
Janus molecules: large second-order nonlinear optical performance, good temporal stability, excellent thermal stability and spherical structure with optimized dendrimer structure†
Pengyu
Chen
a,
Huanyu
Zhang
a,
Mengmeng
Han
a,
Ziyao
Cheng
a,
Qian
Peng
b,
Qianqian
Li
a and
Zhen
Li
 *a
*a
aDepartment of Chemistry, Hubei Key Lab on Organic and Polymeric Opto-Electronic Materials, Wuhan University, Wuhan 430072, China. E-mail: lizhen@whu.edu.cn; lichemlab@163.com
bKey Laboratory of Organic Solids, Beijing National Laboratory for Molecular Science Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
First published on 8th May 2018
Abstract
In this paper, Janus molecules J1 and J2, based on the second-order nonlinear optical (NLO) chromophore of FTC (Fang's thermally stable chromophore), were designed and synthesized, in which the arrangement of chromophore moieties was different on the two sides of the core. Especially, in J2, each FTC piece had similar direction and a more similar spherical structure realized. Janus molecule J2 exhibited good thermal stability and excellent nonlinear optical performance. Thanks to the unique molecular topology, an ultrahigh d33 value of 529 pm V−1 at the wavelength of 1950 nm has been achieved, with 80% of its value retained at the high temperature of 161 °C. Coupled with the good thermal stability and very simple structure, the obtained results are valuable for the further development of NLO materials.
Introduction
Second-order nonlinear optical (NLO) materials have promising applications in many fields, including ultrafast optical switches, high-speed optical modulators, high-density optical data storage and so on. Compared to traditional inorganic crystalline materials, organic ones have numerous advantages, such as better processability, ultrafast response time and superior chemical flexibility; thus, organic NLO materials have attracted many researchers’ attention.1 In this field, making the chromophores well aligned in the non-centrosymmetric arrangement is still a difficult task. Generally, the antiparallel arrangement is the trend of chromophore moieties in the natural state due to the strong dipole–dipole intermolecular interactions, which prevents the NLO polymers from being efficiently poled under an electric field. Besides, the poled polymers are liable to the natural state upon removal of the electric field or increasing temperature (Fig. S1, ESI†).2With the proper design of molecular structures, the dipole–dipole intermolecular interactions among the chromophore moieties could be weakened to a large degree. A lot of work has been conducted about this material by many researchers.3 Based on their excellent work, we have done systematic research on NLO polymers and proposed the concept of “suitable isolation group” (SIG) for the rational design of NLO polymers.4 Correspondingly, as shown in Fig. 1, different types of NLO polymers have been designed to achieve high NLO performance, including linear polymers, hyperbranched polymers, dendrimers, and dendronized hyperbranched polymers.5 Among them, dendrimers demonstrated some more advantages for the perfect and unique structure, regardless of the synthetic difficulty. Also, their spherical structure could contribute much to the large macroscopic NLO effect according to the “site isolation” principle and the SIG concept, with the much decreased dipole–dipole intermolecular interactions of chromophore moieties.6
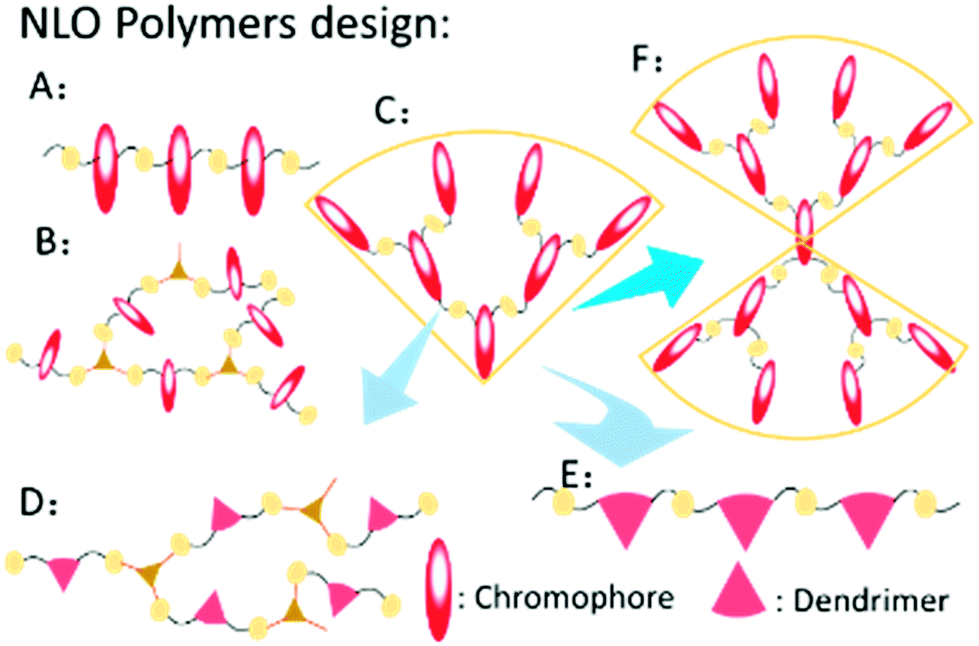 | ||
| Fig. 1 Different types of NLO polymers: (A) linear polymer; (B) hyperbranched polymer; (C) dendrimer; (D) dendronized hyperbranched polymer; (E) dendronized polymer; (F) Janus molecule. | ||
Recently, to improve the NLO performance, a new optimized kind of NLO dendrimer, named Janus dendrimer, was investigated by our group (Fig. 1).7 In comparison with general dendrimers, Janus molecules are more spherical in structure, and the chromophore moieties are partially in an orderly arrangement with the controllable synthesis and more easily achieve the non-centrosymmetric alignment in the poling process. As shown in Fig. 2, two Janus molecules, C2 and D-13N, containing the azo chromophore were synthesized in our previous work, which exhibited very good NLO performance, especially for D-13N, with the d33 value as high as 299 pm V−1.7a This represents the largest NLO efficiency among all NLO molecules based on the azo chromophore, confirming the power and important role of rational molecular design.7a,8 However, to meet practical applications, even larger NLO performance should be achieved, for example, 500 pm V−1. Thus, regardless of the fact that, according to the SIG concept, the NLO performance of the azo chromophore-containing polymers was largely improved from around 40 to 299 pm V−1, further room for improvement might be limited, since the μβ value of the azo chromophore is really too low (580 × 10−48 esu) to realize much higher NLO performance.1,9,10
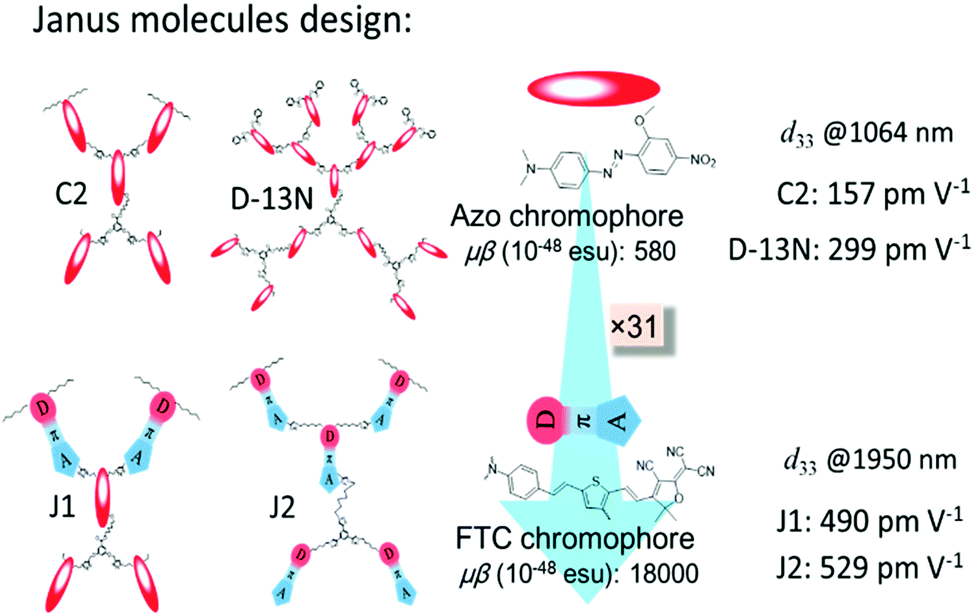 | ||
| Fig. 2 Janus molecules. C2 and D-13N were synthesized in our previous work, and J1 and J2 were synthesized in this paper. | ||
With the above considerations, to obtain even higher NLO performance, a high-μβ chromophore such as FTC (Fang's thermally stable chromophore) with 31 times of that of the azo chromophore (Fig. 2 and Fig. S2, ESI†) should be utilized and preferably introduced to the design of Janus dendrimer molecules.1,9,10 Thus, accordingly, in this paper, FTC was attempted to construct the Janus dendrimer molecule. Excitingly, with very simple structures, Janus molecules J1 and J2, containing three azo and two FTC chromophore (J1) or five FTC chromophore (J2) moieties, demonstrated dramatically enhanced NLO performance of 490 and 529 pm V−1, respectively, once again showing the advantages of the Janus structure. Furthermore, the thermal stability of J2 was excellent, with decomposition temperature up to 367 °C. Unexpectedly, from the UV-vis spectra and theoretical calculations, the topological structure of J1 and J2 was not as simple as expected, thus enriching the related knowledge for the control of dendrimers with special structures. Herein, we present the molecular design, synthesis, characterization, topological structure, and NLO performance of J1 and J2 in detail, in comparison with a reference dendrimer molecule D1.
