF antibody ab46154, anti-CD31 antibody and goat gnti-rabbit IgG H&L (Alexa Fluor® 488) ab150077 were purchased from Abcam company (United Kingdom). All other reagents were of analytical grade.Cells and animals
The SW620, SW480, MCF-7/GFP and 293T/GFP cell lines were purchased from American Type Culture Collection (ATCC, Rockville, MD). The SW620 cells and 293T/GFP cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and grown under humidified air containing 5% CO2 at 37 °C. The SW480 cells and MCF-7/GFP cells were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS) and grown under humidified air containing 5% CO2 at 37 °C. Athymic male mice (BALB/c strain) (5–6 weeks old, 16–18 g) were purchased from Zhejiang University Animal Care and Use Committee, and maintained in a pathogen-free environment under controlled humidity and temperature. The animal experiments were performed in accordance with the CAPN (China Animal Protection Law).Synthesis of PDP
Poly[(2-N,N-diethyl)aminoethyl acrylate] (PDEAEA) was synthesized as reported.37 Briefly, DEAEA (5.00 g, 29.0 mmol) and AIBN (48.0 mg, 0.290 mmol or 24.0 mg, 0.140 mmol) were charged in a 25 mL flask and deoxygenated at room temperature with nitrogen and then polymerized at 65 °C for 24 h. The viscous liquid was dissolved in CH2Cl2, and precipitated in cold n-hexane. The resulting PDEAEA was purified by re-precipitation three times and dried. PDEAEA (20 kDa, 0.300 g, 1.80 mmol tertiary amines) was reacted with 4-(bromomethyl)phenylboronic acid (0.560 g, 2.60 mmol) in 20 mL DMF at room temperature for 24 h.37 The resulting solutions were dialyzed overnight against deionized water using a dialysis bag with a molecular weight cut-off of 3500 Da and then lyophilized to afford PDEAEA-PB (PDP). The 1H-NMR and 11B-NMR spectra and FT-IR of the PDP were used to verify the chemical structures. The chemicals were dissolved in 20.0 mg mL−1 of D2O. 1H-NMR spectra of PDP (20 mg mL−1) was operated after the self-catalyzed hydrolysis at pH 7.4 with H2O2 (100 mM) in D2O at 37 °C.Preparation and characterization of PDP/siRNA polyplexes
PDP was dissolved in HEPES buffer solution (10.0 mM, pH = 7.4) at various concentrations, which were mixed with the siRNA solution of the same volume, and the resultant mixtures were vibrated for one minute to produce the polyplexes at different N/P ratios (N/P ratio, defined as the molar ratio of nitrogen atoms in the polymer to phosphate units of the siRNA). The polyplex solutions were incubated at room temperature for 20 min before characterization. The siRNA binding ability was evaluated by agarose gel retardation assay. The polyplexes were electrophoresed on a 1% agarose gel at 120 V for 20 min. siRNA bands were visualized by staining with ethidium bromide (EB) excited by UV transillumination.Preparation and characterization of Lip/PDP/siRNA
DSPE-PEG-TAT was prepared by coupling the TAT peptide bearing a cysteine on its N-terminus to DSPE-PEG-MAL (2000 Da) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in DMSO at room temperature for 24 h. The reaction mixture was purified by dialysis using a dialysis bag with a molecular weight cutoff of 2000 Da, and then lyophilized. DOPC, DOPA and DSPE-PEG-TAT were dissolved in 3 mL of chloroform and the solution was then dried by rotary evaporation to obtain a thin lipid film (Lip film). The solution of the polyplexes at an N/P ratio of 20 was added to the lipid film under sonication for one minute, then the solution was stirred at room temperature overnight. The particle size and zeta potential of the Lip/PDP/siRNA polyplexes were measured by dynamic light scattering (DLS) at 25 °C using a Zetasizer Nano-ZS (Malvern Instruments, Worcestershire, UK). The data are presented as the mean ± SD (n = 3). The morphology of the polyplexes was visualized using a transmission electron microscope (TEM, HT-7700, Hitachi, Japan), atomic force microscope (AFM, Bruker, Germany) and small-angle X-ray scattering equipment (SAXS, Xenocs, French). The structure of the complexes was determined by small-angle X-ray scattering (Xenocs, French).
:1 molar ratio in DMSO at room temperature for 24 h. The reaction mixture was purified by dialysis using a dialysis bag with a molecular weight cutoff of 2000 Da, and then lyophilized. DOPC, DOPA and DSPE-PEG-TAT were dissolved in 3 mL of chloroform and the solution was then dried by rotary evaporation to obtain a thin lipid film (Lip film). The solution of the polyplexes at an N/P ratio of 20 was added to the lipid film under sonication for one minute, then the solution was stirred at room temperature overnight. The particle size and zeta potential of the Lip/PDP/siRNA polyplexes were measured by dynamic light scattering (DLS) at 25 °C using a Zetasizer Nano-ZS (Malvern Instruments, Worcestershire, UK). The data are presented as the mean ± SD (n = 3). The morphology of the polyplexes was visualized using a transmission electron microscope (TEM, HT-7700, Hitachi, Japan), atomic force microscope (AFM, Bruker, Germany) and small-angle X-ray scattering equipment (SAXS, Xenocs, French). The structure of the complexes was determined by small-angle X-ray scattering (Xenocs, French).
MTT assay
The cytotoxicity of the complexes was evaluated by MTT assay using SW620 and SW480 cells. Generally, the cells were seeded on a 96-well plate at a density of 1 × 104 cells per well in 200 μL of the corresponding medium containing 10% serum for 18 h separately. The culture medium was replaced by a serum-free medium containing serial various concentrations of PDP and different N/P ratios of PDP/siRNA polyplexes for 24 h. MTT solution was added to the wells to achieve a final concentration of 0.5 mg mL−1. After incubation for another 4 h, each well was replaced with 100 μL of DMSO and measured spectrophotometrically on an ELISA plate reader (Model 550, Bio-Rad) at a wavelength of 570 nm. The relative cell growth (%) related to control cells cultured in the medium without the polymer was calculated by the following formula: V% = ([A]experimental − [A]blank)/([A]control − [A]blank) × 100%, where V% is the relative cell viability (%), [A]experimental is the absorbance of the wells culturing the treated cells, [A]blank is the absorbance of the blank, and [A]control is the absorbance of the wells culturing untreated cells.Hemolysis experiment
Red blood cells (RBCs) were isolated from 1 mL of fresh blood collected from healthy female mice by centrifugation at 10000g for 5 min. Then, RBCs were washed several times with PBS until no color was seen in the supernatant. RBCs were suspended in 15 mL of PBS, and 0.50 mL of this RBC suspension was added into 0.50 mL of a suspension containing different concentrations of PDP and Lip/PDP at 1.0, 5.0, 10, 20, 40, and 80 μg mL−1 in HEPES buffer, respectively. A 0.50 mL RBC suspension incubated with 0.50 mL PBS or 0.50 mL distilled water was used as negative and positive controls, respectively. The samples were mixed gently, left at room temperature for 2 h, and then centrifuged at 10000g for 5 min. A total of 100 μL of supernatant was transferred to a 96-well plate and the absorbance of hemoglobin at 540 nm was measured. The percentage of hemolysis was calculated as follows: hemolysis % = ([I]experimental − [I]blank)/([I]control − [I]blank) × 100%. [I]experimental is the absorbance of the samples, [I]blank is the absorbance of the negative control, and [I]control is the absorbance of the positive control.
