
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced crystallinity of CH3NH3PbI3 by the pre-coordination of PbI2–DMSO powders for highly reproducible and efficient planar heterojunction perovskite solar cells†
Jiyong Leea and
Seunghyun Baik *b
*b
aDepartment of Energy Science, Sungkyunkwan University, Suwon 16419, Republic of Korea
bSchool of Mechanical Engineering, Sungkyunkwan University, Suwon 16419, Republic of Korea. E-mail: sbaik@me.skku.ac.kr
First published on 3rd January 2018
Abstract
Solution processable CH3NH3PbI3 has received considerable attention for highly-efficient perovskite solar cells. However, the different solubility of PbI2 and CH3NH3I is problematic, initiating active solvent engineering research using dimethyl sulfoxide (DMSO). Here we investigated the pre-coordination of PbI2–DMSO powders for planar heterojunction perovskite solar cells fabricated by a low-temperature process (≤100 °C). Pre-coordination was carried out by simple mechanical mixing using a mortar and pestle. The composition of PbI2–DMSOx (x = 0, 1, or 2) in the powder mixture was investigated by gradually increasing mechanical mixing time, and a dominant composition of PbI2–DMSO1 was obtained after a 10 min mixing process. The pre-coordinated PbI2–DMSO powders were then blended with CH3NH3I in DMF to make the CH3NH3PbI3 film by toluene-assisted spin-coating and heat treatment. Compared with the one-step blending of CH3NH3I, PbI2, and DMSO in DMF, the pre-coordination method resulted in better dissolution of PbI2, larger grain size, and pinhole-free morphology. Consequently, absorption, fluorescence, carrier lifetime, and charge extraction were enhanced. The average open-circuit voltage (1.046 V), short-circuit current (22.9 mA cm−2), fill factor (73.5%), and power conversion efficiency (17.6%) were increased by 2–12% with decreased standard deviations (13–50%), compared with the one-step blending method. The best efficiency was 18.2%. The simple mechanical pre-coordination of PbI2–DMSO powders was very effective in enhancing the crystallinity of CH3NH3PbI3 and photovoltaic performance.
1. Introduction
Photovoltaic technology that directly converts solar light into electricity has received considerable attention to substitute fossil fuels.1–3 However, the per-watt cost of silicon-based first generation photovoltaic devices is still greater than that of fossil-fuel-based electricity.1–3 The low-cost titanium dioxide (TiO2)-based photovoltaic devices such as dye-sensitized solar cells have been actively investigated to address this hurdle.1–4 Recently, the low-cost and solution processable organometal halide perovskite absorbers, in combination with the double-layered TiO2 structure, have achieved over 20% power conversion efficiency (PCE).5–7 The TiO2 double layers involved a meso-porous or nano-structured TiO2 layer, which was constructed at high temperatures (>550 °C) or by complicated self-assembly methods, on top of a blocking compact TiO2 layer to enhance interfacial coupling with perovskite.5–10 On the other hand, low-temperature processable planar heterojunction perovskite solar cells are of great interest due to the simple single-layered TiO2 structure and cost-competitive solar power applications.2,11–14It is challenging to construct highly crystalline perovskite on the TiO2 layer with high reproducibility by the solution process.11–25 The irregular solvent evaporation rate, different solubility of PbI2 and CH3NH3I, and different crystal growth rate of components in the precursor solution lead to large morphological deviations of perovskite film and photovoltaic performance variations.12–25 The additive-aided dissolution of PbI2 and/or control of solvent evaporation rate have been investigated to enhance reproducibility and PCE.20–25 The solvent engineering using CH3NH3I, PbI2, and dimethyl sulfoxide (DMSO) in N,N-dimethylformamide (DMF) or gamma-butyrolactone was actively investigated.2,21,22,26,27 The adduct of CH3NH3I–PbI2–DMSO was formed due to the interaction between Lewis base (lone pair electrons on oxygen in DMSO and I− in CH3NH3I) and Lewis acid (PbI2) enhancing the solubility of PbI2.22 This approach, together with the DMF evaporation control using nonpolar diethyl ether, enhanced both the reproducibility and PCE (average: 18.3%, best: 19.7%).22 Pham et al. added lead chloride (PbCl2) with the Lewis acid–base adducts for efficient perovskite solar cells (average: 18.1%, best: 19%).27 The PbCl2 additive decreased defects and enhanced crystallinity and PCE.27
The water content in the lead halide was also systematically examined to improve solubility of PbI2 and PCE of CH3NH3PbI3-based solar cells.28 On the other hand, tin-based perovskites have been actively investigated due to the lower toxicity.29 Solvent-coordinated tin halide complexes were investigated as impurity-free precursors for tin halide perovskite-based solar cells.29
Here we investigated the pre-coordination of PbI2–DMSO powders for the planar heterojunction CH3NH3PbI3-based solar cells. The pre-coordination was carried out by the simple mechanical mixing of DMSO and PbI2 particles (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol%) using a mortar and pestle. The composition of PbI2–DMSOx (x = 0, 1, or 2) in the powder mixture was investigated by gradually increasing mechanical mixing time, and the dominant composition of PbI2–DMSO1 was obtained after 10 min mixing process. The PbI2–DMSO powders were then mixed with CH3NH3I in DMF to make CH3NH3PbI3 film by spin-application and heat treatment (100 °C). The planar TiO2 layer (50 nm) was formed on a fluorine-doped tin oxide (FTO) glass substrate using the low-temperature (≤100 °C) TiCl4 bath deposition method.2,13 For comparison, the CH3NH3PbI3 film was also synthesized by one-step blending of CH3NH3I, PbI2, and DMSO in DMF following a previously published protocol.22 The pre-coordination method resulted in better dissolution of PbI2 which was confirmed by X-ray diffraction (XRD) analysis. A larger grain size and smaller grain boundary defects, such as pin holes, were achieved leading to an increase in absorption and photoluminescence. Accordingly, the charge carrier life time was increased and better charge extraction was achieved which was confirmed by time-resolved photoluminescence (TRPL) analysis. Overall, the average open circuit voltage (Voc, 1.046 V), short-circuit current (Jsc, 22.9 mA cm−2), fill factor (FF, 73.5%), and PCE (17.6%) were increased by 2, 3, 6, and 12%, respectively, compared with the one-step blending method. The standard deviations of Voc (±0.009 V), Jsc (±0.4 mA cm−2), FF (±1.2%), and PCE (±0.37%) were decreased by 36, 13, 54, and 50%, compared with the one-step blending method, demonstrating improved reproducibility. The best FF and PCE were 75.5% and 18.2%. This indicated that the simple mechanical pre-coordination of PbI2–DMSO powders was very efficient in enhancing the dissolution of PbI2, crystallinity of CH3NH3PbI3, and photovoltaic performance of planar heterojunction perovskite solar cells prepared by the low-temperature process (≤100 °C).
:1 mol%) using a mortar and pestle. The composition of PbI2–DMSOx (x = 0, 1, or 2) in the powder mixture was investigated by gradually increasing mechanical mixing time, and the dominant composition of PbI2–DMSO1 was obtained after 10 min mixing process. The PbI2–DMSO powders were then mixed with CH3NH3I in DMF to make CH3NH3PbI3 film by spin-application and heat treatment (100 °C). The planar TiO2 layer (50 nm) was formed on a fluorine-doped tin oxide (FTO) glass substrate using the low-temperature (≤100 °C) TiCl4 bath deposition method.2,13 For comparison, the CH3NH3PbI3 film was also synthesized by one-step blending of CH3NH3I, PbI2, and DMSO in DMF following a previously published protocol.22 The pre-coordination method resulted in better dissolution of PbI2 which was confirmed by X-ray diffraction (XRD) analysis. A larger grain size and smaller grain boundary defects, such as pin holes, were achieved leading to an increase in absorption and photoluminescence. Accordingly, the charge carrier life time was increased and better charge extraction was achieved which was confirmed by time-resolved photoluminescence (TRPL) analysis. Overall, the average open circuit voltage (Voc, 1.046 V), short-circuit current (Jsc, 22.9 mA cm−2), fill factor (FF, 73.5%), and PCE (17.6%) were increased by 2, 3, 6, and 12%, respectively, compared with the one-step blending method. The standard deviations of Voc (±0.009 V), Jsc (±0.4 mA cm−2), FF (±1.2%), and PCE (±0.37%) were decreased by 36, 13, 54, and 50%, compared with the one-step blending method, demonstrating improved reproducibility. The best FF and PCE were 75.5% and 18.2%. This indicated that the simple mechanical pre-coordination of PbI2–DMSO powders was very efficient in enhancing the dissolution of PbI2, crystallinity of CH3NH3PbI3, and photovoltaic performance of planar heterojunction perovskite solar cells prepared by the low-temperature process (≤100 °C).
2. Experimental
2.1. Transparent conductive substrate-electron transport layer preparation
All chemicals were of analytical grade and used as received without further purification. The chemical bath deposition method was used to construct a planar TiO2 layer on a fluorine-doped SnO2 (FTO)-coated glass substrate (Pilkington, TEC 8). The cleaned FTO glass was immersed in 0.2 M titanium tetrachloride (TiCl4) aqueous solution at 60 °C for 65 min, washed with de-ionized water, and dried on a hot plate (100 °C, 1 h).1,13 The entire TiO2 deposition process was carried out at low temperature (≤100 °C).2.2. CH3NH3PbI3 precursor solution preparation
The CH3NH3PbI3 film was fabricated on top of the planar TiO2 layer by two different methods; one-step blending and DMSO pre-coordination (DPC) methods. For the DPC method, PbI2 powders (1.22 g, Alpha Aesar) were mechanically mixed with DMSO (207 mg, Aldrich) using a mortar and pestle (1:1 mol%). The mechanical mixing time was varied (3, 6, or 10 min). The CH3NH3I was prepared by stirring methylamine (27.8 mL, 40% in methanol, TCI) with hydroiodic acid (30 mL, 57 wt% in water, Aldrich) in a round-bottom flask (0 °C, 2 h).2 The CH3NH3I precipitate was then recovered by evaporation (50 °C for 5 h), washing with diethyl ether (30 min, 3 times), and drying under vacuum (60 °C for 24 h). Finally, the pre-coordinated PbI2–DMSO powders were stir-mixed (12 h) with CH3NH3I (0.42 g) in DMF (2 mL, Aldrich) to prepare the CH3NH3PbI3 precursor solution. For the one-step blending method, the CH3NH3I powder (0.42 g), PbI2 powder (1.22 g), and DMSO (207 mg) were simultaneously added in DMF (2 mL) (1:1:1 mol%) and stirred at room temperature for 12 h to obtain the CH3NH3PbI3 precursor solution following a previously published protocol.22
2.3. CH3NH3PbI3 film fabrication
The same procedure was used to fabricate the CH3NH3PbI3 film using the precursor solution prepared either by the one-step blending method or DPC method. The CH3NH3PbI3 precursor solution was coated on the planar TiO2 layer the by two-step spinning process (1000 rpm for 3 s and 4500 rpm for 8 s) in a N2 filled glove box. The evaporation rate of DMF was controlled by toluene dripping (0.3 mL) in the final stage (∼2 s) of spinning process.21 The final CH3NH3PbI3 film was obtained by heat treatment (100 °C) at ambient air condition for 20 min.2.4. Hole transport layer-electrode fabrication
The spiro-OMeTAD powder (72.3 mg, Solaronix) was dissolved in chlorobenzene (1.5 mL, Aldrich) with 4-tert-butylpyridine (28.8 μl, Aldrich) and Li-TFSI solution (17.5 μl, 520 mg in 1 mL acetonitrile) for the hole transporting layer precursor solution.2 The spiro-OMeTAD layer was spin-deposited (4000 rpm, 40 s) on the CH3NH3PbI3/TiO2/FTO/glass followed by drying in vacuum for 12 h. Finally, the gold electrode (∼100 nm) was deposited on the spiro-OMeTAD layer by thermal evaporation. The active solar cell area was maintained constant (0.15 cm2) using a photo mask.2.5. Characterization
The pre-coordinated PbI2–DMSO powders were investigated by Fourier transform infrared (FT-IR) spectroscopy (Bruker, IFS-66/S), XRD (Rigaku, D/MAX-2500/PC, Cu Kα radiation, λ = 1.5418 Å), differential scanning calorimetry (DSC, SEIKO, 7020, heating rate = 2 °C min−1), and thermogravimetric analysis (TGA, SEIKO, Exstar 6000). The microstructure was investigated by scanning electron microscopy (SEM, JEOL, JSM-7600F) and XRD. The UV-Vis spectroscopy (Agilent, RSA-HP-8453) was performed on the planar specimen (CH3NH3PbI3/TiO2/FTO/glass). Photoluminescence and TRPL were measured using a confocal microscope system (NTEGRA SPECTRA, NT-MDT) with an objective lens (100×, NA 0.7) and a grating (150 lines per mm, 400 nm blaze). The excitation source was a pulsed laser (405 nm, 20 MHz, 1 μW cm−2). Photoluminescence data were collected using a thermoelectrically-cooled CCD detector. TRPL data were collected with a high-speed photomultiplier tube detector (Photonic Solutions, PMC-100, integration time = 10 s) for a time-correlated single-photon-counting system. The current density–voltage (J–V) characteristics were measured using a digital source meter (Keithley 2400) under the AM 1.5 one sun illumination (Oriel, Sol3ATM, 100 mW cm−2). The voltage scan rate was 0.3 V s−1. The external quantum efficiency (EQE) was also measured (Oriel, IQE 200). The electrochemical impedance spectroscopy (EIS) was performed using a potentiostat/galvanostat (Biologic, SP-240), and the data were analyzed using a Z-fit software (EC-Lab).3. Results and discussion
A schematic of the CH3NH3PbI3 film synthesis process is shown in Fig. 1A. In contrast to the one-step blending method where PbI2, CH3NH3I, DMSO, and DMF are initially mixed at one time,21,22,27 PbI2 powders (2.65 mM) and DMSO (2.65 mM) were mechanically pre-mixed using a mortar and pestle in the DPC method. The mechanical mixing time was gradually increased (3, 6, and 10 min) to systematically investigate PbI2–DMSOx (x = 0, 1, or 2) adduct formation in the powder. The loss of DMSO was negligible during the mortar mixing process due to the low saturation vapor pressure (0.76 kPa at 60 °C) and high boiling point (189 °C).24 The thermogravimetric analysis of the pre-coordinated PbI2–DMSOx powders indicated a 14.4% weight decrease at high temperatures (>140 °C) which was consistent with the initial weight ratio between DMSO and PbI2 powders (ESI Fig. S1†).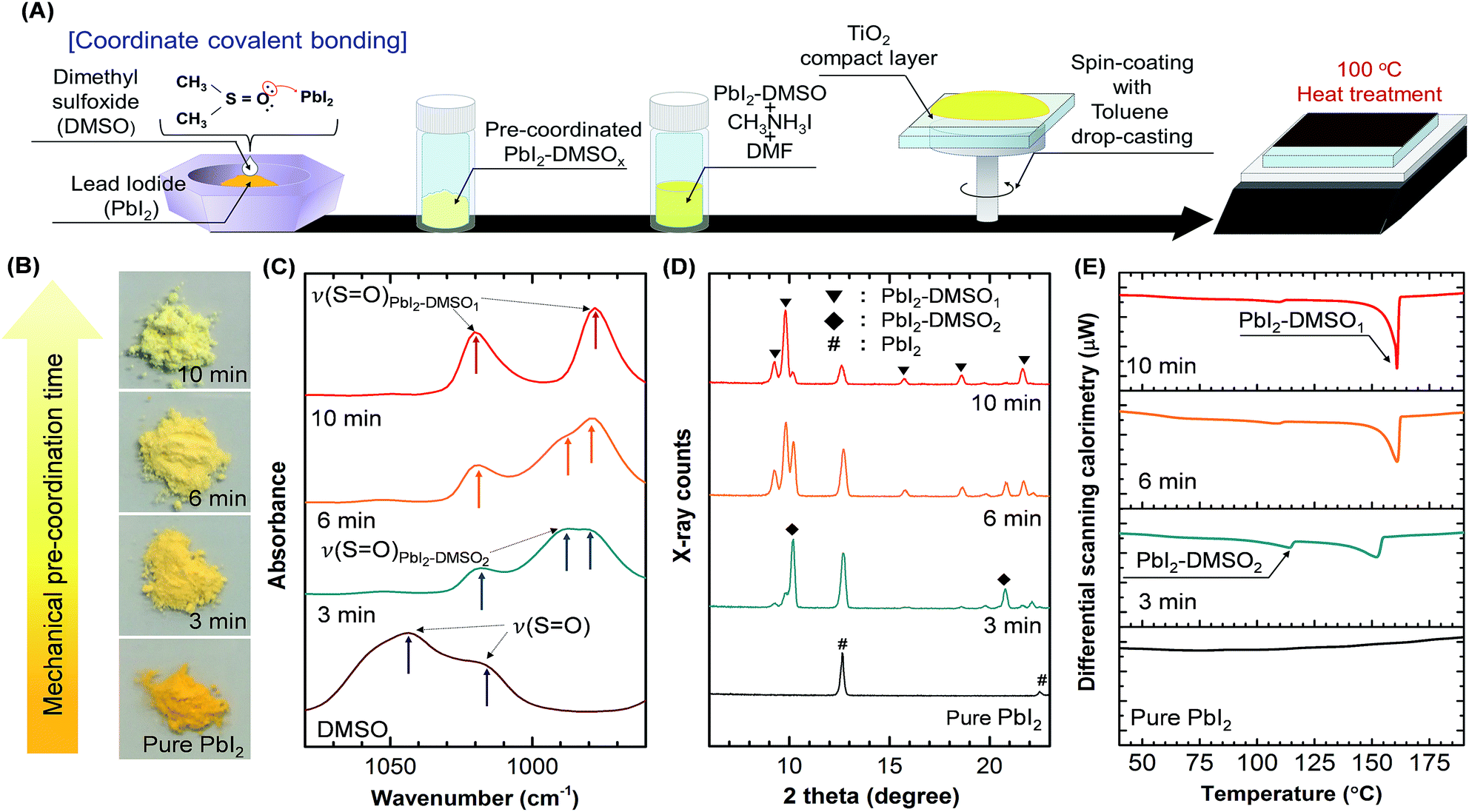 | ||
| Fig. 1 The CH3NH3PbI3 film synthesis on the planar TiO2 by the DMSO pre-coordination (DPC) method. (A) Schematic of the synthesis process. (B–E) The PbI2–DMSOx (x = 0, 1, or 2) adduct formation in the powders was investigated as a function of the mechanical mixing time by optical images (B), FT-IR (C), XRD (D), and DSC (E) analyses. | ||
Fig. 1B shows optical images of as-received PbI2 and PbI2–DMSOx adduct powders. The adduct powder exhibited lighter yellowish color with increasing mechanical mixing time. Fig. 1C compares FT-IR spectra of the powders. The sulfoxide oxygen in DMSO (electron-rich Lewis base) donates an electron pair to PbI2 (electron-deficient Lewis acid) in the PbI2–DMSO adduct formation via Pb–O coordinate covalent bonding.22,26,30 The Pb–O bond formation was confirmed by the red shift of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration.22,26,30 The ν(SO) peaks at 1020 cm−1 and 978 cm−1 correspond to PbI2–DMSO1 and the peak at 989 cm−1 indicates PbI2–DMSO2.30 The PbI2–DMSO2 peak decreased and stoichiometric PbI2–DMSO1 peaks increased as the mechanical mixing time increased. This was consistent with XRD characterization (Fig. 1D). The PbI2–DMSO2 peaks decreased as the mechanical mixing time increased. The PbI2–DMSO1 peaks became dominant after 10 min mixing. DSC analysis also revealed the dominant PbI2–DMSO1 formation as the mechanical mixing time increased (Fig. 1E). The endothermic peak at 114 °C indicates PbI2–DMSO2, and the peak at a higher temperature corresponds to PbI2–DMSO1.31 The different analytical methods consistently revealed the dominant PbI2–DMSO1 adduct formation as the mechanical mixing time increased up to 10 min in the DPC method. This left only a small amount of uncoordinated PbI2 and PbI2–DMSO2 in the powder mixture. The pre-coordinated PbI2–DMSO powders were then mixed with CH3NH3I powders in DMF by stirring. The precursor solution was then coated on a planar TiO2/FTO/glass substrate by the two-step spinning process. The evaporation rate of DMF was controlled by toluene dripping in the final stage (∼2 s) of spinning process.21 The residual solvent was quickly removed by the toluene dripping resulting in spontaneous formation of the CH3NH3I–PbI2–DMSO intermediate phase.21,22 Finally, the CH3NH3PbI3 film was cured at 100 °C in ambient air environment (20 min).12
O stretching vibration.22,26,30 The ν(SO) peaks at 1020 cm−1 and 978 cm−1 correspond to PbI2–DMSO1 and the peak at 989 cm−1 indicates PbI2–DMSO2.30 The PbI2–DMSO2 peak decreased and stoichiometric PbI2–DMSO1 peaks increased as the mechanical mixing time increased. This was consistent with XRD characterization (Fig. 1D). The PbI2–DMSO2 peaks decreased as the mechanical mixing time increased. The PbI2–DMSO1 peaks became dominant after 10 min mixing. DSC analysis also revealed the dominant PbI2–DMSO1 formation as the mechanical mixing time increased (Fig. 1E). The endothermic peak at 114 °C indicates PbI2–DMSO2, and the peak at a higher temperature corresponds to PbI2–DMSO1.31 The different analytical methods consistently revealed the dominant PbI2–DMSO1 adduct formation as the mechanical mixing time increased up to 10 min in the DPC method. This left only a small amount of uncoordinated PbI2 and PbI2–DMSO2 in the powder mixture. The pre-coordinated PbI2–DMSO powders were then mixed with CH3NH3I powders in DMF by stirring. The precursor solution was then coated on a planar TiO2/FTO/glass substrate by the two-step spinning process. The evaporation rate of DMF was controlled by toluene dripping in the final stage (∼2 s) of spinning process.21 The residual solvent was quickly removed by the toluene dripping resulting in spontaneous formation of the CH3NH3I–PbI2–DMSO intermediate phase.21,22 Finally, the CH3NH3PbI3 film was cured at 100 °C in ambient air environment (20 min).12
It is interesting to note that the PbI2–DMSO coordination was maintained even after dissolving the pre-coordinated PbI2–DMSOx powders (DPC-10 min) in DMF. As shown in ESI Fig. S2,† there was negligible difference in the TGA spectra of DPC-10 min powders before and after dissolving in DMF. The fully dissolved DPC-10 min powders in DMF were retrieved by precipitation using toluene and drying in vacuum for the TGA measurement. In contrast, PbI2 powders dissolved in DMF and retrieved by precipitation (PbI2–DMF) shows a decrease in weight concentration at lower temperatures. This clearly indicated the maintenance of PbI2–DMSO coordination in DMF solvent.
The crystallinity of perovskite films was investigated by XRD analysis (Fig. 2A), and corresponding SEM images are shown in ESI Fig. S2.† The CH3NH3PbI3 film prepared without DMSO additive exhibited a residual PbI2 (001) diffraction peak at 12.6°. The PbI2 with low solubility and CH3NH3I with high solubility resulted in the rapid crystal growth of nanorod-like PbI2 with the porous CH3NH3PbI3 morphology during the fast evaporation of solvent (i.e., DMF) as shown in ESI Fig. S2A.†20,22,25 The residual PbI2 peak was reduced in the CH3NH3PbI3 film prepared by the one-step blending method. The film crystallinity was significantly improved although a number of pin-holes were observed (ESI Fig. S2B†). The CH3NH3I–PbI2–DMSO complex enhanced solubility of PbI2 and conversion to CH3NH3PbI3 with more uniform film morphology.21–27 Interestingly, the unconverted residual PbI2 diffraction peak became negligible in the CH3NH3PbI3 film as the mechanical mixing time increased in the DPC method. The number of pin-holes also decreased with the increasing mixing time (ESI Fig. S2C–F†). The (110) peaks of perovskite films were magnified and fitted with Gaussian function (Fig. 2B).11 The full width at half maximum (FWHM) was 0.255° for the one step blending method. The FWHM decreased from 0.253° to 0.230° as the mechanical mixing time increased from 3 to 10 min in the DPC method demonstrating improved crystallinity.11 The XRD analysis indicated that the dominant PbI2–DMSO1 composition in the mechanically mixed powder (Fig. 1) resulted in highly crystalline perovskite film without leaving unconverted PbI2. There was negligible difference in the thickness (∼500 nm) of CH3NH3PbI3 films prepared by the one-step blending method and the DPC method with different mechanical mixing times (see cross-sectional SEM images in ESI Fig. S3†). The enlarged grain size of the CH3NH3PbI3 film synthesized by the DPC method could be clearly observed from SEM images (Fig. 2C–F). The corresponding grain size distribution is shown in Fig. 2G–J. Each grain area was measured from SEM images, and the grain size was defined as a diameter of a circle with an identical area of the grain. The one-step blending method produced the CH3NH3PbI3 grain size distribution from 109 to 288 nm with an average of 179.1 nm. The grain size increased as the mechanical mixing time increased in the DPC method. The average grain size was 356.8 nm with a size distribution from 173 to 506 nm after the 10 min mixing process. Larger grains, reduced grain boundaries, and pin-hole free morphology were achieved by the 10 min pre-coordinated PbI2–DMSO powders. Perovskite films prepared by the 10 min mechanical mixing process were used for the further DPC method analysis unless otherwise stated.
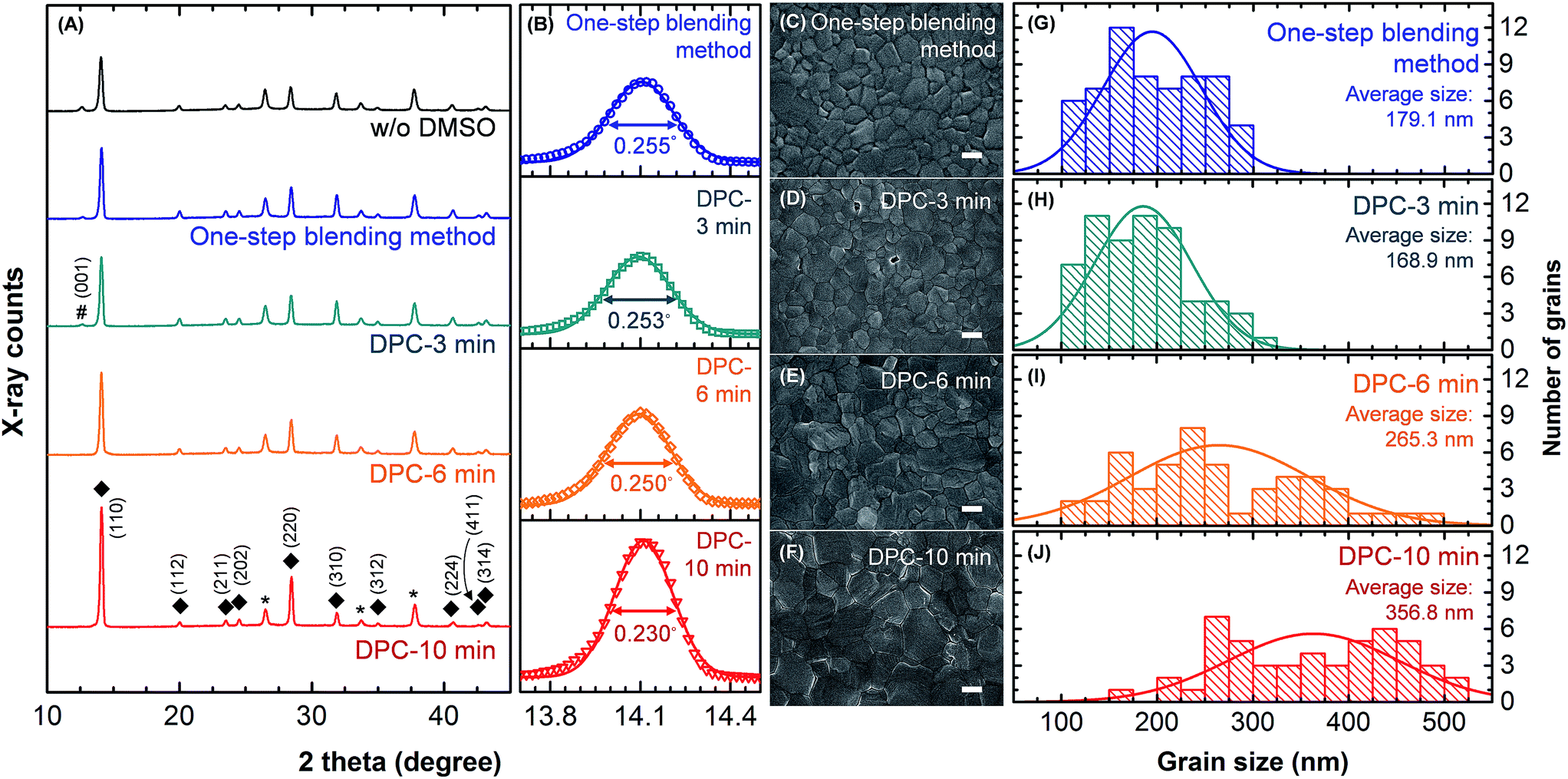 | ||
| Fig. 2 The crystallinity of CH3NH3PbI3 films prepared by the one-step blending method and DPC method (3, 6, and 10 min mechanical mixing). (A) XRD analysis. The film prepared without DMSO was also compared. The PbI2, CH3NH3PbI3, and FTO peaks are denoted by #, ◆, and *, respectively. (B) The (110) peak of CH3NH3PbI3 was magnified and fitted with Gaussian distribution function. (C–F) SEM images of the CH3NH3PbI3 films. The scale bar is 200 nm. (G–J) Histogram of the grain size distribution. | ||
Fig. 3A compares UV-Vis absorption spectra of the CH3NH3PbI3 films synthesized without DMSO, by the one-step blending method, and by the DPC method. The different synthesis method did not affect the film thickness as shown in ESI Fig. S3.† The absorption onset centered at ∼770 nm was observed similar to other CH3NH3PbI3 films.32 The film synthesized without DMSO provided the minimum absorbance. This could be due to the fast growth of nanorod-like PbI2 with surrounding large pin-holes reducing the coverage of CH3NH3PbI3 (ESI Fig. S2†). In contrast, the film synthesized by the DPC method showed the highest absorbance. The pinhole free morphology was observed for the film synthesized by the DPC method (ESI Fig. S2†) increasing the absorbance of the film. The enlarged perovskite grains also reduced the grain boundaries and defective electronic trap sites dominantly located at grain boundaries.6,31 The elimination of non-radiative recombination levels within the band gap, caused by removal of the defective sites at grain boundaries, enhanced photoluminescence intensity (DPC-10 min) as shown in Fig. 3B. The quenching of photoluminescence by the defective sites at grain boundaries was also reported in literature.6,32,34
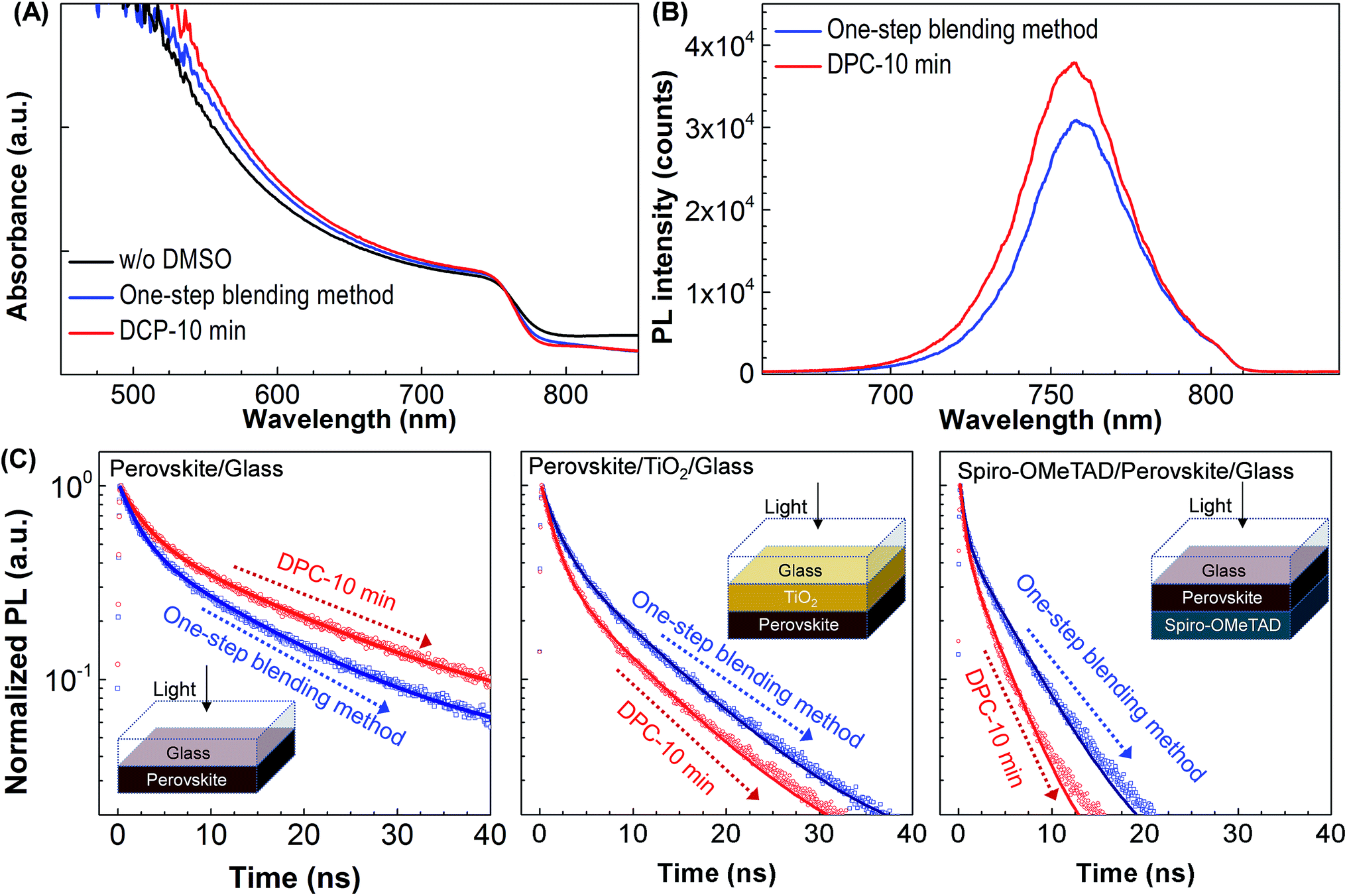 | ||
| Fig. 3 Optical characteristics of the CH3NH3PbI3 films prepared by the one-step blending and DPC methods. (A, B) UV-Vis absorption and photoluminescence spectra (perovskite/TiO2/FTO/glass). (C) TRPL characteristics of perovskite/glass, perovskite/TiO2/glass, and spiro-OMeTAD/perovskite/glass. The specimen was excited from the glass side by a pulsed laser (20 MHz, 407 nm, 1 μW cm−2). | ||
TRPL spectroscopy was employed to investigate the carrier recombination life time which is related with the charge diffusion length.32,35 The TRPL spectra of the perovskite/glass configuration is shown in Fig. 3C.6 The TRPL data were fitted using a bi-exponential decay function to calculate the average life time.6
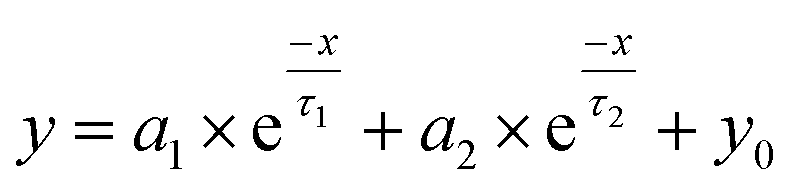 | (1) |
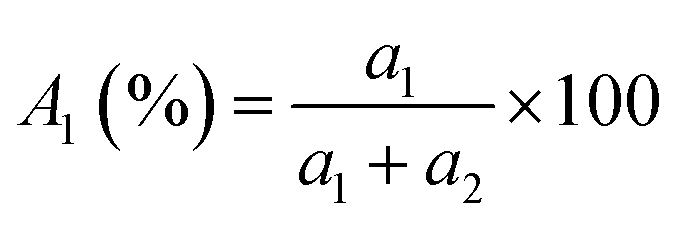 | (2) |
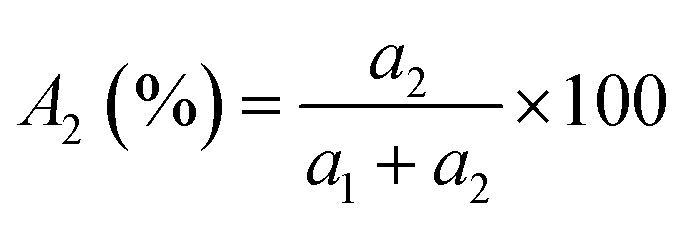 | (3) |
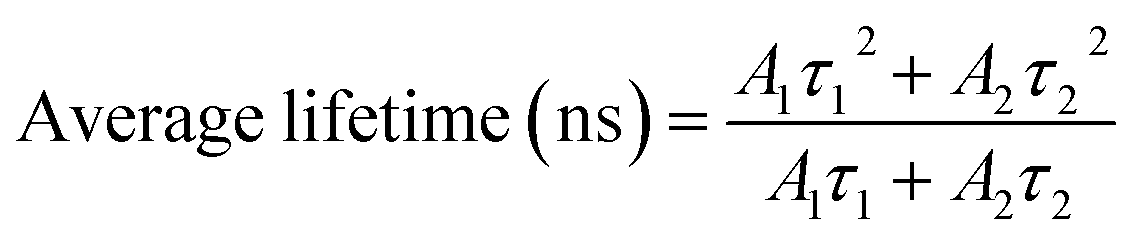 | (4) |
The detailed fitting parameters are provided in ESI Table S1.† The fast decay time constant (τ1) is closely related with the non-radiative recombination by defective charge trap sites at grain boundaries, and the slow decay time constant (τ2) is related with the intrinsic radiative recombination in bulk perovskite crystal.6 The CH3NH3PbI3 film synthesized by the DPC method exhibited a longer τ1 (2.51 ns) implying reduced non-radiative recombination at grain boundaries and a longer τ2 (17.08 ns) due to the improved bulk perovskite crystallinity.6 Overall, the average carrier lifetime (15.34 ns) was 29% longer than that (11.93 ns) of the perovskite film synthesized by the one-step blending method. The perovskite film with an electron transporting layer (perovskite/TiO2/glass) or a hole transporting layer (spiro-OMeTAD/perovskite/glass) showed much faster quenching behavior (Fig. 3C). Compared with the CH3NH3PbI3 film synthesized by the one-step blending method, all the time constants (τ1, τ2, and average life time) were smaller for the perovskite film synthesized by the DPC method either with the electron or hole transporting layer (ESI Table S1†). This indicated a more efficient charge extraction due to the stronger interfacial coupling with the electron and hole transporting layer.30
The increased carrier life time in the perovskite and efficient charge carrier extraction with the transporting layers resulted in enhanced photovoltaic performance as shown in Fig. 4. Fig. 4A compares the best J–V curves of perovskite solar cells synthesized by the one-step blending and DPC methods. The J–V data were measured at reverse scan direction. All the photovoltaic parameters (Jsc = 22.9 mA cm−2, Voc = 1.053 V, FF = 75.5%, and PCE = 18.2%) were improved by the DPC method compared with those (Jsc = 22.3 mA cm−2, Voc = 1.037 V, FF = 72.7%, and PCE = 16.9%) obtained by the one-step blending method. Fig. 4B shows the EQE spectrum of the best performing solar cells. The DPC method possessed a greater integrated Jsc, compared with the one-step blending method, which is consistent with the J–V data in Fig. 4A. The hysteresis between the forward and reverse scan was decreased by the DPC method (ESI Fig. S6†). The decrease in charge trap sites, including pinholes, typically located at grain boundaries contributed to the decrease in hysteresis.33,36,37 The average values and standard deviations of Voc, Jsc, FF, and PCE from 30 devices are compared in Fig. 4C. The DPC method showed a clear enhancement in photovoltaic performance (Voc = 1.046 ± 0.009 V, Jsc = 22.9 ± 0.40 mA cm−2, FF = 73.5 ± 1.2%, and PCE = 17.6 ± 0.37%), compared with the one-step blending method (Voc = 1.027 ± 0.014 V, Jsc = 22.30 ± 0.46 mA cm−2, FF = 69.1 ± 2.6%, and PCE = 15.8 ± 0.74%). The average Voc, Jsc, FF, and PCE values were increased by 2, 3, 6, and 12%, respectively. The standard deviations of Voc, Jsc, FF, and PCE were decreased by 36, 13, 54, and 50% demonstrating enhanced reproducibility. The reduced grain boundary defects including pin holes enabled reproducible photovoltaic performance of perovskite solar cells.21–25
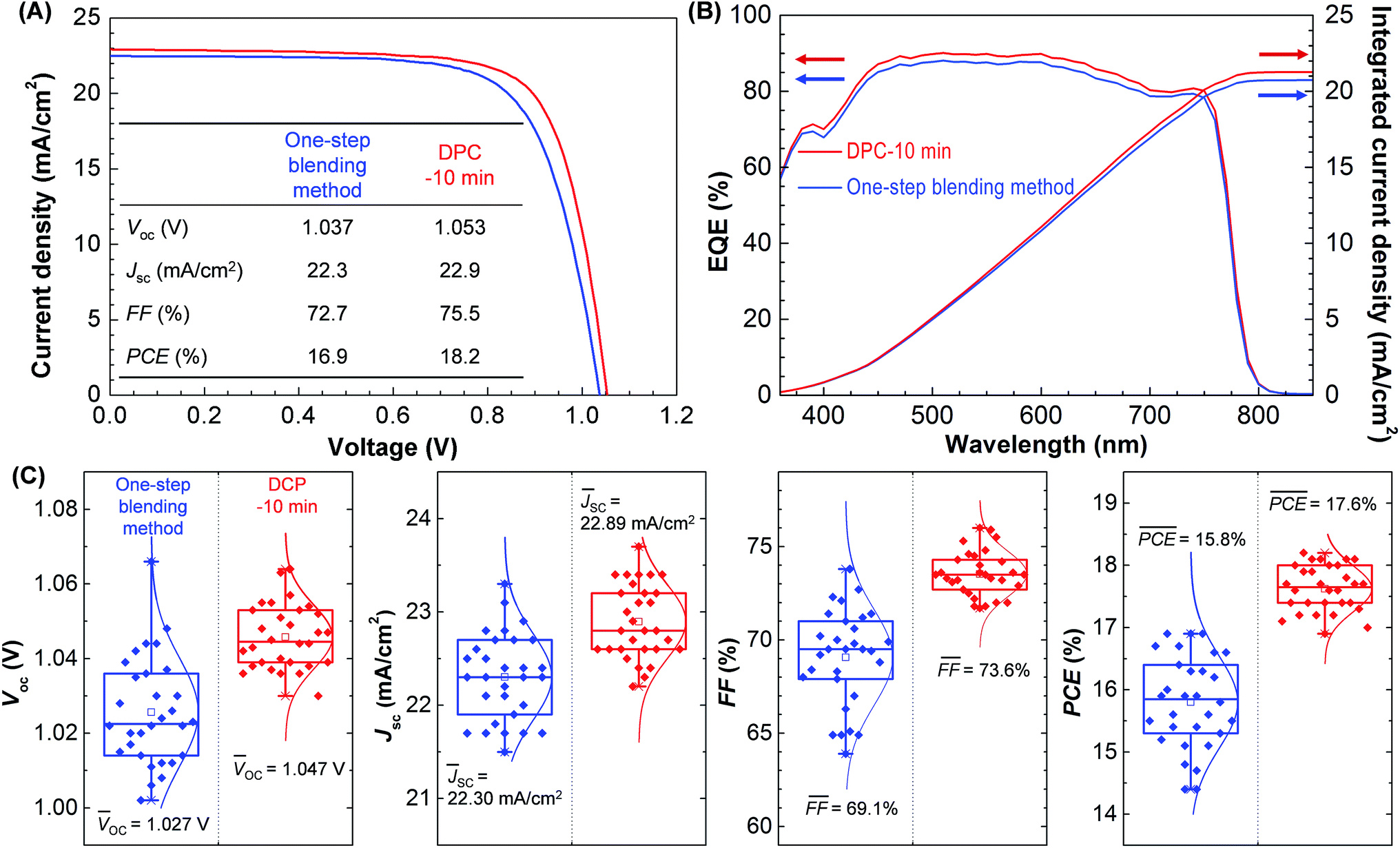 | ||
| Fig. 4 Current density–voltage (J–V) characteristics of perovskite solar cells synthesized by the one-step blending and DPC methods. (A, B) The best J–V curve and external quantum efficiency (EQE) spectrum with integrated Jsc obtained by each method. (C) The average values and standard deviations of Voc, Jsc, FF, and PCE from 30 devices. | ||
The nominal composition of CH3NH3I, PbI2, DMSO, and DMF was same in the precursor solutions for the one-step blending method and DPC method. However, the dominant PbI2–DMSO1 formation, after the mechanical mixing process in the DPC method, left only a small amount of uncoordinated PbI2 and PbI2–DMSO2 in the powder mixture (Fig. 1). This resulted in highly crystalline CH3NH3PbI3 film with negligible residual PbI2, negligible pin-holes, and large grains (Fig. 2). Consequently, the photovoltaic performance was enhanced compared with the one-step method (Fig. 4). Among the solar cells prepared by the DPC method, the photovoltaic performance was systematically enhanced from 15.3 to 18.2% as the mechanical mixing time increased from 3 to 10 min (ESI Fig. S6†). The average PCE also increased with a decrease in standard deviation. A longer mechanical mixing time (15 min) in the DPC method did not further improve the photovoltaic performance. It is interesting to note that an increased stirring time from 12 to 36 hours in the one-step blending method did not further increase the power conversion efficiency (ESI Fig. S7†). This indicated the importance of forming PbI2–DMSO1 in the powder mixture by the DPC method for the enhancement of photovoltaic performance.
As shown in Fig. 5, a decrease in series resistance (Rs) and an increase in shunt resistance (Rsh) were observed with smaller deviations for the solar cells fabricated by the DPC method. Rs and Rsh were calculated using the lumped equivalent circuit model.2,38,39
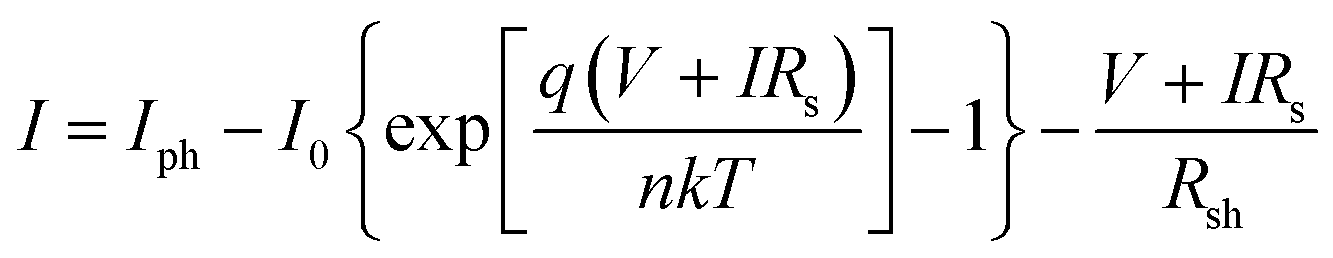 | (5) |
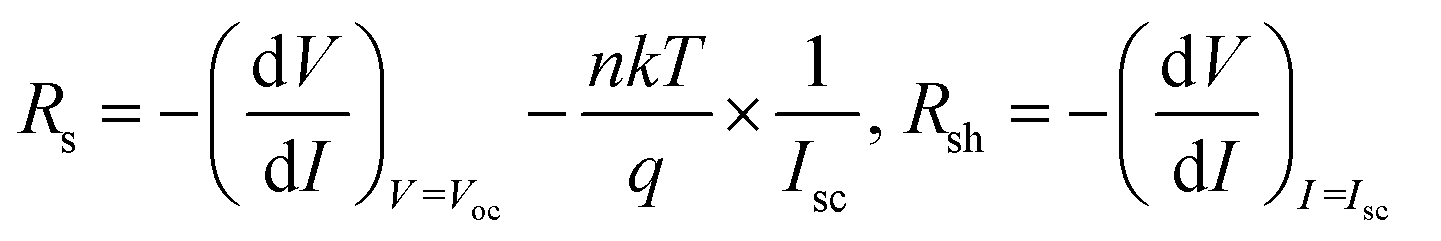 | (6) |
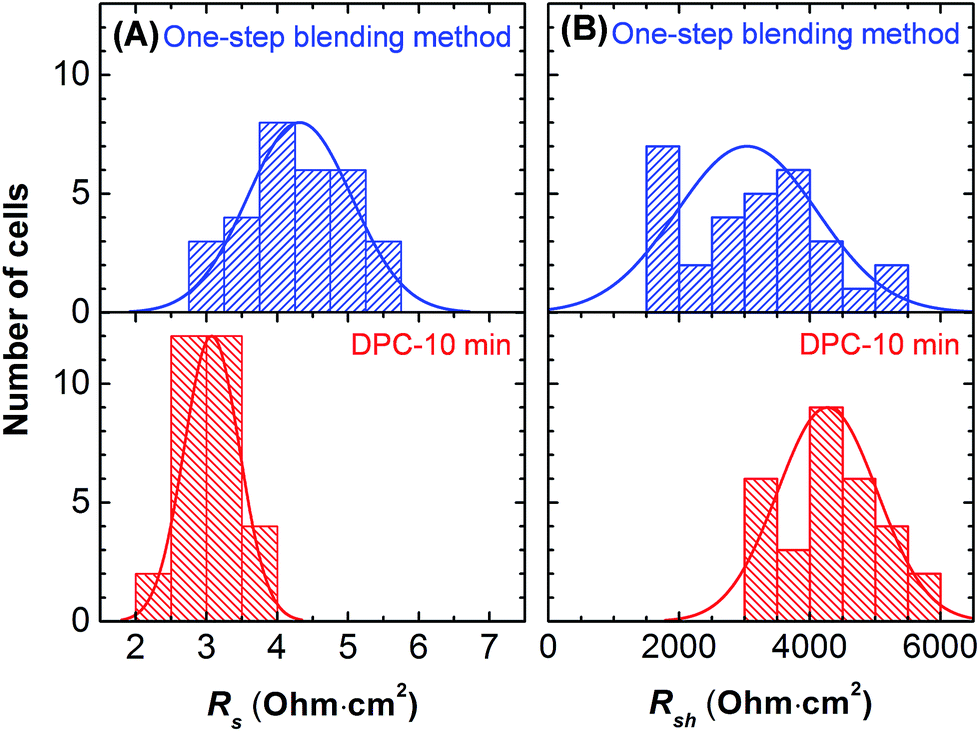 | ||
| Fig. 5 The series (A) and shunt resistance (B) of perovskite solar cells prepared by the one-step blending and DPC methods. Thirty devices were fabricated for each type. | ||
A more efficient charge carrier extraction with the transporting layers (Fig. 3C) resulted in a decrease in Rs. The increase in Rsh was attributed to the reduced grain boundary with pinhole free morphology (Fig. 2 and ESI S2†).37 The dense CH3NH3PbI3 film increased absorbance (Fig. 3a) and reduced recombination sites. Overall, this lead to an impressive increase in FF and PCE.
The EIS analysis was performed to measure the recombination resistance (Rrec).27,41–43 Nyquist plots for the DPC and one-step blending methods were compared at the applied bias of 500, 750, and 1000 mV as shown in Fig. 6A. The frequency range was 1–106 Hz. The arc feature consists of two semicircles. The high frequency semicircle corresponds to the resistance of hole transport materials (RHTM), and the dominant low frequency feature is related with Rrec.27,42,43 The arc decreased as the applied bias increased. The DPC method exhibited a larger arc than the one-step blending method. Rrec was calculated using the equivalent circuit model shown in the inset of Fig. 6A. The same equivalent circuit model has been widely employed to describe the EIS data of perovskite solar cells prepared by the solution process.27,41–43 There was an excellent fitting with the experimental data, and key fitting parameters are provided in Table 1. Compared with the one-step blending method, the DPC method exhibited enhanced Rrec at the entire bias range indicating suppressed charge recombination (Fig. 6B). This contributed to the higher Jsc and Voc in the DPC method.44 Rrec started to decrease over 0.7 V, and the slope was steeper for the DPC method. This indicated a lower diode ideality factor for the DPC method enhancing FF.43,45 Overall, the EIS analysis was consistent with the J–V characteristics (Fig. 4).
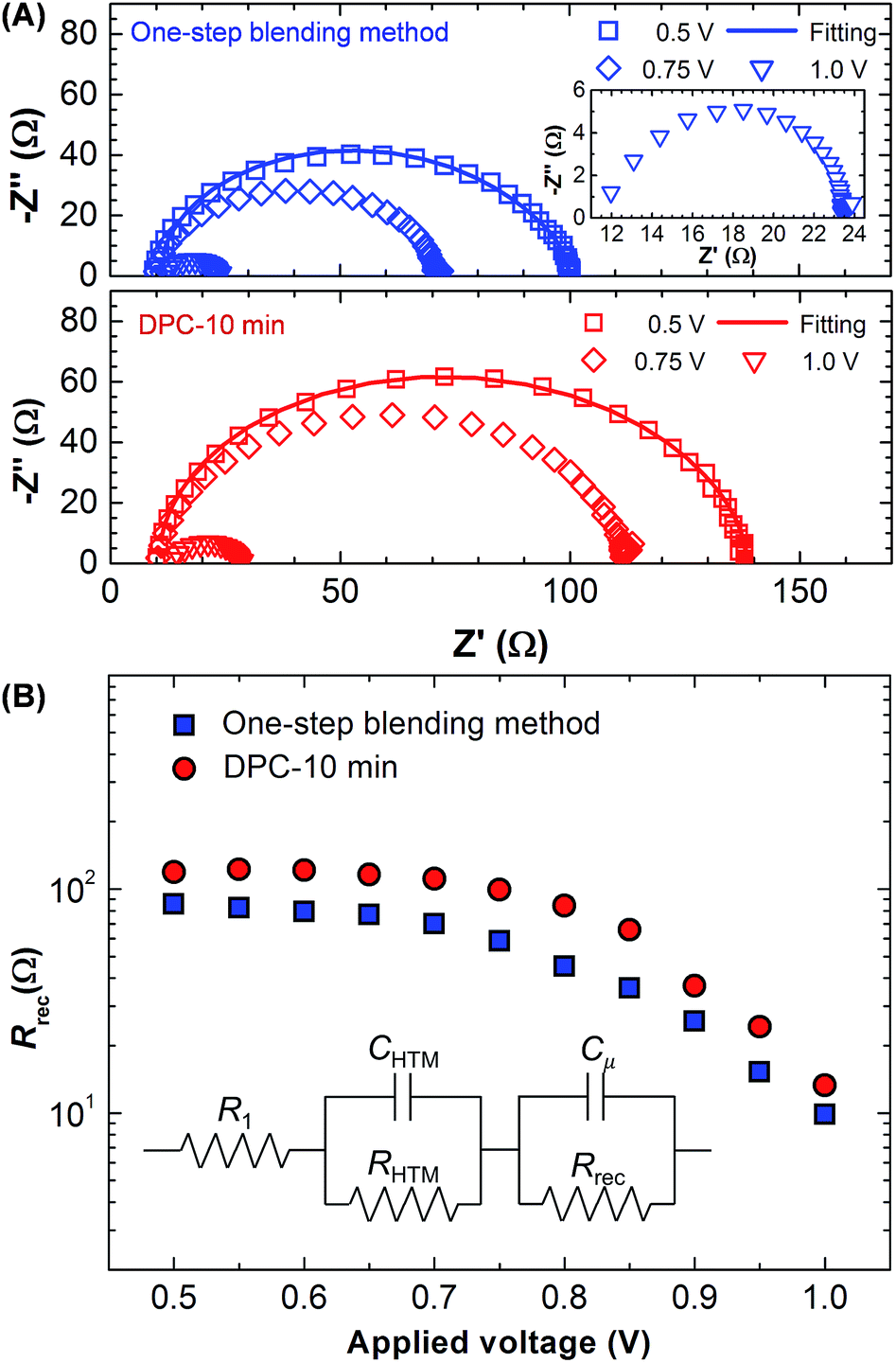 | ||
| Fig. 6 Electrochemical impedance spectroscopy of perovskite solar cells prepared by the one-step blending and DPC methods. (A) Nyquist plots measured at the applied bias of 500, 750, and 1000 mV (100 mW cm−2 illumination). The low impedance region is magnified in the inset. (B) The recombination resistance is shown as a function of the applied bias. The equivalent circuit is provided in the inset. R1 is the series resistance of cell, Cμ is the chemical capacitance, and Rrec is the recombination resistance. CHTM and RHTM are the capacitance and resistance of hole transport materials. | ||
| Method | R1 (Ω) | RHTM (Ω) | CHTM (μF) | Rrec (Ω) | Cμ (μF) |
|---|---|---|---|---|---|
| One-step | 10.85 | 9.79 | 1.31 | 79.1 | 0.03 |
| DPC-10 min | 10.48 | 8.72 | 1.57 | 119.1 | 0.03 |
The statistical photovoltaic performance analysis demonstrated a clear advantage of the pre-coordinated PbI2–DMSO powders prepared by the simple mechanical mixing method. A further optimization of the TiO2 structures, solvents, materials, and detailed processing parameters may enhance the photovoltaic performance of solar cells fabricated by the DPC method in the future.
4. Conclusions
The pre-coordinated PbI2–DMSO powders enhanced the photovoltaic performance of planar heterojunction perovskite solar cells fabricated by the low temperature process (≤100 °C). The pre-coordination was carried out by the simple mechanical mixing of DMSO and PbI2 particles (1:1 mol%) using a mortar and pestle. The dominant composition of PbI2–DMSO1 was obtained after the 10 min mixing process. The PbI2–DMSO powders were then mixed with CH3NH3I in DMF to make the CH3NH3PbI3 film by the toluene-assisted spin-coating and heat treatment. This method resulted in better dissolution of PbI2, larger grain size, and pinhole free morphology. The TRPL analysis revealed an increased charge carrier life time and better charge extraction. Overall, the average Voc (1.046 V), Jsc (22.9 mA cm−2), FF (73.5%), and PCE (17.6%) were enhanced by 2, 3, 6, and 12%, respectively, compared with the one-step mixing method. The standard deviations were decreased indicating improved reproducibility. The simple mechanical pre-coordination of PbI2–DMSO powders effectively enhanced crystallinity of CH3NH3PbI3 leading to the enhanced photovoltaic performance with the best PCE of 18.2%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2017R1A2A1A17069289), Ministry of Trade Industry & Energy (10048884), and the Fundamental Technology Research Program (2014M3A7B4052200) through the NRF grants funded by Korean government (MSIP).Notes and references
- Q. Zheng, H. Kang, J. Yun, J. Lee, J. H. Park and S. Baik, ACS Nano, 2011, 5, 5088–5093 CrossRef CAS PubMed.
- J. Lee, M. M. Menamparambath, J. Y. Hwang and S. Baik, ChemSusChem, 2015, 8, 2358–2362 CrossRef CAS PubMed.
- J. H. Heo, S. H. Im, J. H. Noh, T. N. Mandal, C.-S. Lim, J. A. Chang, Y. H. Lee, H. Kim, A. Sarkar, M. K. Nazeeruddin, M. Grätzel and S. I. Seok, Nat. Photonics, 2013, 7, 486–491 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- D. Y. Son, J. W. Lee, Y. J. Choi, I. H. Jang, S. Lee, P. J. Yoo, H. Shin, N. Ahn, M. Choi, D. Kim and N. G. Park, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- M. Saliba, T. Matsu, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zekeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 CAS.
- J. W. Lee, D. J. Seol, A. N. Cho and N. G. Park, Adv. Mater., 2014, 26, 4991–4998 CrossRef CAS PubMed.
- D. Zhong, B. Cai, X. Wang, Z. Yang, Y. Xing, S. Miao, W. H. Zhang and C. Li, Nano Energy, 2015, 11, 409–418 CrossRef CAS.
- M. S. Seo, I. Jeong, J. S. Park, J. Lee, I. K. Han, W. I. Lee, H. J. Son, B. H. Sohn and M. J. Ko, Nanoscale, 2016, 8, 11472–11479 RSC.
- Q. Chen, H. Zhou, Y. Fang, A. Z. Stieg, T. B. Song, H. H. Wang, X. Xu, Y. Liu, S. Lu, J. You, P. Sun, J. McKay, M. S. Goorsky and Y. Yang, Nat. Commun., 2015, 6, 7269 CrossRef CAS PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T. Song, H. S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- A. Yella, L. P. Heinger, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nano Lett., 2014, 14, 2591–2596 CrossRef CAS PubMed.
- H. Kim, K. G. Lim and T. W. Lee, Energy Environ. Sci., 2016, 9, 12–30 CAS.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- Q. Chen, H. Zhou, Z. Hong, S. Luo, H. S. Duan, H. H. Wang, Y. Liu, G. Li and Y. Yang, J. Am. Chem. Soc., 2014, 136, 622–625 CrossRef CAS PubMed.
- S. Bai, N. Sakai, W. Zhang, Z. Wang, J. T. W. Wang, F. Gao and H. J. Snaith, Chem. Mater., 2017, 29, 462–473 CrossRef CAS.
- G. E. Eperon, V. M. Burlakov, P. Docampo, A. Goriely and H. J. Snaith, Adv. Funct. Mater., 2014, 24, 151–157 CrossRef CAS.
- B. Conings, L. Baeten, C. D. Dobbelaere, J. D'Hean, J. Manca and H. G. Boyer, Adv. Mater., 2014, 26, 2041–2046 CrossRef CAS PubMed.
- Y. J. Jeon, S. Lee, R. Kang, J. E. Kim, J. S. Yeo, S. H. Lee, S. S. Kim, J. M. Yun and D. Y. Kim, Sci. Rep., 2014, 4, 6953 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- N. Ahn, D. Y. Son, I. H. Jang, S. M. Kang, M. Choi and N. G. Park, J. Am. Chem. Soc., 2015, 137, 8695–8699 Search PubMed.
- W. Li, J. Fan, J. Li, Y. Mai and L. Wang, J. Am. Chem. Soc., 2015, 137, 10399–10405 CrossRef CAS PubMed.
- Y. Wu, A. Islam, X. Yang, C. Qin, J. Liu, K. Zhang, W. Peng and L. Han, Energy Environ. Sci., 2014, 7, 2934–2938 CAS.
- X. Cao, C. Li, Y. Li, F. Fang, X. Cui, Y. Yao and J. Wei, Nanoscale, 2016, 8, 19804–19810 RSC.
- X. Li, D. Bi, C. Yi, J. D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58–62 CrossRef CAS PubMed.
- N. D. Pham, V. T. Tiong, P. Chen, L. Wang, G. J. Wilson, J. Bell and H. Wang, J. Mater. Chem. A, 2017, 5, 5195–5203 CAS.
- A. Wakamiya, M. Endo, T. Sasamori, N. Tokitoh, Y. Ogomi, S. Hayase and Y. Murata, Chem. Lett., 2014, 43, 711–713 CrossRef CAS.
- M. Ozaki, Y. Katsuki, J. Liu, T. Handa, R. Nishikubo, S. Yakumaru, Y. Hashikawa, Y. Murata, T. Saito, Y. Shimakawa, Y. Kanemitsu, A. Saeki and A. Wakamiya, ACS Omega, 2017, 2, 7016–7021 CrossRef CAS.
- I. Wharf, T. Gramstad, R. Makhija and M. Onyszchuk, Can. J. Chem., 1976, 54, 3430–3438 CrossRef CAS.
- J. Muhammad, J. H. Heo, C. E. Song, K. Kong, W. S. Shin, J. Lee, S. H. Im and S. Moon, Nano Energy, 2016, 27, 330–339 CrossRef.
- C. Roldán-Carmona, P. Gratia, I. Zimmermann, G. Grancini, P. Gao, M. Grätzel and M. K. Nazeeruddin, Energy Environ. Sci., 2015, 8, 3550–3556 Search PubMed.
- J. Xu, A. Buin, A. H. Ip, W. Li, O. Voznyy, R. Comin, M. Yuan, S. Jeon, Z. Ning, J. J. Mcdowell, P. Kanjanaboos, J. P. Sun, X. Lan, L. N. Quan, D. H. Kim, L. G. Hill, P. Maksymovych and E. H. Sargent, Nat. Commun., 2015, 6, 7081 CrossRef CAS PubMed.
- F. Wang, A. Shimazaki, F. Yang, K. Kanahashi, K. Matsuki, Y. Miyauchi, T. Takenobu, A. Wakamiya, Y. Murata and K. Matsuda, J. Phys. Chem. C, 2017, 121, 1562–1568 CAS.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- W. Nie, H. Tsai, R. Asadpour, J. C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam, H. L. Wang and A. D. Mohite, Science, 2015, 347, 522–525 CrossRef CAS PubMed.
- W. Qiu, T. Merckx, M. Jaysankar, C. M. Huerta, L. Rakocevic, W. Zhang, U. W. Paetzold, R. Gehlhaar, L. Froyen, J. Poortmans, D. Chenyns, H. J. Snaith and P. Heremans, Energy Environ. Sci., 2016, 9, 484–489 CAS.
- J. Lee, H. Kang, J. Y. Hwang, S. W. Kim and S. Baik, Carbon, 2014, 79, 337–345 CrossRef CAS.
- N. Koide, A. Islam, Y. Chiba and L. Han, J. Photochem. Photobiol., A, 2006, 182, 296–305 CrossRef CAS.
- J. H. Im, I. H. Jang, N. Pellet, M. Grätzel and N. G. Park, Nat. Nanotechnol., 2014, 9, 927–932 CrossRef CAS PubMed.
- C. Chen, Y. Zhai, F. Li, F. Tan, G. Yue, W. Zhang and M. Wang, J. Power Sources, 2017, 341, 396–403 CrossRef CAS.
- H. Kim, J. Lee, N. Yantara, P. P. Boix, S. A. Kulkarni, S. Mhaisalkar, M. Grätzel and N. G. Park, Nano Lett., 2013, 13, 2412–2417 CrossRef CAS PubMed.
- J. A. Chritians, R. C. M. Fung and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 758–764 CrossRef PubMed.
- F. Fabregat-Santiago, G. Garcia-Belmonte, I. Mora-Seró and J. Bisquert, Phys. Chem. Chem. Phys., 2011, 13, 9083–9118 RSC.
- Y. Yang, K. Ri, A. Mei, L. Liu, M. Hu, T. Liu, X. Li and H. Han, J. Mater. Chem. A, 2015, 3, 9103–9107 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA characteristics, SEM images, TRPL analysis, and additional photovoltaic performance data. See DOI: 10.1039/c7ra12304c |
| This journal is © The Royal Society of Chemistry 2018 |