
Molecular modulation of fluorene-dibenzothiophene-S,S-dioxide-based conjugated polymers for enhanced photoelectrochemical water oxidation under visible light†
Chunhui
Dai‡
 a,
Xuezhong
Gong‡
b,
Xianglin
Zhu
b,
Can
Xue
*b and
Bin
Liu
*a
a,
Xuezhong
Gong‡
b,
Xianglin
Zhu
b,
Can
Xue
*b and
Bin
Liu
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: cheliub@nus.edu.sg
bSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: cxue@ntu.edu.sg
First published on 30th August 2018
Abstract
Three conjugated polymers (CPs) bearing a common fluorene-dibenzothiophene-S,S-dioxide backbone were designed and synthesized for photoelectrochemical (PEC) water oxidation upon visible-light irradiation (>420 nm). Without the assistance of additional sacrificial agents or co-catalysts, a good photocurrent response of 4.1 μA cm−2 at +0.6 V (vs. Ag/AgCl) was achieved through facile molecular modulation of the polymer, which improved the uniformity and crystallinity of the film and facilitated charge transport among polymer chains. Moreover, C12 was used for preparation of a composite film with TiO2 nanorod arrays via spin-coating to yield an approximately 11-fold higher anodic PEC activity than that of pure TiO2. Our results demonstrated the great potential of solution-processable conjugated polymers for facile construction of highly efficient PEC devices for water splitting.
Introduction
Water splitting via a photoelectrochemical (PEC) approach is receiving increasing attention owing to its great potential for alleviating the global energy crisis though conversion of solar energy into molecular hydrogen and oxygen, which promises to be clean, sustainable and cost-effective.1 Since the first demonstration of PEC activity of n-type TiO2 by Fujishima and Honda in 1972, a wide variety of inorganic photoelectrode materials have been developed, whereas organic semiconductors have been exploited relatively seldomly.2 In fact, organic materials present several advantages: excellent light-harvesting, high charge mobility, flexible nature, and tunable photophysical properties.3 To date, graphitic carbon nitrides (g-C3N4) and their composites have been the most widely studied organic-derived photoelectrode materials, and significant progress has been achieved in this field.4 However, apart from the low-efficiency synthesis at high temperature and limited activity of g-C3N4, the photoelectrodes fabricated by insoluble g-C3N4 suffer from uniform thickness and poor ohmic contact between the polymer layers and conductive substrate, which have greatly limited their PEC performance.4,5 Moreover, like many inorganic catalysts, the insolubility of g-C3N4 makes it rather challenging to process. In this regard, research on the next generation of organic photoelectrode materials with facile fabrication and high efficiency for practical application is of great scientific interest.Solution-processable conjugated polymers (CPs) have been used widely in photoelectronic materials thanks to their easy production and processing, low cost, as well as outstanding optoelectronic properties. Large-scale PEC devices can be prepared readily by low-cost printing and roll-to-roll coating techniques. More generally, the solution processability of CPs should also enable the facile fabrication of nanostructured conjugated polymer films with significantly enhanced photocurrent density as a result of increased junction area and more effective charge transport.1a,6 Based on these advantages, development of photocatalytic electrolysis of water using soluble CPs is very promising. Recently, Ullah and colleagues reported two carbazole-based copolymers with various alkyl chains for PEC water oxidation. However, a poor photocurrent response of 0.31 μA cm−2 (+0.6 V vs. SCE) was achieved under one sun.7
To achieve greater PEC activity, one approach is to improve the charge transport in films by strengthening the π–π packing of the conjugated backbones, which could be accomplished by rational molecular modulation to impact the self-assembly of polymer chains in the film.8 Typically, regular interchain packing could minimize recombination and, thus, promote charge transport between rigid polymer backbones. Inspired by this strategy, three polymers (C6-s, C6-i, and C12) were constructed to balance the stacking of conjugated backbones in film to pursue high photocatalytic efficiency (Scheme 1). Fluorene-dibenzothiophene-S,S-dioxide was selected as the common backbone due to its structural features, such as efficient photoexcited charge generation and separation, good stability, tunable solubility, and crystallinity.9 Moreover, benefiting from the excellent solubility and appropriate energy level of C12, a C12/TiO2 composite film was fabricated by spin-coating to further improve PEC activity.
