
Nano-confinement-driven enhanced magnetic relaxivity of SPIONs for targeted tumor bioimaging†
Tuyen Duong Thanh
Nguyen
ab,
Arunkumar
Pitchaimani
abc,
Colin
Ferrel
bc,
Ravindra
Thakkar
bc and
Santosh
Aryal
 *ab
*ab
aDepartment of Chemistry, Kansas State University, Manhattan, KS 66506, USA. E-mail: saryal@ksu.edu
bNanotechnology Innovation Center of Kansas State (NICKS), Kansas State University, Manhattan, KS 66506, USA
cDepartment of Anatomy and Physiology, Kansas State University, Manhattan, KS 66506, USA
First published on 22nd November 2017
Abstract
Superparamagnetic iron oxide nanoparticles (SPIONs) are highly biocompatible and have a versatile synthetic technique based on coprecipitation, reduction-precipitation, and hydrothermal methods, where Fe3+ and Fe2+ react in aqueous solutions; both these ions are present in our body and have clear metabolic pathways; therefore, they have attracted extensive research interest and development in the field of diagnostic imaging and therapy. However, most SPION-based clinical diagnostic contrast agents are discontinued due to severe pain, low transverse magnetic relaxivity range of 80–180 mM−1 s−1, shorter circulation half-life, and lack of disease specificity. Therefore, in this study, we engineered a bone cancer-targeted hybrid nanoconstruct (HNC) with a high transverse magnetic relaxivity of 625 mM−1 s−1, which was significantly higher than that of clinical contrast agents. The engineered HNC is peripherally decorated with a bone-seeking agent, alendronic acid-conjugated phospholipid, exhibiting a hydrodynamic size of 80 nm with a negative surface potential, −35 mV. The interior skeleton of the HNC is composed of biodegradable and biocompatible poly(L-lactic-co-glycolic acid) (PLGA), in which 5 nm SPIONs are confined. We have successfully tuned the distance between the confined SPIONs from 0.5 to 4 nm, as revealed by transmission electron microscopy (TEM) images and magnetic resonance image (MRI) phantoms. This cluster confinement dramatically enhances magnetic relaxivity possibly due to the increase in net local magnetization due to proximal field inhomogeneity. In an in vitro examination, 80% of HNC is found to bind with hydroxyapatite (HAp), which when characterized by TEM shows a painting of SPIONs over a HAp crystal. HNC is found to accumulate in mouse osteosarcoma tumor (K7M2 tumor model); both MRI and histological examination of the tumor show the potential of HNC as targeting agents for diagnosis of tumor in the bone.
1. Introduction
Engineered magnetic nanoparticles, more specifically superparamagnetic iron oxide nanoparticles (SPIONs), have attracted extensive research interest and development in the field of diagnostic imaging and therapy.1–4 Iron is a major mineral component of a biological system and its biocompatibility is extensively investigated and better understood; hence, it has become a preferred choice for various biomedical applications.5–11 Among them, contrast-enhanced magnetic resonance imaging (ceMRI) is one of the widely explored area as these contrast agents can create extremely large microscopic field gradients that shorten longitudinal and transverse relaxation times (T1 and T2) of protons, thereby producing bright and dark image contrast effects, respectively. SPIONs used as T2 contrast agents primarily increase the rate of T2 relaxation and create dark contrast effects, whereas paramagnetic metal ions such as Gd3+ used as T1 contrast agents accelerate T1 relaxation and produce bright contrast effects in the image.Highly studied paramagnetic ions in MRI are manganese (Mn), chromium (Cr), and gadolinium (Gd). These metallic ions are relatively toxic in free metal ion forms and therefore need to be encapsulated into chelating agents.12–14 In particular, Gd-based MR contrast agents, where Gd3+ is chelated into macromolecular chelating agents such as Gd-BOPTA (gadobenate dimeglumine, Multihance®), GdDTPA (gadopentetate dimeglumine, Magnevist®), and Gd-EOB-DTPA (gadoxetic acid disodium, sold as Primovist® in Europe and Eovist® in the USA), are used in clinical applications with greatest value. However, development of nephrogenic systemic fibrosis (NSF) has been recognized in patients with severe renal impairment.15–17 Reports have emerged regarding the accumulation of gadolinium in various tissues including bone, brain, and kidneys of patients who do not have renal impairment.18–20 Due to this reason, the United States Food and Drug Administration (FDA) published a safety announcement in July 2015 that it is investigating the risk of brain deposits associated with the repeated use of GBCA MRI (FDA Drug Safety Communication 2015).21
On the other hand, SPIONs are highly biocompatible and have versatile synthetic technique based on coprecipitation, reduction-precipitation, and hydrothermal methods, where Fe3+ and Fe2+ react in aqueous solutions.22–24 Both these ions are present in our body and have clear metabolic pathways such as degradation in lysosome due to an acidic environment or elimination via hepatobiliary excretion due to their uptake by the reticuloendothelial system (RES). Owing to the safe and clear excretion pathway of SPION-based contrast agents, there is a huge interest in their development. Unfortunately, commercially available iron oxide MRI contrast agents, such as Feridex® produced by Berlex-USA, Endorem™ produced by Guerbet-France, and Resovist® produced by Bayer Schering Pharma AG-Germany, were discontinued from the manufacturers. These clinical agents consisted of 4–6 nm SPIONs, which were susceptible to rapid renal clearance (renal filtration threshold size is ∼6 nm), and had transverse magnetic relaxivity ranges from 80 to 180 mM−1 s−1 at 3T. Broad applications of these discontinued MRI agents were strictly limited due to rapid uptake by the RES cells viz. Kupffer cells lining the hepatic sinusoids and similar cells in the spleen, lymph nodes, and bone marrow but not in the neoplasms.10,11 Therefore, organ-specific cancer targeted SPIONs with high magnetic relaxivity is highly desirable in clinics for the detection and monitoring of therapeutic response against cancer.
Among the organs susceptible to cancer, bone is a favorable site for tumor growth and a predominant destination for the metastatic cancer cells to reside such as metastatic breast cancer. Despite intensive efforts in the development of therapeutic and diagnostic agents against cancer occurring in the bone, tumors localized in the bone remain an incurable fatal disease due to the fast clearance or the non-specific binding profile of the agents. Among different types of bone targeting ligands, bisphosphonate (e.g. alendronic acid (ALE)) has been long emerging as a bone-seeking agent owing to its strong binding affinity with HAp, a major mineral component in the bone.25–28 However, there exists a knowledge gap regarding the unique design of the system that precisely targets the bone to enhance the diagnostic effect. Herein, we uniquely engineered targeted HNC, composed of PLGA superficially decorated with covalently conjugated ALE, with its inner polymeric core loaded with 5 nm SPIONs in a controlled fashion by tuning the distance between the clustering SPIONs, as demonstrated in Scheme 1. Effect of this cluster confinement on the magnetic properties of HNC was studied in detail, and the possibilities of using the proposed nanoconstruct as an MR contrast agent were also studied to picture the system as a bone tumor imaging contrast agent.