Cellular uptake assay
The cellular uptake and distribution of PDP/siRNA and Lip/PDP/siRNA polyplexes were examined by confocal microscopy using the SW620 and SW480 cell lines. The cells were seeded on 24-well plates and grown for 18 h. The fluorescein-tagged negative-control siRNA (FAM-siRNA) (1.0 μg) was complexed with PDP at a N/P ratio at room temperature for 20 min before use. After culturing for 4 h, the cells were fixed with 400 μL of fresh 4% formaldehyde and then treated with 500 μL of DAPI for 10 min. Alexa Fluor®594 WGA was used to label plasma membranes (red) at a 1/200 dilution for 1 h. The images were acquired on a confocal scanning laser microscope (CLSM, Radiance 2100, Bio-Rad). Line scanning and total fluorescence measurements were analyzed with Image J software as reported previously. In the green fluorescent protein (GFP) silencing study, GFP-expressing 293T cells (293T/GFP) and GFP-expressing MCF-7 cells (MCF-7/GFP) were used as models. The siRNA complex formation and delivery to cells were performed as described above utilizing GFP-siRNA and the negative control. GFP silencing was assessed by flow cytometry after cell fixation and the Geo mean (%) was used to calculate the decrease in the green fluorescence of GFP.Inhibition of VEGF and CD31 expression by the Lip/PDP/siRNA polyplexes
Quantitative reverse transcription (RT)-PCR was used in the analysis of the SW620 and SW480 cell lines’ expression of gene encoding of VEGF and CD31. The cells were treated with the different polyplexes for 4 h. Then the medium was replaced by the corresponding medium containing 10% FBS and the cells were incubated for another 48 h, collected to extract mRNA, and reverse-transcribed into cDNA. The PCR parameters consisted of 5 min of Taq activation at 95 °C, followed by 32 cycles of 95 °C × 30 s, 56 °C × 45 s, and 72 °C × 45 s. The relative gene expression values were determined using the BandScan 4.3 software and the data presenting the VEGF and CD31 expression were normalized to the housekeeping gene GAPDH as the endogenous reference. In the western blotting analysis, the cell protein was extracted after the same treatment as RT-PCR and the total protein was quantified by using the BCA protein assay kit (Promega, USA). An equal amount of protein was separated by SDS-PAGE, transferred onto a nitro-cellulose membrane, blocked, and incubated overnight with monoclonal antibodies against VEGF and CD31. After washing, the membrane was incubated with the HRP-conjugated secondary antibody for 2 h at room temperature. The bands were visualized using the Westzol enhanced chemiluminescence kit (Intron, Sungnam, Korea) and the expression was normalized to the housekeeping gene expression.In vitro immunofluorescence
The SW620 and SW480 cells were seeded on 24-well plates and grown for 18 h. Then the medium was replaced by 600 μL of serum-free medium containing serial dilutions of nanoparticle solutions for 4 h. The cells were fixed (10 min) with 4% PFA and then incubated in 1% BSA/10% normal goat serum/0.3 M glycine in 0.1% PBS-Tween for 1 h to permeate the cells and block non-specific protein–protein interactions. The cells were then incubated with the anti-VEGF antibody ab46154 (1.00 μg mL−1) and anti-CD31 antibody ab28364 (1.00 μg mL−1) overnight at 4 °C. The secondary antibody (green) was Alexa Fluor®488 goat anti-rabbit IgG (H + L) used at a 1/1000 dilution for 1 h. Alexa Fluor®594 WGA was used to label the plasma membranes (red) at a 1/200 dilution for 1 h. DAPI was used to stain the cell nuclei (blue) at a concentration of 1.43 μM for 10 min. The confocal images were acquired on a confocal scanning laser microscope (CLSM, Radiance 2100, Bio-Rad).In vivo tumor therapy
The animal experiments were performed in accordance with the China Animal Protection Law (CAPN), and the protocols were approved by Zhejiang University Animal Care and Use Committee. The SW620 cells were inoculated subcutaneously into the right abdomen of BALB/c mice. When the tumor grew to a diameter of around 90 mm3, the mice were assigned to six groups and treated with PBS, 5-Fu, PDP/siVEGF(low dose, L), PDP/siVEGF (medium dose, M), PDP/siVEGF (high dose, H), and Lip/PDP/siVEGF at 0.30 mg kg−1, 0.60 mg kg−1, and 0.90 mg kg−1 siVEGF (L, M, and H) per mouse via tail vein injection. Each group was composed of eight mice (n = 8) for anti-tumor therapy and three mice (n = 3) for RT-PCR and western blotting analysis. The treatment was performed every three days six times and tumor growth was also monitored using calipers. The tumor volume (V) was calculated by the following formula: tumor volume V (mm3) = π/6 × length (mm) × width (mm)2. In the western blotting analysis, the mice from each group were sacrificed 48 h after the last injection. The tumors were extracted and the other approaches were the same as described above.Photoacoustic (PA) imaging
SW620 tumor xenografted nude mice were anesthetized using 2% isoflurane in oxygen, and placed in the PA imaging system (Visualsonics, 2100 High-Resolution Imaging System). The mice were scanned to determine the endogenous signal of tumors at 750 and 850 nm. A three-dimensional PA image was reconstructed off-line using the data acquired from all 128 transducers at each view and a back-projection algorithm.Detection of VEGF and CD31 protein expression in tumor tissue
Tumor tissues were excised 48 h after the last intra-tumoral injection. For protein analysis, a piece of tumor tissue (≈50 mg) was triturated and detected by western blot analysis using a previously reported method.Histology and immunohistochemistry
Twenty-four hours after the last treatment, the animals were sacrificed and tumor tissues were fixed in 4% formaldehyde for 24 h, then dehydrated in graded ethanol, embedded in paraffin, and cut into 3 μm-thick sections. The fixed sections were deparaffinized and hydrated according to a standard protocol and stained with hematoxylin and eosin (H&E) for microscopic observation. The proliferation of tumor cells was detected using a primary antibody of Ki67 (1:25, ab28364, Abcam, United Kingdom), and then incubated with a secondary antibody of Rb IgG (H + L)/HRP (ZB-2301, ZSGB-BIO, China). The apoptosis of tumor cells following treatments in the mice was determined using the TUNEL method according to the manufacturer's instructions (Roche, Basel, Switzerland).
The immunohistochemistry of tumor tissues was performed as follows: tumor sections were deparaffinized with xylene and alcohol, washed with PBS, and incubated in 10 mM citrate buffer (pH 7.4) for 15 min at 90 °C. The sections were then treated with 0.3% hydrogen peroxide in methanol for 30 min at 4 °C. After blocking with 10% normal horse serum, 2% bovine serum albumin, and 0.5% Triton X-100 for 1 h, the tissues were incubated with primary antibodies for the anti-VEGF antibody ab46154 (1 μg mL−1) and anti-CD31 antibody ab28364 (1 μg mL−1) for 1 h at 37 °C, washed with PBS, and incubated with the secondary antibody of Rb IgG (H + L)/HRP (ZB-2301, ZSGB-BIO, China) for 1 h. The immunoreactivity on the tissue sections was visualized by using the peroxidase substrate diaminobenzidine. All sections were examined on a Leica (DMLB&DMIL) microscope.
Statistical analysis
The data were expressed as mean ± standard deviation. The statistical significance (P < 0.05) was evaluated by the Student t-test or one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test. In all the tests, the statistical significance was set at *P < 0.05 and **P < 0.01.3. Results and discussion
The synthesis and hydrolysis of PDP is shown in Fig. S1 and S2.† The tertiary amine-based polymer poly[2-(N,N-diethylamino)ethyl acrylate] (PDEAEA) was prepared by radical polymerization. Its quaternization reaction with an excess of 4-(bromomethyl)phenylboronic acid produced water-soluble PDEAEA-PB (PDP). The 1H NMR spectrum of PDP showed that the signals at δ 7.2–7.8 ppm were associated with protons in the benzene ring. The chemical shift at 4.3 ppm was attributed to the proton in the vicinity of the alkoxy residue, and the 11B-NMR spectra of PDP also confirmed that the chemical shift at −5.1 ppm was attributed to the boron in the phenylboronic moiety (Fig. 1A). Furthermore, the FT-IR spectrum indicated that the absorption peaks at 1459 cm−1 and 1357 cm−1 were ascribed to the stretching vibration of B–C, whereas the peaks at 634 cm−1 and 633 cm−1 were ascribed to the asymmetric stretching vibration of B–O from the repeating units of the PBA (Fig. S3†). In order to reduce hemolysis to meet the requirement for the intravenous administration, the positive charge of the PDP/siRNA complexes should be shielded with a stealth layer, which can be either a lipid or a polyethylene glycol layer, to minimize the side effects.15,37,45 Since the lipid coating has been widely employed to impart multifunctionalities over a given nanoparticle,46,47 we formulated a layer of lipid film which consists of multiple components including DOPC, DOPA, and DSPE-PEG-TAT (molar ratio: 8:2:1) over the polyplex surface to render a long circulating capacity and membrane fusion feature. Driven by the hydrophobic interaction, DOPC, DOPA and DSPE-PEG-TAT self-assembled into a lipid film where PDP/siRNA complexes can be incorporated simply by sonication (Fig. 1B). The formed lipid layer, termed Lip, is expected to extend the circulation time and to protect the loaded siRNA from enzymatic degradation.
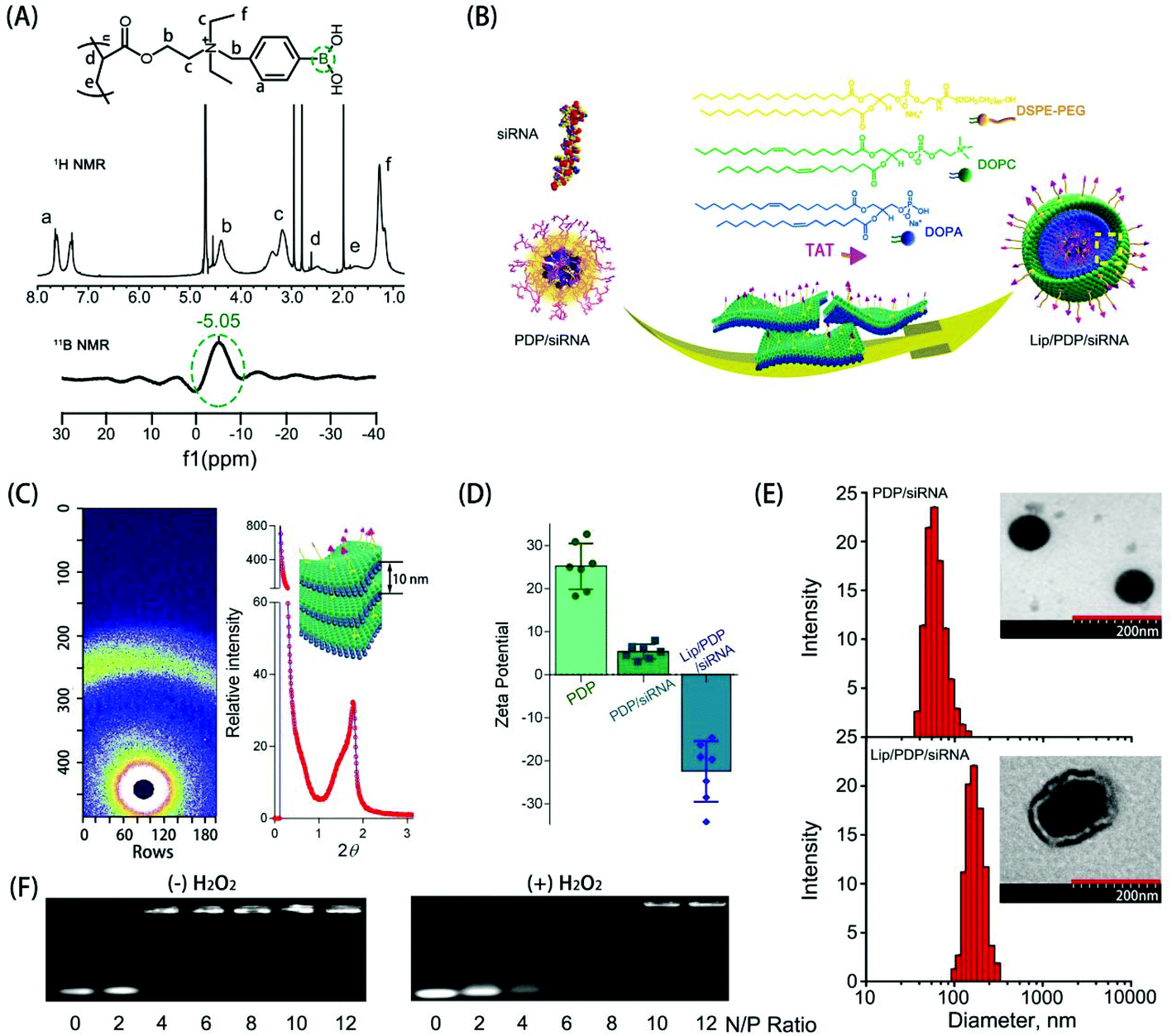 | ||
| Fig. 1 (A) The 1H-NMR and the 11B-NMR spectra of PDP (20 mg mL−1) in D2O at 37 °C. (B) The scheme of the formation process for the Lip/PDP/siRNA complex. (C) SAXS analysis of the Lip/PDP/siRNA. (D) The zeta potential of the PDP, PDP/siRNA and Lip/PDP/siRNA. (E) The size and the TEM micrograph of the PDP/siRNA and Lip/PDP/siRNA complexes. The scale bar represents 200 nm. (F) Agarose gel electrophoresis of PDP/siRNA polyplexes at different N/P ratios in the absence of H2O2 solution (left), and the gel retardation assay of siRNA released from polyplexes at different N/P ratios of 4 h incubation in the presence of H2O2 solution (right). | ||
To reveal the structure of Lip/PDP/siRNA complexes, we subsequently investigated the diffractogram by small angle X-ray scattering (SAXS) (Fig. 1C). The diffraction pattern of the Lip/PDP/siRNA complexes and the Bragg peak indicated that the complexes form a lamellar structure with an interlayer distance related to 2-theta (2θ) of the peak.48 The morphology of the complexes was characterized by transmission electron microscopy (TEM) and atomic force microscopy (AFM), respectively. As shown in Fig. 1E and S4,† the TEM and AFM micrographs indicated that the PDP/siRNA and Lip/PDP/siRNA complexes were able to form spherical nanoparticles with relatively uniform size distribution. The average diameter of the lipid-coated PDP/siRNA complexes was around 220 nm, with a zeta potential of −26 mV. In contrast, the size and zeta potential of the PDP/siRNA polyplexes were around 80 nm and 26 mV, respectively, (Fig. 1D and E). The above evidence clearly indicated that the proposed lipid-decorated PDP/siRNA nanoparticles were prepared successfully. In order to verify the ROS characteristic of the PDP, we also monitored the 1H NMR spectrum of PDP in the presence of H2O2. After 4 h, we observed the deboronation in the side chain due to the release of p-quinone methide, which quickly turned into p-hydroxymethyl-phenol (HMP) in the solution.37 The PDP polymer could effectively bind with siRNA at N/P ratios above 6 through electrostatic interaction (Fig. 1F), and such binding between PDP and siRNA became weak when the PDP/siRNA complexes were exposed to H2O2, suggesting that PDP could potentially release the siRNA under ROS-rich conditions.
The in vitro cytotoxicity was evaluated as a function of concentration or nitrogen/phosphorus (N/P) ratio to examine the biocompatibility of PDP (Fig. 2A). The free PDP polymer showed dose-dependent cytotoxicity, with over 60% of cell viability at the concentration of 100 μg mL−1 towards both SW620 and SW480 cell lines. Similarly, polyplexes at various N/P ratios exhibited low cytotoxicity towards SW620 cells, and exhibited slightly higher cytotoxicity towards SW480 cells only at the N/P ratio of 60. The low cytotoxicity of the PDP polymer was derived from the charge switch from a highly cationic state to a neutral one. The ability of the PDP and Lip/PDP to deliver siRNA and to induce gene silencing was investigated on 293T/GFP cells that stably expressed the green fluorescent protein (GFP). As shown in Fig. 2B and C, the gene silencing ability of the Lip/PDP/GFP-siRNA polyplexes became optimal at the N/P ratio of 20, exhibiting up to 36% of GFP knockdown efficiency. Compared with PDP/GFP-siRNA, the coating of the lipid layer also slightly improved the silencing ability of PDP/GFP-siRNA, as evidenced by fluorescence images after siRNA delivery (Fig. 2C). These results demonstrated that Lip/PDP/FAM-siRNA complexes not only exhibited low cytotoxicity, but also mediated efficient gene silencing.
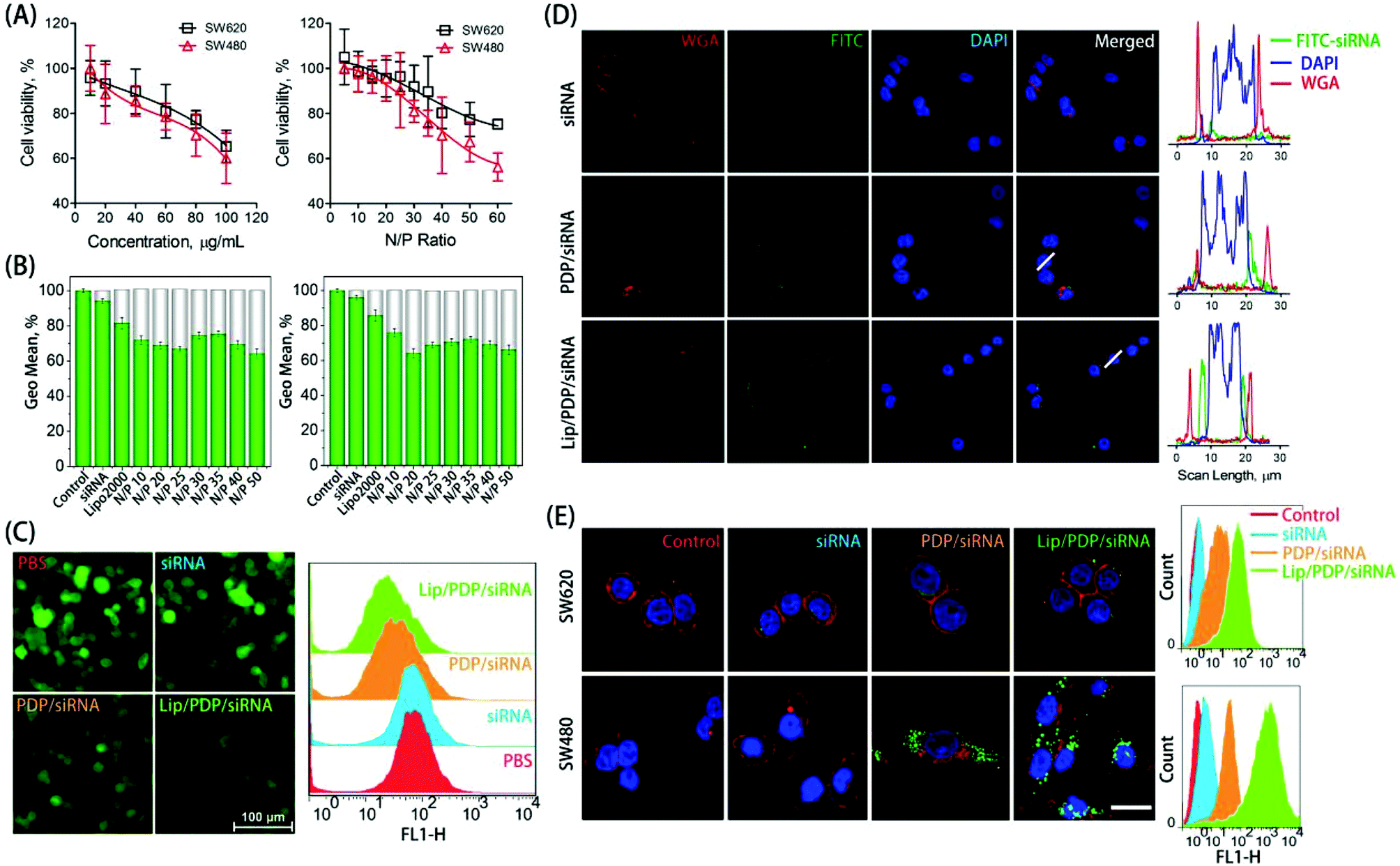 | ||
| Fig. 2 (A) The cytotoxicity assay of PDP towards SW620 and SW480 cells. PDP at the different polymer concentrations, or PDP/siRNA complexes at different N/P ratios were incubated with cells for 24 h. The cytotoxicity was evaluated by MTT assay. (B) GFP knockdown efficiency mediated by PDP/siRNA and Lip/PDP/siRNA complexes at different N/P ratios in 293T/GFP cells. Blank denotes the fluorescence obtained from the cells without the treatment of siRNA polyplexes. siRNA and Lipo2000 were used as the controls. (C) GFP fluorescence intensity, either observed by using a fluorescence microscope, or analyzed by flow cytometry, was investigated after the 293T/GFP cells were incubated with Lip/PDP/GFP-siRNA or PDP/GFP-siRNA for 24 h. The scale bar represents 100 μm. (D) In vitro fluorescence image of SW620 cells transfected with Lip/PDP/siRNA, PDP/siRNA or naked siRNA. siRNA was labeled with FITC (green), the plasma membranes were stained with Alexa Fluor® 647 WGA (red), and cell nuclei were stained with DAPI (blue). The fluorescence intensity was quantified by Image J. The scale bar represents 20 μm. (E) The fluorescence image (observed by CLSM) and the fluorescence intensity (analyzed by flow cytometry) of SW620 and SW480 cells transfected with Lip/PDP/siRNA, PDP/siRNA or naked siRNA. FAM-siRNA was labeled with FITC (green fluorescent), the plasma membranes were stained with Alexa Fluor® 647 WGA (red), and cell nuclei were stained with DAPI (blue). The scale bar represents 20 μm. | ||
Efficient internalization of siRNA into cells is critical for effective RNAi-based clinical therapies.49,50 To verify whether the PDP can efficiently deliver siRNA into cancer cells, the internalization of fluorescein-labeled control siRNA (FAM-siRNA) mediated by PDP or Lip/PDP was investigated. We monitored the cellular uptake behaviour of the complexes in SW620 and SW480 cells with CLSM and flow cytometry. As shown in Fig. 2D and E, green fluorescence was observed mainly in the cytoplasm instead of the nuclei, suggesting that PDP could successfully deliver siRNA into the cytosol. Interestingly, both cell lines treated with Lip/PDP/FAM-siRNA showed higher intracellular accumulation of the green fluorescence signal than those treated with free FAM-siRNA or PDP/FAM-siRNA groups. Quantitative analysis of the fluorescence intensity by flow cytometry indicated that the fluorescence intensity of SW620 and SW480 cells which were treated with Lip/PDP/FAM-siRNA increased dramatically. These results suggested that the Lip/PDP can effectively transport the siRNA into the cytoplasm and may be rapidly degraded by the intracellular H2O2 to release the loaded siRNA.
To obtain further insights into the effectiveness of PDP and Lip/PDP in delivering siRNA, the mRNA and protein levels of vascular endothelial growth factor (VEGF) and CD31 were assessed after the siRNA targeting VEGF (siVEGF) was delivered. VEGF is a well-known angiogenic growth factor and has been shown to enhance angiogenesis, endothelial cell survival and migration, and revascularization in vivo.51–53 As a result, knocking down of the VEGF receptor signaling pathway through RNAi has great potential to prevent angiogenesis and proliferation of tumor cells.54 As shown in Fig. 3A and B, the cells treated with PDP in the presence of negative siRNA (siNC) did not lead to VEGF suppression. We also examined the CD31 expression, the key marker of neovasculature endothelial cell proliferation.55 Similarly, no obvious downregulation of CD31 was observed as well. Nevertheless, the protein levels of VEGF and CD31 were reduced in both SW620 and SW480 cells treated with PDP/siVEGF and Lip/PDP/siVEGF (Fig. 3B). As shown in Fig. 3A, the fluorescence intensity represents the level of mRNA, and we also observed similar trends in the downregulation of VEGF and CD31 mRNA levels in both cell lines. Furthermore, we also carried out immunofluorescence assay to investigate the knockdown level of siVEGF. As shown in Fig. 3C and D, the expression of the VEGF and CD31 protein was downregulated more significantly for the cells treated with Lip/PDP/siVEGF, in comparison with those treated with either naked siVEGF or PDP/siVEGF. Collectively, these results suggested that effective RNAi only occurred after the delivery of siRNA mediated by the proposed carriers. Briefly, after siRNA was delivered to the cytoplasm, it was activated by binding with RISC for ablating VEGF mRNA to fulfil its therapeutic effects.