 | ||
| Scheme 1 Chemical structures of the conjugated polymers synthesized for PEC water splitting. | ||
Results and discussion
Synthesis and characterization of polymers
The synthetic routes for the three polymers are shown in Scheme S1 (ESI†). 3,7-Dibromodibenzothiophene-S,S-dioxide was employed as the common monomer and synthesized according to the literature.10 The three polymers were obtained by Pd-catalyzed coupling of the corresponding fluorene-based monomers via the Suzuki reaction. Polymerization was carried out under similar conditions. To limit the molecular weight and obtain a small amount of soluble polymer (C6-s), the reaction time for C6 (C6-s and C6-i portions) was restricted to 5 h, which was shorter than that for C12 (24 h). The measured number average molecular weight (Mn) and polydispersity index (PDI) of the polymers from gel permeation chromatography (GPC) were Mn = 4810, PDI = 1.52 for C6-s, and Mn = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 040 and PDI = 1.27 for C12. Notably, thermogravimetric analysis (TGA) revealed that all three polymers had high thermal stability with onset decomposition temperatures (Td) exceeding 300 °C at a 5% weight loss under a N2 atmosphere (Fig. S4, ESI†).
040 and PDI = 1.27 for C12. Notably, thermogravimetric analysis (TGA) revealed that all three polymers had high thermal stability with onset decomposition temperatures (Td) exceeding 300 °C at a 5% weight loss under a N2 atmosphere (Fig. S4, ESI†).
Photophysical and electrochemical properties
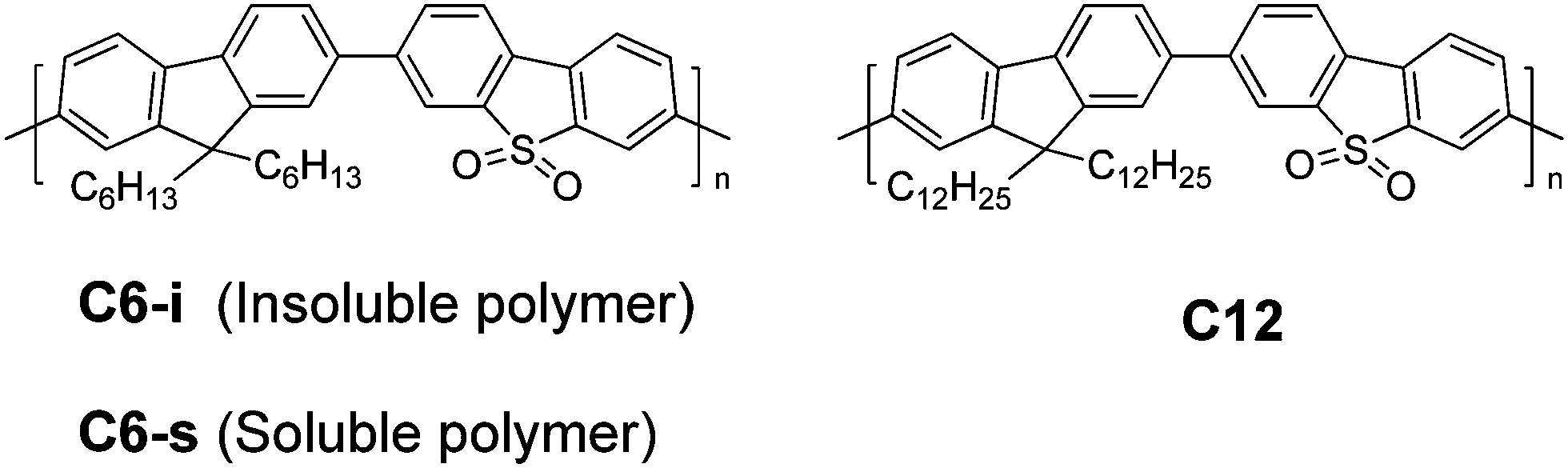
The optical properties of the three polymers were evaluated by UV-vis absorption spectra in CH2Cl2 solution and solid state (Fig. 1). The detailed data are summarized in Table 1.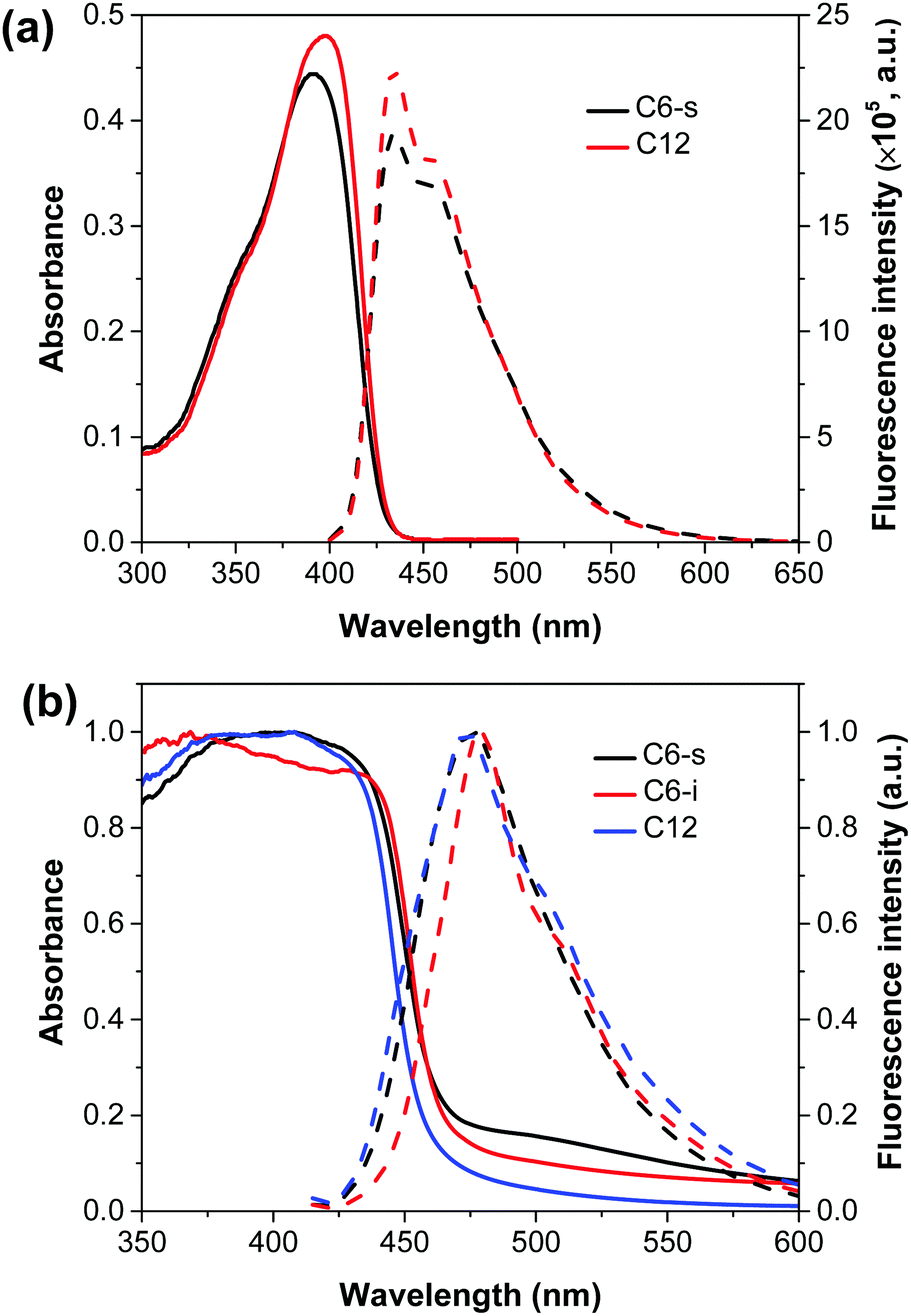 | ||
