
A strategy for the molecular design of aggregation-induced emission units further modified by substituents†
Zhe
Peng‡
 ,
Yingchun
Ji‡
,
Zihan
Huang
,
Bin
Tong
*,
Jianbing
Shi
and
Yuping
Dong
*
,
Yingchun
Ji‡
,
Zihan
Huang
,
Bin
Tong
*,
Jianbing
Shi
and
Yuping
Dong
*
Beijing Key Laboratory of Construction Tailorable Advanced Functional Materials and Green Applications, College of Materials Science and Engineering, Beijing Institute of Technology, 5 South Zhongguancun Street, Beijing, 100081, China. E-mail: tongbin@bit.edu.cn; chdongyp@bit.edu.cn; Tel: +86-10-6891-7390
First published on 10th April 2018
Abstract
Aggregation-induced emission (AIE) molecules with strong luminescence in aggregated states have attracted persistent attention in recent years. The development of new structures of AIE units and their further modification with functional groups to satisfy more specialized applications are important research fields. However, studies on the molecular design associated with the functional modification of AIE units have not been reported to date. Herein, we designed and synthesized 13 aryl-substituted pyrrolo[3,2-b]pyrrole derivatives. Among these compounds, DPP-1CN, DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP with electron-withdrawing groups on the phenyl groups at the 1,4-positions and electron-donating groups on the phenyl groups at the 2,5-positions of pyrrolo[3,2-b]pyrrole core showed AIE characteristics, whereas others showed aggregation-caused quenching (ACQ) characteristics. The absorption and photoluminescence (PL) emission spectra indicated that the AIE compounds exhibited weak intramolecular charge transfer (ICT) absorption and possessed large Stokes shifts, whereas the ACQ derivatives showed obvious ICT absorption. Density functional theory (DFT) calculation results suggested that the HOMOs and LUMOs of the four AIE compounds were spatially isolated that weakened the twisted intramolecular charge transfer (TICT) effect and minimized fluorescence reabsorption in the aggregated states. Single-crystal analysis also confirmed that AIE properties could be realized by the suppression of both the TICT effect and the close π⋯π interactions in the aggregated state. These results are beneficial for understanding the relationship between molecular structure and AIE properties. The resulting structural information provides the basis for the future rationalization of functional modification of the AIE materials.
Introduction
The aggregation-induced emission (AIE) phenomenon has attracted significant attention since the first AIE molecule was reported by the Tang's group.1 These AIE molecules overcome the defect of non-luminescence or weak emission in traditional aggregation-caused quenching (ACQ) materials in aggregated states, which are especially appealing and practically applied in optoelectronic devices,2 luminescent sensors,3 biomedical imaging,4 and smart materials.5 These applications benefit from the molecular design and the exploration of novel AIE units, in particular those with controllable structures and properties.6Many building blocks, such as hexaphenylsilole,7 tetraphenylethene (TPE),8 distyrylanthracene,9 and aryl-substituted pyrrole,10 of AIE luminogens (AIEgens) have been broadly explored and systematically studied. To date, the primarily accepted AIE mechanism is based on the restriction of intramolecular motion (RIM) including the restriction of intramolecular rotations and intramolecular vibrations.11 Despite promising early AIE units, the design and preparation of new AIEgens remained a big challenge. Moreover, two general approaches were adopted to achieve AIEgens: (i) the development of new AIE moieties such as BODIPY compounds,12 Schiff bases,13 and excited-state intramolecular proton transfer compounds14 and (ii) the conversion of ACQ luminophores (ACQphores) to AIEgens. Incorporation of an AIE archetype into an ACQphore-core is an alternative synthetic methodology for converting ACQphores to AIEgens.15
Recently, researchers also modified ACQphores with AIE-inactive functional groups (aldehyde or alkyl group) to simply achieve AIEgens. Konishi et al.16 reported that highly twisted N,N-dialkylamines and dipiperidyls were introduced into anthracene or naphthalene at the para position as AIEgens by prohibiting non-adiabatic transition and internal conversion. Li et al.17 reported the ACQ-to-AIE conversion by changing the flexible chains without disturbing the original π system of naphthalene diimide derivatives. Wong et al.18 demonstrated that terthiophene could present AIE characteristics upon the introduction of terminal aldehyde groups. Our previous study19 showed that aldehyde-decorated anthracene performed ACQ-to-AIE conversion because of steric effects and intermolecular H-bonding interactions of the aldehyde groups. However, systematic investigation of the AIE behaviors by structural modification with different substituents is rarely reported.20
Pyrrole is an important five-membered nitrogen-containing heterocycle with a six π-electron aromatic system. Because of the abundant electron density and high aromaticity, it is a popular building block and shows widespread application in dye-sensitized solar cells21 and second-order nonlinear optical materials.22 Pyrrole is also used to build luminescent materials for chemical23 and biological24 sensors on the basis of facile structural modification. Aryl-substituted pyrrole derivatives initially developed by our group showed satisfactory results in chemical sensing, ion detection, and bioimaging.10
From the past research efforts, we have learned that enlargement of the conjugation and coplanarity of the pyrrole core to a pyrrolo[3,2-b]pyrrole core also leads to the formation of good candidates for AIEgens. Our previous results also indicated that aryl-substituted pyrrolo[3,2-b]pyrrole derivatives showed obvious AIE characteristics with versatile properties and applications such as in chloroform detection, polymorphism, acid response, and temperature monitoring.25 However, there is absence of systematic investigation of the effects of categories and positions of the substituents on the AIE characteristics. Herein, we designed and synthesized a series of aryl-substituted pyrrolo[3,2-b]pyrrole-based derivatives with different electron-withdrawing or electron-donating substituents at different positions. The photoluminescence (PL) properties of these compounds were characterized, and then, the AIE natures of these compounds were clarified by theoretical calculations and single-crystal analysis. These results could be beneficial for providing a strategy for the molecular design of AIE building blocks for future functional modification to satisfy more specialized applications.