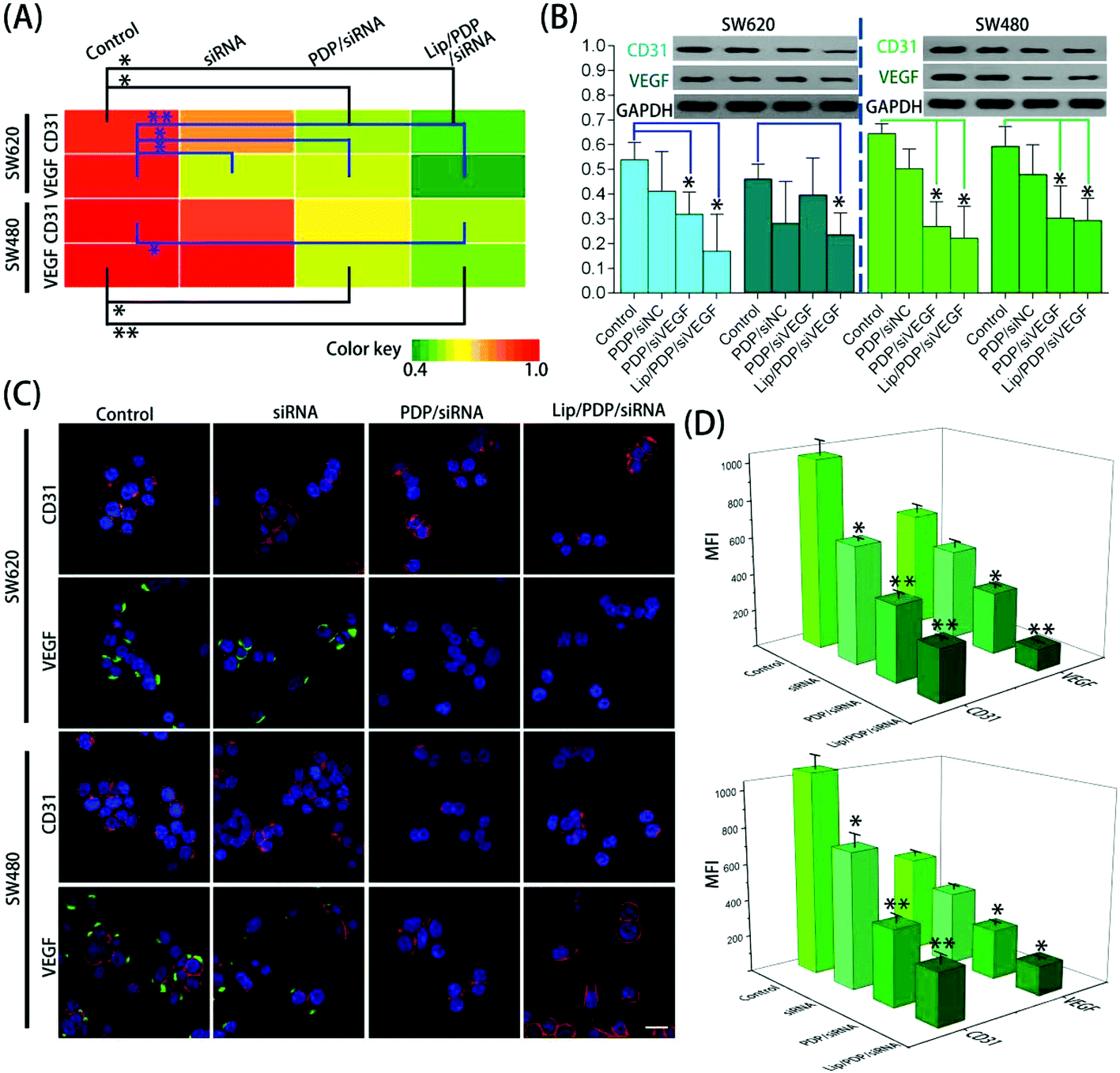 | ||
| Fig. 3 (A) Heat map of VEGF and CD31 mRNA levels in SW620 and SW480 cells after the incubation with the denoted siRNA formulation. The mRNA level was determined by quantitative real-time PCR at 48 h. (B) Representative VEGF and CD31 protein expression determined by western blot analysis in SW620 and SW480 cell lines after the incubation with the indicated siRNA formulation for 48 h, and GAPDH was used as the control. (C) In vitro immunofluorescence image of SW620 and SW480 cells incubated with free siVEGF, PDP/siVEGF or Lip/PDP/siVEGF for 24 h. The VEGF and CD31 were labelled as the corresponding antibody, and the secondary antibody (green) was Alexa Fluor® 488 goat anti-rabbit IgG (H + L), the plasma membranes (red) were stained with Alexa Fluor® 647 WGA, and cell nuclei (blue) were stained with DAPI. The scale bar represents 20 μm. (D) Mean fluorescence intensity of VEGF and CD31 analyzed by Image J. Data represent mean ± SD (n = 3, Student's t-test, *P < 0.05, **P < 0.01). | ||
Blood compatibility is a critical indicator to evaluate whether the proposed delivery system is safe for in vivo applications.56 To study the blood compatibility of native PDP and the lipid-coated PDP, we measured the hemolytic potential. The hemolysis rate represents the percentage of the destroyed membrane of RBCs induced by the carrier. With the concentration increasing from 5 to 80 μg mL−1, PDP caused an increase in the hemolysis rate from 10% to 30%, indicating its poor hemocompatibility (Fig. 4A). The rupture of RBCs induced by PDP, accompanied by significant aggregation, could be clearly observed under the microscope (Fig. S5†). Nevertheless, almost no hemolytic effect was observed for those RBCs treated with Lip/PDP/siRNA polyplexes in the same PDP concentration range, indicating excellent hemocompatibility arising from the lipid modification.
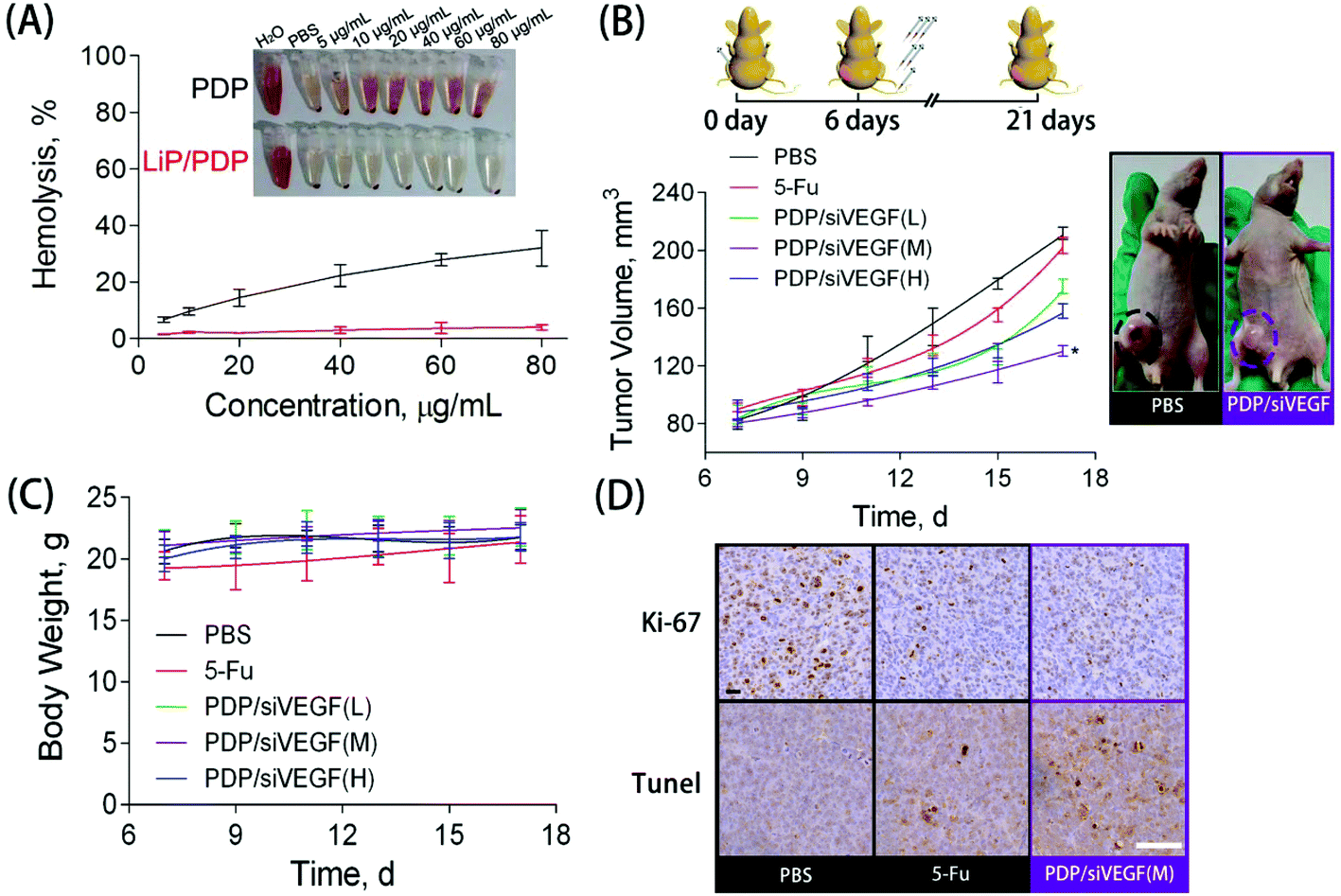 | ||
| Fig. 4 (A) Blood hemolytic effects of PDP and Lip/PDP. Red blood cells were treated with PDP and Lip/PDP at various concentrations from 5 to 80 μg mL−1. Deionized water was used as a positive control and PBS buffer solution was used as a negative control. (B) The treatment regime (upper image): 5 × 106 SW620 cells were subcutaneously injected into the nude mice. Six days after the inoculation, the mice were subjected to i.v. injection on an every-other-day schedule six times. The tumor volumes of the tumor-bearing mice at different time points were measured (lower left). The representative images of the tumor-bearing mice after 21 days since the first treatment was acquired. The image of a mouse treated with PBS (denoted as control in the image) or PDP/siVEGF (medium dose) was obtained. (C) Average body weight of the tumor-bearing mice at different time points after the treatment. (D) Ki67 and TUNEL immunohistochemistry analysis of tumor tissues of murine tumor models with SW620 xenografts after treatment with PBS, 5-Fu, or PDP/siRNA (M). The scale bar represents 40 μm. | ||
To further explore the potential of the Lip/PDP/siVEGF delivery system for in vivo antitumoral effect, we treated SW620 tumor-bearing BALB/c mice via tail-vein injection with the indicated formulations. In order to find out the optimal dosage for siRNA delivery in vivo, we applied a low dose (0.30 mg kg−1), medium dose (0.60 mg kg−1) and high dose (0.90 mg kg−1) of siRNA for the in vivo tumor inhibition study (Fig. 4B–D). We identified that the tumor growth rate and body weight after the treatment with PDP/siVEGF (0.6 mg kg−1) and PDP/siVEGF (0.90 mg kg−1) were similar. Considering that a higher dose may exert other potential side effects, we subsequently chose a medium dose in our further investigations. To test the anti-tumor activity and systemic toxicity, the mice were treated with PBS, 5-Fu, PDP/siVEGF and Lip/PDP/siVEGF, respectively. The first line anti-cancer drug 5-Fu was used as a positive control. As shown in Fig. 5A, rapid tumor growth was observed for the mice treated with PBS. Compared with the groups treated with PBS or 5-Fu, PDP/siRNA exhibited moderate inhibition of tumor growth. Excitingly, the Lip/PDP/siRNA formulation exhibited a more effective anti-tumor effect than other groups. Under this treatment, the tumor growth was very slow and the tumor size only increased ca. 40 mm3 21 days after the first treatment. Apparently, the tumor size decreased dramatically after the mice were treated with Lip/PDP/siVEGF (Fig. S6†), implying that siVEGF systemic delivery mediated by Lip/PDP is efficient in vivo. Compared with the mice administered with 5-Fu, no significant body weight loss was recorded after the treatment with PBS, PDP/siVEGF and Lip/PDP/siVEGF, indicating the low toxicity nature and the minimal side effects of Lip/PDP/siVEGF (Fig. 5A). These results highlighted the significance of the Lip/PDP/siRNA delivery system for in vivo cancer treatment.
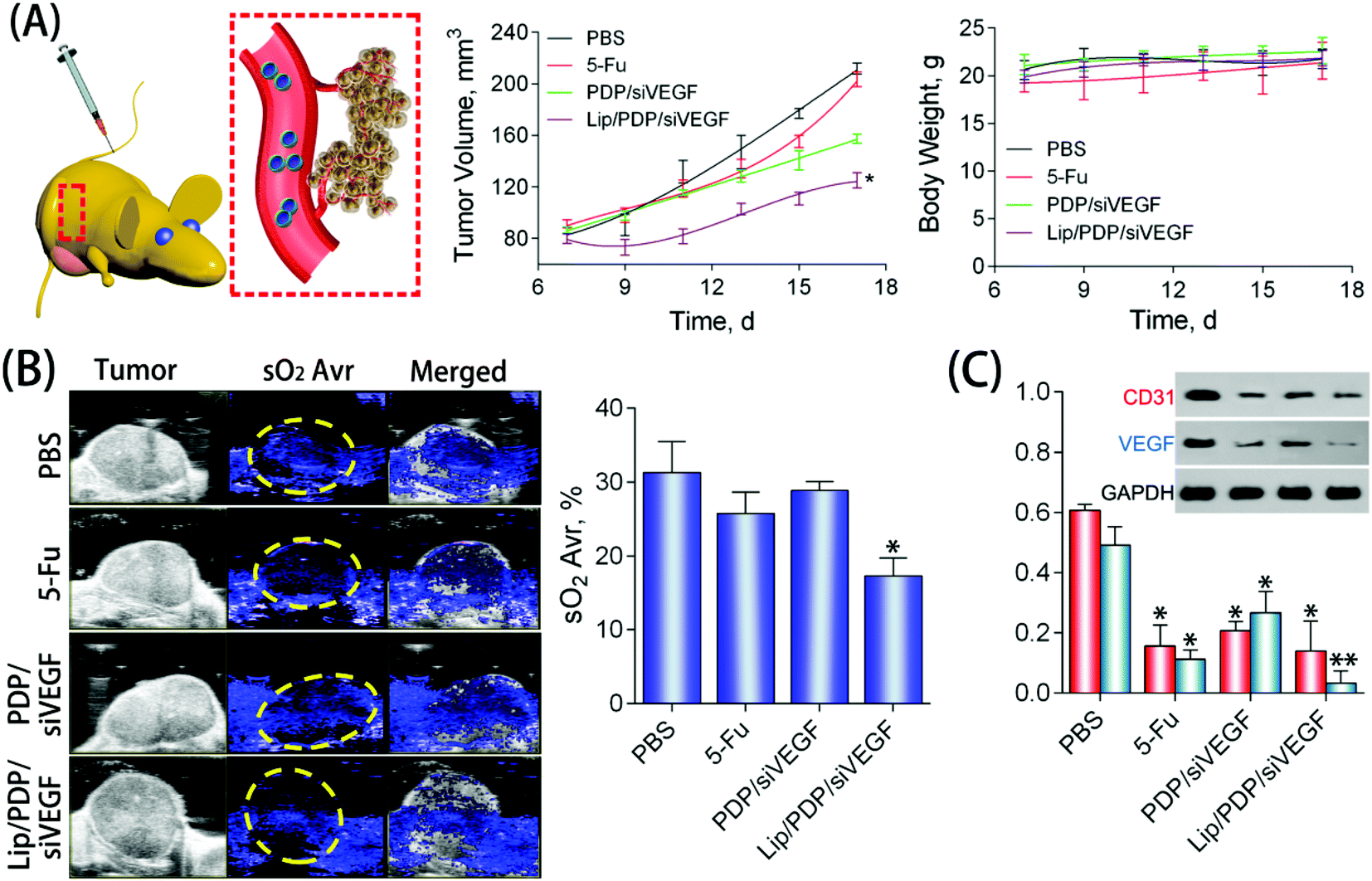 | ||
| Fig. 5 (A) Tumor growth curve of BALB/c nude mice with SW620 xenografts treated with Lip/PDP/siRNA or other formulations: the average body weight of the tumor-bearing mice at different time points. (B) Photoacoustic imaging (left) and the average level of oxyhemoglobin saturation (sO2 Avr) in tumors after the different treatments (right). (C) In vivo western blot analysis of the VEGF and CD31 protein in the tumor after the treatment by different groups. (n = 3, Student's t-test, **P < 0.01 and *P < 0.05). | ||
Cell proliferation and apoptosis in the tumors were analyzed by hematoxylin and eosin (H&E) staining, Ki67 and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay after the treatment (Fig. S7†). The H&E stained sections of tumor tissues in the PBS group appear to be most hypercellular, and exhibited nuclear polymorphism more obviously, suggesting the rapid tumor growth. Among the three therapeutic groups, the tumor tissues from the treatment of Lip/PDP/siVEGF showed the fewest tumor cells with the highest level of tumor necrosis. The Ki67 assay suggested that the Lip/PDP/siVEGF group significantly reduced the percentage of proliferating tumor cells and showed a particularly significant decrease of Ki67-positive tumor cells. As expected, the TUNEL assay confirmed that the treatment with Lip/PDP/siVEGF could induce apoptosis more significantly over other groups, which is in agreement with the results obtained from H&E and Ki67 analyses. Immunohistochemical assays were further performed to detect the expression of the VEGF protein and the blood vessel endothelial cell marker CD31 in tumor tissues. Cell nuclei were stained blue and the brown blots indicated the VEGF and CD31 protein in the tumor tissue.12 As shown in Fig. S7,† VEGF and CD31 protein staining revealed a distinct decrease in the protein density of the tumor slice after the treatment with Lip/PDP/siVEGF formulation when compared with other groups. In collaboration with the in vitro study results, these data collectively demonstrated that the Lip/PDP/siVEGF group presented a strong effect in inhibiting tumor growth in vivo.
