
The regioselective synthesis of 2-phosphinoylindoles via Rh(III)-catalyzed C–H activation†
Huanan
Wang
a,
Shuaiqi
Li
a,
Baiquan
Wang
 abc and
Bin
Li
*a
abc and
Bin
Li
*a
aState Key Laboratory of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: nklibin@nankai.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering, Tianjin 300071, P. R. China
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, shanghai 200032, P. R. China
First published on 22nd September 2017
Abstract
The redox-neutral annulation of N-nitrosoanilines with 1-alkynylphosphine oxides under rhodium catalysis was achieved. This step-economical protocol affords 2-phosphinoylindoles in excellent regioselectivity with good functional group tolerance.
As one of the most important classes of heteroaromatic compounds, indoles are widely found as core structural units in both biologically active natural products and pharmaceuticals, and other functional molecules.1 Recently, indole-based phosphines have attracted much attention due to their unique pharmacological properties and synthetic applications.2 Although methods for the synthesis of indolylphosphonates have been well developed,3 approaches towards indole-based phosphine oxides are still quite limited. Transition-metal-catalyzed direct C–H phosphorylation is a promising method for this purpose, which has been elegantly developed by the groups of Yang and Zou, respectively.4 On the other hand, the route in which an indolyl skeleton is formed with the concomitant incorporation of a phosphorus moiety is particularly attractive since it provides a more step-economical access. In this respect, the Yorimitsu and Oshima group reported a palladium-catalyzed annulation of 2-iodoanilines with 1-alkynylphosphine derivatives to form 2-phosphinoylindoles (Scheme 1a).5 The Tang and Zhao group in 2016 demonstrated the cyclization of N-Ts-2-alkynylanilines with H-phosphine oxides to afford 3-phosphinoylindole derivatives via a silver-mediated cascade phosphinoylation–cyclization–desulfonylation process (Scheme 1b).6 However, these impressive advances still have some drawbacks, such as the utilization of prefunctionalized starting materials, relative low regioselectivity or requirement of large excess of a metal complex as an oxidant. Thus, the development of novel methodologies to indole-based phosphine oxides in a more atom-economical and environmentally friendly manner remains highly desirable.
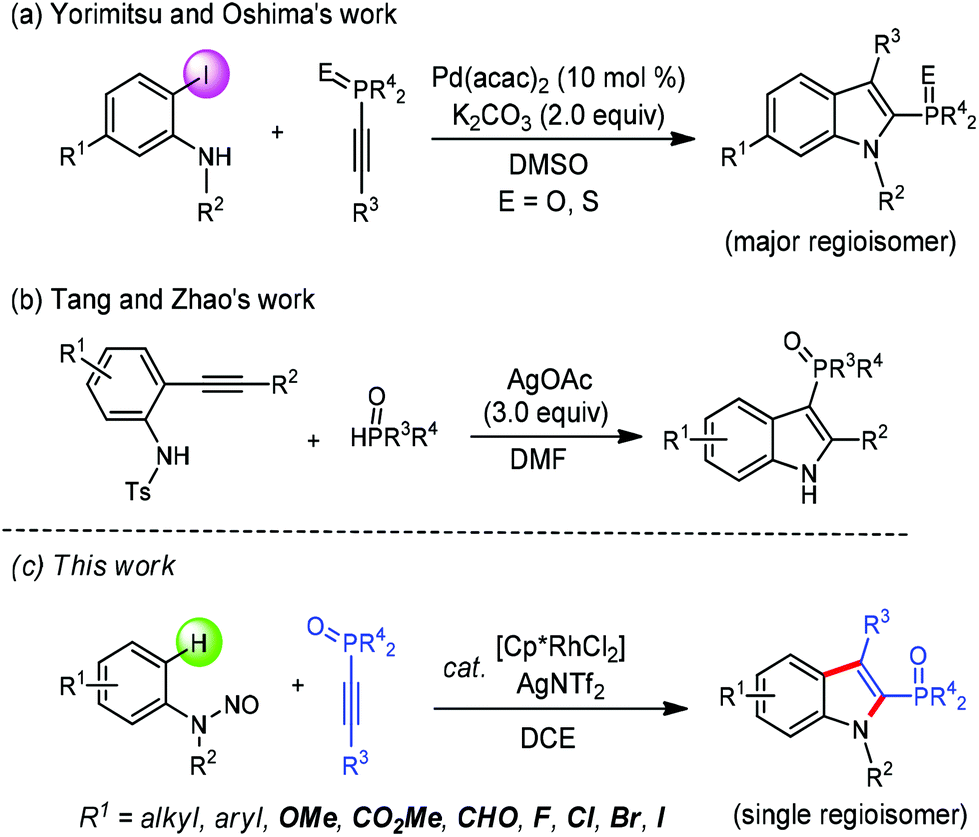 | ||
| Scheme 1 Routes for phosphinoylindole synthesis. | ||
In the past decade, the Rh(III)-catalyzed C–H activation and annulation strategy has provided straightforward and efficient approaches to access various (hetero)cyclic compounds.7 In this regard, oxidizing groups directing Rh(III)-catalyzed C–H annulation towards the indole scaffold synthesis have been realized.8 We envisioned that such a strategy could be employed in the synthesis of 2-phosphinoylindoles via the appropriate choice of coupling partners and reaction conditions. In continuation of our studies on transition-metal-catalyzed heterocycle synthesis through C–H coupling with alkynes and creation of new organophosphine species,9 we here disclosed a redox-neutral annulation of N-nitrosoanilines with 1-alkynylphosphine oxides under rhodium catalysis to afford 2-phosphinoylindole derivatives with excellent regiosectivity (Scheme 1c). The present catalytic system also features atom- and step-economy, broad substrate scope, and good functional-group tolerance.
At the outset of our studies, N-methyl-N-phenylnitrous amide (1a) and diphenyl(phenylethynyl)phosphine oxides (2a) were tested as model substrates for optimization experiments (Table 1). Subjection of 1a (1.5 equiv.) to the reaction with 2a in the presence of [Cp*Rh(MeCN)3][SbF6]2 (10 mol%), AgOAc (0.5 equiv.) in DCE at 80 °C indeed gave the product (1-methyl-3-phenyl-1H-indol-2-yl)diphenylphosphine oxide (3aa) in 39% NMR yield (entry 1). The structure of 3aa was established by 1H and 13C NMR spectroscopy and mass spectrometry, which is consistent with those reported previously.5 We then evaluated the effect of various reaction parameters for this benchmark reaction. It was found that the addition of either acidic or basic additives was detrimental to the reaction (entries 2–4 and Table S2 in the ESI†). Among various solvents screened, DCE was most optimal (entries 5–7 and Table S2†). The addition of 3.0 equiv. of 1a brought about increased product yields (entry 8). The application of [Cp*RhCl2]2/AgNTf2 as the catalyst can further enhance the reactivity (entry 9), thus maintaining the reaction efficiency at lower temperature (entry 10). Other metal catalysts like [Cp*Co(MeCN)3][SbF6]2 and [RuCl2(p-cymene)]2/AgSbF6 displayed no catalytic activity under the present reaction conditions (entries 11 and 12).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additives (equiv.) | Solvent | Yieldb (%) |
| a Reactions in 0.2 mmol scale (0.1 M). b Yield of the crude reaction mixture determined by 31P NMR (internal standard: trimethyl phosphate). c 1a (3.0 equiv.). d 60 °C. e Value in parentheses indicates isolated yield. DCM = dichloromethane, DCE = 1,2-dichloroethane, DMF = N,N-dimethylformamide, L = p-cymene. | ||||
| 1 | [Cp*Rh(MeCN)3][SbF6]2 (10) | AgOAc (0.5) | DCE | 39 |
| 2 | [Cp*Rh(MeCN)3][SbF6]2 (10) | HOAc (2.0) | DCE | 33 |
| 3 | [Cp*Rh(MeCN)3][SbF6]2 (10) | CsOAc (2.0) | DCE | Trace |
| 4 | [Cp*Rh(MeCN)3][SbF6]2 (10) | — | DCE | 64 |
| 5 | [Cp*Rh(MeCN)3][SbF6]2 (10) | — | DCM | 53 |
| 6 | [Cp*Rh(MeCN)3][SbF6]2 (10) | — | DMF | 0 |
| 7 | [Cp*Rh(MeCN)3][SbF6]2 (10) | — | PhMe | 45 |
| 8c | [Cp*Rh(MeCN)3][SbF6]2 (10) | — | DCE | 68 |
| 9c | [Cp*RhCl2]2/AgNTf2 (5/20) | — | DCE | 72 |
| 10c,d | [Cp*RhCl2]2/AgNTf2 (5/20) | — | DCE | 73 (68)e |
| 11c | [Cp*Co(MeCN)3][SbF6]2 (10) | — | DCE | 0 |
| 12c | [LRuCl2]2/AgSbF6 (5/20) | — | DCE | 0 |
After the establishment of the optimal conditions (Table 1, entry 10), the reaction scope with respect to N-nitrosoaniline derivatives was first explored by the adoption of 2a as the coupling partner (Scheme 2). To our delight, the cyclization proceeded smoothly with substrates bearing various arene substituents, affording the desired products 3aa–3ma in moderate to good yields (52–75%). In this respect, the electronic nature of the functional groups was found to little affect the outcome of reaction. For meta-substituted N-nitrosoanilines, annulation occurred at the less steric hindered aromatic C–H bond with exclusive regioselectivity (3la and 3ma), which was confirmed by HMBC spectroscopy of 3ka (see the ESI†). We were pleased to find that several functional groups such as methoxy (3ca and 3ma), ester (3ja), aldehyde (3ka), and moreover halogens including fluoro (3fa), chloro (3ga), bromo (3ha), especially iodo (3ia), remained intact under the present reaction conditions.10 These halides can further participate in cross-coupling reactions to achieve molecular complexity. It is noteworthy that in all cases the transformation occurred with perfect regioselectivity. All products were isolated as a single regioisomer with a diphenyloxyphosphino moiety being installed at the 2-position of indole, as confirmed by X-ray crystallographic analysis of the structure of 3ca (see the ESI†).11
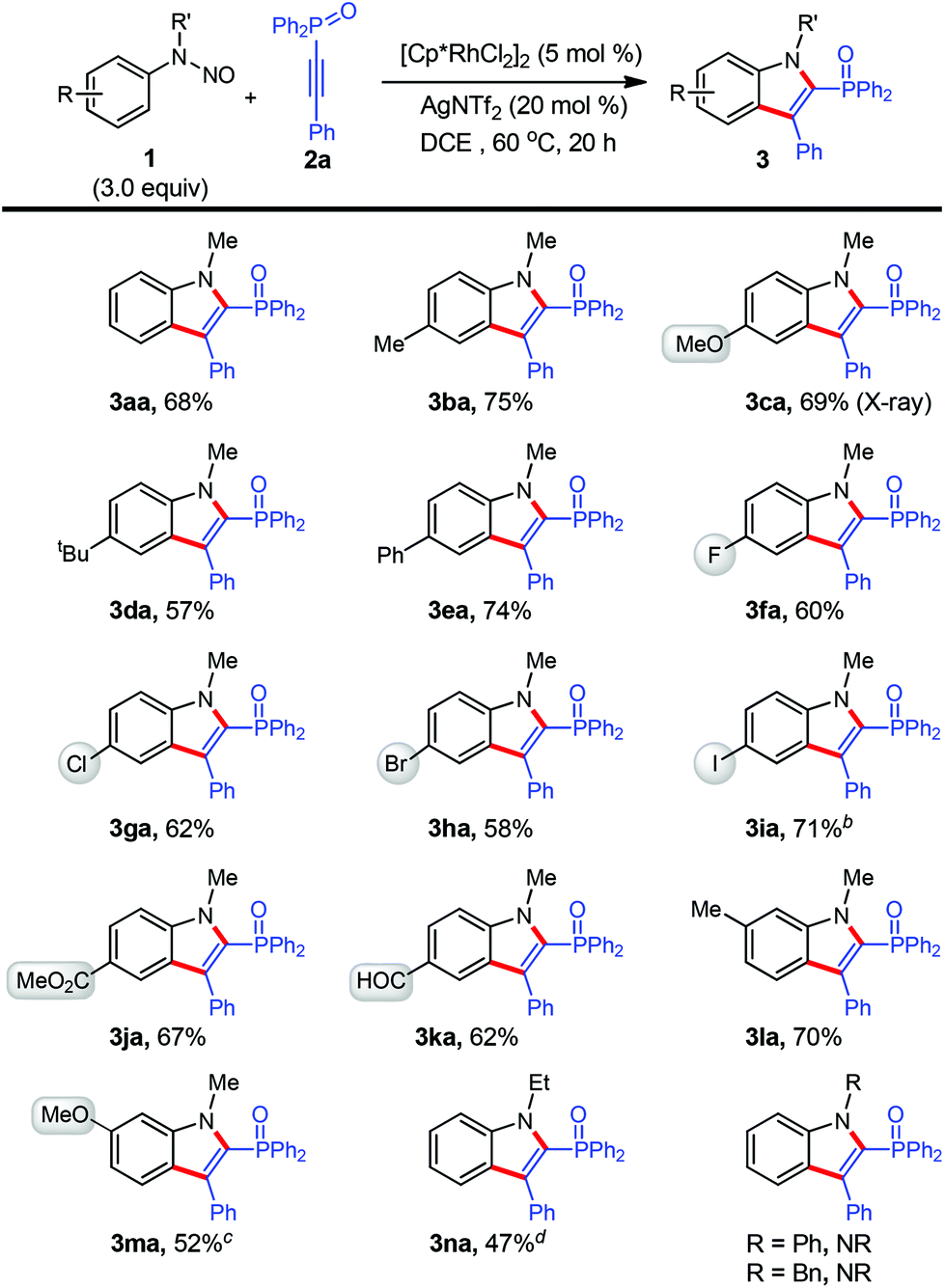 | ||
| Scheme 2 Scope of N-nitrosoanilines. aReactions in 0.2 mmol scale (0.1 M), isolated yield. b30 h. cDCE/TFE (0.5 mL/0.5 mL). TFE = 2,2,2-thifluoroethanol. dYield of the crude reaction mixture determined by 31P NMR (internal standard: trimethyl phosphate). | ||
The effect of different substituents on the N-nitrosoaniline nitrogen atom was also examined (Scheme 2). While an ethyl group was acceptable, product 3na could not be separated from the considerable amount of unreacted 2a and fully characterized. However, no reaction proceeded in cases of utilizing phenyl and benzyl groups.
To further demonstrate the generality of this reaction, various 1-alkynylphosphine oxides were tested. As shown in Scheme 3, irrespective of the electronic nature of arene substituents, 1-alkynylphosphine oxides 2b–e were readily converted into products 3ab–ae (49–66%). Not only were the arylacetylene derivatives 2a–e applicable to the reaction, but also alkyl-substituted substrates 2f and 2e underwent the annulation reactions although a higher reaction temperature was required. Alkyne 2h containing the di(p-toyl)oxyphosphino moiety reacted to furnish the desired indole product 3ah. Gratifyingly, (1-alkynyl)dicyclohexylphosphine oxide 2i was also an active coupling partner for the present annulation protocol, giving rise to the indole product 3ai in high yield. However, 1-alkynylphosphine sulfide as well as terminal alkyne (namely ethynyldiphenylphosphine oxide) failed to work under the current reaction conditions. Again, we were pleased to observe the formation of one single regioisomer for all phosphinoacetylenes employed, showing excellent regioselectivity in this reaction system. Furthermore, the regioselectivity was unambiguously confirmed by NOESY spectroscopy of 3af and X-ray crystallographic analysis of 3ai (see the ESI†).11
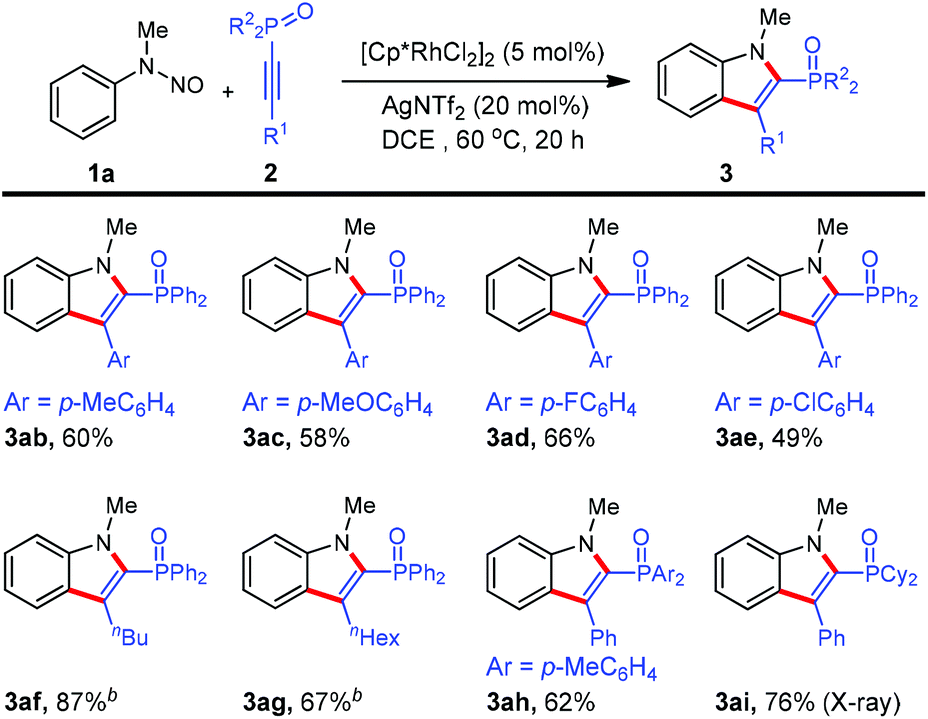 | ||
| Scheme 3 Scope of 1-alkynylphosphine oxides. aReactions in 0.2 mmol scale (0.1 M), isolated yield. b80 °C. | ||
We next conducted preliminary mechanistic studies to gain insights into the mechanism of this catalytic process (Scheme 4). First, H/D exchange experiments were carried out. No matter the absence or presence of phosphinoacetylene 2a, 70% deuterium was incorporated into the recovered 1a at the two ortho-positions of the directing group (Scheme 4a and b), suggesting a reversible of the C–H bond activation process. However, no deuterium incorporation was detected in product 3aa (Scheme 4b). This may arise from a fast cross-coupling with the alkyne prior to the reversible H/D exchange step. Second, a kinetic isotope effect (KIE) of 1.3 was observed from two parallel competition reactions between 1a and 1a-d5 (Scheme 4b), thus demonstrating that the C–H bond cleavage is likely not involved in the rate-determining step.12 Finally, isolated cyclometalated Rh(III) complex 48d successfully catalyzed the annulation reaction of 1f and 2a, which indicated that rhodacycle 4 might be an active species precursor in the catalytic cycle.
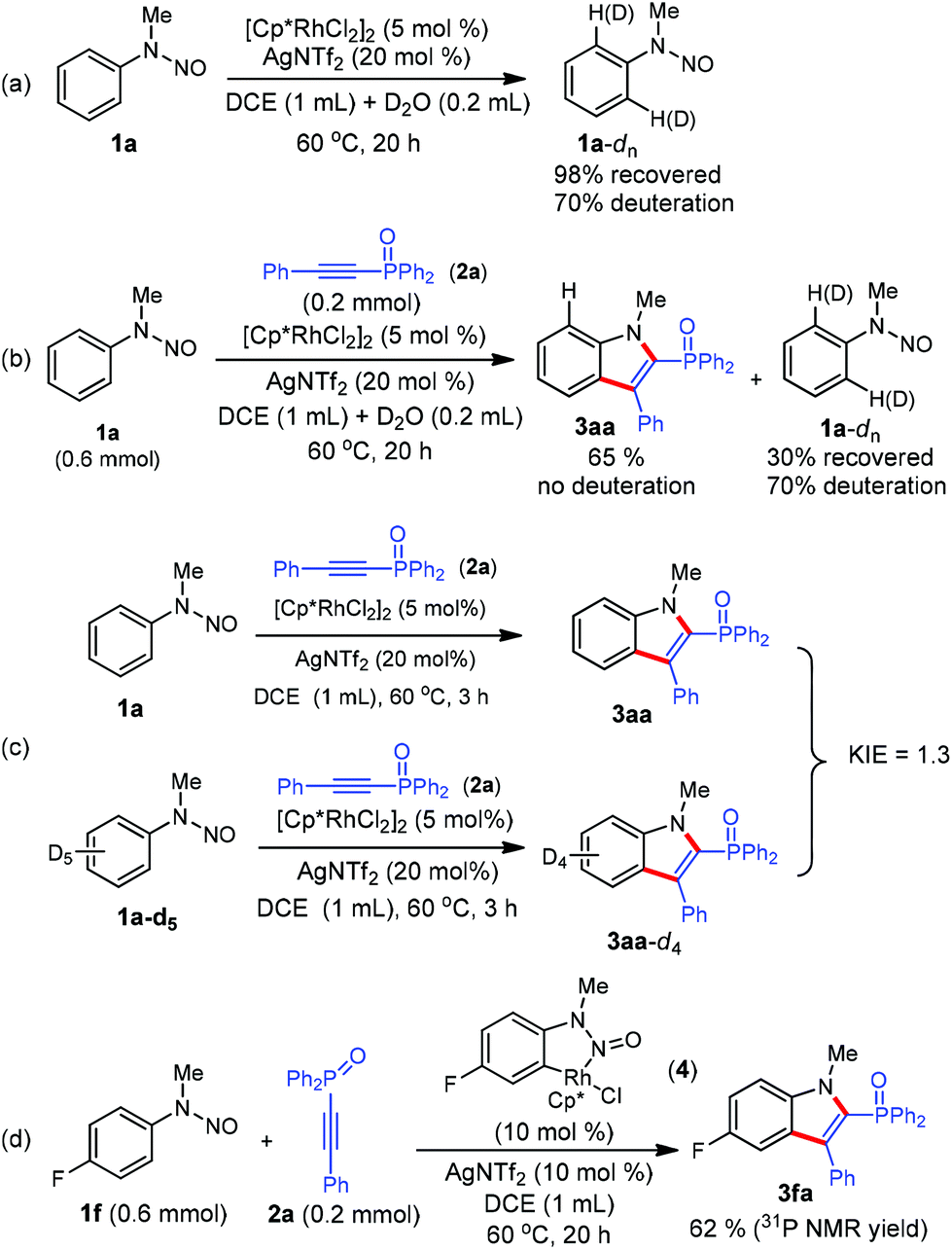 | ||
| Scheme 4 Mechanistic studies. | ||
Based on the above observations and precedent literature studies,8 a tentative reaction mechanism was proposed (Scheme 5). Initially, a reversible C–H activation forms a five-membered cyclometalated species A. After alkyne coordination and migratory insertion, a seven-membered rhodacycle intermediate B is thus generated. Then, the concerted N–N bond cleavage/C–N bond formation process8d occurs to furnish the final product 3aa and concomitantly regenerates the Rh(III) catalyst. The observed insertion outcome could be rationalized by the fact that the interaction of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O motif of 2a with the metal center governs the regioselectivity.
O motif of 2a with the metal center governs the regioselectivity.
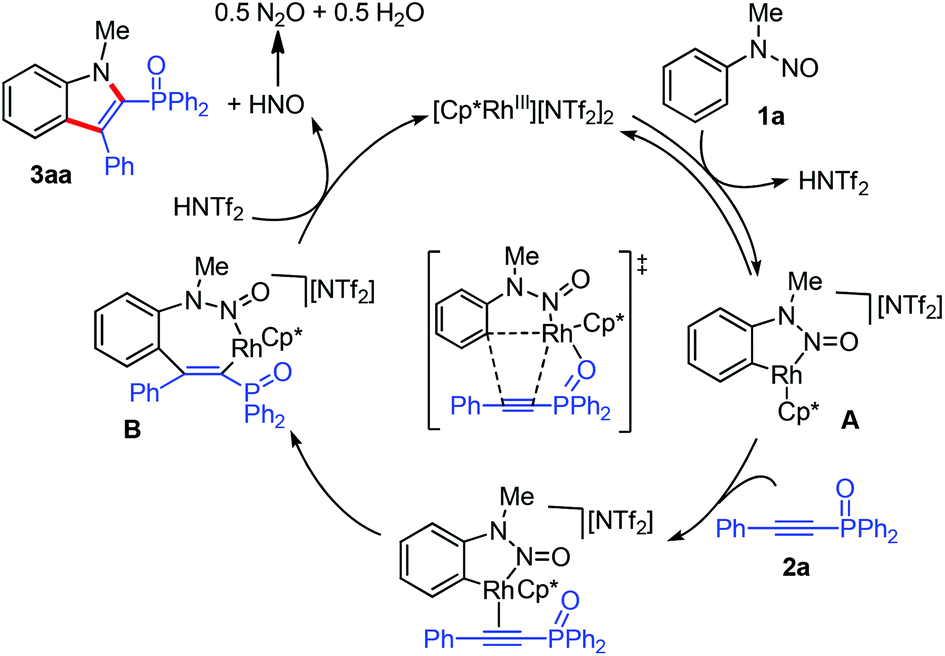 | ||
| Scheme 5 Proposed mechanism. | ||
In summary, we have realized a step-economical route for the synthesis of 2-phosphorylate indoles via Rh(III)-catalyzed redox-neutral C–H bond activation. This annulation approach exhibited excellent regioselectivity, good functional group tolerance and broad substrate scope. Efforts on the application of this catalytic system are underway.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the grants of the National Natural Science Foundation of China (21372121, 21672108, and 21421062) and the Natural Science Foundation of Tianjin (16JCZDJC31700).Notes and references
- For selected reviews, see: (a) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC; (b) S. A. Patil, R. Patil and D. D. Miller, Future Med. Chem., 2012, 4, 2085–2115 CrossRef CAS PubMed; (c) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed; (d) M. Somei and F. Yamada, Nat. Prod. Rep., 2005, 22, 73–103 RSC; (e) T. Kawasaki and K. Higuchi, Nat. Prod. Rep., 2005, 22, 761–793 RSC; (f) T. Eicher and S. Hauptmann, The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, Wiley-VCH, Weinheim, 2nd edn, 2003 Search PubMed; (g) R. J. Sundberg, Indoles, Academic Press, San Diego, 1996 Search PubMed.
- (a) G. L. Regina, A. Coluccia and R. Silvestri, Antiviral Chem. Chemother., 2010, 20, 213–237 CrossRef PubMed; (b) F. R. Alexandre, A. Amador, S. Bot, C. Caillet, T. Convard, J. Jakubik, C. Musiu, B. Poddesu, L. Vargiu, M. Liuzzi, A. Roland, M. Seifer, D. Standring, R. Storer and C. B. Dousson, J. Med. Chem., 2011, 54, 392–395 CrossRef CAS PubMed; (c) W. C. Fu, C. M. So, W. K. Chow, O. Y. Yuen and F. Y. Kwong, Org. Lett., 2015, 17, 4612–4615 CrossRef CAS PubMed; (d) T. Jia, A. Bellomo, K. E. L. Baina, S. D. Dreher and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 3740–3743 CrossRef CAS PubMed; (e) C. M. So and F. Y. Kwong, Chem. Soc. Rev., 2011, 40, 4963 RSC.
- (a) G. A. Russell and C. F. Yao, J. Org. Chem., 1992, 57, 6508–6513 CrossRef CAS; (b) S. Thielges, E. Meddah, P. Bisseret and J. Eustache, Tetrahedron Lett., 2004, 45, 907–910 CrossRef CAS; (c) H. L. Wang, X. C. Li, F. Wu and B. S. Wan, Synthesis, 2012, 941–945 Search PubMed; (d) S. H. Kim, K. H. Kim, J. W. Lim and J. N. Kim, Tetrahedron Lett., 2014, 55, 531 CrossRef CAS; (e) M. Yadav, S. Dara, V. Saikam, M. Kumar, S. K. Aithagani, S. Paul, R. A. Vishwakarma and P. P. Singh, Eur. J. Org. Chem., 2015, 6526–6533 CrossRef CAS.
- (a) A. X. Zhou, L. L. Mao, G. W. Wang and S. D. Yang, Chem. Commun., 2014, 50, 8529–8532 RSC; (b) B. S. Wang, J. F. Xue, G. Y. Zhang, R. S. Zeng, L. T. An, P. Z. Zhang and J. P. Zou, Adv. Synth. Catal., 2016, 358, 1753–1758 CrossRef.
- A. Kondoh, H. Yorimitsu and K. Oshima, Org. Lett., 2010, 12, 1476–1479 CrossRef CAS PubMed.
- Y. Gao, G. Lu, P. Zhang, L. Zhang, G. Tang and Y. Zhao, Org. Lett., 2016, 18, 1242–1245 CrossRef CAS PubMed.
- For recent reviews on Rh(III)-catalyzed C–H activation, see: (a) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212–11222 CrossRef CAS PubMed; (b) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31–41 CAS; (c) S. Chiba, Chem. Lett., 2012, 41, 1554–1559 CrossRef CAS; (d) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (e) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS; (f) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007–1020 CrossRef CAS PubMed; (g) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed; (h) M. Gulías and J. L. Mascareñas, Angew. Chem., Int. Ed., 2016, 55, 11000–11019 CrossRef PubMed.
- (a) K. Muralirajana and C. H. Cheng, Adv. Synth. Catal., 2014, 356, 1571–1576 CrossRef; (b) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426–12429 CrossRef CAS PubMed; (c) L. Zheng and R. Hua, Chem. – Eur. J., 2014, 20, 2352–2356 CrossRef CAS PubMed; (d) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625–16631 CrossRef CAS PubMed; (e) C. Wang and Y. Huang, Org. Lett., 2013, 15, 5294–5297 CrossRef CAS PubMed; (f) Z. Zhang, H. Jiang and Y. Huang, Org. Lett., 2014, 16, 5976–5979 CrossRef CAS PubMed; (g) S. Zhou, J. Wang, F. Zhang, C. Song and J. Zhu, Org. Lett., 2016, 18, 2427–2430 CrossRef CAS PubMed; (h) B. Zhou, J. Du, Y. Yang and Y. Li, Chem. – Eur. J., 2014, 20, 12768–12772 CrossRef CAS PubMed; (i) B. Zhou, Y. Yang, H. Tang, J. Du, H. Feng and Y. Li, Org. Lett., 2014, 16, 3900–3903 CrossRef CAS PubMed.
- (a) B. Li, H. Feng, S. Xu and B. Wang, Chem. – Eur. J., 2011, 17, 12573–12577 CrossRef CAS PubMed; (b) X. Tan, B. Liu, X. Li, B. Li, S. Xu, H. Song and B. Wang, J. Am. Chem. Soc., 2012, 134, 16163–16166 CrossRef CAS PubMed; (c) B. Li, H. Feng, N. Wang, J. Ma, H. Song, S. Xu and B. Wang, Chem. – Eur. J., 2012, 18, 12873–12879 CrossRef CAS PubMed; (d) B. Li, N. Wang, Y. Liang, S. Xu and B. Wang, Org. Lett., 2013, 15, 136–139 CrossRef CAS PubMed; (e) B. Li, H. Xu, H. Wang and B. Wang, ACS Catal., 2016, 6, 3856–3862 CrossRef CAS; (f) B. Li, J. Yang, H. Xu, H. Song and B. Wang, J. Org. Chem., 2015, 80, 12397–12409 CrossRef CAS PubMed; (g) T. Zhou, Y. Wang, B. Li and B. Wang, Org. Lett., 2016, 18, 5066–5069 CrossRef CAS PubMed; (h) H. Wang, B. Wang and B. Li, J. Org. Chem., 2017, 82, 9560–9569 CrossRef CAS PubMed.
- Under the optimal conditions, N-(4-hydroxyphenyl)-N-methylnitrous amide led to no reaction at all.
- CCDC 1563420 (3ai) and 1563421 (3ca)† contains the supplementary crystallographic data for this paper.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1563420 and 1563421. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qo00746a |
| This journal is © the Partner Organisations 2018 |