
Diarylmaleimide-based branched oligomers: strong full-color emission in both solution and solid films†
Jingwei
Wang
a,
Rong
Zheng
a,
Huan
Chen
a,
Huimei
Yao
a,
Liyu
Yan
b,
Jie
Wei
b,
Zhenghuan
Lin
 *ab and
Qidan
Ling
b
*ab and
Qidan
Ling
b
aFujian Key Laboratory of Polymer Materials, Fujian Normal University, Fuzhou 350007, China. E-mail: zhlin@fjnu.edu.cn
bCollege of Chemistry and Materials Science, Fujian Normal University, Fuzhou 350007, China
First published on 30th November 2017
Abstract
Maleimide and benzene are employed as a dendron and a core, respectively, to construct two series of non-conjugate branched oligomers (B3G1 and B1G2) based on diarylmaleimide fluorophores by an alkylation reaction. Surface aryl groups are changed to tune the emissive color of branched oligomers from blue (λem = 480 nm) to red (λem = 651 nm), realizing full-color emission. The investigation on the photophysical properties of the oligomers indicates that they display intense emission in both solution and solid films, due to the suppression of intramolecular rotation and intermolecular interaction. Molecular simulation and natural transition orbital analysis show that the electron transition takes place in the individual arylmaleimide for the non-conjugate linkage of fluorophores in branched oligomers. It can avoid the unpredictability of the luminescence properties caused by the interaction of fluorophores. In addition, the good solubility, thermostability and oxidative stability of the branched oligomers make them have huge potential in the solution-processable photonic application. These results demonstrate that such a design strategy of non-conjugate branched oligomers is a very efficient and constructive method to obtain high-performance light-emitting materials in both solution and solid films.
Introduction
Organic compounds with strong fluorescence have important applications in the fields of chemosensors,1,2 biological probes,3,4 photoelectronic devices,5–7 and fluorescent bioimaging.8,9 Traditional organic fluorescent compounds with a large aromatic system display intense emission in dilute solution, but weak fluorescence in the solid state for long exciton lifetime and aggregation-caused quenching (ACQ) which results from the delocalization of excitons and strong π–π interactions.10–12 The luminescent process in solution offers a benefit of understanding the emission properties at the molecular level. However, the ACQ effect badly impedes the practical applications of fluorescent compounds in devices, like organic light-emitting diodes (OLEDs), organic lasers, etc. Since Tang and his co-workers reported solid-stated fluorescence of siloles and tetraphenylethylenes,13,14 a large number of organic compounds with aggregation-induced emission (AIE) or aggregation-induced emission enhancement (AIEE) have been developed.15–22 Although AIE- and AIEE-active molecules exhibit intense luminescence in aggregated states, they show non-emission or weak emission in the solution for the non-radiative relaxation of excitons mainly caused by the intramolecular rotation. Therefore, dual-state strong emission (DSE) in both solution and the solid state is significant for gaining more comprehensive insight into the organic luminescence mechanisms and properties, as well as its practical utilization. Just because of this, the DSE-active molecules have received considerable attention in recent years.23–27In addition, realizing full-color luminescence by regulation of the photophysical properties of organic molecules is a hot topic for wide applications in the optoelectronic field.27–31 Although a combination of several fluorophores with different emitting colors, such as blue, green and red, can obtain full-color emission, their different stability and luminescence efficiency would cause a big problem in their practical application. It is important to modify the structure of one fluorophore to achieve tunable emission in both solution and the solid state. Whereas, the luminescence of organic compounds is not as easy to be changed in the solid state as in solution due to the huge effect of the packing mode of molecules in the solid state on the luminescence properties.18,32,33 Consequently, it is a big challenge to design new compounds, especially DSE molecules, to realize controllable and tunable fluorescence, even covering the entire visible region.
In this paper, we describe an unprecedented design to construct full-color DSE-active materials by non-conjugately linking a kind of fluorophore into a dendritic structure. The branched structure for DSE molecule design has the following five advantages: (1) the orderly 3D structure can restrain effectively the intermolecular interaction and ACQ for the spatial isolation effect, and consequently improve solid-state emission. (2) The bulky dendritic structure can suppress substantially the intramolecular rotation of the fluorophore, leading to a high quantum yield in solution. (3) The branched structure endows the luminescent materials with good solubility and film-forming ability, which are favorable for solution-processing applications. (4) There are many surface groups on the dendritic structure, thus, it is easy to adjust the emission color of molecules by changing these surface groups. (5) The non-conjugately linkage of dendrons can keep the nature of the fluorophore unchangeable in the branched molecules. Herein, two series of diarylmaleimide-based branched oligomers (Fig. 1) with different surface groups were designed and synthesized by employing benzene ring as the core, and maleimide as the branched units. The branched oligomers display intense full-color emission in both solution and the solid film.
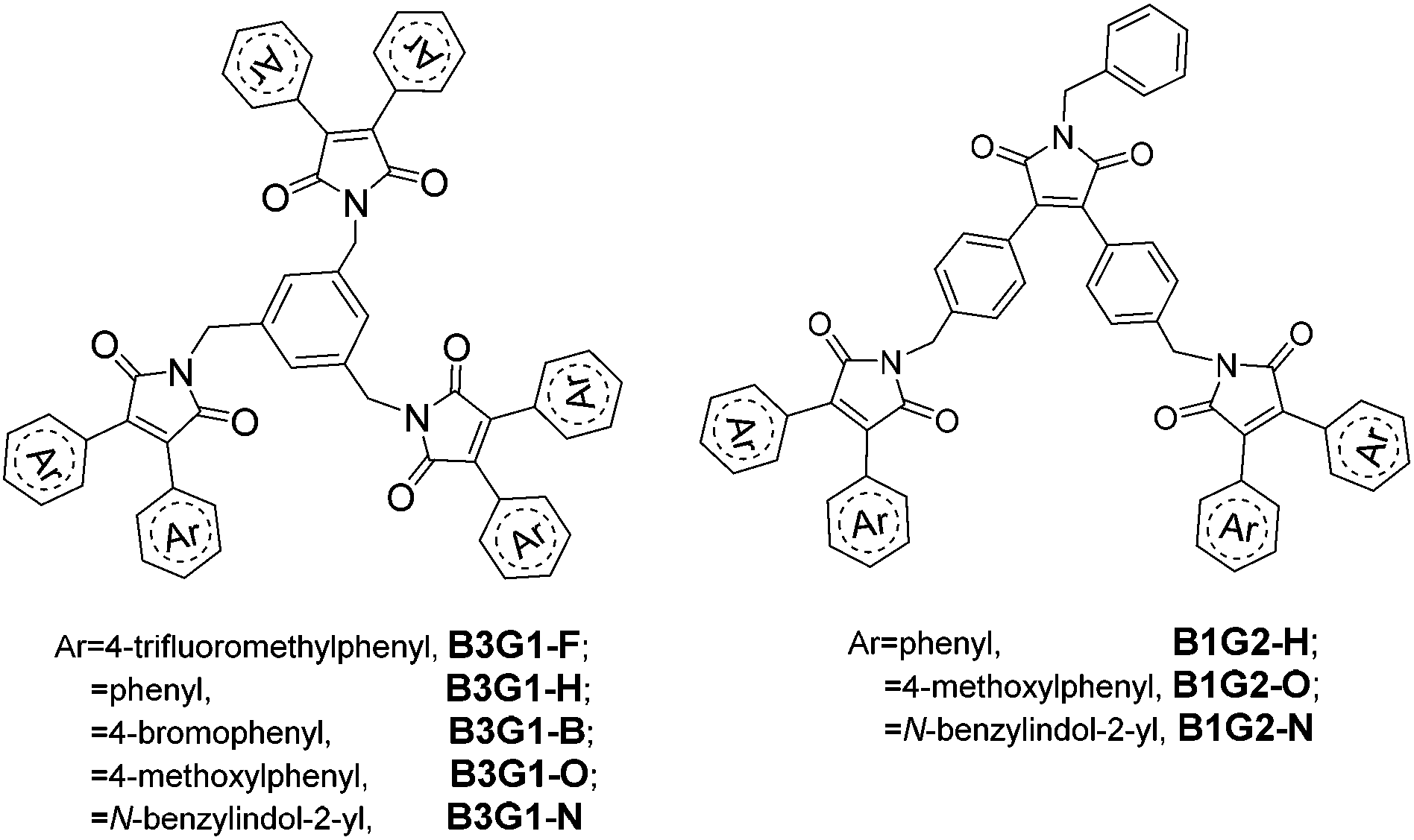 | ||
| Fig. 1 Structure of B3G1 and B1G2 series dendrimers. | ||
Results and discussion
Design and synthesis of branched oligomers
3,4-Disubstituted arylmaleimide was chosen as the fluorophore to design the branched oligomers due to its flexible branched structure, high quantum yield, and tunable emitting colors.17,27,34,35 These advantages render it to be frequently used as a backbone to construct various multifunctional fluorescent materials applied to OLEDs,34,36–40 anion sensing,41,42 and data storage and process.17,18,43 Alkylation of maleimide with benzyl bromide is a common reaction employed to synthesize arylmaleimide derivatives in a high yield. Thus, benzene and maleimide were used as the core and branched units, respectively, to construct the non-conjugate branched oligomers by alkylation. Different surface aryl groups (trifluoromethylbenzene, benzene, bromobenzene, methoxylbenzene and benzylindole) were utilized to adjust the emissive color of the branched oligomers (Fig. 1). The branched oligomers include three-branch-core and one-generation ones (B3G1-F, B3G1-H, B3G1-B, B3G1-O and B3G1-N), and one-branch-core and two-generation ones (B1G2-H, B1G2-O and B1G2-N).The synthetic route of the diarylmaleimide-based branched oligomers is shown in Scheme 1. 3,4-Diphenylaleimides (M1–M4) were obtained according to a reported procedure.35 B3G1-F, B3G1-H, B3G1-B and B3G1-O were synthesized through a substitution reaction of 3,4-diphenylaleimides (M1–M4) with 1,3,5-tris(bromomethyl)benzene (core monomer) under strong alkaline conditions. The core monomer was replaced by monomer M6 to give B1G2-H and B1G2-O. M6 was obtained by the same synthetic reaction of M1–M4, followed by a substitution reaction of benzylbromide and a bromination of NBS (Scheme S1†). The branched oligomers (B3G1-N and B1G2-N) containing a diindolylmaleimide fluorophore were prepared from 3,4-diindolylmaleimide (M5) according to a similar synthetic route (Scheme 1). M5 was firstly turned into 3,4-diindolylmaleic anhydride (M7) in 10% NaOH aqueous solution,27 and then converted into monomer M9 through two-step reactions in a high yield. The resulting B3G1-N and B1G2-N could be obtained through the substitution reaction of M9 with M6 and 1,3,5-tris(bromomethyl)benzene, respectively. It was found that performing the reaction in the dark was favorable to the synthesis of B3G1 and B1G2, due to the instability of the core monomer. For the same reason, the reaction has to be run at a temperature below 100 °C, which, however, results in a low yield of trisubstituted products (resulting molecules). Additionally, the synthesis of B3G1 and B1G2 with different surface aryl groups should choose different alkali/solvent systems. Otherwise, the resulting branched molecules could not be obtained. For example, the strong base NaH was needed in the synthesis of B3G1-F with trifluoromethylbenzene as the surface group, while the combination of the weak base K2CO3 and polar solvent acetone was suitable for the synthesis of B3G1-N and B1G2-N with indole as the surface group. The branched oligomers are soluble in common solvents, such as toluene, dichloromethane, chloroform, THF, DMF, etc.
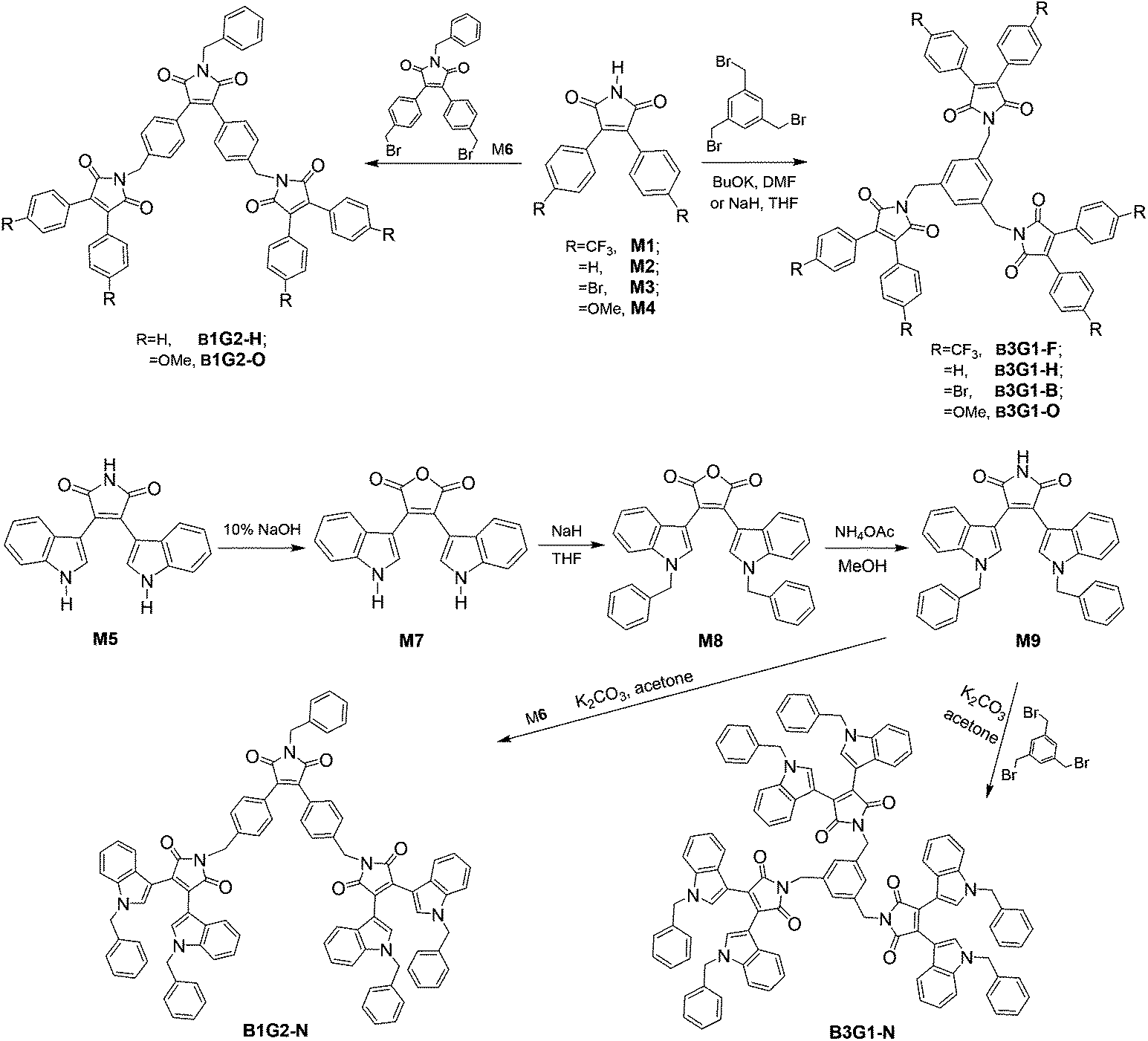 | ||
| Scheme 1 Synthetic route of branched oligomers. | ||
Photophysical properties in solution
The photophysical properties of the branched oligomers in chloroform (10 μM) were investigated first. Fig. 2 shows their absorption and emission spectra which are compared with the spectra of the corresponding arylmaleimide monomers (M1–M5) in Fig. S1.† Due to the non-conjugated linkage between the fluorophores (arylmaleimides), the branched oligomers display optical properties similar to their corresponding monomers. There are mainly two absorption bands between 250 nm and 600 nm in the absorption spectra of the branched oligomers (Fig. 2a). The first band before 325 nm is assigned to the π–π* transition of separating maleimide or aryl units, while the second band after 325 nm should be derived from the π–π* transition involving both maleimide and aryl moieties. For B3G1-O, B3G1-N, B1G2-O and B1G2-N, the second band companied by a shoulder peak shows a red shift in solution. For example, the maximum absorption peak bathochromically shifts from 360 nm of B3G1-F to 482 nm of B3G1-N in chloroform. The red shift should be ascribed to the strong electron-pushing ability of the surface aryl groups, methoxylbenzene and indole, which cause an intramolecular charge transition (ICT) from the aryl groups to maleimide.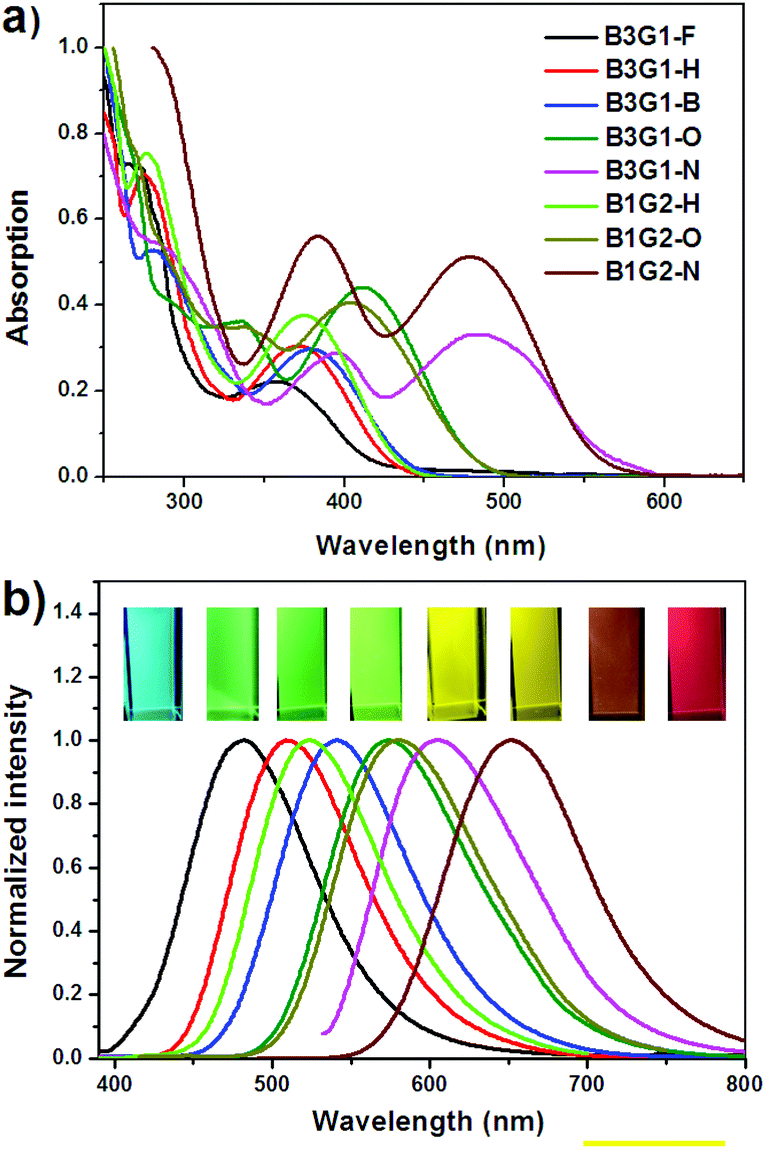 | ||
| Fig. 2 Absorption (a) and emission (b) spectra of oligomers in CHCl3 solution, and their photographs (inset in b) taken under 365 nm UV light. Branched oligomers in (b) arranged from left to right: B3G1-F, B3G1-H, B1G2-H, B3G1-B, B3G1-O, B1G2-O, B3G1-N, and B1G2-N. | ||
The effect of the peripheral aryl groups on the emission of the branched oligomers is more significant than on their absorption. From the emission spectra of the branched oligomers shown in Fig. 2b, it was found that they display a substantial red shift with the increasing electron-pushing ability of the aryl groups, resulting in a full-color emission overlapping from blue of B3G1-F to the red region of B1G2-N. Relative to M2, M4 and M5, the B1G2 type branched oligomers show larger red shifts than the B3G1 type ones (Fig. S2†). It should be ascribed to the space distribution of the three fluorophores. The three arylmaleimides equally disperse around the benzene core, while they concentrate on one side of the core, which makes the environmental polarity of the fluorophore in B1G2 larger than in B3G1. To confirm the polar effect on the luminescence properties of the arylmaleimide fluorophores, their emission spectra in different solvents were investigated (Fig. S3†). These fluorophores indeed show a red shift of the emission band with the increasing polarity of the solvents. Experimental data of the photophysical properties of the branched oligomers and monomers are summarized in Table 1 and Table S1,† respectively.
| λ abs,a (nm) S/F | λ em, (nm) S/F | Φ f, (%) S/F | τ, (ns) S/F | k r,b (ns−1) S/F | k nr,c (ns−1) S/F | |
|---|---|---|---|---|---|---|
| a S = Chloroform solution, F = Film. b Radiative rate constant kr = Φf/τ. c Non-radiative rate constant knr = (1 − Φf)/τ. | ||||||
| B3G1-F | 360/369 | 482/481 | 42/65 | 10.34/10.64 | 0.041/0.061 | 0.056/0.033 |
| B3G1-H | 362/368 | 508/507 | 86/54 | 17.97/10.84 | 0.048/0.050 | 0.008/0.042 |
| B3G1-B | 379/383 | 541/541 | 64/65 | 13.91/3.70 | 0.046/0.176 | 0.026/0.095 |
| B3G1-O | 412/421 | 574/571 | 63/60 | 12.11/5.76 | 0.052/0.104 | 0.031/0.069 |
| B3G1-N | 482/488 | 605/606 | 24/28 | 7.19/2.74 | 0.033/0.102 | 0.106/0.263 |
| B1G2-H | 376/377 | 524/518 | 67/66 | 16.18/8.41 | 0.041/0.078 | 0.020/0.043 |
| B1G2-O | 404/414 | 580/575 | 37/35 | 12.75/6.77 | 0.029/0.052 | 0.049/0.096 |
| B1G2-N | 479/483 | 651/640 | 20/10 | 6.23/2.46 | 0.032/0.041 | 0.128/0.366 |
For the rigid structure, all of the branched oligomers show higher quantum yields (Φf) than their corresponding arylmaleimide monomers in solution. For example, the Φf of B3G1-F and B3G1-N is double that of their corresponding arylmaleimide fluorophores (M1 and M5). The fluorescence lifetimes (τ) of the branched oligomers were measured using the time-resolved single photon counting technique and determined to be in the range of 6.23–17.97 ns in solution. However, the branched oligomers with the same fluorophores, such as B3G1-H and B1G2-H, B3G1-O and B1G2-O, B3G1-N and B1G2-N, display similar lifetimes in solution. It indicates that the photophysical properties of the branched oligomers are closely related to the arylmaleimide fluorophores. The time-resolved transient luminescence decay of the branched oligomers in dilute solution is plotted in Fig. S4.† Consequently, their radiative rate constant (kr) and non-radiative rate constant (knr) can be estimated from the formula kr = Φf/τ and knr = (1 − Φf)/τ, respectively (Table 1). The high radiative rate constant of the branched oligomers results in their high quantum yields.
Theoretical calculation
The above experimental results suggest that each arylmaleimide in the branched oligomers behaves as an isolated fluorophore with little electronic interaction between the dendrons. To gain insight into the photophysical properties of these arylmaleimides and branched oligomers, including their ground-state and excited-state properties, density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations were performed by using B3LYP and cam-6-31G (d) as the functional and the basis set, respectively. The DFT-optimized geometry of the five arylmaleimides (M1–M5) has a non-planar conformation with the twist angle between the maleimide ring and aryl ring at 22–46° (Fig. S5†). The computed vertical absorption properties from TD-DFT given in Table S2† indicate that the first excited-state (S1) transition occurs predominantly between the HOMO and LUMO. The computed electron density distribution of the molecular orbitals of the arylmaleimides is depicted in Fig. S6.† The HOMO is mainly populated on two symmetrically equivalent aryl rings, while the LUMO is concentrated on the central maleimide moiety. The localization degree of electrons is more significant with the increase of the electron-pushing ability of the aryl group. This is an indication of a charge transfer (CT) character for the maximum absorption band which is sensitive to the environmental polarity. It is in agreement with the experimental result in the solution.The DFT-optimized geometry of the branched molecules in the ground state reveals that each fluorophore has similar twisted conformation to their corresponding arylmaleimide monomers (Fig. S7†). The twisted fluorophores linked by a methylene group make branched oligomers present a screwier structure which is helpful to eliminate the intermolecular interplay when aggregated in the condensed phase. The TD-DFT calculated results of the first 6 singlet excited states of the branched oligomers are given in Table S3–S10.† The calculated vertical absorption transition energy (S1) has much better agreement with the experimental ones with an overestimation less than 0.18 eV (Table 2). It was found that all of the branched oligomers have degenerate excited states (S1 and S2 for B1G2-O and B1G2-N, S1, S2 and S3 for other branched oligomers) due to the same separated fluorophores. It is difficult to visualize the location excitons and possible electronic interactions within the branched oligomers due to the presence of such degenerate states and multiple orbital transitions for each electronic transition. Consequently, the natural transition orbitals (NTOs) method was used to give a qualitative description of an electronic excitation. From NTO pairs for the first three excited singlet states of the branched oligomers (Fig. 3, S8–S13†), note that all of the electronic transitions of degenerate states take place in the occupied and virtual orbitals of individual fluorophore, corresponding to their HOMO and LUMO orbitals (Fig. S6†). It indicates that there is no electronic communication between arylmaleimide fluorophores in the branched oligomers, which makes branched oligomers have similar photophysical properties to the isolated fluorophore.
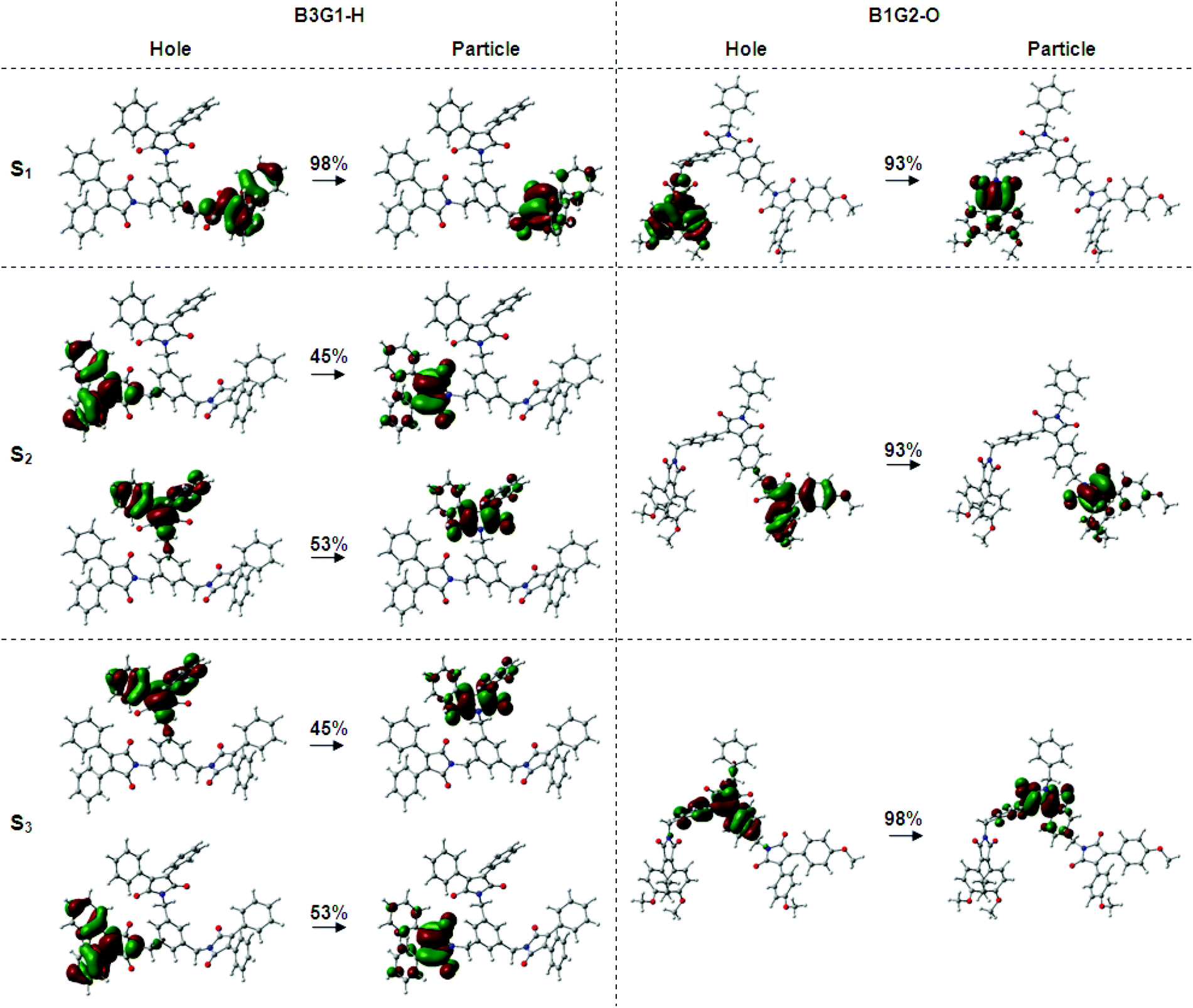 | ||
| Fig. 3 Natural transition orbital pairs for the first three excited singlet states of B3G1-H and B1G2-O. | ||
| Exptl E, eV | Calcd E, eV (f) | |||
|---|---|---|---|---|
| S1 | S2 | S3 | ||
| B3G1-F | 3.44 | 3.52 (0.17) | 3.55 (0.28) | 3.57 (0.10) |
| B3G1-H | 3.44 | 3.45 (0.14) | 3.47 (0.23) | 3.49 (0.11) |
| B3G1-B | 3.27 | 3.36 (0.22) | 3.39 (0.33) | 3.41 (0.16) |
| B3G1-O | 3.01 | 3.16 (0.20) | 3.17 (0.32) | 3.20 (0.19) |
| B3G1-N | 2.57 | 2.75 (0.15) | 2.77 (0.24) | 2.88 (0.17) |
| B1G2-H | 3.30 | 3.40 (0.21) | 3.43 (0.09) | 3.43 (0.25) |
| B1G2-O | 3.07 | 3.13 (0.16) | 3.13 (0.28) | 3.38 (0.26) |
| B1G2-N | 2.59 | 2.67 (0.24) | 2.69 (0.15) | 3.34 (0.18) |
Photophysical properties in films
The photophysical properties of the branched oligomers in the solid films were investigated next. The films were obtained by spin-coating their chloroform solution (10 mg mL−1) at a speed of 1000 r s−1. The absorption and emission spectra of the branched oligomers are shown in Fig. S14† and Fig. 4a, respectively. For a comparison, the spectra properties of monomers (M1–M5) in the solid films were determined and shown in Fig. S15,† and the relevant data are summarized in Table S1.† As mentioned above, the absorption and emission spectra of the dendrimers in solution exhibit similar characteristics to those of their corresponding monomers, due to the non-conjugated linkage between the diarylmaleimides in the branched oligomers. However, the discrepancy of the absorption wavelength (λabs) of the branched oligomers between solution and films is obviously smaller than that of the monomers, which resulted from the specific dendritic structure that can effectively inhibit the intermolecular interaction. For instance, the maximum λabs of M2 bathochromically shifts 22 nm from 361 nm in solution to 383 nm in films. The red shift was found to be 6 nm for B3G1-H and 1 nm for B1G2-H. The same situation was found in the emission spectra of the branched oligomers in the solid film. For instance, the difference of maximum λem between the solution and solid film is more than 10 nm for M1, M3 and M5, but less than 3 nm for B3G1-F, B3G1-B and B3G1-N. Like in solution, branched oligomers in the solid film exhibit full-color emission ranging from blue (481 nm) to red (640 nm).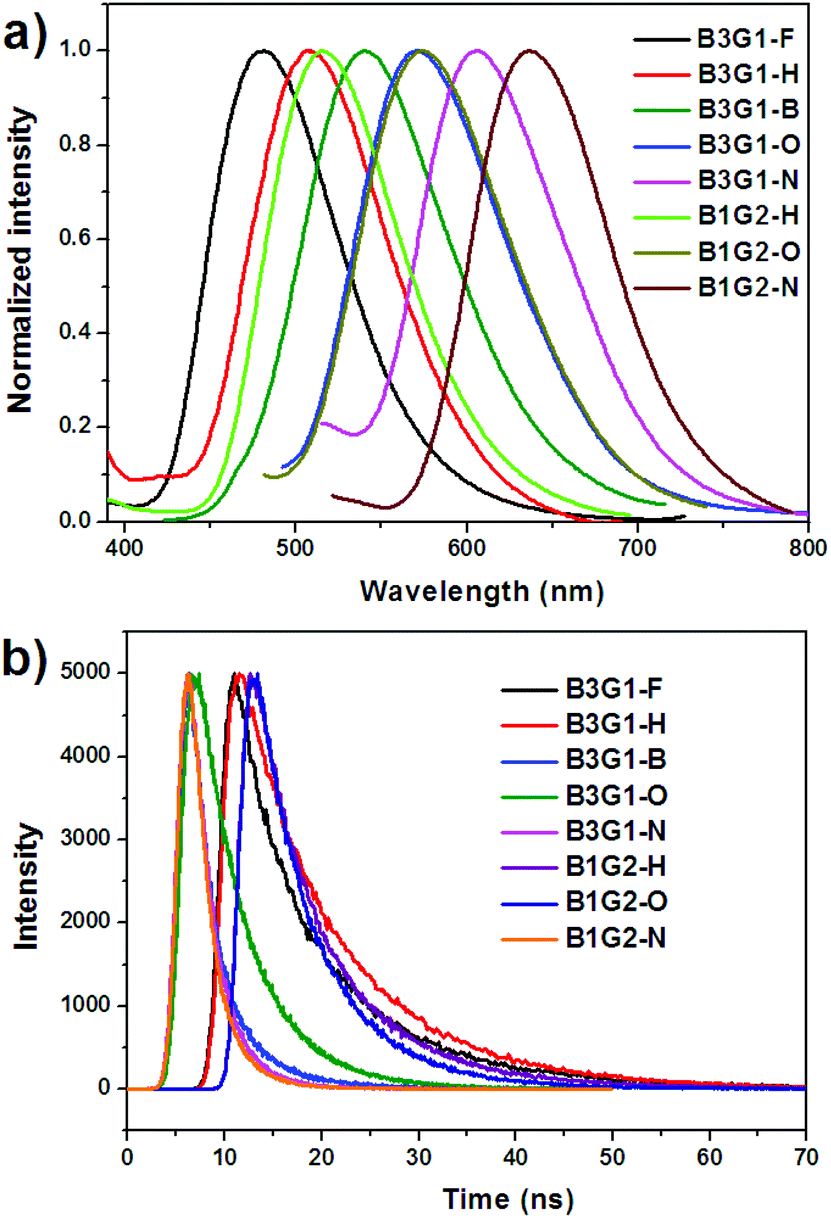 | ||
| Fig. 4 Emission (a) spectra and transient fluorescence decays (b) of branched oligomers in solid films. | ||
Although the spectral properties of the branched oligomers mainly depend on the optical transition of the individual arylmaleimide fluorophores, their quantum yields differ significantly from their corresponding monomers. For the weak intermolecular interaction, the branched oligomers in films show a high emission efficiency and radiative rate constant. The Φf of the branched oligomers in the solid film matches that in solution. In particular, for B3G1-F, B3G1-B and B3G1-N, their Φf in the film exceeds that in solution. It was found that the Φf of most of the monomers in the solid film is lower than that in solution for strong intermolecular interactions, such as π–π stacking and dipole–dipole interaction. In particular, M5 shows a strong ACQ feature. In the case of branched oligomers, the separation and protection of fluorophores in the dendritic structure make them have high emission efficiency in both solution and solid films. Time-resolved transient luminescence decay of branched oligomers in the solid film is plotted in Fig. 4b. After fitting the curves, their fluorescence lifetime was found to range from 2.46 ns to 10.84 ns, which is smaller than that in solution. B3G1-H, B3G1-O and B3G1-N show similar lifetimes to B1G2-H, B1G2-O and B1G2-N, respectively, for the same fluorophore.
Thermal and electrochemical properties
The thermal properties of the polymers were investigated by thermogravimetric analysis (TGA) under a nitrogen atmosphere at a scan speed of 10 °C min−1. From the TGA plots shown in Fig. 5, it can be found that most of the branched oligomers exhibit excellent thermal properties with the decomposed temperature (Td) above 400 °C, except for B3G1-O. However, the 5% weight-loss temperature (T5%) of B3G1-O, B3G1-N, B1G2-O and B1G2-N was found at 277 °C, 282 °C, 212 °C and 272 °C, respectively (Table S11†), which is lower than that of other branched oligomers. It should be related to the deprivation of peripheral methoxy or benzyl groups in B3G1-O, B1G2-O, B3G1-N and B1G2-N at high temperature. In general, the thermal stability of the branched oligomers is good enough to meet their application need for devices.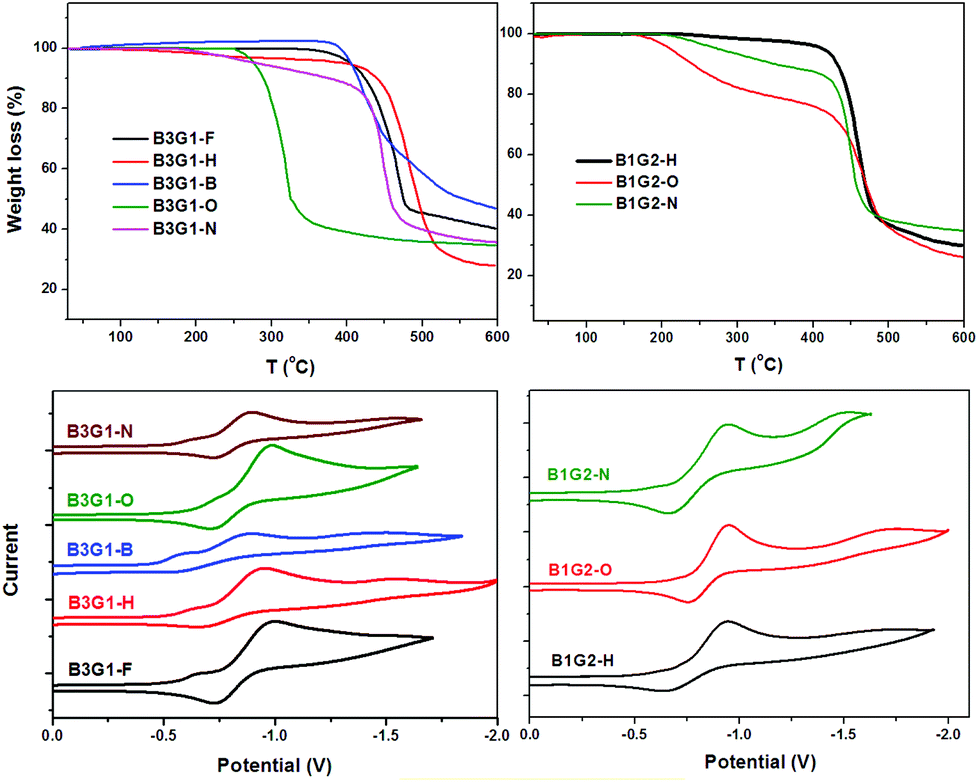 | ||
| Fig. 5 TGA curves (upper) of branched oligomers and their CV curves (bottom) in solid films. | ||
The electrochemical properties of the branched oligomers were determined by cyclic voltammetry (CV) at a scan rate of 100 mV s−1 in a 0.1 M n-Bu4NPF6 solution in acetonitrile with Ag/AgCl as a reference electrode. The CV data and reduction curves of the branched oligomers are given in Table S11† and Fig. 5, respectively. All of the branched oligomers demonstrate mostly a reversible reduction scan with a similar reduction wave at about −0.70 V for their common electron-deficient unit, the maleimide ring. The recorded CV curves were calibrated using a ferrocene/ferrocenium (Fc/Fc+) redox couple (4.8 eV below the vacuum level) as an external standard. The E1/2 of the Fc/Fc+ redox couple was found to be 0.40 V vs. the Ag quasireference electrode. As a result, the LUMO and HOMO energy levels of the branched oligomers can be estimated using the empirical equation ELUMO = −(Eonsetred + 4.40) eV and EHOMO = ELUMO − Eoptg, where Eonsetred and Eoptg stand for the onset potentials of reduction and optical band gap, respectively. The LUMO levels of the branched oligomers lie around −3.70 eV, while their HOMO levels locate in the range of −5.90 ∼ −6.69 eV for the different electron-pushing groups in the fluorophore (Fig. 6). The fact that all of the branched oligomers have oxidation potentials larger than 5.9 eV suggests their stability under ambient conditions. These results will guide the application of the oligomers in photoelectric devices.
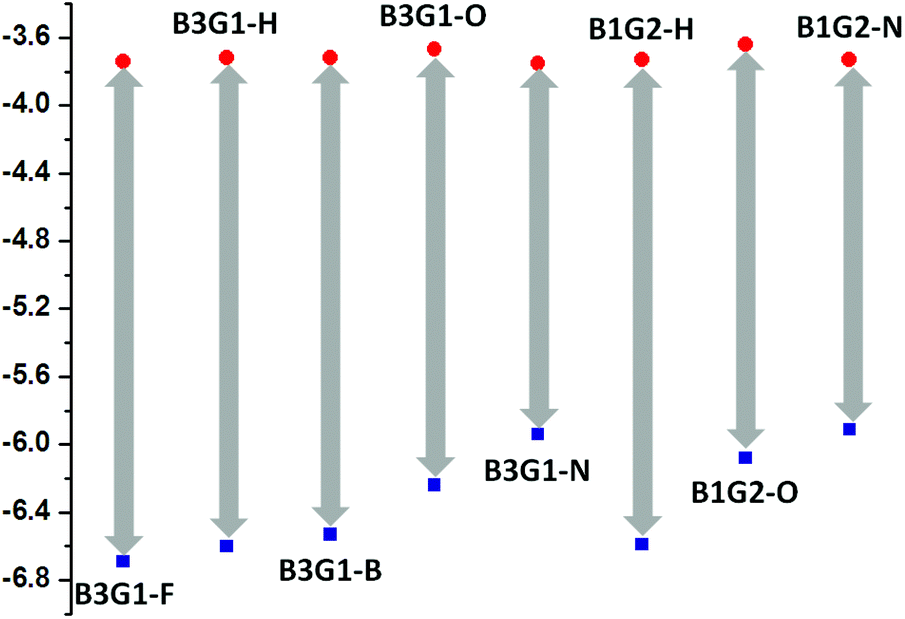 | ||
| Fig. 6 HOMO and LUMO energy levels for branched oligomers in solid films. | ||
Experimental
Materials and measurements
All the reagents and solvents used in the experiments were obtained from commercial suppliers and used as received without further purification, unless otherwise noted. Thin layer chromatography was performed on G254 silica gel plates of Qingdao Haiyang Chemical. Column chromatography was conducted on Yantai Huanghai brand silica gel (200–300 mesh). Diarylmaleimides M1, M2, M3, M4 and M7 were synthesized according to a previously reported method.27,35The low temperature reaction was performed on a Yuhua DFY-5/80 reactive bath. High-resolution MALDI-TOF mass spectra were recorded on a Bruker microflex LRF spectrometer. NMR spectra were recorded in d-CDCl3 or d-DMSO on a Bruker Ascend 400 FT-NMR spectrometer. 1H and 13C chemical shifts were quoted relative to the internal standard tetramethylsilane. UV-vis spectra were obtained on a Shimadzu UV-2600 spectrophotometer. The PL spectra were probed on a Shimadzu RF-5301PC fluorescence spectrophotometer. The fluorescence lifetime and absolute quantum yield values of solution and the solid film were measured using an Edinburgh Instruments FLS920 fluorescence spectrometer with a 6-inch integrating sphere. Thermogravimetric analysis (TGA) was performed by using a Mettler 851e with a heating rate of 10 °C min−1 under flowing nitrogen. Cyclic voltammetry (CV) was performed on a CHI600D electro-chemical analyzer in anhydrous THF containing tetra-n-butyl-ammonium hexafluorophosphate (TBAPF6, 0.1 M) as a supporting electrolyte at 298 K. A conventional three-electrode cell was used with a platinum working electrode and a platinum wire as the counter electrode. The Pt working electrode was routinely polished with a polishing alumina suspension and rinsed with acetone before use. The measured potentials were recorded with respect to the Ag/AgCl reference electrode. All electrochemical measurements were carried out under an atmospheric pressure of nitrogen.
Molecular simulations
Theoretical calculations were carried out with the Gaussian 09 program. The geometry in the ground state was optimized with the density functional theory (DFT) using the B3LYP functional and the 6-31G* basis set. The vertical excited energies were calculated with the time-dependent density functional theory (TD-DFT) at the CAM-B3LYP/6-31G* level.Synthesis of compound M9
3,4-Bisindoylmaleic anhydride (M7) (800 mg, 2.4 mmol) was mixed with NaH (168 mg, 7 mmol) in dried THF (23 ml) under a nitrogen atmosphere, and the solution was stirred at 0 °C for 45 minutes. To the mixture was added benzyl bromide (1.2 mL, 10 mmol), the reaction mixture was kept stirring at reflux under argon for 10 h, and cooled to room temperature. The reaction was diluted with ethyl acetate and successively washed with 1 M dilute hydrochloric acid, water and brine. It was then dehydrated over anhydrous MgSO4 and dried in vacuo to give the crude product of 3,4-bis-(N-benzylindolyl) maleic anhydride (M8) which was used without further purification.3,4-Bis-(N-benzylindolyl) maleic anhydride (600 mg, 1.18 mmol), anhydrous ammonium acetate (2.270 mg, 29.5 mmol) and anhydrous methanol (60 mL) were added to a hydrothermal synthesis reactor. The reaction mixture was placed in an oven at 100 °C overnight, and cooled to room temperature. The reaction mixture was extracted with ethyl acetate. The organic phase was washed with brine. It was then dehydrated over anhydrous MgSO4. After removing the solvent, the crude product was purified by column chromatography with ethyl acetate/petroleum ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) as the eluant, affording compound M9 in 95% yield. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 3.4 Hz, 2H), 7.58 (s, 1H),7.37–7.27 (m, 6H), 7.21 (d, J = 8.3 Hz, 2H), 7.15–7.09 (m, 4H), 7.03 (d, J = 20.4, 13.2,4.5 Hz, 4H), 6.76–6.71 (m, 2H), 5.35 (s, 4H). 13C NMR (100 MHz, DMSO) δ 173.25, 138.11, 136.25, 132.68, 129.03, 128.10, 127.90, 127.31, 126.66, 122.32, 121.53, 120.04, 111.04, 105.80, 49.85.
:4) as the eluant, affording compound M9 in 95% yield. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 3.4 Hz, 2H), 7.58 (s, 1H),7.37–7.27 (m, 6H), 7.21 (d, J = 8.3 Hz, 2H), 7.15–7.09 (m, 4H), 7.03 (d, J = 20.4, 13.2,4.5 Hz, 4H), 6.76–6.71 (m, 2H), 5.35 (s, 4H). 13C NMR (100 MHz, DMSO) δ 173.25, 138.11, 136.25, 132.68, 129.03, 128.10, 127.90, 127.31, 126.66, 122.32, 121.53, 120.04, 111.04, 105.80, 49.85.
Synthesis of compound B3G1-F
M1 (517 mg, 1.35 mmol) and NaH (39 mg, 1.62 mmol) were placed in a 25 mL flask. 8 mL of dry THF was injected into the flask under a nitrogen atmosphere at 0 °C and was vigorously stirred for 40 min. Then, 1,3,5-tris(bromomethyl)benzene (80 mg, 0.224 mmol) in dry THF (2 mL) was added to the reaction mixture, which was heated to refluxed for 12 h. After the reaction mixture was cooled to room temperature, 1 M dilute hydrochloric acid was added and was extracted with ethyl acetate. The organic phase was washed with brine, and then dehydrated over anhydrous MgSO4. After removing the solvent, the crude product was purified by column chromatography with DCM/petroleum ether (1:2) as the eluant, affording light blue solids of B3G1-F. Yield: 41%. 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.3 Hz, 13H), 7.52 (d, J = 8.2 Hz, 11H), 7.45 (s, 3H), 4.82 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 169.37, 137.23, 136.34, 132.31, 131.98, 131.42, 130.24, 128.28, 125.70, 41.85. MALDI MASS m/z [M + Na + H]+ calcd 1293.2058, found 1293.6165.
Synthesis of compound B3G1-H
M2 (0.8 g, 3.2 mmol), 1,3,5-tris(bromomethyl)benzene (141 mg, 0.4 mmol) and potassium tert-butylate (2.17 g, 19.2 mmol) were placed in a 10 mL flask. 6 mL of dry DMF was injected into the flask under a nitrogen atmosphere at 90 °C and was vigorously stirred for 24 h. After the reaction mixture was cooled to room temperature, the reaction mixture was extracted with ethyl acetate. The organic phase was washed with brine. It was then dehydrated over anhydrous MgSO4. After removing the solvent, the crude product was purified by column chromatography with DCM/petroleum ether (1:2) as the eluant, affording green solids of compound B3G1-H. Yield: 48%. 1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 8.0 Hz, 13H), 7.27 (t, J = 7.3 Hz, 7H), 7.24–7.17 (m, 13H), 4.72 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 169.50, 136.63, 135.49, 129.12, 128.88, 127.81, 127.64, 127.27, 40.80. MALDI MASS m/z [M + Na + H]+ calcd 885.2815, found 885.0221.
Synthesis of compound B3G1-B
M3 (1 g, 2.47 mmol) and 1,3,5-tris(bromomethyl)benzene (150 mg, 0.42 mmol) were placed in a 50 mL flask. 5 mL of dry DMF was injected into the flask under a nitrogen atmosphere. Then, a fresh sodium methoxide solution made from sodium (87 mg, 3.8 mmol) and methanol (15 mL) was added to the reaction mixture at 0 °C and vigorously stirred for 40 min, which was heated to 100 °C for 12 h. After the reaction mixture was cooled to room temperature and 1 M HCl aqueous solution was added, it was extracted with ethyl acetate. The organic phase was washed with brine, and then dehydrated over anhydrous MgSO4. After removing the solvent, the crude product was purified by column chromatography with DCM/petroleum ether (1:5) as the eluant, affording green solids of compound B3G1-B. Yield: 37%. 1H NMR (400 MHz, DMSO) δ 7.61–7.55 (m, 12H), 7.27 (dd, J = 10.0, 8.1 Hz, 15H), 4.72 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 169.72, 137.29, 135.42, 132.04, 131.33, 128.12, 127.09, 124.87, 41.68. MALDI MASS m/z [M + K]+ calcd 1373.7045, found 1373.1160.
Synthesis of compound B3G1-O
M4 (507 mg, 1.64 mmol) and NaH (45 mg, 1.968 mmol) were placed in a 25 mL flask. 8 mL of dry THF was injected into the flask under a nitrogen atmosphere at 0 °C and was vigorously stirred for 40 min. Then, 1,3,5-tris(bromomethyl)benzene (60 mg, 0.164 mmol) in dry THF (2 mL) was added to the reaction mixture, which was heated to refluxed for 12 h. After the reaction mixture was cooled to room temperature and 1 M HCl aqueous solution was added, it was extracted with ethyl acetate. The organic phase was washed with brine, and then dehydrated over anhydrous MgSO4. After removing the solvent, the crude product was purified by column chromatography with DCM/petroleum ether (3:1) as the eluant, affording yellow solids of compound B3G1-F. Yield: 38%. 1H NMR (400 MHz, CDCl3) δ 7.37 (t, J = 8.5 Hz, 12H), 7.32 (s, 3H), 6.73 (d, J = 8.8 Hz, 12H), 4.68 (s, 6H), 3.75 (s, 18H). 13C NMR (100 MHz, CDCl3) δ 170.82, 160.60, 137.53, 134.21, 131.51, 127.84, 121.40, 113.99, 55.24, 41.48. MALDI MASS m/z [M + H]+ calcd 1042.3551, found 1041.9077.
Synthesis of compound B3G1-N
M9 (317 mg, 0.624 mmol), 1,3,5-tris(bromomethyl)benzene (35 mg, 0.098 mmol) and potassium carbonate (129 mg, 0.936 mmol) were placed in a 25 mL flask. 6 mL of acetone was injected into the flask under a nitrogen atmosphere and was vigorously stirred for 12 h at 90 °C. After the reaction mixture was cooled to room temperature, the reaction mixture was extracted with ethyl acetate, and washed with brine. It was then dehydrated over anhydrous MgSO4. After removing the solvent, the crude product was purified by column chromatography with ethyl acetate/petroleum ether (1:4) as the eluant, affording red solids of compound B3G1-N. Yield: 48%. 1H NMR (400 MHz, DMSO) δ 7.93 (s, 6H), 7.28 (tt, J = 14.1, 7.7 Hz, 27H), 7.13 (d, J = 7.1 Hz,12H), 6.93 (t, J = 7.7 Hz, 6H), 6.84 (d, J = 8.0 Hz, 6H), 6.53 (t, J = 7.6 Hz, 6H), 5.36 (s,12H), 4.77 (s, 6H). 13C NMR (100 MHz, DMSO) δ 171.68, 138.63, 137.88, 136.24, 132.99, 129.00, 127.90, 127.33, 126.49, 126.18, 122.39, 121.72, 120.17, 111.09, 105.71, 49.85, 41.61. MALDI MASS m/z [M + H]+ calcd 1637.6422, found 1638.1194.
Synthesis of compound B1G2-H and B1G2-O
M2 or M4 (1.14 mmol) and NaH (32 mg, 1.33 mmol) were placed in a 25 mL flask. 6 mL of dry THF was injected into the flask under a nitrogen atmosphere at 0 °C and was vigorously stirred for 40 min. Then, M6 (100 mg, 0.19 mmol) in dry THF (2 mL) was added to the reaction mixture, which was heated to reflux for 12 h. After the reaction mixture was cooled to room temperature and 1 M HCl aqueous solution was added, it was extracted with ethyl acetate. The organic phase was washed with brine. It was then dehydrated over anhydrous MgSO4. After removing the solvent, the crude product was purified by column chromatography with ethyl acetate/petroleum ether (1:4) as the eluant, affording green or yellow solids of compound B1G2-H or B1G2-O.
B1G2-H. Green solids. Yield: 36%. 1H NMR (400 MHz, CDCl3) δ 7.53–7.28 (m, 33H), 4.80 (d, J = 7.8 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 170.37, 138.22, 136.35, 136.28, 135.76, 130.55, 130.22, 129.91, 128.96, 128.86, 128.77, 128.70, 128.58, 128.53, 128.10, 127.89, 42.05, 41.65. MALDI MASS m/z [M + Na + H]+ calcd 885.2815, found 885.2761.
B1G2-O. Green solids. Yield: 33%. 1H NMR (400 MHz, CDCl3) δ 7.49–7.20 (m, 22H), 6.79 (d, J = 8.7 Hz, 7H), 4.70 (s, 6H), 3.75 (s, 12H). 13C NMR (100 MHz, CDCl3) δ 170.90, 170.36, 160.76, 138.42, 136.38, 135.74, 134.14, 131.47, 130.92, 130.18, 128.88, 128.69, 128.00, 127.87, 121.24, 114.11, 55.31, 42.04, 41.49. MALDI MASS m/z [M + Na + H]+ calcd 1005.3237, found 1005.2536.
Synthesis of compound B1G2-N
M9 (510 mg, 1 mmol), M6 (106 mg, 0.2 mmol) and potassium carbonate (207 mg, 1.5 mmol) were placed in a 25 mL flask. 8 mL of dry acetone was injected into the flask under a nitrogen atmosphere and was vigorously stirred for 12 h at reflux. After the reaction mixture was cooled to room temperature, it was extracted with ethyl acetate. The organic phase was washed with brine. It was then dehydrated over anhydrous MgSO4. After removing the solvent, the crude product was purified by plate chromatography with ethyl acetate/petroleum ether (1:4) as the eluant, affording red solids of compound B1G2-N. Yield: 31%. 1H NMR (400 MHz, CDCl3) δ 7.72 (s, 4H), 7.42 (s, 10H), 7.27 (dd, J = 13.8, 6.7 Hz, 15H), 7.19 (d, J = 8.2 Hz, 4H), 7.10 (d, J = 7.1 Hz, 8H), 7.01 (dd, J = 17.1, 8.0 Hz, 8H), 6.72 (t, J = 7.5 Hz, 4H), 5.36–5.22 (m, 8H), 4.83 (d, J = 19.1 Hz, 4H), 4.75 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 171.93, 170.40, 138.85, 136.59, 136.44, 136.36, 135.75, 132.07, 130.17, 128.86, 128.73, 128.67, 127.86, 127.09, 126.83, 126.44, 122.42, 122.28, 120.20, 109.99, 106.42, 50.57, 41.99, 41.50. MALDI MASS m/z [M + Na]+ calcd 1041.5084, found 1041.8810.
Conclusions
An effective design method was presented to construct highly efficient fluorescent materials in both solution and solid films, by non-conjugately linking fluorophores into the branched oligomers. According to this method, two series of branched oligomers, including three-branch-core and one-generation ones (B3G1-F, B3G1-H, B3G1-B, B3G1-O and B3G1-N), and one-branch-core and two-generation ones (B1G2-H, B1G2-O and B1G2-N), were synthesized by employing diarylmaleimide as the fluorophore. Surface aryl groups were changed to tune the luminescent color of the branched oligomers. In order of the electron-pushing ability, they are trifluoromethylbenzene (B3G1-F), benzene (B3G1-H and B1G2-H), bromobenzene (B3G1-B), methoxylbenzene (B3G1-O and B1G2-O) and benzylindole (B3G1-N and B1G2-N). The emissive wavelength of the branched oligomers bathochromically shifts from 480 nm of B3G1-F to 651 nm of B1G2-N with the increasing electron-pushing ability of the surface groups. Theoretical calculation shows that the optical properties of the branched oligomers basically depend on the electronic transition of the individual fluorophore. There are no interactions between the fluorophores in branched oligomers for their non-conjugated structure. However, the emissive intensity of the branched oligomers in both solution and the solid film has been greatly improved, relative to arylmaleimide fluorophores. The good solubility, chemical stability, and thermostability of the branched oligomers make them have huge potential in the solution-processable photonic application.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21374017, 21401023 and 21574021), the Natural Science Foundation of Fujian Province (2017J01684), and the Educational Commission of Fujian Province (JA14068).Notes and references
- L. He, B. Dong, Y. Liu and W. Lin, Chem. Soc. Rev., 2016, 45, 6449–6461 RSC.
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef CAS PubMed.
- E. I. Galanzha, R. Weingold, D. A. Nedosekin, M. Sarimollaoglu, J. Nolan, W. Harrington, A. S. Kuchyanov, R. G. Parkhomenko, F. Watanabe and Z. Nima, Nat. Commun., 2017, 8, 15528 CrossRef CAS PubMed.
- J. Xu, J. Pan, X. Jiang, C. Qin, L. Zeng, H. Zhang and J. F. Zhang, Biosens. Bioelectron., 2016, 77, 725–732 CrossRef CAS PubMed.
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- J.-T. Hou, W. X. Ren, K. Li, J. Seo, A. Sharma, X.-Q. Yu and J. S. Kim, Chem. Soc. Rev., 2017, 46, 2076–2090 RSC.
- P. Reineck and B. C. Gibson, Adv. Opt. Mater., 2017, 5, 1–26 Search PubMed.
- Y.-X. Li, M.-P. Pang, Z.-W. Zhang, G.-B. Li and G.-X. Sun, RSC Adv., 2013, 3, 14950–14953 RSC.
- X. L. Xu, F. W. Lin, W. Xu, J. Wu and Z. K. Xu, Chem. – Eur. J., 2015, 21, 984–987 CrossRef CAS PubMed.
- L. Zong, Y. Xie, C. Wang, J.-R. Li, Q. Li and Z. Li, Chem. Commun., 2016, 52, 11496–11499 RSC.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li and B. Z. Tang, Appl. Phys. Lett., 2007, 91, 011111 CrossRef.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Liu, Y. Lei, F. Li, J. Chen, M. Liu, X. Huang, W. Gao, H. Wu, J. Ding and Y. Cheng, J. Mater. Chem. C, 2016, 4, 2862–2870 RSC.
- P. Mazumdar, D. Das, G. P. Sahoo, G. Salgado-Morán and A. Misra, Phys. Chem. Chem. Phys., 2015, 17, 3343–3354 RSC.
- X. Mei, K. Wei, G. Wen, Z. Liu, Z. Lin, Z. Zhou, L. Huang, E. Yang and Q. Ling, Dyes Pigm., 2016, 133, 345–353 CrossRef CAS.
- X. Mei, G. Wen, J. Wang, H. Yao, Y. Zhao, Z. Lin and Q. Ling, J. Mater. Chem. C, 2015, 3, 7267–7271 RSC.
- X. Wang, Y. Wu, Q. Liu, Z. Li, H. Yan, C. Ji, J. Duan and Z. Liu, Chem. Commun., 2015, 51, 784–787 RSC.
- Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS PubMed.
- B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo and Z. Chi, Chem. Sci., 2015, 6, 3236–3241 RSC.
- S. Xu, T. Liu, Y. Mu, Y. F. Wang, Z. Chi, C. C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed.
- A. S. Abd-El-Aziz, C. Agatemor, N. Etkin and B. Wagner, Macromol. Rapid Commun., 2016, 37, 1235–1241 CrossRef CAS PubMed.
- G. Chen, W. Li, T. Zhou, Q. Peng, D. Zhai, H. Li, W. Z. Yuan, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 4496–4501 CrossRef CAS PubMed.
- M. Huang, R. Yu, K. Xu, S. Ye, S. Kuang, X. Zhu and Y. Wan, Chem. Sci., 2016, 7, 4485–4491 RSC.
- M. Li, Y. Niu, X. Zhu, Q. Peng, H.-Y. Lu, A. Xia and C.-F. Chen, Chem. Commun., 2014, 50, 2993–2995 RSC.
- X. Mei, J. Wang, Z. Zhou, S. Wu, L. Huang, Z. Lin and Q. Ling, J. Mater. Chem. C, 2017, 5, 2135–2141 RSC.
- H. Chen, Y. Tang, H. Shang, X. Kong, R. Guo and W. Lin, J. Mater. Chem. B, 2017, 5, 2436–2444 RSC.
- J. Han, J. You, X. Li, P. Duan and M. Liu, Adv. Mater., 2017, 29, 1606503 CrossRef PubMed.
- H. Tachibana, N. Aizawa, Y. Hidaka and T. Yasuda, ACS Photonics, 2017, 4, 223–227 CrossRef CAS.
- X. Wei, L. Bu, X. Li, H. Agren and Y. Xie, Dyes Pigm., 2017, 136, 480–487 CrossRef CAS.
- Z. Lin, X. Mei, E. Yang, X. Li, H. Yao, G. Wen, C.-T. Chien, T. J. Chow and Q. Ling, CrystEngComm, 2014, 16, 11018–11026 RSC.
- K. Wang, H. Zhang, S. Chen, G. Yang, J. Zhang, W. Tian, Z. Su and Y. Wang, Adv. Mater., 2014, 26, 6168–6173 CrossRef CAS PubMed.
- Z. Lin, Y.-D. Lin, C.-Y. Wu, P.-T. Chow, C.-H. Sun and T. J. Chow, Macromolecules, 2010, 43, 5925–5931 CrossRef CAS.
- H. C. Yeh, W. C. Wu and C. T. Chen, Chem. Commun., 2003, 404–405 RSC.
- Z. Lin, Y.-S. Wen and T. J. Chow, J. Mater. Chem., 2009, 19, 5141–5148 RSC.
- J. Wang, Y. Zhao, K. Wei, G. Wen, X. Li, Z. Lin and Q. Ling, Synth. Met., 2017, 230, 18–26 CrossRef CAS.
- K. Wei, G. Wen, Y. Zhao, Z. Lin, X. Mei, L. Huang and Q. Ling, J. Mater. Chem. C, 2016, 4, 9804–9812 RSC.
- Y. Zhao, Z. Lin, Z. Zhou, H. Yao, W. Lv, H. Zhen and Q. Ling, Org. Electron., 2016, 31, 183–190 CrossRef CAS.
- Y. Zhao, H. Yao, K. Wei, H. Zhen, E. Yang, Z. Lin and Q. Ling, Phys. Chem. Chem. Phys., 2017, 19, 12642–12646 RSC.
- Z. Lin, Y. Ma, X. Zheng, L. Huang, E. Yang, C.-Y. Wu, T. J. Chow and Q. Ling, Dyes Pigm., 2015, 113, 129–137 CrossRef CAS.
- H. Yao, J. Wang, H. Chen, X. Mei, Z. Su, J. Wu, Z. Lin and Q. Ling, RSC Adv., 2017, 7, 12161–12169 RSC.
- R. Zheng, X. Mei, Z. Lin, Y. Zhao, H. Yao, W. Lv and Q. Ling, J. Mater. Chem. C, 2015, 3, 10242–10248 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption and emission spectra of diarylmaleimide fluorophores, theory calculation data, additional experimental details. See DOI: 10.1039/c7ob02446k |
| This journal is © The Royal Society of Chemistry 2018 |