
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePiperazinium-mediated crosslinked polyimide-polydimethylsiloxane (PI-PDMS) copolymer membranes: the effect of PDMS content on CO2 separation†
Hyelim Youab,
Iqubal Hossainab and
Tae-Hyun Kim *ab
*ab
aOrganic Material Synthesis Laboratory, Department of Chemistry, Incheon National University, Incheon, 406-772, Korea. E-mail: tkim@inu.ac.kr; Fax: +82-32-835-0762; Tel: +82-32-835-8232
bResearch Institute of Basic Sciences, Incheon National University, Incheon, 406-772, Korea
First published on 3rd January 2018
Abstract
We synthesized copolymers consisting mostly of physically stable rigid polyimide (PI) and a low content of highly permeable rubbery polydimethylsiloxane (PDMS), that were crosslinked by CO2-philic ionic piperazinium groups attached to the side chains of the copolymers. These crosslinked copolymers (xPI-PDMSs) were fashioned into membranes that showed very high levels of thermochemical stability and excellent CO2 separation performance (PCO2 of 799 Barrer and CO2/N2 permselectivity of 15.7). The inclusion of the piperazinium groups not only endowed these xPI-PDMS membranes with increased selectivity for CO2, but also good resistance to CO2 plasticization. The effect of PDMS content on morphology and CO2 separation properties of xPI-PDMS was also investigated.
Introduction
The concentration of CO2 in the atmosphere is steadily increasing, and this increase could lead to climate change and resulting threats to the environment and the economy. It is hence becoming increasingly important to slow down and even reverse this increase—and developing efficient, reversible, and cost-effective technologies for capturing CO2 is a promising way to reduce its atmospheric concentration.Separation methods based on absorption and on pressure swing adsorption are the predominant methods for capturing CO2.1–5 Nevertheless, separations based on polymer membranes have been attracting much attention as an alternative technology for CO2 separation because this technology does not require high capital costs and it is operationally simple, energy-efficient, and modular in nature and hence easy to scale up; most importantly it is environmentally benign since the mechanisms of these separations rely on physical rather than chemical processes.6–8 Developing polymeric membranes that are highly permeable and that at the same time selectively separate the target gas is, however, challenging due to the generally strong trade-off between permeability and selectivity as formulated by “Robeson's upper bound” for gas pairs.9,10
Several polymers have been investigated for forming membranes to be used in gas separation. Polyimide (PI) glassy polymers are typically used for this purpose, as they display good mechanical and thermal properties,11–14 and exhibit a wide range of permeability levels, accomplished by tuning the polymer structure. In particular, PI membranes based on 4,4′-hexafluoroisopropylidene diphthalic anhydride (6FDA) show good CO2/CH4 separation characteristics.15,16 The rigid structure of PIs, however, generally acts as a barrier in gas transport, causing relatively low gas permeability for practical applications. Furthermore, the selectivity levels of most glassy polymer membranes, including PIs, decrease as the feed pressure is increased above a certain level, due to these membranes becoming plasticized.16 Crosslinking effectively inhibits plasticization;17–21 this technique, however, is frequently accompanied by a loss of free volume elements and resulting significant decrease in permeability, although increased permeability has also been reported in some cases.22 Polydimethylsiloxane (PDMS) is a flexible rubbery polymer with a low glass transition temperature (Tg) and a high free volume for gas transport.23–25 A very high permeability is hence expected for membranes prepared from PDMS, but forming these membranes, or films, is difficult due to the poor mechanical properties of this rubbery polymer.
In order to overcome these limitations of PI and PDMS, several approaches, including crosslinking of PDMS26 or grafting PDMS onto the rigid PI27 have been applied, and were shown to improve the mechanical properties of the highly permeable PDMS. Copolymers between PDMS and PI as the main polymer backbone were also developed,28,29 and shown to exhibit higher free volumes than the pristine PIs due to the flexible nature of the PDMS, leading to enhanced permeability compared to the PIs combined with high amounts of PDMS. A sharp decrease in selectivity, however, inevitably accompanied high loading of PDMS for the PI-PDMS-based copolymers.
In the present work, we prepared PI-PDMS-based copolymers (PI-PDMSs) containing relatively little PDMS to minimize the reduction in the selectivity of the corresponding membranes. In addition, the PI-PDMS-based polymer membranes were crosslinked for the first time with ionic groups, specifically piperazinium groups, which was carried out to compensate for the reduced selectivity and mechanical strength resulting from the inclusion of the rubbery PDMS. The piperazinium groups were introduced onto the side chains of the PI-PDMS copolymers, as pendant ionic groups, to serve as both crosslinking units and CO2-philic functional groups. The resulting novel piperazinium-mediated crosslinked PI-PDMS copolymer product, denoted as “xPI-PDMS”, was shown to indeed have a high affinity for CO2, and hence increased selectivity for CO2. Such increased selectivity has also been previously reported for membranes based on other polymers functionalized with ionic groups.30–38 The xPI-PDMS membranes showed very high levels of thermal and mechanical stability, as well as high resistance to becoming plasticized in the presence of CO2. Most importantly, these membranes were shown to be very effective at separating CO2 from CH4 and from N2. In addition, the PDMS loading level was varied, within a low loading level range, and the gas-separation performances and physical properties of the resulting xPI-PDMS membranes are reported.
Experimental
Materials
4,4′-(Hexafluoroisopropylidene)diphthalic anhydride (6FDA), 2,3,5,6-tetramethyl benzene-1,4-diamine (durene), N-bromosuccinimide (NBS) and N,N′-dimethyl piperazine (piperazine) were purchased from Tokyo Chemical Industry (TCI) Co., Ltd. (Tokyo, Japan) and used as obtained. Ttriethyl amine and acetic anhydride were obtained from Alfa-Aesar (a Jonhson Mathey Co.). Alpha-omega-diamino-functionalized polydimethylsiloxane (PDMS, Mn of 2500 g mol−1) was purchased from Sigma Aldrich. 6FDA and durene were dried under vacuum at 60 °C for 24 h prior to the polymerization. All other chemicals, unless otherwise noted, were obtained from commercial sources and used as received.Synthesis
PI-PDMS-0.05 (3.33 g, 93.8%); δH (400 MHz, CDCl3) 7.90–8.11 (6H, br signal, ArH), 1.94–2.31 (12H, br signal, ArCH3), 0.01–0.09 (0.6H, br signal, CH3SiCH3); GPC (DMF, RI)/Da Mn 4.9 × 104, Mw 7.6 × 104 and Mw/Mn 1.5.
PI-PDMS-0.10 (3.10 g, 87.1%); δH (400 MHz, CDCl3) 7.90–8.12 (6H, br signal, ArH), 1.97–2.31 (12H, br signal, ArCH3), 0.01–0.16 (0.7H, br signal, CH3SiCH3); GPC (DMF, RI)/Da Mn 5.3 × 104, Mw 8.1 × 104 and Mw/Mn 1.5.
PI-PDMS-0.15 (3.10 g, 86.7%); δH (400 MHz, CDCl3) 7.89–8.13 (6H, br signal, ArH), 1.96–2.30 (12H, br signal, ArCH3), 0.01–0.11 (1.0H, br signal, CH3SiCH3); GPC (DMF, RI)/Da Mn 4.0 × 104, Mw 6.8 × 104 and Mw/Mn 1.7.
Br-PI-PDMS-0.05 (0.87 g, 86.1%); δH (400 MHz, CDCl3) 7.90–8.11 (6H, br signal, ArH), 4.45–4.27 (0.6H, br signal, ArCH2), 1.94–2.31 (12H, br signal, ArCH3), 0.01–0.09 (0.48H, br signal, CH3SiCH3).
Br-PI-PDMS-0.10 (0.89 g, 87.8%); δH (400 MHz, CDCl3) 7.90–8.12 (6H, br signal, ArH), 4.60–4.30 (0.6H, br signal, ArCH2), 1.97–2.31 (12H, br signal, ArCH3), 0.01–0.16 (0.53H, br signal, CH3SiCH3).
Br-PI-PDMS-0.15 (0.89 g, 87.4%); δH (400 MHz, CDCl3) 7.89–8.13 (6H, br signal, ArH), 4.50–4.30 (0.52H, br signal, ArCH2), 1.96–2.30 (12H, br signal, ArCH3), 0.01–0.11 (1.0H, br signal, CH3SiCH3).
Membrane preparations
Characterization and measurements
1H NMR spectra were obtained on an Agilent 400-MR (400 MHz) instrument using CDCl3 as a reference or internal deuterium lock. The attenuated total reflection Fourier transform infrared (ATR-FTIR) spectra were recorded using Bruker Vertex 80v, Hyperion2000 ATR-FTIR spectrometer.Molar masses were determined either by comparative spectroscopic methods using 1H NMR or Gel Permeation Chromatography (GPC) using two PL Gel 30 cm × 5 μm mixed C columns at 30 °C running in DFM and calibrated against polystyrene (Mn = 600 × 106 g mol−1) standards using a Knauer refractive index detector.
The densities of the membranes (g cm−3) were determined experimentally using a top-loading electronic Mettler Toledo balance (XP205, Mettler-Toledo, Switzerland) coupled with a density kit based on Archimedes' principle. The samples were weighed in air and a known-density liquid, high purity heptane. The measurement was performed at room temperature by the buoyancy method and the density was calculated as follows:
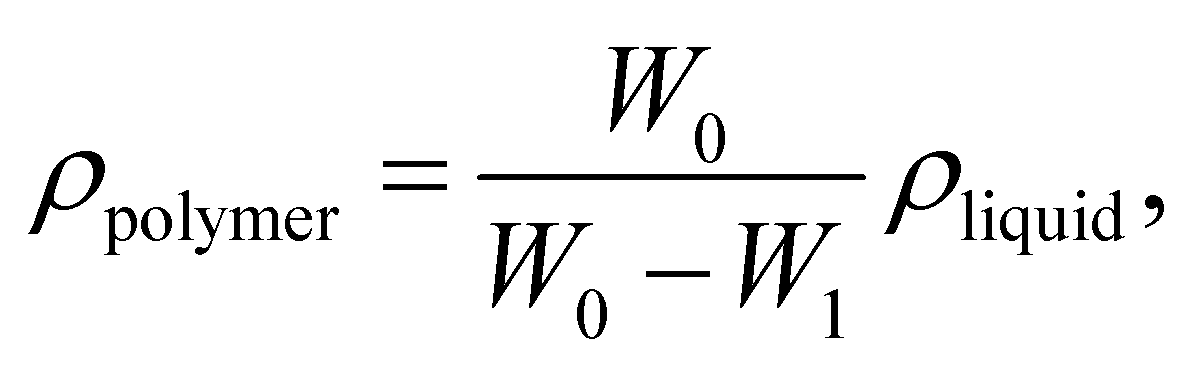
Tapping mode AFM was performed using a Bruker MultiMode instrument. A silicone cantilever with an end radius <10 nm and a force constant of 40 Nm−1 (NCHR, nanosensors, f = 300 kHz) was used to image the samples at ambient temperature.
The X-ray diffraction patterns of the membranes were measured using a Rigaku DMAX-2200H diffractometer by employing a scanning rate of 4° min−1 in a 2θ range from 5° to 30° with a Cu Kα1 X-ray (λ = 0.1540598). The d-spacings were calculated using the Bragg's law (d = λ/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ).
sinθ).
Tensile strength and elongation at break of the membranes were measured on a Shimadzu EZ-TEST E2-L instrument benchtop tensile tester using a crosshead speed of 1 mm min−1 at 25 °C under 50% relative humidity. Engineering stress was calculated from the initial cross-sectional area of the sample and Young's modulus (E) was determined from the initial slope of the stress–strain curve. The membrane samples were cut into a rectangular shape with 80 mm × 8 mm (total) and 80 mm × 4 mm (test area), and five specimens were used for the measurements.
The glass transition temperature (Tg) of each crosslinked membrane was measured using a Perkin-Elmer Pyris-1 DSC from −150 °C to 30 °C with a scan rate of 10 °C min−1 under nitrogen.
The gel fraction of crosslinked membranes was measured by immersing the corresponding membranes in THF for 24 h. The extracted residues were dried at 100 °C for 24 h, and before and after extraction weights were measured to determine the gel fraction by the following equation:
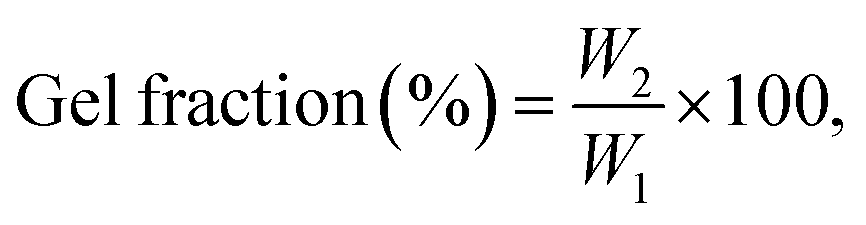
Gas permeation procedure
Permeation measurements of pure gas were taken using a high-vacuum time lag measurement unit based on a constant-volume/variable-pressure method. All of the experiments were performed at a feed pressure of 2 atm and a feed temperature of 30 °C, and further permeability measurements of CO2 and N2 was carried out over the pressure range between 5 and 25 atm with 5 atm increments for the isotherm experiments. Before taking these measurements, both the feed and the permeate sides were thoroughly evacuated to below 10−5 Torr until the readout showed zero values for the removal of any residual gases. The downstream volume was calibrated using a Kapton membrane and was found to be 50 cm3. The upstream and downstream pressures were measured using a Baraton transducer (MKS; model no. 626B02TBE) with a full scale of 10000 and 2 Torr, respectively. Pressure on the permeate side was recorded as a function of time using a pressure transducer and passed to a desktop computer through a shield data cable. The permeability coefficient was determined from the linear slope of the downstream pressure versus time plot (dp/dt) according to the following equation:
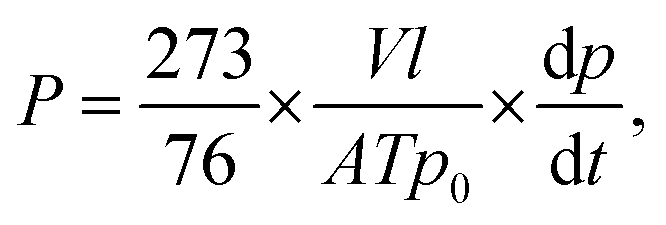 | (1) |
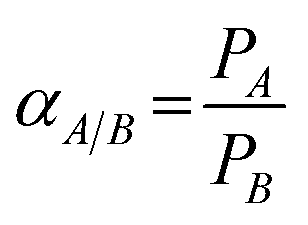 | (2) |
The diffusivity and solubility were obtained from the time-lag (θ) value according to the equations
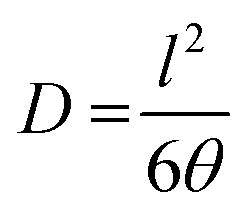 | (3) |
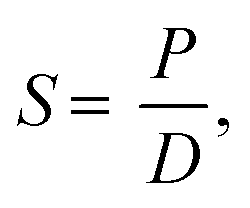 | (4) |
Results and discussion
Syntheses of the polyimide-polydimethylsiloxane (PI-PDMS) copolymers with various relative amounts of PDMS
Three PI-PDMS copolymers, 1, denoted as PI-PDMS-0.05, PI-PDMS-0.10, and PI-PDMS-0.15, and each based on a 6FDA-durene main skeleton, were first synthesized by carrying out polycondensation of 6FDA with durene and 5, 10, and 15 wt% PDMS (relative to durene), respectively (Scheme 1). Of the various possible polyimide structures, 6FDA-durene was chosen as the main skeleton for the polyimide due to its relatively high permeability.15,16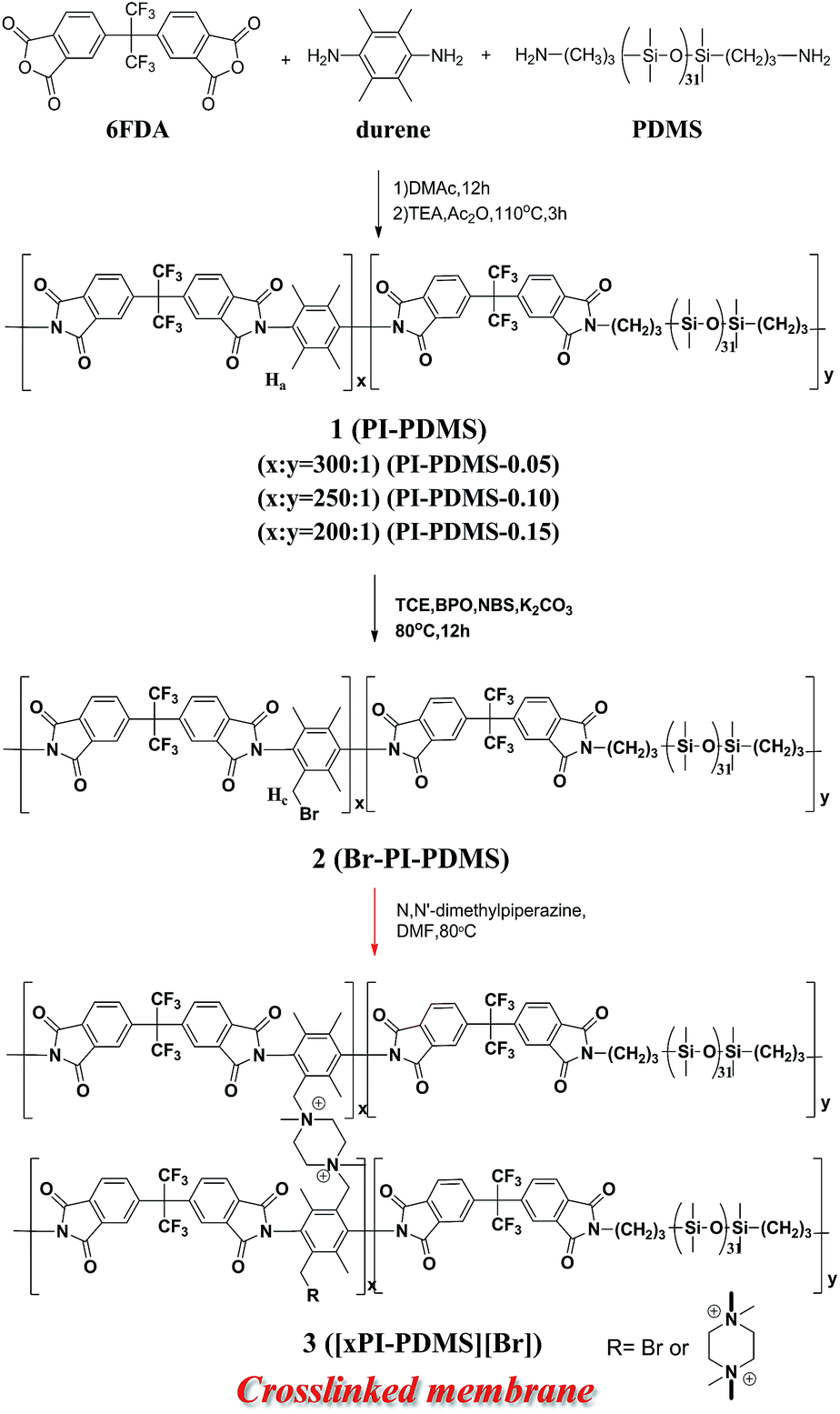 | ||
| Scheme 1 Schematic representation of the preparation of piperizinium-mediated crosslinked PI-PDMSs (xPI-PDMSs). | ||
Low levels of PDMS were included in the PI-PDMS to minimize the reduction in selectivity resulting from the highly permeable PDMS structure. The relative molar ratio of the 6FDA-durene PI to PDMS in each of the PI-PDMS products was determined from the 1H nuclear magnetic resonance (NMR) spectrum of the PI-PDMS, specifically by measuring the integral ratio of the peak corresponding to the benzylic proton (Ha) of durene to that of the methyl proton (Hb) of PDMS; this ratio was found to be about 300:1 for PI-PDMS-0.05, 250:1 for PI-PDMS-0.10, and 200:1 for PI-PDMS-0.15 (Fig. S1 in ESI†). All of the PI-PDMS copolymers were confirmed, using GPC, to have a high molecular weight (Mw = 60 to 80 kDa), despite the low amount of PDMS.
Syntheses of the piperazinium-mediated crosslinked polyimide-polydimethyl siloxane (xPI-PDMS) copolymer membranes 3 with various amounts of PDMS
The pristine PI-PDMS 1 copolymers with various amounts of PDMS were each selectively brominated at their ArCH3 units by using 0.28 equiv. of NBS and catalytic amounts of BPO in a tetrachloroethane solution to produce the corresponding bromobenzylated PI-PDMS (Br-PI-PDMS, 2) copolymers, i.e., Br-PI-PDMS-0.05, Br-PI-PDMS-0.10, and Br-PI-PDMS-0.15. A 1H NMR spectroscopy investigation confirmed the bromination of each PI-PDMS, and specifically indicated the selective bromination of its benzylic group: the intensity of the peak at 2.1 ppm corresponding to the benzylic proton (Ha) of durene was lower for each of the Br-PI-PDMS products than for the corresponding pristine PI-PDMS copolymers, a new peak (Hc) attributed to the bromobenzyl group appeared at 4.5 ppm for each of the products, and there were no changes in the intensities of other aromatic peaks (Fig. S2†).The degree to which each PI-PDMS copolymer was brominated was estimated by measuring the ratio of the integral of the bromobenzylated proton (Hb) peak in the brominated PI-PDMS to that of the benzylic proton (Ha) peak in the corresponding pristine PI-PDMS, and this value was found to be 7% for all PDMS compositions (Fig. S2†).
The piperazinium-mediated crosslinked PI-PDMS membranes (xPI-PDMSs) with the various PDMS contents were simply and efficiently prepared by reacting each Br-PI-PDMS copolymer with piperazine, which was used as a crosslinker (Scheme 1). The crosslinker was dissolved in a DMF solution of Br-PI-PDMS (2), followed by membrane casting and thermal drying. During the thermal drying process, the reactive benzyl bromide group of each 2 rapidly reacted with the piperazine to produce the corresponding piperazinium-mediated crosslinked PI-PDMS membrane with its bromide anion denoted as [xPI-PDMS-0.05][Br], [xPI-PDMS-0.10][Br], or [xPI-PDMS-0.15][Br].
We previously introduced the piperazinium-mediated crosslinked 6FDA-durene PI membrane with its bromide form, denoted as [xPI][Br], in a similar way by reacting piperazine with the bromobenzylated 6FDA-durene PI (Br-PI), which was then prepared from bromination of the 6FDA-durene-based PI.33 The piperazinium-mediated crosslinked [xPI][Br] membrane, with the same polymer backbone but without PDMS unit, was used as a reference to investigate the effect of the flexible PDMS unit.
The xPI-PDMS structures were verified using FT-IR spectroscopy by monitoring the peaks corresponding to the CH2 and CH3 vibration in piperazinium cation at 2860 cm−1 and 3070 cm−1 for all three xPI-PDMS membranes, ([xPI-PDMS-0.05][Br], [xPI-PDMS-0.10][Br], and [xPI-PDMS-0.15][Br]); this analysis indicated the successful incorporation of the ionic group, namely piperazinium (Fig. S3†).35 The characteristic peaks of the PDMS at 1110 cm−1 (Si–O–Si) and 1257 cm−1 (Si–CH3) and the peaks of the PI at 1354 cm−1 (C–N), 1720 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and 1783 cm−1 (CO) were also observed.39
O), and 1783 cm−1 (CO) were also observed.39
Physical properties of the crosslinked PI-PDMS membranes, xPI-PDMSs
[xPI-PDMS-0.05][Br], [xPI-PDMS-0.10][Br], and [xPI-PDMS-0.15][Br] all formed dense membranes, which were suitable for gas permeation testing (Fig. 1). The three membranes containing the piperazine crosslinker were found to be only partially soluble, whereas the pristine PI-PDMS membrane was readily soluble in common organic solvents, including DMAc, DMF, and NMP, indicating that all of the crosslinked membranes were partially crosslinked. About 10% gel fraction was obtained for all xPI-PDMS membranes, and this is because the crosslinking functionality (i.e. degree of bromination) was very low (7%). | ||
| Fig. 1 Photographs of the crosslinked [xPI-PDMS-0.05][Br] (a), [xPI-PDMS-0.10][Br] (b) and [xPI-PDMS-0.15][Br] (c) membranes. | ||
Gas separation properties
The single gas permeabilities and permselectivities of the [xPI-PDMS-0.05][Br], [xPI-PDMS-0.10][Br], and [xPI-PDMS-0.15][Br] membranes were measured at 2 atm and 30 °C using the constant-volume variable-pressure method, and the data were compared with those of the pristine 6FDA-durene and the piperazinium-mediated crosslinked PI without PDMS unit ([xPI][Br]) (Table 1).| Membrane | PCO2 | PN2 | PCH4 | αCO2/N2 | αCO2/CH4 |
|---|---|---|---|---|---|
| a P in barrers, where 1 barrer = 10−10 [cm3 (STP) cm]/(cm2 s cm Hg).b Based on ref. 40.c Based on ref. 33. | |||||
| 6FDA-dureneb | 495 | 41.1 | 37.3 | 12.1 | 13.2 |
| [xPI][Br]c | 475 | 26.4 | 14.2 | 18.0 | 34.5 |
| [xPI-PDMS-0.05][Br] | 629 | 35.3 | 19.7 | 17.8 | 35.3 |
| [xPI-PDMS-0.10][Br] | 799 | 50.9 | 22.2 | 15.7 | 36.0 |
| [xPI-PDMS-0.15][Br] | 377 | 20.9 | 10.6 | 18.1 | 35.5 |
The [xPI-PDMS-0.05][Br] and [xPI-PDMS-0.10][Br] membranes, having 5 wt% and 10 wt% PDMS, showed much higher permeability to CO2 than did the 6FDA-durene-based PI membrane (495 Barrer) and the [xPI][Br] membrane (475 Barrer), and the permeability to all of the tested gases increased with an increase in the relative amount of PDMS up to 10 wt% PDMS ([xPI-PDMS-0.10][Br]) (Table 1). The introduction of the flexible PDMS unit up to 10 wt% may have enhanced the free volume between polymer chains, facilitating diffusion of the gases and hence having increased the permeability. Increasing the amount of PDMS to 15 wt%, however, yielded an [xPI-PDMS-0.15][Br] membrane with dramatically reduced gas permeabilities. Thus, the [xPI-PDMS-0.10][Br] membrane displayed the highest permeability levels, with a value as high as 799 barrer for CO2. These results indicated the existence of a highly permeable PDMS unit-dependent percolation threshold, which effectively transported gases by the [xPI-PDMS-0.10][Br]. Perhaps the relatively high gas permeability of the [xPI-PDMS-0.10][Br] membrane was due to the highly permeable PDMS region and the rigid PI region being most well-mixed when the content of PDMS in the membrane was 10 wt% (see below).
Moreover, all three pipeazinium-mediated crosslinked membranes showed higher permselectivity to CO2 than did the 6FDA-durene-based PI membrane, which showed permselectivity values of only 12.1 for CO2/N2 and 13.2 for CO2/CH4. The enhanced CO2 selectivity level of each of the three xPI-PDMS systems was ascribed to the relatively high CO2-solubility of the piperazinium group crosslinker. The xPI-PDMS and the xPI membranes, both having the piperazinium group as a crosslinker as well as a CO2-solubilizing group, showed similar CO2/N2 and CO2/CH4 selectivities with exception of the very highly permeable [xPI-PDMS-0.10][Br] (Table 1).
As expected, the [xPI-PDMS-0.10][Br] membrane, having the highest permeability, showed the lowest CO2/N2 selectivity among the three xPI-PDMS membranes. In contrast, the [xPI-PDMS-0.10][Br] membrane displayed a slightly higher CO2/CH4 permselectivity value (36.0) than did the other two crosslinked PI-PDMS membranes (35.3 for [xPI-PDMS-0.05][Br] and 35.5 for [xPI-PDMS-0.15][Br]). An increase in the diffusivity selectivity and decrease in the solubility selectivity for CO2/CH4 were observed as the amount of PDMS was increased from 5 wt% to 15 wt% (Table S1 and Fig. S4†). The decrease in solubility selectivity was, however, less than the increase in diffusivity selectivity for the [xPI-PDMS-0.10][Br] membrane, resulting in the higher CO2/CH4 permselectivity for this membrane than the other two crosslinked membranes, i.e., [xPI-PDMS-0.05][Br] and [xPI-PDMS-0.15][Br]. For CO2/N2, both the diffusivity selectivity and solubility selectivity were observed to be lower for the [xPI-PDMS-0.10][Br] membrane than for the other two crosslinked membranes, resulting in the lowest CO2/N2 permselectivity for [xPI-PDMS-0.10][Br].
Morphological analyses using AFM, DSC and XRD
A clear phase separation between PI (bright region) and PDMS (dark region) units was observed using AFM for the [xPI-PDMS-0.05][Br] and [xPI-PDMS-0.15][Br] membranes, i.e., those having 5 and 15 wt% PDMS, respectively, and the [xPI-PDMS-0.15][Br] membrane showed larger dark areas than did the [xPI-PDMS-0.05][Br] membrane (Fig. 2). The soft PDMS may not have mixed well with rigid PI, resulting in a phase separation for the [xPI-PDMS-0.05][Br] membrane, whose PDMS content was low (Fig. 2a and 3a).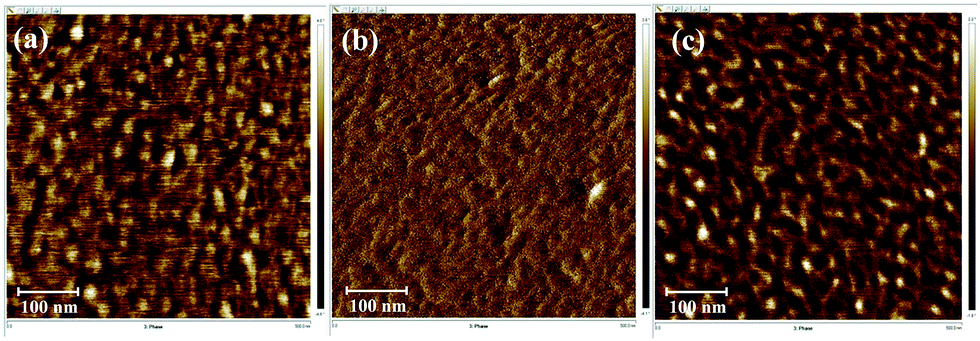 | ||
| Fig. 2 Phase AFM imagesof the [xPI-PDMS-0.05][Br] (a), [xPI-PDMS-0.10][Br] (b) and [xPI-PDMS-0.15][Br] (c) membranes. | ||
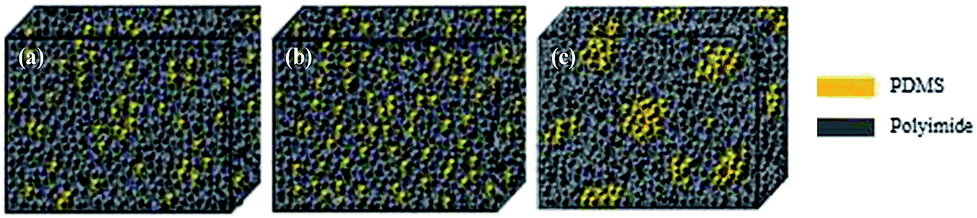 | ||
| Fig. 3 Schematics of the morphologies of the [xPI-PDMS-0.05][Br] (a), [xPI-PDMS-0.10][Br] (b) and [xPI-PDMS-0.15][Br] (c) membranes. | ||
As the amount of PDMS in the membrane was increased to 10%, however, the PDMS region began to mix with PI, and hence a phase separation was not observed for [xPI-PDMS-0.10][Br] by AFM (Fig. 2b and 3b). A miscibility of the highly permeable PDMS region into the rigid PI is believed to be the reason for the permeability for the [xPI-PDMS-0.10][Br] membrane having been higher than the permeabilities of the other membranes (see above). Increasing the amount of PDMS to 15 wt%, on the other hand, seemed to cause an aggregation of the highly permeable PDMS region. Therefore, compared to the [xPI-PDMS-0.05][Br] membrane, the [xPI-PDMS-0.15][Br] membrane showed a more distinct phase separation (Fig. 2c and 3c) and a reduced permeability (Table 1).
Similar results were reported for the morphological analyses of PI-PDMS copolymers with various contents of PDMS:41 when only a small amount of PDMS was included, the PDMS chains were too far apart to aggregate and hence the PDMS mixed well with the hard PI segment; but when more PDMS was included, the PDMS chains aggregated and a clear phase separation is observed. The results for our xPI-PDMS systems indicated 10 wt% to be the optimum amount of PDMS to obtain the least phase separation, or most miscibility, between PDMS and PI.
DSC analysis was carried out to further investigate the effect of the PDMS content on the morphology of the crosslinked xPI-PDMSs by monitoring the glass transition temperature (Tg) of the soft PDMS region (Table 2 and Fig. S5†). No glass transition corresponding to the rigid PI moiety was observed, and the Tg values corresponding to that of the PDMS segment were higher for all three xPI-PDMSs than that of the typical PDMS polymer (−123 °C).42
| Membrane | d-Spacing (Å) | Density (g cm−3) | Tg (°C) |
|---|---|---|---|
| [xPI-PDMS-0.05][Br] | 6.4 | 1.41 | −64 |
| [xPI-PDMS-0.10][Br] | 6.7 | 1.28 | −59 |
| [xPI-PDMS-0.15][Br] | 6.7 | 1.35 | −64 |
Although we expected the increased Tg values of the crosslinked PI-PDMS copolymers, compared to that of the PDMS, as a result of the incorporation of the rigid PI unit, it was noteworthy to observe that the [xPI-PDMS-0.10][Br] membrane, having 10 wt% PDMS, showed a Tg value (−59 °C) higher than those of the membranes with less or more PDMS, i.e., [xPI-PDMS-0.05][Br], and [xPI-PDMS-0.15][Br], respectively. Similar results were also reported for the thermal analyses of PI-PDMS copolymers with various contents of PDMS:41 increased Tg is observed at relatively low PDMS content due to well-miscibility (or homogeneous mixing) between hard PI and soft PDMS; whereas the soft phase comprises only PDMS and tend to aggregate with an increase of PDMS content, lowering Tg of the copolymer. The results from DSC analysis are consistent with those obtained from the morphological analysis by AFM.
A wide-angle X-ray scattering (WAXS) analysis of the xPI-PDMS membranes was also carried out, and showed that the peaks broadened as the content of PDMS was increased, indicating that the xPI-PDMS became amorphous with the increase in the amount of the soft PDMS segment (Fig. S6†). In addition, the intersegmental (d-) spacing between the polymer chains in the membranes increased when more PDMS was included (Table 2), with this result attributed to PDMS having a larger free volume than PI.
The densities of the three xPI-PDMS membranes were also measured. [xPI-PDMS-0.10][Br] showed the lowest density, possibly because PDMS, having a large free volume, mixed best at this content with PI. This result was consistent with the morphological analyses from AFM and DSC, and the highest gas permeability was again expected for this membrane. In addition, the density of the [xPI-PDMS-0.15][Br] membrane was measured to be less than that of the [xPI-PDMS-0.05][Br] membrane (Table 2).
Mechanical properties
Stress–strain curves of the piperazinium-mediated crosslinked PI-PDMS (xPI-PDMS) membranes were acquired at 50% RH (Fig. S7†). As expected, a higher elongation was observed as the relative amount of PDMS was increased (Table 3), due to the flexibility of the PDMS. On the other hand, [xPI-PDMS-0.10][Br], with the intermediary 10 wt% content of PDMS, showed the highest tensile strength, possibly due to the PDMS and PI being most miscible at this PDMS concentration determined using AFM and DSC analyses as described above. Nevertheless, all three crosslinked membranes exhibited excellent tensile strengths of more than 84.6 MPa and Young's modulus values of more than 2.1 GPa, suggesting the suitability of the mechanical properties of the xPI-PDMS membranes we developed for gas separation.| Membrane | Tensile strength (MPa) | Elongation at break (%) | Young's modulus (GPa) |
|---|---|---|---|
| [xPI-PDMS-0.05][Br] | 89.9 | 6.7 | 2.3 |
| [xPI-PDMS-0.10][Br] | 100.1 | 7.0 | 2.5 |
| [xPI-PDMS-0.15][Br] | 84.5 | 9.4 | 2.2 |
Permeability vs. selectivity for xPI-PDMS
The tradeoffs between permeability and selectivity for CO2/CH4 (Fig. 4a) and for CO2/N2 (Fig. 4b) were assessed for the membranes consisting of xPI-PDMS with different PDMS contents, i.e., ([xPI-PDMS-0.05][Br], [xPI-PDMS-0.10][Br] and [xPI-PDMS-0.15][Br]), by producing Robeson plots.9,10 Data from poly (IL)s31,43 and other common crosslinked PIs,44 including our previously developed piperazinium-mediated crosslinked [xPI][Br],33 were also included for comparison.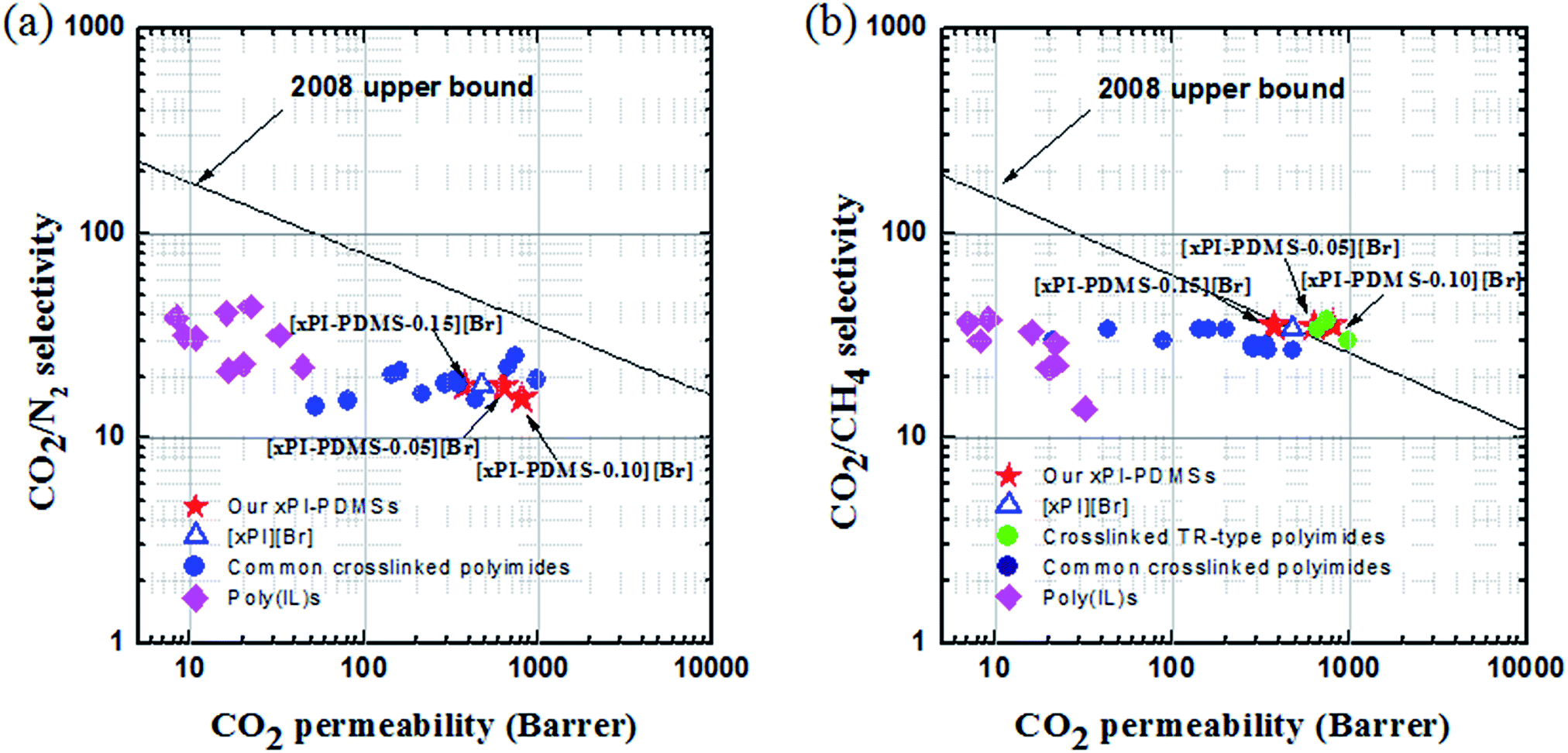 | ||
| Fig. 4 “Robeson upper bound 2008” plot for comparing the CO2/N2 (a) and CO2/CH4 (b) separation performances of the crosslinked PI-PDMS membranes (xPI-PDMSs) with those of other previously reported crosslinked PIs and poly(IL)s whose data were taken from ref. 20, 22, 31, 33, 43 and 44. | ||
Although the values for CO2/N2 tested with all of the xPI-PDMS membranes were found to be below the upper bound line, they were similar to the published data for typical crosslinked PIs and outperformed those for poly(IL)s and even our previously developed [xPI][Br]. Most notably, all of the xPI-PDMS membranes we developed here exhibited outstanding levels of selective permeability for CO2 in the presence of CH4, with these selectivity and permeability values higher than those of other crosslinked PIs and poly(IL)s. In fact, the values for the [xPI-PDMS-0.05][Br] and [xPI-PDMS-0.10][Br] membranes were positioned above the Robeson upper bound of 2008, and those for the [xPI-PDMS-0.15][Br] and our previously developed [xPI][Br] membranes on the upper bound line, for CO2/CH4. According to these results, the piperazinium-mediated crosslinked PI-PDMS polymers have great potential for performing enhanced selective separation of CO2.
We also measured the CO2 permeabilities of the crosslinked PI-PDMS copolymer membranes at various pressure levels up to 25 atm in order to assess their resistance to plasticization (Fig. 5a). An increase in CO2 permeability with increasing feed pressure is indicative of plasticization and typically signals a loss in the ability of the polymer matrix to discriminate particles based on their size and shape.16 None of the three crosslinked PI-PDMS membranes displayed any increase in CO2 permeability with increasing feed pressure up to 25 atm.
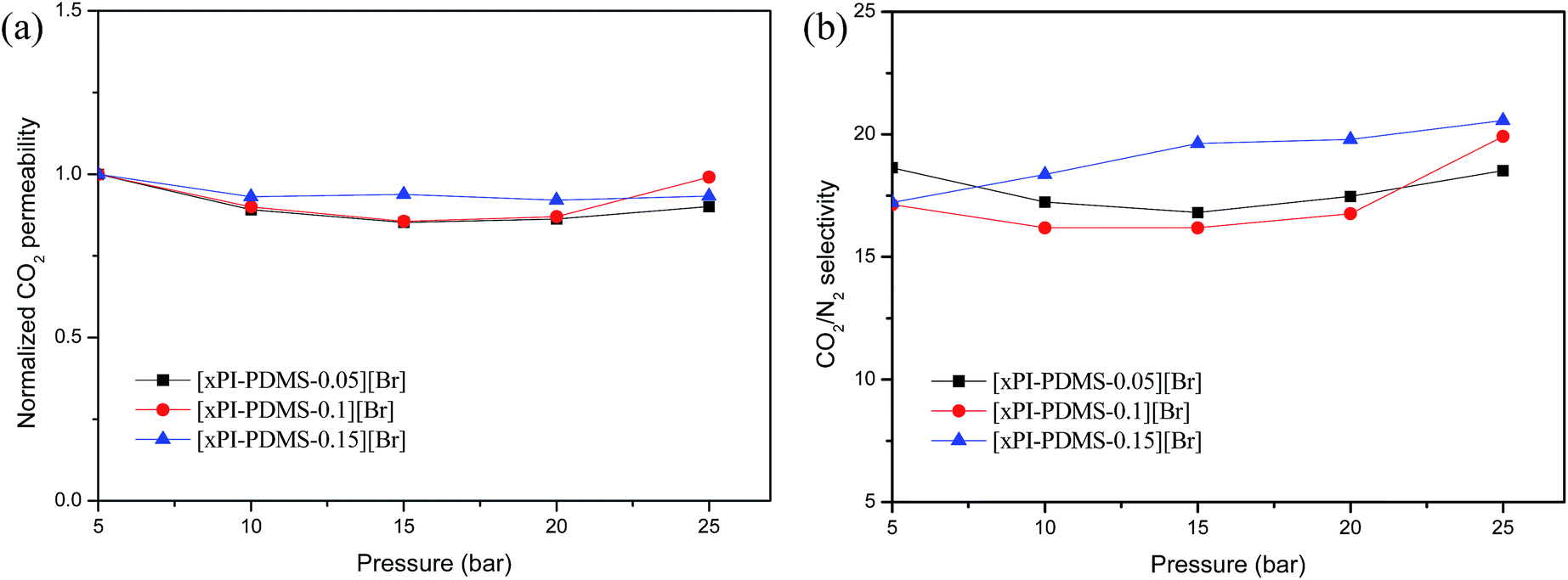 | ||
| Fig. 5 Normalized permeability (a) and CO2/N2 permselectivity (b) of the crosslinked membranes (xPI-PDMSs) as a function of feed pressure. | ||
And, neither did they show any significant changes in CO2/N2 selectivity as the pressure was increased, even at high pressure levels (Fig. 5b). The results suggested that the crosslinked membranes did not show signs of any significant plasticization up to 25 atm. The resistance of these newly prepared crosslinked materials to becoming plasticized at feed pressures as high as 25 atm makes them strong candidate materials for practical applications.
Conclusions
In conclusion, we prepared a novel piperazinium-mediated crosslinked PI-PDMS (xPI-PDMS) with pendant ionic groups, and successfully demonstrated the potential use of membranes made of this copolymer for separating CO2 from other gases. Incorporating ionic groups into a highly permeable and thermomechanically stable polymer backbone for producing a membrane with high CO2-solubility has been attempted for a long time by investigators interested in polymer-membrane-based gas separation. The current work is the first attempt to introduce an ionic group (piperazinium) into the side chain of copolymers consisting of the highly permeable soft PDMS and physically stable rigid PI. By utilizing the ionic pendant group to serve as both a CO2-solubilizing and a crosslinking group, the membranes prepared from the xPI-PDMSs showed an increased selectivity for CO2, as well as good resistance to CO2 plasticization. In addition, the effect of the level of PDMS loading on the morphology and gas separation performance of the resulting membranes prepared from the xPI-PDMSs was investigated; the xPI-PDMS containing 10 wt% PDMS displayed a morphology indicating the best mixing between the highly permeable PDMS and the rigid PI segment, leading to the highest permeability for this membrane. Overall, the newly developed piperazinium-mediated crosslinked PI-PDMS copolymers have high potential as novel polymer membranes for CO2 separation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2017R1A6A1A06015181).Notes and references
- J. C. M. Pires, F. G. Martins, M. C. M. A. Ferraz and M. Simões, Chem. Eng. Res. Des., 2011, 89, 1446–1460 CrossRef CAS.
- R. M. Siqueira, G. R. Freitas, H. R. Peixoto, J. F. do Nascimento, A. P. S. Musse, A. E. B. Torres, D. C. S. Azevedo and M. Bastos-Neto, Energy Procedia, 2017, 114, 2182–2192 CrossRef CAS.
- D. M. D'Alessandro, Chem. Commun., 2016, 52, 8957–8971 RSC.
- N. Du, H. B. Park, M. M. Dal-Cin and M. D. Guiver, Energy Environ. Sci., 2012, 5, 7306–7322 CAS.
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS.
- W. J. Schell, J. Membr. Sci., 1985, 22, 217–224 CrossRef CAS.
- W. J. Koros and G. K. Fleming, J. Membr. Sci., 1993, 83, 1–80 CrossRef CAS.
- P. Bernado, E. Drioli and G. Golemme, Ind. Eng. Chem. Res., 2009, 48, 4638–4663 CrossRef.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- Y. Hirayama, T. Yoshinaga, Y. Kusuki, K. Ninomiya, T. Sakakibara and T. Tamari, J. Membr. Sci., 1996, 111, 169–182 CrossRef CAS.
- T. H. Kim, W. J. Koros, G. R. Husk and K. C. Obrien, J. Membr. Sci., 1988, 37, 45–62 CrossRef CAS.
- D. M. Munoz, E. M. Maya, J. de Abajo, J. G. de la Campa and A. E. Lozano, J. Membr. Sci., 2008, 323, 53–59 CrossRef CAS.
- Y. Xiao, B. T. Low, S. S. Hosseini, T. S. Chung and D. R. Paul, Prog. Polym. Sci., 2009, 34, 561–580 CrossRef CAS.
- K. Tanaka, H. Kita, M. Okano and K. I. Okamoto, Polymer, 1991, 33, 585–592 CrossRef.
- W. Qiu, L. Xu, C. C. Chen, D. R. Paul and W. J. Koros, Polymer, 2013, 54, 6226–6235 CrossRef CAS.
- K. Vanherck, G. Koeckelberghs and I. F. J. Vankelecom, Prog. Polym. Sci., 2013, 38, 874–896 CrossRef CAS.
- H. Eguchi, D. J. Kim and W. J. Koros, Polymer, 2015, 58, 121–129 CrossRef CAS.
- A. Bos, I. G. M. Punt, M. Wessling and H. Strathmann, Sep. Purif. Technol., 1998, 14, 27–39 CrossRef CAS.
- L. Shao, L. Liu, S.-X. Cheng, Y.-D. Huang and J. Ma, J. Membr. Sci., 2008, 312, 174–185 CrossRef CAS.
- Z. Dai, L. Ansaloni, D. L. Gin, R. D. Noble and L. Deng, J. Membr. Sci., 2017, 523, 551–560 CrossRef CAS.
- W. Qiu, C.-C. Chen, L. Xu, L. Cui, D. R. Paul and W. J. Koros, Macromolecules, 2011, 44, 6046–6056 CrossRef CAS.
- J. Schauer, P. Sysel, V. Marousek, Z. Pientka, J. Pokorny and M. Bleha, J. Appl. Polym. Sci., 1996, 61, 1333–1337 CrossRef CAS.
- D. P. Queiroz and M. Norberta de Pinho, Polymer, 2005, 46, 2346–2353 CrossRef CAS.
- H. B. Park, J. K. Kim, S. Nam and Y. M. Lee, J. Membr. Sci., 2003, 220, 59–73 CrossRef CAS.
- T. Hong, Z. Niu, X. Hu, K. Gmernicki, S. Cheng, F. Fan, J. C. Johnson, E. Hong, S. Mahurin, D. E. Jiang, B. Long, J. Mays, A. Sokolov and T. Saito, ChemSusChem, 2015, 8, 3595–3604 CrossRef CAS PubMed.
- Y. B. Lee, H. B. Park, J. K. Shim and Y. M. Lee, J. Membr. Sci., 1999, 74, 965–973 CAS.
- S. Y. Ha, H. B. Park and Y. M. Lee, Macromolecules, 1999, 32, 2394–2396 CrossRef CAS.
- H. B. Park, S. Y. Ha and Y. M. Lee, J. Membr. Sci., 2000, 177, 143–152 CrossRef CAS.
- Z. Dai, R. D. Noble, D. L. Gin, X. Zhang and L. Deng, J. Membr. Sci., 2016, 497, 1–20 CrossRef CAS.
- J. E. Bara, C. J. Gabriel, E. S. Hatakeyama, T. K. Carlisle, S. Lessmann, R. D. Noble and D. L. Gin, J. Membr. Sci., 2008, 321, 3–7 CrossRef CAS.
- J. E. Bara, D. E. Camper, D. L. Gin and R. D. Noble, Acc. Chem. Res., 2010, 43, 152–159 CrossRef CAS PubMed.
- I. Kammakakam, H. W. Yoon, S. Nam, H. B. Park and T.-H. Kim, J. Membr. Sci., 2015, 487, 90–98 CrossRef CAS.
- I. Kammakakam, H. W. Kim, S. Nam, H. B. Park and T.-H. Kim, Polymer, 2013, 54, 3534–3541 CrossRef CAS.
- I. Kammakakam, S. Nam and T.-H. Kim, RSC Adv., 2015, 5, 69907–69914 RSC.
- P. Li, D. R. Paul and T. S. Chung, Green Chem., 2012, 14, 1052–1063 RSC.
- L. C. Tomé and I. M. Marrucho, Chem. Soc. Rev., 2016, 45, 2785–2824 RSC.
- S. C. Kumbharkar, R. S. Bhavsar and U. K. Kharul, RSC Adv., 2014, 4, 4500–4503 RSC.
- P. A. Gurr, J. M. Scorfield, J. Kim, Q. Fu, S. E. Kentish and G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3372–3382 CrossRef CAS.
- C. E. Powell, X. J. Duthie, S. E. Kentish, G. G. Qiao and G. W. Stevens, J. Membr. Sci., 2007, 291, 199–209 CrossRef CAS.
- C. K. Ku and Y. D. Lee, Polymer, 2007, 48, 3565–3573 CrossRef CAS.
- W. Zhao, P. Fonsny, P. Fitzgerald, G. G. Warr and S. Perrier, Polym. Chem., 2013, 4, 2140–2150 RSC.
- J. E. Bara, S. Lessmann, C. J. Gabriel, E. S. Hatakeyama, R. D. Novel and D. L. Gin, Ind. Eng. Chem. Res., 2007, 46, 5397–5404 CrossRef CAS.
- M. Calle, C. M. Doherty, A. J. Hill and Y. M. Lee, Macromolecules, 2013, 46, 8179–8189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, IR, DSC, XRD and stress–strain curve data. See DOI: 10.1039/c7ra10949k |
| This journal is © The Royal Society of Chemistry 2018 |