Experimental section
Materials and instrumentation
Tetrahydrofuran (THF) was dried over and distilled from K–Na alloy under an atmosphere of dry nitrogen. Dichloromethane and N,N-dimethylformamide (DMF) were dried over and distilled from CaH2. Anhydrous ethanol was dried over and distilled from sodium. All other reagents were used as received. The synthesis of the reactants, FTC-1, FTC-2, FTC-3 and compound A, has been previously reported by our group.7a,91H, COSY and 13C NMR spectra were measured on a Bruker Advance III (400 MHz) spectrometer using tetramethylsilane (TMS; δ = 0 ppm) as internal standard. UV-visible spectra were obtained using a Shimadzu UV-2550 spectrometer. The Fourier transform infrared (FTIR) spectra were recorded on a PerkinElmer-2 spectrometer in the region of 3000–500 cm−1. Elemental analyses (EA) were performed by a CARLOERBA-1106 micro-elemental analyzer. Matrix-assisted laser desorption ionization time-of-flight mass spectra were measured on a Voyager-DE-STR MALDI-TOF mass spectrometer (MALDI-TOF MS; ABI, American) equipped with a 337 nm nitrogen laser and a 1.2 m linear flight path in positive ion mode. The thermal transitions were investigated using a METTLER differential scanning calorimeter DSC822e under nitrogen at a scanning rate of 10 °C min−1. The thickness of the films was measured with an Ambios Technology XP-2 profilometer. Thermal analysis was performed on a NETZSCH STA449C thermal analyzer at a heating rate of 10 °C min−1 in nitrogen and a flow rate of 50 cm3 min−1 for thermogravimetric analysis (TGA).
Synthesis
D1. Under an atmosphere of dry nitrogen, a solution of FTC-1 (33 mg, 0.05 mmol), FTC-2 (49 mg, 0.1 mmol), copper sulfate pentahydrate (10% mmol), sodium bicarbonate (20% mmol), and sodium L-ascorbate (20% mmol) in 5/1 mL of THF/H2O was stirred at 28 °C for 3 h. Then, the reaction mixture was poured into water and extracted with CH2Cl2 three times (50 mL × 3). The combined organic solution was dried over anhydrous sodium sulfate and condensed via rotary evaporation. The residue was purified by column spectroscopy on silica gel using the solvents CH2Cl2 and ethyl acetate (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as eluent to give 40 mg of product (yield 48.7%). 1H NMR (400 MHz, THF-d8, 298 K) δ (TMS, ppm): 9.10–9.06 (d, J = 16 Hz, 2H, –CH
:1) as eluent to give 40 mg of product (yield 48.7%). 1H NMR (400 MHz, THF-d8, 298 K) δ (TMS, ppm): 9.10–9.06 (d, J = 16 Hz, 2H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 8.19 (s, 2H, ArH), 7.80–7.76 (d, J = 16 Hz, 1H, –CH), 7.36–7.27 (m, 11H, ArH, –CH), 7.05–6.95 (m, 6H, –CH), 6.63–6.51 (m, 8H, ArH, –CH), 4.36–4.43 (m, 4H, –CH2–), 3.33–3.23 (m, 12H, –CH2–), 1.88–1.87 (m, 4H, –CH2–), 1.70 (s, 12H, –CH3), 1.65 (s, 6H, –CH3), 1.51 (m, 4H, –CH2–), 1.31 (m, 6H, –CH2–), 1.08–1.05 (m, 12H, –CH3). 13C NMR (100 MHz, THF-d8, 298 K), δ (ppm): 153.1, 148.8, 142.4, 139.2, 135.6, 132.1, 129.3, 126.0, 122.8, 97.4, 50.5, 44.5, 29.9, 26.5, 26.2, 12.7. C96H92N18O3S3 (EA) (%, found/calcd): C, 70.22/70.47; H, 5.65/5.82; N, 15.35/15.18; S, 5.86/5.98. MS (MALDI-TOF): m/z for C96H92N18O3S3 (found/calcd): [M + H]+: 1642.4/1641.6.
), 8.19 (s, 2H, ArH), 7.80–7.76 (d, J = 16 Hz, 1H, –CH), 7.36–7.27 (m, 11H, ArH, –CH), 7.05–6.95 (m, 6H, –CH), 6.63–6.51 (m, 8H, ArH, –CH), 4.36–4.43 (m, 4H, –CH2–), 3.33–3.23 (m, 12H, –CH2–), 1.88–1.87 (m, 4H, –CH2–), 1.70 (s, 12H, –CH3), 1.65 (s, 6H, –CH3), 1.51 (m, 4H, –CH2–), 1.31 (m, 6H, –CH2–), 1.08–1.05 (m, 12H, –CH3). 13C NMR (100 MHz, THF-d8, 298 K), δ (ppm): 153.1, 148.8, 142.4, 139.2, 135.6, 132.1, 129.3, 126.0, 122.8, 97.4, 50.5, 44.5, 29.9, 26.5, 26.2, 12.7. C96H92N18O3S3 (EA) (%, found/calcd): C, 70.22/70.47; H, 5.65/5.82; N, 15.35/15.18; S, 5.86/5.98. MS (MALDI-TOF): m/z for C96H92N18O3S3 (found/calcd): [M + H]+: 1642.4/1641.6.
J1. Under an atmosphere of dry nitrogen, a solution of FTC-3 (70 mg, 0.15 mmol), compound A (80 mg, 0.057 mmol), copper sulfate pentahydrate (10% mmol), sodium bicarbonate (20% mmol), and sodium L-ascorbate (20% mmol) in 5/1 mL of THF/H2O was stirred at 28 °C for 3 h. Then, the reaction mixture was poured into water and extracted with CH2Cl2 three times (50 mL × 3). The combined organic solution was dried over anhydrous sodium sulfate and condensed via rotary evaporation. The residue was purified by column spectroscopy on silica gel using the solvents CH2Cl2 and ethyl acetate (1:1) as eluent to give 77 mg of product (yield 51.3%). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 8.73–8.69 (d, J = 16 Hz, 2H, –CH), 8.27–8.25 (d, J = 8 Hz, 4H, ArH), 8.00 (s, 2H, –CH), 7.88–7.81 (m, 13 H, ArH, –CH), 7.60 (m, 3H, –CH, ArH), 7.38–7.35 (d, J = 12 Hz, 4H, ArH), 7.20 (s, 2H, ArH), 7.04–6.91 (m, 5H, –CH, ArH), 6.86–6.83 (d, J = 12 Hz, 2H, ArH), 6.75 (m, 1H, ArH), 6.68–6.66 (d, J = 8 Hz, 4H, ArH), 6.63–6.61 (d, J = 8 Hz, 4H, ArH), 6.36–6.32 (d, J = 16 Hz, 2H, –CH), 5.14 (s, 4H, –CH2–), 4.62–4.58 (m, 8H, –CH2–), 4.29 (m, 2H, –CH2–), 4.22 (m, 2H, –CH2–), 3.92 (m, 8H, –CH2–), 3.32 (m, 8H, –CH2–), 3.22–3.20 (m, 8H, –CH2–), 1.91 (m, 2H, –CH2–), 1.81 (m, 2H, –CH2–), 1.67 (m, 12H, –CH3), 1.60 (m, 12H, –CH2–), 1.33 (m, 24H, –CH2–), 1.09 (m, 6H, –CH3), 0.91 (m, 12H, –CH3). 13C NMR (100 MHz, CDCl3, 298 K), δ (ppm): 173.7, 1.65.9, 159.0, 156.3, 155.5, 153.2, 150.2, 149.3, 147.4, 144.0, 143.6, 132.0, 132.0, 129.2, 126.1, 124.5, 122.6, 111.4, 97.3, 61.9, 54.5, 54.5, 51.0, 47.0, 45.7, 31.6, 26.7, 22.6, 13.9, 12.0. C143H154N14O313S2 (EA) (%, found/calcd): C, 66.24/66.03; H, 5.99/5.78; N, 17.29/17.51; S, 2.47/2.72. MS (MALDI-TOF): m/z for C143H154N14O313S2 (found/calcd): [M + Na]+: 2614.1/2614.1.
J2. Under an atmosphere of dry nitrogen, a solution of compound 7 (88 mg, 0.0413 mmol), FTC-2 (50 mg, 0.0826 mmol), copper sulfate pentahydrate (10% mmol), sodium bicarbonate (20% mmol), and sodium L-ascorbate (20% mmol) in 5/1 mL of THF/H2O was stirred at 28 °C for 3 h. Then, the reaction mixture was poured into water and extracted with CH2Cl2 three times (50 mL × 3). The combined organic solution was dried over anhydrous sodium sulfate and condensed via rotary evaporation. The residue was purified by column spectroscopy on silica gel using the solvents CH2Cl2 and ethyl acetate (1:5) as eluent to give 69 mg of product (yield 50%). 1H NMR (400 MHz, CDCl3, 298 K), δ (TMS, ppm): 8.93–8.74 (m, 3H, –CH), 8.34–8.25 (m, 3H, ArH), 8.01–7.93 (m, 5H, ArH), 7.81–7.59 (m, 2H, ArH), 7.40–7.21 (m, 14H, ArH), 7.08–6.81 (m, 12H, –CH, ArH), 6.66–6.40 (m, 17H, ArH, –CH), 4.48–4.31 (m, 12H, –CH2–), 3.43–3.18 (m, 20H, –CH2–), 2.06–1.50 (m, 56H, –CH2–), 1.46–1.36 (m, 16H, –CH2–), 1.36–1.21 (m, 18H, –CH3), 1.18–1.09 (m, 6H, –CH3), 0.95–0.84 (m, 12H, –CH3). 13C NMR (100 MHz, CDCl3, 298 K), δ (ppm): 149.2, 138.7, 146.9, 137.2, 135.6, 132.1, 131.7, 130.8, 129.0, 128.7, 124,6, 122.6, 120.2, 112.5, 97.4, 50.9, 50.1, 45.0, 37.3, 31.8, 31.6, 29.8, 29.6, 26.7, 22.6, 19.6, 19.0, 13.6, 12.3. C196H208N38O5S5 (EA) (%, found/calcd): C, 70.56/70.53; H, 6.28/6.29; N, 15.95/15.89; S, 4.70/4.46. MS (MALDI-TOF): m/z for C196H208N38O5S5 (found/calcd): [M + Na + K]+: 3395.0/3395.5.
Preparation of thin films
The three molecules were dissolved in THF (concentration, 4 wt%), and the solutions were filtered through syringe filters. Then, the solutions were spin-coated onto the non-conducting face of indium-tin-oxide (ITO)-coated glass substrates, which were cleaned by DMF, acetone, ethyl alcohol, distilled water, and THF sequentially in an ultrasonic bath before use. Residual solvent was removed by heating the films in a vacuum oven at 40 °C for 6 hours.NLO measurement of poled films
A schematic diagram of the poling device is shown in Fig. 4. In this paper, the second-order nonlinear optical (NLO) efficiencies of the compounds were measured by in situ second harmonic generation (SHG) experiment using a closed temperature-controlled oven with optical windows and three needle electrodes. The films were kept at 45° to the incident beam and poled inside the oven, with the conducting planes facing the laser. Then, the laser was lit and the SHG intensity was monitored simultaneously. The poling temperatures of the three films were different, as shown in Table 1. The same poling conditions they shared are as follows: voltage, 7.0 kV at the needle point; gap distance, 0.8 cm. The NLO efficiencies were investigated using 1950 nm laser radiation. The frequency-doubled signals (975 nm) were detected by an Andor's DU420A-BR-DD CCD after the mixed signals passed through the monochromator, and a specially made Y-cut quartz crystal served as the reference.| No. | T g (°C) | T d (°C) | T e (°C) | T 80% (°C) | d 33 (pm V−1) | Solvatochromismf | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1,4-Dioxane | CHCl3 | THF | CH2Cl2 | DMF | DMSO | Δ | ||||||
| a Glass transition temperature. b 5% weight loss temperature. c Best poling temperature. d Temperature at which d33 values decreased to 80%. e d 33 values (NLO performance). f Maximum absorption wavelength (λmax, nm) in different solvents (0.02 mg mL−1), Δ = λmax (in DMSO) − λmax (in dioxane); dielectric constant of solvents: 1,4-dioxane = 2.2; CHCl3 = 4.8; THF = 7.5; CH2Cl2 = 8.9; DMF = 37.6; DMSO = 46.7. | ||||||||||||
| D1 | 80 | 359 | 147.5 | 134 | 342 | 642 | 681 | 644 | 672 | 661 | 668 | 26 |
| J1 | 113 | 269 | 130.0 | 123 | 490 | 452/653 | 454/693 | 462/649 | 458/670 | 476/656 | 483/661 | 31/8 |
| J2 | 108 | 367 | 152.5 | 161 | 529 | 617 | 656 | 617 | 651 | 633 | 644 | 27 |
| Azo | — | — | — | — | — | 452.5 | 454 | 461.5 | 458 | 478 | 487 | 34.5 |
| FTC | — | — | — | — | — | 629 | 687 | 643.5 | 647.5 | 655.5 | 668 | 39 |
Results and discussion
Synthesis and characterization
Fig. 3 demonstrates the synthetic routes for the three compounds D1, J1 and J2, in which J2, with the Janus dendrimer structure, entirely consisted of FTC chromophore moieties. As to J1, it contained two FTC moieties at the head of the molecule and three azo chromophore moieties in the middle and tail, which could be considered as a double Janus structure with the different chromophore moieties on the two sides of the core, in addition to the different alignments relative to the core. For comparison, the reference compound D1 was prepared, which is a dendrimer molecule consisting of three FTC moieties and could be considered as half of J2 (Fig. S1D, ESI†).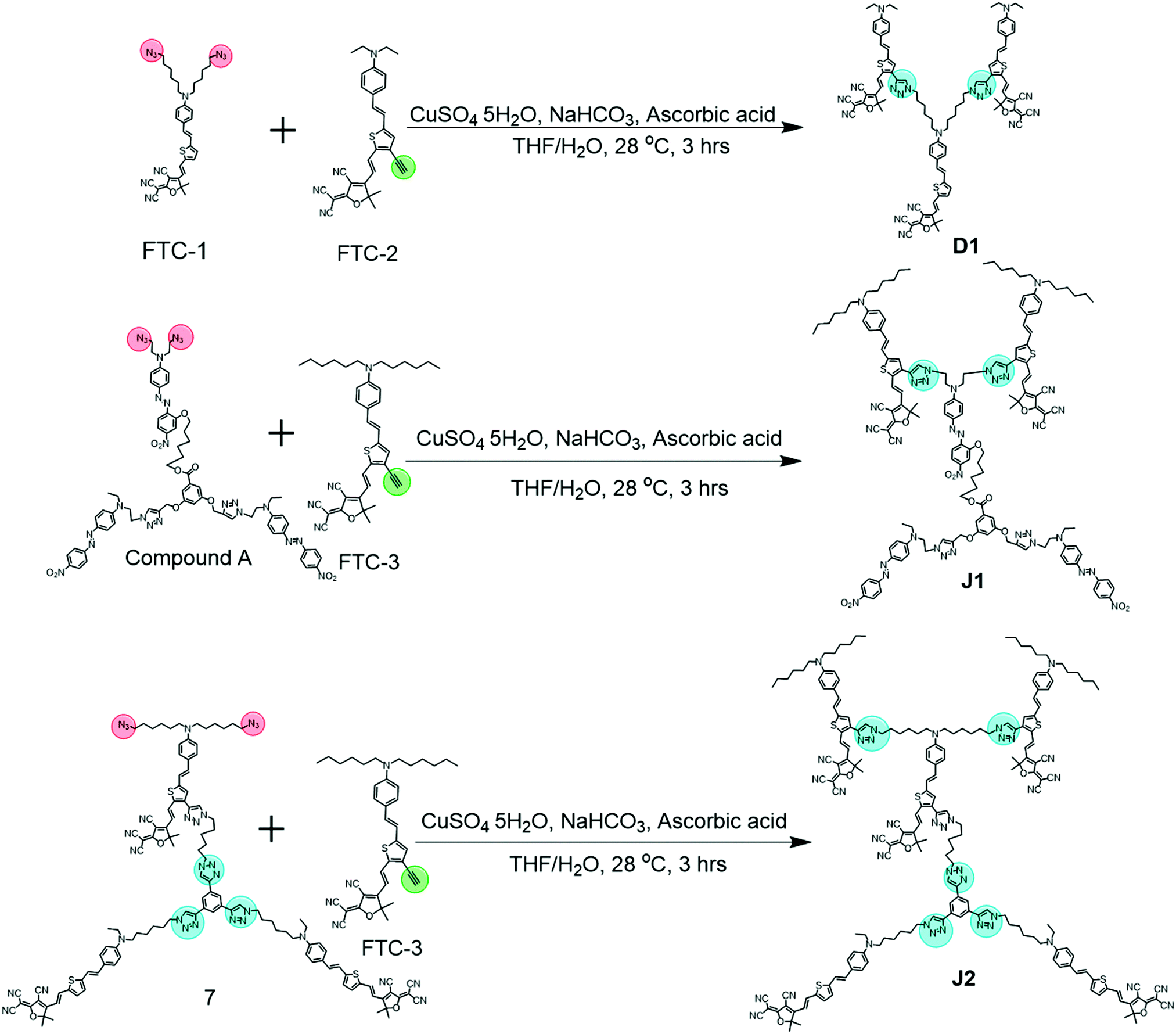 | ||
| Fig. 3 Synthetic routes of D1, J1 and J2. | ||
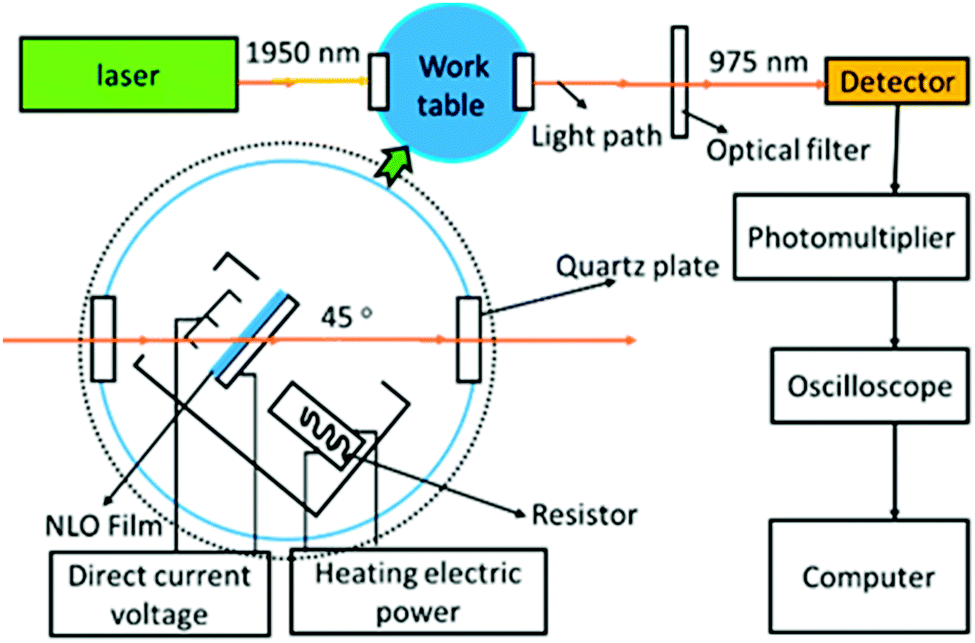 | ||
| Fig. 4 Schematic diagram of the poling device. | ||
D1 and J1 were synthesized through the simple click reaction (Fig. 3). However, the synthesis of J2 was a little difficult. According to our previous work, compound 1 could be easily synthesized, which reacted with compound 2 to give compound 3 in a high yield of 84.3%, through the azido–yne click reaction (Fig. S11, ESI†).9 It should be noted that an excess of compound 2 was added in this reaction to assure that there were two unreacted terminal alkyne groups remaining in compound 3, which reacted with quantitative compound 4 bearing azido reactive groups to yield compound 5. Then, the two bromine atoms in compound 5 were converted to azido moieties of compound 6, which underwent the subsequent Knoevenagel condensation reaction to produce the important reactive intermediate 7. Finally, another click chemistry reaction between compound 7 and FTC-3 gave the product J2 with the satisfactory yield of 50.0%. It should be noted that the bromine atoms should be transformed to azido groups before the Knoevenagel condensation reaction because the reaction conditions of substitution would damage the carbon–carbon double bond formed in the Knoevenagel condensation.
The three molecules, D1, J1 and J2, were well characterized by nuclear magnetic resonance (NMR) (Fig. 5 and Fig. S9, ESI†), infrared spectroscopy (IR) (Fig. 5 and Fig. S4, ESI†), thermogravimetric analysis (TGA) (Fig. 6), ultraviolet-visible (UV-vis) spectroscopy (Fig. 7 and Fig. S5, ESI†), differential scanning calorimetry (DSC) (Fig. S3, ESI†), elemental analyses (EA), and matrix-assisted laser desorption ionization-time of flight-mass spectrometry (MALDI-TOF-MS) (Fig. 5 and Fig. S8, ESI†).
 | ||
| Fig. 5 (A) 1H NMR of intermediate products in the synthesis route of J2; (B) IR spectra of compound 7 and J2; (C) TOF spectra of J1 and J2. | ||
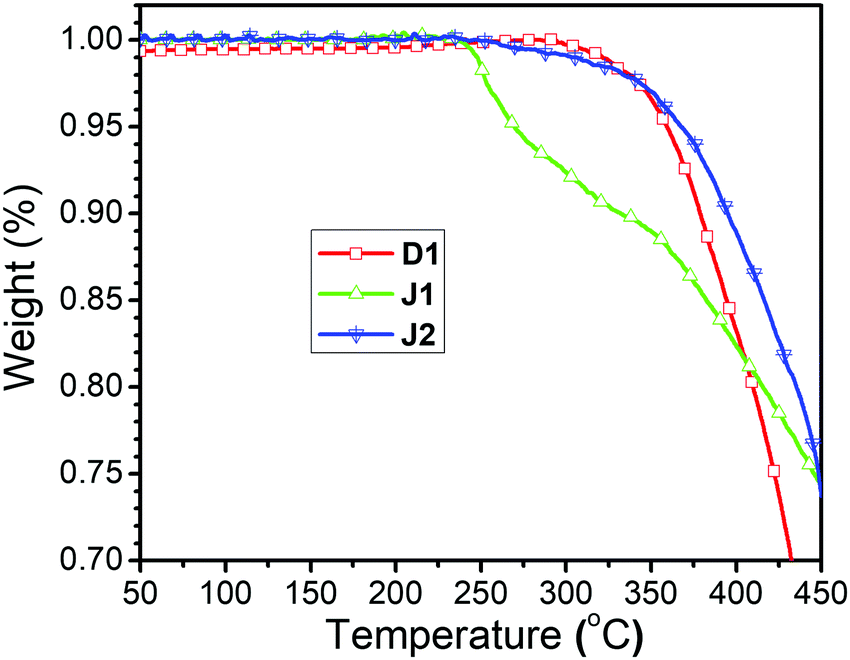 | ||
| Fig. 6 TGA spectra of three compounds. | ||
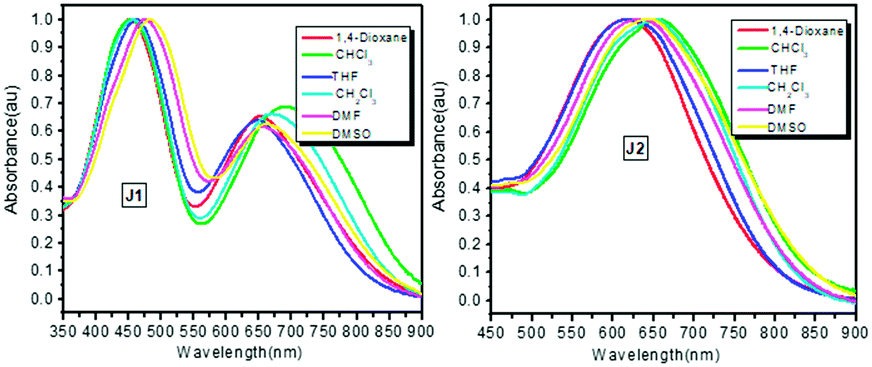 | ||
| Fig. 7 Solvatochromism spectra of J1 and J2. | ||
1H NMR spectrum could be used to detect the reactions by analyzing the change of characteristic peaks. In the synthetic route of J2, compound 3 had one aldehyde group and two terminal alkyne groups, compound 5 had three aldehyde groups, while there were no aldehyde and terminal alkyne groups in compound 7. As shown in Fig. 5A, there was one sharp peak at about 3.2 ppm in the 1H NMR spectrum of compound 3, which should be assigned to the terminal alkyne groups (2 ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH). However, this peak disappeared completely in compound 5, showing the good conversion to the triazole rings through the click chemistry reaction. As shown in Fig. 5A, there was a sharp peak at about 10 ppm in the 1H NMR spectrum of compound 3, which should be assigned to the aldehyde group (1 –CHO), while in the 1H NMR spectrum of compound 5, there were two sharp peaks at about 10 ppm (the area ratio of the two sharp peaks was 1/2), which should also be assigned to the aldehyde groups (the small sharp peak was assigned to the original aldehyde group (1 –CHO) from compound 1, and the large sharp peak was assigned to new aldehyde groups (2 –CHO), from compound 4). The peak of new aldehyde groups in the 1H NMR spectrum of compound 5 proved the successful click chemistry reaction once again. From compound 5 to compound 6, the bromine atoms were substituted by azide groups, which caused the movement of the peak at 3.6 ppm assigned to adjacent methylene (–CH2–) groups. In the Knoevenagel condensation reaction, the aldehyde groups of compound 6 were converted into carbon–carbon double bonds of compound 7, so the two peaks assigned to aldehyde groups disappeared completely in the 1H NMR spectrum of compound 7, and three double-peaks could be found at 9.0, 8.3 and 6.5 ppm in the amplified 1H NMR spectrum of compound 7, as shown in Fig. S9 (ESI†). COSY NMR spectra were conducted to further characterize D1, J1 and J2 (Fig. S10, ESI†).
CH). However, this peak disappeared completely in compound 5, showing the good conversion to the triazole rings through the click chemistry reaction. As shown in Fig. 5A, there was a sharp peak at about 10 ppm in the 1H NMR spectrum of compound 3, which should be assigned to the aldehyde group (1 –CHO), while in the 1H NMR spectrum of compound 5, there were two sharp peaks at about 10 ppm (the area ratio of the two sharp peaks was 1/2), which should also be assigned to the aldehyde groups (the small sharp peak was assigned to the original aldehyde group (1 –CHO) from compound 1, and the large sharp peak was assigned to new aldehyde groups (2 –CHO), from compound 4). The peak of new aldehyde groups in the 1H NMR spectrum of compound 5 proved the successful click chemistry reaction once again. From compound 5 to compound 6, the bromine atoms were substituted by azide groups, which caused the movement of the peak at 3.6 ppm assigned to adjacent methylene (–CH2–) groups. In the Knoevenagel condensation reaction, the aldehyde groups of compound 6 were converted into carbon–carbon double bonds of compound 7, so the two peaks assigned to aldehyde groups disappeared completely in the 1H NMR spectrum of compound 7, and three double-peaks could be found at 9.0, 8.3 and 6.5 ppm in the amplified 1H NMR spectrum of compound 7, as shown in Fig. S9 (ESI†). COSY NMR spectra were conducted to further characterize D1, J1 and J2 (Fig. S10, ESI†).
Some typical groups could be easily detected by infrared spectroscopy, such as azido (2160–2090 cm−1), terminal alkyne (2140–2100 cm−1), and aldehyde groups (1750–1680 cm−1). As shown in Fig. S4 (ESI†), there were no peaks at about 2140–2100 and 2160–2090 cm−1 for D1, J1 and J2, confirming the complete reaction of the terminal alkyne groups and azide groups for the production of compound 7, compound A, FTC-1, FTC-2 and FTC-3. The peaks of aldehyde groups were not observed at 1750–1680 cm−1 for compound 7, demonstrating the complete reaction of the aldehyde groups. Also, the presence of the typical signal of cyano groups at about 2222 cm−1 could confirm the conversion of compound 6 to compound 7, since the cyano groups were assigned to the TCF acceptors in compound 7. As shown in Fig. 3, J2 was synthesized through click chemistry reaction between compound 7 and FTC-3, and all of them had cyano groups, while only compound 7 had azide groups. As shown in Fig. 5B, the absorption band of cyano groups (2222 cm−1) existed in both IR spectra of compound 7 and J2, while the absorption band of azide groups (2098 cm−1) only existed in the IR spectrum of compound 7. These two phenomena proved the success of the click chemistry reaction and Knoevenagel condensation reaction in the synthetic process.
MALDI-TOF-MS is a powerful tool to characterize molecules with high molecular weights. For the large polarity of these three molecules, sodium ionization and potassium ionization were adopted. As shown in Fig. 5C and Fig. S7 (ESI†), the peaks at 2614.1956 and 2629.1663 belong to [M + Na]+ and [M + K]+ of J1, and the peaks at 3358.1414 and 3395.0754 were ascribed to [M + Na]+ and [M + Na + K]+ of J2, which undoubtedly confirm the successful syntheses of J1 and J2.
Thermal properties
The TGA curves of D1, J1 and J2 are shown in Fig. 6. All of them possess very good thermal stability with the decomposition temperatures (Td) (temperatures for 5% weight loss) higher than 260 °C, and the decomposition temperatures (Td) of D1 and J2 even reaching around 360 °C (Table 1). The Td of J2 was much higher than that of J1, possibly due to the influence of ester bond in the azo moieties, similar to our previous case of the FTC derivatives bearing ester bonds.1 Unlike J1, the linkages in D1 and J2 are all triazole groups formed in the click chemistry reaction, which were stable enough against decomposition. Thus, the good thermal stability of J2 gives it good NLO thermal stability after poling. The glass transition temperatures (Tg) of these three compounds were also investigated by using differential scanning calorimetry (DSC), and the glass transition temperatures (Tg) are 80, 113 and 108 °C, respectively (Table 1 and Fig. S3, ESI†).Optical properties
The UV-vis absorption spectra of J1 and J2 in different solvents are shown in Fig. 7, while those of D1 are presented in Fig. S5 (ESI†) for comparison. Their maximum absorption (λmax) wavelengths are summarized in Table 1, and corresponding data for the original FTC and azo chromophores (molecule structures in Fig. S2, ESI†) are also listed for comparison. Analyzing the UV-vis spectra carefully, it could be found that there are two apparent maximum absorption wavelengths for J1. While the λmax at about 460 nm was ascribed to azo moieties, that at ∼660 nm belongs to FTC moieties. As for others, only one maximum absorption wavelength existed between 400–800 nm in the UV-vis absorption spectra. Some interesting results could be found after making a detailed comparison of λmax in different solvents. Theoretically, the λmax should red-shift with the enhancement of the polarity of solvent. Surprisingly, the λmax of azo in CH2Cl2 was much shorter than that in THF, and the λmax of FTC in CHCl3 was the longest among the six solvents (Table 1). Compared with the λmax of the original azo chromophore, the λmax of azo moieties of J1 was almost the same, indicating that each azo moiety was slightly influenced by other moieties. Compared with the λmax of the original FTC chromophore, the λmax of D1 was 13 nm red-shifted in 1,4-dioxane and similar in other high-polarity solvents except CH2Cl2. This indicates the strong influence between FTC moieties, which could be shielded by high-polarity solvents. The λmax of D1 in CH2Cl2 solvent was much more red-shifted than that in DMF, possibly due to the influence of the linkage between FTC moieties. Similar red-shift of λmax of the FTC moieties in CH2Cl2 also existed in our previous work.9 This phenomenon could also be observed in the spectra of J1 and J2. Compared with the λmax of the original FTC chromophore, the λmax of J1 was red shifted in low-polarity solvents and blue-shifted in high-polarity, and all λmaxvalues were very close. In contrast, the λmax of J2 was about 20 nm blue-shifted, which indicated largely improved transparency, possibly derived from the Janus design. Δ value, the difference between the λmax in DMSO and 1,4-dioxane, was introduced to further analyze the structural influence and evaluate the shielding effect on chromophore moieties, since DMSO and 1,4-dioxane had the strongest and weakest polarity in these six solvents. Effectively shielded chromophore moieties exhibited small changes in λmax since the shielded FTC moieties were less affected by the solvent change. As shown in Table 1, the Δ values of the FTC moieties in the original FTC chromophore, D1, J1 and J2 were 39, 26, 8 and 27 nm, respectively. Δ values of D1, J1 and J2 were smaller than that of the original FTC chromophore, indicating the shielding effect of other moieties. J1 got the smallest Δ value, much smaller than others. It could be speculated that the FTC moieties in J1 were best shielded by the bulky dendritic structure and azo moieties. Actually, in our previous work, once the different chromophores were introduced in the same system, the chromophore moieties with lower μβ value could act as “isolation chromophores” to decrease the strong dipole–dipole intermolecular interactions of the chromophore moieties with the higher μβ ones.9,11
Spatial geometry
In order to get some deep understanding of the topological structure of J1 and J2, especially the distribution of FTC moieties in the Janus structure, their spatial structures were calculated through the semi-empirical quantum chemistry method (Fig. 8 and Fig. S6, ESI†). To simplify the calculation, the hexyl and ethyl groups were replaced by methyl groups. As shown in Fig. 8, in J1, unexpectedly, the alignment of the azo and FTC chromophore moieties was a little strange: two FTC moieties covered the other three azo chromophores. Actually, as presented in the introduction, the key point of the Janus molecules was the partially orderly arrangement of the chromophore moieties through the control of the synthetic procedures, contributing to the easy poling process to achieve the non-centrosymmetric chromophore alignment. Thus, under controllable synthesis, in principle, the chromophore moieties of the azo and FTC groups should be partially aligned in order, just as in the cases of C2 and D-13N.7 However, the calculated results were beyond our understanding at first glance. Looking back into the UV-vis spectra, it was speculated that the FTC moieties in J1 were best covered by the bulky dendritic structure and azo moieties. These two results were almost the same. To be exact, J1 was not a correct Janus dendrimer molecule, and the azo chromophore moieties could be treated as isolation groups between two FTC moieties. The upper two FTC moieties showed similar and mutually perpendicular interspersion in space. Theoretically, two FTC moieties trended to the reverse parallel arrangement due to the intramolecular dipole–dipole interaction, partially due to the isolation effect of groups between the two FTC moieties. In J2, FTC moieties had similar direction, which gave J2 a non-centrosymmetric structure of dipole orientation to some degree, as the result of its Janus structure. Compared with J1, J2 was a real Janus molecule. If C2, D-13N and J2 could attain the real Janus structures, why not J1? Possibly, it might be caused by the fact that the chromophore moieties in J1 were different. The two dendrimer structures of J1 consisted of FTC chromophores with high μβ value and azo chromophores with low μβ value. The difference of chromophores caused the two Janus structures to aggregate. So, in Janus structure design, possibly, only one kind of chromophore should be used in order to realize the Janus topology.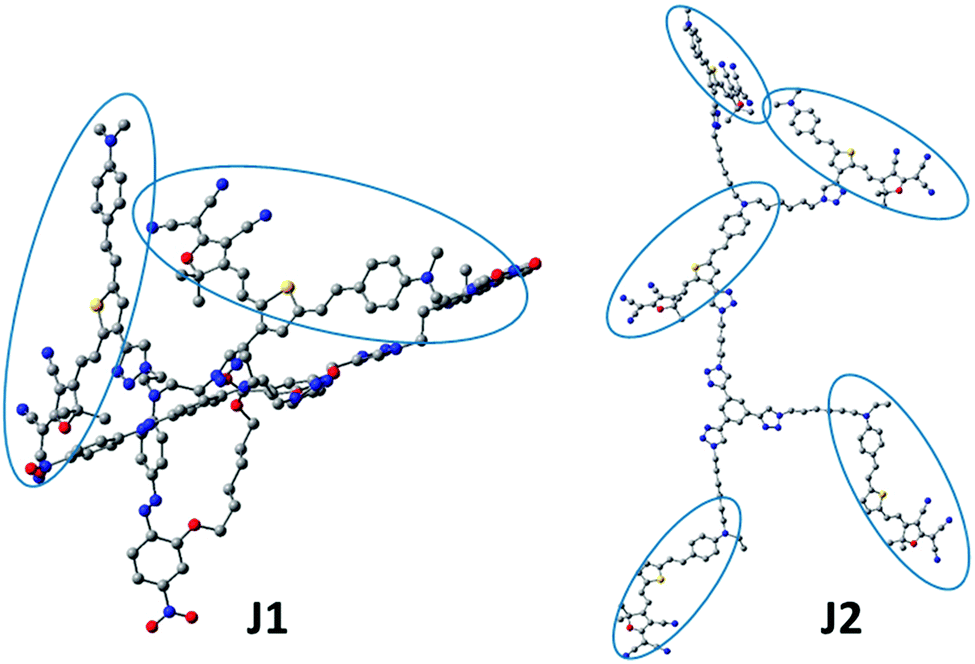 | ||
| Fig. 8 Optimized chemical structure. FTC moieties are encircled. | ||
NLO properties
To evaluate their NLO activity, the poled thin films of D1, J1 and J2 were fabricated through the convenient spin-coating process. Thanks to their good film-forming properties, high-quality thin films were obtained with the thickness in the range of 90 to 150 nm. The most convenient technique to study the second-order NLO activity is to investigate the second harmonic generation (SHG) processes characterized by d33, an SHG coefficient. The method for the calculation of the SHG coefficients (d33) for the poled films has been reported in literature and our previous papers.9 The schematic diagram of the poling device is shown in Fig. 4, and the poling and decay curves of D1, J1 and J2 are shown in Fig. 9.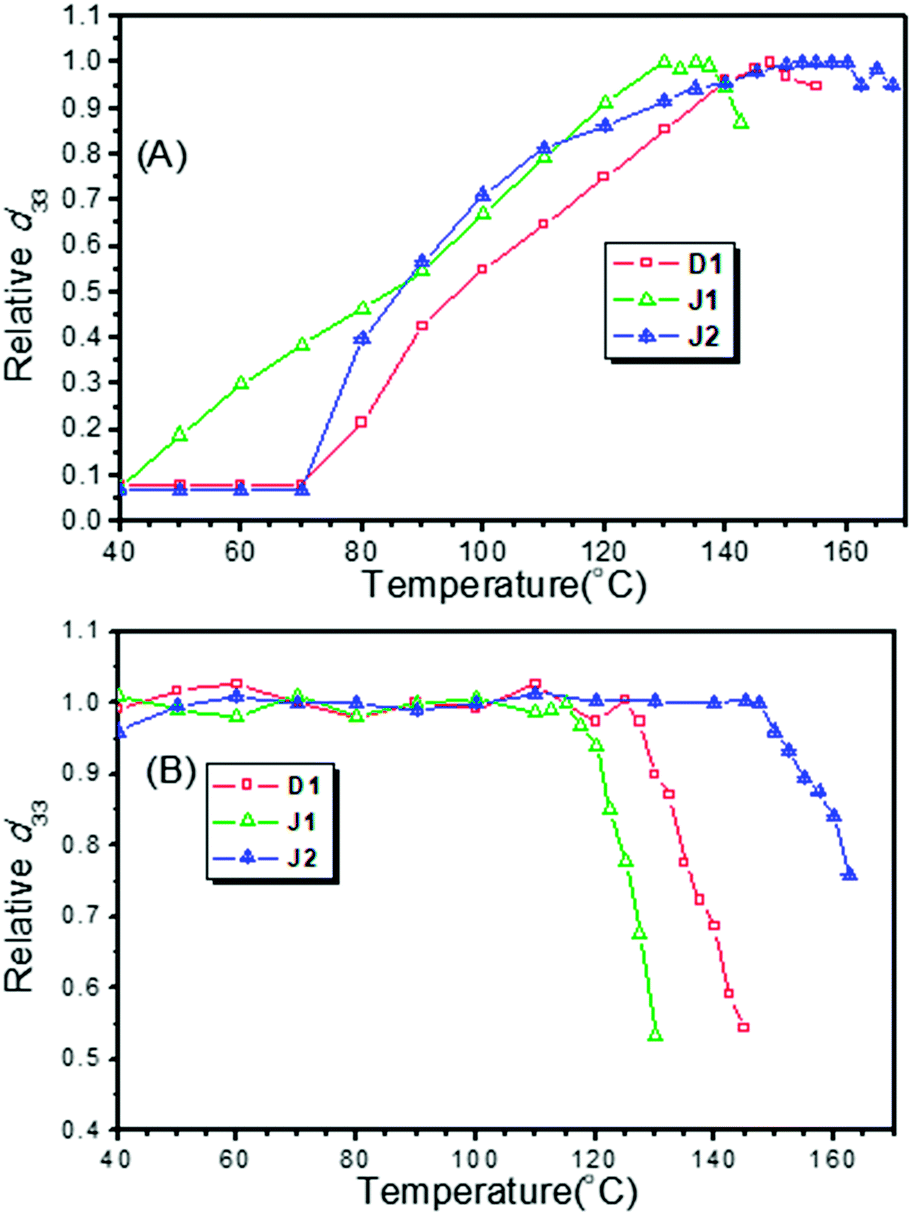 | ||
| Fig. 9 (A) Poling curves of D1, J1 and J2; (B) decay curves of D1, J1 and J2. | ||
From the experimental data, the d33 values of D1, J1 and J2 were calculated to be 342, 490 and 529 pm V−1, respectively, at the fundamental wavelength of 1950 nm. To check the reproducibility, we repeated the measurements at least three times and obtained repeatable results. As shown in Table 1, the best poling temperatures of the three molecules (D1, J1 and J2) were 147.5, 130.0 and 152.5 °C, respectively, and the depoling temperatures (the temperature at which d33 values decrease to 80% of the original) were 134, 123 and 161 °C, respectively. The best poling temperature was used to assess the difficulty of poling. At the best poling temperature, the degree of orientation of chromophore moieties reached the maximum, and the d33 value achieved the maximum. The depoling temperature was used to evaluate the stability of the non-centrosymmetric arrangement of the chromophore moieties in poled films. At the depoling temperature, the chromophore moieties would recover their centrosymmetric arrangement rapidly due to the thermal motion of molecules and strong dipole–dipole interaction.
J1 exhibited very high NLO performance (490 pm V−1), with the d33 value 1.43 times that of D1 (342 pm V−1), while the number of its FTC moieties was 0.66 times that of D1. Thus, the microscopic β values of FTC moieties in J1 could be more effectively translated into the macroscopic NLO activity than those in D1. This high transformation efficiency was due to the good shielding effect of the bulky dendritic structure and azo moieties (Fig. 8 and Table 1), which could be treated as “isolation chromophores” and effectively reduce the dipole–dipole interactions between the two FTC moieties. As J1 was not a strict Janus structure, its two different chromophore moieties located together (Fig. 8). As a result, the temporal stability of J1 was not very good, and its T80% (123 °C) was 38 °C, lower than that of J2. Besides, looking back to the poling curves, it could be found that the d33 value of J1 increased at the beginning of heating (40 °C) and it reached the largest d33 value at 130 °C. Both values are lower than those of D1 (70 °C, 147.5 °C) and J2 (70 °C, 152.5 °C), indicating that J1 is much easier to be poled due to the “isolation chromophore” effect.
J2 obviously demonstrated the best NLO performance, both in d33 value and stability, proving the superiority of Janus design. Its d33 value was the largest and 1.54 times that of D1, while the number of FTC moieties of J2 was 1.66 times that of D1 (Fig. 3). So the dipole–dipole interactions between the two dendrimer structures were decreased effectively. J2 also possessed the highest optimum poling temperature and depoling temperature. As shown in Table 1, the Te of J2 reached up to 152 °C and the T80% of J2 was 27 °C higher than that of D1. Some simple analysis on molecular motion shown in Fig. 10 could be used to give some explanation. The rotation diagram of D1 and J2 is shown in Fig. 10A: in the structure of D1, the dendrimer structure could revolve round the acme but that of J2 could not. The relaxing diagram of D1 and J2 is shown in Fig. 10B. In the process of relaxing, Janus and dendrimer molecules both needed to turn entirely upside down, and the difficulty increased with the size of the molecule. In consideration of the Janus structure of J2, it is reasonable that J2 had the best poling temperature. Due to having the largest molecular weight and Janus molecule structure, which was more like a spherical structure and can reduce the dipole–dipole interaction between chromophores effectively, J2 maintained 80% of its d33 value at very high temperature (161 °C).
 | ||
| Fig. 10 (A) Rotation diagram of D1 and J2; (B) relaxing diagram of D1 and J2. | ||
Conclusions
In this work, three second-order nonlinear optical (NLO) molecules containing FTC chromophores, named D1, J1 and J2, have been designed and synthesized. J2 is a Janus dendrimer structure molecule containing five FTC moieties, and J1 has a similar structure and consists of two FTC moieties and three azo moieties, while D1 is a small dendron containing three FTC moieties for comparison. They all exhibited good thermal stability, with decomposition temperatures higher than 260 °C. The Td of J2 even reached 367 °C. Excitingly, the Janus dendrimer J2 possesses the highest d33 value (529 pm V−1 at 1950 nm), which is 1.54 times that of D1. Besides, the temporal stability of J2 is also very good, and the T80% value was as high as 161 °C. These results confirm the excellence of the Janus dendrimer structure. Unexpectedly, the topological structure of Janus dendrimers is not so simple as considered before, and it was found to be affected not only by the synthetic process but also the electronic nature of the introduced functional groups, such as different chromophore moieties presented in J1. Thus, this study provides more information for the rational design of Janus dendrimers with different functionalities.Conflicts of interest
There are no conflicts to declare on this manuscript.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21734007), Hubei Province (2017CFA002), Special funds for basic scientific research services in central colleges and Universities (2042017kf0247).Notes and references
- (a) D. Yu, A. Gharavi and L. Yu, J. Am. Chem. Soc., 1995, 117, 11680 CrossRef; (b) T. Marks and M. Ratner, Angew. Chem., Int. Ed., 1995, 34, 155 CrossRef; (c) S. Marder, B. Kippelen, A. Jen and N. Peyghambarian, Nature, 1997, 388, 845 CrossRef; (d) Y. Shi, C. Zhang, H. Zhang, J. Bechtel, L. Dalton, B. Robinson and W. Steier, Science, 2000, 288, 119 CrossRef PubMed; (e) J. Luo, M. Haller, H. Ma, S. Liu, T. Kim, Y. Tian, B. Chen, S. Jang, L. Dalton and A. Jen, J. Phys. Chem. B, 2004, 108, 8523 CrossRef; (f) Y. Bai, N. Song, J. Gao, X. Sun, X. Wang, G. Yu and Z. Wang, J. Am. Chem. Soc., 2005, 127, 2060 CrossRef PubMed; (g) Y. Liao, C. Anderson, P. Sullivan, A. Akelaitis, B. Robinson and L. Dalton, Chem. Mater., 2006, 18, 1062 CrossRef; (h) X. Ma, R. Liang, F. Yang, Z. Zhao, A. Zhang, N. Song and Q. Zhang, J. Mater. Chem., 2008, 18, 1756 RSC; (i) J. Luo, X. Zhou and A. Jen, J. Mater. Chem., 2009, 19, 7410 RSC; (j) Z. Li, G. Yu, W. Wu, Y. Liu, C. Ye, J. Qin and Z. Li, Macromolecules, 2009, 42, 3864 CrossRef; (k) L. Dalton, D. Lao, B. Olbricht, S. Benight, D. Bale, J. Davies, T. Ewy, S. Hammond and P. Sullivan, Opt. Mater., 2010, 32, 658 CrossRef; (l) L. Dalton, P. Sullivan and D. Bale, Chem. Rev., 2010, 110, 25 CrossRef PubMed; (m) J. Luo, S. Huang, Z. Shi, B. Polishak, X. Zhou and A. Jen, Chem. Mater., 2011, 23, 544 CrossRef; (n) Z. Li, W. Wu, C. Ye, J. Qin and Z. Li, Polymer, 2012, 53, 153 CrossRef; (o) J. Liu, L. Wang, Z. Zhen and X. Liu, Colloid Polym. Sci., 2012, 290, 1215 CrossRef; (p) J. Luo and A. Jen, IEEE J. Sel. Top. Quantum Electron., 2013, 19, 3401012 CrossRef; (q) J. Wu, J. Luo, N. Cernetic, K. Chen, K. Chiang and A. Jen, J. Mater. Chem. C, 2016, 4, 10286 RSC; (r) D. Chen, P. Chen, L. Zong, Y. Sun, G. Liu, X. Yu and J. Qin, R. Soc. Open Sci., 2017, 4, 171161 CrossRef PubMed; (s) Z. Li, P. Chen, Y. Xie, Z. Li and J. Qin, Adv. Electron. Mater., 2017, 3, 1700138 CrossRef; (t) Y. Yang, J. Liu, H. Xiao, Z. Zhen and S. Bo, Dyes Pigm., 2017, 139, 239 CrossRef; (u) Z. Li and Z. Li, Acta Polym. Sin., 2017, 2, 155 Search PubMed; (v) J. Wu, B. Wu, W. Wang, K. Chiang, A. Jen and J. Luo, Mater. Chem. Front., 2018, 2, 901 RSC.
- (a) B. Robinson, L. Dalton, A. Harper, A. Ren, F. Wang, C. Zhang, G. Todorova, M. Lee, R. Aniszfeld, S. Garner, A. Chen, W. Steier, S. Houbrecht, A. Persoons, I. Ledoux, J. Zyss and A. Jen, Chem. Phys., 1999, 245, 35 CrossRef; (b) L. Dalton, W. Steier, B. Robinson, C. Zhang, A. Ren, S. Garner, A. Chen, T. Londergan, L. Irwin, B. Carlson, L. Fifield, G. Phelan, C. Kincaid, J. Amend and A. Jen, J. Mater. Chem., 1999, 9, 1905 RSC; (c) Z. Li, W. Wu, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2009, 1, 856 CrossRef PubMed; (d) B. Robinson and L. Dalton, J. Phys. Chem. A, 2000, 104, 4785 CrossRef; (e) W. Wu, Q. Huang, G. Qiu, C. Ye, J. Qin and Z. Li, J. Mater. Chem., 2012, 22, 18486 RSC; (f) D. Chen, C. Zhong, Y. Zhao, Y. Liu and J. Qin, Mater. Chem. Front., 2017, 1, 2085 RSC; (g) D. Chen, C. Zhong, Y. Zhao, L. Nan, Y. Liu and J. Qin, J. Mater. Chem. C, 2017, 5, 5199 RSC.
- (a) T. Kim, J. Kang, J. Luo, S. Jang, J. Ka, N. Tucker, J. Benedict, L. Dalton, T. Gray, R. Overney, D. Park, W. Herman and A. Jen, J. Am. Chem. Soc., 2007, 129, 488 CrossRef PubMed; (b) M. Ronchi, A. Biroli, D. Marinotto, M. Pizzotti, M. Ubaldi and S. Pietralunga, J. Phys. Chem. C, 2011, 115, 4240 CrossRef; (c) D. Knorr, S. Benight, B. Krajina, C. Zhang, L. Dalton and R. Overney, J. Phys. Chem. B, 2012, 116, 13793 CrossRef PubMed.
- (a) H. Ma, S. Liu, J. Luo, S. Suresh, L. Liu, S. Kang, M. Haller, T. Sassa, L. Dalton and A. Jen, Adv. Funct. Mater., 2002, 12, 565 CrossRef; (b) Q. Zeng, Z. Li, Z. Li, C. Ye, J. Qin and B. Tang, Macromolecules, 2007, 40, 5634 CrossRef.
- (a) Z. Li, Z. Li, C. Di, Z. Zhu, Q. Li, Q. Zeng, K. Zhang, Y. Liu, C. Ye and J. Qin, Macromolecules, 2006, 39, 6951 CrossRef; (b) W. Wu, Y. Fu, C. Wang, C. Ye, J. Qin and Z. Li, Chem. – Asian J., 2011, 6, 2787 CrossRef PubMed; (c) R. Tang and Z. Li, Chem. Rec., 2017, 17, 71 CrossRef PubMed.
- (a) W. Wu, C. Ye, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2013, 5, 7033 CrossRef PubMed; (b) W. Wu, L. Huang, Y. Fu, C. Ye, J. Qin and Z. Li, Chin. Sci. Bull., 2013, 58, 2753 CrossRef; (c) R. Tang, H. Chen, S. Zhou, W. Xiang, X. Tang, B. Liu, Y. Dong, H. Zeng and Z. Li, Polym. Chem., 2015, 6, 5580 RSC; (d) G. Liu, P. Chen, R. Tang and Z. Li, Sci. China: Chem., 2016, 59, 1561 CrossRef.
- (a) R. Tang, S. Zhou, Z. W. Xiang, Y. Xie, H. Chen, Q. Peng, G. Yu, B. Liu, H. Zeng, Q. Li and Z. Li, J. Mater. Chem. C, 2015, 3, 4545 RSC; (b) R. Tang, S. Zhou, Z. Cheng, G. Yu, Q. Peng, H. Zeng, G. Guo, Q. Li and Z. Li, Chem. Sci., 2017, 8, 340 RSC.
- P. Chen and Z. Li, Chin. J. Polym. Sci., 2017, 35, 793 CrossRef.
- P. Chen, X. Yin, Y. Xie, S. Li, S. Luo, H. Zeng, G. Guo, Q. Li and Z. Li, J. Mater. Chem. C, 2016, 4, 11474 RSC.
- L. Dalton, Opt. Eng., 2000, 39, 589 CrossRef.
- (a) W. Wu, C. Li, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, Chem. – Eur. J., 2012, 18, 11019 CrossRef PubMed; (b) W. Wu and Z. Li, Polym. Chem., 2014, 5, 5100 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00128f |
| This journal is © the Partner Organisations 2018 |