Results and discussion
The synthetic routes and molecular structures of the 13 compounds are illustrated in Scheme 1. All compounds were purified by silica gel column chromatography, and their structures were verified by 1H and 13C NMR spectroscopies and MALDI-TOF-MS analysis (13C NMR of DPP-2CN and DPP-12CN were undesirable due to poor solubility at high concentrations). DPP-2CN and DPP-12CN were moderately soluble in CHCl3 and THF, whereas the other 11 compounds showed good solubility in both CHCl3 and THF, but poor solubility in H2O and hexane. All compounds, except DPP-2MF, DPP-2IP, DPP-1MF-2Me, and DPP-1CN-2MF, could be crystallized via slow evaporation of DMF–hexane or CHCl3–hexane mixtures under ambient conditions. These four compounds failed to produce good quality crystals and discernable X-ray diffraction. The detailed synthesis and characterization of the 13 compounds are described in the ESI† (S2).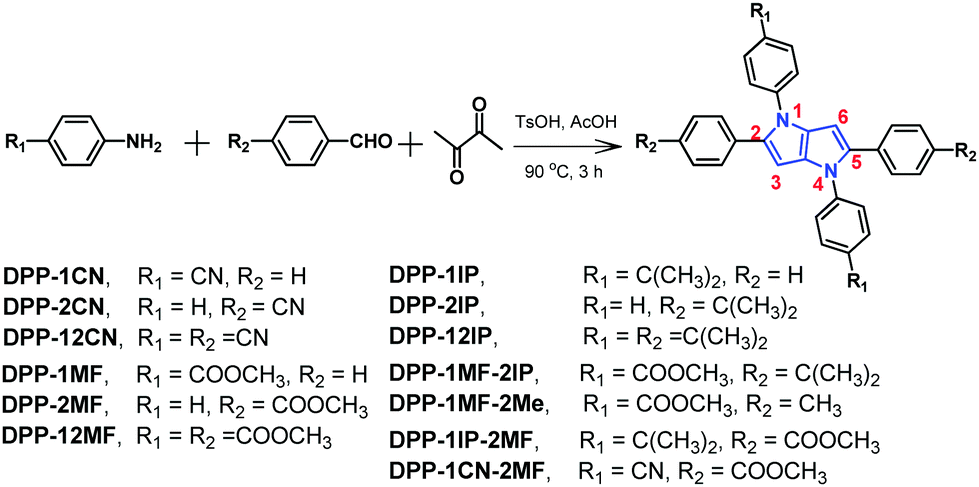 | ||
| Scheme 1 The synthetic routes and molecular structures of the 13 aryl-substituted pyrrolo[3,2-b]pyrrole-based derivatives. | ||
Photophysical properties
The optical properties of these compounds were studied by UV-vis absorption and PL emission spectroscopy. Their normalized absorption spectra in THF solutions (10−5 M) are shown in Fig. 1a. Their basic spectroscopic parameters are summarized in Table S1 (ESI†).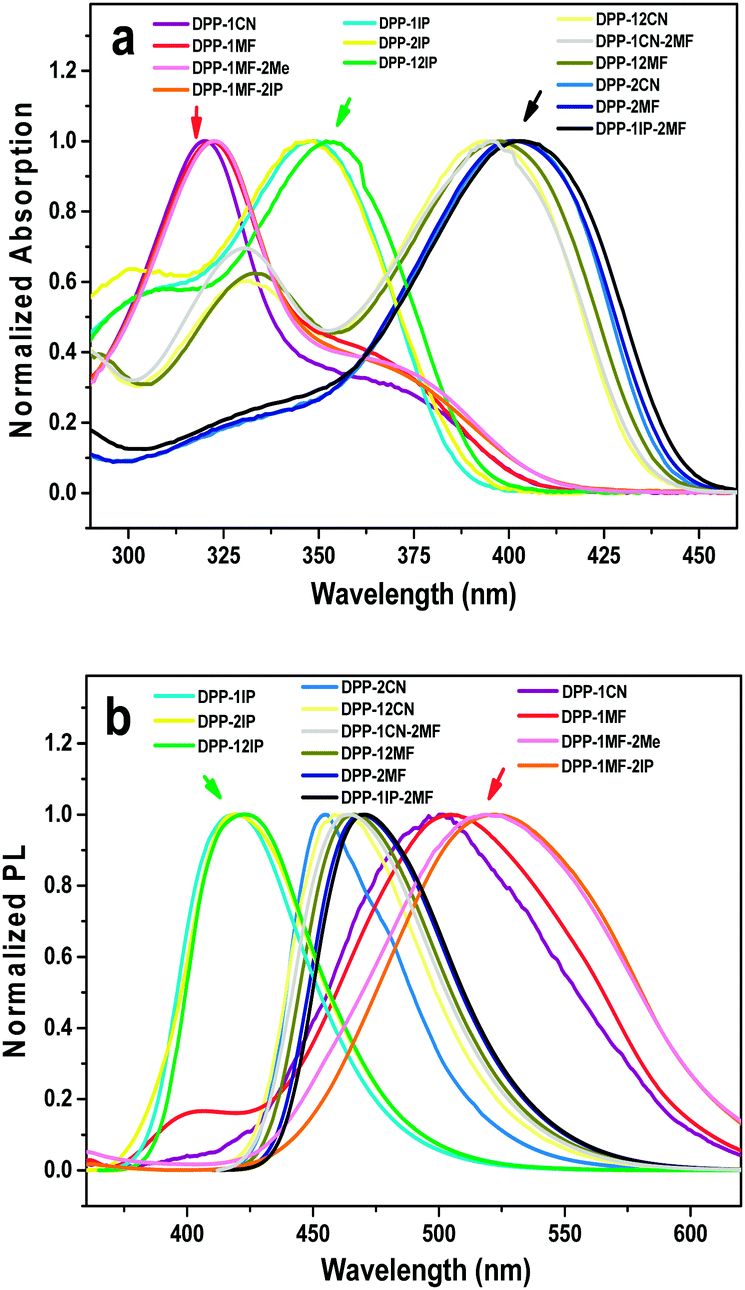 | ||
| Fig. 1 The normalized absorption (a) and emission (b) spectra of 13 aryl-substituted pyrrolo[3,2-b]pyrrole-based derivatives in THF (10−5 M, excitation wavelength (λex): 320 nm for DPP-1CN, DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP; 350 nm for DPP-1IP, DPP-2IP, and DPP-12IP; and 400 nm for DPP-12CN, DPP-1CN-2MF, DPP-12MF, DPP-2CN, DPP-2MF, and DPP-1IP-2MF). | ||
As shown in Fig. 1, three classes of absorption bands were observed between 320 and 410 nm with the molar extinction coefficients of 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–85000 M−1 cm−1. DPP-1CN, DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP, with the pyrrolo[3,2-b]pyrrole core as the donor and the cyan groups or ester groups as acceptors, showed almost similar absorption bands with peaks/shoulder peaks at 321/362, 322/359, 322/363, and 323/363 nm, respectively. The short wavelength bands were attributed to π–π* transitions, and the shoulder peaks should be attributed to intramolecular charge transfer (ICT) transition between the cyan/ester groups and the pyrrolo[3,2-b]pyrrole core.26 The redder shoulder bands were weaker than the π–π* absorption bands due to the significantly twisted structures (illustrated in the following single-crystal analysis). Both DPP-1MF-2IP and DPP-1MF-2Me showed almost the same absorption spectra as DPP-1MF; this indicated that electron-donating groups (EDGs) on the phenyl groups at 2,5-positions contributed negligibly to UV-vis absorption when the electron-withdrawing groups (EWGs) were located on the phenyl groups at the 1,4-positions of the pyrrolo[3,2-b]pyrrole core.
000–85000 M−1 cm−1. DPP-1CN, DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP, with the pyrrolo[3,2-b]pyrrole core as the donor and the cyan groups or ester groups as acceptors, showed almost similar absorption bands with peaks/shoulder peaks at 321/362, 322/359, 322/363, and 323/363 nm, respectively. The short wavelength bands were attributed to π–π* transitions, and the shoulder peaks should be attributed to intramolecular charge transfer (ICT) transition between the cyan/ester groups and the pyrrolo[3,2-b]pyrrole core.26 The redder shoulder bands were weaker than the π–π* absorption bands due to the significantly twisted structures (illustrated in the following single-crystal analysis). Both DPP-1MF-2IP and DPP-1MF-2Me showed almost the same absorption spectra as DPP-1MF; this indicated that electron-donating groups (EDGs) on the phenyl groups at 2,5-positions contributed negligibly to UV-vis absorption when the electron-withdrawing groups (EWGs) were located on the phenyl groups at the 1,4-positions of the pyrrolo[3,2-b]pyrrole core.
DPP-1IP, DPP-2IP, and DPP-12IP with isopropyl as EDGs on phenyl groups at the 1,4/2,5/1,2,4,5-positions of the pyrrolo[3,2-b]pyrrole core, respectively, exhibited intense absorptions around 350 nm, which were attributed to the π–π* transition of the pyrrolo[3,2-b]pyrrole backbone. The UV-vis absorption bands of DPP-1IP, DPP-2IP, and DPP-12IP were quite similar to those reported for 1,2,4,5-tetraphenyl-1,4-dihydropyrrolo[3,2-b]pyrrole25a due to the absence of ICT. This result suggested that the electron-donating substituents had little effect on the UV-vis absorption properties of the pyrrolo[3,2-b]pyrrole-based derivatives.
Moreover, three 2,5-substituted compounds, i.e.DPP-2CN, DPP-2MF, and DPP-1IP-2MF, exhibited weak absorption bands at 330 nm, which were ascribed to π–π* transitions. The strong absorption peaks at 400 nm were assigned to the distinct ICT absorption bands. Similarly, DPP-12CN, DPP-12MF, and DPP-1CN-2MF with quadrupole-like donor–acceptor (D–A) molecular structures showed two absorption bands at ∼330 nm and ∼395 nm, which were also attributed to the π–π* and ICT transitions, respectively. Since all absorption spectra of the DPP-2MF series are quite similar, the optical properties of the DPP-2MF series with different substituents were not correlated with the structural changes. All these results confirmed that the optical property of the D–A type aryl-substituted pyrrolo[3,2-b]pyrrole-based derivatives could be tuned with EWGs in different positions.
The emission spectra of 13 compounds were obtained in THF (10−5 M), as shown in Fig. 1b. The emission spectra of the DPP series showed an emission peak at 420–520 nm with different Stokes shifts from 2900 to 11800 cm−1. The PL emission peaks of DPP-1CN, DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP were at 500, 504, 519, and 522 nm, respectively, which exhibited the largest Stokes shifts, larger than 11100 cm−1 (Δλ > 180 nm). The PL emission peaks of DPP-1IP, DPP-2IP, and DPP-12IP were around 420 nm with minor Stokes shifts (Δλ ≈ 70 nm). The PL emissions of DPP-2CN, DPP-2MF, DPP-1IP-2MF, DPP-12CN, DPP-12MF, and DPP-1CN-2MF peaked between 455 and 471 nm with a moderate Stokes shift (Δλ ≤ 75 nm).
Aggregation-induced emission characteristics
As deionized water was a poor solvent for all compounds, the fluorescence spectra in THF/H2O mixtures were obtained to investigate the fluorescence properties in the aggregated states. For DPP-1CN, a weak emission peak at 507 nm was observed in a dilute THF solution (Fig. 2a). In the THF/water mixture, its emission first underwent quenching with water fraction (fw) between 10 and 60%. This phenomenon probably originated from the twisted internal charge transfer (TICT) effect,27 which was verified by the red-shifted and decreased emission of DPP-1CN with an increase in solvent polarity. Then, the fluorescence intensity increased with an increase in water fraction (fw > 60%), and ∼2.2-fold emission enhancement was observed with fw = 99% (Fig. 2b). The Tyndall effect of DPP-1CN in THF/water (fw = 99%) demonstrated the formation of aggregates (inset of Fig. 2b). The quantum yields (QYs) in pure THF and in solid powder were 5.04% and 13.6%, respectively. Clearly, the PL and QY results demonstrated that DPP-1CN exhibited the AIE characteristic.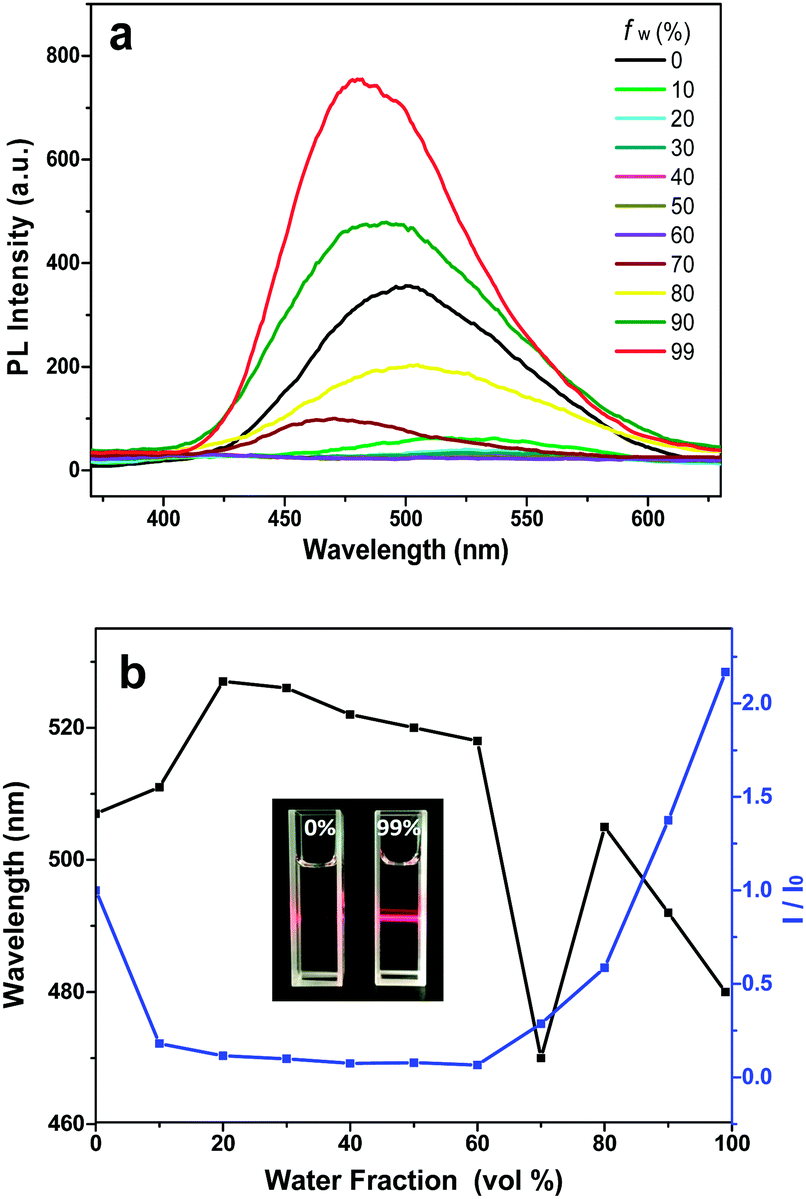 | ||
| Fig. 2 (a) Fluorescence spectra of DPP-1CN in THF/H2O with different water fractions. (b) A plot of wavelength and ratio of maximum fluorescence intensity of DPP-1CNvs. fraction of water (inset: the Tyndall effect of fw = 99%). (Concentration: 10−5 M, λex = 320 nm.) | ||
Interestingly, the PL emission of DPP-1CN with fw = 70% was unstable. The PL intensity of this system gradually increased with increasing time and achieved 12-fold enhancement in 30 min (Fig. S1a, ESI†). This phenomenon was not monitored in other systems with different water fractions in the DPP-1CN solution. The particle sizes of DPP-1CN in 70% water at 2 and 30 min were measured by dynamic light scattering. The average particle sizes (shown in Fig. S1b and c, ESI†) were 113 nm at 2 min and 131.8 nm at 30 min, which showed that the change in PL intensity originated from variation of the aggregates formed.
The PL emission spectra of DPP-2CN—an isomer of DPP-1CN—in THF/H2O mixtures are shown in Fig. S2a and b (ESI†). The PL emission of DPP-2CN remained constant at a low water content (fw < 60%) and decreased rapidly at fw > 70%. The ϕPL of DPP-2CN in the THF solution and solid powder were 95.59% and 24.7%, respectively. Both the PL emission spectra and ϕPL demonstrated that DPP-2CN showed a typical ACQ characteristic. A similar optical property was observed from DPP-12CN (Fig. S2c and d, ESI†). These results indicate that AIE/ACQ properties can be tuned by simple structural modification without changing the emissive unit.
The emission spectra of DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP in THF and THF/H2O mixtures are shown in Fig. 3a, b, and c, respectively. DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP exhibited similar cyan fluorescence with maximum wavelengths at 505, 519, and 522 nm, respectively. When the water fraction increased, the emission dropped slightly. This phenomenon is typical of many fluorophores with TICT properties, where the emission is red-shifted and weakened in high polarity solvents. Dramatic increases and blue shift in emission were observed when the water fraction was over 80% for DPP-1MF, 70% for DPP-1MF-2Me, and 60% for DPP-1MF-2IP, indicating that all three compounds showed a typical AIE characteristic (Fig. 3d). The aggregation of DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP was confirmed in a high water fraction solution, as shown in Fig. S3 (ESI†). Due to the increase in steric hindrance from DPP-1MF, DPP-1MF-2Me to DPP-1MF-2IP, the AIE became more obvious (Fig. 3d). The maximum PL emission of DPP-1MF-2IP reduced slightly at high water fractions (fw > 80%), and the reason could be that the luminogen molecules might quickly agglomerate in a random manner to form less emissive particles. The maximal emission enhancements of DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP were higher than that of DPP-1CN; this indicated that the methyl ester group as an EWG at the para position of the phenyl group at the 1,4-positions of the pyrrolo[3,2-b]pyrrole core induced a stronger AIE effect than the cyan group.
 | ||
| Fig. 3 Fluorescence spectra of DPP-1MF (a), DPP-1MF-2Me (b), and DPP-1MF-2IP (c) in THF/water with different water fractions; (d) a plot of the ratio of maximum fluorescence intensity of DPP-1MF/DPP-1MF-2Me/DPP-1MF-2IPvs. fraction of water. (Concentration: 10−5 M, λex = 320 nm.) | ||
The PL spectra of DPP-2MF, DPP-12MF, DPP-1CN-2MF, and DPP-1IP-2MF in THF and THF/H2O mixtures are obtained, as shown in Fig. S4 (ESI†). DPP-2MF, DPP-12MF, DPP-1CN-2MF, and DPP-1IP-2MF in pure THF showed strong blue-green fluorescence with maximum wavelengths at 469, 466, 464, and 470 nm, respectively. Upon continuous addition of water as a poor solvent (fw < 60% for DPP-2MF and DPP-12MF and fw < 70% for DPP-1CN-2MF and DPP-1IP-2MF), their PL emission intensities decreased, and emission wavelength maxima red-shifted due to the increase of mixed solvent polarity (i.e., TICT nature for D–A type molecules).
Slight emission enhancements were observed when the water fractions were 60% ≤ fw ≤ 80% for DPP-2MF and DPP-12MF and 70% ≤ fw ≤ 80% for DPP-1CN-2MF and DPP-1IP-2MF, indicating that these compounds were weak AIE but predominant TICT. When fw was over 80%, their PL were quenched and bathochromically shifted. The PL intensities in aggregated states were lower than the initial PL intensities; this showed that the ACQ effect played a dominant role in their photophysical processes.
In addition to those of EWGs as substituents, the emission spectra of pyrrolo[3,2-b]pyrrole-based derivatives with EDGs were obtained. As shown in Fig. S5 (ESI†), the PL intensities of DPP-1IP, DPP-2IP, and DPP-12IP dropped dramatically when the water fraction was higher than 60%, exhibiting typical ACQ characteristics.
From the UV and emission results, we can conclude that the AIE-active compounds DPP-1CN, DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP show intense π–π* absorption and weak ICT transition absorption with large Stokes shifts (Δλ > 180 nm). Generally, Stokes shifts over 80 nm are desirable for minimizing molecular reabsorption,28i.e., DPP-1CN, DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP with large Stokes shift can suppress the self-absorption effect and are favorable for AIE. Moreover, reduction of the non-radiative pathway and restriction of the formation of the TICT state can enhance emission in the aggregated state.
DFT calculations
To gain a better understanding of the correlation between molecular structure and emission property of these aryl-substituted pyrrolo[3,2-b]pyrrole derivatives, DFT calculations were conducted using Gaussian 09 programs at the B3LYP/6-31G(d) level. DFT calculations were performed on the basis of the single-crystal structures of DPP-1CN, DPP-2CN, DPP-12CN, DPP-1MF, DPP-12MF, DPP-1IP, DPP-12IP, DPP-MF-2IP, and DPP-1IP-2MF, and the molecular conformations of DPP-2MF, DPP-2IP, DPP-1MF-2Me, and DPP-1CN-2MF were optimized by the abovementioned program (Fig. 4). The highest occupied molecular orbitals (HOMOs) of all the compounds were localized on the pyrrolo[3,2-b]pyrrole core and phenyl groups at the 2,5-positions, which had a significant influence on the electronic structures and the optical properties.29 However, the distributions of the lowest unoccupied molecular orbitals (LUMOs) were different. The detailed discussion of the LUMOs is as follows.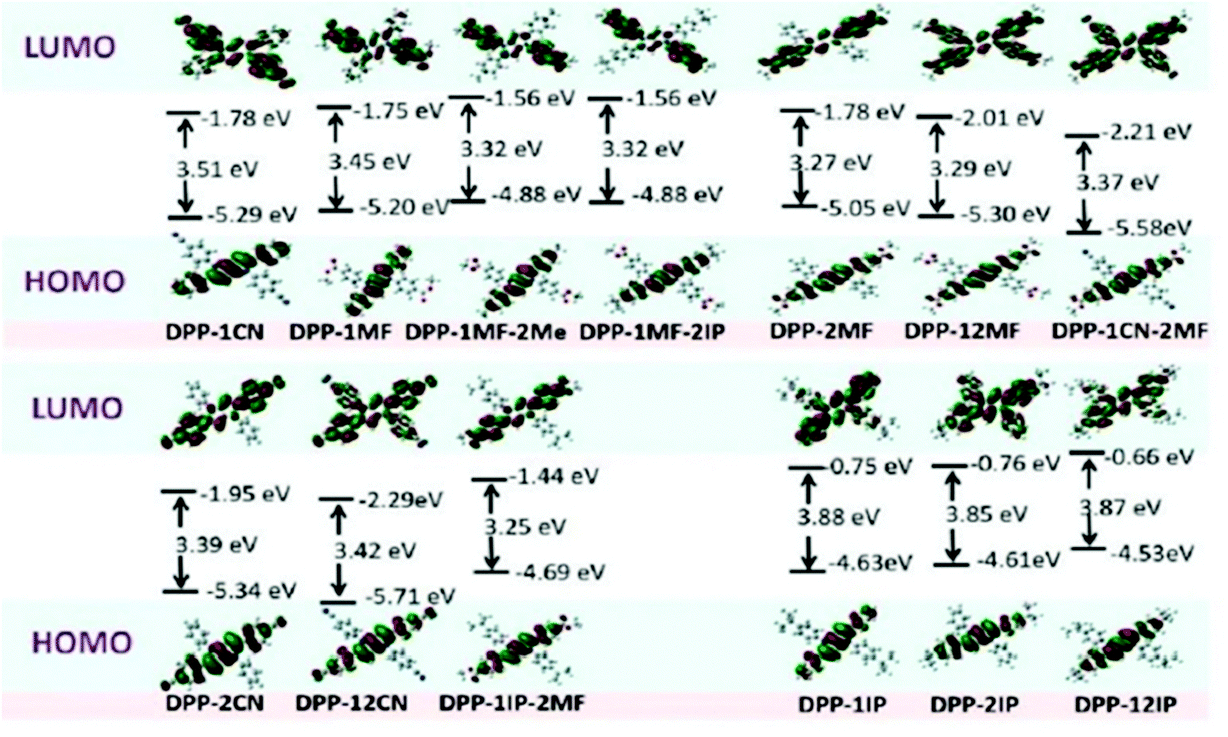 | ||
| Fig. 4 Molecular orbital and energy levels of 13 aryl-substituted pyrrolo[3,2-b]pyrrole-based derivatives, calculated at the B3LYP/6-31G level. | ||
First, the LUMOs of the four AIE-active compounds were mainly localized on the phenyl groups at the 1,4-position of the pyrrolo[3,2-b]pyrrole core (electron acceptor moiety). Their HOMOs and LUMOs were completely separated, resulting in weak ICT transition absorption bands at around 363 nm. Therefore, introduction of EWGs into phenyl groups at the 1,4-positions for the isolation of HOMO and LUMO is beneficial to the AIE property.17
Second, the LUMOs of DPP-2CN, DPP-2MF, and DPP-1IP-2MF were mainly located on the pyrrolo[3,2-b]pyrrole core as well as the phenyl groups at the 2,5-positions, which showed the highest overlap with the HOMOs. All these molecules showed intense ICT absorption bands at around 400 nm and exhibited ACQ effects because the electrons in the excited state could easily relax back to the ground state via non-radiative transition.
Third, the LUMOs of DPP-12CN, DPP-12MF, DPP-1CN-2MF, DPP-1IP, DPP-2IP, and DPP-12IP were mainly located on the pyrrolo[3,2-b]pyrrole core, and the four peripheral phenyl groups. Similar molecular HOMO–LUMO orbital distribution as in DPP-12CN, DPP-12MF, and DPP-1CN-2MF with quadrupole-like D–A molecular structures caused similar biabsorption bands at around 330 and 395 nm. DPP-1IP, DPP-2IP, and DPP-12IP without EWGs showed similar absorption bands.
DPP-1IP, DPP-2IP, and DPP-12IP showed high-lying HOMO and LUMO energy levels and large energy gaps (3.85–3.87 eV). The HOMO and LUMO energy levels of the other ten pyrrolo[3,2-b]pyrrole-based derivatives decreased upon introducing EWGs at different positions into the pyrrolo[3,2-b]pyrrole core. Compared with DPP-1IP, DPP-1IP-2MF showed a lower LUMO energy level, but equal HOMO energy. However, both the HOMO and LUMO energies of DPP-1MF-2IP were much lower than those of DPP-2IP due to the increase in the electron-accepting ability. Therefore, the HOMO and LUMO energies decreased gradually (DPP-2MF > DPP-1MF > DPP-1CN > DPP-2CN > DPP-12MF > DPP-1CN-2MF > DPP-12CN). The energy band values of the compounds DPP-2MF, DPP-1MF, DPP-1CN, DPP-2CN, DPP-12MF, DPP-1CN-2MF, DPP-12CN, DPP-1IP-2MF, DPP-1MF-2IP, and DPP-1MF-2Me were 3.27, 3.45, 3.51, 3.39, 3.29, 3.37, 3.42, 3.25, 3.32, and 3.32 eV, respectively (Fig. 4). The experimental results were consistent with these DFT calculations for DPP-1IP (ΔEg-cal (eV)/1240/λonsetabs (eV) − 3.88/3.19), DPP-12IP (3.87/3.17), DPP-1CN (3.51/3.06), DPP-1MF (3.45/3.05), DPP-1MF-2IP (3.32/3.03), DPP-12MF (3.29/2.83), DPP-2MF (3.27/2.80), and DPP-1IP-2MF (3.25/2.79), but not for DPP-2IP (3.85/3.21), DPP-1ME-2IP (3.32/3.00), DPP-12CN (3.42/2.87), DPP-1CN-2MF (3.37/2.84), and DPP-2CN (3.39/2.82).
Although the emission wavelengths of 13 pyrrolo[3,2-b]pyrrole-based derivatives in pure THF solution varied from 419 to 522 nm (Fig. 1b), the energy band gaps did not exactly correlate with the wavelength of fluorescence emission in solution (Table S1, ESI†) because the HOMO and LUMO were calculated based on the ground states rather than the excited states.30
Single-crystal structural analysis
The single-crystal analyses of DPP-1CN and DPP-2CN are shown in Fig. 5. Both DPP-1CN and DPP-2CN showed similar symmetrical structures and propeller-shaped conformations, but their dihedral angles between the phenyl and the pyrrolo[3,2-b]pyrrole core, packing patterns, and intermolecular interactions were quite different.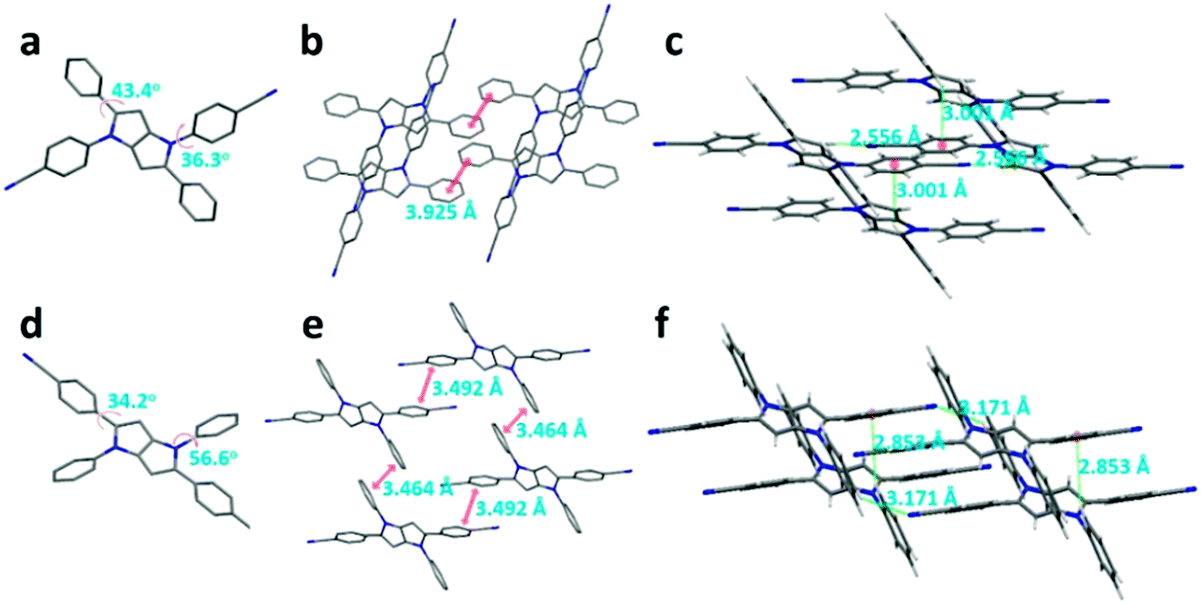 | ||
| Fig. 5 The crystal structure, packing patterns, and intermolecular interactions of DPP-1CN (a, b, and c) and DPP-2CN (d, e, and f), respectively. (Hydrogen omitted for clarity (a, b, d, and e).) | ||
The dihedral angle between the phenyl groups at the 1,4-position and the pyrrolo[3,2-b]pyrrole core (ψ1) of DPP-1CN was 36.3°, which was smaller than that of DPP-2CN. However, the dihedral angle between phenyl groups at the 2,5-position and the pyrrolo[3,2-b]pyrrole core (ψ2) of DPP-1CN was larger than that of DPP-2CN (Fig. 5a and d). Both DPP-1CN and DPP-2CN molecules showed a parallel-slipped stack. No obvious π⋯π interactions of DPP-1CN were observed. The two phenyl groups at the 2,5-positions of DPP-2CN showed effective π⋯π surface overlap with a distance of 3.492 Å, the same as the two phenyl groups at 1,4-positions with a distance of 3.464 Å. The CN⋯H and CH⋯π distances of intermolecular DPP-1CN are respectively 2.556 and 3.001 Å, and those of DPP-2CN are respectively 3.171 and 2.853 Å. Both multiple intermolecular CN⋯H and CH⋯π interactions restrict intramolecular rotation, and the lack of π⋯π interaction enhances the emission of DPP-1CN in the aggregated state. In contrast, the ACQ effect of DPP-2CN was caused by π⋯π interactions.
As shown in Fig. 5, ψ2 of DPP-2CN (34.2°) was smaller than that of DPP-1CN (43.4°). DPP-2CN showed higher planarity leading to longer absorption band than that in DPP-1CN. A similar trend was observed in the solid emission peaks.
The single-crystal structures of DPP-1MF and DPP-1MF-2IP are presented in Fig. 6. ψ1 and ψ2 of DPP-1MF were 33.6° and 55.2°, respectively. The DPP-1MF molecules adopted a zigzag packing pattern along the a axis, which was mainly dominated by intermolecular CH⋯π interaction with a distance of 3.256 Å. The distance between adjacent pyrrolo[3,2-b]pyrrole cores was 4.230 Å, indicating the lack of intermolecular π⋯π interactions. In addition, intermolecular CH⋯π interactions were observed with distances of 2.898 and 2.957 Å, and the distances of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H interactions were 2.709, 2.840, and 2.912 Å. The four different dihedral angles between peripheral phenyl groups and the central pyrrolo[3,2-b]pyrrole core of DPP-1MF-2IP were different, which should be ascribed to the steric hindrance of isopropyl on phenyl groups at the 2,5-positions. The planes of the pyrrolo[3,2-b]pyrrole core in DPP-1MF-2IP were aligned parallel to the interplane distances of 4.408 and 4.049 Å along the b axis in the crystal. Multiple intermolecular CH⋯π interactions were observed in DPP-1MF-2IP with distances of 3.093, 3.152, and 3.353 Å, and the distances of CO⋯H interactions were 2.633, 3.045, 3.223, 3.378, and 3.647 Å. These interactions were helpful to lock the DPP-1MF-2IP molecules and restrict the rotation in the aggregated state.
O⋯H interactions were 2.709, 2.840, and 2.912 Å. The four different dihedral angles between peripheral phenyl groups and the central pyrrolo[3,2-b]pyrrole core of DPP-1MF-2IP were different, which should be ascribed to the steric hindrance of isopropyl on phenyl groups at the 2,5-positions. The planes of the pyrrolo[3,2-b]pyrrole core in DPP-1MF-2IP were aligned parallel to the interplane distances of 4.408 and 4.049 Å along the b axis in the crystal. Multiple intermolecular CH⋯π interactions were observed in DPP-1MF-2IP with distances of 3.093, 3.152, and 3.353 Å, and the distances of CO⋯H interactions were 2.633, 3.045, 3.223, 3.378, and 3.647 Å. These interactions were helpful to lock the DPP-1MF-2IP molecules and restrict the rotation in the aggregated state.
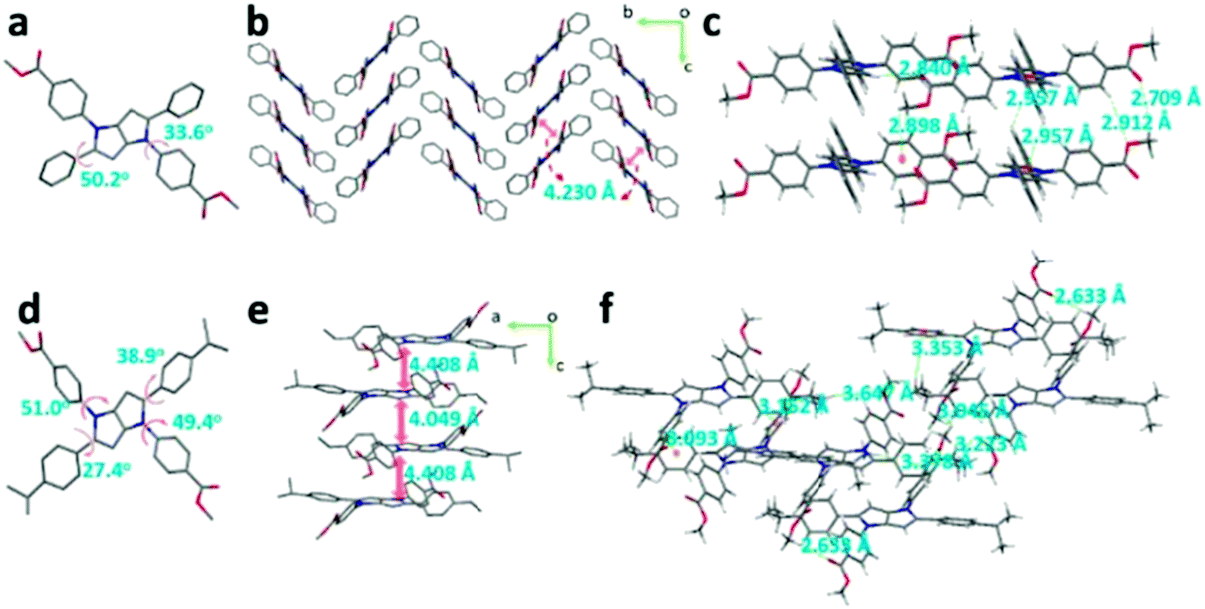 | ||
| Fig. 6 The crystal structure, packing patterns, and intermolecular interactions of DPP-1MF (a, b, and c) and DPP-1MF-2IP (d, e, and f), respectively. (Hydrogen omitted for clarity (a, b, d, and e).) | ||
These results indicated that the AIE-active DPP-1CN, DPP-1MF, and DPP-1MF-2IP adopted rigid molecular configurations to suppress non-radiative process and boosted emission in the condensed states. Therefore, as the amounts of intermolecular H-bonding and CH⋯π interactions for DPP-1CN, DPP-1MF, and DPP-1MF-2IP increased, their maximal enhancement of PL gradually increased.
It is worth noting that similarity of molecular structure can result in similar electronic structures (including HOMOs and LUMOs) among DPP-1CN, DPP-1MF, and DPP-1MF-2IP. For example, ψ2 decreased from DPP-1MF (50.2°) to DPP-1CN (43.4°) to DPP-1MF-2IP (38.9°/27.4°). The coplanarity of the molecular structures increased accordingly. Similar trends were observed for their absorption and solid emission bands.
The single-crystal structure of DPP-12MF with weak AIE characteristic showed no π⋯π interaction. DPP-12MF existed in two different conformations with different dihedral angles (Fig. S6a, ESI†). The planes of two different conformational pyrrolo[3,2-b]pyrrole cores in DPP-12MF were severally aligned parallel with interspersed distances of 8.852 and 8.944 Å in one column (Fig. S6b, ESI†). Multiple intermolecular CH⋯π interactions between the two different conformational molecules were also observed (Fig. S6c, ESI†), accounting for the AIE characteristic of DPP-12MF. However, the molecule eventually showed ACQ characteristic due to the dominant ICT effect.
The single-crystal structures of the ACQ-active molecules DPP-12CN, DPP-1IP-2MF, DPP-1IP, and DPP-12IP are presented in Fig. S7 (ESI†). DPP-12CN and DPP-2CN showed similar molecular packing. DPP-12CN showed π⋯π interaction between adjacent phenyl groups at the 2,5-position with a distance of 2.748 Å.
The ψ2 of DPP-1IP-2MF showed a low dihedral angle in all nine crystalline compounds with the angle of 23.7° indicating excellent planarity in the lateral molecular configuration. This high planarity also led to strong π⋯π interactions between adjacent phenyl groups at the 2,5-positions with a distance of 2.464 Å. π⋯π interactions were also observed in DPP-12CN, DPP-1IP, and DPP-1IP-2MF, which caused ACQ features in the condensed states. Interestingly, DPP-12IP without π⋯π interactions in crystal structures also show the ACQ characteristic. This was because DPP-12IP molecules adopted the malposed packing style with two columns along the a axis and distance of 7.743 Å. The distances of three adjacent molecules were 10.451 and 12.144 Å (Fig. S8, ESI†). This kind of packing style had enough spaces to allow the phenyl ring to freely rotate to consume the excited energy in the solid state.
Conclusions
In summary, a series of new propeller-like luminophores consisting of pyrrolo[3,2-b]pyrrole cores and peripheral rotatable phenyls with different electronic characteristic groups was synthesized. The effects of different electronic characteristic groups on the AIE/ACQ properties were investigated using UV-vis absorption spectra, PL emission spectra, DFT calculations, and analyses of single crystal structures. DPP-1CN, DPP-1MF, DPP-1MF-2Me, and DPP-1MF-2IP bearing strong EWGs on the phenyl groups at the 1,4-positions of the pyrrolo[3,2-b]pyrrole core showed obvious AIE characteristics. DPP-2CN, DPP-12CN, and DPP-1IP-2MF with strong EWGs on the phenyl groups at the 2,5-positions of the pyrrolo[3,2-b]pyrrole core and DPP-1IP, DPP-2IP and DPP-12IP with EDGs on the phenyl groups at the 1,4- or 2,5-positions of the pyrrolo[3,2-b]pyrrole core showed ACQ characteristics.The absorption and PL emission spectra of the 13 compounds could be evidently divided into three groups. The AIE compounds exhibited weak ICT absorption and possessed the largest Stokes shifts, whereas the ACQ derivatives showed obvious ICT absorption. In addition, the HOMOs and LUMOs of the four AIE compounds are spatial isolated, which suppressed both the TICT effect and π⋯π stacking interactions in the aggregated state. These results not only are beneficial for the molecular design of the aryl-substituted pyrrolo[3,2-b]pyrrole, but also provide basic knowledge for further functional modification of other kinds of AIE units to obtain new AIE materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the support provided by the National Basic Research Program of China (973 Program: 2013CB834704) and the National Natural Scientific Foundation of China (No. 51673024, 51328302, 21404010).Notes and references
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (b) F. Hu, G. X. Zhang, C. Zhan, W. Zhang, Y. L. Yan, Y. S. Zhao, H. B. Fu and D. Q. Zhang, Small, 2015, 11, 1335–1344 CrossRef CAS PubMed.
- (a) X. Z. Yan, M. Wang, T. R. Cook, M. M. Zhang, M. L. Saha, Z. X. Zhou, X. P. Li, F. H. Huang and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 4580–4588 CrossRef CAS PubMed; (b) S. Samanta, U. Manna, T. Ray and G. Das, Dalton Trans., 2015, 44, 18902–18910 RSC.
- (a) J. Y. Xiang, X. L. Cai, X. D. Lou, G. X. Feng, X. H. Min, W. W. Lou, B. R. He, C. C. Goh, L. G. Ng, J. Zhou, Z. J. Zhao, B. Liu and B. Z. Tang, ACS Appl. Mater. Interfaces, 2015, 7, 14965–14974 CrossRef CAS PubMed; (b) Z. F. Chang, L. M. Jing, B. Chen, M. S. Zhang, X. Cai, J. J. Liu, Y. C. Ye, X. D. Lou, Z. J. Zhao, B. Liu, J. L. Wang and B. Z. Tang, Chem. Sci., 2016, 7, 4527–4536 RSC; (c) Y. L. Wang, M. Chen, N. Alifu, S. W. Li, W. Qin, A. J. Qin, B. Z. Tang and J. Qian, ACS Nano, 2017, 11, 10452–10461 CrossRef CAS PubMed.
- (a) C. Y. Y. Yu, R. T. K. Kwok, J. Mei, Y. Hong, S. Chen, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2014, 50, 8134–8136 RSC; (b) Y. X. Guo, S. Z. Gu, X. Feng, J. N. Wang, H. W. Li, T. Y. Han, Y. P. Dong, X. Jiang, T. D. James and B. Wang, Chem. Sci., 2014, 5, 4388–4393 RSC.
- (a) Z. J. Zhao, B. R. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC; (b) J. Yang, J. Huang, Q. Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 2663–2684 RSC.
- (a) G. N. Zhao, B. Tang, Y. Q. Dong, W. H. Xie and B. Z. Tang, J. Mater. Chem. B, 2014, 2, 5093–5099 RSC; (b) S. Xue, L. M. Meng, R. S. Wen, L. Shi, J. W. Lam, Z. Y. Tang, B. S. Li and B. Z. Tang, RSC Adv., 2017, 7, 24841–24847 RSC.
- (a) C. F. A. Gomez-Duran, R. R. Hu, G. X. Feng, T. Z. Li, F. Bu, M. Arseneault, B. Liu, E. Peña-Cabrera and B. Z. Tang, ACS Appl. Mater. Interfaces, 2015, 7, 15168–15176 CrossRef CAS PubMed; (b) S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. L. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed; (c) N. Sinha, L. Stegemann, T. T. Y. Tan, N. L. Doltsinis, C. A. Strassert and F. EkkehardtHahn, Angew. Chem., Int. Ed., 2017, 56, 2785–2789 CrossRef CAS PubMed.
- (a) J. T. He, B. Xu, F. P. Chen, H. J. Xia, K. P. Li, L. Ye and W. J. Tian, J. Phys. Chem. C, 2009, 113, 9892–9899 CrossRef CAS; (b) Y. J. Dong, B. Xu, J. B. Zhang, X. Tan, L. J. Wang, J. L. Chen, H. G. Lv, S. P. Wen, B. Li, L. Ye, B. Zou and W. J. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS PubMed; (c) J. L. Chen, S. Q. Ma, J. B. Zhang, B. Li, B. Xu and W. J. Tian, ACS Photonics, 2015, 2, 313–318 CrossRef CAS; (d) J. B. Zhang, S. Q. Ma, H. H. Fang, B. Xu, H. B. Sun, I. Chan and W. J. Tian, Mater. Chem. Front., 2017, 1, 1422–1429 RSC.
- (a) X. Feng, B. Tong, J. B. Shen, J. B. Shi, T. Y. Han, L. Chen, J. G. Zhi, P. Lu, Y. G. Ma and Y. P. Dong, J. Phys. Chem. B, 2010, 114, 16731–16736 CrossRef CAS PubMed; (b) T. Y. Han, J. W. Y. Lam, N. Zhao, M. Gao, Z. Y. Yang, E. G. Zhao, Y. P. Dong and B. Z. Tang, Chem. Commun., 2013, 49, 4848–4850 RSC; (c) W. Y. Li, D. D. Chen, H. Wang, S. S. Luo, L. C. Dong, Y. H. Zhang, J. B. Shi, B. Tong and Y. P. Dong, ACS Appl. Mater. Interfaces, 2015, 7, 26094–26100 CrossRef CAS PubMed; (d) G. G. Liu, D. D. Chen, L. W. Kong, J. B. Shi, B. Tong, J. G. Zhi, X. Feng and Y. P. Dong, Chem. Commun., 2015, 51, 8555–8558 RSC.
- J. Mei, Y. N. Hong, J. W. Y. Lam, A. J. Qin, Y. H. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- (a) X. Q. Wang, Q. S. Liu, F. Qi, L. Li, H. D. Yu, Z. P. Liu and W. Huang, Dalton Trans., 2016, 45, 17274–17280 RSC; (b) Y. Ooyama, M. Hato, T. Enoki, S. Aoyama, K. Furue, N. Tsunoji and J. Hshita, New J. Chem., 2016, 40, 7278–7281 RSC.
- (a) T. Han, X. G. Gu, J. W. Y. Lam, A. C. S. Leung, R. T. K. Kwok, T. Y. Han, B. Tong, J. B. Shi, Y. P. Dong and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 10430–10434 RSC; (b) X. P. Gan, G. J. Liu, M. J. Chu, W. G. Xi, Z. L. Ren, X. L. Zhang, Y. P. Tian and H. P. Zhou, Org. Biomol. Chem., 2017, 15, 256–264 RSC; (c) Q. Feng, Y. Y. Li, L. L. Wang, C. Li, J. M. Wang, Y. Y. Liu, K. Li and H. W. Hou, Chem. Commun., 2016, 52, 3123–3126 RSC.
- (a) S. Samanta, U. Manna and G. Das, New J. Chem., 2017, 41, 1064–1072 RSC; (b) P. Singh, H. Singh, R. Sharma, G. Bhargava and S. Kumar, J. Mater. Chem. C, 2016, 4, 11180–11189 RSC; (c) P. S. Zhang, X. Z. Nie, M. Gao, F. Zeng, A. J. Qin, S. Z. Wu and B. Z. Tang, Mater. Chem. Front., 2017, 1, 838–845 RSC.
- (a) Z. J. Zhao, P. Lu, J. W. Y. Lam, Z. M. Wang, C. Y. K. Chan, H. H. Y. Sung, I. D. Williams, Y. G. Ma and B. Z. Tang, Chem. Sci., 2011, 2, 672–675 RSC; (b) Q. L. Zhao, K. Li, S. J. Chen, A. J. Qin, D. Ding, S. Zhang, Y. Liu, B. Liu, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 15128–15135 RSC.
- (a) S. Sasaki, S. Suzuki, W. M. C. Sameera, K. Igawa, K. Morokuma and G.-i. Konishi, J. Am. Chem. Soc., 2016, 138, 8194–8206 CrossRef CAS PubMed; (b) S. Sasaki, K. Igawa and G.-i. Konishi, J. Mater. Chem. C, 2015, 3, 5940–5950 RSC.
- L. Y. Zong, Y. J. Xie, C. Wang, J. R. Li, Q. Q. Li and Z. Li, Chem. Commun., 2016, 52, 11496–11499 RSC.
- J. Cheng, X. Z. Liang, Y. X. Cao, K. P. Guo and W. Y. Wong, Tetrahedron, 2015, 71, 5634–5639 CrossRef CAS.
- Z. Peng, Z. Wang, B. Tong, Y. C. Ji, J. B. Shi, J. G. Zhi and Y. P. Dong, Chin. J. Chem., 2016, 34, 1071–1075 CrossRef CAS.
- B. Chen, H. Zhang, W. W. Luo, H. Nie, R. R. Hu, A. J. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 960–968 RSC.
- (a) Q. Q. Li, J. Shi, H. Y. Li, S. Li, C. Zhong, F. L. Guo, M. Peng, J. L. Hua, J. G. Qin and Z. Li, J. Mater. Chem., 2012, 22, 6689–6696 RSC; (b) H. Y. Li, Y. Q. Hou, Y. Z. Yang, R. L. Tang, J. N. Chen, H. Wang, H. W. Han, T. Y. Peng, Q. Q. Li and Z. Li, ACS Appl. Mater. Interfaces, 2013, 5, 12469–12477 CrossRef CAS PubMed.
- Q. Q. Li, C. G. Lu, J. Zhu, E. Q. Fu, C. Zhong, S. Y. Li, Y. P. Cui, J. G. Qin and Z. Li, J. Phys. Chem. B, 2008, 112, 4545–4551 CrossRef CAS PubMed.
- (a) Y. D. Hang, J. Wang, T. Jiang, N. N. Lu and J. L. Hua, Anal. Chem., 2016, 88, 1696–1703 CrossRef CAS PubMed; (b) Y. B. Ding, X. Li, T. Li, W. H. Zhu and Y. S. Xie, J. Org. Chem., 2013, 78, 5328–5338 CrossRef CAS PubMed.
- (a) L. W. He, W. Y. Lin, Q. Y. Xu and H. P. Wei, ACS Appl. Mater. Interfaces, 2014, 6, 22326–22333 CrossRef CAS PubMed; (b) Y. Li, Y. H. Zhang, H. J. Niu, C. Wang, C. L. Qin, X. D. Bai and W. Wang, New J. Chem., 2016, 40, 5245–5254 RSC.
- (a) Z. Peng, X. Feng, B. Tong, D. D. Chen, J. B. Shi, J. G. Zhi and Y. P. Dong, Sens. Actuators, B, 2016, 232, 264–268 CrossRef CAS; (b) Z. Peng, Y. C. Ji, Z. Wang, B. Tong, J. B. Shi and Y. P. Dong, Acta Chim. Sin., 2016, 74, 942–948 CrossRef CAS; (c) Y. C. Ji, Z. Peng, B. Tong, J. B. Shi, J. G. Zhi and Y. P. Dong, Dyes Pigm., 2017, 139, 664–671 CrossRef CAS.
- Y. L. Lin, G. Chen, L. F. Zhao, W. Z. Yuan, Y. M. Zhang and B. Z. Tang, J. Mater. Chem. C, 2015, 3, 112–120 RSC.
- J. F. Araneda, W. E. Piers, B. Heyne, M. Parvez and R. McDonald, Angew. Chem., Int. Ed., 2011, 50, 12214–12217 CrossRef CAS PubMed.
- (a) H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2016, 9, 83–87 Search PubMed; (b) H. G. Lu, Y. D. Zheng, X. W. Zhao, L. J. Wang, S. Q. Ma, X. Q. Han, B. Xu, W. J. Tian and H. Gao, Angew. Chem., Int. Ed., 2016, 55, 155–159 CrossRef CAS PubMed.
- (a) T. Zhang, Y. Q. Jiang, Y. L. Niu, D. Wang, Q. Peng and Z. G. Shuai, J. Phys. Chem. A, 2014, 118, 9094–9104 CrossRef CAS PubMed; (b) H. Nie, B. Chen, C. Y. Quan, J. Zhou, H. Y. Qiu, R. R. Hu, S. J. Su, A. J. Qin, Z. J. Zhao and B. Z. Tang, Chem. – Eur. J., 2015, 21, 8137–8147 CrossRef CAS PubMed.
- Y. L. Zhang, J. Li, B. Z. Tang and K. S. Wong, J. Phys. Chem. C, 2014, 118, 26981–26986 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1544597–1544605. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00096d |
| ‡ Zhe Peng and Yingchun Ji contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |