
Aerobic oxidative acylation of nitroarenes with arylacetic esters under mild conditions: facile access to diarylketones†
Jiang-Sheng
Li
 *a,
Qian
Yang‡
a,
Fan
Yang‡
ba,
Guo-Qin
Chen
a,
Zhi-Wei
Li
a,
Yin-Jie
Kuang
a,
Wei-Jing
Zhang
a and
Peng-Mian
Huang
a
*a,
Qian
Yang‡
a,
Fan
Yang‡
ba,
Guo-Qin
Chen
a,
Zhi-Wei
Li
a,
Yin-Jie
Kuang
a,
Wei-Jing
Zhang
a and
Peng-Mian
Huang
a
aHunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, School of Chemistry and Biological Engineering, Changsha University of Science & Technology, Changsha 410114, P.R. China. E-mail: jslichem@gmail.com; Fax: +86-731-85258733
bSINOPEC Baling Petrochemical Company, Yueyang 414014, P.R. China
First published on 1st December 2017
Abstract
A facile and regioselective base-mediated aerobic oxidative acylation of nitroarenes to access diarylketones under mild conditions has been developed. It features the use of bench-stable and readily available arylacetates as acyl surrogates, and the absence of transition-metals and synthetic oxidants. This protocol involves a cascade CDC/oxidative decarboxylation process.
Introduction
The cross-dehydrogenative coupling (CDC) reaction, which readily enables the efficient construction of C-based chemical bonds, especially the C–C bonds, is of fundamental importance to the organic synthetic community.1 Due to the superior sustainability and environmental compatibility, significant progress in CDC chemistry has flourished mostly in the past decade.2 These reactions can undergo catalysis using transition-metals (TMs) such as copper3 or iron salts4 combined with synthetic oxidants or elemental oxygen. Nevertheless, one of the challenging difficulties encountered in practical industrial applications is to efficiently remove trace amounts of transition-metal residues from coupled products, especially pharmaceuticals. Accordingly, tremendous efforts have been devoted to developing novel TM-free CDC reactions,5 but aerobic CDC reactions without TM catalysts are still very limited.5c–k Most of them relate to the acid-catalysed aerobic C–C bond-forming reactions centring on the benzylic sp3 C–H bonds in xanthenes,5e acridines,5e tetrahydroisoquinolines5e,f and glycine derivatives5g in the absence of metal catalysts. Very recently, the TM-free aerobic CDC reactions have been shown to accomplish the C–C bond formation of the nitroarene sp2 C–H bonds mediated by bases such as tertiary butoxides.5h–j In 2013, the group of Ess reported a t-BuONa-mediated TM-free aerobic CDC alkylation of nitroarenes with ketones, allowing the regiospecific synthesis of mono-α-arylated ketones under open-to-air conditions (Scheme 1).5h Later, Kumar et al. developed a t-BuOK-mediated TM-free aerobic CDC indolylation of nitroarenes with indoles under open flask conditions, enabling facile access to β-(2/4-nitroaryl)-indoles under mild conditions (Scheme 1).5i Following this work, they further presented the synthesis of unsymmetrical diaryl acetamides through the TM-free CDC reactions of the nitroarene sp2 C–H and amide sp3 C–H bonds (Scheme 1).5j However, there seems to be few reports concerning the direct CDC reaction of nitroarenes with arylacetates under TM-free and aerobic conditions.6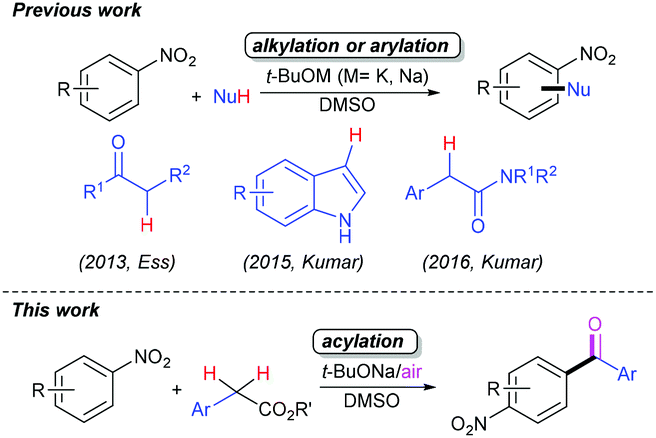 | ||
| Scheme 1 Previous and current studies. | ||
The incorporation of an acyl substituent into arenes is of central importance in organic transformations due to the extensive applications of arylketones in syntheses, materials and pharmaceuticals.7 Thus numerous methods have been developed.7a,8 Remarkably, the direct acylative functionalization of electron-deficient arenes including nitroarenes is a great challenge. To our knowledge, the direct and efficient acylation reactions of the electron-deficient nitroarene sp2 C–H bonds without any directing group have not been reported. Continuing our effort of t-BuONa-mediated TM-free autoxidative carbonylation of diarylmethylenes,9 herein, we present a TM-free aerobic formal acylation reaction of simple nitroarenes with readily available and bench-stable arylacetic esters under very mild conditions through a cascade CDC/oxidative decarboxylation process (Scheme 1).
Results and discussion
Our initial investigation into this acylation chemistry commenced by the treatment of nitrobenzene 1a with methyl phenylacetate 2a using the strong base NaOtBu (2.0 equiv.) in dry DMSO under open air atmosphere at room temperature (entry 1, Table 1). Delightedly, the acylation product 4-nitro-diphenylketone 3aa was formed regioselectively and isolated in 70% yield, with a concomitant normal CDC product 2-arylated phenylacetic ester. Decreasing the reaction temperatures diminished the yields of 3aa, while elevating the temperatures significantly increased those of 3aa (entries 2, 3 vs. 4, 5, Table 1). However, the higher temperature of 60 °C gave rise to the dramatic fall in the yield (entry 6, Table 1). Therefore, the reaction at 45 °C was preferred, wherein the ketone 3aa was isolated in 80% yield. Other inorganic bases such as K2CO3, KOtBu, NaH, KOH, and NaOH were screened (entries 7–11, Table 1). It was found that the strong bases delivered better yields, while the weak base K2CO3 could not initiate the reaction with the materials recovered. Likewise, the organic base DBU was ineffective for this chemistry (entry 12, Table 1). Typically, DMSO has been used as the best solvent in the base-mediated autoxidative reactions reported previously.5h–j,9 Nevertheless, the other solvents were further considered, including CH3CN, DMF, CH3OH, CH2Cl2, THF, and EtOAc (entries 13–18, Table 1). The results showed that all the aforementioned solvents furnished worse efficiencies than DMSO. Noteworthy, the protic solvent CH3OH afforded no product, due to its strong acidity disfavoring the formation of the carbanion 2a (entry 15, Table 1). Similarly, a trace amount of water in the undried DMSO imposed an adverse effect on the reaction yield (entry 19, Table 1). Additionally, performing the reaction under an O2 balloon instead of open air didn't improve the yield of the desired ketone 2a (entry 20, Table 1). After further optimization with respect to the equivalents of base, nitrobenzene and the concentration (please see the ESI†), the optimum acylation conditions were established: NaOtBu (2.0 equiv.)-mediated acylation of 1a (2.0 equiv.) with 2a (0.2 mmol) in dry DMSO (0.5 ml) under open flask conditions at 45 °C, providing 3aa in the isolated 80% yield.|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | T (°C) | Yieldb (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a The reaction was performed using 1a (0.4 mmol), 2a (0.2 mmol), base (0.4 mmol), and dry solvent (0.5 mL) in an open flask unless otherwise stated. b Isolated yield. c DMSO was not dried. d Under an O2 balloon. N.R. = no reaction. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | NaOtBu | DMSO | 12 | 25 | 70 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | NaOtBu | DMSO | 12 | 15 | 65 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | NaOtBu | DMSO | 12 | 10 | 50 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | NaOtBu | DMSO | 8 | 35 | 77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | NaOtBu | DMSO | 3 | 45 | 80 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | NaOtBu | DMSO | 3 | 60 | 56 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | K2CO3 | DMSO | 8 | 45 | N.R. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | KOtBu | DMSO | 3 | 45 | 75 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | NaH | DMSO | 3 | 45 | 65 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | KOH | DMSO | 3 | 45 | 20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | NaOH | DMSO | 3 | 45 | 50 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | DBU | DMSO | 8 | 45 | N.R. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | NaOtBu | CH3CN | 3 | 45 | 25 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | NaOtBu | DMF | 3 | 45 | 35 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | NaOtBu | CH3OH | 8 | 45 | N.R. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | NaOtBu | CH2Cl2 | 3 | 45 | 10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | NaOtBu | THF | 8 | 45 | 40 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18 | NaOtBu | EtOAc | 3 | 45 | 10 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 19c | NaOtBu | DMSO | 3 | 45 | 65 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20d | NaOtBu | DMSO | 2 | 45 | 76 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
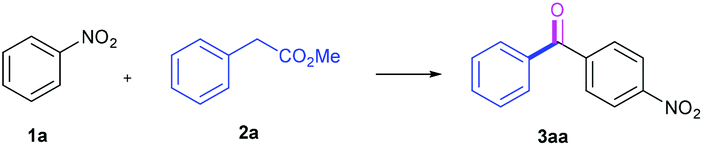
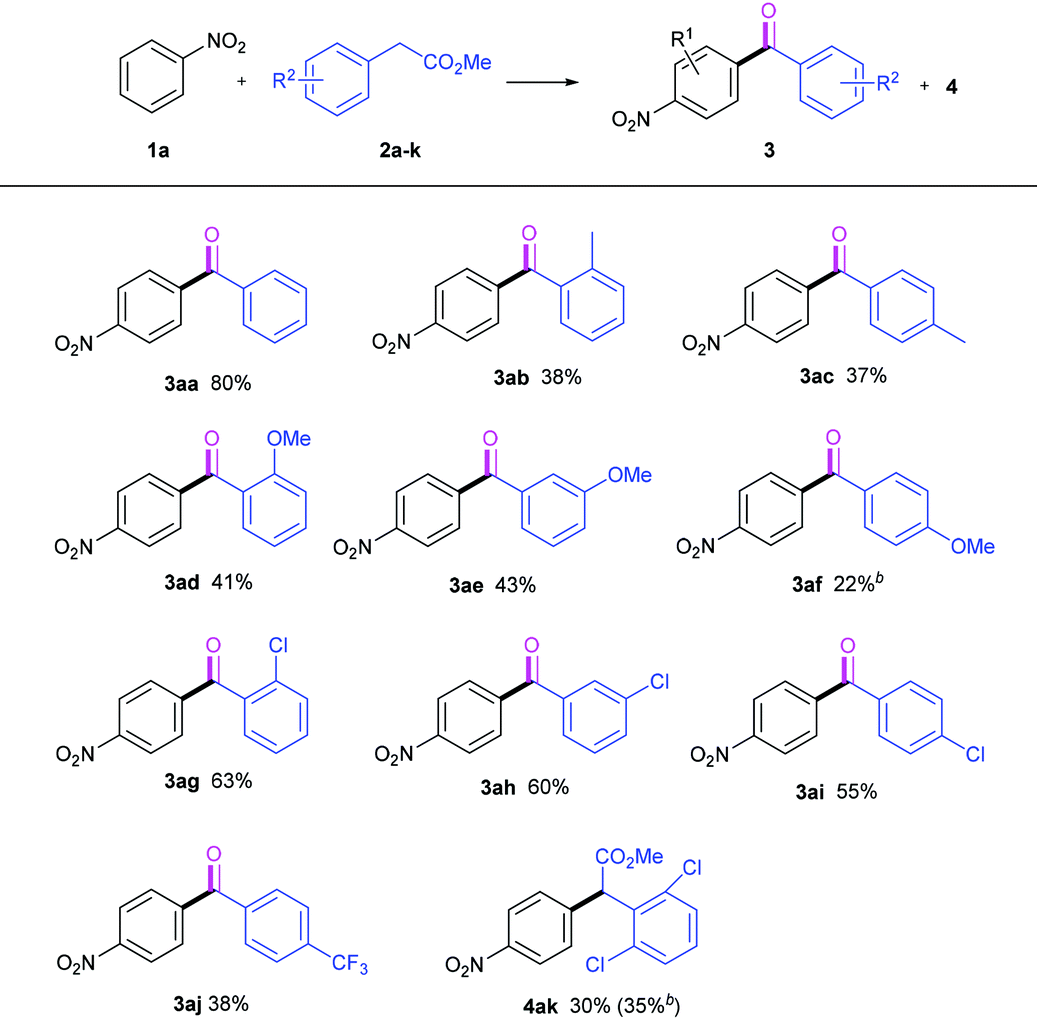
With the optimized reaction conditions in hand, we set out to study the scope of the methyl arylacetic esters used in this TM-free base-mediated autoxidative acylation reaction, and the results are listed in Table 2. In general, all the methyl arylacetates tested except 2k smoothly underwent the oxidative acylation to give their corresponding ketones 3 in moderate to good yields when nitrobenzene 1a was used as the reaction partner (3aa–3aj). The introduction of the substituent to the benzene ring of the arylacetate 2 remarkably resulted in efficiency loss (3ab–3aj). Moreover, the electron-donating groups (–Me, –OMe) seemed to disfavor the acylation occurrence more than did the electron-withdrawing one (–Cl). However, the CF3 group delivered only 38% yield (3aj). The steric effects of the substituents on the reaction were observed. Surprisingly, the steric hindrance of the group accelerated this oxidative acylation to some extent. For example, groups such as –Me, –OMe and –Cl, at the ortho-position resulted in higher yields than at the para-position. It should be pointed out that 2f led to the formation of a complex system under standard conditions, and yielded and isolated the acylation product 3af (22%) at 0 °C. Besides, methyl 2,6-dichlorophenylacetate 2k generated only the CDC ester 4ak, either at 45 °C (30% yield) or at 0 °C (35% yield). This may be due to the difficulties in the formation of the carbanion 4ak by the hindered base NaOtBu, and in the autoxidative transformation with molecular O2.
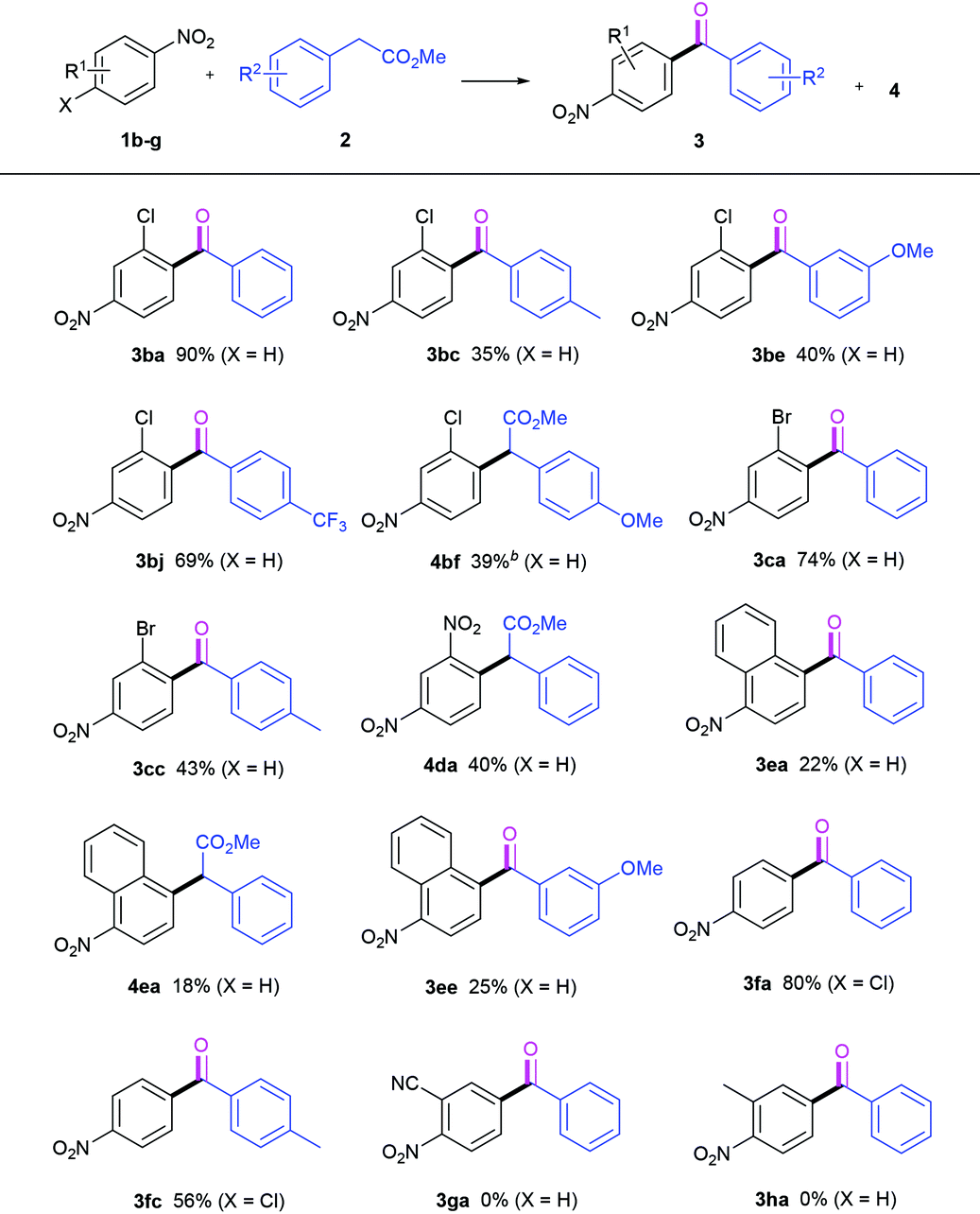
Next, we turned our attention to the nitroarene reaction partners, as shown in Table 3. 3-Chloro-4-nitrobenzene 1b was found to produce its expected acylation products 3. Its reaction with 2a afforded the desired ketone 3ba in 90% yield, and with 2j delivered 3bj in 69% yield. In the two cases of 3ba and 3bj, the efficiencies are higher compared to those of 3aa and 3aj. With regard to 2f, the CDC product 4bf was obtained in 39% yield at 0 °C. Likewise, 3-bromo-4-nitrobenzene 1c readily reacted with 2a and 2c, providing the expected ketones 3ca and 3cc in 74% and 43% yields, respectively. However, the reaction of 1,3-dinitrobenzene 1d with 2a gave only the CDC ester 4da in 40% yield under standard conditions. 1-Nitronaphthalene 1e is an effective coupling partner. Its reaction with 2a generated our desired product 3ea, along with the intermediate 4ea unconverted even for the prolonged time, while with 2e only the ketone 3ee was isolated in 25% yield. When 4-chloronitrobenzene 1f was used instead, no ortho-nitro ketones were detected. A competitive SN-Ar/oxidative decarboxylative carbonylation reaction preferably occurred by the replacement of the Cl moiety instead of the hydrogen atom, whereby the same products with those derived from 1a were obtained in moderate to good yields (3fa & 3fc). It is disappointing to find that 2-nitrobenzonitrile 1g and 2-nitrotoluene 1h are bad substrates. 1g was almost fully converted, but no corresponding ketone was detected. 1h was recovered quantitatively after workup.
Surprisingly, when it came to 2-CF3-nitrobenzene 1i, the cascade oxidative reaction yielded an abnormal product 3iavia the subsequent nucleophilic displacement of the nitro group by the –OMe group generated in the oxidative decarboxylation step (Scheme 2, 1i).10 In the case of 3-nitrotoluene 1j, the abnormal reaction occurred in the same manner, affording the 4-methoxylated ketone 3ja in 34% yield (Scheme 2, 1j). More interestingly, the use of 4-nitroanisole 1k gave rise to 51% yield of a denitro-coupling product 3ka, where the nitro group was formally displaced by an acyl group (Scheme 2, 1k).
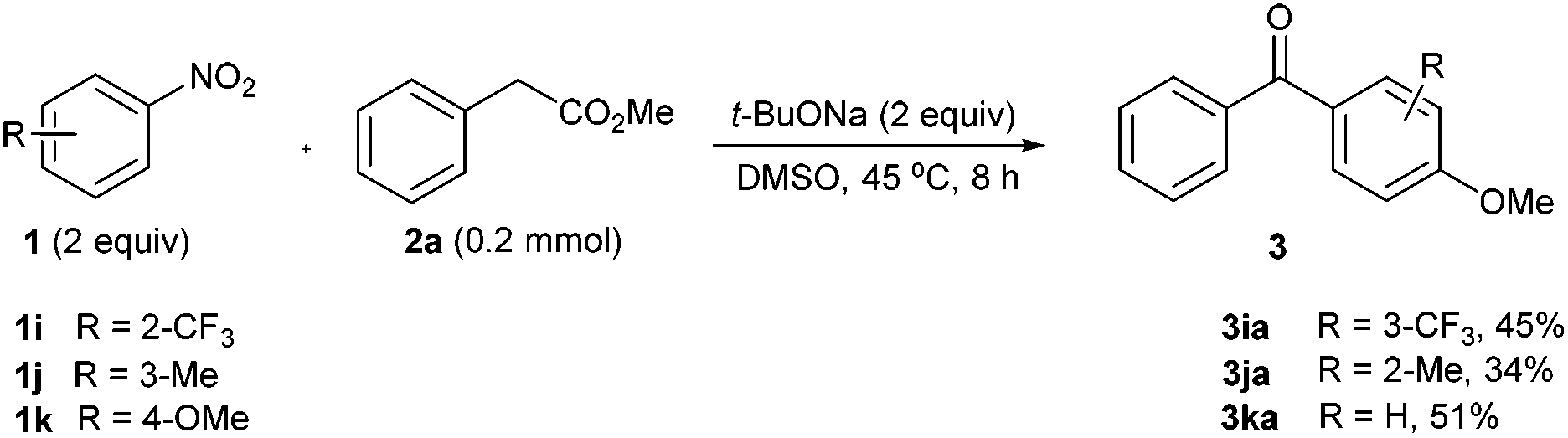 | ||
| Scheme 2 Exceptional examples related to the nitro displacement. | ||
Besides, we changed the alkyl groups in phenylacetates used in this transformation, to scrutinize their effect on oxidative acylation efficiency (Scheme 3). It was found that the ethyl ester 2l led to the notably lower yield of 3aa than the methyl version 2a, and moreover the iso-propyl and tert-butyl variants (2m & 2n) can't realize such acylation reaction under the standard conditions.
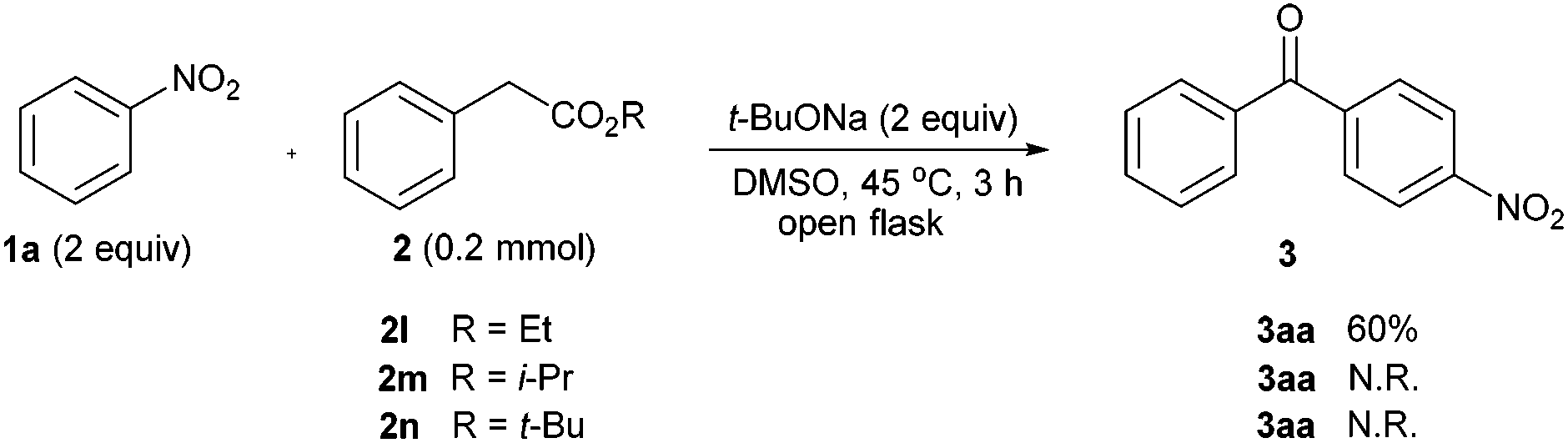 | ||
| Scheme 3 The effect of the alkyl groups in phenylacetates (N.R. = no reaction). | ||
To figure out the mechanism of this oxidative acylation chemistry, a variety of control experiments were carried out, as shown in Scheme 4. From the experiments for reaction temperature optimization, we realized that the reaction probably underwent the CDC process and that the low reaction temperature may favour the formation of the CDC product without further conversion to the acylation product. Thus, we strived to obtain 4aa by performing the reaction at 0 °C wherein 4aa was isolated in 30% yield. Then the CDC product 4aa as precursor was fully converted under the standard conditions, and the ketone 3aa was isolated in 92% yield (Scheme 4a). Furthermore, 4aa was observed and isolated in 28% yield when the mixture of 1a and 2a was treated at 45 °C under an argon atmosphere after five freeze–pump–thaw cycles (Scheme 4b). This suggests that DMSO could play a role as a hydrogen acceptor in the CDC coupling step (please see the ESI, section 5†). However, the reaction of 4aa as precursor under argon afforded nothing even for the prolonged time (8 h), but 4aa was fully recovered after neutralization under argon. This indicates that the followed oxidative decarboxylation process necessitates the involvement of molecular oxygen (O2). All these results confirmed our hypothesis that the first step of this chemistry involved a CDC reaction between nitroarenes and arylacetic esters and the second step implicated an aerobic oxidative decarboxylation mediated by a base.
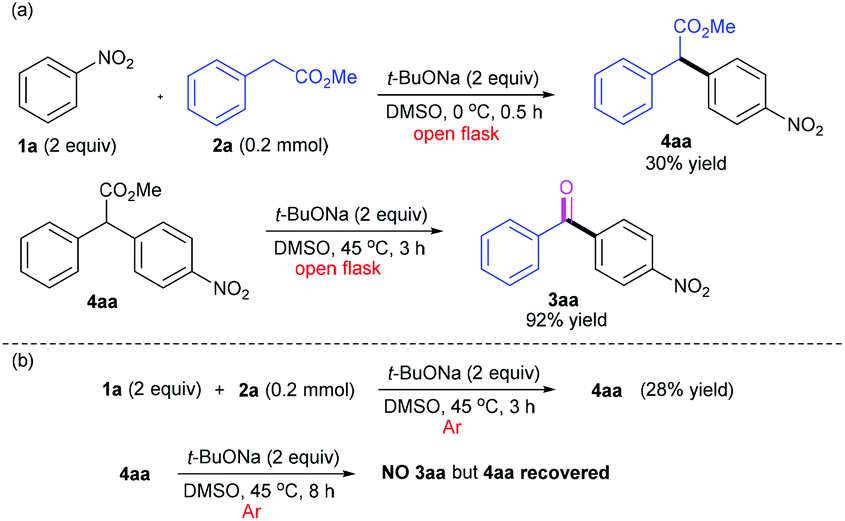 | ||
| Scheme 4 Control experiments. | ||
Based on our experimental observations and previous reports,5h,i,11 a tentative mechanism is proposed for this cascade oxidative acylation reaction (Scheme 5). Firstly, 2a is deprotonated by t-BuONa to generate a carbanion, which undergoes sequential nucleophilic addition to 1a, O2-mediated H-atom abstraction from II, radical combination and HOO− dissociation to furnish a CDC product 4aa with a concomitant hydroperoxide anion.5h In this stage (II to 4aa), DMSO as an oxidant can't be ruled out. Secondly, the intermediate 4aa is oxidized into a peroxide anion VII in the presence of a base and O2. Then two possible pathways may exist in the next transformation.11a,b One possible route involves a sequential hydrolysis−decarboxylation−hydroperoxy bond cleavage process11a,b (pathway A). This may explain why the reaction gave better yield under open air than under pure O2. This may be because a trace amount of water from the environment helps to promote the base-catalysed hydrolysis of IV. The minor competitive alternative implicates the intramolecular ester exchange of VII to in situ generate a dioxetanone IX, and subsequent decarboxylation to generate the product 3aa, along with peroxide cleavage11a (pathway B).
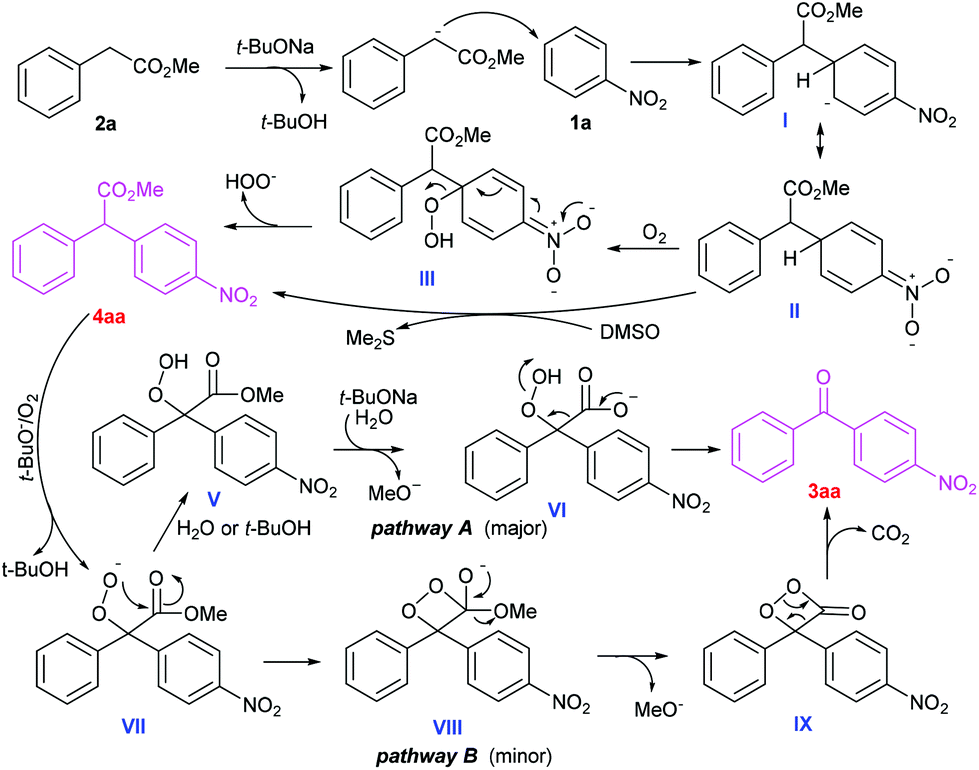 | ||
| Scheme 5 Proposed pathways for the cascade oxidative reaction. | ||
Conclusions
In conclusion, we have demonstrated a facile TM-free base-mediated aerobic oxidative acylation of nitroarenes with bench-stable and readily available arylacetic acid esters under mild conditions. This approach allows for the facile and regioselective synthesis of 4-nitro-diarylketones, and obviates the use of harsh conditions, TMs and Lewis acids. This chemistry involves the in situ generation of 2-arylated arylacetic acid esters via the CDC reaction, followed by the aerobic oxidative decarboxylation mediated by a base, and will supplement the classical Friedel–Crafts acylation chemistry. In view of its numerous advantages, it will provide an alternative and powerful tool to access ketones, particularly those containing the nitro group, in organic synthesis and pharmaceutical fields.Experimental
General information
All the solvents and reagents were purchased from the suppliers and used without further purification unless otherwise stated. The yields reported are for isolated yields unless otherwise stated. 1H NMR (400 MHz) and 13C NMR (101 MHz) spectra were recorded in CDCl3 at room temperature on a Bruker Avance III 400 spectrometer using TMS as the internal reference. MS spectra were recorded on an Agilent 6890/5973 GC-MS (EI) or Waters UPLC_Xevo_TQD (ESI) system. Elemental analyses were performed on a PerkinElmer 2400 series analyser. The melting point (m.p.) was determined using an open glass tube. TLC analyses were performed on silica gel plates and column chromatography was conducted over silica gel (mesh 200–300) at increased pressure.General procedure for the synthesis of diarylketones
To a predried 10 mL round-bottom flask was sequentially added phenylacetates 1 (0.2 mmol), nitroarenes 2 (0.4 mmol), dry DMSO (0.5 mL), and t-BuONa (0.4 mmol). The reaction system became dark purple on adding the base. The resulting mixture was stirred at 45 °C for the specific time (normally 3 h). Then it was cooled to room temperature; aqueous HCl was added (1 mol L−1) to pH = 6–7, and the resulting mixture was extracted with ethyl acetate, dried over anhydrous Na2SO4, and concentrated with rotary evaporation. Then the resulting residue was subjected to column chromatography on silica gel using a co-solvent (ethyl acetate/petroleum ether = 1/40, v/v) as an eluent to give the corresponding diarylketones 3. The representative ketones are listed here.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21202010), the Hunan Provincial Natural Science Foundation of China (2017JJ2275 & 2015JJ3012), the Scientific Research Fund of Hunan Provincial Education Department (16B003), the Hunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation (2015CL05), and Changsha University of Science & Technology, P. R. China.Notes and references
- (a) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed; (b) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed; (c) B. J. Li and Z. J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC.
- (a) C. Li and Z. Li, Pure Appl. Chem., 2006, 78, 935 CAS; (b) C. J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (c) C. J. Scheuermann, Chem. – Asian J., 2010, 5, 436 CrossRef CAS PubMed; (d) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (e) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (f) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138 CrossRef CAS PubMed.
- (a) H. Z. Zhi, S. P. M. Ung, Y. Liu, L. Zhao and C. J. Li, Adv. Synth. Catal., 2016, 358, 2553 CrossRef CAS; (b) C. A. Correia and C. J. Li, Adv. Synth. Catal., 2010, 352, 1446 CrossRef CAS; (c) O. Basle and C. J. Li, Chem. Commun., 2009, 4124 RSC; (d) C. Zhang, X. L. Zong, L. R. Zhang and N. Jiao, Org. Lett., 2012, 14, 3280 CrossRef CAS PubMed.
- (a) Z. P. Li, L. Cao and C. J. Li, Angew. Chem., Int. Ed., 2007, 46, 6505 CrossRef CAS PubMed; (b) F. Jia and Z. Li, Org. Chem. Front., 2014, 1, 194 RSC; (c) T. Wang, W. Zhou, H. Yin, J. A. Ma and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 10823 CrossRef CAS PubMed.
- (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed; (b) P. Finkbeiner and B. J. Nachtsheim, Synthesis, 2013, 45, 979 CrossRef CAS; (c) Q. Lu, H. Yi and A. Lei, Acta Chim. Sin., 2015, 73, 1245 CrossRef CAS; (d) K. Luo, Y. Z. Chen, L. X. Chen and L. Wu, J. Org. Chem., 2016, 81, 4682 CrossRef CAS PubMed; (e) A. Pinter, A. Sud, D. Sureshkumar and M. Klussmann, Angew. Chem., Int. Ed., 2010, 49, 5004 CrossRef CAS PubMed; (f) H. Ueda, K. Yoshida and H. Tokuyama, Org. Lett., 2014, 16, 4194 CrossRef CAS PubMed; (g) C. Huo, Y. Yuan, M. Wu, X. Jia, X. Wang, F. Chen and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 13544 CrossRef CAS PubMed; (h) Q.-L. Xu, H. Gao, M. Yousufuddin, D. H. Ess and L. Kurti, J. Am. Chem. Soc., 2013, 135, 14048 CrossRef CAS PubMed; (i) S. Kumar, V. Rathore, A. Verma, C. D. Prasad, A. Kumar, A. Yadav, S. Jana, M. Sattar, Meenakshi and S. Kumar, Org. Lett., 2015, 17(1), 82–85 CrossRef CAS PubMed; (j) V. Rathore, M. Sattar, R. Kumar and S. Kumar, J. Org. Chem., 2016, 81, 9206 CrossRef CAS PubMed; (k) S. Song, Y. Q. Zhang, A. Yeerlan, B. C. Zhu, J. Z. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487 CrossRef CAS PubMed; (l) P. M. Wang, F. Pu, K. Y. Liu, C. J. Li, Z. W. Liu, X. Y. Shi, J. Fan, M. Y. Yang and J. F. Wei, Chem. – Eur. J., 2016, 22, 6262 CrossRef CAS PubMed.
- M. Makosza, K. Kamienska-Trela, M. Paszewski and M. Bechcicka, Tetrahedron, 2005, 61, 11952 CrossRef CAS.
- (a) R. C. Larock, Comprehensive organic transformations: a guide to functional group preparations, Wiley-vch, New York, 1999 Search PubMed; (b) S.-B. Wu, C. Long and E. J. Kennelly, Nat. Prod. Rep., 2014, 31, 1158 RSC; (c) S. K. Vooturi, C. M. Cheung, M. J. Rybak and S. M. Firestine, J. Med. Chem., 2009, 52, 5020 CrossRef CAS PubMed; (d) S. Y. Lee, T. Yasuda, Y. S. Yang, Q. Zhang and C. Adachi, Angew. Chem., Int. Ed., 2014, 53, 6402 CrossRef CAS PubMed; (e) J. S. Li, Y. Xue, Z. W. Li, W. D. Liu, C. H. Lu and P. X. Zhao, Synlett, 2013, 24, 2003 CrossRef CAS; (f) J. S. Li, F. F. Cai, Z. W. Li, W. D. Liu, J. Simpson, Y. Xue, H. L. Pang, P. M. Huang, Z. Cao and D. L. Li, RSC Adv., 2014, 4, 474 RSC.
- (a) G. Sartori and R. Maggi, Advances in Friedel-Crafts acylation reactions: catalytic and green processes, CRC Press, 2009 Search PubMed; (b) S. Sharma, N. Kumar Mishra, Y. Shin and I. Su Kim, Curr. Org. Chem., 2016, 20, 471 CrossRef CAS; (c) X.-F. Wu, Chem. –Eur. J., 2015, 21, 12252 CrossRef CAS PubMed; (d) C. Pan, X. Jia and J. Cheng, Synthesis, 2012, 44, 677 CrossRef CAS.
- J.-S. Li, F. Yang, Q. Yang, Z.-W. Li, G.-Q. Chen, Y.-D. Da, P.-M. Huang, C. Chen, Y. Zhang and L.-Z. Huang, Synlett, 2017, 28, 994 CrossRef CAS.
- (a) J. P. Idoux, M. L. Madenwald, B. S. Garcia, D. L. Chu and J. T. Gupton, J. Org. Chem., 1985, 50, 1876 CrossRef CAS; (b) J. H. Gorvin and D. P. Whalley, J. Chem. Soc., Perkin Trans. 1, 1979, 1364 RSC; (c) N. Kornblum, L. Cheng, R. C. Kerber, M. M. Kestner, B. N. Newton, H. W. Pinnick, R. G. Smith and P. A. Wade, J. Org. Chem., 1976, 41, 1560 CrossRef CAS; (d) J. H. Gorvin, Chem. Ind., 1967, 1525 CAS.
- (a) Y. Sawaki and Y. Ogata, J. Org. Chem., 1977, 42, 40 CrossRef CAS; (b) M. Avramoff and Y. Sprinzak, J. Am. Chem. Soc., 1963, 85, 1655 CrossRef CAS.
- S. Barroso, G. Blay, L. Cardona, I. Fernández, B. García and J. R. Pedro, J. Org. Chem., 2004, 69, 6821 CrossRef CAS PubMed.
- M. Cai, J. Peng, W. Hao and G. Ding, Green Chem., 2011, 13, 190 RSC.
- J. Mahdi, H. Ankati, J. Gregory, B. Tenner and E. R. Biehl, Tetrahedron Lett., 2011, 52, 2594 CrossRef CAS.
- M. Makosza and J. Winiarski, J. Org. Chem., 1984, 49, 1494 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and copies of NMR spectra. See DOI: 10.1039/c7ob02865b |
| ‡ Both have equal contributions. |
| This journal is © The Royal Society of Chemistry 2018 |