| Fig. 1 (a) UV-vis absorption (solid lines) and photoinduced fluorescence (dashed lines) spectra of C6-s and C12 in CH2Cl2 solution (10 μM; λex = 380 nm). (b) UV-vis diffuse reflectance spectra (solid lines) and steady-state fluorescence spectra of the polymer powders (dashed lines; λex = 380 nm). | ||
| Polymers | λ abs (nm) | λ em (nm) | λ onset (nm) | E g,opt (eV) | E g,ec (eV) | E LUMO (eV) | E HOMO (eV) |
|---|---|---|---|---|---|---|---|
| a In CH2Cl2 solution. b Polymer powder. c Estimated from the absorption onset (Eg = 1240/λonset). d Results from cyclic voltammetry. | |||||||
| C6-s | 393 | 475 | 466 | 2.66 | 2.63 | −2.78 | −5.41 |
| C6-i | — | 479 | 465 | 2.67 | 2.66 | −2.77 | −5.43 |
| C12 | 398 | 474 | 458 | 2.71 | 2.70 | −2.78 | −5.48 |
From the onset of the UV-vis diffuse reflectance spectra of the polymers, C6-s and C6-i exhibited a quite close bandgap (2.66 eV for C6-s and 2.67 eV for C6-i). By attaching the longer dodecyl side chains, an increased optical bandgap of ∼2.71 eV for C12 was obtained. Cyclic voltammetry (CV) was done to evaluate the electrochemical energy levels of the three polymers. Similar values (C6-s, 2.63 eV; C6-i, 2.66 eV; and C12, 2.70 eV) were found for the three polymers (Fig. S5, ESI†). Meanwhile, the emission of C12 (474 nm) was slightly blue-shifted in comparison with those for C6-s (475 nm) and C6-i (479 nm). Introduction of longer side chains should increase the dihedral angles between fluorene and the dibenzothiophene-S,S-dioxide unit, thereby interrupting the π-conjugation system and leading to blue-shifted absorption and emission in the solid state.11 In addition, C6-s and C12 showed similar absorption and emission spectra in CH2Cl2. Therefore, the three polymers maintained similar electronic properties, which allowed us to focus on the specific influence of polymer self-assembly in film on their PEC performances.
