
Ionic liquids and plastic crystals with a symmetrical pyrrolidinium cation†
Ruhamah
Yunis
 a,
Timothy W.
Newbegin
a,
Anthony F.
Hollenkamp
b and
Jennifer M.
Pringle
*a
a,
Timothy W.
Newbegin
a,
Anthony F.
Hollenkamp
b and
Jennifer M.
Pringle
*a
aInstitute for Frontier Materials, Deakin University, Melbourne, Victoria 3125, Australia. E-mail: jenny.pringle@deakin.edu.au
bCommonwealth Scientific and Industrial Research Organisation (CSIRO), Energy Flagship, Clayton, 3168, VIC, Australia
First published on 9th May 2018
Abstract
Solid and liquid salts utilising the N-methyl-N-alkyl pyrrolidinium cation, [Cnmpyr]+, and their use in electrochemical devices, are well established. However, new materials with enhanced properties, such as higher conductivity, lower viscosity or more favourable thermal phase behaviour, are still required. Here we report the synthesis and characterisation of new ionic liquids and plastic crystals using the N,N-diethylpyrrolidinium cation ([C2epyr]+) with six different anions. With the fluorosulfonyl(trifluoromethanesulfonyl)imide ([FTFSI]−) and dicyanamide ([DCA]−) anions, room temperature ionic liquids with low viscosity are formed. With the bis(trifluoromethanesulfonyl)imide ([NTf2]−), bis(fluorosulfonyl)imide ([FSI]−), hexafluorophosphate ([PF6]−) and tetrafluoroborate ([BF4]−) anions, organic ionic plastic crystals are produced. Of the new solid salts, [C2epyr][FSI] has the highest conductivity, higher than the well-established methyl-substituted analogue, giving 1.9 × 10−5 S cm−1 at 30 °C. Thermal analysis, conductivity and the Walden relationship are used to compare the new N,N-diethylpyrrolidinium salts across the different anions and with some previously reported N-methyl-N-alkylpyrrolidinium salts.
Introduction
There is increasing demand to replace the current flammable liquid battery electrolytes with less volatile or solidified materials, particularly for applications such as electric vehicles where safety is paramount.1,2 Ionic liquids (ILs) and organic ionic plastic crystals (OIPCs) generally show high decomposition temperatures, non-volatility and negligible vapour pressure, making them potentially safer electrolytes for applications such as batteries, fuel cells and solar cells.3–5 Both materials are composed entirely of ions; ILs that are liquid at room temperature are often referred to as room temperature ionic liquids (RTILs), while OIPCs are a specific type of salt structurally analogous to ILs but solid at room temperature and with significant disorder within the crystal lattice.6–8 The term “plastic crystal”, described in detail by Timmermans in the 1960s with respect to the molecular species,9 indicates that these materials have rotational, translational and/or conformational motions that allow them to flow under stress. The most plastic phase of OIPCs, denoted as phase I (the phase immediately before the melt), is reached via one or more solid–solid phase transition. In electrochemical applications, the plasticity of OIPCs can provide better contact with electrodes during volume change compared to brittle solid electrolytes, while also preventing the leakage problems associated with liquid electrolytes. OIPCs can be used to produce relatively conductive solid-state electrolytes, particularly when doped with an additional salt, e.g. lithium salts for use in Li batteries,6,10 or sodium salts for Na batteries,11 and so forth depending on the target ion required for the application. Examples of high conductivity OIPCs include diethyl(methyl)(isobutyl)phosphonium thiocyanate, [P1224][SCN], with a solid state conductivity approaching 10−3 S cm−1 at 40 °C,12 and the tetraethylphosphonium fluorohydrogenate salt, [P2222][(FH)2F], with a conductivity of 3 × 10−5 S cm−1 at 25 °C (phase II).13Although ILs have been known for more than 100 years, and OIPCs for more than a decade, the exact structure–property relationships that define the thermal and transport behaviour of these salts are still poorly understood. In comparison to the plethora of cations and anions previously explored to make ionic liquids, the range of known OIPC-forming ions is significantly smaller. The most commonly used cations include pyrrolidinium, tetraalkyl ammonium and phosphoniums, combined with anions such as dicyanamide ([DCA]−), bis(trifluoromethanesulfonyl)imide ([NTf2]−), hexafluorophosphate ([PF6]−), bis(fluorosulfonyl)imide ([FSI]−)14 and tetrafluoroborate ([BF4]−).12,15–19 Alternative OIPC cations include trialkylsulfonium,20 and pyrazolium.21 Relatively new anions include (fluorosulfonyl)(trifluoromethanesulfonyl)imide ([FTFSI]−),22 and the six-membered cyclic N(SO2CF2)2CF2 anion.23
Many combinations of these cations and anions form RTILs; to make OIPCs, cations substituted by relatively short alkyl chains are normally used, which elevates the melting point to above room temperature and can lower the energy required for rotational disorder of the cation. Thus, for example, the majority of research into pyrrolidinium-based salts has focused previously on cations with one methyl substituent and either methyl or ethyl as the second substituent, i.e. the [Cnmpyr]+ cations where n = 1 or 2. For example, [C2mpyr][FSI] is a plastic crystal with two solid–solid phase transitions, at −72 °C and −22 °C, and a melt at 203 °C.14 This OIPC has recently been demonstrated to be a very promising solid-state electrolyte for lithium batteries, achieving good stability and cycling performance.24,25
As an alternative to the methyl-substituted pyrrolidinium salt, it is proposed that increased ion symmetry through use of two ethyl substituents could lead to increased crystal packing efficiency (thus higher melting points) while at the same time enabling rotational disorder of the cation. Both of these effects are predicted to encourage the formation of rotationally disordered OIPCs and hence our interest in the lesser-used [C2epyr] cation. Here we have investigated the synthesis of highly conductive ILs and OIPCs combining the [C2epyr]+ cation with a range of different anions. These salts were prepared by anion metathesis of [C2epyr]Br with the respective lithium, potassium or silver salts of the anion. The [C2epyr]Cl salt26 was first synthesized in 1944, and later [C2epyr]Br27,28 and [C2epyr]I29,30 were reported. There are some prior reports on the use of this cation for synthesis of salts with more charge diffuse anions, as summarised below, but without any thorough investigation of the physical and thermal properties. Synthesis of the [C2epyr][BF4] salt by anion exchange of the bromide salt in the presence of hydrogen peroxide and tetrafluoroboric acid was previously reported, plus the electrochemical characterisation of this salt in a solution of propylene carbonate.31 The [C2epyr][(FH)2.0F] salt has been shown to exhibit two solid–solid transitions before melting at 358 K, indicative of plastic crystal behaviour.27 [C2epyr][NTf2] has also been synthesized by microwave irradiation of equimolar quantities of diethylamine and 1,4-dibromobutane in the presence of K2CO3 at 120 °C, followed by anion exchange with LiNTf2. The characterisation reported was the melting point and decomposition temperatures, at 92–94 °C and 317 °C, respectively.32
Here we report the thermal behaviour of salts based on the [C2epyr] cation with anions [DCA]−, [NTf2]−, [FSI]−, [PF6]−, [BF4]− and [FTFSI]−. These anions were chosen in order to utilise the predicted benefits of the relatively small and symmetrical cation, with respect to forming rotationally disordered OIPCs or low-viscosity ILs, and to probe the influence of different anions on the thermal and physical properties of the new salts.
Results and discussion
Thermal properties
The N,N-diethylpyrrolidinium salts were synthesized in good yield by the direct reaction of N-ethylpyrrolidine with ethylbromide, as shown in Fig. 1. The thermal behaviour of the synthesized salts, analysed by differential scanning calorimetry (DSC), is shown in Fig. 2 and Table 1. There is a clear dependence of thermal behaviour on the nature of the anion with, for example, melting points ranging from below room temperature to over 100 °C. Two new RTILs have been formed through use of the symmetrical cation: [C2epyr][FTFSI] shows two solid–solid phase transitions at low temperature before melting at 8 °C, and [C2epyr][DCA] melts at 9 °C. | ||
| Fig. 1 Reaction scheme for the synthesis of [C2epyr]+ salts, and the structure and abbreviations of the anions (X) used. | ||
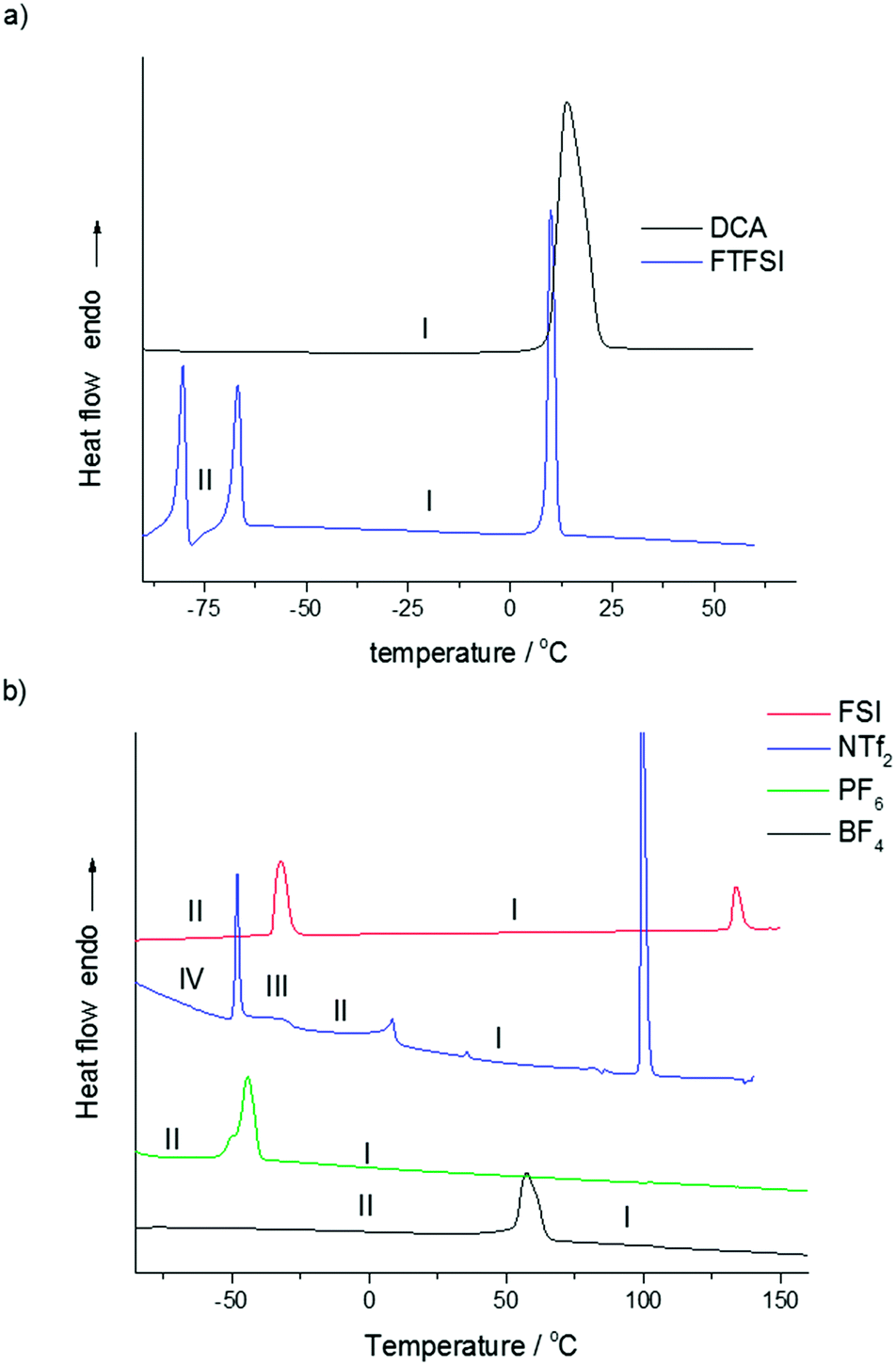 | ||
| Fig. 2 Differential scanning calorimetry (DSC) traces for the [C2epyr] salts (a) RTILs, and (b) OIPCs. Y-Axis scaled for comparison. | ||
| Phase IV–III | Phase III–II | Phase II–I | Melt | |||||
|---|---|---|---|---|---|---|---|---|
| T/°C ±1 | ΔS/J K−1 mol−1 ±10% | T/°C ±1 | ΔS/J K−1 mol−1 ±10% | T/°C ±1 | ΔS/J K−1 mol−1 ±10% | T/°C ±1 | ΔS/J K−1 mol−1 ±10% | |
| a Sample decomposed before clear melting point. Onset temperatures are reported for all transitions. Melting points were also confirmed using visual melting point apparatus. | ||||||||
| [C2epyr][FTFSI] | −82 | 19 | −69 | 13 | 8 | 20 | ||
| [C2epyr][DCA] | 9 | 84 | ||||||
| [C2epyr][NTf2] | −50 | 13 | −45 | 2 | 5 | 7 | 98 | 33 |
| [C2epyr][FSI] | −35 | 33 | 131 | 9 | ||||
| [C2epyr][PF6] | −54 | 41 | —a | |||||
| [C2epyr][BF4] | 53 | 23 | —a | |||||
A number of the salts show multiple solid–solid phase transitions, indicative of plastic crystal behaviour;7,9 by convention the highest temperature phase is denoted as phase I, with lower temperature phases denoted as phase II, III and so on. Such multiple solid phases are understood to arise from motions of both the cation and anion, which become increasingly more complex as the temperature is increased. Both [C2epyr][BF4] and [C2epyr][PF6] show one solid–solid phase transition (phase II to phase I), at 53 °C and −54 °C respectively, before a combined melt/decomposition (as evidenced by discolouration of the samples after the DSC analysis) at around 300 °C. Fewer solid–solid phase transitions are observed in the new ethyl salts than in the methyl analogues [C2mpyr][BF4] and [C2mpyr][PF6].17,33 While this may be consistent with reduced degrees of freedom of the more symmetrical cation, the OIPC phase behaviour reflects the combined motions of both types of ion and thus the rotational and translation motion of the anion must also be taken into consideration when understanding the OIPC phase behaviour. This makes isolating the effect of changing the anion or cation very complex. For example, the tetraalkylammonium and tetraalkylphosphonium BF4 and PF6 salts characterised by Matsumoto et al.34 shows no single anion trend across the range of different cations. The same is true for comparison of [Cnmpyr][BF4] and [PF6] salts,17,33 where n = 1–3. Further analysis of the [C2epyr] salts, with techniques such as solid state NMR and X-ray diffraction,35 will be used to further understand the complex molecular motions within the different materials.
For the use of OIPCs as solid-state electrolytes it is most advantageous to have the material in phase I over the temperature range of application as this is the most conductive phase. This is the case for the PF6 and BF4 salts reported here. However, the relatively high melting temperature is not ideal as it suggests less disorder in the material at ambient temperature than might be present in a lower melting analogue; this is consistent with the relatively low conductivity of these materials, as discussed below.
More advantageous thermal behaviour is displayed by the [C2epyr][NTf2] and [C2epyr][FSI] salts. The [C2epyr][NTf2] exhibits three solid–solid phase transitions, which is beneficial for increasing the disorder of the salt, before melting at 98 °C. An interesting feature of this salt is the unusual plateau region immediately following the first solid–solid phase transition, provisionally labelled phase III. We have recently observed a similar feature in a phosphonium OIPC with the FSI− anion (triethyl(methyl)phosphonium bis(fluorosulfonyl)imide).36 As here, the feature appeared immediately after a solid–solid phase transition, and spanned between −55 to −30 °C. This feature was assigned to a second-order displacive phase transition, attributed to the FSI anion undergoing progressive transformation from a less energetically favourable trans orientation to cis and assisted by cooperative motion of the cation. The NTf2 anion can also exist in a number of different conformers. For example, the DSC trace of [C2mpyr][NTf2] shows multiple solid–solid transitions, including one that appears to be second order,18 and thus it is proposed that the unusual feature in the DSC trace of [C2epyr][NTf2] is again primarily the result of conversion between the two conformers of the anion. Work is underway to analyse further this low temperature feature.
Of the new OIPCs reported here, the [C2epyr][FSI] salt displays what appears to be the most ideal thermal behaviour for practical application. It has a large low-temperature solid–solid phase transition, at −35 °C, and melts at 131 °C; above the temperature range of most applications but arguably low enough to ensure that the material is relatively disordered at ambient temperature. This significant disorder in phase I is consistent with the high conductivity, discussed further below. This salt also has the lowest entropy of melt of the series, at only 9 J K−1 mol−1. A low entropy of fusion of <20 J K−1 mol−1 is consistent with Timmermans’ criterion for plastic crystal behaviour for molecular plastic crystals,9 although for OIPCs plasticity has been observed even with higher entropies of melt.7
Comparison of the melting points of the new [C2epyr] salts with those of the methyl-substituted analogues reported previously highlights the complexity of factors that dictate this property. The melting point of [C2epyr][DCA] is higher than that of [C2mpyr][DCA]37 (9 °C and −10 °C respectively), and similarly the melting point of [C2epyr][NTf2] is a little higher than that of [C2mpyr][NTf2]16 (98 °C and 91 °C, respectively). However, moving to the ethyl substituent in the FSI family produces a surprising reduction in melting point: from 205 °C,14 to 131 °C. This shows that it is not just the symmetry of the cation that affects the melting point – in addition, changes to the anion symmetry (such as possible differences in the relative populations of the different conformers), and different crystal packing of the anion conformer(s) with the new cation, may impact the melting temperature.
Transport properties
The conductivity of an electrolyte is a key parameter that determines its suitability for electrochemical applications, and achieving suitable transport properties is an on-going challenge for solid-state electrolytes. The conductivities of OIPCs vary significantly, depending on the nature of the cation and anion that dictate the thermal and transport properties; both the cation and anion may be mobile and contribute to the total ionic conductivity. To allow their use in Li batteries, these materials are doped with Li salts, typically with the same anion as the OIPC, and this can give rise to orders of magnitude improvements in conductivity.6,10 Here, we compare the properties of neat salts to allow assessment of the effect of different anions on the transport of the pure OIPCs. However, it is important to note that to enable their practical application the OIPCs would be doped with target ions, thereby further increasing the conductivity.The ionic conductivities of the new [C2epyr]+ salts, over a range of temperatures, are shown in Fig. 3a and the activation energies for conduction given in Table 2. The solid-state conductivity is affected by both the nature of the anion and the thermal behaviour, with the highest conductivity observed in phase I (the phase prior to melting). The most conductive of the new solid salts is [C2epyr][FSI], at 1.9 × 10−5 S cm−1 at 30 °C, which also displays the lowest activation energy. This is among the highest OIPC conductivity found to-date and higher than the previously reported [C2mpyr][FSI] (1.23 × 10−6 S cm−1 at 25 °C),14 which has been successfully used in lithium metal batteries.24,25
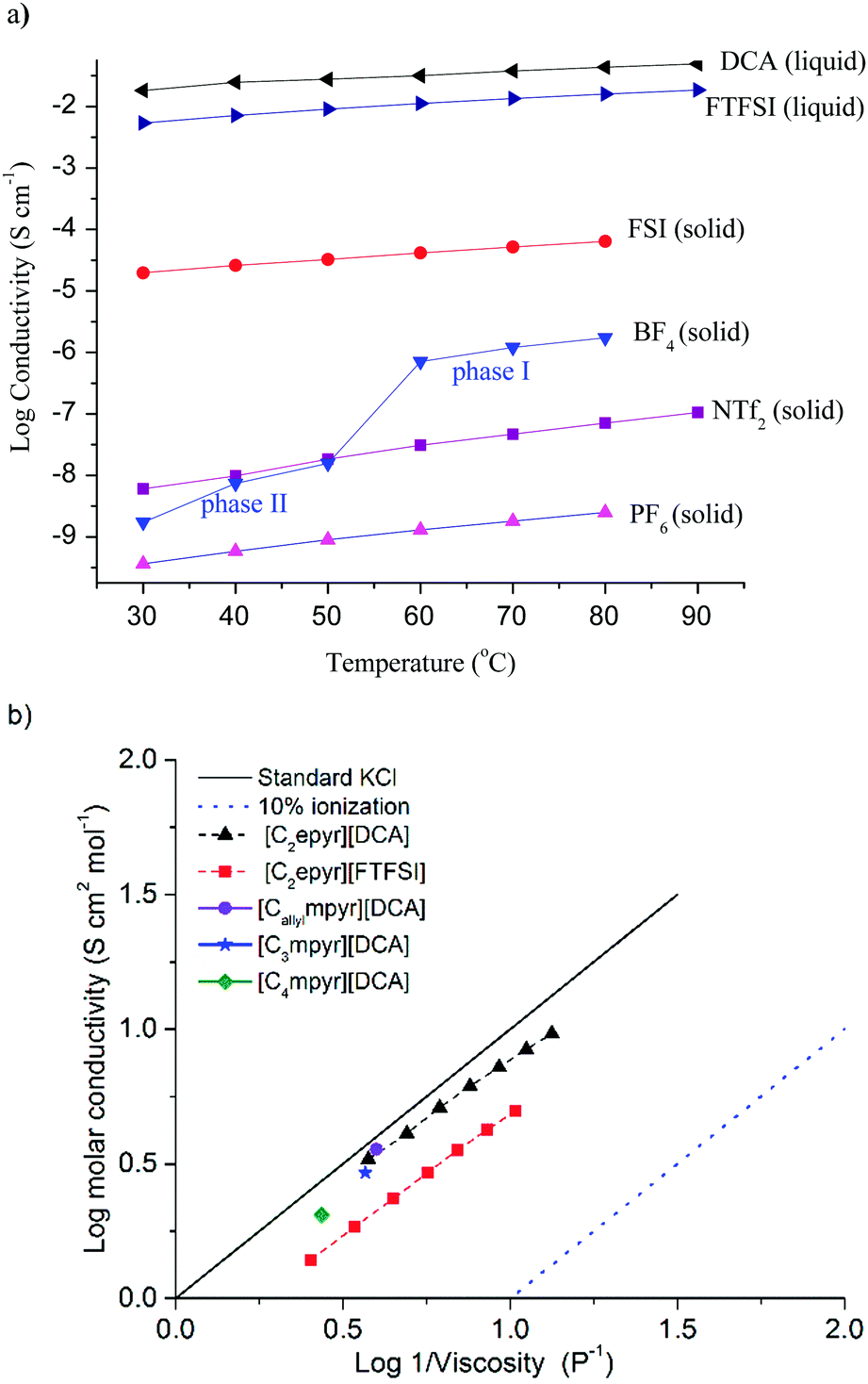 | ||
| Fig. 3 (a) Conductivity of the [C2epyr] RTILs and OIPCs, and (b) Walden plot for the two RTILs, including comparison with three previously reported pyrrolidinium DCA salts.38,39 | ||
| Conductivity/S cm−1 (±5%) | Viscosity/mPa s | E a/kJ mol−1 | |
|---|---|---|---|
| [C2epyr][FTFSI] | 5.4 × 10−3 | 40 | 18 |
| [C2epyr][DCA] | 1.8 × 10−2 | 27 | 15 |
| [C2epyr][NTf2] | 8.5 × 10−9 | — | 44 |
| [C2epyr][FSI] | 1.9 × 10−5 | — | 27 |
| [C2epyr][PF6] | 3.6 × 10−10 | — | 34 |
| [C2epyr][BF4] | 1.7 × 10−9 | — | Phase II: 90 |
| Phase I: 44 |
It is also notably more conductive than the new [C2epyr][NTf2] salt (Table 1). This is consistent with the higher entropy of fusion of the latter (Table 1), which indicates that the material is more ordered in phase I. Thus, the [C2epyr][FSI] appears to be very promising as a new electrolyte material.40
When OIPCs are heated they undergo one or more solid–solid phase transitions, which are often correlated with a step-increase in conductivity as a result of the onset of additional rotational and/or translational motions.41 The new [C2epyr][BF4] displays such behaviour, with a significant increase in conductivity at the phase II to I transition at around 55 °C, and also a significant decrease in the activation energy of conduction. The conductivity of [C2epyr][BF4] becomes higher than that of [C2epyr][NTf2] after the phase II–I transition. As for the thermal behaviour, the effect on solid-state conductivity of changing from a methyl to an ethyl group on the cation is not predictable. For the NTf2 and BF4 salts, the ambient temperature conductivity of the ethyl analogue is lower than the methyl analogue (the latter of which are ca. 10−8 and 10−6 S cm−1 respectively).16,42 However, as noted above, use of the ethyl-substituted cation with the FSI anion results in significant enhancement in conductivity.
The ionic conductivities of the new RTILs [C2epyr][DCA] and [C2epyr][FTFSI], over a range of temperatures, are shown in Fig. 3a and the viscosity is given in Table 2. The [C2epyr][DCA] has a lower viscosity than [C2epyr][FTFSI], and both are relatively low compared to many other ionic liquids with larger or less charge-diffuse anions.43 Low viscosities have also been reported for other DCA-based RTILs, such as with the [C4mpyr] or [C3mpyr],37,38 and [Callylmpyr][DCA].39 The ionic conductivity of [C2epyr][DCA] is higher than [C2epyr][FTFSI], attributed to the lower viscosity that results from the small, charge-diffuse anion, and also to less ion pairing in the former as shown by the Walden plot.
The Walden rule correlates molar conductivity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Λ) to viscosity (logη−1), and standard KCl can be used as a reference (i.e. as an ideal salt with completely dissociated ions), shown in Fig. 3b and eqn (1).
Λ) to viscosity (logη−1), and standard KCl can be used as a reference (i.e. as an ideal salt with completely dissociated ions), shown in Fig. 3b and eqn (1).
| C = Ληα | (1) |
Experimental
Materials and methods
N-Ethylpyrrolidine (97%, Sigma Aldrich, Australia), bromoethane (98%, Sigma Aldrich, Australia), lithium bis(trifluoromethanesulfonyl)imide (>99.9%, Solvay, Canada), potassium bis(fluorosulfonyl)imide (>99.9%, Suzhou Fluolyte Co, China), silver tetrafluoroborate (99%, Oakwood, USA), lithium(fluorosulfonyl)trifluoromethanesulfonylimide (99%, PROVISCO CS, Czech Republic), potassium hexafluorophosphate (≥99%, Sigma Aldrich, Australia), sodium dicyanamide (≥99%, Sigma Aldrich, Australia) and silver nitrate (≥99%, Sigma Aldrich, Australia) were used without further purification.The structures, abbreviations and synthetic route is shown in Fig. 1. The synthesis was predominantly performed using a nitrogen Schlenk line. Milli-Q water was used in all the syntheses. 1H, 13C and 19F NMR spectra were collected on Bruker Avance III instrument operating at 400 MHz, 100 MHz and 375 MHz respectively, in CD3OD by referencing the solvent peak. Mass spectrometry was performed on an Agilent 1200 series HPLC system. All samples were dried for at least 72 hours on a Schlenk line. All samples were sent to The Campbell Microanalytical Laboratory, New Zealand for elemental analysis. Bromide, silver and potassium contents were determined by an Ionode IJ-Ag or Br selective electrode, or Hanna potassium selective electrode, respectively, after calibration with 10 and 100 ppm solutions of the respective ion. Water contents were measured using a Metrohm 831 Karl-Fisher Coulometer.
Quantitation of lithium was carried out using an inductively coupled plasma-mass spectrometer (ICP-MS; NexION 350X, PerkinElmer, USA). The internal standards Sc (200 ppb) and Rh (20 ppb) in 1% aqua regia were used for correction of matrix effects. The internal standard solution was mixed prior to the nebulizer using a T-piece in a 1:1 ratio. Calibration standards for lithium (Perkin Elmer, Lithium standard 1000 ppm in 2% HNO3) were prepared at 0.1, 1, 10, 50, 100 and 500 ppb with 2% HNO3 in each. The mass spectrometer was operated in kinetic energy discrimination mode (KED) with 50 ms dwell times, 20 sweeps, one reading and three replicates. The plasma source conditions were: nebulizer gas flow 1.02 L min−1, auxiliary gas flow 1.2 L min−1, plasma gas flow 15 L min−1, ICP RF power 1500 W. Data analysis was carried out using Syngistix (PerkinElmer) software. Signal responses were normalized to the scandium internal standard.
Viscosity (2.5 mm tube) and density were measured on a dual Lovis 2000 M/ME from 20 to 90 °C. Before performing DSC, the melting points were determined visually using a Gallenkamp melting point apparatus. DSC was measured on a Mettler Toledo DSC STARe with a scan rate of 10 °C min−1 for both the heating and cooling cycles. Three heating and cooling cycles were performed, using a sample size of 4–10 mg in an aluminium pan under inert atmosphere. The temperature ranges used were from −173 K, up to 333 K for DCA and FTFSI salts, 423 K for TFSI and FSI salts, and 573 K for the BF4 and PF6 salts. The data reported is from the second heating run unless otherwise specified. The cooling traces are shown in the ESI.† Before measurement the heat flow and temperature was calibrated using cyclohexane.
The ionic conductivity was measured by electrochemical impedance spectroscopy using Solartron Modulab (Solartron Analytical, Ametek). The solid samples were pressed into a pellet with 13 mm diameter. The pellet was placed between two stainless steel electrodes (with spacer) in a dry barrel cell in an inert atmosphere. The thickness of the pellet was measured before and after measurement. In the case of [C2epyr][FSI] the sample was manually pressed between two stainless steel plates, without additional applied pressure, as this material was very soft. The conductivities were measured from 30 to 80 °C with a temperature settling time of 40 minutes, frequency range from 1 MHz to 0.1 Hz with an amplitude of 100 mV. For liquid samples the conductivities were measured using a dip cell with platinum electrodes, with a temperature range from 30 to 90 °C. The cell constant was measured using 1 mM KCl solution at 30 °C. The conductivities of the samples were determined from the real axis intercept in the Nyquist plot of the impedance data. The activation energies were calculated using fits to the Arrhenius equation, which gave linear relationships between lnσ and 1/T.
Synthetic procedures
[C2epyr]Br (5.356 g, 26 mmol) and freshly prepared AgDCA (11.734 g, 64 mmol) were added to water (55 mL) and the resulting mixture was stirred at room temperature overnight in the dark. The reaction mixture was then filtered under vacuum and the filtrate was refrigerated for 3 hours. Solid AgBr was removed by filtration, followed by centrifugation at 15000 rpm at 0 °C for 10 minutes and the filtration through a 0.2 μm syringe filter. The resulting filtrate was dried in vacuo to obtain [C2epyr][DCA] (4.331 g, 88%). 1H NMR (400 MHz, CD3OD): 1.36 (tt, JHH = 7.2, 2.0 Hz, 6H, CH2CH3), 2.20 (m, 4H, NCH2CH2), 3.39 (q, JHH = 7.2 Hz, 4H, CH2NCH2), 3.53 (m, 4H, NCH2CH3) ppm; 13C NMR (100 MHz, CD3OD): 7.46 (CH2CH3), 21.43 (NCH2CH2), 54.10 (NCH2CH3), 61.62 (CH2NCH2) ppm. ES+m/z 129.2 (C8H18N1)+, ES−m/z 65.9 (DCA)−. Anal. calculated for C10H21N4O1.5; C, 54.28; H, 9.57; N, 25.32. Found: C, 54.8; H, 9.7; N, 25.8. Bromide content (ISE) is 378 ppm. Silver (ICP-MS) 23 ppm. Water content less than 300 ppm.
[C2epyr]Br (2.265 g, 11 mmol) and AgPF6 (3.59 g, 11 mmol) were added to a pre-dried flask. After the addition of dry acetonitrile (30 mL) via syringe, the solution was stirred under an inert atmosphere (N2) for an hour in the dark. The solution was filtered, followed by centrifugation for 15 min (4000 rpm at 4 °C). The solution was then filtered through a syringe filter (0.22 μm, PTFE). The organic solvent was removed in vacuo. The white solid was dissolved in water (10 mL) and kept in the fridge overnight, followed by filtration through sintered funnel, washing with diethyl ether and drying to obtain a white powder of [C2epyr][PF6] (1.50 g, 30% yield). 1H NMR (400 MHz, CD3OD): 1.44 (tt, JHH = 7.2, 1.8 Hz, 6H, CH2CH3), 2.29 (m, 4H, NCH2CH2), 3.57 (q, JHH = 7.2 Hz, 4H, CH2NCH2), 3.72 (m, 4H, NCH2CH3) ppm; 13C NMR (100 MHz, CD3OD): 8.05 (CH2CH3), 21.70 (NCH2CH2), 54.30 (NCH2CH3), 61.83 (CH2NCH2) ppm; 19F NMR (376.5 MHz, CD3OD): −72.51 (d, JFP = 712 Hz), −81.61 (d, JFP = 941 Hz) ppm. ES+m/z 127.7 (C8H18N1)+, ES−m/z 144.9 (PF6)−. Anal. calculated for C8H18N1F6P1; C, 35.17; H, 6.64; N, 5.13. Found: C, 35.28; H, 6.77; N, 5.3. Bromide content (ISE) 80 ppm. Silver content (ISE) 380 ppm.
Conclusions
The N,N-diethylpyrrolidinium cation has been used to synthesise two new room temperature ionic liquids and four organic ionic plastic crystals. Both of the RTILs, [C2epyr][DCA] and [C2epyr][FTFSI], exhibit low viscosities. The organic ionic plastic crystals, utilising anions [NTf2]−, [FSI]−, [BF4]− or [PF6]−, display a variety of interesting thermal and transport behaviour. Of these materials, the [C2epyr][FSI] is particularly promising as a new solid-state electrolyte as it displays among the highest ionic conductivity to-date for OIPCs, at 1.9 × 10−5 S cm−1 at 30 °C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council through Discovery Grant DP170101087. The authors also thank Dr Damien Callahan at Deakin University for his help with the ICP-MS measurements.Notes and references
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587 CrossRef CAS.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154 CrossRef CAS PubMed.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- M. Barghamadi, A. S. Best, A. I. Bhatt, A. F. Hollenkamp, M. Musameh, R. J. Rees and T. Ruther, Energy Environ. Sci., 2014, 7, 3902 CAS.
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994 CrossRef CAS PubMed.
- D. R. MacFarlane, J. Huang and M. Forsyth, Nature, 1999, 402, 792 CrossRef CAS.
- D. R. MacFarlane, P. Meakin, N. Amini and M. Forsyth, J. Phys.: Condens. Matter, 2001, 13, 8257 CrossRef CAS.
- D. R. MacFarlane, M. Kar and J. M. Pringle, Fundamentals of Ionic Liquids: From Chemistry to Applications, Wiley, 2017 Search PubMed.
- J. Timmermans, J. Phys. Chem. Solids, 1961, 18, 1 CrossRef CAS.
- L. Jin, P. C. Howlett, J. M. Pringle, J. Janikowski, M. Armand, D. R. MacFarlane and M. Forsyth, Energy Environ. Sci., 2014, 7, 3352 CAS.
- F. Makhlooghiazad, P. C. Howlett, X. Wang, M. Hilder, D. R. MacFarlane, M. Armand and M. Forsyth, J. Mater. Chem. A, 2017, 5, 5770 CAS.
- V. Armel, M. Forsyth, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2011, 4, 2234 CAS.
- T. Enomoto, S. Kanematsu, K. Tsunashima, K. Matsumoto and R. Hagiwara, Phys. Chem. Chem. Phys., 2011, 13, 12536 RSC.
- M. Yoshizawa-Fujita, E. Kishi, M. Suematsu, T. Takekawa and M. Rikukawa, Chem. Lett., 2014, 43, 1909 CrossRef.
- J. M. Pringle, Phys. Chem. Chem. Phys., 2013, 15, 1339 RSC.
- D. R. MacFarlane, P. Meakin, J. Sun, N. Amini and M. Forsyth, J. Phys. Chem. B, 1999, 103, 4164 CrossRef CAS.
- S. Forsyth, J. Golding, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2001, 46, 1753 CrossRef CAS.
- W. A. Henderson, V. G. Young, S. Passerini, P. C. Trulove and H. C. De Long, Chem. Mater., 2006, 18, 934 CrossRef CAS.
- Q. Zhou, W. A. Henderson, G. B. Appetecchi, M. Montanino and S. Passerini, J. Phys. Chem. B, 2008, 112, 13577 CrossRef CAS PubMed.
- H.-B. Han, J. Nie, K. Liu, W.-K. Li, W.-F. Feng, M. Armand, H. Matsumoto and Z.-B. Zhou, Electrochim. Acta, 2010, 55, 1221 CrossRef CAS.
- Y. Abu-Lebdeh, A. Abouimrane, P.-J. Alarco and M. Armand, J. Power Sources, 2006, 154, 255 CrossRef CAS.
- H. Matsumoto, N. Terasawa, T. Umecky, S. Tsuzuki, H. Sakaebe, K. Asaka and K. Tatsumi, Chem. Lett., 2008, 37, 1020 CrossRef CAS.
- M. Moriya, T. Watanabe, W. Sakamoto and T. Yogo, RSC Adv., 2012, 2, 8502 RSC.
- X. Li, Z. Zhang, S. Li, K. Yang and L. Yang, J. Mater. Chem. A, 2017, 5, 21362 CAS.
- Y. Zhou, X. Wang, H. Zhu, M. Armand, M. Forsyth, G. W. Greene, J. M. Pringle and P. C. Howlett, Phys. Chem. Chem. Phys., 2017, 19, 2225 RSC.
- M. S. Kharasch and C. F. Fuchs, J. Org. Chem., 1944, 09, 359 CrossRef CAS.
- R. Taniki, K. Matsumoto and R. Hagiwara, Chem. Lett., 2013, 42, 1469 CrossRef CAS.
- F. F. Blicke and E. B. Hotelling, J. Am. Chem. Soc., 1954, 76, 5099 CrossRef CAS.
- B. M. Lowe and H. M. Rendall, Trans. Faraday Soc., 1971, 67, 2318 RSC.
- B. M. Lowe and H. M. Rendall, J. Chem. Soc., Faraday Trans., 1972, 68, 2191 RSC.
- M. Ue, K. Ida and S. Mori, J. Electrochem. Soc., 1994, 141, 2989 CrossRef CAS.
- A. J. Ward, A. F. Masters and T. Maschmeyer, RSC Adv., 2014, 4, 23327 RSC.
- J. Golding, N. Hamid, D. R. MacFarlane, M. Forsyth, C. Forsyth, C. Collins and J. Huang, Chem. Mater., 2001, 13, 558 CrossRef CAS.
- K. Matsumoto, U. Harinaga, R. Tanaka, A. Koyama, R. Hagiwara and K. Tsunashima, Phys. Chem. Chem. Phys., 2014, 16, 23616 RSC.
- L. Jin, K. M. Nairn, C. M. Forsyth, A. J. Seeber, D. R. MacFarlane, P. C. Howlett, M. Forsyth and J. M. Pringle, J. Am. Chem. Soc., 2012, 134, 9688 CrossRef CAS PubMed.
- L. Jin, K. M. Nairn, C. D. Ling, H. Zhu, L. A. O’Dell, J. Li, F. Chen, A. F. Pavan, L. A. Madsen, P. C. Howlett, D. R. MacFarlane, M. Forsyth and J. M. Pringle, J. Phys. Chem. B, 2017, 121, 5439 CrossRef CAS PubMed.
- D. R. MacFarlane, J. Golding, S. Forsyth, M. Forsyth and G. B. Deacon, Chem. Commun., 2001, 1430 RSC.
- C. Wolff, S. Jeong, E. Paillard, A. Balducci and S. Passerini, J. Power Sources, 2015, 293, 65 CrossRef CAS.
- N. Cai, J. Zhang, D. Zhou, Z. Yi, J. Guo and P. Wang, J. Phys. Chem. C, 2009, 113, 4215 CAS.
- D. Al-Masri, R. Yunis, A. F. Hollenkamp and J. M. Pringle, Chem. Commun., 2018, 54, 3660 RSC.
- S. J. Pas, J. Huang, M. Forsyth, D. R. MacFarlane and A. J. Hill, J. Chem. Phys., 2005, 122, 064704 CrossRef PubMed.
- P. C. Howlett, F. Ponzio, J. Fang, T. Lin, L. Jin, N. Iranipour and J. Efthimiadis, Phys. Chem. Chem. Phys., 2013, 15, 13784 RSC.
- K. Paduszyński and U. Domańska, J. Chem. Inf. Model., 2014, 54, 1311 CrossRef PubMed.
- P. Walden, Z. Phys. Chem., Stoechiom. Verwandtschaftsl., 1906, 55, 207 CAS.
- W. Xu, E. I. Cooper and C. A. Angell, J. Phys. Chem. B, 2003, 107, 6170 CrossRef CAS.
- D. R. MacFarlane, M. Forsyth, E. I. Izgorodina, A. P. Abbott, G. Annat and K. Fraser, Phys. Chem. Chem. Phys., 2009, 11, 4962 RSC.
- K. Ueno, H. Tokuda and M. Watanabe, Phys. Chem. Chem. Phys., 2010, 12, 1649 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00016f |
| This journal is © the Partner Organisations 2018 |