
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomimetic silica deposition promoted by sub-5 μm complexes of dicarboxylic acids/polyethyleneimine microballs: a new approach to tuning silica structures using messenger-like dicarboxylic acids†
Daiki Soma and
Ren-Hua Jin *
*
Department of Material and Life Chemistry, Kanagawa University, 3-2-7 Rokkakubashi, Yokohama 221-8686, Japan. E-mail: rhjin@kanagawa-u.ac.jp
First published on 20th December 2017
Abstract
Acid–base complexes prepared from sub-5 μm polyethyleneimine (PEI) microballs and dicarboxylic acids such as adipic acid (AA), succinic acid (SA), meso-tartaric acid (m-TA) as well as mucic acid (MA) were used as catalytic templates in hydrolytic condensation of tetramethoxy silane (TMOS). By means of FT-IR, 13C NMR, 29Si NMR, XRD, SEM, TGA and nitrogen sorption isotherms, we thoroughly investigated the effects of the dicarboxylic acids complexed with PEI microballs on the resultant silica structures. We found that in this silica deposition process, the presence of the dicarboxylic acids of adipic acid (AA) and succinic acid (SA), which do not have alcoholic groups, could adapt to deposit high content of silica than the alcoholic acids of meso-tartaric acid (m-TA) and mucic acid (MA). More interestingly, the surface structures of the resultant silica microballs were different with different types of acids. The presence of m-TA and MA with meso-type alcoholic structures produced nanofibre or nanoplate-covered silica microballs, while the presence of AA and SA led to the formation of nanoparticles-covered silica microballs. Accompanying these structural features, the BET surface areas of the four types of acid-mediated silica microballs after calcination at 800 °C appeared remarkably different with the order of 615 > 430 > 133 > 96 m2 g−1, respectively, corresponding to the contribution of MA, m-TA, SA and AA. These results evidently indicate that the structures of the dicarboxylic acids associated with PEI microballs play a messenger-like role to tune the silica structures.
Introduction
Silica is one of the major inorganic metal oxides, which is widely used for its designed and structured morphologies for various applications, such as catalytic supports, adsorbents, drug vehicles, coatings and optical materials.1 Control of silica structure, studied from biosilica, has attracted much attention in the field of material chemistry.2 Sumper et al. revealed that silaffin isolated from diatom biosilica has two functional groups of phosphoric acid and a short alkyl amine chain which act as a catalyst to promote rapid silica deposition under neutral and arbitrary conditions.3 Wallace et al. reported that silica deposition from silica sources favourably progresses on an acid–base interface formed between the surfactants of alkyl amine and alkyl carboxylic acid than a single basic surface of alkyl amine.4 Such acid–base cooperated effect also can be observed in case of polymers-templated silica deposition. Kuno et al. compared the silica deposition ability of alternative copolypeptide and block copolypeptide possessing different side groups of the carboxylic acid and the amine, and confirmed that the copolypeptide with an alternative arrangement of –COOH/–NH2 is more active than that with a block structure of polyacid/polyamine.5 They called this phenomenon as an electric charge relay effect. It appears that the presence of an interface or an ordered domain of acid–base is extremely important in promoting silica deposition.Previously, we reported that polyethyleneimines (PEI) with only secondary amine in the main chain serves as catalytic templates to obtain tube-, ribbon-, fibre-, and sheet-like nanostructured silica in silicification.6–8 Furthermore, we also found that PEI complexed with tartaric acid and glucarate enantiomers produced chiral silica nanofibres by transferring chiral information of tartaric acid and glucarate to the silica frame.9 Of course, without the help of carboxylic acids, PEI itself is sufficient to catalyze the silica mineralization of silica sources. However, the finding that chiral tartaric acid and glucarate promote the formation of chiral structures in silica frame makes us believe that the role of the acid-components in complexes of PEI/acids is not only limited to participation of polymerization of siloxane monomers as co-catalysts, but also in transfer of structural information (structure carrier), such as structural “messengers”. Unfortunately, there are few reports on the relations between the formation of silica and the structures of acidic compounds in acid–base complexes consisting of polyamines and organic acids.
Soluble linear macromolecules of PEI with different structural dimensions are effective candidates to design and direct inorganic materials of various micro-scale bundles and morphologies, but with precise unit nanostructures since the PEIs easily self-organize in the employed mediation conditions affording catalytic templates for hydrolytic condensation of silica and titania sources.9b,10 However, in many cases, the crystallization-driven self-assemblies of the soluble PEIs grow to form irregular larger bundles from aggregation of nano-objects. The formation of such bundles is disadvantageous in the tailor-made regular and definite size silica or titania with an internal nanostructure. We recently reported a unique sub-5 μm polystyrene microgel grafting PEI brush (μ-PSt-g-PEI), from which silica microballs with different internal structures were templated.11 This microgel of μ-PSt-g-PEI is tightly filled with numerous PEI chains, which are also able to form a crystalline domain in the restricted sub-5 μm spherical space. In other words, the PEI chains in the sub-5 μm spherical space could self-assemble on the internal spheres. In particular, in any case, the self-assembly on the internal spheres does not exceed the spherical sub-5 μm boundary and thus can act as shape-spherical and size-definite templates to direct various targeting inorganic products. In the silica deposition using only μ-PSt-g-PEI, we found that different mediation conditions of solvents resulted in silica micro-balls with different internal structures indicating the influence of mediation conditions on the silica microballs. We think that the restricted space of sub-5 μm basic microballs is an ideal micro place to judge the structure-directing factors and to design controllable silica microballs. In the biomimetic silica deposition, carboxylates associated with polyamines are usually considered as co-catalysts with polyamines. However, directing silica structures is also an important role of carboxylates as indicated in our chiral silica studies using asymmetric tartaric acid and glucarate.9a,9d If the carboxylic acids entrapped into the space-confined basic microballs form complexes with PEI chains, two possible complexes can be expected. One is a crystalline complex formed between PEI and carboxylic acids; the other is a non-crystalline complex possessing a certain conformational structure. Nevertheless, the complexed microballs would be capable of being used as catalytic templates in silica deposition to tune the resultant silica. In order to understand the effect of carboxylic acids on the silica deposition on the restricted micro space of μ-PSt-g-PEI, herein, we prepared acid–base complexes by complexation of sub-5 μm microballs of μ-PSt-g-PEI with four types of dicarboxylic acids (adipic acid (AA), succinic acid (SA), meso-tartaric acid (m-TA) and mucic acid (MA)) and used them as catalytic templates for silica mineralization via hydrolytic condensation of tetramethoxysilane (TMOS). The silica microballs obtained as-synthesized and after calcined states were characterized, from which a new insight for prompting silica structures was presented.
Experimental
Materials
4-Chloromethylstyrene (CMS, >90%, TCI) was purified by column chromatography with basic aluminum oxide (Aldrich) before use. 2-Methyl-2-oxazoline (MOZ, 98%, Aldrich) was distilled from sodium hydroxide under atmospheric pressure and then stored under nitrogen atmosphere prior to use. Divinylbenzene (DVB, m- and p-mixture, >50%, TCI) was extracted from sodium hydroxide aqueous solution (1 M) to remove polymerization inhibitor. Azobis(isobutyronitrile) (AIBN, >98%, TCI) was crystallized from ethanol. Potassium iodide (KI, >99.5%, Wako), tetramethoxysilane (TMOS, >98%, TCI), dimethylacetamide (DMAc, super dehydrated, >99.5%, Wako), dimethylformamide (DMF, >99.5%, Wako), polyvinylpyrrolidone K25 (PVP, Wako), meso-tartaric acid monohydrate (>98%, TCI), mucic acid (>98%, TCI), succinic acid (>98%, TCI), adipic acid (>98%, TCI), hydrochloric acid (5 M, Wako), ammonium solution (28 vol%, Wako) and other reagents were used without further purification. Commonly used solvents were obtained from commercial resources.Preparation of poly(chloromethylstyrene-divinylbenzene) microgel (μ-PStCl)
According to our previous report, the dispersion polymerization was performed as follows. To a 100 mL three-neck round-bottom flask equipped with a condenser, ethanol (solvent, 50 mL), DMAc (co-solvent, 3 mL), and polyvinylpyrrolidone (PVP, surfactant, 0.6 g) were added and stirred at room temperature until complete dissolution of PVP. Then, divinyl benzene (DVB, cross-linker, 25 μL) and chloromethylstyrene (CMS, monomer, 1.4 g) were added, followed by nitrogen bubbling through the solution for 30 min to exclude the dissolved oxygen. Following this, after being heated to 60 °C, AIBN (initiator, 0.03 g) dissolved in 2 mL of ethanol was added and then, the solution stirred for 24 h. After polymerization, the obtained suspension products were collected by centrifugation (4000 rpm, 5 min), washed with methanol to remove the excess amount of PVP and CMS, and dried under reduced pressure at 50 °C for 6 h. White powders of the μ-PStCl (0.8 g) were obtained (yield: 57%).Synthesis of microballs grafting poly(2-methyl-2-oxazoline) (μ-PSt-g-PMOZ)
Polymerization of MOZ was performed using μ-PStCl as a solid initiator as follows. μ-PStCl 0.2 g (–CH2Cl unit: 1.2 mmol), KI (0.6 mg), and DMAc (15 mL) were placed into a two neck round-bottom flask and stirred at 100 °C for 20 min under nitrogen atmosphere. Then, MOZ (5.5 mL, 65 mmol) was added and stirred at 100 °C for 24 h. After polymerization, the precipitate products were collected by centrifugation (4000 rpm, 5 min), and washed with water and methanol, and then dried under reduced pressure. White powders of the μ-PSt-g-PMOZ were obtained in 1.8 g.Synthesis of microballs grafting PEI brush (μ-PSt-g-PEI)
The microballs of μ-PSt-g-PMOZ (1.8 g) were dispersed into 5 M HCl (50 mL), and stirred at 100 °C for 8 h. After hydrolysis, the brown precipitates were washed with methanol by centrifugation. Then, the separated precipitates were mixed with ammonium solution, and stirred at room temperature for 24 h. The products were corrected by filtration (0.45 μm), and washed by distilled water and acetone, and then dried under atmosphere. Yield: 1.5 g (white powders).Complexation of μ-PSt-g-PEI with various kinds of dicarboxylic acids
Typically, μ-PSt-g-PEI (50 mg, nitrogen content ca. 0.6 mmol) was dispersed in a mixture of water (5 mL) and DMF (5 mL) at 80 °C for 1 h. To this dispersion, meso-tartaric acid (1.5 mmol) dissolved in a mixture of water (5 mL) and DMF (5 mL) was added, followed by stirring at 80 °C for 3 h. Then, the complexed microballs (μ-PSt-g-PEI/m-TA) were collected by centrifugation (4000 rpm, 5 min), washed with distilled water and acetone and finally dried under ambient conditions.By replacing meso-tartaric acid, other organic acids such as succinic acid and adipic acid (1.5 mmol) were also used for preparation of complexes of μ-PSt-g-PEI/SA and μ-PSt-g-PEI/AA in the same manner.
Complexation of μ-PSt-g-PEI with mucic acid
For complexation of μ-PSt-g-PEI with mucic acid, μ-PSt-g-PEI (50 mg, nitrogen content ca. 0.6 mmol) was dispersed in 0.5 M HCl (10 mL) at 80 °C. To this dispersion, mucic acid (1.5 mmol) was first completely dissolved using 1 M ammonium solution (5 mL) and then, water (10 mL) was added and stirred at 80 °C for 3 h. Following this, the complexed microballs (μ-PSt-g-PEI/MA) were collected by centrifugation (4000 rpm, 5 min), and washed with distilled water and acetone, and finally dried under ambient conditions.Silica hybridization on μ-PSt-g-PEI/m-TA
Typically, TMOS (1.0 mL) was added into a dispersion of μ-PSt-g-PEI/m-TA (50 mg) in water (20 mL) and stirred at room temperature for 3 h. Then, the precipitates were collected by centrifugation (4000 rpm, 5 min), and washed with distilled water and acetone, and dried under air atmosphere, due to which white powders of silica hybrids μ-PSt-g-PEI/m-TA@SiO2 were obtained.The other complexes were also hybridized in the same manner to obtain silica hybrids microballs of μ-PSt-g-PEI/SA@SiO2, μ-PSt-g-PEI/AA@SiO2, and μ-PSt-g-PEI/MA@SiO2.
Calcination of μ-PSt-g-PEI/m-TA@SiO2
Typically, the microballs of μ-PSt-g-PEI/m-TA@SiO2 were calcined at 800 °C for 2 h (rising time 1 h and keeping time 1 h) to remove the polymeric components from the hybrid balls, and the resultant silica microballs were subjected to the structural characterizations.The other silica hybrids microballs were also treated in the same manner to remove all the organic components.
Etching the calcined silica microballs by APS
According to our previously reported method,12 four types of silica microballs after calcination were etched as follows. The calcined silica (25 mg), aminopropyl trimethoxysilane (APS, 1 mL) and distilled water (7.5 mL) were added into a 20 mL bottom-rounded flask, and the mixture was stirred at 80 °C for 8 h. Then, the precipitates obtained were collected by centrifugation, washed with water and acetone, and dried at room temperature overnight. Finally, 5 mg of the white powders were recovered.Characterization
Fourier transform infrared (FT-IR) spectra were recorded on a JASCO FT-IR-4600 spectrometer by diffusing the samples in KBr pellets. 13C cross polarization magic angle spinning (CP/MAS) NMR spectra and 29Si CP/MAS NMR spectra were recorded on JEOL ECA-400 spectrometer operating at 400 MHz. Scanning electron microscopy (SEM) observations were performed with a Hitachi SU-8010 microscope at 5 kV after the samples were sputtered with Pt particles. X-ray diffraction (XRD) measurement was conducted on a Rigaku Rint-Ultime III X-ray diffractometer with Cu Kα radiation (λ = 0.1540 nm) operating at 40 kV and 40 mA. TGA was performed on a Seiko Instruments EXSTER 6000 at a heating rate of 10 °C min−1 in the range of 30–900 °C under air conditions. N2 adsorption/desorption isotherms were recorded with a Micrometrics Tristar-3000 instrument at liquid-nitrogen temperature; all samples were degassed at 120 °C under vacuum for 3 h prior to analysis.Results and discussion
The preparation process, including the formation of acid–base complexes, silica deposition and calcination, is illustrated in Fig. 1. We prepared four types of complexes, μ-PSt-g-PEI/m-TA, μ-PSt-g-PEI/MA, μ-PSt-g-PEI/SA and μ-PSt-g-PEI/AA, by mixing μ-PSt-g-PEI (50 mg, N-content ca. 0.6 mmol) and excess of organic acids (1.5 mmol), m-TA, MA, SA, and AA, in mixed aqueous solvents at 80 °C for 3 h. Subsequently, these complexes were investigated for silica mineralization using tetramethoxysilane (TMOS) as the silica source at room temperature for 3 h in water. Finally, the silica hybrids obtained were calcined at 800 °C to remove the organic components.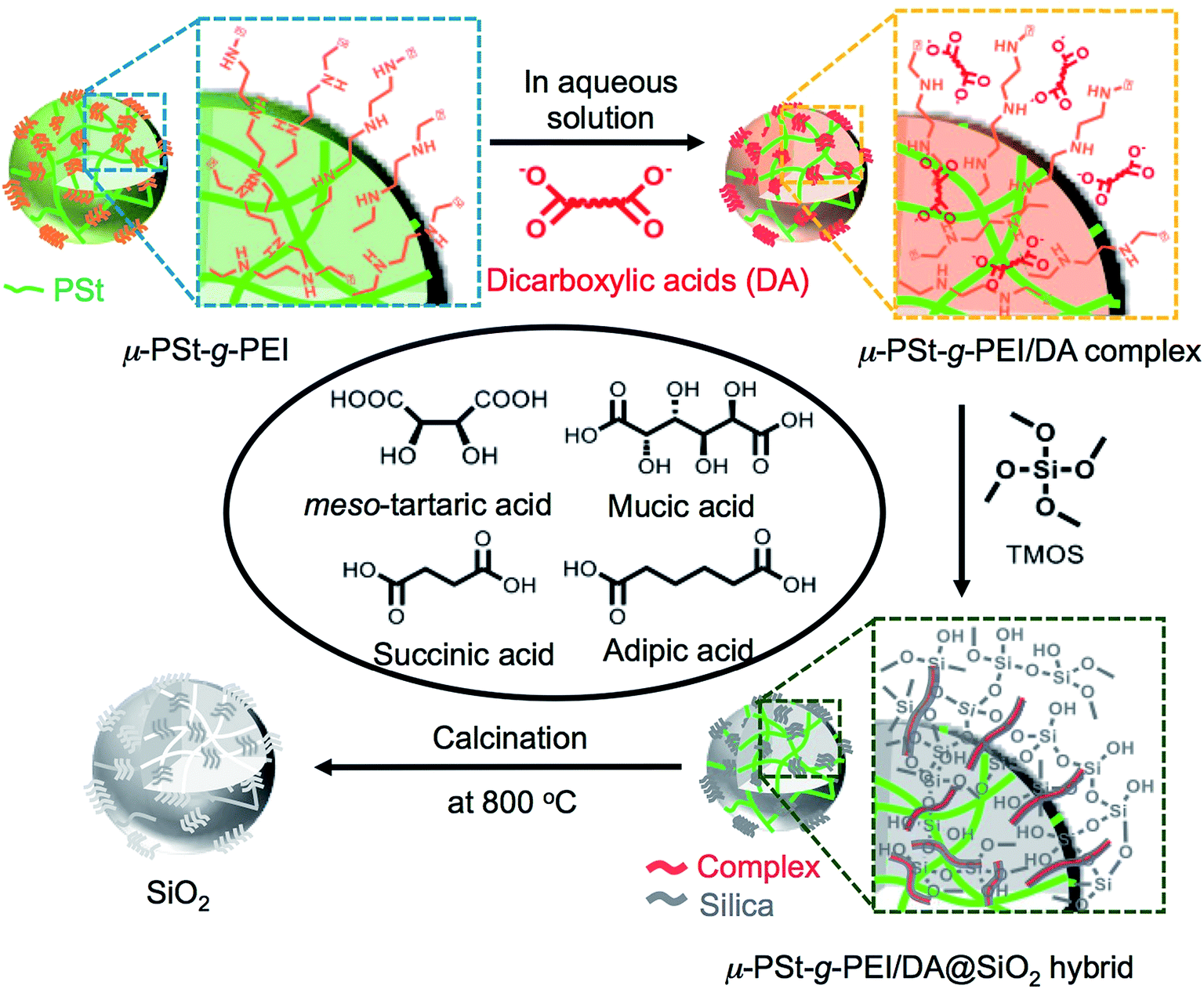 | ||
| Fig. 1 Schematic representation of the process from complexation to silica calcination. | ||
PEI-microballs complexed with dicarboxylic acids
First, we performed elemental analysis (EA) and SEM observation for the samples before and after complexation of the μ-PSt-g-PEI with dicarboxylic acids. As shown in in Table 1, it is apparent that the nitrogen content decreased after complexation in the presence of excess dicarboxylic acids. From the nitrogen values, we estimated the loading amount of the dicarboxylic acids on the microballs by assuming that one mol of nitrogen combines with 1/2 mol dicarboxylic acids and found that the loaded acidic component was about 100% (m-TA), 100% (MA), 88.6% (AA) and 76.4 (SA). Structurally, the dicarboxylic acids of m-TA and SA have the same butanedioic form with four carbons, while the former is a di-alcoholic type; MA and AA have the same hexanedioic form with six carbons, but the former is a tetra-alcoholic type. It appears that alcoholic dicarboxylic acids are easier to load on the μ-PSt-g-PEI than the non-alcoholic dicarboxylic acids. As shown in the SEM images (Fig. 2), upon loading the dicarboxylic acids on the microballs, the diameters tend to slightly increase from the initial 3.2 (±0.3) μm (before complexation) to 3.5 (±0.2), 3.8 (±0.3), 3.8 (±0.3) and 3.7 (±0.5) μm, respectively, after complexation with m-TA, MA, AA and SA. This result indicates that the mass increment due to the loading of dicarboxylic acids causes volume enlargement of the microballs.| Complex sample | Measurement value (%) | Acids loading (%) |
|---|---|---|
C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H:N :H:N |
||
| μ-PSt-g-PEI | 40.1:9.6:16.7 |
— |
| μ-PSt-g-PEI/m-TA | 39.9:9.3:12.9 |
100 |
| μ-PSt-g-PEI/MA | 39.3:7.7:9.8 |
100 |
| μ-PSt-g-PEI/AA | 42.6:8.4:9.9 |
86.6 |
| μ-PSt-g-PEI/SA | 39.9:6.6:9.7 |
76.4 |
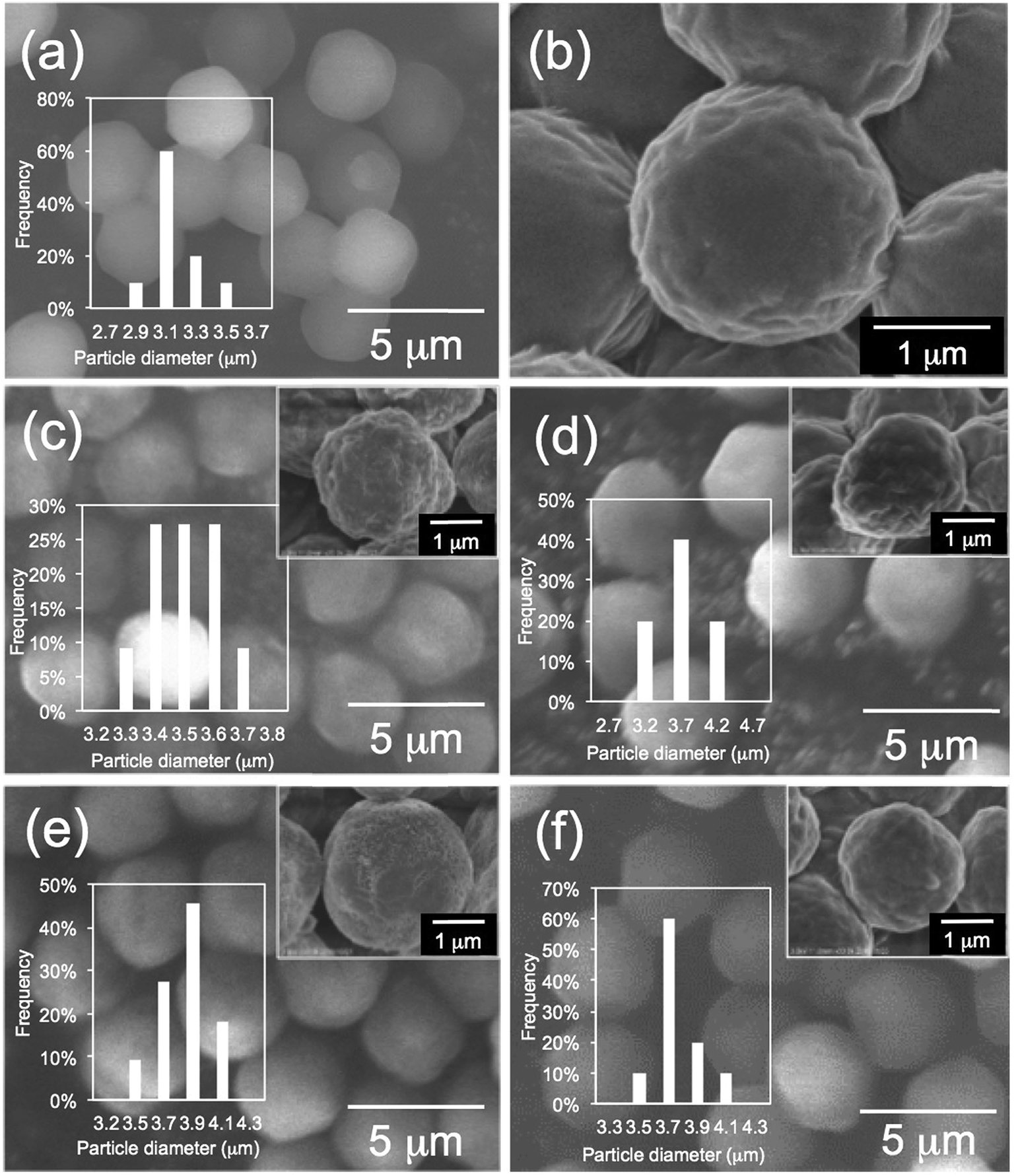 | ||
| Fig. 2 SEM images and histograms of (a, b) μ-PSt-g-PEI (c) μ-PSt-g-PEI/m-TA, (d) μ-PSt-g-PEI/SA, (e) μ-PSt-g-PEI/MA, (f) μ-PSt-g-PEI/AA. | ||
We reported that polymers composed of PEI easily retain PEI-based crystallinity even when they have different dimensional structures, such as star- and comb-like or the state of cross-linked gel.7,8,13 In case of the microballs of μ-PSt-g-PEI, the PEI grafted on the polystyrenic network backbone also showed crystallinity, although the spheres only have nearly 3 μm diameters. We also know that complexes prepared via complexation of PEI with D- and L-tartaric acid have a crystalline feature showing specific diffraction patterns different from the PEI.9a To investigate the physical properties of the complexes of μ-PSt-g-PEI/dicarboxylic acids, herein, the complexes composed of μ-PSt-g-PEI and the four types of dicarboxylic acids were subjected to XRD measurement. As shown in Fig. S1,† μ-PSt-g-PEI itself exhibited remarkable XRD diffraction strength, but the crystalline pattern disappeared completely after complexing with dicarboxylic acids. The complex associated with MA showed very weak reflection patterns, which would be due to the insufficient growth of crystalline structures. The other complexes from m-TA, SA and AA showed nearly amorphous halo patterns without convincing crystalline features. As a set of control experiments, we tried complexing the four acids with comb-like polymers, such as cPEI with a polystyrenic backbone in aqueous solution (see details in the ESI†). It should be noted here that the four types of acids and the powders of cPEI are crystalline solids and they can be soluble in water under certain conditions. It was found that mixing the solutions of MA or m-TA with the solution of the comb cPEI afforded precipitates while SA and AA retained the solution state without any precipitates. From the XRD measurement of the precipitates of cPEI/MA and cPEI/m-TA, we could not find convincing crystalline information although there appeared very weak reflection around 2θ at 20–25 degrees (see Fig. S2;† probably, the sizes of crystallites were too small to be effectively detected by XRD). It appears that the complexation of PEI-grafted comb-like polymer (cPEI) with dicarboxylic acids of MA and m-TA did not prompt the growth of size-efficient crystallites although precipitates were easily obtained from their aqueous solutions. This trend might also appear in the complexations of μ-PSt-g-PEI microballs with the acids because the PEI chains in microballs are also grafted on the polystyrenic backbones.
FT-IR spectroscopy is a simple tool to assess the complexes of μ-PSt-g-PEI/carboxylic acids. As shown in Fig. 3, compared to the μ-PSt-g-PEI, the four complexes of μ-PSt-g-PEI/m-TA, μ-PSt-g-PEI/MA, μ-PSt-g-PEI/SA and μ-PSt-g-PEI/AA showed a remarkable new peak near at 1650 cm−1, which can be assigned to the –COO− stretching vibration from the residues of the dicarboxylic acids. In addition, the 13C CP/MAS NMR spectra of μ-PSt-g-PEI/dicarboxylic acid complexes with meso-tartaric acid and mucic acid are presented in Fig. 4. A set of three signals are clearly observed at 43, 74 and 178 ppm, which are assigned to ethyleneimine carbon (43 ppm) of the main chain of PEI and to carbons of the –CH2OH (74 ppm) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (178 ppm) groups for acidic component, respectively, indicating the presence of the two components of PEI and the dicarboxylic acids in the complexed microballs.
O (178 ppm) groups for acidic component, respectively, indicating the presence of the two components of PEI and the dicarboxylic acids in the complexed microballs.
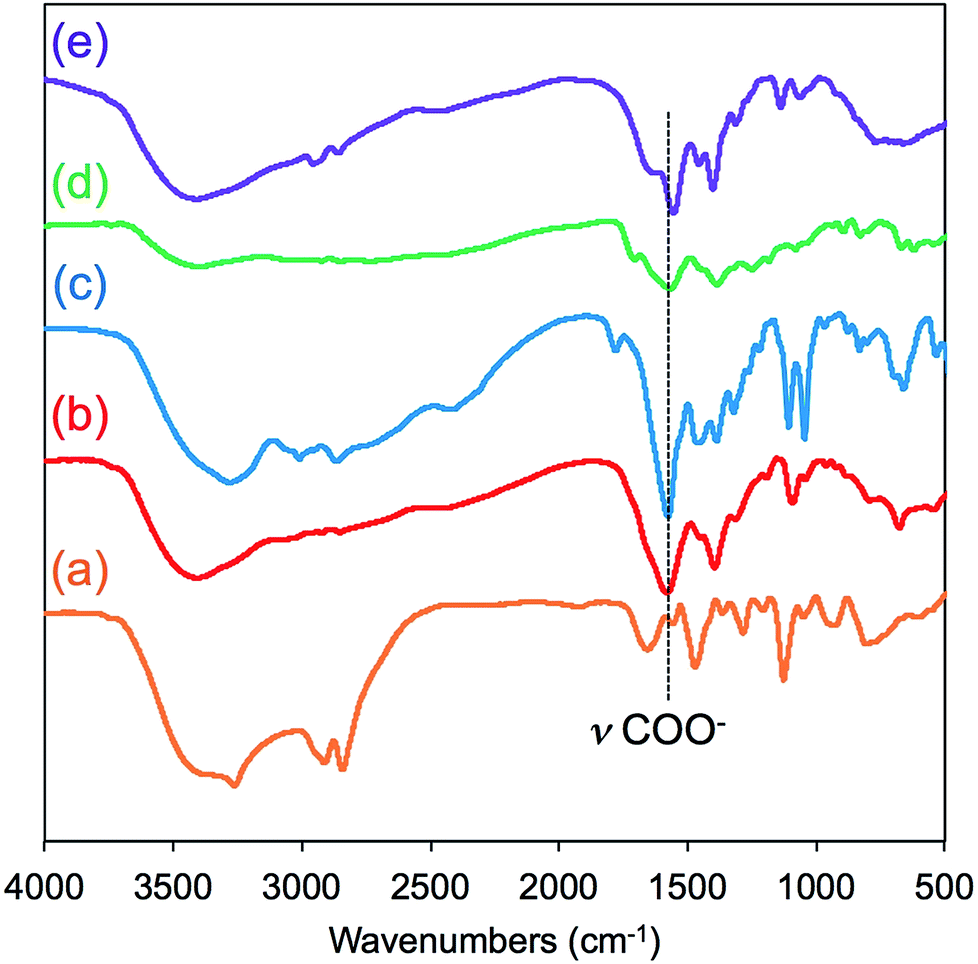 | ||
| Fig. 3 FT-IR spectra of (a) μ-PSt-g-PEI, (b) μ-PSt-g-PEI/m-TA, (c) and μ-PSt-g-PEI/MA, (d) μ-PSt-g-PEI/SA and (e) μ-PSt-g-PEI/AA. | ||
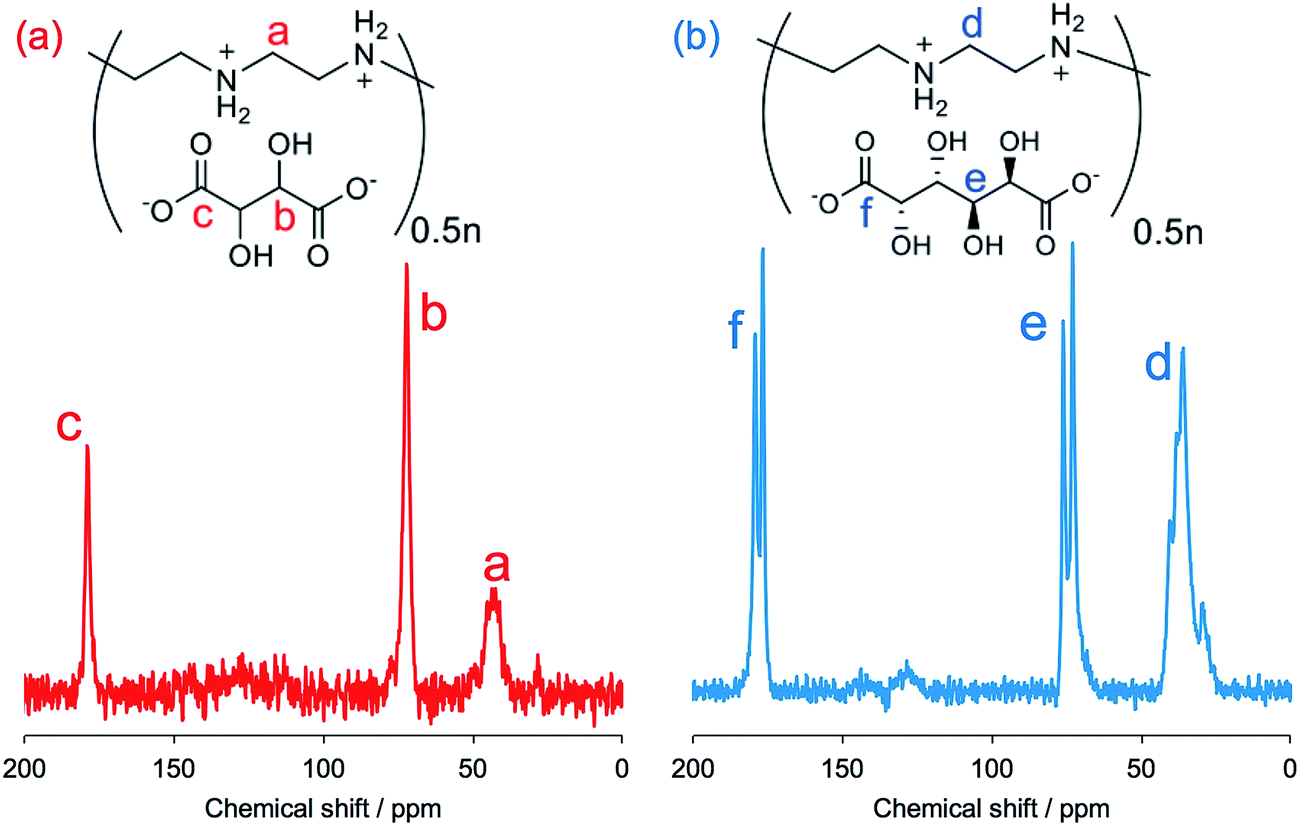 | ||
| Fig. 4 13C NMR spectra of (a) μ-PSt-g-PEI/m-TA and (b) μ-PSt-g-PEI/MA. | ||
Silica mineralization on the acid–base complexed microballs
Using the PEI-microballs complexed with the four types of dicarboxylic acids, we performed the silica mineralization by mixing the complexes and TMOS in water at room temperature for 3 h. The silica hybrid microballs and their calcined (at 800 °C) forms were characterized by FT-IR, 29Si CP/MAS NMR, TGA, SEM, and N2 absorption–desorption isotherm.Fig. S3 and S4† show the FT-IR spectra of a series of silica hybrids and calcined silica mediated in the presence of dicarboxylic acids. All the vibrational spectral lines after silica deposition still showed stretching vibration near 1650 cm−1 due to the presence of –COO− groups, similar to the vibrations appearing in the samples of μ-PSt-g-PEI/dicarboxylic acids (Fig. 1) and new remarkable absorption at 1050 cm−1 assigned to the Si–O–Si stretching vibration (Fig. S2†). After calcination, the peak at 1650 cm−1, ascribed to carbonyl group, tended to disappear, while the Si–O–Si vibration peak is still present (Fig. S3†). Furthermore, the 29Si CP/MAS NMR spectra of the calcined samples from μ-PSt-g-PEI/dicarboxylic acid@SiO2 are indicated in Fig. S5.† All the calcined samples showed three peaks at −90, −100, and −110 ppm, which are assigned to the unit bonding structures of Q2 {(SiO)2Si(OH)2}, Q3 {(SiO)3Si(OH)} and Q4 {Si(OSi)4} representing the silica framework.
TGA was conducted on the four hybrids samples to estimate the thermoproperty and the contents of silica deposition. As shown in the TGA chart (Fig. 5), the thermal weight loss underwent two ranges of heating temperature. The thermal weight loss from 200 to 300 °C could be due to thermal decomposition of the parts of acid–base complexes by PEI (–CH2CH2NH–) and organic acids, while the thermal weight loss from 400 to 700 °C was due to thermal decomposition of the parts of cross-linked polystyrene backbones and a part of dehydration between silanols. The mass finally remaining at 700 °C should be attributed to the amount of the deposited silica. It is found that the amount of silica deposited on the acid–base complexes showed apparent difference when the associated acid changed. For instance, the silica deposition increased in the order of 32 < 45 < 68 < 71 wt%, respectively, for m-TA, MA, SA and then AA. It appears that the non-alcoholic type dicarboxylic acid (adipic acid and succinic acid) associated with PEI microballs has higher silica deposition ability than the meso-type dicarboxylic acid (meso-tartaric acid and mucic acid). This indicates that the silica deposition ability of the complexes μ-PSt-g-PEI/dicarboxylic acid depended strongly on the structure of the dicarboxylic acid.
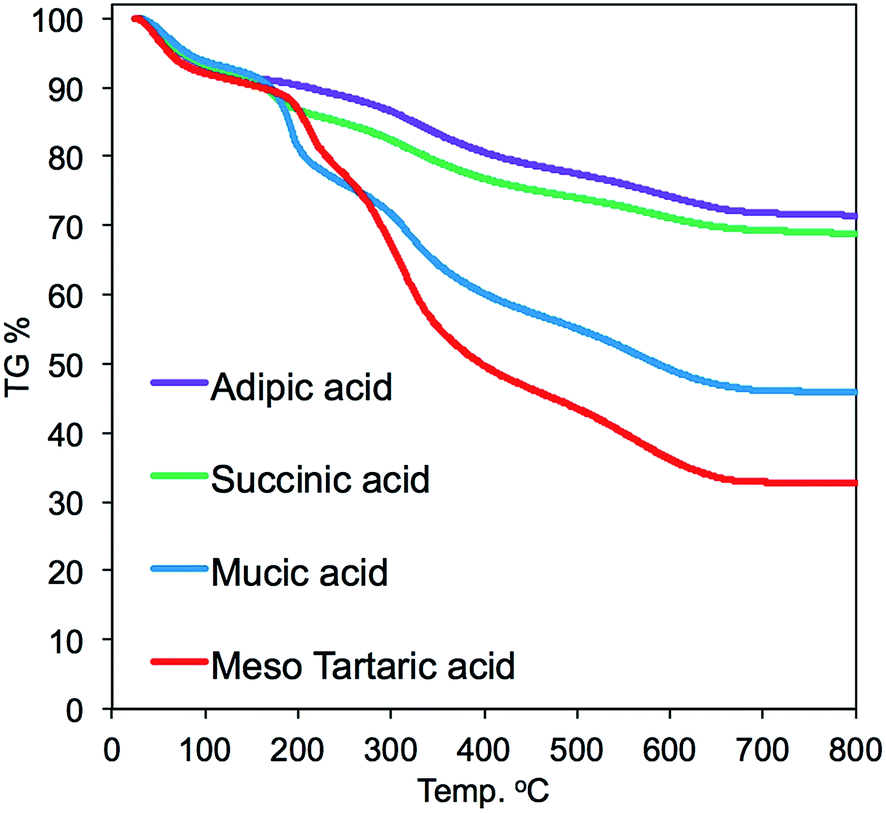 | ||
| Fig. 5 TGA curves of μ-PSt-g-PEI/dicarboxylic acid@SiO2 mediated by four kinds of dicarboxylic acids. | ||
Fig. 6 and 7 show SEM images of a series of the as-prepared hybrids silica. Herein, we first compared the surface of the hybrids microballs obtained from the PEI complexes associated with four-carbon dicarboxylic acids of m-TA and SA. Interestingly, the surface of the hybrid silica microballs with m-TA was densely covered with numerous nanofibres with thickness about 30 nm (see Fig. 6) and these characteristic structures remained unchanged after calcination at 800 °C (see Fig. S6a and b†). It appears that meso-tartaric acid is particularly important for the formation of specific nanofibres-covered silica structures on the PEI-balls of sub-5 μm. In comparison, the surface of the hybrid silica microballs with SA was densely packed by rough nanoparticle (about 30 nm) aggregates and this structure also remained unchanged after calcination (see Fig. S6c and d†). Such differences also can be seen in cases of microballs associated with MA and AA. In case of MA association, the surface of the hybrid microballs displayed a short-haircut-like surface, which was covered densely with numerous nanoplates (see Fig. 7a and c), while the surface related to AA was roughly composed of nanoparticles aggregates (Fig. 7b and d). In addition, these structures did not change even after being calcined at 800 °C (see Fig. S6e–h†). Evidently, the formation of surface structures on the μ-PSt-g-PEI/dicarboxylic acid strongly depended upon the acidic structures; both the meso-type alcoholic acids of m-TA and MA favour the formation of nanofibres or nanoplates-coated surfaces, but the non-alcoholic acids of SA and AA resulted in nanoparticles-filled surfaces. None of the four types of hybrids microballs showed significant changes in size after calcination, indicating no marked volume shrinkage in the calcination process. This is apparently different than our former results, in which the free-base form of μ-PSt-g-PEI without acids association produced silica microballs with a smooth surface and hollow internal structure under the same aqueous conditions.10 In terms of structure, μ-PSt-g-PEI microballs resemble 3-dimensional cross-linked comb-like PEI. In order to further understand the reason for the formation of the different surface structures on the microballs, we performed control experiments of silica deposition under the same conditions using the complexes (as mentioned above in the section about complexation of comb-like PEI), which consisted of comb-polymers cPEI and 4 types of acids. The silica templated by the 4 types of complexes, cPEI/m-TA (powders dispersion), cPEI/MA (powders dispersion), cPEI/SA (aqueous solution) and cPEI/AA (aqueous solution), showed sharp distinction in their morphologies (see Fig. S7†). The complex of cPEI/m-TA resulted in nanofibrous (diameter < 20 nm) silica bundles, while the complex of cPEI/MA afforded largely gathered aggregates of silica nanoplates (ca. 20 nm thickness). In comparison, both complexes cPEI/SA and cPEI/AA, which were soluble complexes in aqueous phase, resulted in agglomerates of silica nanospheres (diameters: 25–30 nm). The formation of silica with nanofibres and nanoplates morphologies from cPEI systems would be related to the defined morphologies of the complexes of cPEI/m-TA and cPEI/MA in spite of lack of sufficient growth of crystallites for the complexes. From the comparison of silica structures directed by μ-PSt-g-PEI and cPEI, evidently, the behavior and/or action of μ-PSt-g-PEI microballs after association with four types of acids in silica deposition is the same as the complexes of comb-like PEI molecules, although the PEI chains in microballs are confined in the sub-5 μm spherical space. In particular, association of both alcoholic acids MA and m-TA with PEI chains on the surface of μ-PSt-g-PEI gave morphology-defined templating-sites, which could result in unique surfaces covered by a unit structure of nanofibre or nanoplate, similar to the case of the cPEI system. To visualize the inner structure of the calcined silica microballs, we performed silica etching for the 4 types of silica microballs by means of our new finding of etching technique (mixing and stirring silica with 3-aminopropyltrimethoxysilane (APS) in water at 100 °C)10,12 and subjected the etched samples to SEM observation. As shown in Fig. S8,† the silica microballs mediated from PEI/m-TA and PEI/MA have loose internal packing, while the silica microballs mediated from PEI/SA and PEI/AA are densely packed by silica nanoparticles.
 | ||
| Fig. 6 SEM images (of magnified area) of (a, c) μ-PSt-g-PEI/m-TA@SiO2, (b, d) μ-PSt-g-PEI/SA@SiO2. | ||
 | ||
| Fig. 7 SEM images (of magnified area) of (a, c) μ-PSt-g-PEI/MA@SiO2, (b, d) μ-PSt-g-PEI/AA@SiO2. | ||
Furthermore, we compared N2 adsorption/desorption isotherms and the pore size distribution of the four calcined samples. As shown in Fig. 8a, the calcined silica mediated from PEI/m-TA showed a type-II isotherm curve almost without a hysteresis loop with the average pore size of 3.5 nm in diameter. The BET surface area was 430 m2 g−1 (see Table S1†). In contrast, the calcined silica mediated from PEI/SA exhibited a type-IV isotherm curve with hysteresis loop in the relative pressure (P/P0) between 0.4 and 0.8. The average pore size was 4 nm in diameter and the BET surface area was 133.3 m2 g−1 (Fig. 8b and Table S1†). In the case of both the calcined silica mediated from PEI/MA and PEI/AA (see Fig. S9 and Table S1†), there appeared the same type-IV isotherms but the former had larger BET surface area (615.0 m2 g−1, pore size with 3.9 nm) than the later (96.7 m2 g−1, pore size with 3.8 nm). From these results combining the deposition amount and surface structural images, it can be concluded that the silica microballs with a relatively lower silica deposition amount and a dense nanofibres/nanoplates-covered surface structure tend to occupy a high BET surface area, while those with higher silica deposition and nanoparticles-covered surface exhibited low BET surface area. Such differences are dominated by the structures of dicarboxylic acids used in complexation with the μ-PSt-g-PEI. In the silica deposition on the PEI/acids complexes microballs, the role of the alcohol type acids is extremely different than that of the non-alcoholic acids although the exact structural reason is not clear at present (it needs further study). Undoubtedly, the dicarboxylic acids associated with PEI play a structural messenger-like role in silica formation.
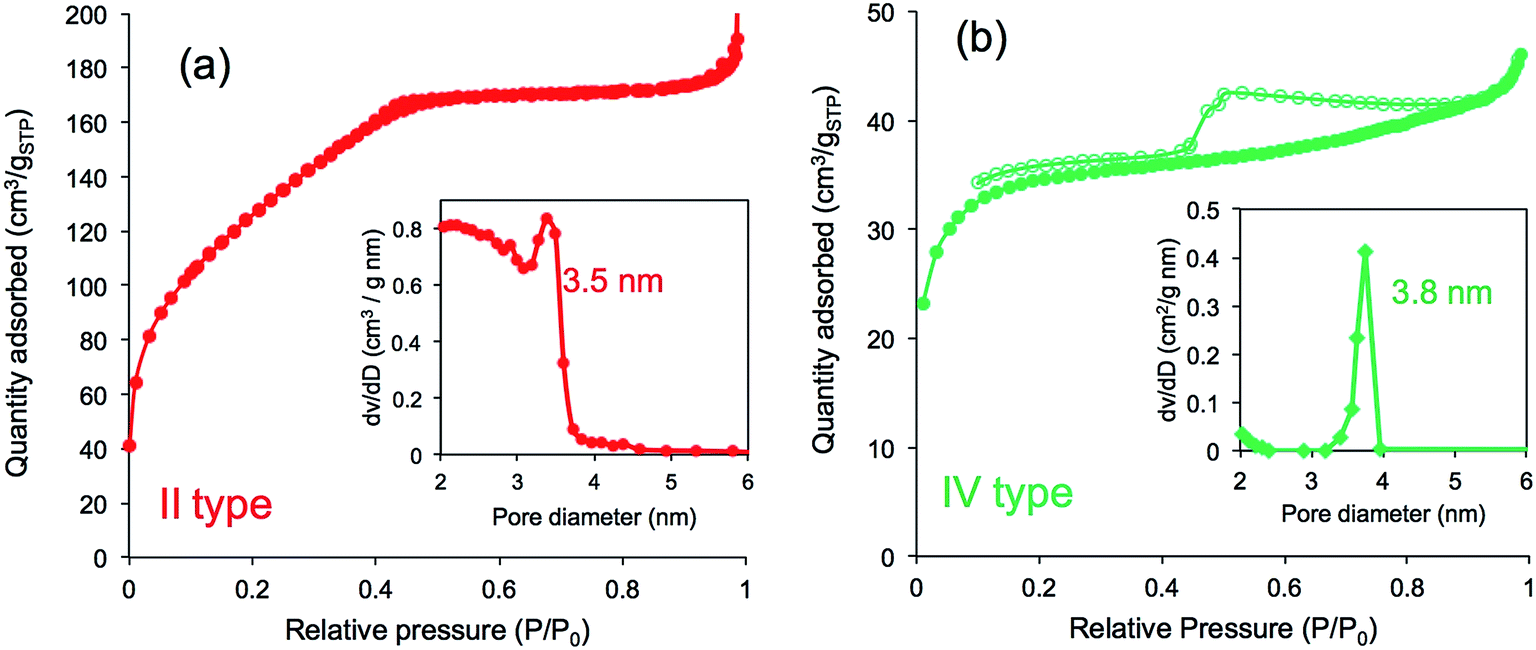 | ||
| Fig. 8 N2 adsorption desorption isotherms and pore size distribution after calcination of (a) μ-PSt-g-PEI/m-TA@SiO2 (b) μ-PSt-g-PEI/SA@SiO2. Open dots: desorption; close dots; adsorption. | ||
Conclusions
In this study, acid–base complexes formed from sub-5 μm PEI-microballs and dicarboxylic acids such as adipic acid (AA), succinic acid (SA), meso-tartaric acid (m-TA) as well as mucic acid (MA) were used as catalytic templates in silica deposition for understanding the role of the dicarboxylic acids, which complexed with PEI-microballs. Interestingly, we found that changing the structures of dicarboxylic acids could effectively influence the silica structure formed through PEI-microballs. When PEI microballs carried multi-alcoholic diacids such as meso-tartaric acid and mucic acid, the complexes could promote the formation of microballs with unique surface structures covered densely by nanofibres or nanoplates. Similar dicarboxylic acids with the same number of carbons but without an alcoholic group, however, specifically directed different surface structures, in which the nanoparticles densely but roughly filled around the microballs. In addition, the BET surface areas were also greatly influenced by the structures of dicarboxylic acids, being changeable in the range of 90–600 m2 g−1. In this sense, we can say that the dicarboxylic acids exactly play a messenger-like role to prompt the structures of silica in the system of polyethyleneimine-dominated silica deposition. Our finding in this study suggests that the structures of silica microballs in the sub-5 μm space can be conveniently custom-made by choice of suitable carboxylic acids as long as the polyamines-microballs are prepared. This would provide a new insight into the chemistry of silica to design silica in the restricted small sizes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the MEXT Supported Program for the Strategic Research Foundation at Private Universities: “Creation of new fusion materials by integration of highly-ordered nano-inorganic materials and ultra-precisely controlled organic polymers” (2013–2017), no. S1311032.References
- (a) K. Y. Ho, G. McKay and K. L. Yeung, Langmuir, 2003, 19, 3019–3024 CrossRef CAS; (b) Z.-J. Jiang, C.-Y. Liu and L.-W. Sun, J. Phys. Chem. B, 2005, 109, 1730–1735 CrossRef CAS PubMed; (c) Y.-F. Han, F. Chen, Z. Zhong, K. Ramesh, L. Chen and E. Widjaja, J. Phys. Chem. B, 2006, 110, 24450–24456 CrossRef CAS PubMed; (d) H. Zhu, C. Liang, W. Yan, S. H. Overbury and S. Dai, J. Phys. Chem. B, 2006, 110, 10842–10848 CrossRef CAS PubMed; (e) W. Huang, J. N. Kuhn, C.-K. Tsung, Y. Zhang, S. E. Habas, P. Yang and G. A. Somorjai, Nano Lett., 2008, 7, 2027–2034 CrossRef PubMed; (f) C.-W. Yen, M.-L. Lin, A. Wang, S.-A. Chen, J.-M. Chen and C.-Y. Mou, J. Phys. Chem. C, 2009, 113, 17831–17839 CrossRef CAS; (g) J. Aguado, J. M. Arsuaga, A. Arencibia, M. Lindo and V. Gascon, J. Hazard. Mater., 2009, 163, 213–221 CrossRef CAS PubMed; (h) J. Choi and M. Tsapatsis, J. Am. Chem. Soc., 2010, 132, 448–449 CrossRef CAS PubMed; (i) Z. Liang, W. Shi, Z. Zhao, T. Sun and F. Cui, Colloids Surf., A, 2017, 513, 250–258 CrossRef CAS; (j) E. S. Sanz-Pérez, T. C. M. Dantas, A. Arencibia, G. Calleja, A. P. M. A. Guedes, A. S. Araujo and R. Sanz, Chem. Eng. J., 2017, 308, 1021–1033 CrossRef; (k) A. Shajkumar, B. Nandan, S. Sanwaria, V. Albrecht, M. Libera, M.-H. Lee, G. Auffermann, M. Stamm and A. Horechyy, J. Colloid Interface Sci., 2017, 491, 246–254 CrossRef CAS PubMed.
- (a) M. Sumper, Science, 2002, 295, 240–2432 CrossRef PubMed; (b) V. C. Sunder, A. D. Yablon, J. L. Grazul, M. Ilan and J. Aizenberg, Nature, 2003, 424, 899–990 CrossRef PubMed; (c) J. Aizenberg, J. C. Weaver, M. S. Thanawala, V. C. Sundar, D. E. Morse and P. Fratzl, Science, 2005, 309, 275–278 CrossRef CAS PubMed; (d) M. Sumper and E. Brunner, Adv. Funct. Mater., 2006, 16, 17–26 CrossRef CAS; (e) M. Sumper and E. Brunner, ChemBioChem, 2008, 9, 1187–1194 CrossRef CAS PubMed; (f) M. Hildebrand, Chem. Rev., 2008, 108, 4855–4874 CrossRef CAS PubMed; (g) S. V. Patwardhan, Chem. Commun., 2011, 47, 7567–7582 RSC; (h) N. Kroger and E. Brunner, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 615–627 CrossRef PubMed.
- (a) N. Kroger, R. Deutzmann and M. Sumper, Science, 1999, 286, 1129–1131 CrossRef CAS PubMed; (b) N. Kroger, S. Lorenz, E. Brunner and M. Sumper, Science, 2002, 298, 584–586 CrossRef PubMed; (c) M. Sumper and N. Kroger, J. Mater. Chem., 2004, 14, 2059–2065 RSC.
- A. F. Wallace, J. J. DeYoreo and P. M. Dove, J. Am. Chem. Soc., 2009, 20, 5244–5250 CrossRef PubMed.
- T. Kuno, T. Nonoyama, K. Hirao and K. Kato, Langmuir, 2011, 27, 13154–13158 CrossRef CAS PubMed.
- (a) P.-X. Zhu, N. Fukazawa and R.-H. Jin, Small, 2007, 3, 394–398 CrossRef CAS PubMed; (b) R.-H. Jin and J.-J. Yuan, Polym. J., 2007, 39, 464–470 CrossRef CAS; (c) D. Noda, Y. Arai, D. Souma, H. Nagashima and R.-H. Jin, Chem. Commun., 2014, 50, 10793–10796 RSC.
- (a) R.-H. Jin and K. Motoyoshi, J. Porphyrins Phthalocyanines, 1999, 3, 60–64 CrossRef CAS; (b) R.-H. Jin and J.-J. Yuan, Chem. Mater., 2006, 18, 3390–3396 CrossRef CAS.
- (a) D.-D. Yao and R.-H. Jin, Polym. Chem., 2015, 6, 2255–2263 RSC; (b) D.-D. Yao, H. Kubosawa, D. Soma and R.-H. Jin, Polymer, 2016, 86, 120–128 CrossRef CAS.
- (a) H. Matsukizono and R.-H. Jin, Angew. Chem., Int. Ed., 2012, 51, 5862–5865 CrossRef CAS PubMed; (b) R.-H. Jin, D.-D. Yao and R. Levi, Chem.–Eur. J., 2014, 20, 7196–7214 CrossRef CAS PubMed; (c) D.-D. Yao, H. Murata, S. Tsunega and R.-H. Jin, Chem.–Eur. J., 2015, 21, 15667–15675 CrossRef CAS PubMed; (d) H. Matsukizono, H. Murata and R.-H. Jin, Chem.–Eur. J., 2014, 20, 1134–1145 CrossRef CAS PubMed.
- R.-H. Jin and J.-J. Yuan, in Advances in Biomimetics, ed. A. George, InTech, 2011, ch. 8, pp. 159–184 Search PubMed.
- D. Soma and R.-H. Jin, RSC Adv., 2017, 7, 36302–36312 RSC.
- X.-L. Liu, S. Tsunega and R.-H. Jin, ACS Omega, 2017, 2, 1431–1440 CrossRef CAS.
- D. Soma and R.-H. Jin, Colloid Polym. Sci., 2017, 295, 1585–1594 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12413a |
| This journal is © The Royal Society of Chemistry 2018 |