
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent incorporation of tobacco mosaic virus increases the stiffness of poly(ethylene glycol) diacrylate hydrogels†
A. Southan *a,
T. Langa,
M. Schweikertb,
G. E. M. Tovarac,
C. Weged and
S. Eiben*d
*a,
T. Langa,
M. Schweikertb,
G. E. M. Tovarac,
C. Weged and
S. Eiben*d
aInstitute of Interfacial Process Engineering and Plasma Technology IGVP, University of Stuttgart, Nobelstr. 12, 70569 Stuttgart, Germany. E-mail: alexander.southan@igvp.uni-stuttgart.de; Tel: +49 711 68568162
bDepartment of Biobased Materials, Institute of Biomaterials and Biomolecular Systems, University of Stuttgart, Pfaffenwaldring 57, 70569 Stuttgart, Germany
cFraunhofer Institute for Interfacial Engineering and Biotechnology IGB, Nobelstr. 12, 70569 Stuttgart, Germany
dDepartment of Molecular Biology and Plant Virology, Institute of Biomaterials and Biomolecular Systems, University of Stuttgart, Pfaffenwaldring 57, 70569 Stuttgart, Germany. E-mail: sabine.eiben@bio.uni-stuttgart.de
First published on 25th January 2018
Abstract
Hydrogels are versatile materials, finding applications as adsorbers, supports for biosensors and biocatalysts or as scaffolds for tissue engineering. A frequently used building block for chemically cross-linked hydrogels is poly(ethylene glycol) diacrylate (PEG-DA). However, after curing, PEG-DA hydrogels cannot be functionalized easily. In this contribution, the stiff, rod-like tobacco mosaic virus (TMV) is investigated as a functional additive to PEG-DA hydrogels. TMV consists of more than 2000 identical coat proteins and can therefore present more than 2000 functional sites per TMV available for coupling, and thus has been used as a template or building block for nano-scaled hybrid materials for many years. Here, PEG-DA (Mn = 700 g mol−1) hydrogels are combined with a thiol-group presenting TMV mutant (TMVCys). By covalent coupling of TMVCys into the hydrogel matrix via the thiol-Michael reaction, the storage modulus of the hydrogels is increased compared to pure PEG-DA hydrogels and to hydrogels containing wildtype TMV (wt-TMV) which is not coupled covalently into the hydrogel matrix. In contrast, the swelling behaviour of the hydrogels is not altered by TMVCys or wt-TMV. Transmission electron microscopy reveals that the TMV particles are well dispersed in the hydrogels without any large aggregates. These findings give rise to the conclusion that well-defined hydrogels were obtained which offer the possibility to use the incorporated TMV as multivalent carrier templates e.g. for enzymes in future studies.
Introduction
Hydrogels based on poly(ethylene glycol) (PEG) constitute an interesting class of materials for different applications such as drug delivery,1,2 tissue engineering,3,4 3D printing,5–7 or sensing.8,9 Hydrogels for these applications have to be functional materials with defined chemical, biological, or physical properties. For the preparation of PEG-based hydrogels, poly(ethylene glycol) diacrylate (PEG-DA) is frequently used as a building block due to its low toxicity10 and excellent water-solubility. Additionally, the mechanical properties, the water content and the permeability for small molecular compounds of PEG-DA hydrogels can be easily adjusted by using different molecular weights and concentrations in the hydrogel precursor solutions.11–13 Hydrogel formation using PEG-DA can be achieved e.g. by radical polymerisation of the acrylate end groups14,15 or by Michael addition chemistry.16,17However, pure PEG-DA hydrogels provide rather limited chemical or biological functionality. The PEG backbone renders the hydrogels protein repellent and generally unsuitable for biological applications such as cell culture.17,18 Chemical modification of cured PEG-DA hydrogels is also difficult without deteriorating the network structure due to the low reactivity of PEG. Therefore, functional PEG-DA hydrogels are often obtained by co-polymerisation of PEG-DA with functional building blocks19 or by prior modification of one PEG-DA chain end by Michael addition of functional thiols20–22 or amines.23
Tobacco mosaic virus (TMV) is a versatile nano-scaled scaffold with the possibility to present more than 2000 functional groups per particle. This plant virus, composed of a genomic RNA and about 2100 coat protein (CP) subunits, has a tube-like structure 300 nm in length with inner and outer diameters of 4 nm and 18 nm, respectively.24 TMV has been investigated as a carrier scaffold for example for enzymes, or as template for mineralisation for more than twenty years.25,26 Virus-like particles (VLPs) can be obtained with artificially produced RNAs,27,28 thus changing the length or form of the virus, as well as with altered CPs presenting genetically encoded specific coupling groups such as primary amines or thiols.29 In addition, we have recently achieved to produce VLPs with highly defined longitudinal domains.30 With regard to hydrogels, TMV by itself and in combination with alginate hydrogels has been shown to have a positive effect on osteogenesis without provoking adverse immune reactions in vivo.31–33 For these alginate gels, the impact of TMV particles on their mechanical properties has been characterised, demonstrating that TMV rods induced non-linear responses to compression at high strains.31 Additionally, Wang and co-authors have fabricated TMV-interlinked hydrogels from methacrylated hyaluronan (MeHA) and were able to demonstrate improved in vitro chondrogenesis of bone marrow mesenchymal stem cells (BMSCs) in these matrices, which have also been characterised for their internal structure, stability and TMV retention.34 Lewis et al. produced hybrid hydrogel microparticles from PEG-DA (Mn = 700 g mol−1) and functionalized TMV and showed a regular distribution of TMV within the hydrogels at the microscale by fluorescence microscopy.35 However, they did not investigate the impact of TMV on the mechanical properties of the hydrogels or virus distribution at the nanoscale.
As described above, PEG-DA hydrogels can be adapted as a versatile platform for functional hydrogels. Therefore, we aimed at both covalent and non-covalent integration of TMV nanoparticles into PEG-DA hydrogels, and an in-depth physical characterisation of these hybrid materials. In particular, we wanted to assess the impact of covalent and non-covalent integration of TMV into PEG-DA hydrogels on the physical properties of the hydrogels, such as mechanical properties and equilibrium water content (EWC). For covalent integration, a cysteine-containing TMV mutant (TMVCys) was used. On the other hand, wildtype TMV (wt-TMV) was used for non-covalent integration. We hypothesised that due to the rod-like structure of TMV with the length of 300 nm, a covalent integration of TMVCys into the PEG-DA hydrogel network would result in a stiffer hydrogel compared to a non-covalent integration of wt-TMV, provided that the TMVCys is evenly distributed throughout the hydrogel volume.
Experimental
Materials
Potassium dihydrogen phosphate and silver nitrate were obtained from Merck (Darmstadt, Germany). Di-sodium hydrogen phosphate, acetone, sodium dodecyl sulfate, tris(hydroxymethyl)aminomethane, acetic acid, sodium carbonate, ammonium persulfate, tetramethylenediamine, sodium chloride, sodium thiosulfate and poly(ethylene glycol) (PEG, Mn = 6000 g mol−1) were obtained from Carl Roth (Karlsruhe, Germany). Uranyl acetate and acrylamide/bis solution, 37.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were purchased from Serva (Heidelberg, Germany). Spurr Low Viscosity Embedding Kit was obtained from Polysciences (Washington, USA). Poly(ethylene glycol) diacrylate (PEG-DA, Mn = 700 g mol−1) and formaldehyde were obtained from Sigma-Aldrich (Merck; Darmstadt, Germany). The radical photoinitiator 1-[4-(2-hydroxyethoxy)phenyl]-2-hydroxy-2-methyl-1-propan-1-one (Irgacure 2959) was a kind gift from Bodo Möller Chemie GmbH (Offenbach, Germany). A Micro BCA Protein Assay Kit was obtained from Thermo Scientific (Waltham, USA). All chemicals and reagent were used as received. Stock solutions containing 28 mg Irgacure 2959 in 3.972 g SPP buffer were obtained by gentle heating with a heat gun at 100 °C until all material was dissolved, and subsequent cooling to room temperature. Water was withdrawn from a Barnstead GenPure xCAD water purification system (Thermo Scientific, Waltham, USA).
:1 were purchased from Serva (Heidelberg, Germany). Spurr Low Viscosity Embedding Kit was obtained from Polysciences (Washington, USA). Poly(ethylene glycol) diacrylate (PEG-DA, Mn = 700 g mol−1) and formaldehyde were obtained from Sigma-Aldrich (Merck; Darmstadt, Germany). The radical photoinitiator 1-[4-(2-hydroxyethoxy)phenyl]-2-hydroxy-2-methyl-1-propan-1-one (Irgacure 2959) was a kind gift from Bodo Möller Chemie GmbH (Offenbach, Germany). A Micro BCA Protein Assay Kit was obtained from Thermo Scientific (Waltham, USA). All chemicals and reagent were used as received. Stock solutions containing 28 mg Irgacure 2959 in 3.972 g SPP buffer were obtained by gentle heating with a heat gun at 100 °C until all material was dissolved, and subsequent cooling to room temperature. Water was withdrawn from a Barnstead GenPure xCAD water purification system (Thermo Scientific, Waltham, USA).
Instrumentation and methods
For UV induced curing of the hydrogels, the UV chamber SOL2/400F (Dr Hönle, Gräfelfing, Germany) was used. The irradiation intensity was approx. 50 mW cm−2. The storage moduli (G′) and loss moduli (G′′) of swollen hydrogels were measured at 20 °C on a Physica MCR 301 rheometer (Anton Paar, Ostfildern, Germany) using a parallel plate geometry with a diameter of 20 mm and a normal force of 1 N. Amplitude sweeps were carried out at a frequency of 10 Hz and deformations between 0.01% and 10%. Frequency sweeps were carried out at amplitudes of 0.1% and frequencies between 0.5 Hz and 100 Hz. For comparison of different hydrogels, G′ and G′′ were extracted from the measurements at a frequency of 10 Hz and a deformation of 0.1%. The equilibrium water content (EWC) of the hydrogels was determined gravimetrically and is defined as follows (mdry = mass of the dried hydrogel, mswollen = mass of the swollen hydrogel):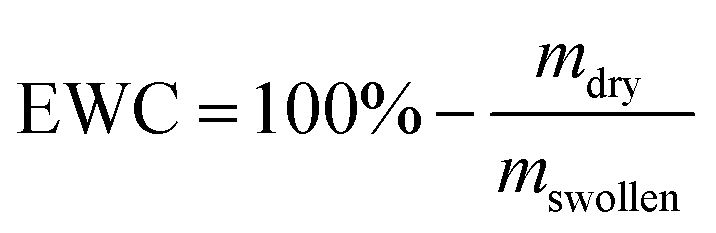
The value of mdry is determined after drying the hydrogels in a vacuum chamber at a pressure of 30 mbar at 50 °C for 24 h. ATR-FT-IR spectra were recorded on a Bruker Equinox 55 spectrometer using a DTGS detector. The concentration of TMV in the washing buffer of the hydrogels was determined by a Micro BCA Assay according to the supplier's protocol (Thermo Scientific). Diluted solutions of the respective TMV-variants were applied for calibration. Each sample was measured in triplicates and at least washing buffers of three different gels per condition were investigated.
Preparation of TMV and coupling to PEG-DA
Viruses, wt-TMV and the CP mutant TMVCys, bearing an exchange of serine at position three to cysteine,29 were propagated in Nicotiana tabacum ‘Samsun’ nn plants in a greenhouse. They were purified according to the protocol of Chapman using PEG precipitation and ultracentrifugation steps.36 The viruses were stored at a concentration of 10 mg mL−1 in 10 mM sodium–potassium-phosphate buffer (SPP) pH 7.4 at 4 °C. To obtain TMV at higher concentrations, the virus particles were pelleted by ultracentrifugation at 34000 rpm (134000g) for 1.5 h at 4 °C in a 45 Ti rotor (Beckman Coulter, Brea, USA) and resuspended in one third of the initial volume, resulting in a TMV concentration of 20–33 mg mL−1. In a first series of experiments the coupling efficiencies of PEG-DA to TMVCys at varying conditions were determined by discontinuous sodium dodecyl sulfate poly-acrylamide gel electrophoresis (SDS-PAGE).37 The reaction conditions deviated from the hydrogel preparations described below, as for this experiment the TMV concentration was fixed to 1 μg μL−1 TMV while the PEG-DA concentration was varied between molar ratios of acrylate groups to coupling sites on TMV of 1:3333 and 1:1000. A ratio of 3333:1 with a TMVCys concentration of 1 μg μL−1 corresponds to a solution containing 20 wt% PEG-DA and 0.3 wt% TMVCys. Reactions were performed in 10 mM SPP pH 7.4. Samples were withdrawn at different time points between 5 min and several days and analysed by standard SDS-PAGE followed by silver staining.
Hydrogel preparation
As an example, the preparation of a PEG-DA hydrogel containing 20 wt% PEG-DA and 1 wt% TMVCys is described. All other hydrogels were prepared accordingly. For hydrogels containing no TMV, instead of TMV stock solution SPP buffer was added. For each hydrogel sample, a separate solution was prepared. For detailed concentrations of the different components in the hydrogels, see Table 1.| Sample | cPEG-DA [wt%] | cTMVCys [wt%] | cwt-TMV [wt%] |
|---|---|---|---|
| TMVCys0.1 | 20.0 | 0.1 | — |
| TMVCys0.3 | 20.0 | 0.3 | — |
| TMVCys1.0 | 20.0 | 1.0 | — |
| TMVCys2.0 | 20.0 | 2.0 | — |
| wt-TMV0.1 | 20.0 | — | 0.1 |
| wt-TMV0.3 | 20.0 | — | 0.3 |
| wt-TMV1.0 | 20.0 | — | 1.0 |
| wt-TMV2.0 | 20.0 | — | 2.0 |
| PEG-DA0.1 | 20.1 | — | — |
| PEG-DA0.3 | 20.3 | — | — |
| PEG-DA1.0 | 21.0 | — | — |
| PEG-DA2.0 | 22.0 | — | — |
150 mg of PEG-DA were mixed with 107 mg of the Irgacure 2959 stock solution (containing 0.75 mg Irgacure 2959, 0.5% of PEG-DA mass) and 144 mg of SPP buffer as well as 349 mg of the TMVCys stock solution (containing 7.5 mg TMVCys) were added. The mixture was shaken until clear. Then, the solution was left to stand at room temperature for 1 h in order to achieve coupling of the TMVCys to the PEG-DA by thiol-Michael reaction. Subsequently, the solution weighing 750 mg in total was transferred into a cylindrical aluminum mold (diameter: 30 mm, height: 1 mm) and covered with a quartz glass pane. Radical-cross-linking was achieved by irradiation in the UV chamber for 7.5 min. The hydrogels were washed three times in SPP buffer solution (10 mL) for 24 h and swollen to equilibrium prior to characterisation.
Transmission electron microscopy (TEM) of the hydrogels
Hydrogels with and without TMV were embedded in epoxy resin (Spurr) after dehydration of the gels through a solvent series using different acetone:water mixtures (30%, 50%, 75%, 90%). The gels were incubated 20 min at each step. This was followed by staining with 2% uranyl acetate in acetone for one hour and two additional incubations in 100% acetone for 20 min. The samples were then immersed in a solution containing 33% Spurr and 67% acetone overnight without closing the vessels to allow the acetone to evaporate. Before the gels were put into embedding capsules and cured for at least 24 h at 60 °C, the solutions were twice exchanged with 100% Spurr for 1–2 h. The epoxide blocks were trimmed to the gel and ∼80 nm thin sections were prepared using a Leica Ultracut UCT ultramicrotome assembled with a diamond knife (Diatome, Nidau, Switzerland). The sections were placed on Pioloform covered 400 mesh copper grids and investigated using a Tecnai G2 Sphera FEI TEM (FEI, Eindhoven, Netherlands) at 200 kV. TEM images were recorded by a 16 megapixel camera (TemCam-F416 [4k × 4k], TVIPS, Gauting, Germany).
Statistical evaluation of data
Significant effects on hydrogel properties (EWC, G′) and the amount of washed out TMV were evaluated using a one-way analysis of variance (ANOVA). In case of significant effects, significant differences between individual mean values were evaluated using the Holm–Bonferroni correction. Effects were considered to be significant for p < 0.05. Statistical analysis was performed using the software OriginPro 9.1 (OriginLab Corporation).Results and discussion
Reaction of TMVCys and wt-TMV with PEG-DA
In this study, two different TMV derivatives were applied for integration in PEG-DA hydrogels. A cysteine mutant, TMVCys,29 was used for covalent immobilisation of TMV particles in hydrogels and a wild-type, wt-TMV, was used for non-covalent immobilisation. The thiol groups available through the cysteine moiety in every CP subunit of TMVCys should be able to participate in manifold chemical reactions due to their rich redox chemistry and extremely high nucleophilicity.38 With PEG-DA, a chemical coupling with the thiols in aqueous media should be possible by the thiol-Michael reaction which is known to proceed in neutral to slightly basic conditions without any catalyst (Scheme 1).39 Thus, it should be possible to functionalise TMVCys with acrylic double bonds, provided that one PEG-DA end group reacts with one TMVCys CP, leaving the other PEG-DA end group unaltered. The so formed double bond functionalised TMV particles (TMV-PEG-A) should, in a second step, be able to participate in a subsequent radical cross-linking reaction which forms a PEG-DA hydrogel. A reaction of both PEG-DA end groups with CPs is to be avoided, if possible, as it would leave the resulting moiety with no residual acrylic end group for a subsequent hydrogel cross-linking reaction. For wt-TMV no chemical reaction is expected with PEG-DA.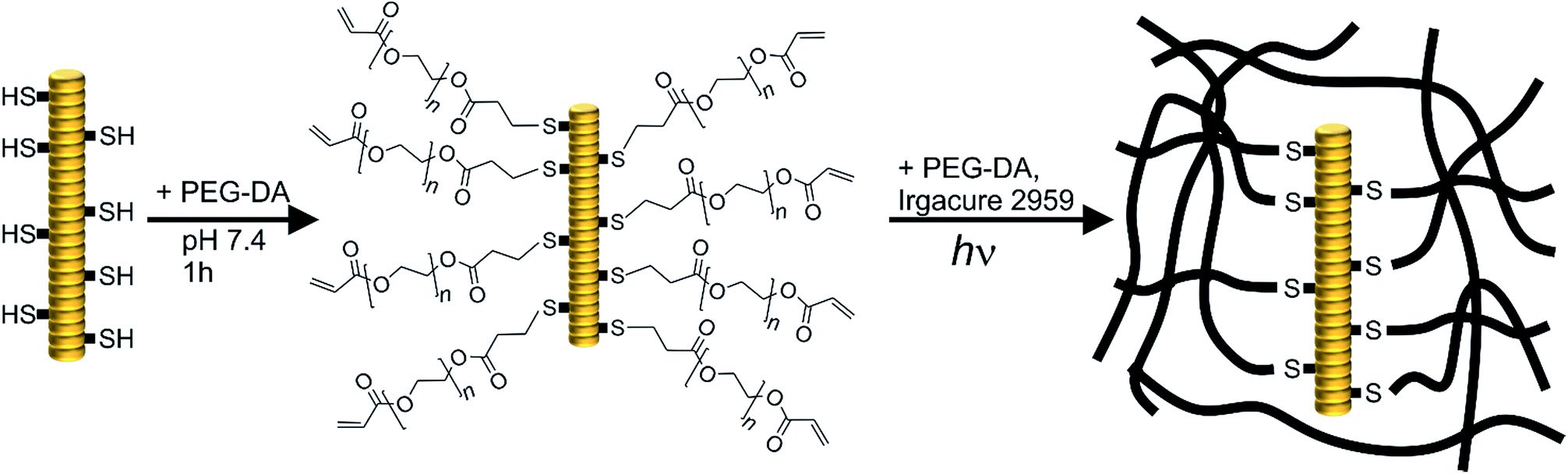 | ||
| Scheme 1 Schematic representation of the coupling of PEG-DA to TMVCys by thiol-Michael reaction resulting in double bond functionalised TMV particles (TMV-PEG-A, left) and subsequent photopolymerisation of the attached acrylate functionalities with more PEG-DA to hydrogels (right). Thus, covalent incorporation of TMVCys into the hydrogels is possible. In the case of wt-TMV, no free cysteine moieties are present and no covalent coupling with PEG-DA is expected. | ||
In order to investigate the reaction of TMVCys and wt-TMV with PEG-DA, we prepared mixtures of TMVCys and wt-TMV, respectively, with PEG-DA in SPP buffer at pH 7.4 with different molar ratios of TMV CPs and PEG-DA acrylate end groups. The mixtures were then reacted for different times. In case of a coupling reaction between a TMVCys CP and PEG-DA, an increase of molecular weight of the CP is expected. Therefore, the molecular weights of the reaction products were assessed by SDS-PAGE. With this method, the TMV particles are disassembled so that single CPs can be analysed. The resulting silver stained SDS-PAGE image is shown in Fig. 1.
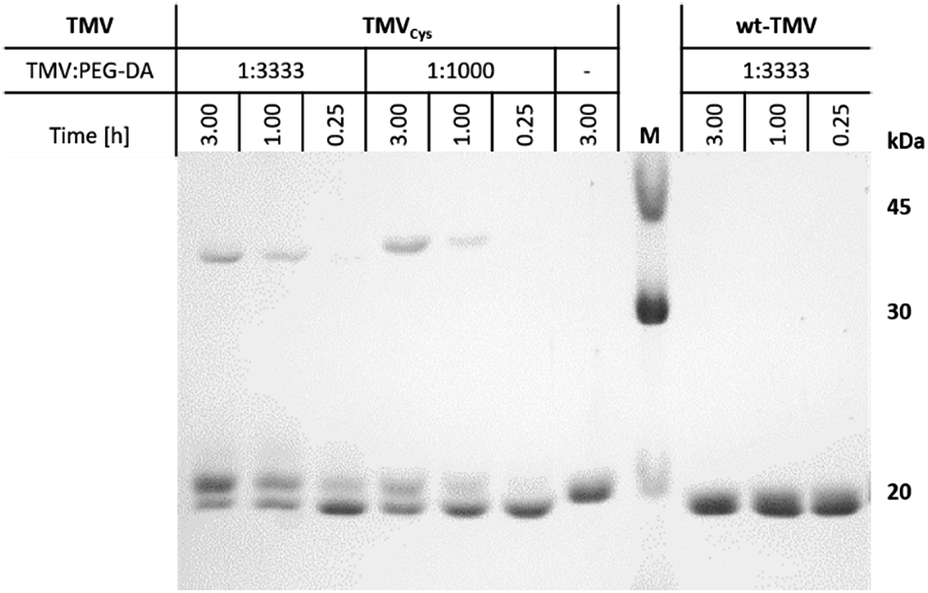 | ||
| Fig. 1 SDS-PAGE of the different coupling products between the CPs of TMVCys with PEG-DA, and corresponding preparations with wt-TMV, at different reaction times and functional group ratios of TMV CP and PEG-DA, as indicated. | ||
For wt-TMV, no additional band of the wt-CP was observed, indicating that indeed no chemical reaction took place between wt-TMV and PEG-DA. This result can be explained by the absence of reactive groups on the wt-TMV surface. On the other hand for TMVCys, depending on the reaction time and the ratio of functional groups, generally three different product species were identified after the reaction. One of them is the unmodified CP of TMVCys, showing that reaction of PEG-DA with the CPs of TMVCys did not proceed quantitatively. However, also one reaction product with a slightly larger molecular weight than the CP was observed. This was attributed to a coupling of one PEG-DA molecule (Mn = 700 g mol−1) with one CP, leading to CP-PEG-A adducts. This would result in TMV-PEG-A particles as depicted in Scheme 1. Additionally, a reaction product was formed which had approx. twice the molecular weight of one CP. This can be explained by a reaction of two CPs with one PEG-DA molecule, stemming from CP-PEG-CP adducts. This can involve either two CPs of the same TMVCys, resulting in a loop on the TMVCys surface, or two CPs of two different TMVCys, resulting in a coupling of two TMVCys-containing particles. In these cases, both end groups of PEG-DA are converted and no residual acrylic end group can participate in a subsequent hydrogel cross-linking reaction. Therefore, this double reaction of PEG-DA is not desirable for the covalent integration of TMVCys into PEG-DA hydrogels. The slight shift in migration distance of wt-CP and CPCys in the presence of PEG-DA in comparison with CPCys without PEG-DA is due to the influence of the polymer on the general migration behaviour, which also hinders the SDS-PAGE analysis of higher PEG-DA ratios to TMV (e.g. 10000:1).
In order to have as many reactive acrylic groups as possible on the TMVCys surface, we aimed to maximise the percentage of CP-PEG-A and to minimise the percentage of CP-PEG-CP. For this, we quantified the respective amounts under different reaction conditions by ImageJ analysis of Fig. 1.40 It was found that after one hour, at least one third of the CPs had reacted only once with PEG-DA corresponding to approx. 700 reactive acrylates per TMV particle. An increase in reaction time resulted especially for the lowest PEG-DA:TMV ratio in an increase of double-reacted PEG-DA (Table 2). As a compromise between maximised CP-PEG-A content and minimised CP-PEG-CP content, for the hydrogel preparation experiments described below the coupling was allowed to proceed for one hour.
| TMV:PEG-DA |
1:3333 |
1:1000 |
||||
| Time [h] | 3.00 | 1.00 | 0.25 | 3.00 | 1.00 | 0.25 |
| Free CP, 17.6 kDa | 34 | 39 | 56 | 41 | 57 | 68 |
| CP-PEG-A, 18.3 kDa | 44 | 42 | 33 | 35 | 30 | 23 |
| CP-PEG-CP, 35.9 kDa | 22 | 19 | 11 | 24 | 13 | 9 |
Hydrogel preparation and characterisation
Next, both the TMV-PEG-A particles and wt-TMV were incorporated into respective PEG-DA hydrogels. The corresponding formulations contained PEG-DA, the photo-initiator Irgacure 2959 as well as wt-TMV or TMV-PEG-A, respectively. For all hydrogels prepared with one type of the respective TMV particles, the PEG-DA concentration cPEG-DA (20 wt%) and Irgacure 2959 concentration (0.1 wt%) were kept constant in order to evaluate the effect of TMV particle addition to the formulations on the hydrogel properties. For this, the respective TMV particles were simply added to the PEG-DA containing hydrogel formulations at concentrations of 0.1 wt%, 0.3 wt%, 1.0 wt%, and 2.0 wt% and were allowed to react with the PEG-DA for 1 h. With the molecular weight of one TMV CP of 17.6 kDa, the ratio of TMV CPs and PEG-DA acrylate groups thus was 10000, 3333, 1000, and 500, respectively, which should lead mainly to TMV-PEG-A in the case of TMVCys, whereas wt-TMV would remain unaltered. Subsequently, the formulations were cured by radical photo-polymerisation of the PEG-DA end groups induced by UV irradiation. As reference samples, pure PEG-DA hydrogels with solid contents of 20.1 wt%, 20.3 wt%, 21 wt%, and 22 wt% were prepared, thus having the same solid content as the TMV containing samples. The resulting compositions of all hydrogel formulations in this study are shown in Table 1. The sample names denote which kind of compound is added to the basic hydrogel formulation as well as the additional amount of added compound in wt%. For example, sample TMVCys1.0 contained 20 wt% PEG-DA, 0.1 wt% Irgacure 2959 and 1.0 wt% TMVCys in SPP buffer.
For all samples, preparation of hydrogels by UV irradiation was possible with high efficiency. Upon curing, the mobile liquids were all converted into well-defined solids. Also with the eye, no differences between the hydrogels could be observed (photos in ESI†). All hydrogels could be handled easily without breakage and were slightly opaque. In order to evaluate if the presence of wt-TMV or TMVCys had an effect on the double bond conversion of the PEG-DA, ATR-FT-IR spectra of dried hydrogel samples were collected (Fig. 2). Compared to unreacted PEG-DA, we observed the disappearance of the double bond stretching vibration at 1635 cm−1 and the in-plane scissoring vibration at 1408 cm−1.41,42 This was the case for all samples, indicating that all samples were fully cured and that the addition of wt-TMV or TMV-PEG-A to the hydrogel formulations did not hamper the radical polymerisation of the acrylate groups.
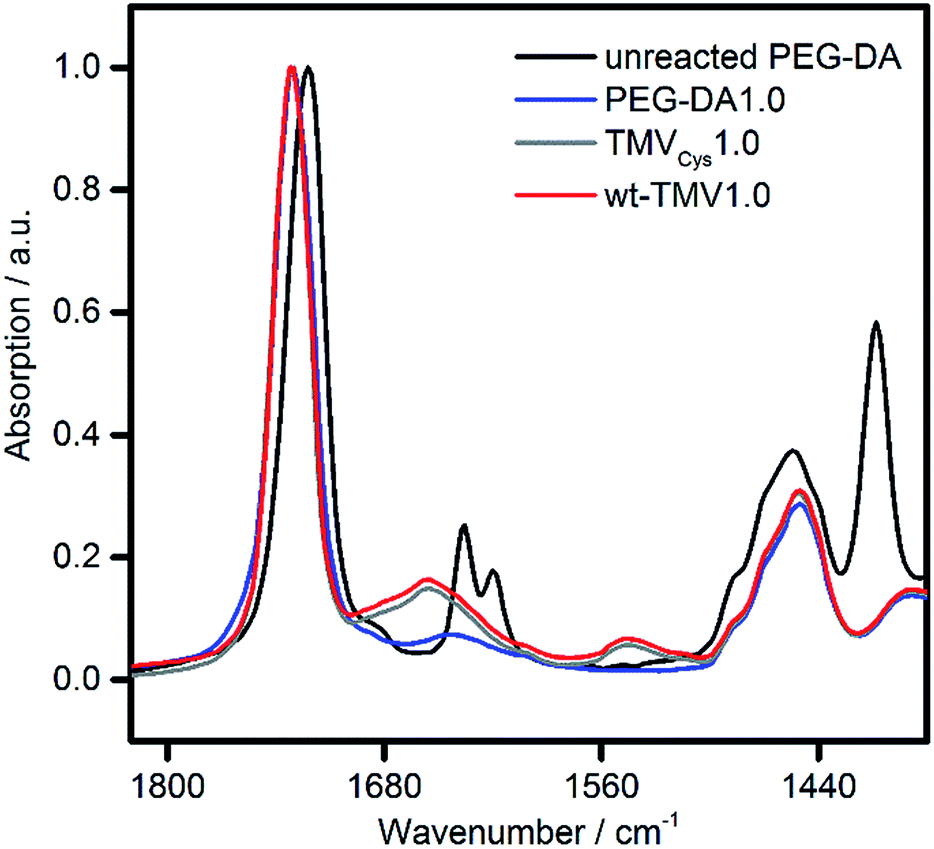 | ||
| Fig. 2 ATR-FT-IR spectra of unreacted PEG-DA and of dried hydrogel samples containing 20 wt% PEG-DA supplemented with either 1 wt% PEG-DA (PEG-DA1.0), TMVCys (TMVCys1.0) or wt-TMV (wt-TMV1.0). | ||
Due to the high double bond conversion in all samples after curing, it can be expected that for the samples containing TMV-PEG-A, the TMV particles are covalently bound into the hydrogel polymer network as depicted in Scheme 1. In order to investigate the effect of the TMV particle addition on the hydrogel properties, we assessed the mechanical properties of the hydrogels by oscillatory shear rheology. The values for the storage moduli G′ and the loss moduli G′′ are shown in Fig. 3. For all samples, G′ was two to three orders of magnitude larger than G′′, revealing the mainly elastic behaviour of the hydrogels. This observation is in accordance with the ATR-FT-IR data confirming successful cross-linking. Looking at the storage moduli of pure PEG-DA hydrogels, a significant (p < 0.01), nearly linear increase of G′ with increasing cPEG-DA was observed from 225 kPa (PEG-DA0.1) to 234 kPa (PEG-DA0.3), 269 kPa (PEG-DA1.0), and 290 kPa (PEG-DA2.0). This behaviour can be explained by the increasing cross-link density with increasing PEG-DA content and is in accordance with the literature.43
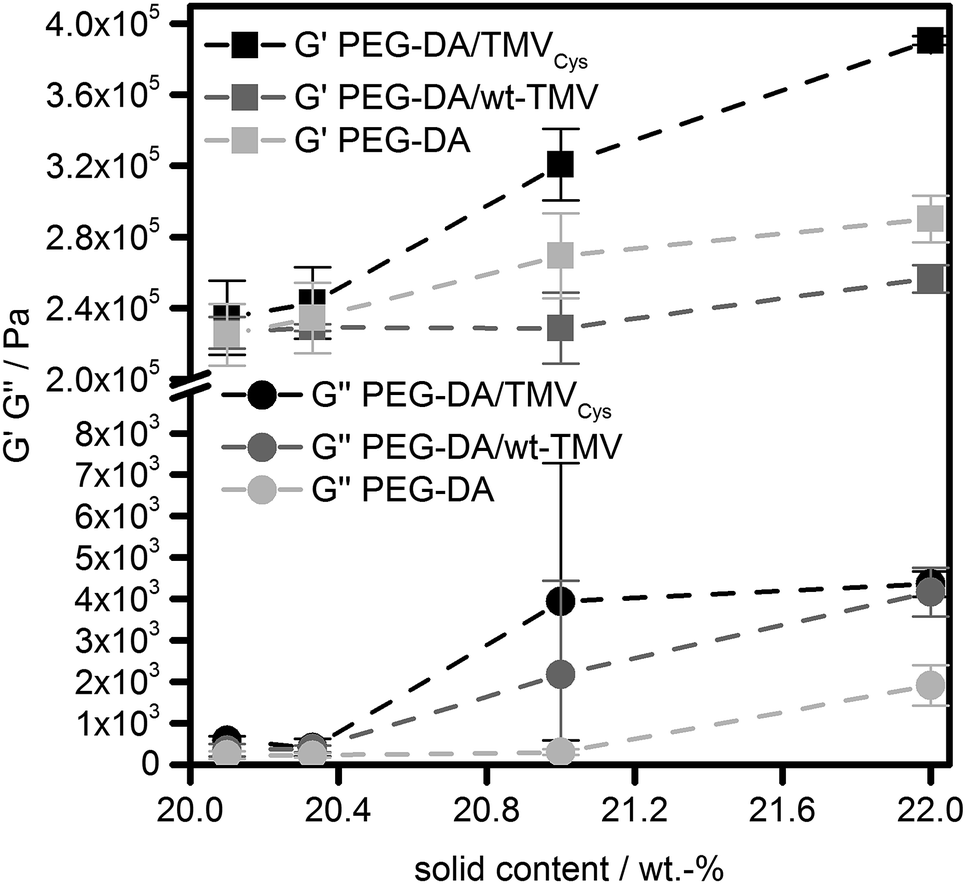 | ||
| Fig. 3 Storage moduli (G′) and loss moduli (G′′) of PEG-DA hydrogels containing TMVCys, wt-TMV or no TMV, respectively. The dashed lines are for the guidance of the eye only. | ||
Looking at the wt-TMV-containing samples, a different behaviour was observed. The addition of wt-TMV did not lead to a significant change of G′, leading to G′ values of 226 kPa (wt-TMV0.1), 229 kPa (wt-TMV0.3), 229 kPa (wt-TMV1.0) and 256 kPa (wt-TMV2.0). This observation is in accordance with the assumption that the wt-TMV particles are not covalently bound to the hydrogel polymer network upon photo-polymerisation and also do not interact with the polymer network significantly in any other way. This can be explained by the lack of functional groups on the wt-TMV surface which can participate in the radical cross-linking.
This was not the case for hydrogels which contained TMVCys. As described above, the thiols present on the TMV surface readily undergo thiol-Michael reactions to yield acrylate functionalized TMV-PEG-A. These can participate in the radical cross-linking and can be incorporated covalently into the hydrogel matrix. The effect can be seen from the G′ values of the respective hydrogels which were generally higher than the G′ of PEG-DA hydrogels with an identical solid content. Thus, with increasing concentration of TMVCys, G′ increased significantly (p < 0.01) from 235 kPa (TMVCys0.1) to 243 kPa (TMVCys0.3), 321 kPa (TMVCys1.0), and 391 kPa (TMVCys2.0). All mean values were significantly different from the other mean values (p < 0.01), except for the results of TMVCys0.1 and TMVCys0.3. The larger effect of TMVCys addition compared to PEG-DA addition seems to confirm the different architecture of the network points resulting from TMV-PEG-A (Scheme 1) as compared to the PEG-DA hydrogels. Comparing the different sample types (without TMV, with wt-TMV, with TMVCys) at the same solid content, the general trend in G′ as a measure for hydrogel stiffness can be summarised as follows: G′TMVCys > G′PEG-DA > G′wt-TMV. At smaller solid contents (20.1 wt%, 20.3 wt%), this trend is not significant, however at larger solid contents (21.0 wt%, 22.0 wt%), it becomes significant (p < 0.05 and p < 0.01).
Interestingly, the EWC of the hydrogels was practically constant for all formulations tested (Fig. 4). All hydrogels contained between 81% and 82% of water. However, there was a small, significant effect (p < 0.01) on the EWC with the solid content for all sample types which lead to a decrease of EWC with the solid content. Between the different sample types (without TMV, with wt-TMV, with TMVCys), no significant differences in EWC were observed. This leads to the conclusion that the water uptake capacity of the hydrogels was obviously dominated by the PEG-DA in all cases. This is due to the small fraction of TMV particles in the solid content of the hydrogels. Even at the highest concentrations tested, only 9.1% of the solid content were TMV particles, given by the ratio of 2 wt% TMV particles and the total solid content of 22 wt%. Since the wt-TMV particles do not interact strongly with the polymer network, a large influence on the EWC was not expected. However, also the covalent integration of TMV-PEG-A into the hydrogels showed only a very small effect on the EWC.
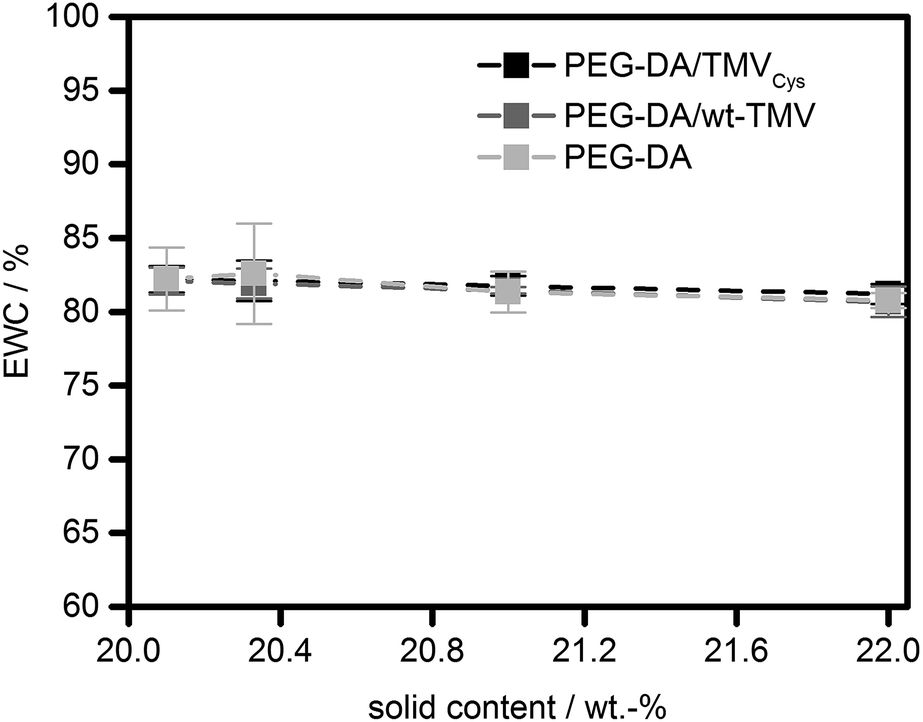 | ||
| Fig. 4 Equilibrium water contents (EWC) of PEG-DA hydrogels containing TMVCys, wt-TMV or no TMV, respectively. | ||
In order to explain the different impact of TMV addition on G′ and the EWC, a closer look at the molecular structure of the hydrogel networks is necessary. In order to estimate if unbound TMV particles can diffuse through the hydrogel networks, the EWCs of the pure PEG-DA hydrogels were used to calculate the molecular weight Mc between cross-links in the hydrogels using the Flory–Rehner equation in its modified version by Merrill and Peppas.44,45 With the Flory–Huggins interaction parameter with water (0.426),46 the density (1.12 g cm−3),47 and the number average molecular weight (700 g mol−1) of PEG-DA, an Mc of approx. 200 g mol−1 is estimated for the pure PEG-DA hydrogels. Following the reasoning of Canal and Peppas,48 with the average bond length (0.146 nm),49 the molecular weight of the repeating unit (44.05 g mol−1), and the characteristic ratio of PEG (4.1),49 this translates into average mesh sizes of approx. 2 nm. Therefore, it should be impossible for intact TMV particles with an outer diameter of 18 nm50 to diffuse through the hydrogel network. As soon as the network is formed, the TMV particles are trapped inside the hydrogel independent of the presence of covalent bonds between the TMV particles and the network. Therefore, the amide I and amide II vibrations at 1655 cm−1 and 1545 cm−1 are visible in the ATR-FT-IR spectra both in the wt-TMV and TMVCys containing hydrogels also after washing, without any notable difference between the two TMV particle types (Fig. 2).
The comparison between the average mesh sizes in the hydrogels and the size of the TMV particles with a length of 300 nm also gives insight into the impact of TMV-PEG-A addition to PEG-DA hydrogels on their molecular structure. The TMV nanorods penetrate through the length of approx. 150 meshes of the PEG-DA network. Due to the stiff nature of the TMV particles, this results in stiffening of relatively large regions of the hydrogel, also explaining the increase in G′ with larger TMV-PEG-A concentrations. On the other hand, meshes adjacent to the TMV containing meshes and further away – which are the majority of meshes – are not changed much on the molecular level, resulting in very little impact on the EWC. The addition of wt-TMV therefore has no significant effect on the mechanical properties. However, it can be expected that the TMV particles occupy a large space compared to the average mesh size and thus cause defects in the polymer networks much bigger than the average mesh size. Without the covalent bonds which link TMV-PEG-A to the networks, with wt-TMV the PEG-DA network can obviously slide over the TMV surfaces, thereby overriding the stiffening effect on the hydrogel. However, these defects are not accessible to water due to the presence of the TMV particles, explaining the low effect of wt-TMV addition on the EWC.
Interestingly, our results on the mechanical properties of the TMV containing PEG-DA hydrogels are not reflected by results published so far for hydrogels made from other polymers. Luckanagul et al. added 0.1% of wt-TMV into porous alginate hydrogels and found an increase of compressive modulus in unconfined uniaxial compression compared to samples containing no wt-TMV.31 We assume that the reason for this different behaviour can be found in more non-covalent interactions which are possible between alginate and wt-TMV CP compared to the interactions between the rather unfunctional PEG backbone of PEG-DA and wt-TMV. On the other hand, Maturavongsadit et al. reported that in hyaluronan hydrogels prepared from methacrylated hyaluronan, the shear modulus of hydrogels in which covalently bound cysteine-containing TMV was present (0.1%) was slightly lower than for hydrogels without cysteine-containing TMV.34 However, their samples were very soft with a G′ in the range of 50 Pa to 100 Pa and the difference in sample properties was small. These data suggest a very low degree of cross-linking and a large mesh size in general, especially when compared to other hyaluronan hydrogels which were prepared by thiol-Michael addition cross-linking.51 Additionally, the degree of hyaluronan conjugation to the cysteine-containing TMV was not determined in this study and thus the impact of TMV on the hydrogel structure cannot directly be compared. From our results it can be assumed that in their case the conjugation between hyaluronan and cysteine-containing TMV did not lead to substantially increased cross-linking density because they did not observe a stiffening effect.
To further support the above-mentioned conclusion that TMVs cannot diffuse through the hydrogel network, we quantified the TMV concentrations in the three washing buffers of the hydrogels with a micro BCA assay. The washing buffer was exchanged every 24 h so that the three measurements allow determining the kinetics with which TMV is washed out of the hydrogels over 72 h. As a prerequisite for this experiment, we verified the possibility to quantify the concentration of wt-TMV and TMVCys in suspension by measuring a calibration curve using TMV suspensions with defined concentrations (ESI†). The micro BCA assay was then performed with the serial washing buffers, and the respective TMV concentration was calculated using the calibration function determined before. For both the wt-TMV and TMVCys-containing hydrogels, only the first of the three washing buffers, collected after 24 h washing time, contained detectable concentrations of wt-TMV and TMVCys, respectively. The relative amount of the TMV derivatives found in these washing buffers compared to the total amount of TMV in the hydrogels is shown in Fig. 5.
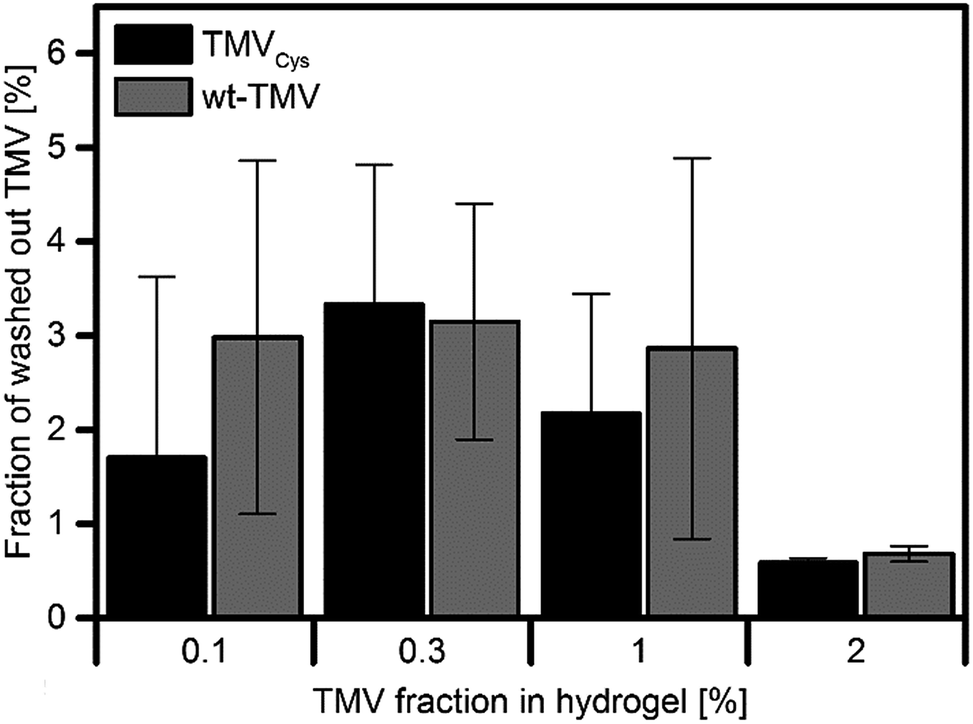 | ||
| Fig. 5 Fraction of initially applied TMV released in the first washing step (24 h). In the subsequent two wash solutions, no further TMV was detected. | ||
It is evident that the washed out TMV varied between approx. 1% and 3% of the initial amount, independent of the TMV derivative used. No significant differences between the samples were observed. The fact that TMV was found only in the first washing buffer suggests that it derived mainly from TMV adsorbed on the hydrogel surface, but not from TMV diffused out of the interior of the hydrogel.
Investigation of TMV structure in the hydrogels
Depending on the solvent conditions, TMV particles can either be solvated as individual virus rods, or form nematic crystals, or aggregate to bigger virus bundles.52 As PEG of higher molecular weight (Mn = 6000 g mol−1) was used for precipitating TMV, it cannot be completely excluded that a similar effect took place in PEG-DA solutions. Therefore, it was interesting to investigate the distribution of the virus particles within the hydrogels. So far most structural investigations on hydrogel preparations have been performed by (E-)SEM analysis of (freeze-)dried hydrogels with a resolution in the 100 nm to 1 μm range not sensitive enough to see features as small as TMV.15 Also changes in hydrogel morphology can be expected to occur during the drying process. Therefore, the TMV containing hydrogels were embedded in epoxy resin, stained with uranyl acetate and sectioned into 60–100 nm thick slices. TMV particles in the hydrogels appear as 10 to 20 nm thick lines of different lengths (100–1000 nm, see Fig. 6).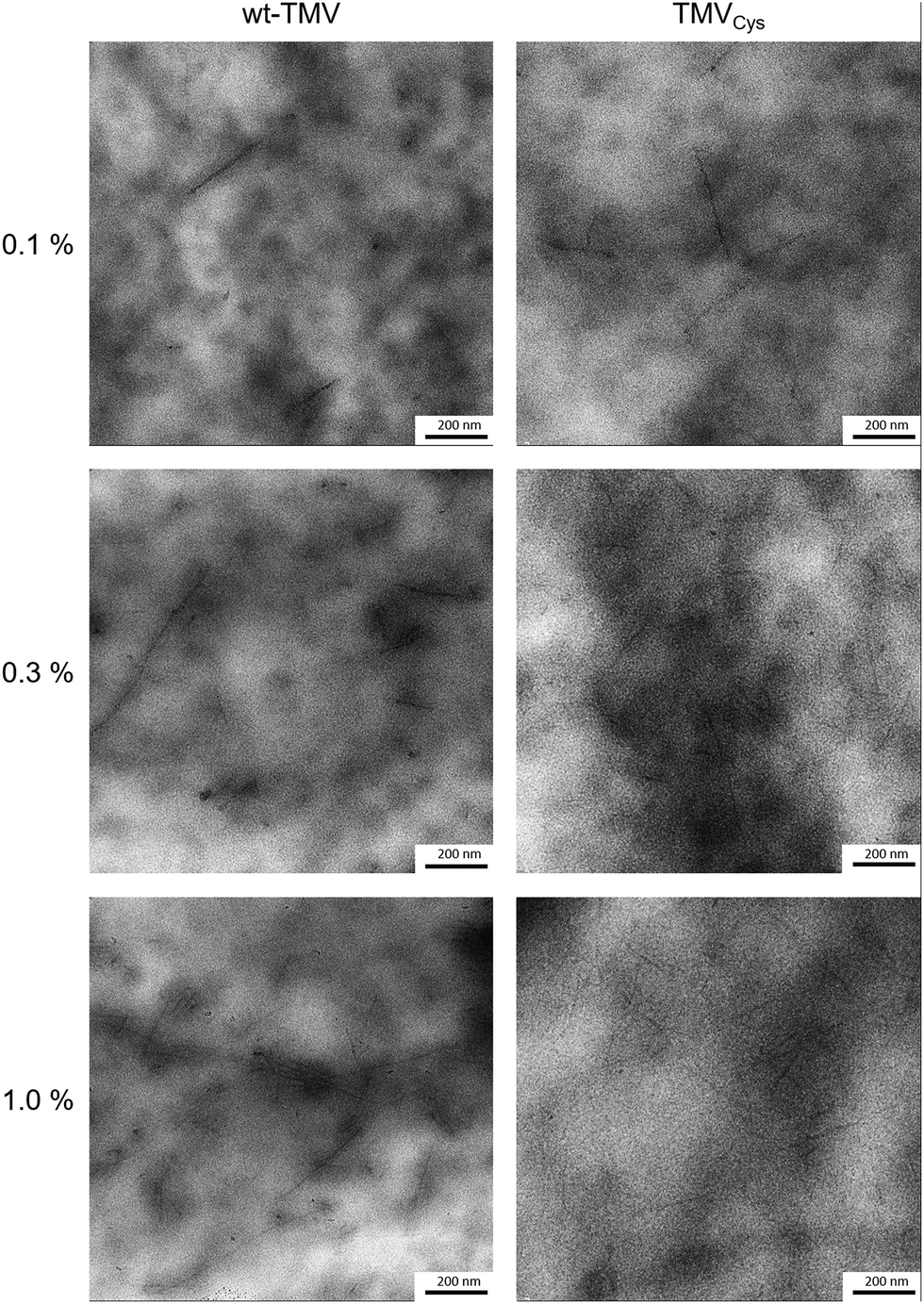 | ||
| Fig. 6 TEM images of TMV containing hydrogels after resin embedding and sectioning. TMV particles can be seen as dark, uranyl acetate stained lines upon the cloudy hydrogel background. | ||
This variation in length is due to two effects; first, the virus particles are embedded with varying orientations and second, they tend to form so-called head-to-tail attachments so that five TMV attached in this way would result in a 1.5 μm long particle.53 For both TMV variants, TMV particles could be detected at all concentrations. There was also no significant difference in the amount of TMV for wt-TMV in comparison to TMVCys visible, supporting the conclusion that the viruses are confined within the hydrogels due to the small mesh size. The viruses were also relatively homogeneously distributed within the hydrogels, thus it can be concluded that PEG-DA does not have an aggregating effect on TMV. The absence of larger TMV aggregates in the hydrogels containing TMVCys also indicates that the formation of CP-PEG-CP during the thiol-Michael reaction takes place prior to cross-linking preferentially by forming PEG loops on a single TMV particle rather than by linking CPs of two different TMVs to each other. This is in contrast to the results of Wu et al.,54 they combined TMV at 1.5 wt% with alginate and observed immediate clouding of the solution while TMV-PEG-DA solutions stayed clear upon mixing. In addition, the corresponding TEM images showed clusters of laterally aligned TMV particles, oriented by the application of shear forces, within an unstained alginate background.
Conclusions
The TMVCys mutant was successfully functionalised with acrylate groups by coupling thiol-presenting CPCys subunits with PEG-DA via the thiol-Michael reaction. The thus obtained TMV-PEG-A particles were integrated covalently into PEG-DA hydrogels, as evidenced by the increased G′ of the hydrogels with increasing TMV-PEG-A concentration. On the other hand, wt-TMV did not have a significant effect on G′ due to the missing covalent interactions. The presence of the TMV derivatives had no effect on the curing efficiency of PEG-DA or on the EWC. Due to the small mesh size of the hydrogels, both TMV derivatives were immobilised in the hydrogels. They were distributed quite homogeneously throughout the whole volume. The results give rise to the conclusion that TMV particles offer a way to add functionality into otherwise non-functional PEG-DA hydrogels, extending earlier findings for alginate and hyaluronan gels and adding a detailed characterisation to the initial promising results obtained for TMV-containing PEG-DA gels.35 Especially the thiol groups of the TMVCys mutant or functional groups of other TMV mutants not tested in this study might be used for future coupling of further functional moieties such as growth factors or enzymes into the hydrogels, e.g. for uses as cell culture or tissue engineering matrices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank David Brodbeck for the initial TMV/PEG-DA coupling studies and Holger Jeske for fruitful discussions about TEM sample preparation. We gratefully acknowledge generous financial support by the Carl Zeiss Foundation and the University of Stuttgart within the Projekthaus NanoBioMater. S. E. acknowledges financial support by the Deutsche Forschungsgemeinschaft SPP1569.Notes and references
- S. Lee, X. Tong and F. Yang, Acta Biomater., 2014, 10, 4167–4174 CrossRef CAS PubMed.
- M. B. Mellott, K. Searcy and M. V. Pishko, Biomaterials, 2001, 22, 929–941 CrossRef CAS PubMed.
- J. P. Mazzoccoli, D. L. Feke, H. Baskaran and P. N. Pintauro, J. Biomed. Mater. Res., Part A, 2010, 93, 558–566 Search PubMed.
- X. Zhang, B. Xu, D. S. Puperi, A. L. Yonezawa, Y. Wu, H. Tseng, M. L. Cuchiara, J. L. West and K. J. Grande-Allen, Acta Biomater., 2015, 14, 11–21 CrossRef CAS PubMed.
- F. Markus, F. Dreher, S. Laschat, S. Baudis, G. E. M. Tovar and A. Southan, Polymer, 2017, 108, 21–28 CrossRef CAS.
- G. Kraut, L. Yenchesky, F. Prieto, G. E. M. Tovar and A. Southan, J. Appl. Polym. Sci., 2017 DOI:10.1002/APP.45083.
- A. Hiller, K. Borchers, G. E. M. Tovar and A. Southan, Addit. Manuf., 2017, 18, 136–144 CrossRef CAS.
- Z. Gao, C. S. Kim and D. B. Henthorn, IEEE Sens. J., 2013, 13, 1690–1698 CrossRef CAS.
- F. Kivlehan, M. Paolucci, D. Brennan, I. Ragoussis and P. Galvin, Anal. Biochem., 2012, 421, 1–8 CrossRef CAS PubMed.
- S. D. Esha, Y. T. Mary, E. R. Amy and A. G. Richard, Biomed. Mater., 2012, 7, 024108 CrossRef PubMed.
- H. Zhang, L. Wang, L. Song, G. Niu, H. Cao, G. Wang, H. Yang and S. Zhu, J. Appl. Polym. Sci., 2011, 121, 531–540 CrossRef CAS.
- X. Dai, X. Chen, L. Yang, S. Foster, A. J. Coury and T. H. Jozefiak, Acta Biomater., 2011, 7, 1965–1972 CrossRef CAS PubMed.
- G. Majer and A. Southan, J. Chem. Phys., 2017, 146, 225101 CrossRef PubMed.
- P. Malo de Molina, S. Lad and M. E. Helgeson, Macromolecules, 2015, 48, 5402–5411 CrossRef CAS.
- N. Karaca, G. Temel, D. Karaca Balta, M. Aydin and N. Arsu, J. Photochem. Photobiol., A, 2010, 209, 1–6 CrossRef CAS.
- A. Metters and J. Hubbell, Biomacromolecules, 2005, 6, 290–301 CrossRef CAS PubMed.
- A. Southan, E. Hoch, V. Schönhaar, K. Borchers, C. Schuh, M. Müller, M. Bach and G. E. M. Tovar, Polym. Chem., 2014, 5, 5350–5359 RSC.
- E. K. U. Larsen, M. B. L. Mikkelsen and N. B. Larsen, Biomicrofluidics, 2014, 8, 064127 CrossRef PubMed.
- F. Tan, X. Xu, T. Deng, M. Yin, X. Zhang and J. Wang, Biomed. Mater., 2012, 7, 055009 CrossRef PubMed.
- M. Peter and P. Tayalia, RSC Adv., 2016, 6, 40878–40885 RSC.
- B. Yañez-Soto, S. J. Liliensiek, C. J. Murphy and P. F. Nealey, J. Biomed. Mater. Res., Part A, 2013, 101, 1184–1194 CrossRef PubMed.
- J. S. Miller, C. J. Shen, W. R. Legant, J. D. Baranski, B. L. Blakely and C. S. Chen, Biomaterials, 2010, 31, 3736–3743 CrossRef CAS PubMed.
- R. A. McBath and D. A. Shipp, Polym. Chem., 2010, 1, 860–865 RSC.
- Y. Okada, Philos. Trans. R. Soc. Lond. Ser. B: Biol. Sci., 1999, 354, 569–582 CrossRef CAS PubMed.
- C. Koch, F. J. Eber, C. Azucena, A. Forste, S. Walheim, T. Schimmel, A. M. Bittner, H. Jeske, H. Gliemann, S. Eiben, F. C. Geiger and C. Wege, Beilstein J. Nanotechnol., 2016, 7, 613–629 CrossRef CAS PubMed.
- A. Calo, S. Eiben, M. Okuda and A. M. Bittner, Jpn. J. Appl. Phys., 2016, 55(3S2), 03DA01 CrossRef.
- F. J. Eber, S. Eiben, H. Jeske and C. Wege, Nanoscale, 2015, 7, 344–355 RSC.
- Z. Wu, A. Mueller, S. Degenhard, S. E. Ruff, F. Geiger, A. M. Bittner, C. Wege and C. E. Krill 3rd, ACS Nano, 2010, 4, 4531–4538 CrossRef CAS PubMed.
- F. C. Geiger, F. J. Eber, S. Eiben, A. Mueller, H. Jeske, J. P. Spatz and C. Wege, Nanoscale, 2013, 5, 3808–3816 RSC.
- A. Schneider, F. J. Eber, N. L. Wenz, K. Altintoprak, H. Jeske, S. Eiben and C. Wege, Nanoscale, 2016, 8, 19853–19866 RSC.
- J. Luckanagul, L. A. Lee, Q. L. Nguyen, P. Sitasuwan, X. M. Yang, T. Shazly and Q. Wang, Biomacromolecules, 2012, 13, 3949–3958 CrossRef CAS PubMed.
- P. Sitasuwan, L. A. Lee, P. Bo, E. N. Davis, Y. Lin and Q. Wang, Integr. Biol., 2012, 4, 651–660 RSC.
- J. A. Luckanagul, L. A. Lee, S. J. You, X. M. Yang and Q. Wang, J. Biomed. Mater. Res., Part A, 2015, 103, 887–895 CrossRef PubMed.
- P. Maturavongsadit, J. A. Luckanagul, K. Metavarayuth, X. Zhao, L. M. Chen, Y. Lin and Q. Wang, Biomacromolecules, 2016, 17, 1930–1938 CrossRef CAS PubMed.
- C. L. Lewis, Y. Lin, C. X. Yang, A. K. Manocchi, K. P. Yuet, P. S. Doyle and H. Yi, Langmuir, 2010, 26, 13436–13441 CrossRef CAS PubMed.
- S. N. Chapman, Methods Mol. Biol., 1998, 81, 123–129 CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS PubMed.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- P. Carmona and J. Moreno, J. Mol. Struct., 1982, 82, 177–185 CrossRef CAS.
- R. P. Witte, A. J. Blake, C. Palmer and W. J. Kao, J. Biomed. Mater. Res., Part A, 2004, 71, 508–518 CrossRef PubMed.
- J. A. Beamish, J. Zhu, K. Kottke-Marchant and R. E. Marchant, J. Biomed. Mater. Res., Part A, 2010, 92, 441–450 Search PubMed.
- P. J. Flory and J. Rehner, J. Chem. Phys., 1943, 11, 521–526 CrossRef CAS.
- N. A. Peppas and E. W. Merrill, J. Appl. Polym. Sci., 1977, 21, 1763–1770 CrossRef CAS.
- E. W. Merrill, K. A. Dennison and C. Sung, Biomaterials, 1993, 14, 1117–1126 CrossRef CAS PubMed.
- G. M. Cruise, D. S. Scharp and J. A. Hubbell, Biomaterials, 1998, 19, 1287–1294 CrossRef CAS PubMed.
- T. Canal and N. A. Peppas, J. Biomed. Mater. Res., 1989, 23, 1183–1193 CrossRef CAS PubMed.
- J. E. Mark and P. J. Flory, J. Am. Chem. Soc., 1965, 87, 1415–1423 CrossRef CAS.
- X. Z. Fan, E. Pomerantseva, M. Gnerlich, A. Brown, K. Gerasopoulos, M. McCarthy, J. Culver and R. Ghodssi, J. Vac. Sci. Technol., A, 2013, 31, 4816584 Search PubMed.
- V. Hagel, M. Mateescu, A. Southan, S. V. Wegner, I. Nuss, T. Haraszti, C. Kleinhans, C. Schuh, J. P. Spatz, P. J. Kluger, M. Bach, S. Tussetschläger, G. E. M. Tovar, S. Laschat and H. Boehm, Sci. Rep., 2013, 3, 2043 CrossRef PubMed.
- J. D. Bernal and I. Fankuchen, J. Gen. Physiol., 1941, 25, 111–U118 CrossRef CAS PubMed.
- W. Shenton, T. Douglas, M. Young, G. Stubbs and S. Mann, Adv. Mater., 1999, 11, 253–256 CrossRef CAS.
- Y. H. Wu, Z. Y. Jiang, X. J. Zan, Y. Lin and Q. Wang, Colloids Surf., B, 2017, 158, 620–626 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10364f |
| This journal is © The Royal Society of Chemistry 2018 |