
A highly sensitive and selective two-photon fluorescent probe for real-time sensing of cytochrome P450 1A1 in living systems†
Jing
Ning‡
a,
Zhenhao
Tian‡
b,
Bo
Wang‡
a,
Guangbo
Ge
c,
Yue
An
a,
Jie
Hou
a,
Chao
Wang
 a,
Xinyu
Zhao
a,
Yannan
Li
a,
Xiangge
Tian
a,
Zhenlong
Yu
a,
Xiaokui
Huo
a,
Chengpeng
Sun
a,
Lei
Feng
*abd,
Jingnan
Cui
b and
Xiaochi
Ma
*a
a,
Xinyu
Zhao
a,
Yannan
Li
a,
Xiangge
Tian
a,
Zhenlong
Yu
a,
Xiaokui
Huo
a,
Chengpeng
Sun
a,
Lei
Feng
*abd,
Jingnan
Cui
b and
Xiaochi
Ma
*a
aCollege of Pharmacy, Academy of Integrative Medicine, The National & Local Joint Engineering Research Center for Drug Development of Neurodegenerative Disease, Second Affliated Hospital, Dalian Medical University, Lvshun South Road No. 9, Dalian 116044, China. E-mail: leifeng@mail.dlut.edu.cn; maxc1978@163.com
bState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, China
cShanghai University of Traditional Chinese Medicine, Shanghai 201210, P. R. China
dInstitute of Functional Materials and Molecular Imaging, College of Emergency and Trauma, Hainan Medical University, Haikou, 571199, China
First published on 7th September 2018
Abstract
Cytochrome P450 1A1 (CYP1A1), a heme-containing monooxygenase, is of particular importance for human health because of its vital role in the metabolic activation of pro-carcinogenic compounds to their ultimate carcinogens. However, CYP1A1 protein levels are extraordinarily low in normal and cancer tissues. Thus, a practical method for ultra-sensitive and real-time monitoring of CYP1A1 activity in complex biological systems is highly sought after. In the present study, we developed a highly specific and sensitive two-photon fluorescent probe for monitoring CYP1A1 activity on the basis of the substrate preferences of this key enzyme. A panel of O-alkylated derivatives was designed and synthesized using HBN as the basic fluorophore. After screening and optimization, the derivative iPrBN was selected for further study as it displayed excellent specificity, high sensitivity and fast turn-on response to CYP1A1 relative to other human CYP isoforms. The detection limit of iPrBN for CYP1A1 was 0.036 nM, suggesting that it would be sensitive and versatile enough to detect endogenous CYP1A1 activity. Indeed, we successfully applied iPrBN to the real-time monitoring of CYP1A1 activity in human cancer cell lines and performed high-throughput screening of CYP1A1 modulators. iPrBN was also applied for the first time to the two-photon imaging of intracellular CYP1A1 in living cancer tissues and zebrafish, and our results showed that iPrBN exhibited high imaging resolution and fast response towards CYP1A1. These findings suggest that this probe is capable of accurately sensing CYP1A1 activity in complex biological systems, which will facilitate further investigations of CYP1A1-associated physiological and pathological processes.
Introduction
Cytochrome P450s (CYPs) are a superfamily of heme-containing monooxygenase enzymes that have a ubiquitous presence throughout the biological kingdoms.1 In mammals, the CYPs play a crucial role in the oxidative metabolism of a large number of structurally diverse chemicals, which include carcinogens, environmental chemicals, drugs, and endogenous compounds such as steroids and fatty acids.2,3 CYP1A1, a major member of the CYP1A subfamily, is of particular importance for human health because of its vital role in the metabolic bioactivation of pro-carcinogenic chemicals and toxins, such as polycyclic aromatic hydrocarbons (PAHs), heterocyclic aromatic amines/amides, and aminoazo dyes.4–6 Oxygenation of a pro-carcinogen can produce arene oxides, diol epoxides, and other electrophilic reactive species that form DNA and protein adducts, leading to tumor formation and organ toxicity.4,7,8 Many studies have demonstrated that an individual's susceptibility to lung cancer is strongly associated with the bioactivation of environmental carcinogens such as PAHs, particularly benzo[a]pyrene (B[a]P).9 Apart from its role in the metabolic activation of pro-carcinogenic chemicals, CYP1A1 is also involved in the metabolism of clinical drugs such as phenacetin, caffeine, warfarin, and other therapeutic agents.4,5 Thus, many studies on CYP1A1 activity and function have a profound influence on broad areas of biomedical science including pharmacology and toxicology, and provide important data on the metabolism of therapeutic agents and environmental chemicals.In the human body, CYP1A1 is generally expressed in extra-hepatic tissues, such as the lung epithelium, skin, and placenta. The expression of CYP1A1 in these tissues is extremely low but it is highly inducible by exogenous substances such as environmental chemicals and drugs.10,11 Several studies have reported that CYP1A1 is highly expressed in a wide range of tumor types and may greatly contribute to the development and progression of cancer.12 Epidemiological studies have confirmed the correlation between the aberrant high expression of CYP1A1 and the pathogenesis of neoplasms.13,14 Therefore, CYP1A1 has been regarded as a biomarker for cancer diagnosis and prevention.
In the past few decades, fluorescent probes have been widely used to monitor key enzymes in the human biological processes owing to their inherent advantages. These features include non-destructiveness, ease of use, and applicability to high-throughput screening and in situ imaging.15–21 Although several fluorescent probes for CYP1A enzymes have been reported and applied in drug metabolism-related fields, the isoform-specific probes for the highly sensitive sensing of CYP1A1 activity in complex biological systems have rarely been reported.22–25 Recently, a fluorescent probe (NBCeN) was reported to detect CYP1A1 activity in living cells and tissues.26 However, its LOD value (2.5 nM) and unsatisfactory selectivity towards other CYP isoforms limited its application to real-time monitoring or in situ imaging of CYP1A1, because of the extremely low levels of CYP1A1 in the extrahepatic tissues of living animals. Therefore, a practical fluorescent probe capable of specific, sensitive and real-time monitoring of CYP1A1 activities is urgently needed.
Herein, a practical two-photon fluorescent probe was developed for sensing CYP1A1 in complex biological systems, including human tissue preparations, live cells, and even zebrafish. For this purpose, 4-hydroxy-7H-benzo[de]benzo[4,5]imidazo[2,1-a] isoquinolin-7-one (HBN) was selected as the basic fluorophore because of its polycyclic aromatic skeleton and desirable photophysical properties such as high photostability, excellent fluorescence quantum yield, and two-photon absorbability.27,28 A panel of O-alkylated derivatives of HBN was then designed on the basis of the substrate preferences of CYP1A1 and the principle of intramolecular charge transfer (ICT).29 The potential of these HBNO-alkylated derivatives as CYP1A1 substrates was carefully investigated using reaction phenotyping assays. Among the substrates tested, 4-isopropyl-7H-benzo[de]benzo[4,5]imidazo[2,1-a] isoquinolin-7-one (iPrBN) displayed high sensitivity and selectivity towards CYP1A1 relative to the other CYP isoforms. The isopropyl group of iPrBN could be selectively cleaved by CYP1A1, with CYP1A1-mediated iPrBNO-deisopropylation triggering marked fluorescence emission around 525 nm (Scheme 1). The probe iPrBN also displayed beneficial optical properties, such as two-photon absorption behavior and sharp increases in fluorescence emission upon catalysis. We also demonstrated the applicability of an iPrBN-based fluorescence detection system for real-time monitoring and biological imaging of intracellular CYP1A1 activity in complex biological systems.
 | ||
| Scheme 1 Proposed response mechanism of iPrBN for sensing CYP1A1. | ||
Experimental section
Materials and instruments
The active cDNA-expressed recombinant human cytochrome P450 isoforms (rhCYP), including rhCYP1A1, 1A2, 1B1, 2A6, 2A13, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2J2, 3A4, 3A5, 4F2 and 4F3B, which were derived from baculovirus-infected insect cells coexpressing NADPH-P450 reductase, were purchased from Cypex (Dundee, UK). The human liver microsomes from individuals were obtained from the Research Institute for Liver Diseases (Shanghai, China). Glucose-6-phosphate dehydrogenase, NADPNa2 and D-glucose-6-phosphate were purchased from Sigma-Aldrich (MO, USA). Resveratrol, quercetin and myricetin were purchased from Shanghai Boyle Chemical Company (Shanghai, China). All other reagents, fine chemicals and liquid chromatography (LC) solvents were of the highest grade commercially available and were obtained from J&K Chemical Ltd (Shanghai, China) and Tedia (OH, USA).The human A549 and MCF-7 cell lines were obtained from ATCC (Manassas, VA). Small interfering RNAs (siRNA) against human CYP1A1 were obtained from Shanghai GenePharma Co. (Shanghai, China). Lipofectamine 2000 Reagent was purchased from Invitrogen (NY, USA). All other chemicals and reagents were of analytical grade or the highest quality commercial grade.
1H NMR and 13C NMR spectra were recorded on a Bruker Avance II (500 MHz). Accurate mass detection was performed with a Hybrid Ion Trap-Orbitrap Mass Spectrometer (LTQ Orbitrap XL, Thermo). Absorption spectra and fluorescence emission/excitation spectra were measured on a Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek).
Synthesis of iPrBN
The detailed synthesis and structural characterization of iPrBN and other derivatives of HBN are described in the ESI† (Scheme S-1 and Fig. S-1–S-4).General procedures for measuring CYP1A1 activity
The incubation mixture, with a total volume of 200 μL, consisted of 100 mM potassium phosphate buffer (pH 7.4), an NADPH-generating system (4 mM MgCl2, 1 unit per mL of glucose-6-phosphate dehydrogenase, 10 mM glucose-6-phosphate, and 1 mM NADP+), and a human tissue preparation or recombinant human CYP. In all experiments, iPrBN (dissolved in acetonitrile previously) was serially diluted to the required concentrations and the final concentration of acetonitrile did not exceed 1% (v/v) in the mixture. After pre-incubation at 37 °C for 3 min, the reaction was initiated by adding the NADPH-generating system and further incubated at 37 °C in a shaking water bath. The reaction was terminated by the addition of ice-cold acetonitrile. The mixture was kept on ice until it was centrifuged at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 20 min at 4 °C. Aliquots of the supernatants were taken for further fluorescence analysis. Control incubations without the NADPH-generating system or without substrate or without CYP enzyme sources were carried out to investigate whether the formation of metabolite(s) was enzyme (CYP)- and NADPH-dependent. Data are expressed as mean standard deviation (SD) of three independent measurements.
000 × g for 20 min at 4 °C. Aliquots of the supernatants were taken for further fluorescence analysis. Control incubations without the NADPH-generating system or without substrate or without CYP enzyme sources were carried out to investigate whether the formation of metabolite(s) was enzyme (CYP)- and NADPH-dependent. Data are expressed as mean standard deviation (SD) of three independent measurements.
Enzyme kinetic analysis
Briefly, CYP1A1 (1 nM) was incubated with iPrBN (0–2 μM) in 200 μL of the incubation mixture, which consisted of 100 mM potassium phosphate buffer (pH 7.4) and an NADPH-generating system (1 unit per mL of glucose-6-phosphate dehydrogenase, 4 mM MgCl2, 10 mM glucose-6-phosphate, and 1 mM NADP+). Prior to kinetic analysis, incubation time and protein concentration were optimized to obtain a linear range response. The reaction was initiated by adding NADP+ to a pre-incubated enzyme mixture at 37 °C. After 20 min of incubation, the reaction was terminated by adding an equal volume of icy acetonitrile. The formation of metabolites was determined by measuring the fluorescence intensity of HBN at 525 nm. Kinetic parameters (Km and Vmax) were determined by nonlinear regression analysis by GraphPad Prism 6.0.Determination of the detection of limit
The detection limit was determined based on the method reported in the previous literature.26,30 A mixture of iPrBN, NADPH-generating system, and CYP1A1 (0.0075 nM to 0.25 nM) in potassium phosphate buffer (pH 7.4, total volume 200 μL) was incubated for 30 min at 37 °C. The fluorescence emission spectrum of iPrBN was recorded five times and the standard deviation of the blank measurement was obtained. The fluorescence intensity at 525 nm was plotted as the concentration of CYP1A1. The detection limit was calculated by using 3σ/κ: where σ is the standard deviation of the blank measurement, and κ is the slope between the fluorescence intensity versus the CYP1A1 concentration.CYP1A1 inhibition assay
A mixture of iPrBN (10 μM), CYP1A1 (1 nM), the CYP1A1 selective inhibitor quercetin (0–100 μM) or myricetin (0–100 μM), and NADPH-generating system, in potassium phosphate buffer (pH 7.4) was incubated for 20 min at 37 °C. The IC50 values were determined by incubating iPrBN with different concentrations of quercetin or myricetin. The inhibitory effects were expressed as the percent decrease in the fluorescence intensities. Data were fit to log (inhibitor) vs. normalized response using the variable slope equation in GraphPad Prism 6.0 (San Diego, CA).Cytotoxicity assays
Cell viability was investigated by cholecystokinin octapeptide (CCK-8) assay. A549 and MCF-7 were seeded in a 96-well plate at a concentration of 5 × 104 mL−1 in 200 μL of Roswell Park Memorial Institute-1640 (RPMI-1640) medium with 10% FBS and maintained at 37 °C in a 5% CO2 incubator for 24 h. Then the cells were incubated with different concentrations of iPrBN and HBN for 24 h. The cells were washed once with PBS at 37 °C and a blank medium (serum-free medium) containing a final concentration of 0.5 mg mL−1 CCK-8 was added. The plates were shaken gently for 1 h and the absorbance was measured at 450 nm. The absorbance of the treated cells was compared with the absorbance of the controls, in which cells were exposed only to the vehicle and were considered to have 100% viability value.Cell culture and live cell imaging
A549 and MCF-7 cells were grown in RPMI-1640 culture medium (containing 10% FBS). Cells were seeded at a density of 1 × 105 cells per dish (Φ 20 mm) and incubated in a humidified incubator containing 5% CO2 at 37 °C overnight. The adherent cells were washed twice with FBS-free culture medium, and the stock solution of the probe iPrBN in DMSO was diluted into the cell culture media (FBS free) to a final concentration of 1 μM. The cells were then incubated at 37 °C for another 60 min, followed by rinsing with PBS (pH = 7.4) three times to remove the extracellular probe, and imaged under a confocal microscope (Olympus, FV1000). Images were acquired under 800 nm excitation and a fluorescence emission window of 520–560 nm.siRNA design and transfection
Two small interfering (si) RNAs against human CYP1A1 were designed by Shanghai GenePharma Co., with the following sequences: 5′-GCU AUC GAC AAG GUG UUA ATT-3′, and 5′-UUA ACA CCU UGU CGA UAG CTT-3′. Cells were transiently transfected with CYP1A1 siRNAs using Lipofectamine 2000 Reagent, followed by Western blotting or other experiments.In vivo imaging in zebrafish
Zebrafish were maintained in embryo media (15 mM NaCl, 0.5 mM KCl, 1 mM MgSO4, 1 mM CaCl2, 0.15 mM KH2PO4, 0.05 mM Na2HPO4 and 0.7 mM NaHCO3; pH = 7.4). In fluorescence imaging experiments, seven day old zebrafish were incubated with 1 μM iPrBN in E3 embryo medium for 60 min at 28 °C. After washing with PBS (pH = 7.4) to remove the remaining iPrBN, the zebrafish were imaged on a confocal microscope (Olympus FV1000). To ensure that the fluorescence signal was probe dependent, control incubations were performed simultaneously, which excluded iPrBN. The fish were rinsed with embryo medium three times to remove the unabsorbed probe and imaged under a confocal microscope (Olympus, FV1000). Images were acquired under 800 nm excitation and the fluorescence emission window of 520–560 nm.Results and discussion
Design and synthesis of iPrBN
Considering that the active site cavity of CYP1A1 is larger than that of CYP1A2 and that CYP1A1 prefers to dealkylate larger O-alkyl groups, a panel of O-alkylated HBN derivatives were purposely designed and then evaluated for potential as substrates of CYP1A1 via reaction phenotyping assays. The structures and chemical characteristics of these derivatives are provided in Fig. S-1 (ESI†), which depicts the introduction of substituents comprising methyl (1 and 8), ethyl (2 and 9), propyl (3 and 10), isopropyl (4 and 11), benzyl (5 and 12), chloroethyl (6 and 13), and chlorobutyl (7 and 14) groups. The detailed synthetic procedures for iPrBN are provided in Scheme S-1 (ESI†), and the chemical structure of iPrBN was fully characterized by HRMS, 1H NMR and 13C NMR spectroscopy (Fig. S-2–S-4, ESI†). Reaction phenotyping assays using a panel of human CYPs revealed that isoforms of the CYP1 subfamily displayed good activity towards O-alkylated HBN derivatives (1)–(7), but most of these derivatives could also be rapidly O-dealkylated by other CYPs including isoforms of the CYP2C and CYP3A subfamilies. In addition, all of the CYP isoforms, including the CYP1A isoforms, showed negligible activity towards the O-dealkylation of HBN derivatives (8)–(14). Among these derivatives, 4-isopropoxy-7H-benzo[de]benzo[4,5]imidazo[2,1-a]isoquinolin-7-one (4, iPrBN) displayed excellent reactivity and high specificity towards CYP1A1 relative to the other CYP isoforms, including CYP1B1 and CYP1A2 (Fig. S-5, ESI†). The isopropyl group of iPrBN was selectively cleaved by CYP1A1, leading to the release of 4-hydroxy-7H-benzo[de]benzo[4,5]imidazo[2,1-a]isoquinolin-7-one (HBN). The response mechanism of iPrBN for sensing CYP1A1 is depicted in Scheme 1.Spectral response of iPrBN towards CYP1A1
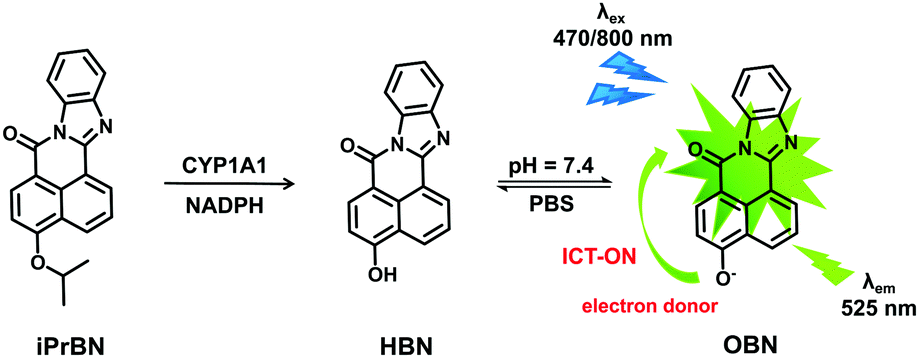
To test the validity of this probe, the spectral response of iPrBN towards CYP1A1 was investigated. Upon excitation at 470 nm, a marked fluorescence emission peak was observed at 525 nm (Fig. 1A). The fluorescence intensity increased linearly when CYP1A1 was added at progressively increasing concentrations from 0 to 6 nM (R2 = 0.9973, Fig. 1B). This distinct fluorescence response confirmed that CYP1A1-mediated deisopropylation of iPrBN liberated the oxygen atom as a strong electron donor in the D–π–A structure, thereby increasing the intramolecular charge transfer (ICT). The formation of HBN was identified by comparing the LC retention time and MS spectra with those of a standard compound (Fig. S-6, ESI†). The reaction was time-, NADPH-, and enzyme-dependent. The effects of pH on the fluorescence intensity of iPrBN and its metabolite HBN were also investigated and the results are shown in Fig. S-7 (ESI†). iPrBN was quite stable in the pH range from 7.0 to 12.0, as these pH values did not affect the fluorescence intensity of either iPrBN or HBN. Hence, iPrBN can be used to detect the CYP1A1 activity under physiological conditions (pH 7.4).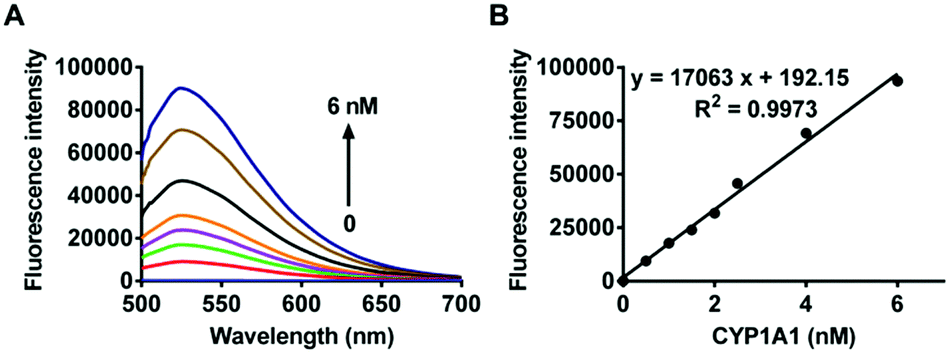 | ||
| Fig. 1 (A) Fluorescence spectra of iPrBN (10 μM) upon the addition of increasing concentrations of CYP1A1 (0–6 nM) in the reaction system incubated for 30 min. λex = 470 nm. (B) Fluorescence intensity of iPrBN at 525 nm upon the addition of increasing concentrations of CYP1A1 (0–6 nM) in the reaction system incubated for 30 min. | ||
Selectivity and sensitivity of iPrBN
The selectivity of iPrBN towards CYP1A1 was then examined under physiological conditions (pH 7.4 at 37 °C). A series of CYP isoforms were used to evaluate their participation in the formation of HBN. As shown in Fig. 2, among all the CYP isoforms examined, only CYP1A1 triggered a marked fluorescence emission. There was at least a 60-fold difference in the fluorescence intensity obtained with CYP1A1 when compared with the other two members of the CYP1 subfamily (including CYP1A2 and CYP1B1). This high specificity of iPrBN for CYP1A1 should enable the accurate detection of CYP1A1 activity in the complex biosamples. The selectivity of iPrBN for CYP1A1 over various potential interfering analytes was then investigated. As shown in Fig. S-8 (ESI†), iPrBN also exhibits high selectivity for CYP1A1 over the other factors examined, which may be ascribed to the specific oxidation of iPrBN by CYP1A1. These results indicated that iPrBNO-deisopropylation was selectively catalyzed by CYP1A1.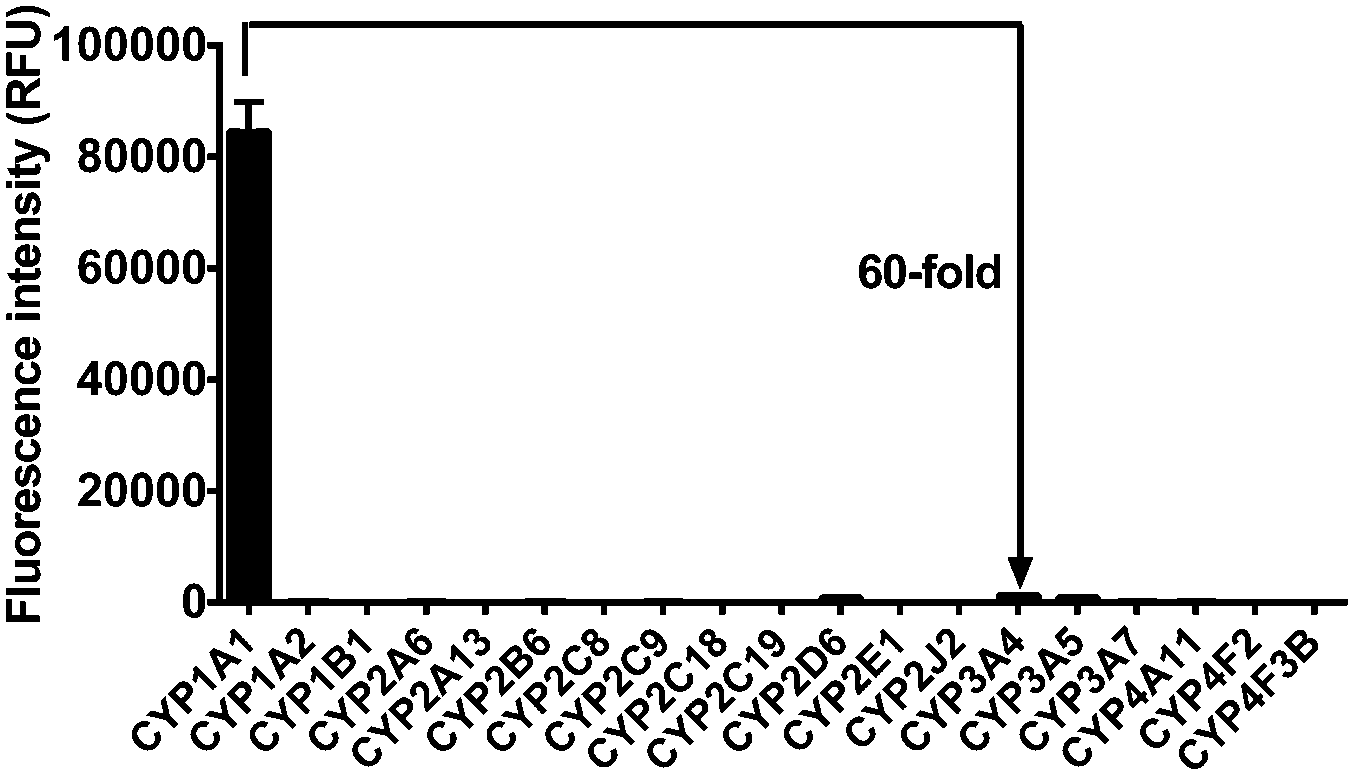 | ||
| Fig. 2 Fluorescence response of iPrBN (10 μM) at 525 nm (λex = 470 nm) upon the addition of various CYP isoforms in human. The spectra were measured in the reaction system incubated for 30 min. | ||
After confirming the high selectivity of iPrBN towards CYP1A1, we next focused on the sensitivity of CYP1A1 detection. Upon the addition of CYP1A1 at decreasing concentrations, the fluorescence intensity at 525 nm gradually decreased with a CYP1A1 detection limit of 0.036 nM (Fig. S-9, ESI†). This revealed the high sensitivity of our probe to changes in CYP1A1 expression. It is very noteworthy that the detection limit of iPrBN for CYP1A1 was almost 100-fold lower than that of the previously reported probe NBCeN (detection limit: 2.5 nM).26 The expression of CYP1A1 is usually extraordinarily low (around 2.27 fmol per μg tissue), which places greater demands on the activatable probe in terms of sensitivity.31 The apparent substrate affinity constant (Km) of iPrBN in the CYP1A1-catalyzed reaction was calculated to be 0.29 μM, which was lower than that of the previously reported probe NBCeN (0.84 μM) and indicated a higher affinity of iPrBN towards CYP1A1 (Fig. S-10, ESI†). The excellent selectivity and sensitivity of the present fluorescence-based assay fully suggested that iPrBN would enable the rapid and quantitative detection of CYP1A1 in biological samples.
Quantification of CYP1A1 in human tissue preparations
We next assessed the catalytic activity of CYP1A1 in individual human liver microsomal (HLM) samples. As shown in Fig. 3A, the activity for deisopropylation of CYP1A1 in a panel of HLM samples was measured by using iPrBN as the molecular probe. More than 3-fold variation in the catalytic activity was observed. To determine whether the rate of HBN formation reflected the catalytic activity of CYP1A1 in these individual samples, correlation analysis between the formation rates of HBN and the protein levels of CYP1A1 (determined with a proteomics-based approach) was carried out. As expected, a strong correlation was observed between the formation rate of HBN and the level of CYP1A1 in the panel of individual HLM samples (Fig. 3B). These results confirmed that iPrBN-O-deisopropylation exhibited excellent selectivity toward CYP1A1. These findings strongly suggest that iPrBN could serve as a novel tool for detecting trace amounts of CYP1A1 in human samples. | ||
| Fig. 3 (A) The catalytic activities of CYP1A1 in 13 individual human liver microsomes (HLM) using iPrBN as the probe substrate. (B) Correlation analysis between iPrBN (10 μM) O-deisopropylation and the level of CYP1A1 in HLM from individuals. The activities for iPrBN were expressed by the formation rate of HBN (λex/λem = 470/525 nm). | ||
High-throughput screening of CYP1A1 inhibitor
Recent studies have uncovered the important roles for CYP1A1 in the pathogenesis of a variety of human neoplasms,12–14,32 which indicates that the selective inhibition of CYP1A1 may be a promising therapeutic strategy for cancer.33,34 We subsequently employed iPrBN-O-deisopropylation as the probe reaction to rapidly screen CYP1A1 inhibitors. To this end, two known CYP1A1 inhibitors including quercetin and myricetin were used, while their inhibitory effects on CYP1A1-mediated iPrBN-O-deisopropylation were assayed in a 96-well microplate format. As shown in Fig. S-11 (ESI†), the IC50 values of quercetin and myricetin towards CYP1A1 were 4.12 and 2.79 μM, respectively. Notably, the inhibitory capacity of these two inhibitors as assessed with iPrBN was highly consistent with that determined by 7,8-diol-B[a]P epoxidation, a representative toxic reaction mainly catalyzed by CYP1A1.35,36 These results clearly showed that our probe iPrBN could be used as the detector for high-throughput screening of CYP1A1 inhibitors. Our assay may provide a much simpler alternative to the traditional in vitro assays using B[a]P analogs that mainly rely upon mass spectrometry, or high-performance liquid chromatography for detection.Bioimaging of CYP1A1 in living cells
We further explored whether iPrBN could be used for the tracking and imaging of endogenous CYP1A1 activity in living cells. Because of their high-level expression of CYP1A1, A549 and MCF-7 were chosen as the model cell lines for this experiment. To verify the selectivity of iPrBN for CYP1A1 in cell imaging, CYP1A1-specific siRNA was introduced into the cells by employing a RNA transfection method to down-regulate CYP1A1. The results of the cytotoxicity assays indicated that both iPrBN and its metabolite HBN had weak toxicity with superior biocompatibility towards the cultured cell lines (Fig. S-13 and S-14, ESI†). Incubation of the A549 and MCF-7 cells with 1 μM iPrBN for 1 h resulted in robust intracellular fluorescence signals as monitored by fluorescence microscopy (Fig. 4), strongly confirming the high basal activity of CYP1A1 in A549 and MCF-7 cells. We found that the RNA interference knockdown of CYP1A1 significantly blocked the fluorescence signal, which indicated that this fluorescence specifically occurred following iPrBNO-deisopropylation by the cellular CYP1A1. Furthermore, the dramatic change in fluorescence intensity between the control cells and the corresponding CYP1A1 knockdown cells was consistent with the changes in CYP1A1 protein expression observed in the cell lysates as determined by immunoblot analysis (Fig. 4B and E). Collectively, these results confirmed that iPrBN can be used to specifically measure endogenous CYP1A1 levels in living cells.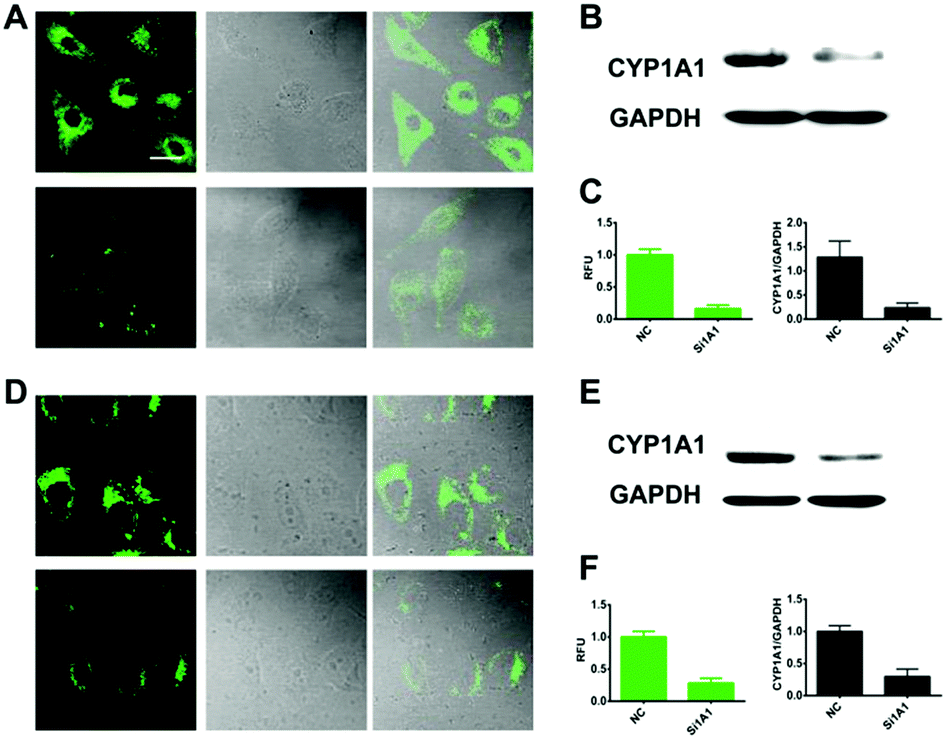 | ||
| Fig. 4 Two-photon confocal fluorescence images and graphical quantification of average fluorescence intensity of CYP1A1 activities by iPrBN in living human cancer cells. Confocal images of A549 (A-top) and MCF-7 (D-top) cells and the corresponding CYP1A1 knockdown cells (A-down and D-down) incubated with iPrBN (1 μM) for 60 min. Fluorescence images stained with iPrBN (1 μM), bright field images and overlay images are shown from left to right, respectively. The scale bar is 25 μm. Excitation, 800 nm; semiconductor laser emission, 520–560 nm. (B and E) The protein level of CYP1A1 in A549 and MCF-7 cells and the corresponding CYP1A1 knockdown cells analyzed by western blot. (C and F) Graphical quantification of average fluorescence intensity of CYP1A1 activities by iPrBN and gray analysis of CYP1A1 protein level in A549 and MCF-7 cells and the corresponding CYP1A1 knockdown cells. | ||
Encouraged by the good performance of iPrBN in living cells, we next sought to investigate its utility for screening of CYP1A1 inducer in the A549 cell model. Three different concentrations of dibenz[α,h]anthracene (DBA), which is a classical inducer of CYP1A1,34,37 were used to stimulate the expression of CYP1A1 in living cells. After pretreatment with DBA, the A549 cells showed a strong increase in fluorescence corresponding to increased CYP1A1 protein levels (Fig. 5). These findings clearly showed that iPrBN can be used to screen CYP1A inducers and confirmed the superior reliability of this iPrBN-based CYP1A1 measurement system.
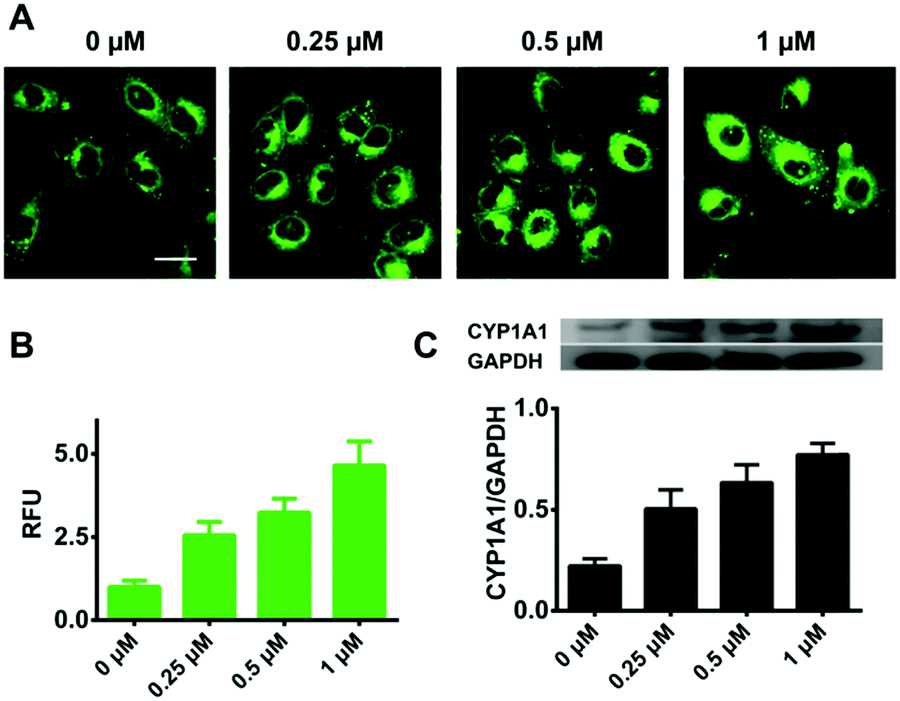 | ||
| Fig. 5 Two-photon confocal fluorescence images of DBA pretreated A549 cells. (A) Fluorescence images of A549 cells that were pretreated with 0, 0.25, 0.5, and 1 μM DBA and then incubated with iPrBN (1 μM) for 60 min. (B) Graphical quantification of the average fluorescence intensity of CYP1A1 activities by iPrBN in DBA pretreated A549 cells. (C) The protein level and gray analysis of CYP1A1 in DBA pretreated A549 cells analysed by western blot. Two photon images were acquired using 800 nm excitation and the fluorescence emission window was 520–560 nm. Scale bar: 25 μm. | ||
Bioimaging of CYP1A1 in human tumor sections
Epidemiological studies have shown that the over-expression of CYP1A1 results in a significant increase in the incidence and pathogenesis of neoplasms.13,14,38 Therefore, CYP1A1 has become a biomarker and a target for cancer therapies, and the sensitive and specific detection of CYP1A1 would prove to be very helpful for cancer imaging and diagnosis. Inspired by our results, we next investigated the utility of iPrBN for imaging of CYP1A1 in resected tumor tissues using two-photon microscopy. After a frozen section of metastatic breast carcinoma was subjected to iPrBN staining for 10 min, marked fluorescence images were observed upon excitation at 800 nm (Fig. 6). It should be noted that only a short incubation period (10 min) for iPrBN staining was required to obtain a signal strong enough for measuring the activity of CYP1A1 in the tumor tissues. The two-photon fluorescence emission and rapid response to CYP1A1 make iPrBN a suitable tool for detecting CYP1A1, with the further advantages of simplicity, rapidity and high sensitivity. These findings strongly suggest that iPrBN would be a valuable intraoperative tool for imaging CYP1A1 in tissue sections, and it could be used for the surgical diagnosis of cancers.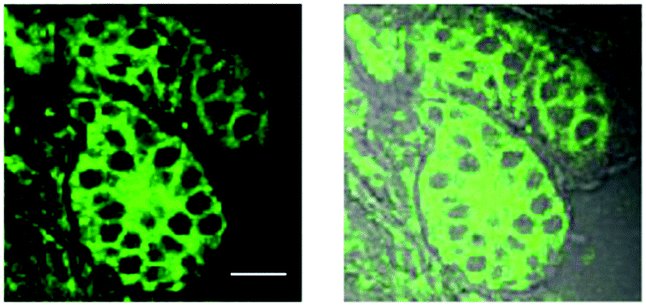 | ||
| Fig. 6 Two-photon confocal fluorescence images of the CYP1A1 in tumor tissue slices. Fluorescence images (left) of CYP1A1 in the frozen section of metastatic breast carcinoma, after staining with 1 μM iPrBN for 10 min. The overlay images of fluorescence and bright field (right). Excitation, 800 nm; semiconductor laser emission collected, 520–560 nm. The scale bar is 50 μm. | ||
In vivo imaging of CYP1A1 in zebrafish
Based on the excellent performance of iPrBN, we subsequently evaluated the CYP1A1 activity tracking ability of iPrBN in zebrafish. As shown in Fig. 7, zebrafish larvae displayed a strong, well-defined region of fluorescence, which precisely corresponded to the position of the liver.39,40 Our present results accurately reflected the specific expression sites and functional activity of CYP1A1 in living zebrafish. This fluorescence was greatly attenuated by pretreatment with CYP1A1 inhibitor resveratrol.41 All of the data, therefore, clearly demonstrated the specificity of iPrBN towards CYP1A1 in vivo. It should be noted that zebrafish-based small molecule screening has become a widely recognized and accepted method for drug evaluation, research, and development because this in vivo model closes the gap between high-throughput cell-based in vitro assays and low-throughput, expensive, and ethically controversial mammalian in vivo tests.42 Therefore, by using the newly developed two-photon probe for CYP1A1 (iPrBN), more rapid and sensitive quantitative detection of CYP1A1 may be achieved, which would facilitate in situ sensing of CYP1A1 and image-based screening of CYP1A1 modulators.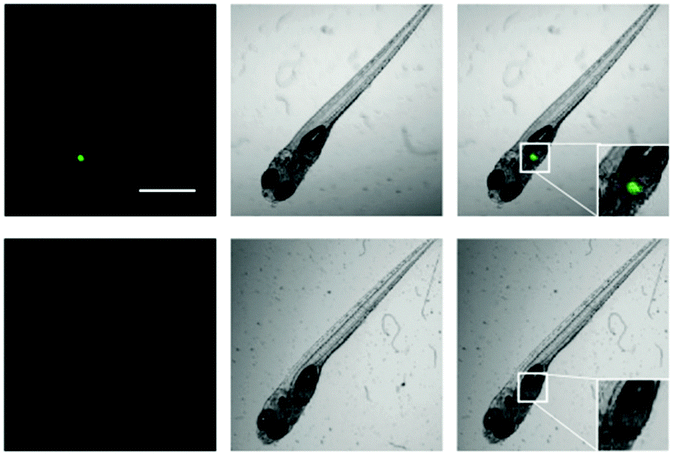 | ||
| Fig. 7 Two-photon confocal fluorescence images of CYP1A1 with iPrBN in living zebrafish. Fluorescence (left); brightfield (middle); merged (right). Zebrafish were treated with iPrBN (1 μM) for 60 min (top). Zebrafish were pretreated with resveratrol (10 μM) and then incubated with iPrBN (1 μM) for 60 min (down). Excitation, 800 nm; semiconductor laser emission collected, 520–560 nm. The scale bar is 100 μm. | ||
Conclusions
We developed a highly sensitive and selective two-photon fluorescent probe, iPrBN, for the real-time detection of human cytochrome P450 1A1. iPrBN was designed based on a local modification of the reaction site in the fluorophore HBN, and the differences in the catalytic cavity and substrate preference between CYP1A1 and other CYP isoforms. Among the various probes initially designed and tested, only the isopropyl-modified probe iPrBN displayed a fast response, high sensitivity, excellent selectivity, and marked fluorescence emission in the presence of CYP1A1. iPrBN could be used as an effective molecular tool to sensitively monitor CYP1A1 activity with a LOD of 0.036 nM, thereby providing a fluorescence off–on readout for the high-throughput screening of CYP1A1 modulators in complex biological systems. Two-photon confocal fluorescence imaging confirmed the CYP1A1 detection ability of iPrBN at the cellular level, the tissue level, and even at the level of the whole organism. Using this newly developed two-photon probe for CYP1A1, we have developed a more sensitive quantitative method for detecting CYP1A1, which will promote the future understanding of CYP1A1 biological functions, in situ sensing of CYP1A1, and image-based screening of CYP1A1 modulators.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 81622047, 81503201, 81473334 and 21572029), the Dalian Outstanding Youth Science and Technology Talent program (2015J12JH201) and the Distinguished Professor of Liaoning Province program for financial support, as well as the State Key Laboratory of Fine Chemicals (KF1603).Notes and references
- D. Fanni, R. Ambu, C. Gerosa, S. Nemolato, M. Castagnola, P. Van Eyken, G. Faa and V. Fanos, Int. J. Immunopathol. Pharmacol., 2014, 27, 5–13 CrossRef PubMed.
- T. Shimada, E. M. Gillam, P. Sandhu, Z. Guo, R. H. Tukey and F. P. Guengerich, Carcinogenesis, 1994, 15, 2523–2529 CrossRef PubMed.
- F. P. Guengerich, M. R. Waterman and M. Egli, Trends Pharmacol. Sci., 2016, 37, 625–640 CrossRef PubMed.
- A. H. Conney, Cancer Res., 1982, 42, 4875–4917 Search PubMed.
- D. Kim and F. P. Guengerich, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 27–49 CrossRef PubMed.
- V. P. Androutsopoulos, A. M. Tsatsakis and D. A. Spandidos, BMC Cancer, 2009, 9, 187 CrossRef PubMed.
- H. V. Gelboin, Physiol. Rev., 1980, 60, 1107–1166 CrossRef PubMed.
- K. P. Miller and K. S. Ramos, Drug Metab. Rev., 2001, 33, 1–35 CrossRef PubMed.
- R. N. Proctor, Nat. Rev. Cancer, 2001, 1, 82–86 CrossRef PubMed.
- Q. Ma, Curr. Drug Metab., 2001, 2, 149–164 CrossRef PubMed.
- L. E. Wohak, A. M. Krais, J. E. Kucab, J. Stertmann, S. Øvrebø, A. Seidel, D. H. Phillips and V. M. Arlt, Arch. Toxicol., 2016, 90, 291–304 CrossRef PubMed.
- Q. Ma and A. Y. Lu, Chem. Res. Toxicol., 2003, 16, 249–260 Search PubMed.
- G. I. Murray, J. Pathol., 2000, 192, 419–426 CrossRef PubMed.
- R. D. Bruno and V. C. Njar, Bioorg. Med. Chem., 2007, 15, 5047–5060 CrossRef PubMed.
- H. Zhang, J. Fan, J. Wang, S. Zhang, B. Dou and X. Peng, J. Am. Chem. Soc., 2013, 135, 11663–11669 CrossRef PubMed.
- J. Zhang, H. W. Liu, X. X. Hu, J. Li, L. H. Liang, X. B. Zhang and W. Tan, Anal. Chem., 2015, 87, 11832–11839 CrossRef PubMed.
- X. Wu, L. Li, W. Shi, Q. Gong and H. Ma, Angew. Chem., Int. Ed. Engl., 2016, 55, 14728–14732 CrossRef PubMed.
- X. F. Hou, J. Peng, F. Zeng, C. M. Yu and S. Z. Wu, Mater. Chem. Front., 2017, 1, 660–667 RSC.
- X. Zhang, Y. C. Yan, Q. Peng, J. Wang, Y. D. Hang and J. L. Hua, Mater. Chem. Front., 2017, 1, 2292–2298 RSC.
- T. Liu, J. Ning, B. Wang, B. Dong, S. Li, X. Tian, Z. Yu, Y. Peng, C. Wang, X. Zhao, X. Huo, C. Sun, J. Cui, L. Feng and X. Ma, Anal. Chem., 2018, 90, 3965–3973 CrossRef PubMed.
- J. Hu, K. Cheng, Q. Wu, D. S. Ding, C. G. Li and Z. Li, Mater. Chem. Front., 2018, 2, 1201–1206 RSC.
- D. M. Stresser, S. D. Turner, A. P. Blanchard, V. P. Miller and C. L. Crespi, Drug Metab. Dispos., 2002, 30, 845–852 CrossRef.
- A. Schiwy, M. Brinkmann, I. Thiem, G. Guder, K. Winkens, K. Eichbaum, L. Nüßer, B. Thalmann, S. Buchinger, G. Reifferscheid, T. B. Seiler, B. Thoms and H. Hollert, Nat. Protoc., 2015, 10, 1728–1741 CrossRef PubMed.
- Z. R. Dai, G. B. Ge, L. Feng, J. Ning, L. H. Hu, Q. Jin, D. D. Wang, X. Lv, T. Y. Dou, J. N. Cui and L. Yang, J. Am. Chem. Soc., 2015, 137, 14488–14495 CrossRef PubMed.
- X. X. Zhang, Y. Zhou, X. F. Gu, Y. Cheng, M. X. Hong, L. Q. Yan, F. L. Ma and Z. J. Qi, Talanta, 2018, 186, 413–420 CrossRef PubMed.
- Z. R. Dai, L. Feng, Q. Jin, H. Cheng, Y. Li, J. Ning, Y. Yu, G. B. Ge, J. N. Cui and L. Yang, Chem. Sci., 2017, 8, 2795–2803 RSC.
- J. Cui, G. Q. Gao, H. Z. Zhao, Y. Z. Liu, H. L. Nie and X. L. Zhang, New J. Chem., 2017, 41, 11891–11897 RSC.
- Y. Y. Chen, L. T. Zhao, H. H. Fu, C. H. Rao, Z. Y. Li and C. X. Liu, New J. Chem., 2017, 41, 8734–8738 RSC.
- W. Tassaneeyakul, D. J. Birkett, M. E. Veronese, M. E. McManus, R. H. Tukey, L. C. Quattrochi, H. V. Gelboin and J. O. Miners, J. Pharmacol. Exp. Ther., 1993, 265, 401–407 Search PubMed.
- K. Zhang, J. Zhang, Z. Xi, L. Y. Li, X. Gu, Q. Z. Zhang and L. Yi, Chem. Sci., 2017, 8, 2776–2781 RSC.
- E. Miyauchi, M. Tachikawa, X. Declèves, Y. Uchida, J. L. Bouillot, C. Poitou, J. M. Oppert, S. Mouly, J. F. Bergmann, T. Terasaki, J. M. Scherrmann and C. Lloret-Linares, Mol. Pharmacol., 2016, 13, 2631–2640 CrossRef PubMed.
- A. Luch, Nat. Rev. Cancer, 2005, 5, 113–125 CrossRef PubMed.
- H. P. Ciolino and G. C. Yeh, Br. J. Cancer, 1999, 79, 1340–1346 CrossRef PubMed.
- Y. J. Chun, S. Y. Ryu, T. C. Jeong and M. Y. Kim, Drug Metab. Dispos., 2001, 29, 389–393 Search PubMed.
- H. Doostdar, M. D. Burke and R. T. Mayer, Toxicology, 2000, 144, 31–38 CrossRef PubMed.
- D. Schwarz, P. Kisselev and I. Roots, Eur. J. Cancer, 2005, 41, 151–158 CrossRef PubMed.
- K. Skupinska, I. Misiewicz and T. Kasprzycka-Guttman, Arch. Toxicol., 2007, 81, 183–200 CrossRef PubMed.
- A. Al-Dhfyan, A. Alhoshani and H. M. Korashy, Mol. Cancer, 2017, 16, 14 CrossRef PubMed.
- C. B. Kimmel, W. W. Ballard, S. R. Kimmel, B. Ullmann and T. F. Schilling, Dev. Dyn., 1995, 203, 253–310 CrossRef PubMed.
- D. M. Parichy, M. R. Elizondo, M. G. Mills, T. N. Gordon and R. E. Engeszer, Dev. Dyn., 2009, 238, 2975–3015 CrossRef PubMed.
- M. F. Yueh, M. Kawahara and J. Raucy, Toxicol. In Vitro, 2005, 19, 275–287 CrossRef PubMed.
- A. Hill, N. Mesens, M. Steemans, J. J. Xu and M. D. Aleo, Drug Metab. Rev., 2012, 44, 127–140 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures, tables, and synthetic routes. See DOI: 10.1039/c8qm00372f |
| ‡ Contributed equally to this manuscript. |
| This journal is © the Partner Organisations 2018 |