
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and kinetics of disassembly for silyl-containing ethoxycarbonyls using fluoride ions†
Eugene Camerinoa,
Grant C. Danielsb,
James H. Wynne b and
Erick B. Iezzi*b
b and
Erick B. Iezzi*b
aAmerican Society for Engineering Education, Chemistry Division, U.S. Naval Research Laboratory, Washington, DC 20375, USA
bChemistry Division, U.S. Naval Research Laboratory, Washington, DC 20375, USA. E-mail: erick.iezzi@nrl.navy.mil
First published on 9th January 2018
Abstract
In this study, a series of silyl-containing ethoxycarbonates and ethoxycarbamates on electron poor anilines and phenols were synthesized and their kinetics of disassembly determined in real-time upon exposure to fluoride ion sources at room temperature. The results provide a greater understanding of stability and kinetics for silyl-containing protecting groups that eliminate volatile molecules upon removal, which will allow for simplification of orthogonal protection in complex organic molecules.
Silicon-based groups have demonstrated wide utility in organic syntheses, ranging from orthogonal protection of amine, hydroxyl and acid functionalities in complex drug molecules1–3 to utilization as a trigger in self-immolative polymers and dendrimers.4–6 One of the commonly used silicon-based protecting groups is 2-(trimethylsilyl)ethoxycarbonyl (TEOC),7 which was originally developed and later optimized for N-protection of amino acids to prevent formation of oligopeptides, but has since been utilized for protection of other functionalities.8 The uniqueness of TEOC is that it can be preferentially removed with a mild de-blocking agent, such as fluoride ion, in the presence of other base-sensitive groups to produce low molecular weight and volatile by-products via cascading bond cleavage, thereby easing purification of complex mixtures and enabling greater product yields.
During our course of utilizing TEOC to protect amine and hydroxyl functionality on aliphatic oligomers we discovered there was a severe lack of kinetic data in the literature for removal of TEOC using fluoride ion, as most papers generally stated the times for removal as occurring within hours at elevated temperatures7 to days at room temperature.9,10 Furthermore, similar silyl-containing protecting groups, such as (2-phenyl-2-trimethylsilyl)ethoxycarbonyl (PSOC)11 and 2-(triphenylsilyl)ethoxycarbonyl (TPSEOC),12 were reported to be removed under milder fluoride ion conditions and/or faster times than TEOC due to a more readily formed fluoro-containing penta-coordinated intermediate as a result of phenyl group incorporation. However, while these groups were reported to be removed within minutes to hours at room temperature the studies provide minimal details on the actual kinetics compared to TEOC. As a result, we aimed to generate quantitative kinetic data for the removal of TEOC and other silyl-containing ethoxycarbonyl groups from functionality by performing disassembly experiments with fluoride ion sources at room temperature, as this information is relevant to those utilizing orthogonal protecting group chemistry in multi-step synthesis and potentially in the processing of pharmaceuticals.
Herein, we report the kinetics of disassembly (i.e., rate constants (k) for removal) for a series of silyl-terminated ethoxycarbonyl protecting groups, such as TEOC, 2-(methyldiphenylsilyl)ethoxycarbonyl (MDPSEOC), and several extended chain versions of these molecules, from electron poor hydroxyl and amine functionalities. To do this, we utilized non-substituted and para-substituted phenols and anilines, as these molecules provided a UV-visible chromophore for real-time detection during removal of the protecting groups. We also synthesized and determined the kinetics of disassembly for several novel silyl-centered bis(ethoxycarbonyls), as we were interested in studying the reaction of multiple fluoride ions with silicon and their influence on the resulting kinetics.
Our study of these molecules commenced by attempting to determine the rate constants of disassembly for 4-nitrophenyl (2-(trimethylsilyl)ethyl) carbonate (1a), a commercially available control,13 and synthesized 2-(trimethylsilyl)ethyl (4-nitrophenyl)carbamate (1b), using solutions of different fluoride salts in organic solvents and water. It was initially found that tetrahydrofuran (THF), dichloromethane, and acetonitrile provided good solubility of these organic molecules, with water and water/solvent combinations providing limited solubility. Furthermore, fluoride ion sources, such as tetrabutylammonium fluoride trihydrate (TBAF·3H2O) and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of TBAF/CsF, provided good solubility in the aforementioned solvents and several solvent/water combinations, whereas solubility of cesium fluoride (CsF), sodium fluoride (NaF), potassium fluoride (KF), and stannous fluoride (SnF2) were limited. It should be noted that a 1:1 TBAF/CsF mixture in THF provided nearly identical rate constants of disassembly during initial studies as TBAF in THF. However, to maintain fluoride ion solution consistency it was determined that all kinetic experiments would be conducted in THF using commercially available TBAF in THF.
:1 molar ratio of TBAF/CsF, provided good solubility in the aforementioned solvents and several solvent/water combinations, whereas solubility of cesium fluoride (CsF), sodium fluoride (NaF), potassium fluoride (KF), and stannous fluoride (SnF2) were limited. It should be noted that a 1:1 TBAF/CsF mixture in THF provided nearly identical rate constants of disassembly during initial studies as TBAF in THF. However, to maintain fluoride ion solution consistency it was determined that all kinetic experiments would be conducted in THF using commercially available TBAF in THF.
Kinetic experiments for 1a and 1b were conducted as 0.1 mM solutions in THF using 1 ml cuvettes and an Agilent 8453 UV-Visible Spectrophotometer. To initiate disassembly, 4 molar equivalents of 1.0 M TBAF in THF were added separately to 1a and 1b at room temperature (Scheme 1), then the absorbance of the solutions were recorded up to 120 minutes. The cuvettes were not stirred upon addition of the fluoride salt solution in effort to prevent an increase in the rate of disassembly due to turbidity. Silyl-terminated carbonate 1a, which has a maximum absorbance (λmax) at 270 nm, decreased within minutes as a new absorbance at λmax of 423 nm appeared, corresponding to 4-nitrophenolate (2a).14 Ethylene and carbon dioxide were detected via gas chromatography/mass spectrometry (GC-MS), thereby supporting the proposed mechanism of disassembly as shown in Scheme 1.7 The rate constant of disassembly for 1a was found to be 2196 ± 1380 M−1 min−1 for the second order reaction (Table 1). For silyl-terminated carbamate 1b, addition of 1.0 M TBAF in THF resulted in rapid formation of an absorbance at λmax of 444 nm, which corresponded to 4-nitrophenylamide (2b),15 although this peak quickly decreased as a new absorbance at λmax of 366 nm was formed. The latter corresponded to 4-nitroaniline (2c),16 the conjugate acid of 2b, and is proposed to have resulted from beta-elimination of a proton on the tetrabutylammonium cation, thereby forming tributylamine and 1-butene as side products via a Hofmann elimination.17 1H-NMR and GC-MS analysis of the solution after disassembly supports this hypothesis. The rate constant of disassembly for 1b was determined to be 38.5 ± 0.3 M−1 min−1, and the slower rate compared to 1a is consistent with substituted anilines possessing a larger pKa value than substituted phenols. 2-(methyldiphenylsilyl)ethyl (4-nitrophenyl) carbonate 1c and 2-(methyldiphenylsilyl)ethyl (4-nitrophenyl)carbamate 1d were subsequently synthesized and their rate constants of disassembly determined. Although similar to the triphenylsilyl and 2-phenyl-2-trimethylsilyl ethoxycarbonyl protecting groups reported in the literature,11,12 we chose the methyldiphenylsilyl group because it was a hybrid of the aforementioned groups, the kinetics of phenyl-containing silyl groups had not been reported, and the lone methyl group provided a basis for larger aliphatic groups, which are discussed in subsequent paragraphs.
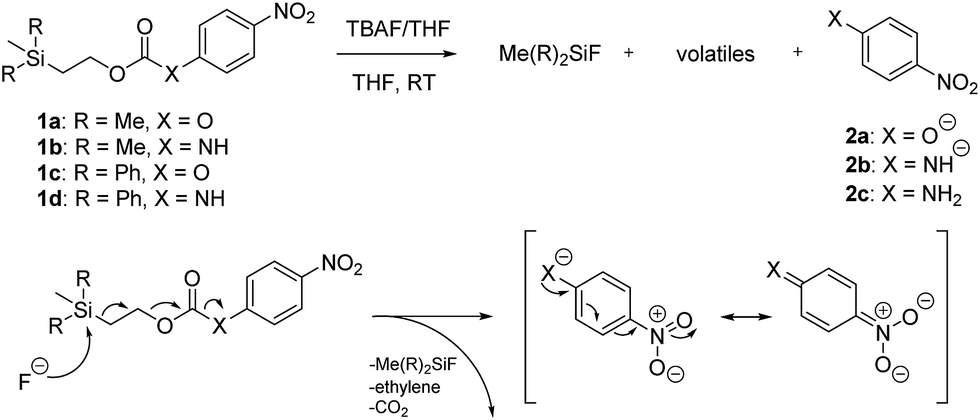 | ||
| Scheme 1 Products and proposed mechanism of disassembly for reaction of silyl-terminated carbonates and carbamates with TBAF. | ||
| Entry | Structure | k (M−1 min−1) | Approx. % remaining |
|---|---|---|---|
| 1a | 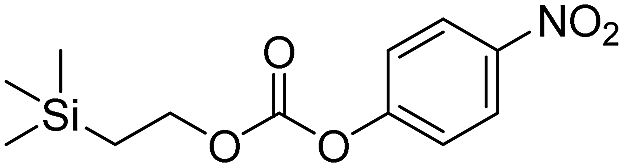 |
2196 ± 1380 | 0 |
| 1b | 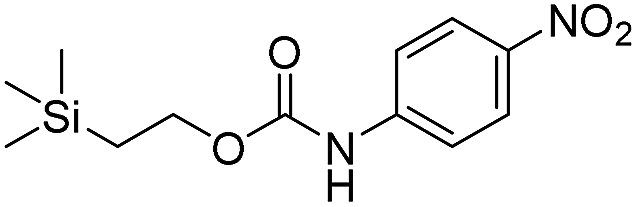 |
38.5 ± 0.3 | 68 |
| 1c | 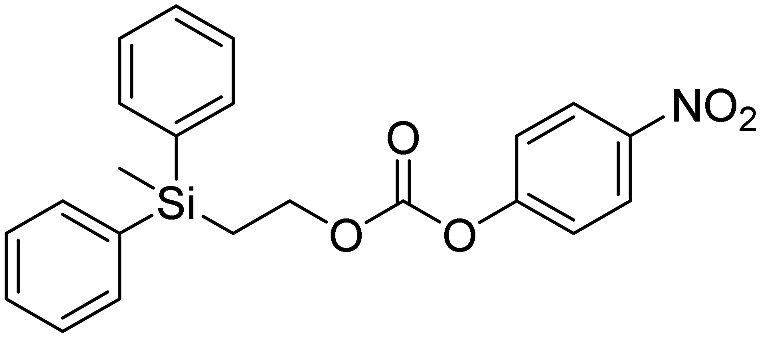 |
355 ± 28 | 41 |
| 1d | 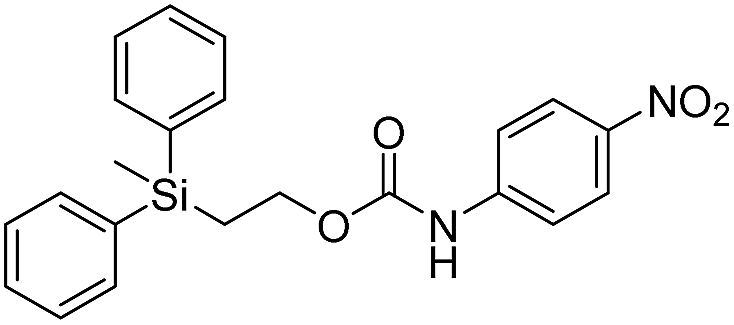 |
77.1 ± 0.2 | 27 |
The rate constants of 1c and 1d were found to be 355 ± 28 M−1 min−1 and 77.1 ± 0.2 M−1 min−1 (Table 1), respectively, with 1d first forming 4-nitrophenylamide (2b) at 444 nm before protonating to form 4-nitroaniline (2c) at 366 nm. These results are consistent with the greater electron withdrawing nature of substituted phenols compared to anilines, in addition to the reported increase in electrophilicity of the silicon atom, and hence faster removal, due to phenyl group attachment.12 However, the rate constant of 1c was slower than 1a, which contradicted this rationale. To address the issue, we synthesized 1a and compared it to the rate constant of disassembly for the commercially available version, yet we found no change in the results. After viewing the first few seconds of the absorbance spectra we noticed the absorbance of 1a was decreasing slightly, even before addition of TBAF, which signified molecule instability at room temperature. This resulted in a faster rate constant compared to 1b when calculated via Excel plot. We currently do not have the ability to study the kinetics at reduced temperatures, but the instability supports literature examples for the use of 1a at 0 °C to protect organic functionality. On the contrary, 1c was stable at room temperature for weeks, and thus may be useful as an alternative protecting group for orthogonal chemistry.
Once changes in the UV-visible spectra no longer appeared to occur, thus signifying that disassembly had slowed or ceased, the lowest absorbance of the silyl-terminated carbonates and carbamates was used to calculate the concentration of non-disassembled starting material. This, in-turn, could be represented as an approximate percentage of remaining starting material as shown in Table 1. For instance, 1a completely disassembled within 5 minutes of treatment with 1.0 M TBAF in THF, whereas about 68% of 1b remained in solution after 120 minutes. The percentage of 1c remaining after 16 minutes was about 41%, whereas about 73% of 1d remained after 120 minutes. As discussed in the aforementioned paragraph, the complete disassembly of 1a resulted from an instability at room temperature. However, all other molecules likely ceased to disassemble within the measured time period due to reduced collisions with fluoride ion as a result of not stirring the solutions. For these examples, the percentage of arene chromophores (2a–2c) generated during disassembly was equivalent to the percentage of disassembled starting materials.
Based on the success with 4-nitrophenol and 4-nitroaniline as leaving groups we decided to synthesize and investigate the change in rate constants for silyl-terminated ethoxycarbonates and ethoxycarbamates using alterative substituents, such as 4-methoxy, 3-trifluoromethyl, 4-methyl, and 4-hydrogen, on phenol and aniline. However, in all cases, the absorbance of the disassembled phenolate or amide significantly overlapped with those of the starting materials, thus the rate constants could not be accurately determined via UV-visible spectroscopy.
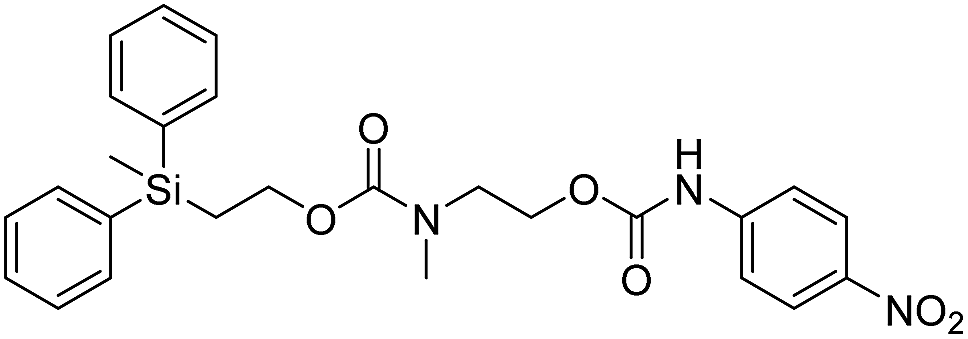
We next aimed to determine the rate constants of disassembly for silyl-terminated molecules with extended chains (3a–3h), as we were interested to learn if the molecules would fully disassembly and eliminate cyclic oxizolidinones (Scheme 2), and if so, how their rates differed from the non-extended molecules (1a–1d). We also aimed to confirm whether or not these extended molecules would provide an increased rate constant of disassembly when phenyl groups were attached to the silicon atom. To accomplish this, we developed a synthetic route to insert ethanolamine or N-methyl ethanolamine between the ethoxycarbonyl and aromatic chromophore (Scheme S1 of ESI†). Upon addition of 4 molar equivalents of 1.0 M TBAF in THF, silyl-terminated molecules 3a and 3b resulted in complete disassembly with rate constants of 42638 ± 1720 M−1 min−1 and 46049 ± 1860 M−1 min−1, respectively, whereas molecules 3c and 3d resulted in complete disassembly with rate constants of 118711 ± 14000 M−1 min−1 and 123825 ± 4000 M−1 min−1, respectively (Table 2). All reactions generated ethylene, carbon dioxide and a single new absorbance at λmax of 423 nm, corresponding to 4-nitrophenolate (2a), and in all cases the central linker was ejected via intramolecular cyclization to form oxizolidin-2-one (4a) or 3-methyl-oxizolidin-2-one (4b), thus supporting the proposed mechanism shown in Scheme 2 and demonstrating that the rates were significantly enhanced by the entropic increase.18,19 Detection of the oxizolidinones were confirmed by GC-MS analysis of the reaction solutions after disassembly. The rate constants for the molecules containing N-methyl linkages (3b and 3d) were found to be greater than those with non-N-alkylated linkages (3a and 3c), which is consistent with the greater nucleophilicity of the resulting secondary amide ion (i.e., after loss of CO2) compared to those of primary amine ions. Furthermore, molecules with methyldiphenylsilyl groups (3c and 3d) proved to have faster rates of disassembly than those with trimethylsilyl groups (3a and 3b), which, as previously mentioned, is consistent with literature reports for enhanced reactivity at the silicon atom upon phenyl group attachment.
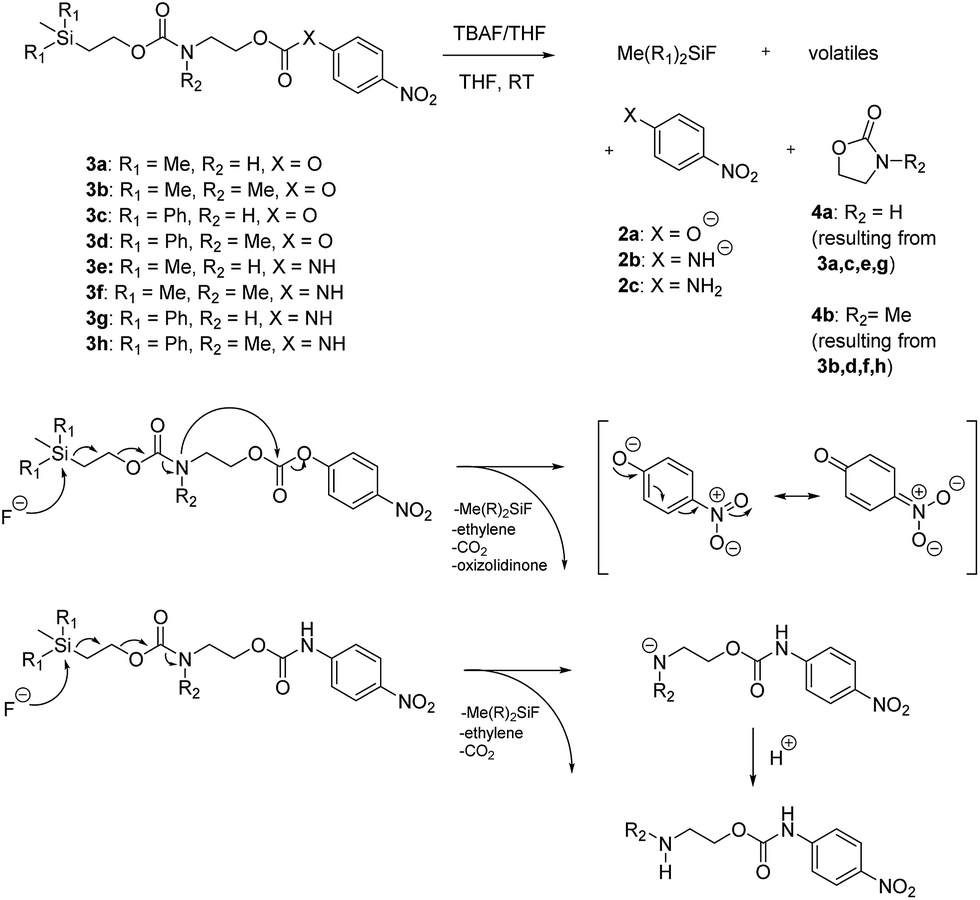 | ||
| Scheme 2 Products and proposed mechanisms of disassembly for reaction of silyl-terminated extended chain carbonates and carbamates with TBAF. | ||
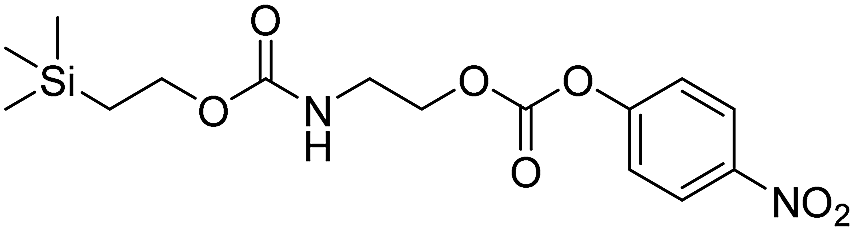
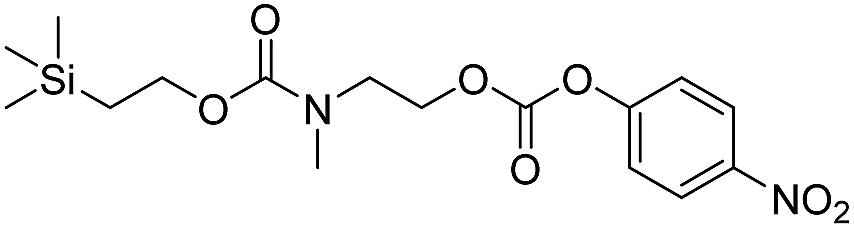
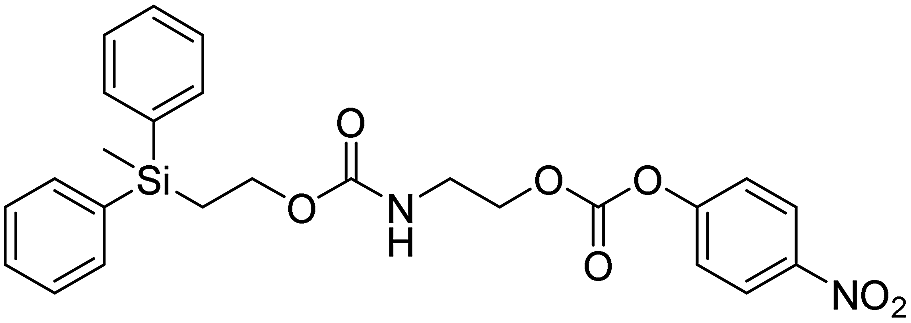
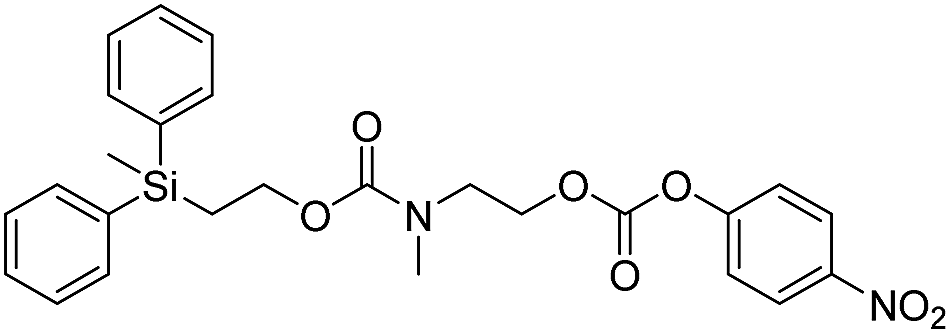
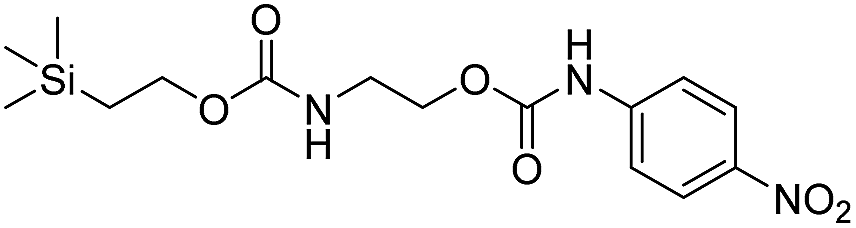
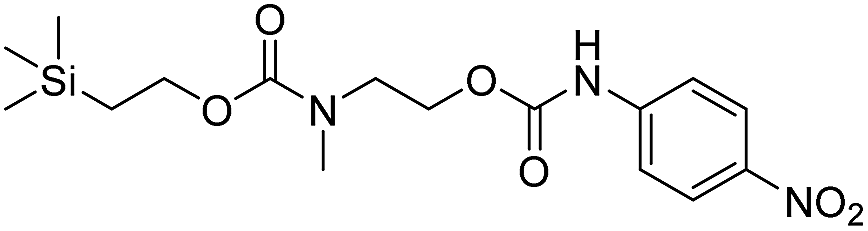
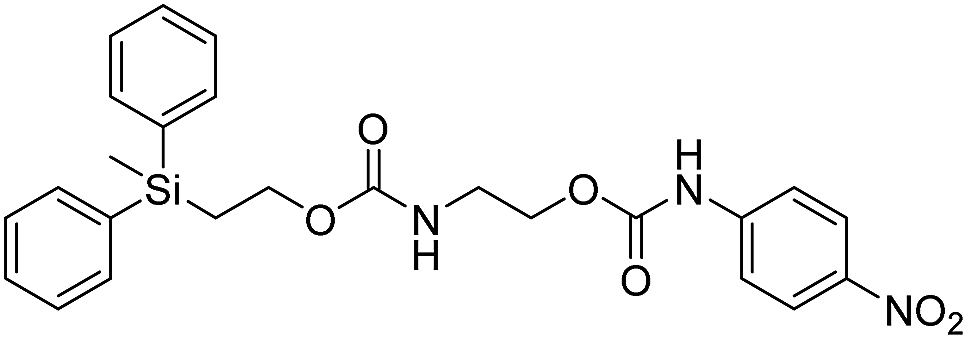
As shown in Table 2, approximately 13–15% of extended carbonates 3a–3d remained in solution after monitoring the reaction with TBAF for 10 minutes. Because the UV-vis and GC-MS data supports the proposed mechanism shown in Scheme 2 the percentage of 4-nitrophenolate (2a) produced during disassembly was likely equivalent to the percentage of starting material disassembled.
Following the results of the extended carbonates we decided to synthesize and determined the rate constants of disassembly for several silyl-terminated extended chain carbamates (3e–3h, Schemes 2 and S1 of ESI†). However, when each of these carbamates (λmax at 321 nm) were treated with 4 molar equivalents of 1.0 M TBAF in THF we observed the minor formation of a species with a similar absorption to 4-nitrophenylamide (2b, λmax of 445 nm), but we did not observe a peak for 4-nitroaniline (2c, λmax of 366 nm) or the appearance of any other peaks. Furthermore, the absorbance peak for the starting materials never decreased beyond 70%, even after monitoring the reaction for 24 hours, and treatment with 100 equivalents of TBAF did not influence the decrease in the starting materials. Although extend chain carbamates 3e–3h did not fully disassemble, we report the rates for loss of the starting materials during the first minutes of exposure to TBAF (Table 2).
To understand why the complete disassembly of 3e–3h ceased to proceed we performed additional experiments using 3e as the representative carbamate. For instance, after addition of TBAF to 3e in THF, GC-MS analysis showed peaks for oxazolidin-2-one (4a) and 4-nitroaniline (2c) (see ESI†). This suggested that complete disassembly was possible, although the elevated temperature at the inlet likely facilitated disassembly. Furthermore, while the addition of TBAF at room temperature showed a new spot via TLC, we were unable to purify and analyse the by-product. Thus, we assume that fluoride ion attack at room temperature resulted in cleavage of the silyl group and elimination of ethylene and carbon dioxide, but that formation of the oxazolidinone ring did not occur due to the greater thermal barrier to cyclization at the carbamate linkage (i.e., compared to similar carbonates). As a result, protonation of the amide anion may have occurred, possibly by reaction with tetrabutylammonium ion via a Hofmann elimination. Protonation of the amide anion would also explain why the starting material never fully decreased, as the by-product would have a similar absorbance as the starting material.
Akin to the extended chain carbonates, the absorbance of extended chain carbamates 3e–3h after 10 minutes could be used to calculate an approximate percentage of non-disassembled starting material. However, unlike the carbonates, the carbamates did not completely disassemble, thus the percentage of generated arene chromophores could not be determined.
Our focus then turned to the synthesis of novel silyl-centered molecules, such as bis(ethoxycarbamates) (5a and 5b) and bis(ethoxycarbonates) (5c and 5d) (Scheme S2 of ESI†). Upon treatment of (dimethylsilanediyl)bis(ethane-2,1-diyl) bis((4-nitrophenyl)carbamate) (5a, Scheme 3) with 8 molar equivalents of 1.0 M TBAF in THF we detected 4-nitrophenolate (2b), followed by the formation of 2c, and calculated a rate constant of disassembly of 967 ± 14 M−1 min−1 (Table 3). This rate was about a 25-fold increase when compared to that of 38.5 M−1 min−1 for trimethylsilyl-terminated ethoxycarbamate 1b. The significant increase in rate implied that once the first fluoride ion bonded with silicon the resulting fluorinated silyl group increased in electrophilicity, thereby increasing the rate of attack by a second fluoride ion as shown for the proposed mechanism in Scheme 3. Silyl-centered molecule (diphenylsilanediyl)bis(ethane-2,1-diyl) bis((4-nitrophenyl)carbamate) (5b), when treated with 8 molar equivalents of TBAF in THF, demonstrated a rate constant of 1733 ± 30 M−1 min−1, which followed a similar trend for silyl-terminated molecules where the electrophilicity of silicon was increased upon phenyl group attachment. It is important to note that 8 molar equivalents of TBAF were used in order to maintain a ratio of 4 molar equivalents per UV-detectable end group. However, when 4 molar equivalents of TBAF were utilized the rates for 5a and 5b were 50% slower, thus confirming the second order reaction. Nevertheless, these rates were still faster when compared to silyl-terminated molecules 1b and 1d.
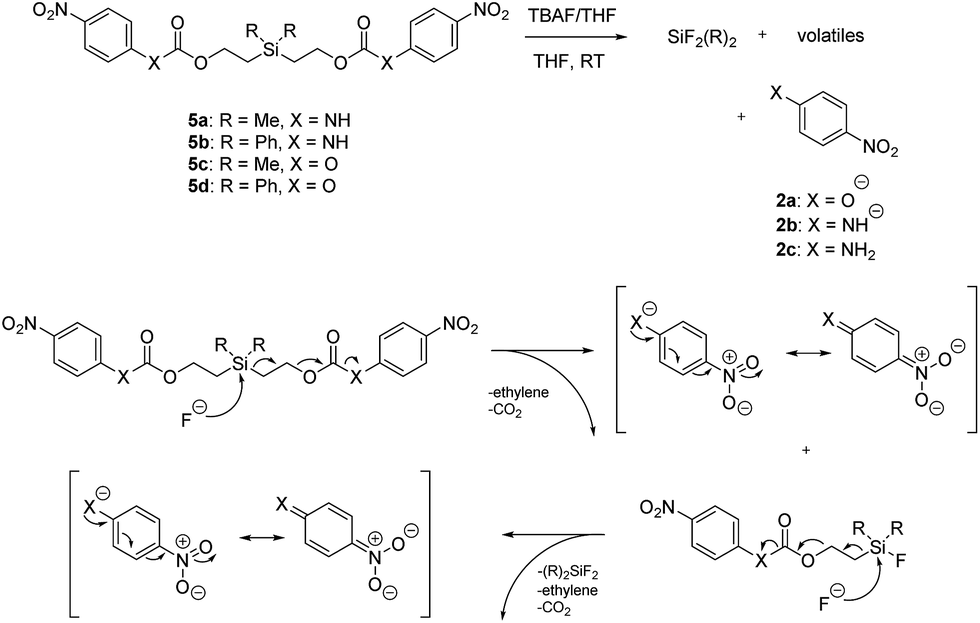 | ||
| Scheme 3 Products and proposed mechanism of disassembly for reaction of silyl-centered bis(ethoxycarbonyls) with TBAF. | ||
| Entry | Structure | k (M−1 min−1) | Approx. % remaining |
|---|---|---|---|
| 5a | 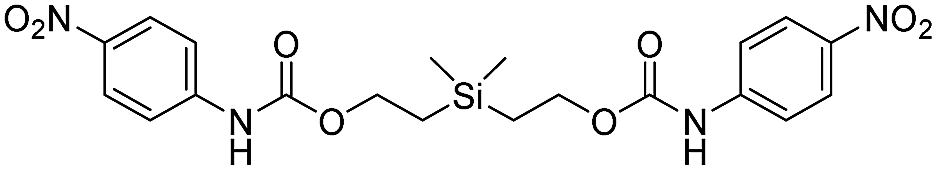 |
967 ± 14 | 35 |
| 5b | 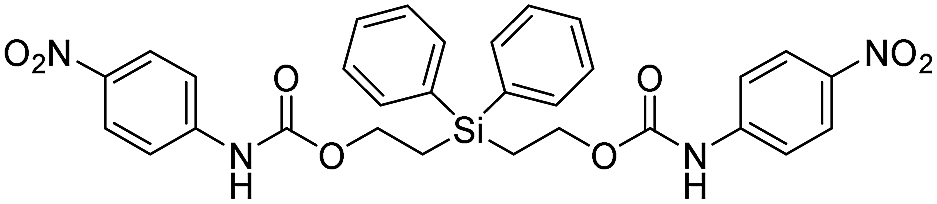 |
1733 ± 30 | 34 |
| 5c | 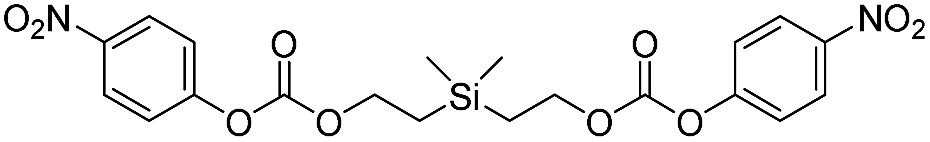 |
38358 ± 2200 |
31 |
| 5d | 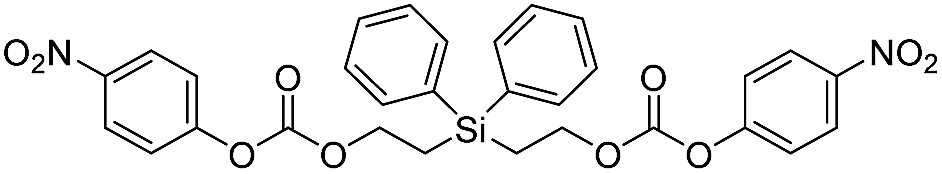 |
404699 ± 64100 |
16 |
In contrast to the bis(ethoxycarbamates), silyl-centered bis(ethoxycarbonates) (5c and 5d) demonstrated a significant increase in their rates of disassembly compared to silyl-terminated carbonates. For instance, the rate constant for 5c was 38358 ± 2200 M−1 min−1, about a 39-fold increase compared to 5a. However, the rate for 5d was 404699 ± 64100 M−1 min−1, about a 233-fold increase compared to 5b and about a 10-fold increase compared to 5c (Table 3). The large increase in rate for 5d is most likely a cumulative effect due to the increased electrophilicity at the silyl group as a result of phenyl group inclusion and the first reaction with fluoride ion, the increased amount of TBAF present, and the greater electron withdrawing nature of the leaving group.
The rates of disassembly for the silyl-centered molecules were significantly faster than those for all silyl-terminated molecules presented in this paper. However, disassembly significantly slowed around 15–20 minutes for the bis(ethoxycarbamates) and after 10–20 seconds for the bis(ethoxycarbonates). For 5a, the percentage of approximate starting material remaining was 35% after 20 minutes of reaction with TBAF, whereas approximately 34% remained for 5b after 14 minutes of reaction (Table 3). Even after 120 minutes of reaction the approximate percentages remaining for 5a and 5b were 31% and 26%, respectively. For 5c and 5d, the approximate percentages of starting material remaining after about 20 seconds was 31% and 16%, respectively. After 10 minutes of reaction the percentages had decreased to only 26% for 5c and 15% for 5d. The percentage of arene chromophores produced during disassembly was likely equivalent to the percentage of starting material disassembled based on the proposed mechanism and lack of other peaks in the UV-visible absorbance spectra.
In conclusion, this study is the first comprehensive report demonstrating that TEOC alternatives, such as silyl-terminated extended chain ethoxycarbonyls, provide significantly faster rates of disassembly, and hence removal from hydroxyl and amine-terminated molecules, when treated with fluoride ion. The reactions were not stirred during disassembly, and thus the rates would likely increase upon stirring. These new alternatives were found to possess increased stability at room temperature, thereby supporting their use as orthogonal protecting groups for complex organic molecules. The increased stability and increased rates of removal will also provide for increased yields and ease of purifications. The addition of phenyl rings on silicon consistently increased the rates of disassembly, likely due to an electronic effect. Likewise, the greater acidity of 4-nitrophenol compared to 4-nitroaniline led to increased rates of disassembly. Silyl-terminated ethoxycarbonates resulted in complete formation of the arene chromophore, whereas disassembly of the carbamates was halted after loss of volatiles and formation of the amide ion. Finally, the silyl-centered bis(ethoxycarbonates) had significantly faster rates compared to all silyl-terminated molecules due to increased electrophilicity at silicon after addition of the first fluoride ion. These molecules may have applications in self-immolative materials for detecting fluoride ion concentrations in water.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Naval Research Laboratory Base Program. E. C. would like to thank financial support from the American Society for Engineering Education (ASEE).References
- L. A. Carpino and G. Y. Han, J. Am. Chem. Soc., 1970, 92, 5748–5749 CrossRef CAS.
- S. E. Denmark, C. S. Regens and T. Kobayashi, J. Am. Chem. Soc., 2007, 129, 2774–2776 CrossRef CAS PubMed.
- B. M. O'Keefe, D. M. Mans, D. E. Kaelin and S. F. Martin, Tetrahedron, 2011, 67, 6524–6538 CrossRef PubMed.
- M. E. Roth, O. Green, S. Gnaim and D. Shabat, Chem. Rev., 2016, 116, 1309–1352 CrossRef CAS PubMed.
- M. G. Olah, J. S. Robbins, M. S. Baker and S. T. Phillips, Macromolecules, 2013, 46, 5924–5928 CrossRef CAS.
- A. M. DiLauro, H. Zhang, M. S. Baker, F. Wong, A. Sen and S. T. Phillips, Macromolecules, 2013, 46, 7257–7265 CrossRef CAS.
- L. A. Carpino, J. H. Tsao, H. Ringsdorf, E. Fell and G. Hettrich, Chem. Commun., 1978, 358–359 RSC.
- H. Liang, L. Hu and E. J. Corey, Org. Lett., 2011, 13, 4120–4123 CrossRef CAS PubMed.
- S. Bjorkman and J. Chattopadhyaya, Chem. Scr., 1982, 20, 201–202 Search PubMed.
- C. Gioeli, N. Balgobin, S. Josephson and J. B. Chattopadhyaya, Tetrahedron Lett., 1981, 22, 969–972 CrossRef CAS.
- M. Wagner, S. Heiner and H. Kunz, Synlett, 2000, 1753–1756 CAS.
- M. Golkowski and T. Ziegler, Molecules, 2011, 16, 4695–4718 CrossRef CAS PubMed.
- Sigma Aldrich, #92748.
- M.-B. S. Kirketerp, M. A. Petersen, M. Wanko, L. A. E. Leal, H. Zettergren, F. M. Raymo, A. Rubio, M. B. Nielsen and S. B. Nielsen, ChemPhysChem, 2009, 10, 1207–1209 CrossRef PubMed.
- R. A. Ando, A. C. Borin and P. S. Santos, J. Phys. Chem. A, 2007, 111, 7194–7199 CrossRef CAS PubMed.
- 4-Nitroaniline, http://www.webbook.nist.gov/chemistry/, accessed 2/24/2017.
- H. R. Sun and S. G. DiMagno, J. Am. Chem. Soc., 2005, 127, 2050–2051 CrossRef CAS PubMed.
- R. A. McBride and E. R. Gillies, Macromolecules, 2013, 46, 5157–5166 CrossRef CAS.
- A. Alouane, R. Labruere, T. Le Saux, F. Schmidt and L. Jullien, Angew. Chem., Int. Ed., 2015, 54, 7492–7509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, characterization data, NMR spectra, UV-visible spectra, rate plots and GC-MS chromatograms. See DOI: 10.1039/c7ra07876e |
| This journal is © The Royal Society of Chemistry 2018 |