The deoxygenated hemoglobin (Hb) and oxygenated hemoglobin (HbO2) in the blood could be detected by photoacoustic imaging, which can reflect the location of blood vessels inside the tumors to further estimate the anti-angiogenesis effect.57,58 As shown in Fig. 5B, the blue signal represented the Hb, and the signal was also quantified as the oxyhemoglobin saturation (sO2 Avr). The PBS group showed a strong signal intensity over the tumor area, suggesting the high density of blood vessels in the tumor tissue. Nevertheless, mice treated with the Lip/PDP/siVEGF formulation showed a relatively lower intensity of the signal over the tumor area, suggesting poor formation of blood vessels in the tumor tissue. These results suggested that the Lip/PDP/siVEGF group could effectively retard tumor growth through a mechanism of RNAi-mediated anti-angiogenesis. The VEGF and CD31 protein in the tumor tissues were also evaluated by western blotting. Compared with the PBS group, obvious downregulation of the VEGF protein was detected after 5-Fu, PDP/siVEGF or Lip/PDP/siVEGF treatment (Fig. 5C). The Lip/PDP/siVEGF group is most obvious in terms of the downregulation of both VEGF and CD31 proteins in the tumor tissue, which further confirmed that Lip/PDP/siVEGF is effective for cancer treatment through the RNAi-based therapeutic mechanism.
We finally validated the tumor targeting efficiency by investigating the accumulation of Lip/PDP/siRNA complexes in the tumor tissue in vivo. Cy5-labeled siRNA was delivered by Lip/PDP and the fluorescence intensity was investigated in the tissue slice to evaluate the tumor targeting and deep tissue penetration mediated by Lip/PDD. As shown in Fig. S8,† almost no red fluorescence was detected in the case of either PBS or the siRNA group 24 hours post-injection. Faint red fluorescence was observed in the tumor slice when the siRNA delivery was mediated by PDP. Excitingly, strong red fluorescence was detected after Lip/PDP/Cy5-siRNA was delivered for 24 hours. The high level of accumulation of Lip/PDP/Cy5-siRNA complexes was due to the non-fouling nature of the enhanced permeation and retention (EPR) effect and the negatively charged nature of the complexes, which can synergistically enhance the tumor accumulation of therapeutic complexes in the tumor tissue.
4. Conclusion
In conclusion, we developed a lipid-coated, ROS-responsive vector for systemic siRNA delivery, which was demonstrated to significantly improve the efficacy of cancer therapy through efficient RNAi. The efficiency of siRNA delivery largely relied on the quick ROS responsiveness of the PDP polymer to mediate timely siRNA release, the PEG-functionalized lipid layer to yield a low fouling surface as well as the ability of phenylboronic moieties to stabilize siRNA. This multifunctional delivery system provides the efficient systemic transportation of siRNA to the tumor site for the effective knockdown of the VEGF, which may be derived from its high ROS sensitivity, long circulation time and good targeting ability, together with enhanced siRNA stability. The current study defines a new systemic delivery strategy for siRNA by cooperatively integrating multifunctional lipid coatings with a ROS-responsive boronic polymer, which may potentially benefit RNAi-based therapy in the dawning era of precision nanomedicine for cancer therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge the National Basic Research Program (2014CB931900), the National Natural Science Foundation of China (51573161), the Thousand Talents Plan, and the Hundred Talents Program of Zhejiang University (0017058) for the financial support of this work.References
- W. Viricel, S. Poirier, A. Mbarek, R. M. Derbali, G. Mayer and J. Leblond, Nanoscale, 2017, 9, 31–36 RSC.
- J. Chen, H. Tian, X. Dong, Z. Guo, Z. Jiao, F. Li, A. Kano, A. Maruyama and X. Chen, Macromol. Biosci., 2013, 13, 1438–1446 CrossRef CAS PubMed.
- A. Fire, S. Q. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
- A. de Fougerolles, H. P. Vornlocher, J. Maraganore and J. Lieberman, Nat. Rev. Drug Discovery, 2007, 6, 443–453 CrossRef CAS PubMed.
- C. T. Greco, V. G. Muir, T. H. Epps 3rd and M. O. Sullivan, Acta Biomater., 2017, 50, 407–416 CrossRef CAS PubMed.
- X. Liu, J. Zhou, T. Yu, C. Chen, Q. Cheng, K. Sengupta, Y. Huang, H. Li, C. Liu, Y. Wang, P. Posocco, M. Wang, Q. Cui, S. Giorgio, M. Fermeglia, F. Qu, S. Pricl, Y. Shi, Z. Liang, P. Rocchi, J. J. Rossi and L. Peng, Angew. Chem., Int. Ed., 2014, 53, 11822–11827 CrossRef CAS PubMed.
- C. Ornelas-Megiatto, P. R. Wich and J. M. Frechet, J. Am. Chem. Soc., 2012, 134, 1902–1905 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- J. Kurreck, Angew. Chem., Int. Ed., 2009, 48, 1378–1398 CrossRef CAS PubMed.
- Y. C. Tseng, S. Mozumdar and L. Huang, Adv. Drug Delivery Rev., 2009, 61, 721–731 CrossRef CAS PubMed.
- X. Liu, Y. Wang, C. Chen, A. Tintaru, Y. Cao, J. Liu, F. Ziarelli, J. Tang, H. Guo, R. Rosas, S. Giorgio, L. Charles, P. Rocchi and L. Peng, Adv. Funct. Mater., 2016, 26, 8594–8603 CrossRef CAS.
- K. Wang, Q. Hu, W. Zhu, M. Zhao, Y. Ping and G. Tang, Adv. Funct. Mater., 2015, 25, 3380–3392 CrossRef CAS.
- B. J. Hong, A. J. Chipre and S. T. Nguyen, J. Am. Chem. Soc., 2013, 135, 17655–17658 CrossRef CAS PubMed.
- C. Y. Sun, S. Shen, C. F. Xu, H. J. Li, Y. Liu, Z. T. Cao, X. Z. Yang, J. X. Xia and J. Wang, J. Am. Chem. Soc., 2015, 137, 15217–15224 CrossRef CAS PubMed.
- N. Qiu, X. Liu, Y. Zhong, Z. Zhou, Y. Piao, L. Miao, Q. Zhang, J. Tang, L. Huang and Y. Shen, Adv. Mater., 2016, 28, 10613–10622 CrossRef CAS PubMed.
- M. Naito, T. Ishii, A. Matsumoto, K. Miyata, Y. Miyahara and K. Kataoka, Angew. Chem., Int. Ed., 2012, 51, 10751–10755 CrossRef CAS PubMed.
- R. Mo, T. Jiang, R. DiSanto, W. Tai and Z. Gu, Nat. Commun., 2014, 5, 3364 Search PubMed.
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2016, 2, 16075 CrossRef.
- C.-C. Song, F.-S. Du and Z.-C. Li, J. Mater. Chem. B, 2014, 2, 3413–3426 RSC.
- C. Tapeinos and A. Pandit, Adv. Mater., 2016, 28, 5553–5585 CrossRef CAS PubMed.
- J. Liang and B. Liu, Bioeng. Transl. Med., 2016, 1, 239–251 CAS.
- K. E. Broaders, S. Grandhe and J. M. Frechet, J. Am. Chem. Soc., 2011, 133, 756–758 CrossRef CAS PubMed.
- A. Napoli, M. Valentini, N. Tirelli, M. Muller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS PubMed.
- G. C. Van de Bittner, E. A. Dubikovskaya, C. R. Bertozzi and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21316–21321 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2012, 134, 4112–4115 CrossRef CAS PubMed.
- B. Liu, D. Wang, Y. Liu, Q. Zhang, L. Meng, H. Chi, J. Shi, G. Li, J. Li and X. Zhu, Polym. Chem., 2015, 6, 3460–3471 RSC.
- D. S. Wilson, G. Dalmasso, L. Wang, S. V. Sitaraman, D. Merlin and N. Murthy, Nat. Mater., 2010, 9, 923–928 CrossRef CAS PubMed.
- M. S. Shim and Y. Xia, Angew. Chem., Int. Ed., 2013, 52, 6926–6929 CrossRef CAS PubMed.
- J. Liu, Y. Pang, Z. Zhu, D. Wang, C. Li, W. Huang, X. Zhu and D. Yan, Biomacromolecules, 2013, 14, 1627–1636 CrossRef CAS PubMed.
- W. Cao, Y. Gu, T. Li and H. Xu, Chem. Commun., 2015, 51, 7069–7071 RSC.
- C.-K. Lim, Y.-D. Lee, J. Na, J. M. Oh, S. Her, K. Kim, K. Choi, S. Kim and I. C. Kwon, Adv. Funct. Mater., 2010, 20, 2644–2648 CrossRef CAS.
- Y.-D. Lee, C.-K. Lim, A. Singh, J. Koh, J. Kim, I. C. Kwon and S. Kim, ACS Nano, 2012, 6, 6759–6766 CrossRef CAS PubMed.
- S. H. Lee, T. C. Boire, J. B. Lee, M. K. Gupta, A. L. Zachman, R. Rath and H.-J. Sung, J. Mater. Chem. B, 2014, 2, 7109–7113 RSC.
- Z. Deng, Y. Qian, Y. Yu, G. Liu, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2016, 138, 10452–10466 CrossRef CAS PubMed.
- Q. Sun, Z. Kang, L. Xue, Y. Shang, Z. Su, H. Sun, Q. Ping, R. Mo and C. Zhang, J. Am. Chem. Soc., 2015, 137, 6000–6010 CrossRef CAS PubMed.
- A. Matsumoto, K. Kataoka and Y. Miyahara, Polym. J., 2014, 46, 483–491 CrossRef CAS.
- X. Liu, J. Xiang, D. Zhu, L. Jiang, Z. Zhou, J. Tang, X. Liu, Y. Huang and Y. Shen, Adv. Mater., 2016, 28, 1743–1752 CrossRef CAS PubMed.
- D. Liu, C. Poon, K. Lu, C. He and W. Lin, Nat. Commun., 2014, 5, 4182 CAS.
- C. He, D. Liu and W. Lin, Biomaterials, 2015, 36, 124–133 CrossRef CAS PubMed.
- H. Wang, Y. Li, H. Bai, J. Shen, X. Chen, Y. Ping and G. Tang, Adv. Funct. Mater., 2017, 27, 1700339 CrossRef.
- G. J. Wang, T. T. Jia, X. X. Xu, L. Chang, R. Zhang, Y. Fu, Y. L. Li, X. Yang, K. Zhang, G. G. Lin, Y. X. Han and J. M. Li, Oncotarget, 2016, 7, 59402–59416 Search PubMed.
- I. Nakase, H. Akita, K. Kogure, A. Graslund, U. Langel, H. Harashima and S. Futaki, Acc. Chem. Res., 2012, 45, 1132–1139 CrossRef CAS PubMed.
- S. Wang, P. Huang and X. Chen, ACS Nano, 2016, 10, 2991–2994 CrossRef CAS PubMed.
- E. Jin, B. Zhang, X. Sun, Z. Zhou, X. Ma, Q. Sun, J. Tang, Y. Shen, E. Van Kirk, W. J. Murdoch and M. Radosz, J. Am. Chem. Soc., 2013, 135, 933–940 CrossRef CAS PubMed.
- Q. Sun, X. Sun, X. Ma, Z. Zhou, E. Jin, B. Zhang, Y. Shen, E. A. Van Kirk, W. J. Murdoch, J. R. Lott, T. P. Lodge, M. Radosz and Y. Zhao, Adv. Mater., 2014, 26, 7615–7621 CrossRef CAS PubMed.
- C. Poon, C. He, D. Liu, K. Lu and W. Lin, J. Controlled Release, 2015, 201, 90–99 CrossRef CAS PubMed.
- C. Luo, L. Miao, Y. Zhao, S. Musetti, Y. Wang, K. Shi and L. Huang, Biomaterials, 2016, 102, 239–248 CrossRef CAS PubMed.
- H. Wang, Y. Li, M. Zhang, D. Wu, Y. Shen, G. Tang and Y. Ping, Adv. Healthcare Mater., 2017, 6, 1601293 CrossRef PubMed.
- C. A. Hong, A. A. Eltoukhy, H. Lee, R. Langer, D. G. Anderson and Y. S. Nam, Angew. Chem., Int. Ed., 2015, 54, 6740–6744 CrossRef CAS PubMed.
- Y. Li, H. Wang, K. Wang, Q. Hu, Q. Yao, Y. Shen, G. Yu and G. Tang, Small, 2017, 13, 1602697 CrossRef PubMed.
- S. Lee, C. M. Valmikinathan, J. Byun, S. Kim, G. Lee, N. Mokarram, S. B. Pai, E. Um, R. V. Bellamkonda and Y. S. Yoon, Biomaterials, 2015, 63, 158–167 CrossRef CAS PubMed.
- T. Kofidis, D. Nolte, A. R. Simon, A. Metzakis, L. Balsam, R. Robbins and A. Haverich, Angiogenesis, 2002, 5, 87–92 CrossRef CAS.
- S. Bellou, G. Pentheroudakis, C. Murphy and T. Fotsis, Cancer Lett., 2013, 338, 219–228 CrossRef CAS PubMed.
- A. Egorova, A. Shubina, D. Sokolov, S. Selkov, V. Baranov and A. Kiselev, Int. J. Pharm., 2016, 515, 431–440 CrossRef CAS PubMed.
- X. L. Liu, L. D. Liu, S. G. Zhang, S. D. Dai, W. Y. Li and L. Zhang, Genet. Mol. Res., 2015, 14, 13496–13503 CrossRef CAS PubMed.
- T. Wei, C. Chen, J. Liu, C. Liu, P. Posocco, X. X. Liu, Q. Cheng, S. D. Huo, Z. C. Liang, M. Fermeglia, S. Pricl, X. J. Liang, P. Rocchi and L. Peng, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2978–2983 CrossRef CAS PubMed.
- X. Guan, Z. Guo, L. Lin, J. Chen, H. Tian and X. Chen, Nano Lett., 2016, 16, 6823–6831 CrossRef CAS PubMed.
- W. Song, Z. Tang, D. Zhang, H. Yu and X. Chen, Small, 2015, 11, 3755–3761 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr06689a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |