
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcridinedione as selective flouride ion chemosensor: a detailed spectroscopic and quantum mechanical investigation†
Nafees Iqbala,
Syed Abid Alia,
Iqra Munira,
Saima Khanb,
Khurshid Ayub b,
Mariya al-Rashidac,
Muhammad Islamd,
Zahid Shafiqdg,
Ralf Ludwigef and
Abdul Hameed*a
b,
Mariya al-Rashidac,
Muhammad Islamd,
Zahid Shafiqdg,
Ralf Ludwigef and
Abdul Hameed*a
aH. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan. E-mail: abdul_hameed8@hotmail.com; abdul_hameed@iccs.edu; Fax: +92-21-3481901; Tel: +92-21-99261701-2
bDepartment of Chemistry, COMSATS Institute of Information Technology, Abbottabad, KPK, Pakistan 22060
cDepartment of Chemistry, Forman Christian College, A Chartered University, Ferozepur Road, Lahore, Pakistan
dInstitute of Chemical Sciences, Bahauddin Zakariya University, Multan, Pakistan
eLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
fDepartment of Physical Chemistry, University of Rostock, Dr.-Lorenz-Weg 1, 18059 Rostock, Germany
gDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan
First published on 9th January 2018
Abstract
The use of small molecules as chemosensors for ion detection is rapidly gaining popularity by virtue of the advantages it offers over traditional ion sensing methods. Herein we have synthesized a series of acridine(1,8)diones (7a–7l) and explored them for their potential to act as chemosensors for the detection of various anions such as fluoride (F−), acetate (OAc−), bromide (Br−), iodide (I−), bisulfate (HSO4−), chlorate (ClO3−), perchlorate (ClO4−), cyanide (CN−), and thiocyanate (SCN−). Acridinediones were found to be highly selective chemosensors for fluoride ions only. To investigate in detail the mechanism of selective fluoride ion sensing, detailed spectroscopic studies were carried out using UV-visible, fluorescence and 1H NMR spectroscopy. Fluoride mediated (NH) proton abstraction of acridinedione was found to be responsible for the observed selective fluoride ion sensing. Quantum mechanical computational studies, using time dependent density functional theory (TDDFT) were also carried out, whereupon comparison of acridinedione interaction with fluoride and acetate ions explained the acridinedione selectivity for the detection of fluoride anions. Our results provide ample evidence and rationale for further modulation and exploration of acridinediones as non-invasive chemosensors for fluoride ion detection in a variety of sample types.
1. Introduction
In the realm of sensors, the application of small molecules as chemosensors has emerged as an exciting area of research with vast and diverse applications in the areas of biological, environmental, clinical and industrial research.1 A variety of small molecules have found applications as ion sensors, where both cations and anions can be precisely and selectively determined. Moreover, the structure of these small molecules can be easily tailored or “custom-made” to make them more suitable for specific applications, for example such molecules can be easily made more or less water soluble or membrane permeable. The use of chemosensors offers non-invasive and non-destructive methods of ion analysis; these are particularly desirable traits for ion sensing in biological samples such as cells and tissues. The mechanism of ion sensing by these chemosensors usually involves a modulation of some measurable property (such as absorbance or fluorescence) of the chemosensor (receptor or ligand), upon binding with ions. Anion detection with small molecules generally occurs via addition of anions to the receptor,2,3 or removal of acidic protons from the receptor resulting in visible colorimetric changes.4,5A range of different compounds such as acridinium,2 quinolone,6 and urea/thiourea-based7 receptors have been reported as chemosensors in anion detection via addition, substitution, or photo-induced electron transfer mechanisms.1 Ion detection for anions such as halides (F−, Cl−, Br−, I−), acetate (OAc−), sulfate, chlorate, cyanide and thiocyanate carries much analytical importance. Among the pool of anions, fluoride detection possesses significant importance in osteoporosis, which is characterized by fluoride accumulation in bones.8 In recent years, selective fluoride detection has received considerable attention due to its consumption in variety of products such as toothpastes, pesticides, fertilizers etc. Compounds bearing amino or hydroxyl groups have been known to act as chemosensors for fluoride detection either via deprotonation of acidic protons, or via hydrogen bond formation with fluoride.1,9 Previously rhodamine based molecules were found to act as selective sensors for fluoride ions, moreover they were also successfully applied for imaging of fluoride content in live cells.10 Similarly receptors containing thiosemicarbazide moieties also act as ion-sensors towards different anions. In this case, the acidic protons of thiosemicarbazide moiety act as binding sites and can selectively bind fluoride ions.11,12 Related to this class of sensors, azo dye-semicarbazones have also been known to act as anion sensors via related mechanism.13 N-Monoalkylated 1,4-diketo-3,6-diphenylpyrrolo[3,4-c]-pyrroles have been developed as efficient and highly selective fluorescent probes for fluoride ions14 Recently naphthodipyrrolidones were found to act as sensitive and selective probes for fluoride ions.15 In the present study, we explored phenyl acridine(1,8)diones as chemosensors for a range of different anions and found them to be selective only for fluoride detection.
Acridine(1,8)diones are a well-known class of compounds and have found many interesting applications. They exhibit excellent photo-physical properties,16 making them ideal candidates to be used as laser dyes.17 Acridine(1,8)diones are used as photosensitizers18 and as photoinitiators in polymerisation reactions.19 These molecules can exist in neutral, protonated and a deprotonated forms.20 Phenyl acridine(1,8)dione contains a 1,4-dihydropyridine scaffold in the center of the molecule. 1,4-Dihydropyridine (DHP) belongs to a well-known class of compounds with significant biological activity21 such as antitumor,22 antitubercular activity,23 antihypertensive,24 and anti-inflammatory activity.25 Previously acridine(1,8)dione derivatives have been used for metal ion sensing, for example aza-crown ether acridinedione-functionalized quantum dots and aza-crown ether acridinedione-functionalized gold nanorods have been used to detect Ca2+ and Mg2+ ions.26 Other molecules such as dibutylaniline- and/or pyridine-containing 2,6,9,10-tetrarylvinylanthracene cruciforms are also known as highly effective chemosensors for metal ions.27 Similarly acridinedione-based heteroditopic hosts containing oxyethylene linkages have been used for metal ion detection as well as for fluoride ion detection.28 Acridinedione functionalized with thiourea moiety was found to be non-selective anion sensor via PET (Photoinduced Electron Transfer) mechanism.29 In continuation of our work on development of novel anion chemosensors3 we decided to synthesize a series of 9-phenyl-acridine-1,8-diones and explore their potential as anion chemosensors Fig. 1.
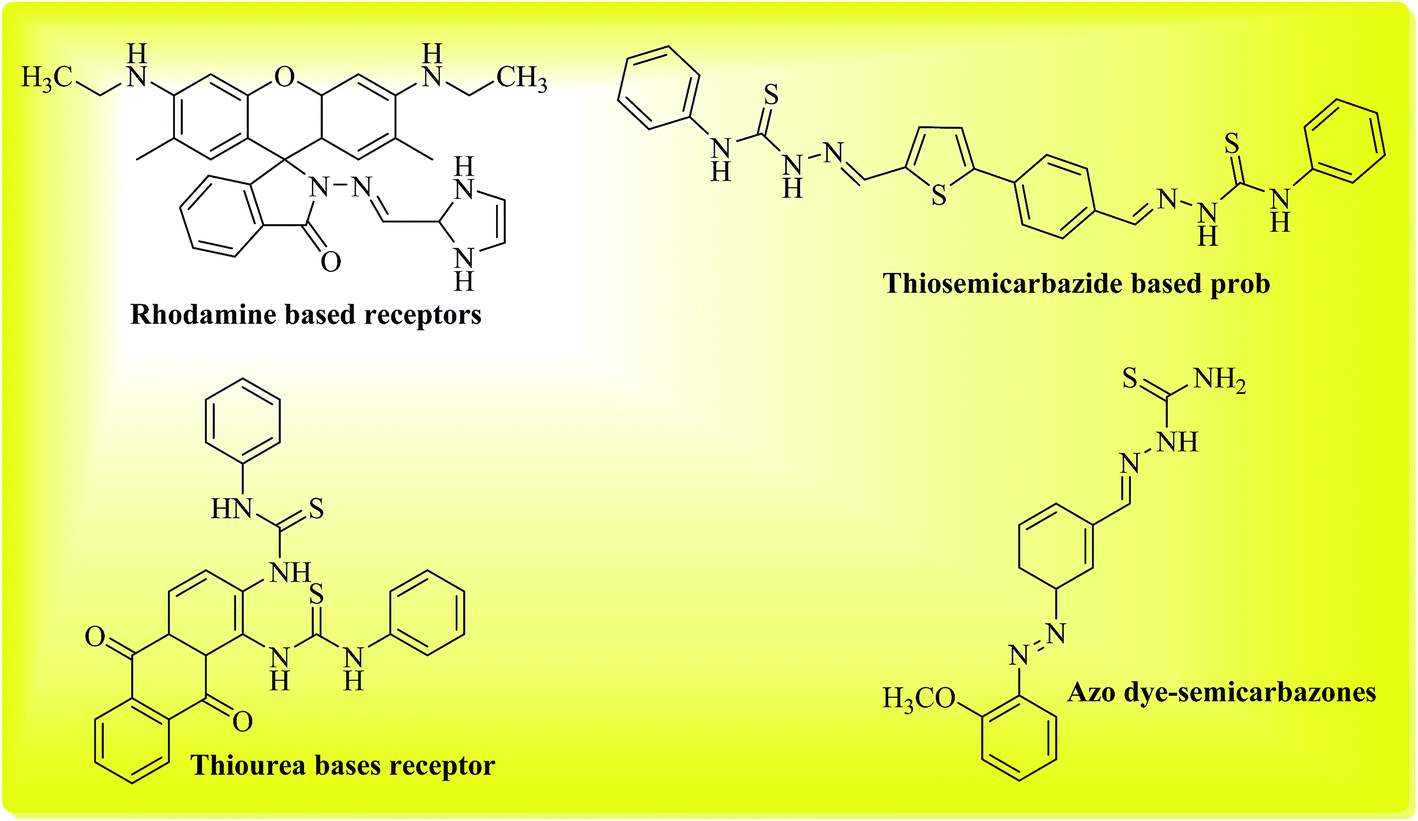 | ||
| Fig. 1 Structures of some reported molecules as selective probes/chemosensors for fluoride ion. | ||
2. Results and discussion
2.1. Chemistry
The synthesis of a series of 9-phenyl-acridine-1,8-diones (7a–7l) was carried out under solvent-free conditions using newly synthesized novel morpholinium-based ionic liquids (3–6), that were also synthesized in our lab. Ionic liquids (ILs) serve as an excellent alternative for traditionally volatile organic solvents. In contrast to typically obnoxious organic solvents, ILs provide alternative eco-friendly, green method of syntheses.30,31 Among the pool of ionic liquids, imidazole or pyridine-based ionic liquids have been employed as catalysts in organic reactions, however the inert nature of imidazole, and high toxicity and volatility of pyridine are major obstacles.32–34 N-Alkyl morpholine, being a less toxic compound, provides an easy access to inexpensive morpholinium cation for ionic liquid preparation (Fig. 2). Although morpholine as a base has been diversely employed in organic reactions, the use of N-alkylated morpholinium-based ionic liquids has been less exploited in organic synthesis.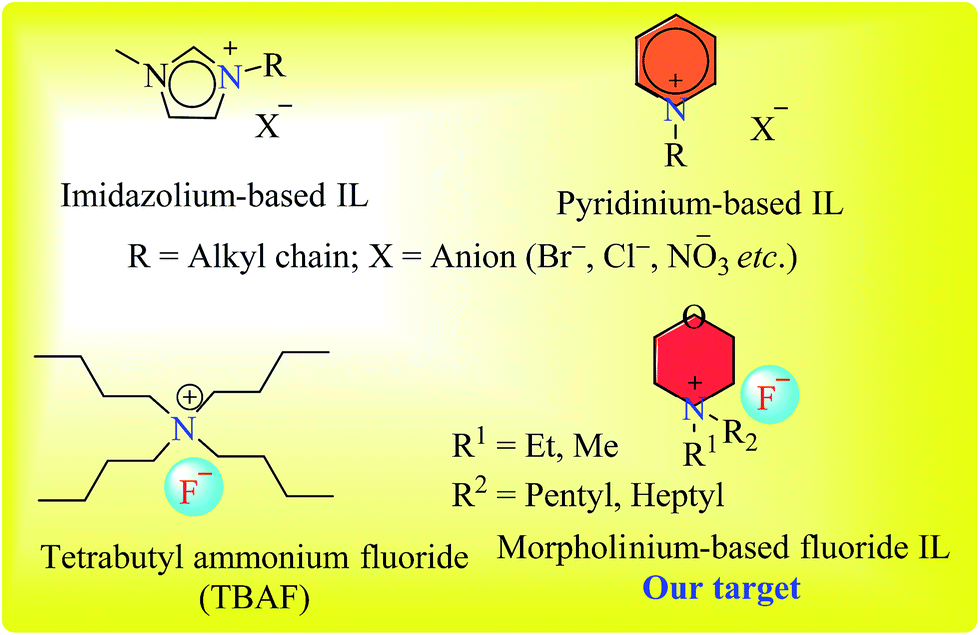 | ||
| Fig. 2 Structures of imidazolium, pyridinium, TBAF and morpholinium-based ILs. | ||
Synthesis of morpholinium-based ionic liquids (ILs) is given in Scheme 1. The N-alkyl (Me, Et) morpholine was treated with corresponding alkyl bromides (C5 and C7) in toluene at 118–120 °C until the complete consumption of starting materials as monitored by TLC. In the next step, the counter anion (Br−) was exchanged with fluoride (F−) by using silver fluoride, since fluoride ion exhibits overlying catalytic efficiency and mild basic characteristics in mediating organic reactions.35,36 Thus, morpholinium-based ionic liquids with fluoride counter anion could potentially serve as better alternate to TBAF (tetrabutyl ammonium fluoride, a commercially available IL) in organic synthesis due to easy and rapid preparation of morpholinium-based ionic liquids from cost effective commercially available N-alkylated morpholine Scheme 2.
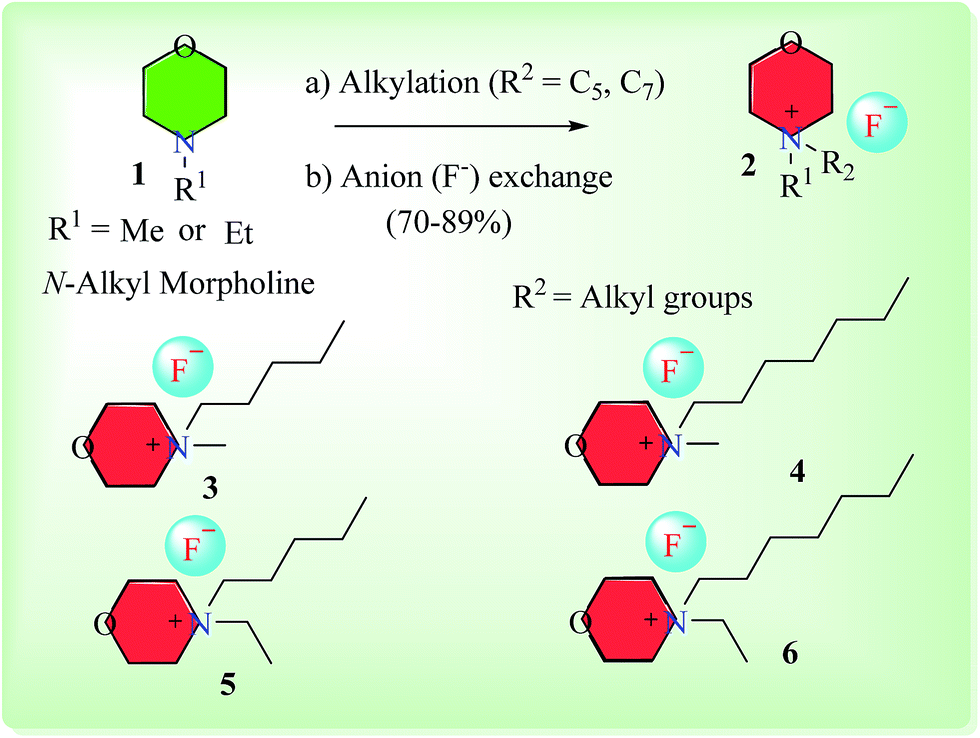 | ||
| Scheme 1 Synthesis of morpholinium-based ionic fluoride liquids (3–6). (a) Alkyl bromides, toluene, 118–120 °C; (b) aqueous silver fluoride solution. | ||
 | ||
| Scheme 2 Synthesis of 9-phenyl-acridine-1,8-diones 7a–l. | ||
These newly synthesized morpholinium-based ionic liquids (3–6) were employed as reaction media and catalyst under solvent-free conditions for the synthesis of 9-phenyl-acridine-1,8-diones (7a–7l). One-pot multicomponent reaction between dimidone (2 equiv.), benzaldehyde (1 equiv.) and ammonium acetate (1 equiv.) was successfully carried out using morpholinium-based ionic liquids (3–6) as catalysts (1.0 equiv.) at 115–120 °C. Synthesis of one of the acridinedione derivative (7f) was carried out under these above optimized conditions using different ILs (3–6), the percentage yields of product (7f) obtained with these different ILs were compared, and were not found to be very different from one another (Table 1). In order to signify the catalytic efficiency of morpholinium-based ionic liquids in reactions demanding high temperatures, thermal studies of IL 3 (as model IL) were studied in detail using differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), and derivative thermogravimetry (DTG) (data given in Fig. S5a and b†).
| Morpholinium-ILs | Equiv. | Product (7f) yield (%) |
|---|---|---|
| a Conditions: morpholinium-ILs (1.0 equiv.), heat (115–120 °C), 16–18 h. | ||
| 3 | 1.0 | 74 |
| 4 | 1.0 | 73 |
| 5 | 1.0 | 73 |
| 6 | 1.0 | 71 |
2.2. Anion sensing study
Anion sensing by small molecules as chemosensors is a rapidly growing area of research. Among the anions of biological significance, fluoride occupies a unique place due to its utilization in dental care creams and over-accumulation in bones, which leads to osteoporosis. Accordingly, we investigated the potential of acridinediones in anion sensing for the possible detection of fluoride (F−), acetate (OAc−), bromide (Br−), iodide (I−), bisulfate (HSO4−), chlorate (ClO3−), perchlorate (ClO4−), cyanide (CN−), and thiocyanate (SCN−) ions, by using naked eye, UV-visible, fluorescence and NMR spectroscopic techniques. In this study, the acridinediones 7i and 7l were selected as model compounds to act as chemosensor in anion sensing study. Stock solutions of acridinedione receptor (chemosensor) and anions were freshly prepared in CH3CN or DMSO (in case of KCN and KSCN) and then diluted in CH3CN for analysis. Solutions of receptors 7i and 7l were treated with different anions F−, OAc−, Br−, I−, HSO4−, ClO3−, ClO4−, CN−, and SCN−, and were observed for any change in color with naked eye, and under UV irradiation. Only addition of fluoride ions caused a conspicuous change in color of receptors 7i and 7l from colorless to yellow, indicating that acridinediones 7i and 7l are selective chemosensors for fluoride ion detection (red line curve). Fig. 4 (top) shows, (a) solution of 7i without any fluoride ion as observed by naked eye, (b) solution of 7i (1 equiv.) with added fluoride ions (10 equiv.) as observed by naked eye, and (c) same solution of 7i with added fluoride ion as observed under UV irradiation (254 nm). These observations were confirmed via detailed UV-visible studies shown in Fig. 3a (7i) and Fig. 3b (7l), fluorescence, and 1H-NMR studies with 7i. | ||
| Fig. 3 Different substituted 9-phenyl-acridine-1,8-diones 7a–l. | ||
The UV-visible spectroscopic studies were carried out with 7i and 7l (1 equiv.) by adding 10 equiv. of different ions (F−, OAc−, Br−, I−, HSO4−, ClO3−, ClO4−, CN−, and SCN−) in CH3CN/DMSO and then further diluting with CH3CN solvent. The UV spectra of both receptors (7i and 7l) showed λmax at 370 nm which is attributed to intermolecular charge transfer (ICT) transition between nitrogen atom of acridine scaffold and carbonyl group.37 After titration with different anions, the both probes showed a new shift with λmax at 460 nm when titrated against fluoride anion (Fig. 4). As the anion sensing pattern of both receptors was found to be similar, only receptor 7i has been taken into account for further discussion. With receptor 7i, a conspicuous change in color from colorless to yellow was observed upon the addition of fluoride ion. The compound 7i has also been treated with mixture of anions without (TBAF) and in the presence of F− sources. A change in UV spectra showed the interaction of prob 7i with fluoride anion (Fig. S6†). Upon gradual addition of fluoride ion (up to 30 equiv.) a new absorption band started to appear at 460 nm with a concomitant decrease in intensity of absorption at 370 nm, as shown in UV-visible spectra in Fig. 5a. These results indicate the interaction between receptor and fluoride ion which lead to abstraction of proton from NH group of acridinone scaffold. The changes induces the ICT transition between nitrogen and carbonyl group which resulted in new red shift in absorption (λmax 460) and emission (770 nm) spectra.37 The UV-absorption pattern of receptor 7l, upon the gradual addition of fluoride anion, was found to be similar to that of 7i (Fig. S3†). Further the absorption at 460 nm was used to determine binding affinity of receptor and F− anion. A Job's plot (Fig. S1†) at total concentration of receptor-anion (7i and F−) showed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding ratio. The binding constant was calculated using Benesi–Hildebrand equation38 and was found to be 8.47 × 102 M−1 with limit of detection equal to 5.49 × 10−5 M (Fig. S2†). In case of receptor 7l, the binding constant was found to be 1.14 × 103 M−1 and limit of detection was 9.29 × 10−5 M (Fig. S4†). The larger binding constant value of 4-fluoro substituted receptor 7i as compared to 4-chloro 7l may be due to higher electronegativity of fluoro group which enhance the interaction between receptor and anion via deprotonation of NH group. Moreover, upon diluting the receptor-fluoride ion solution in methanol, absorption intensity at 360 nm was restored via re-protonation of NH group in the presence of protic medium (MeOH) (Fig. S1†).
:1 binding ratio. The binding constant was calculated using Benesi–Hildebrand equation38 and was found to be 8.47 × 102 M−1 with limit of detection equal to 5.49 × 10−5 M (Fig. S2†). In case of receptor 7l, the binding constant was found to be 1.14 × 103 M−1 and limit of detection was 9.29 × 10−5 M (Fig. S4†). The larger binding constant value of 4-fluoro substituted receptor 7i as compared to 4-chloro 7l may be due to higher electronegativity of fluoro group which enhance the interaction between receptor and anion via deprotonation of NH group. Moreover, upon diluting the receptor-fluoride ion solution in methanol, absorption intensity at 360 nm was restored via re-protonation of NH group in the presence of protic medium (MeOH) (Fig. S1†).
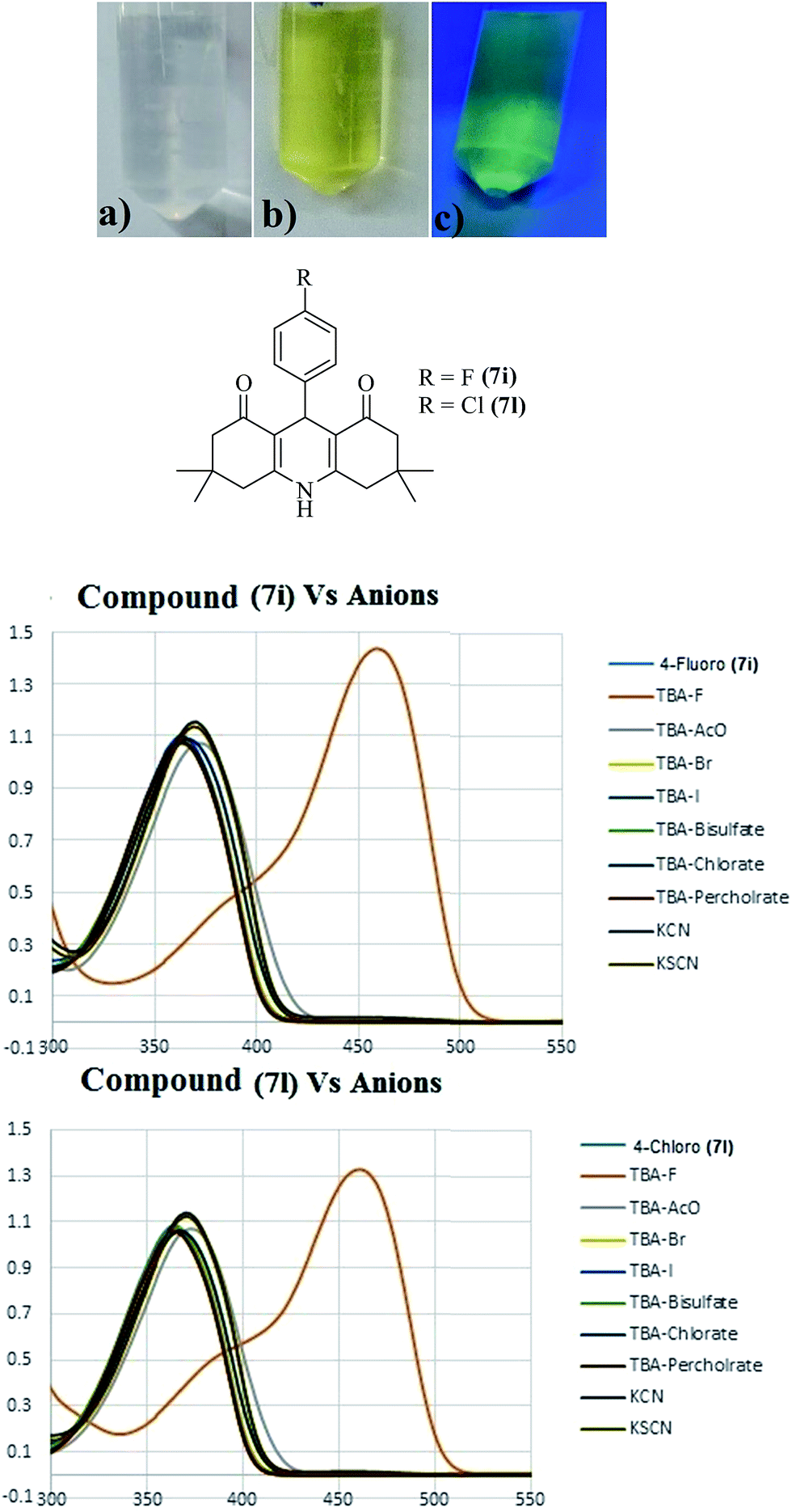 | ||
| Fig. 4 Fluoride detection by chemosensor 7i (a) without fluoride anion, (b) naked eye and (c) under UV-light (254 nm); UV-absorption spectra of 7i and 7l with different anions. | ||
 | ||
| Fig. 5 (a) Absorption spectra of 7i upon gradual increase of fluoride addition; (b) fluorescence spectra (excitation at 380 nm) in DMSO as solvent. | ||
The step-wise addition of fluoride ion (up to 10 equiv.) was also studied using fluorescence spectroscopy. In the absence of any anions, the fluorescence spectrum of chemosensor (7i), upon excitation at 380 nm, showed two emission bands at 450 nm and at 770 nm (Fig. 5b). The emission intensity of band at 450 nm was found to decrease with increasing concentration of fluoride ions. However, the emission intensity of the band at 770 nm first increases with the addition of fluoride ions from 0.5 to 4.0 equiv., upon further addition of fluoride ions, from 6 to 10 equiv., the emission intensity decreases (albeit slightly) and eventually becomes constant (Fig. 5b).
In order to further understand the nature and mechanism of highly selective interaction between the chemosensor 7i and fluoride ions, 1H NMR titrations were performed and spectra were recorded before and after adding fluoride ions in different concentrations (1, 2, and 5 equiv.) (Fig. 6). The detailed 1H-NMR spectrum of 7i (along with structural assignment of peaks) is given in the experimental section. Fig. 5 shows zoomed-in area of interest (3–10 ppm) in the 1H-NMR spectrum of 7i. Prior to addition of any fluoride ions, the 1H NMR spectrum for 7i, in the area selected, shows a sharp singlet for –CH proton at 4.78 ppm, aromatic protons (≈6.7–7.2 ppm) are also visible in this region, the NH proton appears as a singlet at 9.28 ppm (Fig. 6a). The most noticeable change after addition of just 1 equiv. of fluoride ions, is a drastic decrease in the signal for NH proton (Fig. 6b); the signal for NH proton completely disappears after addition of 2 equiv. of fluoride ions (Fig. 6c). Similar observations were made after addition of 5 equiv. of fluoride ions. The signal for CH proton at 4.78 ppm remains unaltered. A minute (insignificant) up-field chemical shift for aromatic protons was observed with increasing addition of fluoride ions. The disappearance of characteristic signal for NH proton as a result of fluoride ion addition, clearly indicates proton abstraction by fluoride ions acting as base. This proton abstraction by fluoride ions seems to be a highly selective process for fluoride ions only (among other anions studied here). Accordingly this proton abstraction mechanism seems to be the reason for selective fluoride detection by chemosensor 7i.
 | ||
| Fig. 6 Study of anion (F−) addition to probe 7i via 1H NMR spectra; (a) without F− anion and (b) 1 equiv. (c) 2 equiv. (d) 5 equiv. of F−source (TBAF) in DMSO-d6 as solvent. | ||
Quantum mechanical Density Functional Theory (DFT) calculations have also been performed to gain further insight into the structural and spectroscopic changes during complex formation. As a model, we have optimized the structure of probe 7i and its fluoride (7i–F−) and acetate (7i–OAc−) complexes. We considered only acetate ion for comparative study with fluoride ion, mainly due to the fact that acetate ion is more basic than all other anions against which 7i is not responsive. The optimized geometries are shown in Fig. 7.
 | ||
| Fig. 7 Optimized geometries of 7i and its complexes with acetate and fluoride ion, optimized at B3LYP/6-31G(d,p). | ||
The optimized geometries reveal that the N–H bond in 7i has a bond length of 1.00 Å, which is slightly elongated to 1.06 Å upon interaction with acetate ion. The change in N–H bond distance of the probe on complexation with acetate ions is not significant enough to cause corresponding changes in electronic and spectroscopic properties of the chemosensor (vide infra). The interaction of fluoride ion with N–H proton of the chemosensor is quite strong. The fluoride ion pulls the proton toward itself and the N–H bond is significantly elongated to 1.49 Å (Fig. 8). The F–H bond length is 1.0 Å. The negative charge generated on the nitrogen in 7i–F− (as a result of fluoride interaction) is somewhat delocalized on the neighboring alkene bonds. The delocalization of charge also affects the N–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths. The N–C and CC bond lengths in 7i–F− are 1.37 Å and 1.43 Å compared to 1.40 Å and 1.42 Å for the parent compound 7i.
C bond lengths. The N–C and CC bond lengths in 7i–F− are 1.37 Å and 1.43 Å compared to 1.40 Å and 1.42 Å for the parent compound 7i.
 | ||
| Fig. 8 Overlay UV-vis absorption spectrum of 7i and fluoride complex 7i–F−, calculated at TD-B3LYP/6-31G(d,p). | ||
The strong proton abstraction ability of F− can be attributed to the high charge density on fluoride ion. The fluoride has −1 charge which is completely localized on it, whereas the negative charge on acetate ion is more delocalized. The Mulliken charge analysis reveals that the charge on the oxygen of acetate is −0.64, which is considerably smaller than −1 on fluoride. This difference in charge is most likely to be responsible for abstraction of proton of acridinedione chemosensor by the fluoride ion.
Binding energies are also calculated in order to rationalize the stabilities of these complexes. The free energies of interaction of 7i with fluoride and acetate ion are −95.35 and −36.50 kcal mol−1. The higher interaction energy of 7i with fluoride ion provides the driving force for transfer of proton to the latter. The interaction energy of fluoride ion with 7i is comparable to a covalent bond (also supported from geometric analysis) whereas the interaction with acetate ion is in the range of non-bonding interaction.
Finally, the UV-vis spectra are calculated for 7i–F− and 7i–OAc− complexes, and compared to the UV-vis spectrum of probe 7i. All complexes show several weak transitions in visible and near infra-red region but their oscillator strengths are less than 0.05, therefore, these transitions are believed not to play any significant role in the observable spectrum. The λmax of 7i is calculated at 322 nm with an oscillator strength of 0.2. Another peak is present at 326 nm but the oscillator strengths is relatively low (0.0695). These peaks are red shifted to 333 and 349 nm, respectively for 7i–F−. The red shifting of the λmax is consistent with the experimental observation where addition of fluoride ion leads to red shift with tail extending to 450 nm (compare Fig. 5a with Fig. 8). The experimental UV-vis spectrum shows relatively strong peak in the region of 400–500 nm upon adding 2–10 equivalent of fluoride ions. The appearance of strong absorption band in this region at higher equivalents of F− suggests complexes with more than one fluoride per phenyl dihydropyridine unit. Addition of acetate ion to 7i, on the other hand, leads to transitions with very low oscillator strength. The theoretical findings are also consistent with the experimental observation where addition of acetate ion leads to quenching of UV-vis absorption. Our theoretical and experimental results corroborate with each other and provide conclusive evidence that the interaction of fluoride ion with 7i is different than that for other anions.
3. Conclusion
In conclusion, a series of acridinediones (7a–l) has been synthesized using new morpholinium-based ionic fluoride liquids under solvent-free conditions. The acridinediones 7i and 7l were explored as chemosensors for detection of different anions such as F−, OAc−, Br−, I−, HSO4−, ClO3−, ClO4−, CN−, and SCN−. A conspicuous change in color from colorless to yellow, upon addition of fluoride ions to acridindione shows selectivity towards fluoride ion detection. This was further investigated and confirmed via detailed UV-visible, fluorescence and 1H-NMR spectroscopic techniques. The ICT mechanism responsible for these observed changes was found to be fluoride ion mediated abstraction of NH proton of dihydropyridine part of acridinedione. Further insight was gained via computational studies using DFT (B3LYP/6-31G(d,p)), whereupon comparison of acridinedione interaction with fluoride and acetate ion explained why (and how) it is selective for fluoride ion. Our results provide a strong basis for further modulation and exploration of acridinediones as non-invasive chemosensors for fluoride ion detection in a variety of sample types (possibly) including biological samples as well.4. Material and methods
N-Methyl morphiline (≥98.0% (GC)) and N-ethyl morpholine (≥97.0% (GC)), n-pentyl bromide, n-heptyl bromide, 5,5-dimethyl-1,3-cyclohexanedione (95%), substituted benzonitriles, trimethylsilyl azide, and silver fluoride (≥99%) were purchased from Sigma Aldrich and used directly for carrying our reaction unless otherwise stated. Toluene (analytical grade), methanol and distill or Milli-Q water were used as solvents. Thin layered chromatography was carried out with silica gel 60 aluminium-backed plates 0.063–0.200 mm. Analytical grade solvents such as ethyl acetate (EtOAc), diethyl ether, hexane and methanol etc. were used. Round bottomed flasks (5, 10, 25, 50, 100, 250 mL) and screw capped micro reaction vessels (3–5 mL) were used. For TLC spots visualization, in UV lamp a radiation of 254 nm was used. In addition, different staining mixtures, such as basic potassium permanganate or vanillin were also used. Infrared (IR) spectra were recorded on Bruker Vector-22 spectrometer. The 1H NMR spectra were recorded using Bruker spectrometers at 300 MHz, 400 MHz, 500 MHz, and 600 MHz, while 13C NMR spectra were recorded at 75 MHz, 100 MHz, 125 MHz, 150 MHz in the appropriate deuterated solvent. The chemical shifts were recorded on the δ-scale (ppm) using residual solvents as an internal standard (DMSO; 1H 2.50, 13C 39.43 and CHCl3; 1H 7.26, 13C 77.16). Coupling constants were calculated in Hertz (Hz) and multiplicities were labelled s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet) and the prefixes br (broad) or app (apparent) were used. Mass spectra (EI+ and FAB) were recorded on Finnigan MAT-321A, Germany. Melting points of solids were determined using a Stuart™ melting point SMP3 apparatus.5. Computational methodology
All calculations were performed with Gaussian 09 suit of programs.39 Geometries of phenyl dihydropyridine 7i and its complexes with fluoride and acetate ions are optimized without any symmetry constraints at B3LYP method of density functional theory at 6-31G(d,p) basis set.40 The B3LYP method consists of three parameter hybrid functional of Becke41 in conjunction with the correlation function of Lee et al.42 The B3LYP method provides a nice balance between cost and accuracy, and it is known to perform reasonably well for the prediction of geometries of a number of synthetic,43–46 natural products47–50 and conjugated systems.51,52 The optimized structures are confirmed as true minima through frequency calculations. Absence of any imaginary frequencies confirmed as true minima. The interaction energies of dihydropyridine with fluoride and acetate ions are calculated by the following equation Eint = Ecomplex − (Edhp + Eanion).Where Ecomplex is the energy of complex formed between acridinedione and anion whereas Edhp and Eanion are the energies of isolated acridinedione and anion, respectively. The reported energies of interaction are based on Gibbs free energies.
The spectroscopic properties are calculated in DMSO solvent. The solvent effect is introduced through IEFPCM approach, as implemented in Gaussian 09. Time dependent DFT calculations are performed to calculate the excited state properties (UV-vis spectra) of these complexes. TD-B3LYP/6-31G(d,p) method is employed for calculation of UV-vis spectra. A total of 20 states are calculated with 50:50 singlet:triplet states. 1H NMR of the optimized geometries is calculated at B3LYP/6-311+G(2d,p) through GIAO formalism. The 1H NMR values reported in this study are with reference to TMS.
6. General synthetic procedure for morpholinium-based ionic liquids (MoFs) (3–6)
An oven dried round bottom flask was charged with N-alkyl (Me/Et) morpholine (1 g, 10 mmol, 1 equiv.) and toluene (5 mL). Subsequently, corresponding alkyl (C5, C7) bromide (1.5 equiv.) was added to the solution and heated the resulting mixture at reflux (115–120 °C). The progress of reaction was monitored by TLC analysis until the complete consumption of starting material. The reaction mixture was cooled to room temperature and solvent was decanted off. Further, some more cooled toluene was added, shaken well and removed to get a thick ionic liquid which was used without purification in the next counter anion exchange step. The crude mixture of corresponding ionic liquids was dissolved in water (5 mL) and added to the aqueous solution of AgF (5 mL, 1 equiv.). Subsequently, the precipitates of silver bromide were removed and water was evaporated via freeze drying. The residue dissolved in chloroform, filtered and evaporated by using rotary evaporator. The obtained material was occasionally washed with cooled toluene to get the final desired product. The final ionic morpholinium-based ionic liquids 3–6 were characterized with 1H, 13C-NMR, IR spectroscopy and mass spectrometry.6.1. 4-Methyl-4-pentylmorpholin-4-ium fluoride (3)
Light brown liquids, (1.70 g, 89%), IR (νmax, cm−1): (solution, CHCl3) 3405, 2955, 2870, 1630, 1472, 1160. 1H NMR (400 MHz, DMSO): δH 3.90 (4H, app brs, CH2–O–CH2), 3.45–3.39 (6H, m, CH2–N–CH2 & N–CH2), 3.12 (3H, s, N–CH3), 1.71–1.63 (2H, m, CH2), 1.36–1.23 (4H, m, CH2–CH2), 0.88 (3H, t, J = 7.2 Hz, CH3); 19F NMR (400 MHz, DMSO) −126.4; 13C NMR (75 MHz, DMSO): δC 63.8 (CH2), 59.8 (2CH2), 58.9 (2CH2), 46.07 (CH3), 27.9 (CH2), 21.6 (CH2), 20.5 (CH2), 13.8 (CH3). 1H NMR (400 MHz, DMSO): MS-MALDI m/z (%), 172.2 (M+ − F−, 100).6.2. 4-Methyl-4-heptylmorpholin-4-ium fluoride (4)
Brown liquids, (1.90 g, 88%), IR (νmax, cm−1): (solution, CHCl3) 3416, 2928, 2863, 1632, 1471, 1119. 1H NMR (400 MHz, DMSO): δH 3.90 (4H, app brs, CH2–O–CH2), 3.45–3.39 (6H, m, CH2–N–CH2 & N–CH2), 3.12 (3H, s, N–CH3), 1.68–1.64 (2H, m, CH2), 1.26 (8H, m, (CH2)4), 0.88 (3H, t, J = 7.2 Hz, CH3); 13C NMR (100 MHz, DMSO): δC 63.8 (CH2), 59.8 (2CH2), 58.9 (2CH2), 46.03 (CH3), 31 (CH2), 28.2 (CH2), 25.7 (CH2), 21.9 (CH2), 20.7 (CH2), 13.9 (CH3). MS-MALDI m/z (%), 199.5 (M+ − F−, 100).6.3. 4-Ethyl-4-pentylmorpholin-4-ium fluoride (5)
Light brown liquids, (1.30 g, 73%), IR (νmax, cm−1): (solution, CHCl3) 3451, 2957, 2872, 1635, 1473, 1123. 1H NMR (400 MHz, DMSO): δH 3.90 (4H, app d, J = 2.4 Hz CH2–O–CH2), 3.48 (2H, q, J = 7.2 Hz, CH2–CH3), 3.43–3.35 (6H, m, CH2–N–CH2 & N–CH2), 1.61–1.57 (2H, m, CH2), 1.35–1.26 (8H, m, (CH2)2), 1.18 (3H, t, J = 7.2 Hz, CH2–CH3), 0.88 (3H, t, J = 7.2 Hz, CH3); 13C NMR (100 MHz, DMSO): δC 61.4 (2CH2), 58.9 (2CH2 & CH2), 54.9 (CH2), 29.7 (CH2), 23.5 (CH2), 22.1 (CH2), 15.7 (CH3), 8.64 (CH3). MS-MALDI m/z (%), 186.2 (M+ − F−, 100).6.4. 4-Ethyl-4-heptylmorpholin-4-ium fluoride (6)
Light brown liquids, (1.43 g, 71%), IR (νmax, cm−1): (solution, CHCl3) 3403, 2929, 2864, 1666, 1595, 1454. 1H NMR (300 MHz, DMSO): δH 3.88 (4H, app brs, CH2–O–CH2), 3.48 (2H, q, J = 7.2 Hz, CH2–CH3), 3.43–3.35 (6H, m, CH2–N–CH2 & N–CH2), 1.58 (2H, m, CH2), 1.35–1.26 (8H, m, (CH2)4), 1.18 (3H, t, J = 7.2 Hz, CH2–CH3), 0.88 (3H, t, J = 7.2 Hz, CH3); 19F NMR (400 MHz, DMSO) −126.9. MS-EI m/z (%), 214.1 (M+ − F−, 100).7. General procedure for acridinedione synthesis
In an oven dried flask commercially available dimedone (280 mg, 2 mmol), ammonium acetate (77.08 mg, 1 mmol), synthetic morpholinium-IL 3 (191 mg, 1 mmol) were added to corresponding benzaldehyde derivatives (1 mmol). The reaction mixture was heated under 125-135 °C until the complete disappearance of starting material monitored by thin layer chromatography (TLC) using ethyl acetate and n-hexane as a solvent system at different ratio. The reaction mixture was cooled to room temperature and resultant pure compound was precipitated by washing crude reaction mixture in ethyl acetate (2 × 20 mL). While some compounds need column chromatography for their purification direct after crude mixture. The resultant dihydropyridines (7a–7l) were yellow to off-white solids in different yields (48–78%).7.1. 9-(4′-Methylphenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7a)
133 mg (73%); IR (νmax, cm−1): (solid, KBr) 3280, 3065, 2954, 2925, 1651, 1607, 1493, 1365, 1220, 1146. 1H NMR (400 MHz, DMSO): δH 9.22 (1H, brs, NH), 7.01 (2H, d, J = 8 Hz, ArH), 6.93 (2H, d, J = 8 Hz, ArH), 4.74 (1H, brs, CH-9), 2.42 (2H, d, J = 16.8 Hz, CH-4a/5a), 2.29 (2H, d, J = 16.8 Hz, CH-4b/5b), 2.17 (3H, s, ArCH3), 2.14 (2H, d, J = 15.2, CH-2a/7a), 1.95 (2H, d, J = 16.4 Hz, CH-2b,7b), 0.99 (6H, s, (CH3)2) 0.84 (6H, s, (CH3)2); 13C NMR (125 MHz, DMSO): δC 194.3 (2CO), 149.1 (2C), 144.3 (C), 134.2 (C), 128.1 (2CH), 127.5 (2CH), 111.6 (2C), 50.2 (2CH2), 39.6 (2CH2), 32.4 (CH), 32.1 (2C), 29.1 (2CH3), 26.4 (2CH3), 20.5 (ArCH3). MS-EI m/z 363.3 (M+). The data is identical to those previously reported.537.2. 9-(4′-Hydroxyphenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7b)53
132 mg, (82%); IR (νmax, cm−1): (solid, KBr) 3276, 3198, 2956, 1613, 1471, 1372, 1223, 1141. 1H NMR (300 MHz, DMSO): δH 9.17 (1H, brs, NH), 8.96 (1H, brs, ArOH), 7.01 (2H, d, J = 8.4 Hz, ArH), 6.93 (2H, d, J = 8.4 Hz, ArH), 4.69 (1H, brs, CH-9), 2.42 (2H, d, J = 16.8 Hz, CH-4a/5a), 2.28 (2H, d, J = 16.8 Hz, CH-4b/5b), 2.14 (2H, d, J = 16.2 Hz, CH-2a/7a), 1.95 (2H, d, J = 16.2 Hz, CH-2b,7b), 0.99 (6H, s, (CH3)2) 0.86 (6H, s, (CH3)2); 13C NMR (125 MHz, DMSO): δC 194.3 (2CO), 154.2 (C), 148.87 (2C), 137.9 (C), 128.4 (2CH), 114.2 (2CH), 111.8 (2C), 50.1 (2CH2), 39.5 (2CH2), 32.1 (2C), 31.6 (CH), 29.1 (2CH3), 26.4 (2CH3). MS-EI m/z 365.2 (M+); EI-HRMS (M+) C23H27O3N found 365.2001, calculated 365.1985.7.3. 9-(3′-Hydroxyphenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7c)
139 mg, (86%); IR (νmax, cm−1): (solid, KBr) 3283, 3207, 3072, 2960, 2929, 1620, 1482, 1365, 1223, 1143. 1H NMR (400 MHz, DMSO): δH 9.23 (1H, brs, NH), 9.02 (1H, brs, ArOH), 6.90 (1H, t, J = 7.6 Hz, ArH), 6.61 (1H, brs, ArH), 6.55 (1H, d, J = 7.6 Hz, ArH), 6.40 (1H, d, J = 8 Hz, ArH), 4.70 (1H, brs, CH-9), 2.42 (2H, d, J = 17.2 Hz, CH-4a/5a), 2.28 (2H, d, J = 17.2 Hz, CH-4b/5b), 2.14 (2H, d, J = 16 Hz, CH-2a/7a), 1.95 (2H, d, J = 16.4 Hz, CH-2b/7b), 0.99 (6H, s, (CH3)2) 0.87 (6H, s, (CH3)2); MS-EI m/z 365.2 (M+); EI-HRMS (M+) C23H27O3N found 365.1962, calculated 365.1985.7.4. 9-(3′-Ethoxy-4′-hydroxyphenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7d)
146 mg, (71%); IR (νmax, cm−1): (solid, KBr) 3273, 3202, 3071, 2961, 1617, 1481, 1366, 1223, 1149. 1H NMR (400 MHz, DMSO): δH 9.19 (1H, brs, NH), 8.47 (1H, brs, ArOH), 6.68 (1H, s, ArH), 6.53 (1H, d, J = 8.4 Hz, ArH), 6.49 (1H, d, J = 8.4 Hz, ArH), 4.69 (1H, brs, CH-9), 3.87 (2H, q, J = 7.2 Hz, CH2–CH3), 2.42 (2H, d, J = 17.2 Hz, CH-4a/5a), 2.28 (2H, d, J = 16.8 Hz, CH-4b/5b), 2.14 (2H, d, J = 16 Hz, CH-2a/7a), 1.95 (2H, d, J = 16 Hz, CH-2b/7b), 1.27 (3H, t, J = 7.2 Hz, CH2–CH3), 0.99 (6H, s, (CH3)2) 0.87 (6H, s, (CH3)2); 13C NMR (100 MHz, DMSO): δC 194.4 (2CO), 148.9 (2C), 145.6 (C′), 144.7 (C), 138.4 (C), 119.8 (CH), 114.8 (CH), 113.8 (CH), 111.7 (2C), 63.8 (CH2), 50.3 (2CH2), 39.5 (2CH2), 32.1 (2C), 31.8 (CH), 29.1 (2CH3), 26.3 (2CH3), 14.7 (CH3). MS-EI m/z 409.3 (M+); EI-HRMS (M+) C25H32O4N found 409.2258, calculated 409.2248.7.5. 9-(4′-Methoxyphenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7e)53
140 mg, (73%); IR (νmax, cm−1): (solid, KBr) 3277, 3207, 2956, 2930, 1644, 1607, 1481, 1366, 1224, 1141. 1H NMR (400 MHz, DMSO): δH 9.22 (1H, brs, NH), 7.03 (2H, d, J = 8.8 Hz, ArH), 6.69 (2H, d, J = 8.4 Hz, ArH), 4.73 (1H, brs, CH-9), 3.64 (3H, s, OCH3), 2.42 (2H, d, J = 17.2 Hz, CH-4a/5a), 2.28 (2H, d, J = 17.2 Hz, CH-4b/5b), 2.14 (2H, d, J = 16 Hz, CH-2a/7a), 1.95 (2H, d, J = 16 Hz, CH-2b/7b), 0.99 (6H, s, (CH3)2) 0.86 (6H, s, (CH3)2); 13C NMR (125 MHz, DMSO): δC 194.3 (2CO), 157.1 (C), 148.9 (2C), 139.5 (C), 128.5 (2CH), 112.9 (2CH), 111.7 (2C), 54.8 (OCH3), 50.2 (2CH2), 39.5 (2CH2), 32.1 (2C), 31.9 (CH), 29.1 (2CH3), 26.4 (2CH3). MS-EI m/z 379.1 (M+); EI-HRMS (M+) C24H29O3N found 379.2155, calculated 379.2142.7.6. 9-(3′-Bromo-4′-methoxyphenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7f)
170 mg, (74%); IR (νmax, cm−1): (solid, KBr) 3282, 3213, 2962, 2925, 1650, 1610, 1486, 1363, 1218, 1136. 1H NMR (300 MHz, DMSO): δH 9.29 (1H, brs, NH), 7.26 (1H, d, J = 2.1 Hz, ArH), 7.06 (1H, dd, J = 8.4, 2.1 Hz, ArH), 6.90 (1H, d, J = 8.4 Hz, ArH), 4.71 (1H, brs, CH-9), 3.64 (3H, s, OCH3), 2.42 (2H, d, J = 17.1 Hz, CH-4a/5a), 2.30 (2H, d, J = 17.1 Hz, CH-4b/5b), 2.16 (2H, d, J = 16.2 Hz, CH-2a/7a), 1.95 (2H, d, J = 16 Hz, CH-2b/7b), 0.99 (6H, s, (CH3)2) 0.87 (6H, s, (CH3)2); 13C NMR (125 MHz, DMSO): δC 194.3 (2CO), 153.2 (C), 149.3 (2C), 141 (C), 131.9 (CH), 127.8 (CH), 111.9 (CH), 111.1 (2C), 109.6 (C), 55.9 (OCH3), 50.2 (2CH2), 39.5 (2CH2), 32.1 (2C), 31.9 (CH), 29.1 (2CH3), 26.4 (2CH3). MS-EI m/z 458.1 (M+); EI-HRMS (M+) C24H28O3NBr found 457.1240, calculated 457.1247.7.7. 9-(2′-Methoxyphenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7g)
128 mg, (67%); IR (νmax, cm−1): (solid, KBr) 3271, 3174, 3058, 2952, 1643, 1601, 1365, 1218, 1142. 1H NMR (400 MHz, DMSO): δH 9.16 (1H, brs, NH), 7.15 (1H, d, J = 6.4 Hz, ArH), 6.99 (1H, t, J = 7.2 Hz, ArH), 6.78 (1H, d, J = 8.4 Hz, ArH), 6.71 (1H, t, J = 7.2 Hz, ArH), 4.90 (1H, brs, CH-9), 3.65 (3H, s, OCH3), 2.40 (2H, d, J = 16.8 Hz, CH-4a/5a), 2.21 (2H, d, J = 16.8 Hz, CH-4b/5b), 2.09 (2H, d, J = 16.4 Hz, CH-2a/7a), 1.87 (2H, d, J = 16 Hz, CH-2b/7b), 0.99 (6H, s, (CH3)2) 0.87 (6H, s, (CH3)2); 13C NMR (125 MHz, DMSO): δC 194 (2CO), 157.6 (C), 149.6 (2C), 133.8 (C), 131.4 (CH), 126.6 (CH), 119.1 (CH), 110.8 (CH), 110.1 (2C), 55 (OCH3), 50.4 (2CH2), 39.5 (2CH2), 31.9 (2C), 31.4 (CH), 29.2 (2CH3), 25.9 (2CH3). MS-EI m/z 379.3 (M+); EI-HRMS (M+) C24H29O3N found 379.2162, calculated 379.2142.7.8. 9-(2′,3′,4′-Trimethoxyphenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7h)
172 mg, (78%); Mp 246–250 °C, IR (νmax, cm−1): (solid, KBr) 3306, 3190, 3069, 2954, 1640, 1609, 1224, 1097. 1H NMR (400 MHz, DMSO): δH 9.14 (1H, brs, NH), 6.79 (1H, d, J = 8.8 Hz, ArH), 6.54 (1H, t, J = 8.8 Hz, ArH), 4.88 (1H, brs, CH-9), 3.80 (3H, s, OCH3), 3.68 (3H, s, OCH3), 3.63 (3H, s, OCH3), 2.38 (2H, d, J = 17.2 Hz, CH-4a/5a), 2.24 (2H, d, J = 17.2 Hz, CH-4b/5b), 2.09 (2H, d, J = 16 Hz, CH-2a/7a), 1.87 (2H, d, J = 16 Hz, CH-2b/7b), 0.98 (6H, s, (CH3)2) 0.88 (6H, s, (CH3)2); 13C NMR (75 MHz, DMSO): δC 194.1 (2CO), 151.5 (C), 151.3 (C), 149.2 (2C), 133.8 (C), 132.5 (C), 124.8 (CH), 110.1 (2C), 106.4 (CH), 60.3 (OCH3), 59.8 (OCH3), 55.6 (OCH3), 50.4 (2CH2), 39.5 (2CH2), 32 (2C), 29.6 (CH-9), 29.2 (2CH3), 25.9 (2CH3). MS-EI m/z 439.3 (M+); EI-HRMS (M+) C26H33O5N found 439.2333, calculated 439.2353.7.9. 9-(4′-Fluorophenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7i)
137 mg, (75%); IR (νmax, cm−1): (solid, KBr) 3528, 3346, 3275, 3164, 3045, 2956, 1613, 1486, 1366, 1221, 1146. 1H NMR (400 MHz, DMSO): δH 9.29 (1H, brs, NH), 7.14 (2H, dd, J = 8, 6 Hz, ArH), 6.96 (2H, t, J = 8.8 Hz, ArH), 4.78 (1H, brs, CH-9), 2.42 (2H, d, J = 17.2 Hz, CH-4a/5a), 2.31 (2H, d, J = 17.2 Hz, CH-4b/5b), 2.18 (2H, d, J = 16 Hz, CH-2a/7a), 1.87 (2H, d, J = 16 Hz, CH-2b/7b), 0.99 (6H, s, (CH3)2) 0.88 (6H, s, (CH3)2); 13C NMR (75 MHz, DMSO): δC 194.2 (2CO), 161.8/158.6 (C), 149.3 (2C), 143.3 (C), 129.2/129.1 (2CH), 114.3/114.9 (2CH), 111.3 (2C), 50.2 (2CH2), 39.5 (2CH2), 32.2 (CH-9), 32.1 (2C), 29.2 (2CH3), 25.9 (2CH3). MS-EI m/z 367.3 (M+); EI-HRMS (M+) C23H26O2NF found 367.1921, calculated 367.1942.7.10. 9-(3′-Nitrophenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7j)53
124 mg, (63%); IR (νmax, cm−1): (solid, KBr) 3176, 2926, 2857, 1647, 1607, 1532, 1399, 1222, 1145. 1H NMR (400 MHz, DMSO): δH 9.45 (1H, brs, NH), 7.95 (1H, s, ArH), 7.93 (1H, d, J = 12.4 Hz, ArH), 7.60 (1H, d, J = 7.6 Hz, ArH), 7.49 (1H, t, J = 8 Hz, ArH), 4.91 (1H, brs, CH-9), 2.46 (2H, d, obscured by DMSO signal), 2.35 (2H, d, J = 17.2 Hz, CH-4b/5b), 2.18 (2H, d, J = 16 Hz, CH-2a/7a), 1.87 (2H, d, J = 16 Hz, CH-2b/7b), 1.01 (6H, s, (CH3)2) 0.85 (6H, s, (CH3)2); MS-EI m/z 394.2 (M+); EI-HRMS (M+) C23H26O2N2 found 394.1891, calculated 394.1887.7.11. 9-(3′-Cyanophenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7k)
127 mg, (67%); IR (νmax, cm−1): (solid, KBr) 3300, 3232, 3088, 2956, 2892, 2233, 1644, 1486, 1365, 1221, 1141. 1H NMR (400 MHz, DMSO): δH 9.39 (1H, brs, NH), 7.52–7.46 (3H, m, ArH), 7.39 (1H, app t, J = 8.4 Hz, ArH), 4.82 (1H, brs, CH-9), 2.46 (2H, d, obscured by DMSO signal), 2.36 (2H, d, J = 17.2 Hz, CH-4b/5b), 2.18 (2H, d, J = 16.4 Hz, CH-2a/7a), 1.87 (2H, d, J = 16.4 Hz, CH-2b/7b), 1.00 (6H, s, (CH3)2) 0.85 (6H, s, (CH3)2); 13C NMR (75 MHz, DMSO): δC 194.3 (2CO), 149.9 (2C), 148.4 (C), 132.5 (2CH), 131 (2CH), 129.4 (CH), 128.9 (CH), 118.9 (C), 110.5 (2C), 110.4 (C), 50 (2CH2), 39.5 (2CH2), 33.7 (CH), 33.3 (2C), 29.2 (2CH3), 25.9 (2CH3). MS-EI m/z 375.3 (M+); EI-HRMS (M+) C24H26O2N2 found 374.1978, calculated 374.1989.7.12. 9-(4′-Chlorophenyl)-3,3,6,6-tetramethyl-3,4,6,7,9,10-hexahydroacridine-1,8(2H,5H)-dione (7l)53
150 mg, (78%); IR (νmax, cm−1): (solid, KBr) 3276, 3177, 3059, 2875, 1647, 1609, 1491, 1365, 1221, 1145. 1H NMR (500 MHz, DMSO): δH 9.34 (1H, brs, NH), 7.20 (2H, d, J = 8.5 Hz, ArH), 7.13 (2H, d, J = 8.5 Hz, ArH), 4.77 (1H, brs, CH-9), 2.42 (2H, d, J = 17 Hz, CH-4a/5a), 2.31 (2H, d, J = 17 Hz, CH-4b/5b), 2.18 (2H, d, J = 16 Hz, CH-2a/7a), 1.87 (2H, d, J = 16 Hz, CH-2b/7b), 0.99 (6H, s, (CH3)2) 0.84 (6H, s, (CH3)2); 13C NMR (125 MHz, DMSO): δC 194.4 (2CO), 149.5 (2C), 146.1 (C), 129.6 (C), 129.5 (2CH), 127.6 (2CH), 111.1 (2C), 50.2 (2CH2), 39.5 (2CH2), 32.6 (CH), 32.1 (2C), 29.2 (2CH3), 25.9 (2CH3). MS-EI m/z 382.9 (M+); EI-HRMS (M+) C23H26O2NCl found 383.1629, calculated 383.1647.Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
We are thankful to Higher Education Commission (HEC) with grant number 20-4732, Pakistan for providing financial support for this project.References
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC.
- Y.-C. Lin and C.-T. Chen, Org. Lett., 2009, 11, 4858–4861 CrossRef CAS PubMed.
- M. Ishtiaq, I. Munir, M. al-Rashida, K. Ayub, J. Iqbal, R. Ludwig, K. M. Khan, S. A. Ali and A. Hameed, RSC Adv., 2016, 6, 64009–64018 RSC.
- S. A. Kumar, M. S. Kumar, P. Sreeja and A. Sreekanth, Spectrochim. Acta, Part A, 2013, 113, 123–129 CrossRef PubMed.
- T. Anand, G. Sivaraman, M. Iniya, A. Siva and D. Chellappa, Anal. Chim. Acta, 2015, 876, 1–8 CrossRef CAS PubMed.
- U. N. Yadav, P. Pant, D. Sharma, S. K. Sahoo and G. S. Shankarling, Sens. Actuators, B, 2014, 197, 73–80 CrossRef CAS.
- X. Shang, Z. Yang, J. Fu, P. Zhao and X. Xu, Sensors, 2015, 15, 28166–28176 CrossRef PubMed.
- A. Wiegand and T. Attin, Oral Health Prev. Dent., 2003, 1, 245–253 Search PubMed.
- S. Peckham and N. Awofeso, Sci. World J., 2014, 2014, 1–10 CrossRef PubMed.
- G. Sivaraman and D. Chellappa, J. Mater. Chem. B, 2013, 1, 5768–5772 RSC.
- M. M. M. Raposo, B. García-Acosta, T. Ábalos, P. Calero, R. n. Martínez-Máñez, J. V. Ros-Lis and J. Soto, J. Org. Chem., 2010, 75, 2922–2933 CrossRef CAS PubMed.
- L. E. Santos-Figueroa, M. E. Moragues, M. M. M. Raposo, R. M. Batista, S. P. Costa, R. C. M. Ferreira, F. Sancenón, R. Martínez-Máñez, J. V. Ros-Lis and J. Soto, Org. Biomol. Chem., 2012, 10, 7418–7428 CAS.
- W. Radchatawedchakoon, W. Sangsuwan, S. Kruanetr and U. Sakee, Spectrochim. Acta, Part A, 2014, 121, 306–312 CrossRef CAS PubMed.
- C. Yang, M. Zheng, Y. Li, B. Zhang, J. Li, L. Bu, W. Liu, M. Sun, H. Zhang and Y. Tao, J. Mater. Chem. A, 2013, 1, 5172–5178 CAS.
- Q. K. Sun, M. S. Chen, Z. W. Liu, H. C. Zhang and W. J. Yang, Tetrahedron Lett., 2017, 58, 2711–2714 CrossRef CAS.
- V. Karunakaran, P. Ramamurthy, T. Josephrajan and V. Ramakrishnan, Spectrochim. Acta, Part A, 2002, 58, 1443–1451 CrossRef CAS.
- N. Srividya, P. Ramamurthy and V. Ramakrishnan, Spectrochim. Acta, Part A, 1998, 54, 245–253 CrossRef.
- J.-P. Fouassier, F. Morlet-Savary, J. Lalevée, X. Allonas and C. Ley, Materials, 2010, 3, 5130–5142 CrossRef CAS PubMed.
- P. Xiao, F. Dumur, M.-A. Tehfe, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Polymer, 2013, 54, 3458–3466 CrossRef CAS.
- B. Venkatachalapathy, P. Ramamurthy and V. Ramakrishnan, J. Photochem. Photobiol., A, 1997, 111, 163–169 CrossRef CAS.
- A. Madaan, R. Verma, V. Kumar, A. T. Singh, S. K. Jain and M. Jaggi, Arch. Pharm., 2015, 348, 837–860 CrossRef CAS PubMed.
- K. Sirisha, G. Achaiah and V. M. Reddy, Arch. Pharm., 2010, 343, 342–352 CrossRef CAS PubMed.
- A. R. Trivedi, D. K. Dodiya, B. H. Dholariya, V. B. Kataria, V. R. Bhuva and V. H. Shah, Bioorg. Med. Chem. Lett., 2011, 21, 5181–5183 CrossRef CAS PubMed.
- H. Haller, Int. J. Clin. Pract., 2008, 62, 781–790 CrossRef CAS PubMed.
- S. Bahekar and D. Shinde, Acta Pharm., 2002, 52, 281–287 CAS.
- R. Velu, N. Won, J. Kwag, S. Jung, J. Hur, S. Kim and N. Park, New J. Chem., 2012, 36, 1725–1728 RSC.
- M. Zheng, M. Sun, D. Zhang, T. Liu, S. Xue and W. Yang, Dyes Pigm., 2014, 101, 109–115 CrossRef CAS.
- P. Ashokkumar, V. Ramakrishnan and P. Ramamurthy, J. Phys. Chem. B, 2010, 115, 84–92 CrossRef PubMed.
- V. Thiagarajan and P. Ramamurthy, Spectrochim. Acta, Part A, 2007, 67, 772–777 CrossRef PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- A.-G. Ying, L.-M. Wang, H.-X. Deng, J.-H. Chen, X.-Z. Chen and W.-D. Ye, ARKIVOC, 2009, xi, 288–298 Search PubMed.
- X. Chen and A. Ying, in Ionic Liquids: Applications and Perspectives, ed. P. A. Kokorin, InTech, 2011 Search PubMed.
- T. P. Thuy Pham, C.-W. Cho and Y.-S. Yun, Water Res., 2010, 44, 352–372 CrossRef PubMed.
- J. H. Clark, Chem. Rev., 1980, 80, 429–452 CrossRef CAS.
- A. Hameed, R. D. Alharthy, J. Iqbal and P. Langer, Tetrahedron, 2016, 72, 2763–2812 CrossRef CAS.
- V. Thiagarajan, P. Ramamurthy, D. Thirumalai and V. T. Ramakrishnan, Org. Lett., 2005, 7, 657–660 CrossRef CAS PubMed.
- U. N. Yadav, P. Pant, D. Sharma, S. K. Sahoo and G. S. Shankarling, Sens. Actuators, B, 2014, 197, 73–80 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. . Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- V. Specowius, F. Bendrath, M. Winterberg, K. Ayub and P. Langer, Adv. Synth. Catal., 2012, 354, 1163–1169 CrossRef CAS.
- V. O. Iaroshenko, D. Ostrovskyi, K. Ayub, A. Spannenberg and P. Langer, Adv. Synth. Catal., 2013, 355, 576–588 CAS.
- M. N. Arshad, A. Bibi, T. Mahmood, A. M. Asiri and K. Ayub, Molecules, 2015, 20, 5851–5874 CrossRef CAS PubMed.
- M. N. Arshad, A. M. Asiri, K. A. Alamry, T. Mahmood, M. A. Gilani, K. Ayub and A. S. Birinji, Spectrochim. Acta, Part A, 2015, 142, 364–374 CrossRef CAS PubMed.
- M. A. Hashmi, A. Khan, K. Ayub and U. Farooq, Spectrochim. Acta, Part A, 2014, 128, 225–230 CrossRef CAS PubMed.
- H. Ullah, A. Rauf, Z. Ullah, M. Anwar, G. Uddin and K. Ayub, Spectrochim. Acta, Part A, 2014, 118, 210–214 CrossRef CAS PubMed.
- T. U. Rahman, G. Uddin, R. U. Nisa, R. Ludwig, W. Liaqat, T. Mahmood, G. Mohammad, M. I. Choudhary and K. Ayub, Spectrochim. Acta, Part A, 2015, 148, 375–381 CrossRef CAS PubMed.
- T. U. Rahman, M. Arfan, T. Mahmood, W. Liaqat, M. A. Gilani, G. Uddin, R. Ludwig, K. Zaman, M. I. Choudhary and K. F. Khattak, Spectrochim. Acta, Part A, 2015, 146, 24–32 CrossRef PubMed.
- M. Kamran, H. Ullah, A. S. Anwar-ul-Haq, S. Bilal, A. A. Tahir and K. Ayub, Polymer, 2015, 72, 30–39 CrossRef CAS.
- H. Ullah, A.-u.-H. A. Shah, S. Bilal and K. Ayub, J. Phys. Chem. C, 2014, 118, 17819–17830 CAS.
- A. Amoozadeh, S. Golian and S. Rahmani, RSC Adv., 2015, 5, 45974–45982 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary information contains the NMR spectra of the synthesized compounds. See DOI: 10.1039/c7ra11974g |
| This journal is © The Royal Society of Chemistry 2018 |