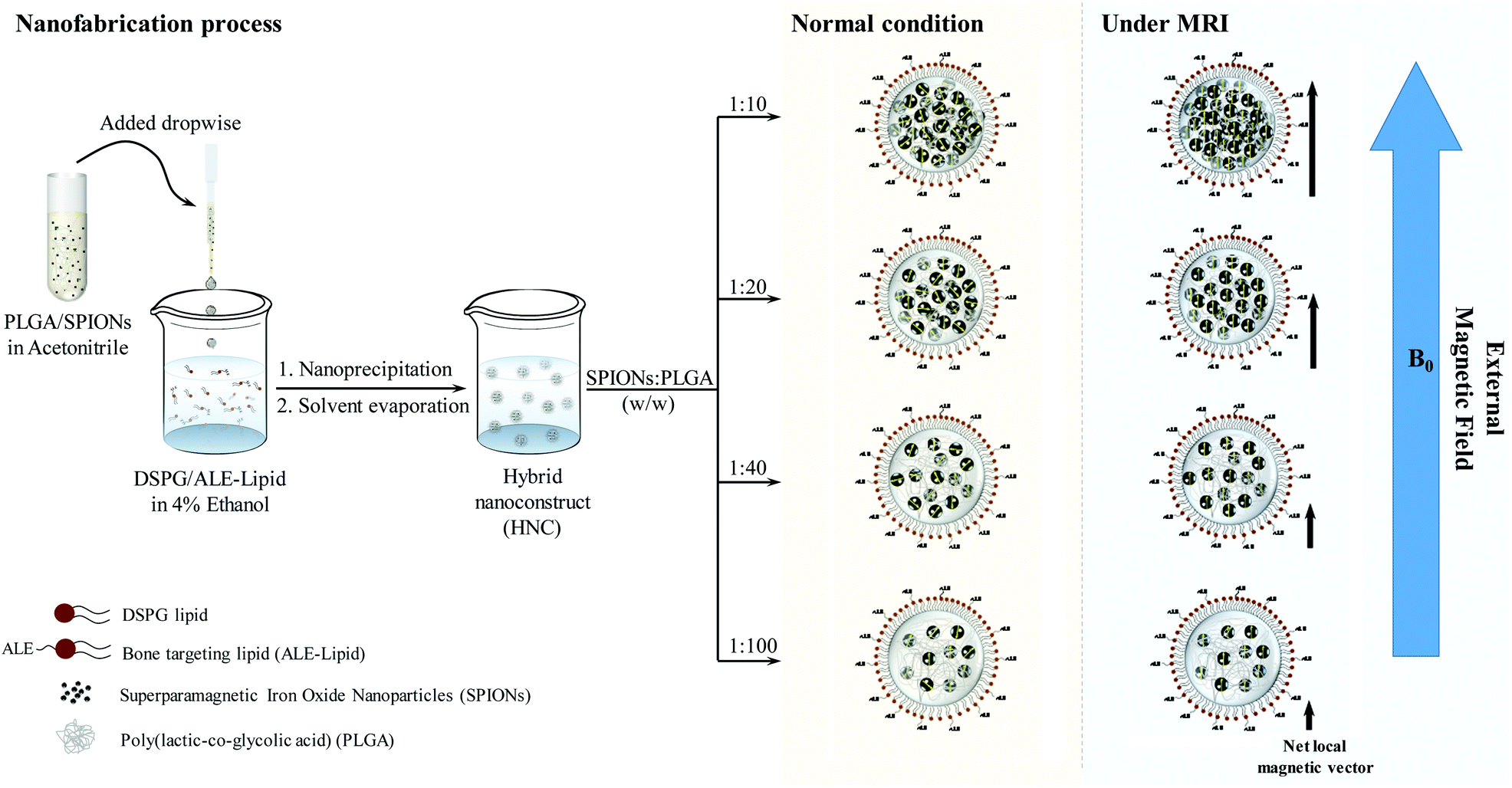 | ||
Scheme 1 Schematic represents the fabrication process of HNC using nanoprecipitation. The clustering degree of HNC is tunable by changing the ratio (w/w) of SPIONs and polymeric matrix PLGA in the organic phase. In the designed experiment, the amount of PLGA was kept constant at 1 mg, while the amount of SPIONs was varied at 10, 25, 50, and 100 μg, corresponding to SPIONs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PLGA (w/w) ratio of 1:100, 1:40, 1:20, and 1:10, respectively. As a consequence, the interparticle distant of SPIONs reduced and they confined within the polymeric matrix. Such cluster confinement dramatically enhanced the magnetic relaxivity, which could be attributed to the increase in net local magnetization due to proximal field inhomogeneity when an external magnetic field (Bo) is applied. :PLGA (w/w) ratio of 1:100, 1:40, 1:20, and 1:10, respectively. As a consequence, the interparticle distant of SPIONs reduced and they confined within the polymeric matrix. Such cluster confinement dramatically enhanced the magnetic relaxivity, which could be attributed to the increase in net local magnetization due to proximal field inhomogeneity when an external magnetic field (Bo) is applied. | ||
2. Results and discussion
Design, synthesis, and characterization of targeted HNC
ALE is bisphosphonate that has a high binding affinity toward bone cancer.25 Structurally, ALE is a zwitterion compound functionalized with phosphate, primary amine, and tertiary hydroxyl that has high electron density and is capable of undergoing a coupling reaction with carboxylic acid. However, due to acidic functionality (pKa2 = 2.6, pKa3 = 6.73, pKa4 = 11.51, and pKa5 = 12.44), the reaction capability of the primary amine of ALE is severely hindered.29 Therefore, it is critical to use a certain kind of base (e.g., triethylamine) to neutralize and further expose the lone electron pair present in the primary amine of ALE to drive the coupling reaction. Previously, we have established a protocol to attach ALE to the hydrophilic head of a phospholipid using triethylamine as a base to deprotonate the primary amine of ALE and increase the solubility of this compound in water.30 A two-step coupling reaction was performed, as outlined in Fig. S1.† The obtained phospholipid conjugation was characterized by its proton environment to confirm the conjugation, as can be seen from the change in the neighboring proton at δ 2.75 (m) and δ 3.29 (m), which corresponds to –CC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2C2CH2NHCO– and –CCH2CH2C2NHCO–, respectively.
2C2CH2NHCO– and –CCH2CH2C2NHCO–, respectively.
With the confirmation of the formation of the ALE–lipid conjugate, we next synthesized HNC in which 5 nm SPIONs (Fig. S2†) were embedded and confined into the core of the nanoconstruct. Nanoprecipitation of SPION and the polymer mixture in the lipid suspension resulted in spherical SPION clusters entrapped in a polymeric matrix and decorated peripherally with a bone targeting ligand (ALE–lipid), as demonstrated in Scheme 1. HNCs exhibited unimodal size distribution with Z-average hydrodynamic diameter of 80 ± 6 nm regardless to SPION number and density of confinement (Table S1†), while the average core size of the SPION clusters in HNC was 50 nm (Fig. 1C), as shown by transmission electron micrographs (TEM). This significant difference between hydrodynamic and TEM sizes indicates that the ALE–lipid conjugate provides a significantly thick hydrating layer over the surface of the nanoparticle. Such a dense hydrating layer could enhance the nanoparticle stability by minimizing steric attraction between nanoparticles, which minimizes the protein corona formation in the blood stream, and can prevent the nanoconstruct from being recognized as a foreign material by the immune system.30,31 Indeed, over a long period of incubation in ionic and serum conditions, the size of HNC remained constant (Fig. 1D and Fig. S3†). The stability of the nanoconstruct, being coated by the bone targeting ALE–lipid conjugate would be advantageous not only for prolonging circulation time in the biological condition and enhancing bone tumor targeting but also facilitating interaction with the surrounding water for MRI contrast enhancement.
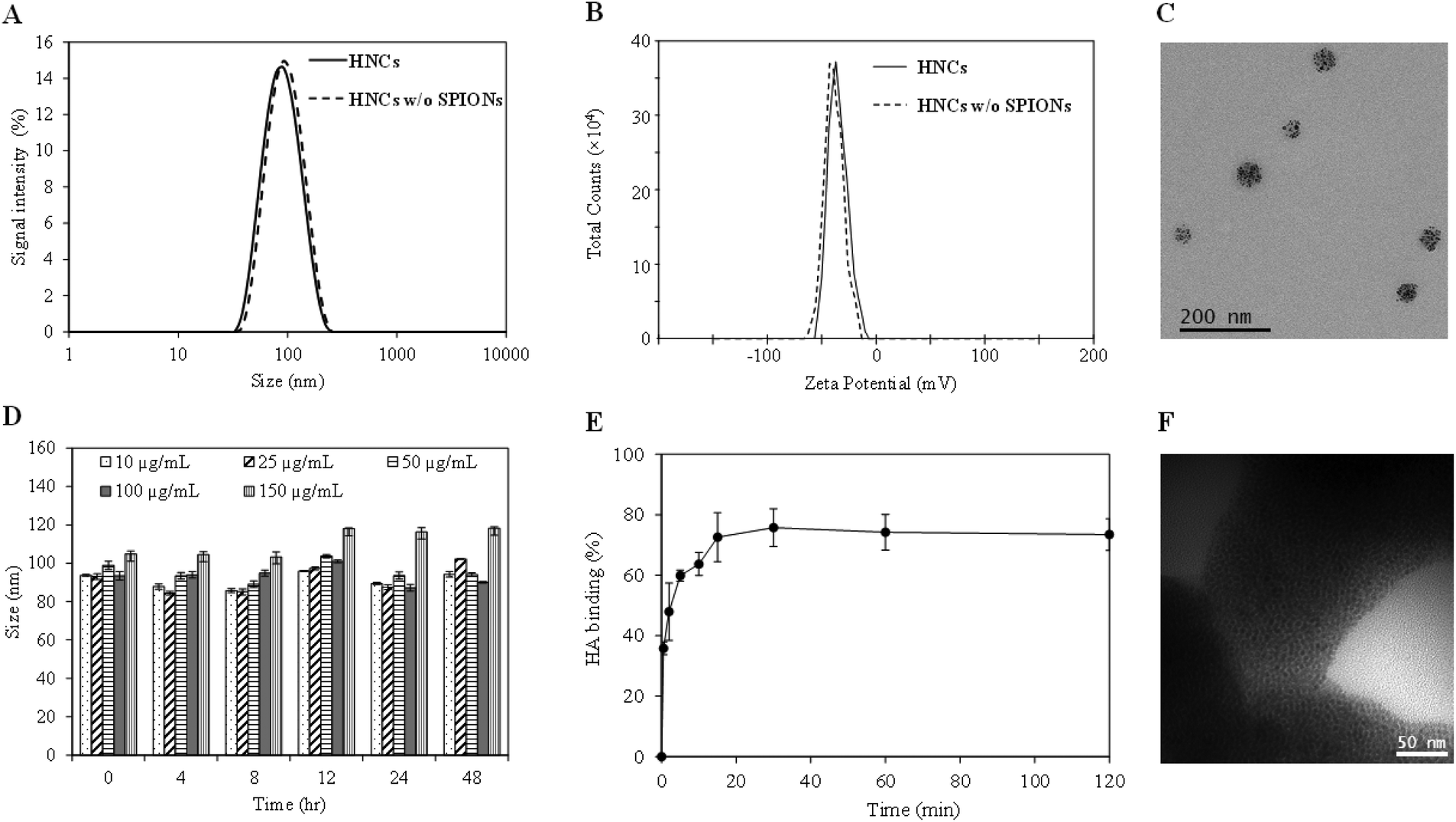 | ||
| Fig. 1 Physicochemical characterization of targeted HNC. (A) Hydrodynamic size distribution of nanoparticles with and without SPION loading. (B) Zeta potential showing consistent negative surface charge property of HNCs before and after loading of SPION nanocluster. (C) Representative transmission electron micrograph demonstrates SPION clustering in the core of the targeted nanoconstruct. (D) Stability of HNCs with different SPION loading in medium mimicking biological conditions (10% FBS in PBS). (E) Representative binding profile of HNCs with biomimetic bone hydroxyapatite (HAp) crystal. (F) TEM showing the interaction between HAp and HNC with abundance of SPION aggregates on the surface of HAp. Values are represented mean ± s.d., n = 3. | ||
In the bone microenvironment, the prevalence of osteosarcoma cells subsequently leads to abnormal activity of osteoblasts and osteoclasts, which consequently causes the release of bone minerals, predominantly in the form of calcium ions for tumor persistence.32 Previous studies have shown that the released calcium ions at bone cancer sites can be selectively targeted by using ALE in the form of free molecules or nanoformulations.33,34 Hence, by formulating a nanoconstruct with an ALE-decorated surface to target the areas with high bone turnover could be advantageous for bone-targeted bioimaging. In this study, HAp, a biomimetic bone mineral, was used for preliminary evaluation of the in vitro binding affinity of targeted HNC toward the bone cancer. The binding affinity was measured by quantifying the amount of iron that binds with HAp crystals using inductively coupled plasma-mass spectroscopy (ICP-MS). The result demonstrated rapid binding kinetics in which more than 80% HNCs bound to the surface of HAp within 10 min of incubation (Fig. 1E). Imaging of the HAp crystals under TEM further confirmed their targeting ability, which showed the presence of SPIONs (dark spots) on the surface of HAp (Fig. 1F). Note: as no staining agents were used, the polymer was not visualized under TEM.
Controlling SPION cluster density and tuning magnetic relaxivity
Clustering of SPIONs is an interesting phenomenon that governs r2 relaxivity of SPIONs.35–38 In general, T2 relaxation of water protons depends on the water diffusion rate, interaction time between water protons and local magnetic moment of nanoclusters.35,38–40 Therefore, to simplify and fully understand the confinement effect, in this study, we controlled the nanofabrication process such that the obtained HNCs exhibited a consistent size distribution regardless to SPION confinement (Scheme 1). The overall hydrodynamic and TEM size distribution of HNCs with different SPION loadings were well ordered by consistently following the same ratio of polymer and lipid suspension during the preparation process. Meanwhile, the SPION loading density was tuned by changing SPION feed amount in the polymer pre-coating step. As shown in Fig. 2A, the degree of clustering increased when the feed amount of SPIONs increased from 10 μg to 100 μg per 1 mg PLGA. As a result, the inter-SPION distance inside the polymer core became shorter, leading to greater confinement. Specifically, the average interparticle distance (distance between the confined SPIONs) inside the polymer core obtained from TEM images by measuring 100 HNCs in each formulation indicated that the average gap between two individual SPIONs was successfully tuned from 5, 3, 2, to 0.4 nm in accordance with initial feed amount of 10, 25, 50, and 100 μg SPIONs, respectively (Fig. 2B). The Fe loading efficiency in these formulations was investigated by ICP-MS, in which a plateau was observed at 100 μg mL−1 input with the maximum loading of 7% (w/w) SPIONs per 1 mg polymer (Fig. 2C). When the feed amount of Fe increases up to 150 μg per 1 mg PLGA, the nanoconstruct started to aggregate due to the destabilization initiated by the excess amount of hydrophobic SPIONs in the suspension (Fig. S3†). Therefore, we used the feed amount of 10, 25, 50, and 100 μg SPIONs per 1 mg PLGA for further investigations.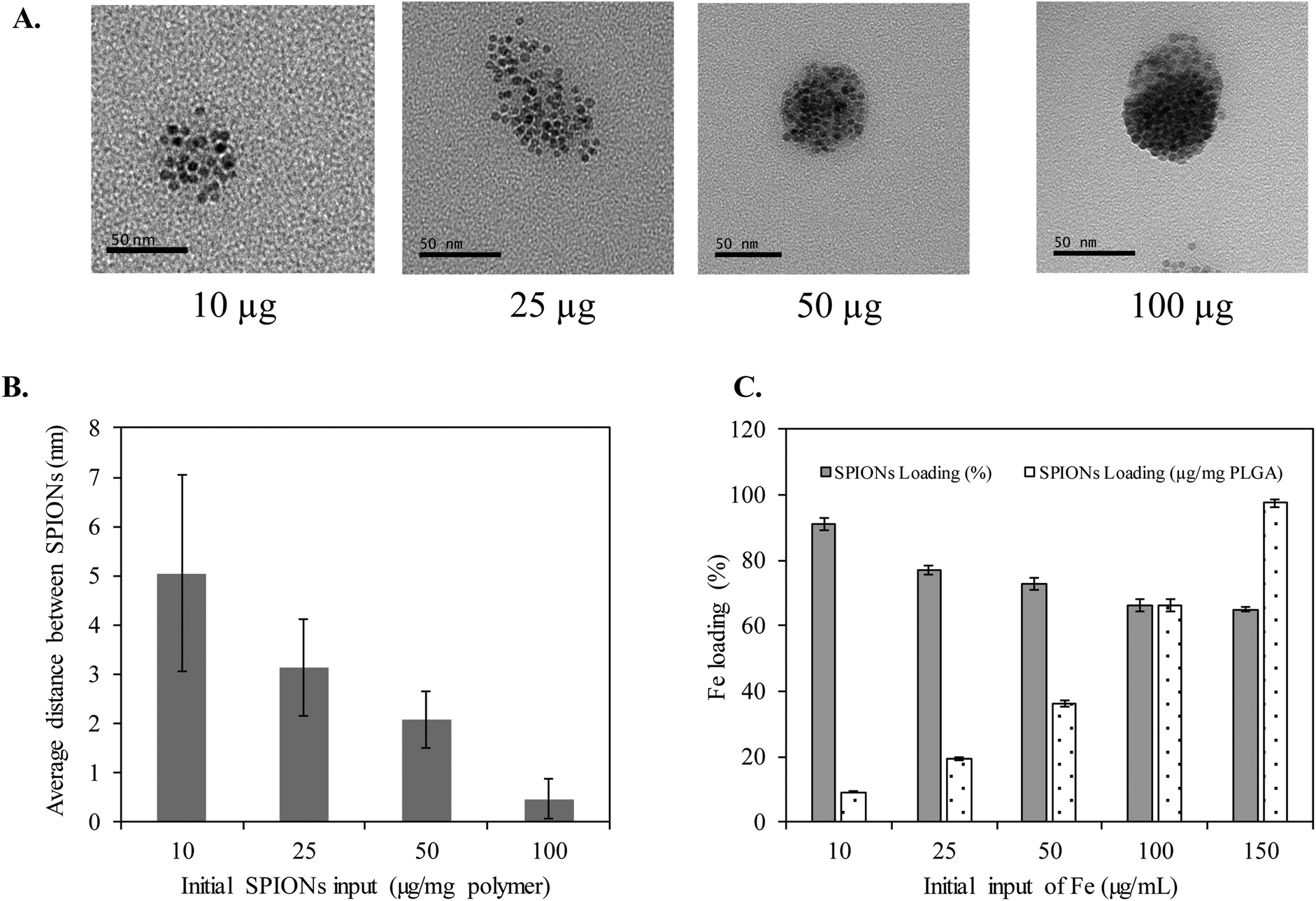 | ||
| Fig. 2 Controlling SPION cluster density. (A) TEM images showing tunable SPION packing density. (B) Average distance between two SPIONs in various packing nanoclusters (data obtained from 100 HNCs of each formulation using TEM images). (C) Iron loading efficiency measured by ICP-MS (n = 6). | ||
To further investigate the effect of SPION confinement on MR relaxivity, a serial dilution of each HNC samples, corresponding to 0.4, 2, 3, and 5 nm interparticle distance, was carried out to obtain constant Fe concentration. The resulting diluted samples were subjected to T2 measurement using a Bruker 600 MHz (14.1 T) Avance III with microimaging capability. The magnetic properties of HNCs were first investigated by measuring the signal intensity of water protons in the presence of different formulations with echo time (TE) varying from 0 to 240 ms at a constant repetition time of 1500 ms. Surprisingly, even though the Fe concentration in the 4 different HNCs was kept constant at 0.4 mM, the signal intensity of water protons dramatically decreased (became darker) when interparticle distance decreased (Fig. 3A). Compared to 5 nm interparticle (SPIONs) confined clusters, the 0.4 nm clusters exhibited 5.5-fold shorter T2 relaxation time and ∼40 times quicker T2 relaxation time than pure water. To further investigate the dependence of MR relaxation rate on clustering density, we measured T2 relaxation time at two more Fe concentrations in HNCs (0.1 and 0.2 mM) for each formulation (Fig. S4†). T2 relaxation time obtained for 0.1 and 0.2 mM Fe concentration was converted to s−1 (1/T2) and plotted against Fe concentration in mM. The transverse relaxation rates exhibited a linear relationship with respect to iron concentration in bulk solution in all cases (Fig. 3A) with the coefficient factor (R2) greater than 0.98. In our measurements, the confined structure that has interparticle distance of 0.4 nm was measured to exhibit an r2 of 641.2 s−1 mM−1 [Fe], while the less-dense confined nanoconstruct had r2 of 335.3, 162.6, and 124.2 s−1 mM−1 [Fe] for interparticle distances of 2, 3, and 5 nm, respectively (Fig. 3B). The results showed that a denser packing of SPIONs in HNCs could serve as an excellent T2 contrast agent. This phenomenon was also depicted by T2-weighted images of different HNC samples with equivalent iron concentrations, as shown in Fig. 3C; water was used as a control. Our results demonstrated that the sample with 0.4 nm interparticle distance always showed a darker contrast than other samples at the same iron concentration. Hence, MR relaxivity of the designed nanoconfinement displayed SPION density-dependent behavior, characterized by greatly increased relaxivity with higher packing density.
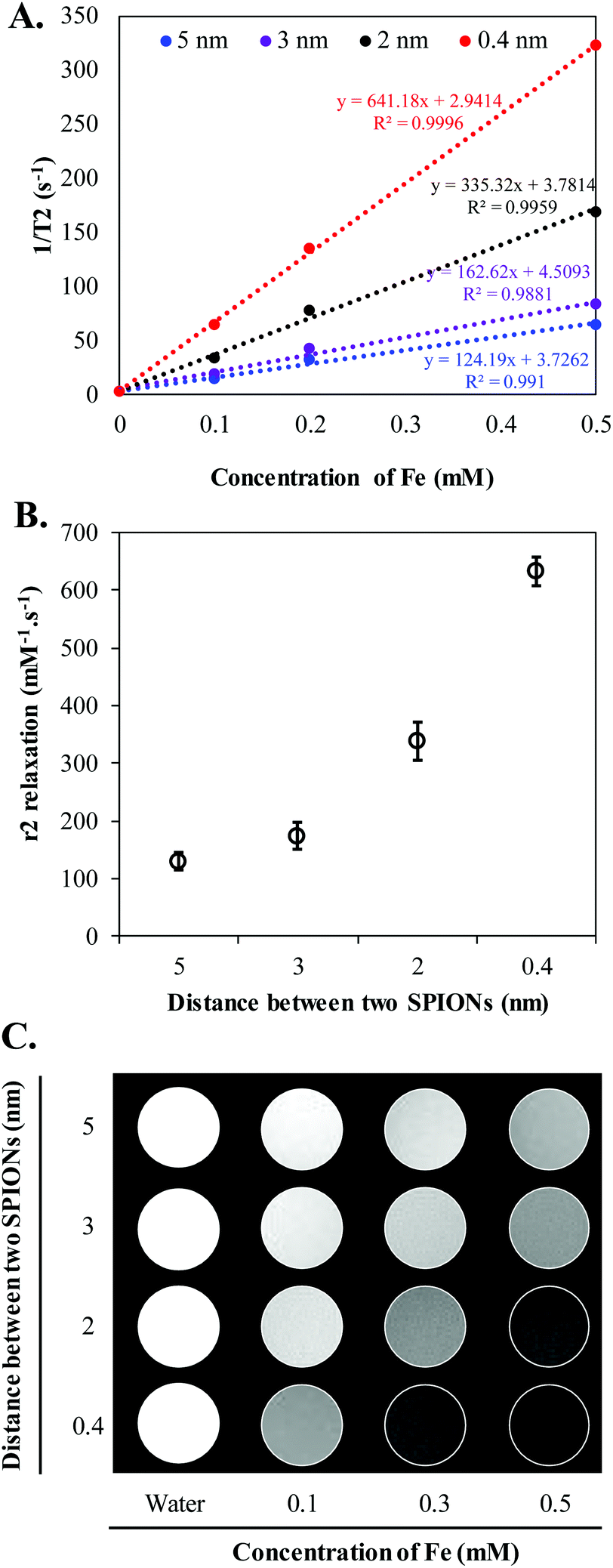 | ||
| Fig. 3 Confinement driven relaxivity enhancement. (A) Plot of 1/T2vs. the concentration of iron in aqueous solution measured in a 14.1 T MRI system at 25 °C. (B) r2 relaxation rate (s−1 mM−1) with different SPION packing density. (C) T2-Weighted MR images of HNC aqueous suspensions with different concentrations and packing densities. The T2-weighted MRI phantoms were taken at TR = 1500 ms, TE = 15 ms, and slice thickness = 1 mm. | ||
The relaxivity of iron oxide nanocluster with a hydrophilic coating is governed by the following factors: net magnetic moment, interaction time between water protons and magnetic fields, and water diffusion rate. Of the different design considerations to restore the relaxivity of SPIONs, packing individual SPIONs into a cluster is one of the effective ways reported so far.35–41 Clusters of different sizes are created by encapsulating SPIONs into self-assembled amphiphilic polymers. This is based on the principle that an increase in the sizes of SPIONs increases their transverse relaxivity. The core of densely packed SPIONs generates high relaxivity due to the resulting high magnetic moment of the particles, and if the envelope is hydrophilic, it enhances the interaction with water protons, thereby boosting the relaxivity. An approach has been proposed by Paquet et al., where the authors encapsulated the cluster of SPIONs into the core of a hydrogel, which resulted in 2 to 3 times enhancement in relaxivity.38 It has also been reported that clinically used contrast agents, such as Resovist® or Endorem™, when encapsulated into the liposome exhibited a high degree of particle aggregation and therefore high r2 relaxivity, ranging from 150 to 200 mM−1 s−1.42,43 Similarly, Lin et al. demonstrated the encapsulation of iron oxide nanoparticles into the chitosan matrix showing an 8-fold increase in magnetic relaxivity and assumed this was due to the effect of the cluster of iron oxide nanoparticles.36 In another study, an oil-in-water emulsion technique was used to encapsulate SPIONs into poly(isobutylene-alt-maleic anhydride) polymer to enhance the relaxivity and observed maximum r2 of ∼430 mM−1 s−1.44 A number of theoretical studies have illustrated that such an enhancement in relaxivity is due to the fact that the decrease in the diffusion coefficient of water at the vicinity of the clustering particles increases the interaction time of water protons and the magnetic field generated by the particles. In the efforts above, although attention has been given to the enhancement in T2 relaxivity of SPIONs, less attention has been given to the distance induced confinement of individual SPIONs and the influence of confinement on T2 relaxivity.
In the present study, a densely packed SPION core generating a cooperative magnetic effect in each particle led to an increase in magnetic moment, resulting in high relaxivity.45 The ALE–lipid conjugate with phosphate moiety on the outer layer trapped water molecules via hydrogen bonding, creating a bulk water proton environment surrounding magnetic fields near the SPION core, which further increased the possibility of the interaction between water and magnetic particles. Similarly, the PLGA polymeric matrix contributed in arresting water, which in turn slowed down the water diffusion rate and as a consequence extended the interaction time of the water protons and contrast agents. This result was also observed in a similar system where chitosan was used as the polymeric matrix.36 As a result, these three components together generated a synergistic effect to shorten T2 relaxation times, resulting in relaxivity enhancement of the nanoconstruct. Therefore, by controllably confining SPIONs, the r2 relaxivity of HNCs could be easily tuned and used as a technological platform for designing T2 MRI contrast agents.
In vitro uptake of HNCs in osteosarcoma cells
Before evaluating the cellular targeting capability of HNCs, we first accessed the cellular biocompatibility of HNCs in vitro by incubating them with K7M2 for 24, 48, and 72 h with a wide range of concentration from 10 to 200 μg mL−1. As shown in Fig. 4A, there was no dose-dependent cytotoxicity in HNCs over a long period of incubation, suggesting biocompatibility of HNCs in the cellular environment. To confirm the cellular interaction of HNCs with K7M2, a cellular Prussian blue staining technique was adopted. By using this technique, the ferric ions present in the cell underwent a histochemical reaction with potassium ferrocyanide to form ferric ferricyanide, exhibiting blue colour. For this experiment, cells were incubated with 100 μg mL−1 PEGylated SPIONs (control nanoparticles) or HNCs (targeted nanoparticles) in culture media at 37 °C for 3 h. Fig. 4B showed a higher accumulation of iron in the K7M2 cells treated with HNCs, as indicated by the blue staining of a large number of cells, whereas few cells treated with control PEGylated SPION nanoclusters showed accumulation. Although the Prussian blue staining technique could give an idea about the presence of iron in the cells, intracellular distribution of HNCs was not clearly understood. Therefore, to study the cellular colocalization of HNCs, we peripherally labeled HNCs with red fluorescent dye (Rhodamine B (RhB)) and investigated the intracellular distribution of HNCs using a confocal laser scanning microscope (CLSM). The CLSM technique is capable of providing continuous slices along the z-axis for acquiring an image of the exterior and interior compartment of the cell body.46 The representative middle slice of the cell image is shown in Fig. 4C, a blue channel for DAPI stained nuclei and RhB labeled targeted HNCs in the red channel, which indicated the relative distribution of HNCs throughout cell body. A control experiment with non-targeted SPION loaded PEGylated NPs was also carried out to evaluate the HNCs. As shown in Fig. 4C, it is clear that the cellular accumulation of HNCs is an order of magnitude higher than that of non-targeted NPs over the period of incubation. The quantitative evaluation of cellular internalization was also conducted by measuring the RhB fluorescence intensity using Image-J software, which supported our hypothesis of targeting (Fig. 4D). In addition, the z-stack images of single cells presented in Fig. S5† further assured that these NPs were inside the cell body. The sequential slices taken horizontally from a stack of images indicated that targeted HNCs were predominantly distributed in the cell, implying the targeting property of HNCs towards K7M2. This observation was further quantitatively supported by measuring cellular iron concentration using ICP-MS (Fig. S6†). The results revealed that targeted HNCs were associated with cell more efficiently than the control PEGylated NPs, giving us strong evidence of the targeting property of HNCs.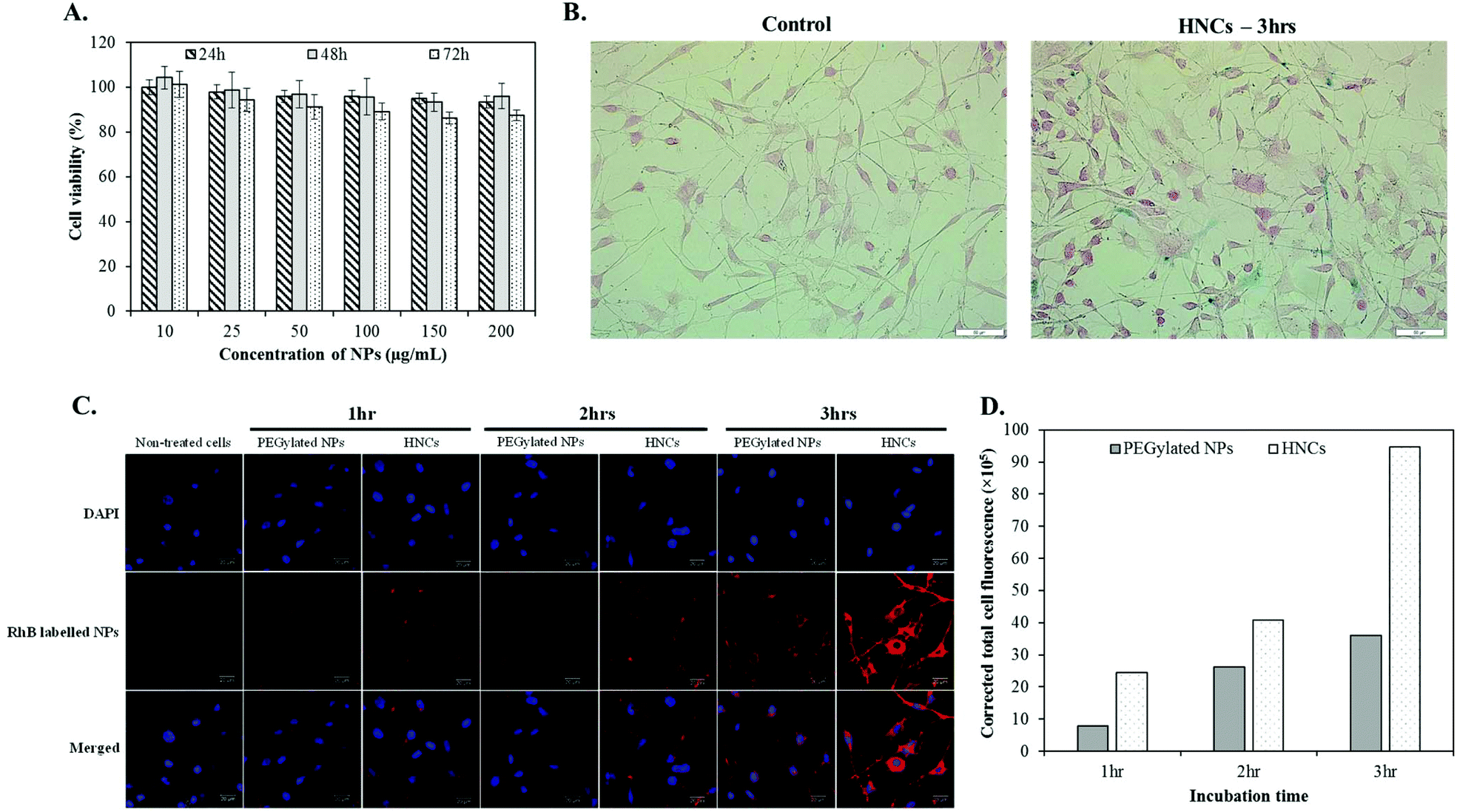 | ||
| Fig. 4 In vitro cellular studies. (A) Biocompatibility of HNCs. The result is represented as mean ± SD (n = 8). (B) Prussian blue cellular staining showing the presence of intracellular SPIONs. (C) Confocal images of K7M2 cells incubated with RhB labeled NPs over the period of incubation at 37 °C. The cell nuclei were stained by DAPI (blue). (D) A quantitative cellular NP uptake by measuring RhB fluorescence intensity in panel C using Image-J software. | ||
Ex vivo imaging study and tumor histological analysis
To further examine the tumor targeting ability of HNCs for potential MRI application, we injected 100 μL of HNCs (0.25 mg kg−1 of iron) into immunodeficient female NU/NU nude mice bearing K7M2 tumors systematically via the tail vein. Two hours post-injection, the mice were euthanized and immediately taken for T2-weighted MR imaging acquisition. Since HNCs showed significant enhancement in transverse relaxation, the dose of iron used in our experiment was lower than the previously reported dose as well as the clinical dosage of Feridex® (0.56 mg kg−1).47 Representative T2-weighted MR images taken at TE/TR = 21/1500 ms along the z-axis are shown in Fig. 5A. As can be seen in the figure, a significant reduction in the MR signal was observed (negative contrast) not only from around the periphery of the tumor but also from the different depths of tumor slice, and this signal dramatically reduced at the center of the tumor region. The results indicated the effective accumulation and penetration of HNCs into the deep tumor. On the other hand, less T2 contrast enhancement was observed in the case of non-targeted SPION loaded PEGylated NPs (Fig. S7A†). The darker contrast enhancement of HNCs was further confirmed using Image-J software to quantitatively evaluate the signal intensity distribution (negative contrast distribution) throughout the tumor (Fig. S7B†). Such a significant contrast enhancement was probably caused by the selective accumulation of HNCs driven by ALE that targets the bone tumor lesion. ALE can distinctively chelate released Ca2+ at the tumor site, thereby increasing the possibility of HNC accumulation in K7M2 cells, as demonstrated in in vitro studies. To further verify the HNC accumulation in the tumor, after imaging, the tumor tissue was subjected to histological analysis to study the distribution of SPIONs in the tumor. It is well known that Prussian blue dye has high affinity to iron and can be used to detect the presence of iron in hard tissues. As expected, a large number of blue-stained iron oxide NPs were observed in the histological slice of the tumor, augmenting the distribution data of NPs (Fig. 5B). This further supports the accumulation potential of HNC in tumor tissues for tumor diagnosis.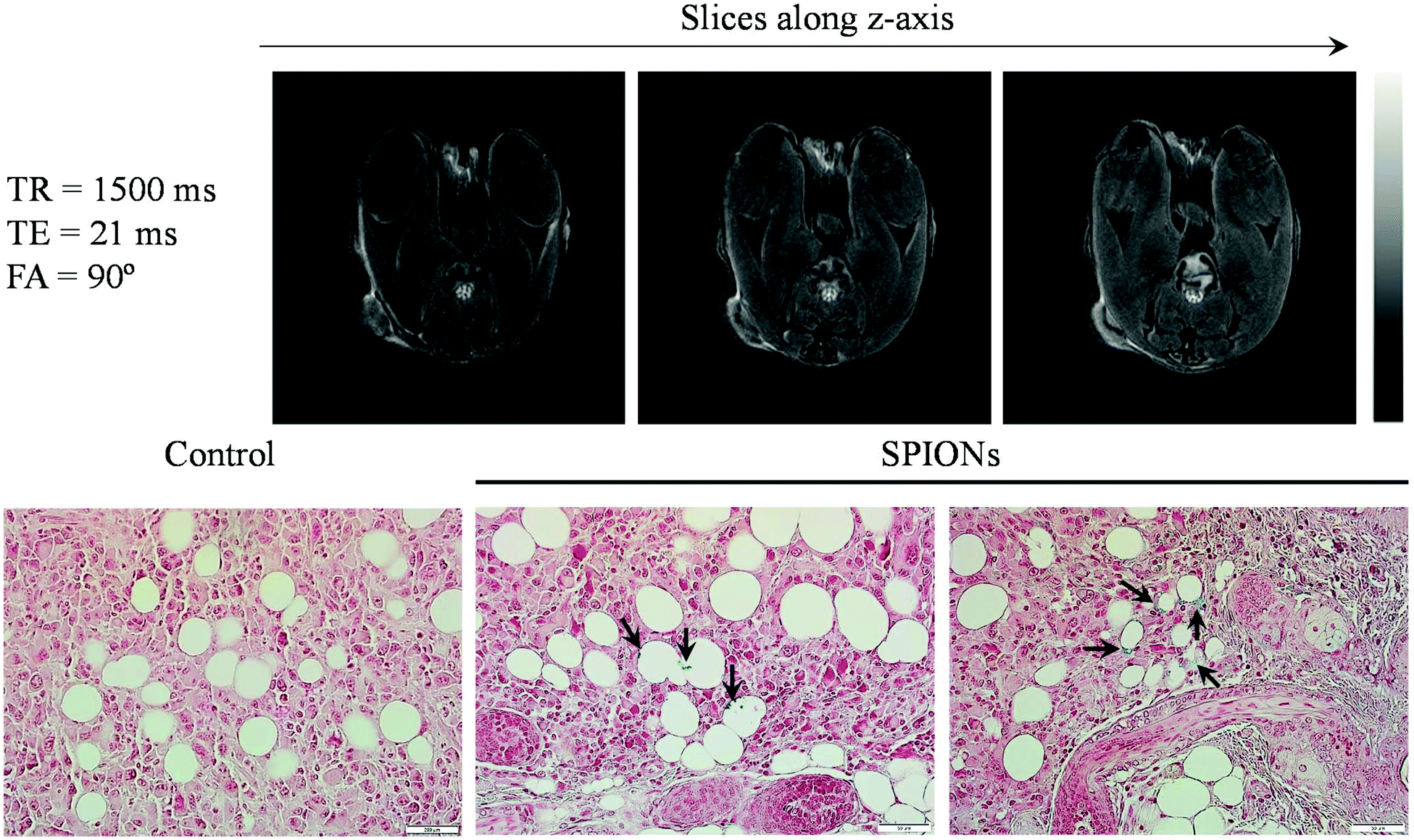 | ||
| Fig. 5 Ex vivo study. (A) MR images of a tumor bearing mouse at different z-planes with plane thickness of 1 mm, TE/TR = 21/1500 ms, flip angle = 90°. (B) Histology of tumor section of HNC treated animals stained with Prussian blue indicates the presence of SPIONs at the tumor site. | ||
3. Experimental
Chemicals and reagents
Alendronic acid was purchased from TCI America. 1,2-Dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(succinyl) (sodium salt) (16:0 Succinyl PE), 1,2-distearoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt), and L-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (Ammonium Salt) (egg-transphosphatidylated, Chicken) (Egg Liss Rhod PE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Iron oxide nanocrystals with an oleic acid coating (5 nm) were purchased from Ocean NanoTech (San Diego, CA, USA). 50:50 poly(D,L-lactide-co-glycolide) carboxylate end group (0.55–0.75 dL g−1) was purchased from DURECT Corporation (Birmingham, AL, USA). Osteosarcoma cell line K7M2 was purchased from ATCC and maintained according to the manufacturer's recommendation. All other chemicals and solvents were purchased from Sigma-Aldrich (Milwaukee, WI, USA) and used as received.
Animal and tumor model
All animal experiments and protocols were performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and approved by the Institutional Animal Care and Use Committee (IACUC) and Institutional Biosafety Committee (IBC), Kansas State University, Manhattan, Kansas. Six-week-old female NU/NU nude mice were purchased from Charles River Laboratories International, Inc. and used for the study after 7 days of adaptation. For mice bearing K7M2 tumor models, 1 × 106 K7M2 cells in 1× PBS were subcutaneously injected into the hind rear flank region of the mice. Tumor size and body weight were monitored periodically.Synthesis of ALE–lipid conjugate
The reaction scheme for bone targeting ligand modified phospholipid is shown in Fig. S1† and generally followed a previously reported procedure using EDC/NHS coupling reaction.30 To prepare head group modified phospholipids, 50 mg of succinyl-PE lipid was hydrated in 6 mL deionized water at 60 °C until a clear viscous solution was obtained. After the mixture was cooled down, the lipid was activated by 67.2 mg EDC and 58 mg of NHS under a vigorous stirring condition at room temperature for 30 min. To this reaction mixture, 120 mg of ALE dissolved in 2 mL water containing 15% TEA was added and stirred for an additional 24 h at room temperature. At the end of reaction time, the reaction mixture was transferred into a cellulose dialysis bag (MWCO ∼500 Da), and dialyzed against water for 24 h at room temperature; the water was freshly changed every 6 h. The samples were lyophilized to obtain a dry powder and stored at −20 °C for further use. The obtained product was 35 mg (90% yield). The formation of lipid bisphosphonate (ALE–lipid conjugate) was confirmed by FT-IR and 1H-NMR.Preparation of nanoparticles
Bone-targeted hybrid nanoconstruct (HNC) was prepared using one-step nanoprecipitation. Briefly, SPIONs were precoated with PLGA polymer by mixing 1 mg PLGA with different amounts of hydrophobic SPIONs (10, 25, 50, 100, and 150 μg) in chloroform and dried under vacuum. The iron oxide nanoparticles entrapped in the polymeric film was dissolved in 400 μL acetonitrile. The mixture was then added drop-wise to 3 mL of 4% ethanol containing 200 μg ALE–lipid conjugate and 260 μg DSPG under a magnetic stirring condition at 60 °C. At the end of the nanoprecipitation process, 1 mL of Mili-Q water was added to cool down the reaction mixture. The mixture was stirred continuously for an additional 1 h to facilitate the formation of nanoparticles and evaporation of the organic solvent at room temperature. The obtained nanoparticles were further purified using an Amicon Ultra-4 centrifugal filter (Millipore, MA) with a molecular weight cut-off of 10 kDa and stored at 4 °C for further use. Nanoparticles labeled with RhB-lipid were prepared following a similar protocol with the introduction of 20 μg RhB-lipid in the lipid mixture prior to nanoparticle preparation. The controlled PEGylated nanoparticles were also prepared following a similar protocol using DSPE-PEG-COOH as an alternative to the ALE–lipid conjugate.Characterization of HNC
The morphology of the prepared nanoparticle loaded SPION cluster was analyzed using a transmission electron microscope (TEM, Tecnai G2, Spirit Bio TWIN). TEM samples were prepared by drop casting and evaporation using formvar coated copper grid (400 mesh). TEM images were analyzed by a GATAN digital imaging system (GATAN, Inc.). The hydrodynamic size and zeta potential measurements of the prepared nanoconstructs were analyzed by dynamic light scattering (DLS) using a Zeta sizer Nano ZSP (Malvern, Worcestershire, UK). Brownian motion and the Smoluchowski equation were used to calculate the average hydrodynamic size and zeta potential value, respectively. The colloidal stability of HNCs was investigated in both ionic and serum conditions to mimic biological environment. In brief, 1 mg HNC was dispersed in PBS (pH 7.4) or 10% FBS. The change in size was recorded at a predetermined time using DLS. All data represent the average of triplicate measurements of samples prepared in different preparations. The amount of iron loaded into the HNC was determined using inductively coupled plasma mass spectrometry (ICP-MS, PerkinElmer, NEXion 350X). For ICP-MS, the HNC samples were digested with 2.0 ml of concentrated HNO3 for 5 h. After chemical digestion, 100 μl of the sample was diluted with 10 ml of 2% HNO3 and analyzed using ICP-MS.In vitro bone targeting assay
The binding affinity of HNCs to HAp was determined directly by measuring the amount of iron oxide nanoparticles present in HAp pellets and comparing it with the initial concentration of iron oxide nanoparticles by ICP-MS. Briefly, 100 μL of HNC (100 μg mL−1) was incubated with 1 mL HAp (1 mg mL−1) in 1 ml Eppendorf tubes for varying periods (0.5, 2, 5, 10, 15, 30, 60, and 120 min) at 37 °C. At the end of the incubation, the samples were centrifuged at 1500 rpm for 5 min to obtain HAp crystals and the HNC bound to them. Pellets were substantially washed 3 times with Milli-Q water, digested with 70% HNO3, and quantified using ICP-MS.In vitro magnetic properties
The MRI relaxivity of HNC was performed using a 14.1 T MRI system (Bruker Avance III, WB, 600 MHz NMR-MRI). The transverse relaxation time (T2) of different formulations of the HNCs (10, 25, 50 and 100 μg mL−1 initial input concentration) in Milli-Q water was determined using turbo spin echo sequence. T2 relaxation was calculated using six measurements acquired with a constant repetition time (TR) of 1500 ms by varying echo time (TE) from 8 ms to 240 ms. The transverse relaxivity (r2) was determined from the T2 relaxation time and the concentration of iron (in mM). The corresponding T2 weighted MRI phantoms were recorded using a turbo spin echo sequence with TR = 1500 ms, TE = 15 ms and slice thickness = 1 mm. The confinement effect of the SPION nanocluster in the polymeric core of nanoparticles was confirmed by evaluating the r2 relaxation time of different SPION cluster compactness at the same iron concentration.In vitro cellular uptake study
To detect the presence of intracellular SPIONs, we performed cellular histology using the Prussian blue staining procedure. K7M2 mouse osteosarcoma cells were seeded onto poly-D-lysine coated glass coverslips at the density of 2 × 105 cells per coverslip and incubated for 24 h for adherence. Then cells were treated with 100 μg mL−1 non-targeted or targeted NPs for 3 h at 37 °C. At the end of the incubation time, cells were washed twice in PBS, fixed with 4% paraformaldehyde for 30 min followed by Prussian blue staining (5% potassium ferrocyanide II in 5% hydrochloric acid (ratio 1:1)) for 1 h at 37 °C. Then, the cells were washed with PBS and subsequently counterstained with a 0.1% (w/v) nuclear fast red solution in distilled water for 1 min. After washing with distilled water, the images of the cell were obtained through an inverted microscope.
To further quantify the amount of HNC taken up by the cells, ICP-MS was adapted. In brief, cells were seeded in a 12-well plate with a density of 6.5 × 105 cell per well and incubated for 24 h. When cell confluency reached 80%, the cell medium was replaced with 1 mL of 10 μg mL−1 of HNCs or PEGylated NPs suspended in culture media and incubated for 3, 6 and 12 h. At the end of the predetermined time, the cells were washed 3 times with PBS, trypsinized, and collected by centrifugation at 5000 rpm for 10 min. Cellular uptake of HNCs and PEGylated NPs was further processed by sonicating with 2 mL HNO3 at 70 °C for 60 min. The digested cell was diluted and subjected to ICP-MS for iron concentration analysis. Cells without HNC treatment was taken as the control.
Confocal study
Confocal laser scanning microscopy (CLSM) with Z-stack was used to confirm the intracellular distribution of HNCs. Non-treated cells and non-targeted SPION loaded PEGylated NPs were used as the control. In brief, cells were seeded in poly-D-lysine-coated 8 chamber slides at a density of 20000 cells per well and incubated for 24 h. Then, the cells were treated with 50 μg mL−1 RhB-labeled NP suspension and incubated over varying periods period (1, 2, and 3 h). After incubation, the treated cells were washed twice with 1× PBS (pH 7.4), fixed with 4% paraformaldehyde for 30 min at room temperature, stained with DAPI for an additional 10 min and imaged using CLSM (Carl Ziess, LSM-700). The cellular uptake was accessed by measuring RhB fluorescence intensity using Image-J software (National Institute of Health).
Cellular cytotoxicity studies
The in vitro biocompatibility of HNCs was investigated on osteosarcoma K7M2 using MTT assay. In brief, 2 × 104 cells per well in DMEM were seeded in a 96-well plate and incubated for 24 h. To evaluate the biocompatibility of NPs, the media were replaced with different NP concentrations (10, 25, 50, 100, 150 and 200 μg mL−1) and incubated for an additional 24, 48 and 72 h. Control cells were also maintained without any NP treatment (n = 6). After the completion of incubation, MTT was added to each well and further incubated for 3 h according to the manufacturer recommendation. The insoluble formazan crystals were solubilized using DMSO and the absorbance was recorded at 570 nm using a microplate reader (BioTek, Synergy H1 hybrid reader).Ex vivo magnetic resonance imaging study
A pilot MRI study was conducted in female Nu/Nu mice bearing subcutaneous mouse osteosarcoma (K7M2) tumors. When the tumor size reached around 40–80 mm3, the animals were treated with HNCs or non-targeted SPION-loaded PEGylated NPs containing 0.25 mg of Fe per kg of body weight in 100 μL volume via the tail vein. Two hours post-injection, the animals were euthanized to simplify the MRI pilot experiment and immediately imaged using a Bruker WB 600 MHz NMR-MRI (14.1 tesla). The MR images were obtained using a QTR 30 mm coil at 25 °C with a FLASH (fast slow angle shot) protocol. The T2 weighted images were taken with fat suppression parameters viz. TE/TR = 21/1500 ms, slice thickness = 1 mm, flip angle = 90°, image size 256 × 256, FOV = 30 × 30, and the total acquisition time = 16 min. The signal intensity of the tumor area was further analyzed using Image-J software (National Institute of Health) to access signal distribution within the tumor region quantitatively.Histological analysis
After imaging, the animals were euthanized and the tumors were dissected and fixed in formalin for histology analysis. The tumor samples were embedded in paraffin wax and sections were made of around 6 μm thickness. Further, the tissue sections were stained with H&E and Prussian blue to locate iron distribution in the tumor.4. Conclusions
In summary, a targeted hybrid nanoconstruct was successfully engineered using a bone targeting ligand, ALE, and hydrophobic PLGA polymeric core. The core of the nanoconstruct was loaded with SPIONs in a controlled fashion by tuning the distance between the clustering SPIONs. The designed HNC was found to be of 80 ± 5 nm size and showed enhanced r2 relaxivity from 124 to 641 mM−1 s−1 at 14.1 T, which is one order of magnitude higher than that of iron oxide-based clinically used magnetic contrast agents, such as Feridex® (r2 = 120 mM−1 s−1, 3 T) and Supravist® (r2 = 57 mM−1 s−1, 3 T). Moreover, the results from a binding study using HAp as a bone model revealed that HNC had strong binding affinity to the bone. In an in vitro cellular study against K7M2, HNC showed biocompatibility over a range of concentrations and exhibited higher cellular uptake than conventional PEGylated nanoparticles. Due to its higher targeting affinity and magnetic relaxivity, the MRI of the bone tumor-bearing mice showed an enhanced contrast capable of distinguishing the surrounding soft tissue. Therefore, the targeting and nanoconfinement strategy presented herein could be a promising design to enhance the contrast ability of SPION based contrast agents in the diagnosis and treatment of cancer.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the support from the Department of Chemistry and the Innovative Research Award from Johnson Cancer Research Center (JCRC), Kansas State University (KSU), Manhattan, Kansas. The authors also thank the Confocal Core supported by CVM-KSU.References
- A. Gizzatov, J. Key, S. Aryal, J. Ananta, A. Cervadoro, A. L. Palange, M. Fasano, C. Stigliano, M. Zhong, D. Di Mascolo, A. Guven, E. Chiavazzo, P. Asinari, X. Liu, M. Ferrari, L. J. Wilson and P. Decuzzi, Adv. Funct. Mater., 2014, 24, 4584–4594 CrossRef CAS PubMed.
- S. Aryal, J. Key, C. Stigliano, M. D. Landis, D. Y. Lee and P. Decuzzi, Small, 2014, 10, 2688–2696 CrossRef CAS PubMed.
- A. Cervadoro, M. Cho, J. Key, C. Cooper, C. Stigliano, S. Aryal, A. Brazdeikis, J. F. Leary and P. Decuzzi, ACS Appl. Mater. Interfaces, 2014, 6, 12939–12946 CAS.
- S. Aryal, J. Key, C. Stigliano, J. S. Ananta, M. Zhong and P. Decuzzi, Biomaterials, 2013, 34, 7725–7732 CrossRef CAS PubMed.
- G.-P. Yan, L. Robinson and P. Hogg, Radiography, 2007, 13, e5–e19 CrossRef.
- Y. Imai, T. Murakami, S. Yoshida, M. Nishikawa, M. Ohsawa, K. Tokunaga, M. Murata, K. Shibata, S. Zushi, M. Kurokawa, T. Yonezawa, S. Kawata, M. Takamura, H. Nagano, M. Sakon, M. Monden, K. Wakasa and H. Nakamura, Hepatology, 2000, 32, 205–212 CrossRef CAS PubMed.
- M. G. Harisinghani and R. Weissleder, PLoS Med., 2004, 1, e66 Search PubMed.
- T. Ichikawa, A. S. Arbab, T. Araki, K. Touyama, H. Haradome, J. Hachiya, M. Yamaguchi, H. Kumagai and S. Aoki, Am. J. Roentgenol., 1999, 173, 207–213 CrossRef CAS PubMed.
- P. Reimer and T. Balzer, Eur. Radiol., 2003, 13, 1266–1276 Search PubMed.
- E. Senéterre, P. Taourel, Y. Bouvier, J. Pradel, B. Van Beers, J. P. Daures, J. Pringot, D. Mathieu and J. M. Bruel, Radiology, 1996, 200, 785–792 CrossRef PubMed.
- Y. Imai, T. Murakami, M. Hori, K. Fukuda, T. Kim, T. Marukawa, H. Abe, M. Kuwabara, H. Onishi, K. Tsuda, Y. Sawai, M. Kurokawa, N. Hayashi, M. Monden and H. Nakamura, Hepatol. Res., 2008, 38, 147–158 Search PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- A. Pitchaimani, T. D. T. Nguyen, L. Maurmann, J. Key, S. H. Bossmann and S. Aryal, J. Biomed. Nanotechnol., 2017, 13, 417–426 CrossRef CAS.
- A. Pitchaimani, T. D. T. Nguyen, H. Wang, S. H. Bossmann and S. Aryal, RSC Adv., 2016, 6, 36898–36905 RSC.
- J. Kurtkoti, T. Snow and B. Hiremagalur, Nephrology, 2008, 13, 235–241 CrossRef PubMed.
- M. A. Perazella, Clin. J. Am. Soc. Nephrol., 2007, 2, 200–202 CrossRef CAS PubMed.
- A. Kribben, O. Witzke, U. Hillen, J. Barkhausen, A. E. Daul and R. Erbel, J. Am. Coll. Cardiol., 2009, 53, 1621–1628 CrossRef CAS PubMed.
- A. Myrissa, S. Braeuer, E. Martinelli, R. Willumeit-Römer, W. Goessler and A. M. Weinberg, Acta Biomater., 2017, 48, 521–529 CrossRef CAS PubMed.
- M. Rogosnitzky and S. Branch, BioMetals, 2016, 29, 365–376 CrossRef CAS PubMed.
- R. J. McDonald, J. S. McDonald, D. F. Kallmes, M. E. Jentoft, D. L. Murray, K. R. Thielen, E. E. Williamson and L. J. Eckel, Radiology, 2015, 275, 772–782 CrossRef PubMed.
- C. for D. E. and Research, Drug Safety and Availability – FDA Drug Safety Communication, https://www.fda.gov/Drugs/DrugSafety/ucm455386 (accessed August 16, 2017).
- J. Mürbe, A. Rechtenbach and J. Töpfer, Mater. Chem. Phys., 2008, 110, 426–433 CrossRef.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- F.-Y. Cheng, C.-H. Su, Y.-S. Yang, C.-S. Yeh, C.-Y. Tsai, C.-L. Wu, M.-T. Wu and D.-B. Shieh, Biomaterials, 2005, 26, 729–738 CrossRef CAS PubMed.
- M. T. Drake, B. L. Clarke and S. Khosla, Mayo Clin. Proc. Mayo Clin., 2008, 83, 1032–1045 CrossRef CAS PubMed.
- A. L. Boskey, BoneKEy Rep., 2013, 2, 447 Search PubMed.
- C. Rey, C. Combes, C. Drouet and M. J. Glimcher, Osteoporos. Int., 2009, 20, 1013–1021 CrossRef CAS PubMed.
- D. B. Burr and O. Akkus, in Basic and Applied Bone Biology, Academic Press, San Diego, 2014, pp. 3–25 Search PubMed.
- M. Meloun, Z. Ferenčíková, L. Netolická and T. Pekárek, J. Chem. Eng. Data, 2011, 56, 3848–3854 CrossRef CAS.
- T. D. T. Nguyen, A. Pitchaimani and S. Aryal, Sci. Rep., 2016, 6, 36707 CrossRef CAS PubMed.
- J. Israelachvili and H. Wennerström, Nature, 1996, 379, 219–225 CrossRef CAS PubMed.
- Y.-C. Chen, D. M. Sosnoski and A. M. Mastro, Breast Cancer Res., 2010, 12, 215 CrossRef CAS PubMed.
- S. I. Thamake, S. L. Raut, Z. Gryczynski, A. P. Ranjan and J. K. Vishwanatha, Biomaterials, 2012, 33, 7164–7173 CrossRef CAS PubMed.
- P. Liu, L. Sun, D. Zhou, P. Zhang, Y. Wang, D. Li, Q. Li and R.-J. Feng, Sci. Rep., 2015, 5, 17387 CrossRef CAS PubMed.
- C. E. Smith, D. Ernenwein, A. Shkumatov, N. E. Clay, J. Lee, M. Melhem, S. Misra, S. C. Zimmerman and H. Kong, Biomaterials, 2015, 69, 184–190 CrossRef CAS PubMed.
- Y. Lin, S. Wang, Y. Zhang, J. Gao, L. Hong, X. Wang, W. Wu and X. Jiang, J. Mater. Chem. B, 2015, 3, 5702–5710 RSC.
- R. R. T. Ragheb, D. Kim, A. Bandyopadhyay, H. Chahboune, B. Bulutoglu, H. Ezaldein, J. M. Criscione and T. M. Fahmy, Magn. Reson. Med., 2013, 70, 1748–1760 CrossRef CAS PubMed.
- C. Paquet, H. W. de Haan, D. M. Leek, H.-Y. Lin, B. Xiang, G. Tian, A. Kell and B. Simard, ACS Nano, 2011, 5, 3104–3112 CrossRef CAS PubMed.
- S. S. Ray, P. R. Rajamohanan, M. V. Badiger, I. Devotta, S. Ganapathy and R. A. Mashelkar, Chem. Eng. Sci., 1998, 53, 869–877 CrossRef CAS.
- S. Tong, S. Hou, Z. Zheng, J. Zhou and G. Bao, Nano Lett., 2010, 10, 4607–4613 CrossRef CAS PubMed.
- N. A. Keasberry, M. Bañobre-López, C. Wood, G. J. Stasiuk, J. Gallo and N. J. Long, Nanoscale, 2015, 7, 16119–16128 RSC.
- C. Lorenzato, A. Cernicanu, M.-E. Meyre, M. Germain, A. Pottier, L. Levy, B. D. de Senneville, C. Bos, C. Moonen and P. Smirnov, Contrast Media Mol. Imaging, 2013, 8, 185–192 CrossRef CAS PubMed.
- G. Mikhaylov, U. Mikac, A. A. Magaeva, V. I. Itin, E. P. Naiden, I. Psakhye, L. Babes, T. Reinheckel, C. Peters, R. Zeiser, M. Bogyo, V. Turk, S. G. Psakhye, B. Turk and O. Vasiljeva, Nat. Nanotechnol., 2011, 6, 594–602 CrossRef CAS PubMed.
- E. S. G. Choo, X. Tang, Y. Sheng, B. Shuter and J. Xue, J. Mater. Chem., 2011, 21, 2310–2319 RSC.
- Y. X. Wang, S. M. Hussain and G. P. Krestin, Eur. Radiol., 2001, 11, 2319–2331 CrossRef CAS PubMed.
- M. Sugita and Y. Tenjin, Nihon Rinsho Jpn. J. Clin. Med., 1993, 51, 1108–1113 CAS.
- Feridex – FDA prescribing information, side effects and uses, https://www.drugs.com/pro/feridex.html (accessed September 4, 2017).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr07035g |
| This journal is © The Royal Society of Chemistry 2018 |