Fabrication and characterization of polymer films
For fair comparison, photoelectrodes were fabricated by drop-coating the polymer solution/suspension (0.5 mg mL−1) on the surface of fluorine-doped tin oxide (FTO) glass and then dried in air. The microstructures of the polymer films were studied by field emission scanning electron microscope (SEM) (Fig. 2a–c). Obviously, compared with C6-i, the films of C6-s and C12 on FTO glass showed a much more uniform and smooth surface due to better solubility. The crystallinity of the three polymer films was examined by X-ray diffraction (XRD), which revealed formation of an amorphous structure of C6-i (Fig. S6, ESI†) and C12, but C6-s was semi-crystalline, indicating more efficient packing of C6-s, which is favourable for charge transfer in films (Fig. 2d).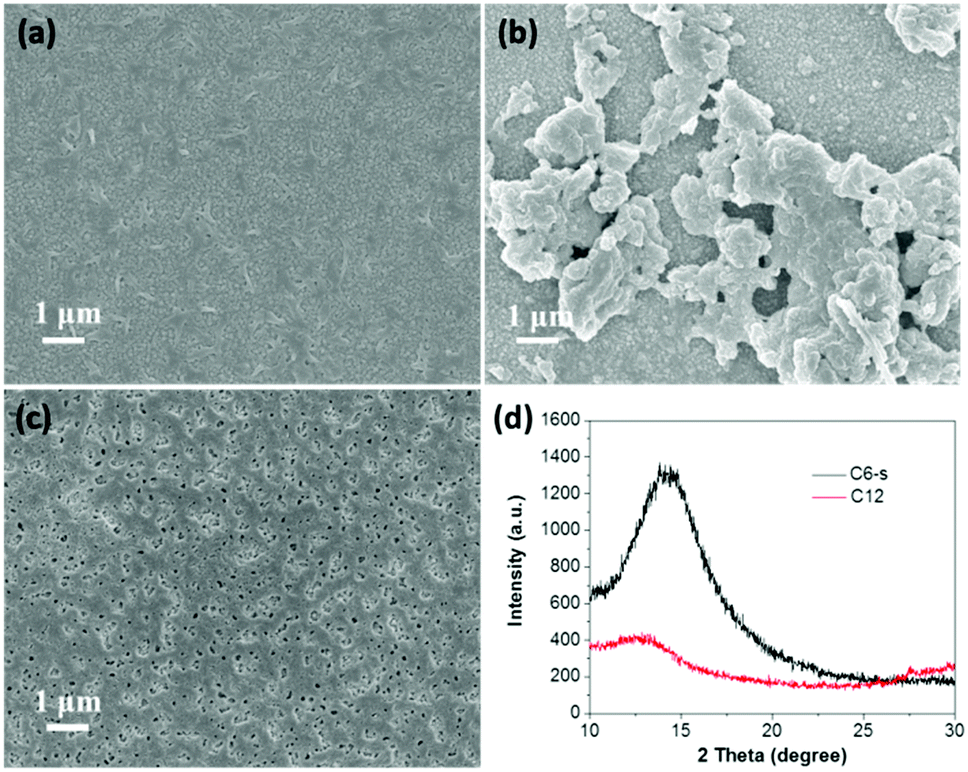 | ||
| Fig. 2 SEM images of C6-s (a), C6-i (b) and C12 (c) films on FTO substrate. (d) Powder X-ray diffraction pattern of C6-s and C12 films on FTO substrate. | ||
Photoelectrochemical performance
The PEC performance of the as-prepared polymer photoanodes was measured using a three-electrode system in 0.5 M of Na2SO4 (pH ∼ 7.0). The photoanode was used as the working electrode, whereas the counter electrode and reference electrode were Pt wire and Ag/AgCl electrode, respectively. Fig. 3a shows the current density–potential (i–V) curves of the polymers, and the corresponding current densities at +0.6 V vs. Ag/AgCl are displayed in Fig. 3b. C6-s electrode generated the highest photocurrent density (∼4.1 μA cm−2 at +0.6 V) vs. Ag/AgCl (1.20 V vs. RHE), which remained constant under chopped light illumination. In comparison, C6-i and C12 presented close but obviously lower photocurrent (1.1 μA cm−2 for C12, 1.2 μA cm−2 for C6-i). These results indicated the impact of the uniform and crystallinity nature of polymer films in PEC water splitting. | ||
| Fig. 3 Linear sweep voltammetry (LSV) curves (a) and transient photocurrent densities (b) of the three polymers in 0.5 M of Na2SO4 electrolyte under irradiation by visible light (λ > 420 nm). Nyquist plots (c) and Mott–Schottky plots (d) of the three polymer films. | ||
To understand the separation and transport of electron–hole pairs in the polymer electrode, we undertook electrochemical impedance spectroscopy on these photoelectrodes. The radii of the semicircles for C6-i and C12 were very close and smaller than C6-s (Fig. 3c), indicating more effective generation and transport of electron–hole pairs in C6-s film relative to those of C6-i and C12.
Mott–Schottky curves (Fig. 3d) showed that the flat-band potentials of C6-s, C6-i and C12 to be −1.15, −1.25 and −1.20 V vs. Ag/AgCl (−3.95, −3.85 and −3.90 eV vs. vacuum), respectively. Accordingly, we could construct a composite film utilizing the three polymers because of the matched conduction band level between the polymer layer and TiO2 (−4.2 eV vs. vacuum).5b The photoexcited electrons could migrate readily from the conduction band of the polymer to TiO2. The holes were transported from TiO2 to the polymer layer for water oxidation (Fig. 4).
 | ||
| Fig. 4 Photocatalysis of C12 on the surface of TiO2 nanorods under visible-light irradiation (schematic). | ||
Although C6-s presented the highest PEC efficiency, unfortunately its solubility was limited in tetrahydrofuran (THF), so processing it into a satisfactory composite with TiO2 was not possible. Therefore, C12 was chosen instead for its excellent solubility to allow processing by spin-coating (10 mg mL−1 in THF). The composite film was expected to combine the strong visible light-harvesting of C12 and excellent charge-transfer ability of TiO2.2a,4c,12a,b Hence, we first prepared TiO2 nanorods with a diameter of ∼250 nm by simple hydrothermal growth on the surface of FTO (Fig. 5a and b).12c Then, spin-coating of C12 on the TiO2 layer was done to form a composite film (Fig. 5c and d). The composite photoelectrode showed a photocurrent of 32.5 μA cm−2 at +0.6 V vs. Ag/AgCl (Fig. 5e), which is 11 times that of the bare TiO2 film, 29.5 times that of the pure C12 film, and 4 times that of bulk g-C3N4/TiO2. The tremendously increased PEC activity compared with C12 was probably due to improved charge separation at the contact surface of the materials, and allowed more efficient oxidation of water.13
 | ||
| Fig. 5 Morphology of the top view and lateral section of a TiO2/FTO film (a and b) and C12/TiO2/FTO film (c and d). (e) Transient photocurrent densities of C12/TiO2, pristine TiO2, and bulk g-C3N4/TiO2 (reference). | ||
Conclusions
We demonstrated molecular modulation of dibenzothiophene-S,S-dioxide-based alternating polymers to impact polymer self-assembly in films for PEC water oxidation. Replacement of hexyl with dodecyl resulted in a higher molecular weight and better solubility, but triggered a critical change of crystallinity in thin films, resulting in a dramatic drop in PEC performance. Moreover, we prepared a composite film of C12 and TiO2, which gave significantly improved photocurrent intensity of 32.5 μA cm−2 compared with C12 and TiO2 without additional sacrificial agents or a co-catalyst (λ > 420 nm). Our study offers a useful and important guideline for rational design of soluble semiconducting polymers for high-performance photoelectrodes.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are very grateful for the financial support from the National University of Singapore (R279-000-482-133), Singapore National Research Foundation (R279-000-444-281 and R279-000-483-281), and Singapore MOE AcRF-Tier1 (RG 12/15).Notes and references
- (a) M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef PubMed; (b) T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC; (c) D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef PubMed.
- (a) J. Sun, D. K. Zhong and D. R. Gamelin, Energy Environ. Sci., 2010, 3, 1252–1261 RSC; (b) H. M. Chen, C. K. Chen, R.-S. Liu, L. Zhang, J. Zhang and D. P. Wilkinson, Chem. Soc. Rev., 2012, 41, 5654–5671 RSC; (c) D. Kang, T. W. Kim, S. R. Kubota, A. C. Cardiel, H. G. Cha and K.-S. Choi, Chem. Rev., 2015, 115, 12839–12887 CrossRef PubMed; (d) Y. Zhao, N. Hoivik and K. Wang, Nano Energy, 2016, 30, 728–744 CrossRef; (e) S. Shen, S. A. Lindley, X. Chen and J. Z. Zhang, Energy Environ. Sci., 2016, 9, 2744–2775 RSC; (f) S. S. M. Bhat and H. W. Jang, ChemSusChem, 2017, 10, 3001–3018 CrossRef PubMed; (g) C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- (a) Y. Li and Y. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef; (b) J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC; (c) E. K. Lee, M. Y. Lee, C. H. Park, H. R. Lee and J. H. Oh, Adv. Mater., 2017, 29, 1703638 CrossRef PubMed.
- (a) F. Yang, V. Kuznietsov, M. Lublow, C. Merschjann, A. Steigert, J. Klaer, A. Thomas and T. Schedel-Niedrig, J. Mater. Chem. A, 2013, 1, 6407–6415 Search PubMed; (b) M. Basu, Z.-W. Zhang, C.-J. Chen, T.-H. Lu, S.-F. Hu and R.-S. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 26690–26696 CrossRef PubMed; (c) J. Su, L. Zhu and G. Chen, Appl. Catal., B, 2016, 186, 127–135 CrossRef; (d) J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef; (e) J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC; (f) H. Li, F. Zhao, J. Zhang, L. Luo, X. Xiao, Y. Huang, H. Ji and Y. Tong, Mater. Chem. Front., 2017, 1, 338–342 RSC.
- (a) A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23406–23433 RSC; (b) Q. Gu, X. Gong, Q. Jia, J. Liu, Z. Gao, X. Wang, J. Long and C. Xue, J. Mater. Chem. A, 2017, 5, 19062–19071 RSC.
- P. Bornoz, M. S. Prévot, X. Yu, N. Guijarro and K. Sivula, J. Am. Chem. Soc., 2015, 137, 15338–15341 CrossRef PubMed.
- M. Mansha, I. Khan, N. Ullah, A. Qurashi and M. Sohail, Dyes Pigm., 2017, 143, 95–102 CrossRef.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef.
- (a) I. I. Perepichka, I. F. Perepichka, M. R. Bryce and L.-O. Pålsson, Chem. Commun., 2005, 3397–3399 RSC; (b) J. Liu, J. Zou, W. Yang, H. Wu, C. Li, B. Zhang, J. Peng and Y. Cao, Chem. Mater., 2008, 20, 4499–4506 CrossRef; (c) C.-Y. Hsu, M.-T. Hsieh, M.-K. Tsai, Y.-J. Li, C.-J. Huang, Y.-K. Su and T.-J. Whang, Tetrahedron, 2012, 68, 5481–5491 CrossRef.
- K. Kawabata, M. Takeguchi and H. Goto, Macromolecules, 2013, 46, 2078–2091 CrossRef.
- K. T. Kamtekar, H. L. Vaughan, B. P. Lyons, A. P. Monkman, S. U. Pandya and M. R. Bryce, Macromolecules, 2010, 43, 4481–4488 CrossRef.
- (a) J. S. Salafsky, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 10885–10894 CrossRef; (b) X. Fan, T. Wang, B. Gao, H. Gong, H. Xue, H. Guo, L. Song, W. Xia, X. Huang and J. He, Langmuir, 2016, 32, 13322–13332 CrossRef PubMed; (c) E. S. Aydil, J. Am. Chem. Soc., 2009, 131, 3985–3990 CrossRef PubMed.
- X. Zhou, B. Jin, L. Li, F. Peng, H. Wang, H. Yu and Y. Fang, J. Mater. Chem., 2012, 22, 17900–17905 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00275d |
| ‡ C. H. D. and X. Z. G. contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |