
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile construction of dual-targeting delivery system by using lipid capped polymer nanoparticles for anti-glioma therapy†
Shengfeng Wangab,
Chuantong Zhaoa,
Peng Liua,
Zhe Wanga,
Jinsong Ding*a and
Wenhu Zhou *a
*a
aXiangya School of Pharmaceutical Sciences, Central South University, Changsha, Hunan 410013, China. E-mail: zhouwenhuyaoji@163.com
bDepartment of Pharmacy, The Third Xiangya Hospital of Central South University, Changsha, Hunan 410013, China
First published on 20th December 2017
Abstract
Recent progress has suggested that dual targeting drug delivery systems, which are functionalized with two active ligands targeting the blood brain barrier (BBB) and brain tumor cells respectively, provide a promising strategy to improve the chemotherapeutic efficacy for gliomas. The key to successfully developing such systems is to functionalize nanoparticles (NPs) with active ligands, though they usually suffer from several issues such as NP aggregation, drug leakage and complicated chemical synthesis. Herein we report a facile way of constructing a dual targeting delivery system by simply using lipid capped polymer NPs. The NPs were constructed with a poly lactic-co-glycolic acid (PLGA) core for hydrophobic doxorubicin (DOX) loading, and a lipid (lecithin and DSPE–PEG2000–COOH) shell. The DSPE–PEG2000–COOH provides an excellent anchor for ligand conjugation. As a proof of concept, two active ligands of angiopep-2 peptide and AS1411 aptamer were covalently linked to NPs through a single step synthesis, with little effect on particle size, stability, drug loading and drug release performance. The accumulative drug release of the NPs in PBS (pH 7.4) over 144 h is ∼50%, suggesting a sustained drug release profile. The NPs showed significantly enhanced uptake by brain capillary endothelial cells (BCECs) and C6 glioma cells as indicated by fluorescence microscopy and quantitative flow cytometry, demonstrating the dual targeting efficiency. The BBB penetration ability is validated by using an in vitro BBB model, and the in vitro cell inhibition study showed increased cytotoxicity of the NPs to glioma cells.
1 Introduction
Gliomas are the most frequent form of primary malignant tumor in the brain, accounting for ∼80% of the incidence of central nervous system tumors.1,2 These tumors, especially high-grade gliomas and glioblastoma, grow invasively and carry a high rate of morbidity and mortality.3,4 Current standard treatment for gliomas is surgical resection, while it usually has poor outcomes because of the diffuse invasion of gliomas into nearby brain tissues, resulting in relapse after treatment.5 Due to this issue, chemotherapy presents an essential auxiliary treatment for patients with glioma after surgery. The efficacy of chemotherapy, however, is also reported to be unsatisfactory with high side-effects.6,7 This can be ascribed to the non-specific bio-distribution of current therapeutic drugs and their inadequate delivery to the tumor.For brain tumors chemotherapy, a major challenge is the blood–brain barrier (BBB), which is a diffusion barrier formed by brain capillary endothelial cells (BCECs) along with other perivascular cells. This barrier is featured by the absence of fenestrations and by extensive tight junctions. This unique structure prevents nearly 100% large-molecule and more than 98% of small-molecule drugs from transporting into the brain,8,9 which is due to their low permeability and the high level of drug efflux by active transporters.10 With the recent progress of nano-medicine, it has been shown that nanoparticles (NPs) functionalized with bioactive ligands is a promising tool to mediate the transportation of drugs across BBB.11 This strategy is inspired by a biological process called receptor-mediated transcytosis (RMT),12 through which many nutritional bio-macromolecules are able to actively transport the BBB barrier via specific receptors, such as insulin receptor,13 the transferrin receptor (TfR),14 and the low-density lipoprotein receptor-related protein (LRP).15 In principle, any ligand that is capable of binding to these receptors can facilitate the transportation of NPs into brain through this RMT process and thereby enhance the therapeutic effects.16,17
After being delivered into brain, the NPs have to selectively accumulate into the tumor tissue to exert the therapeutic effect while the access to surrounding healthy tissues should be minimized, thus increasing the therapeutic index.18 Towards this goal, the NPs need to be equipped with another targeting ligand that can promote the drug accumulation into tumor cells. Indeed, this so-called cascade dual-targeting strategy has been demonstrated to be a viable approach for glioma therapy, where the first-stage targeting ligand circumvents the BBB barrier and the second one targets to brain cancer cells for selective drug delivery.19–22 So far, various materials and active ligands have been employed to fabricate such NPs system.23–32 For this design, the targeting ligands are the crucial component. Based on the choice of targeting ligands, the dual targeting delivery system can be generally categorized into two types: (1) targeting BBB and tumor cells using different ligands, in which each ligand binds distinct receptor at different sites; and (2) dual targeting with one ligand, where the ligand can directly target both the BBB and brain tumor because its receptor is over-expressed on both types of cells.
The key of construction dual targeting NPs is to attach the active ligands on NPs surface. Two methods are currently available for ligands attachments. For example, the ligands can be directly conjugated to the surface of NPs. While several successful examples have been reported via this method,28,33,34 a few technique challenges have been met, such as NP aggregation, drug leakage and difficulty of precisely tuning the ratio of each ligand on particle surface. Alternatively, the target ligands can be linked to the hydrophilic fragment of the polymer materials, and these modified materials were used as building block for the NPs assembly. While this method can bypass the above-mentioned issues, extensively chemical synthesis steps are usually required.35
To address this dilemma, herein we reported a general way of construction dual targeting delivery system by simply using lipid capped polymer nanoparticles, with poly lactic-co-glycolic acid (PLGA) as the core and lipids (lecithin and DSPE–PEG2000–COOH) as the shell. The PLGA core is able to load hydrophobic drugs with high efficiency, and the DSPE–PEG2000–COOH provides a carboxyl group for ligand conjugation. The most notable merit of such strategy is that the ligands can be directly conjugated to DSPE–PEG2000–COOH fragment before or after NPs formulation, with a single synthesis step.
As a proof of concept, the angiopep-2 (ANG-2) was used as the first-stage targeting ligand to facilitate the transportation of NPs into brain. ANG-2 is a peptide derived from the Kunitz domain of aprotinin,36 which can bind LRP that expresses on the membrane of BBB.37,38 Because of this, it has been used to construct the BBB transportation NPs by several research groups.23,26,39–41 In addition, we also utilized a second binding ligand of a DNA aptamer called AS1411 to achieve accurate glioma cells targeting. Aptamer (Apt) is a type of artificial single-stranded oligonucleotide that can bind to a wide range of targets with high specificity and affinity.42–44 AS1411 is a G-rich DNA aptamer that binds to nucleolin, a protein which is located in the nucleus and highly expressed in the plasma membrane of many cancer cells such as glioma cells.45–48
In this study, we established a cascade dual-targeting NPs platform by using lipid capped polymer nanoparticles. Both ANG-2 and aptamer AS1411 were used as binding ligands for NPs functionalization, and the doxorubicin (DOX) as a model drug to evaluate the anti-cancer effect. The resulting dual targeting nanoparticles showed receptors-mediated cellular uptake, significantly enhanced BBB penetration and anti-proliferation effect in vitro. This work provides a facile way of construction dual targeting delivery system for chemotherapy of gliomas.
2 Materials and methods
2.1 Materials
Carboxyl-terminated PLGA (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, Mw: 20000 Da, inherent viscosity 0.21 dL g−1) was purchased from Shandong institute of medical instrument (Shandong, China). Soybean lecithin consisting of 90–95% phosphatidylcholine was obtained from the LIPOID group (Ludwigshafen, Germany). DSPE–PEG2000–COOH (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-carboxy (polyethyleneglycol) 2000) was obtained from NANOCS Inc. (Boston, USA). N-Hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), polyvinyl alcohol (PVA, 87–90% hydrolyzed, MW of 30–70 kDa) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Doxorubicin (DOX) was obtained from Biochempartner Co., Ltd. (Shanghai, China). 5′ amino functionalized AS1411 aptamer (sequence: 5′-GGTGGTGGTGGTTGTGGTGGTGGTGG-3′) was synthesized by Sangon Biotech (Shanghai, China). Angiopep-2 (TFFYGGSRGKRNNFKTEEY) was synthesized by Chinapeptides Co., Ltd. (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS) and 0.25% (w/v) trypsin solution were purchased from Gibco BRL (Grand Island, NY, USA). Penicillin–streptomycin solution and 4′,6-diamidino-2-phenylindole (DAPI) were provided by Solarbio Biotech, Co.,Ltd. (Beijing, China). All other reagents of analytical pure grade and chromatographic pure grade were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
:50, Mw: 20000 Da, inherent viscosity 0.21 dL g−1) was purchased from Shandong institute of medical instrument (Shandong, China). Soybean lecithin consisting of 90–95% phosphatidylcholine was obtained from the LIPOID group (Ludwigshafen, Germany). DSPE–PEG2000–COOH (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-carboxy (polyethyleneglycol) 2000) was obtained from NANOCS Inc. (Boston, USA). N-Hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), polyvinyl alcohol (PVA, 87–90% hydrolyzed, MW of 30–70 kDa) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Doxorubicin (DOX) was obtained from Biochempartner Co., Ltd. (Shanghai, China). 5′ amino functionalized AS1411 aptamer (sequence: 5′-GGTGGTGGTGGTTGTGGTGGTGGTGG-3′) was synthesized by Sangon Biotech (Shanghai, China). Angiopep-2 (TFFYGGSRGKRNNFKTEEY) was synthesized by Chinapeptides Co., Ltd. (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS) and 0.25% (w/v) trypsin solution were purchased from Gibco BRL (Grand Island, NY, USA). Penicillin–streptomycin solution and 4′,6-diamidino-2-phenylindole (DAPI) were provided by Solarbio Biotech, Co.,Ltd. (Beijing, China). All other reagents of analytical pure grade and chromatographic pure grade were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
2.2 Cell lines and culture conditions
C6 glioma cell line (from rat), brain capillary endothelial cells (BCECs, from rat) and astrocyte cells (from rat) were obtained from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). The cells were routinely cultured in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin solution. All the cells were cultured in incubators maintained at 37 °C under a humidified atmosphere containing 5% CO2. All experiments were performed on cells in the logarithmic phase of growth.2.3 Preparation of NPs
The DOX free base was obtained by reacting DOX hydrochlorate with 3 molar equivalents of triethylamine (TEA) in 12.5% (v/v) methanol in chloroform solution overnight.49 The DOX-loaded NPs were prepared by the emulsion/solvent evaporation method.45,50 In a typical procedure, 20 mg of PLGA and 1 mg of DOX were dissolved in 2 mL of 12.5% (v/v) methanol in chloroform solution. The resultant organic solution was emulsified in 6 mL of aqueous solution containing 1% PVA (w/v), 2.1 mg soybean lecithin and 0.9 mg DSPE–PEG2000–COOH to form an oil-in-water emulsion by sonication (120 W) for 2 min on ice bath using probe sonication (Branson, USA). The emulsion was stirred overnight at room temperature on a magnetic stir plate to allow evaporation of the organic solvent and the formation of NPs. NPs were centrifuged at 20000 rpm for 20 min at 4 °C (TGL 20M, Hunan, China), and washed 3 times with deionized water to remove unbound PVA and free DOX. Then, the NPs were re-dispersed in deionized water for further use.
The conjugation of ANG-2 and Apt to the surface of NPs were performed by pre-functionalization strategy using an EDC/NHS technique. Briefly, the carboxyl unit of DSPE–PEG2000–COOH was activated by EDC and NHS for 0.5 h and then reacted with amino-modified ANG-2 or AS1411 (the molar ratio of DSPE–PEG2000–COOH to ANG-2 or AS1411 was 1:1) in 1 mL of PBS (pH 7.4) for 4 h in the dark. The conjugates were transferred into an ultraltration tube (MWCO is 10 kD and 5 kD for AS1411 and ANG-2 conjugation, respectively) and centrifuged at 3000 rpm for 20 min, and then washed with deionized water until there was no absorption in the filtrate to obtain pure DSPE–PEG2000–ANG-2 and DSPE–PEG2000–Apt. AS1411 modified NPs (Apt–NPs/DOX) and ANG-2 modified NPs (ANG–NPs/DOX) were prepared with the same emulsion/solvent evaporation method as described above using the respectively modified DSPE–PEG2000. To prepare ANG-2 and AS1411 dual-functionalized NPs (AA–NPs/DOX) a mixture of DSPE–PEG2000–ANG-2 and DSPE–PEG2000–Apt (at 1:1 ratio) were used.
2.4 Characterization of NPs
| DL (%) = amount of loaded DOX in the NPs/weight of NPs × 100 |
| EE (%) = amount of loaded DOX in the NPs/total amount of DOX added × 100 |
2.5 In vitro cellular uptake studies
2.6 Construction of in vitro BBB model and transendothelial transport experiments
The in vitro BBB model was established by using the co-culture of BCECs and astrocyte cells as previously described.51 Transwell (12-well, pore size 0.4 μm, Corning, New York, USA) was used to establish BBB model. Astrocyte cells (5 × 105 cells per mL) were added to the underside of the Transwell chamber and incubated for 24 h to adhere to the wall. The Transwell chamber was carefully turned upright and placed in a 12-well plate. When the astrocyte cells reached 80% confluence under an inverted microscope, BCECs (5 × 105 cells per mL) were seeded on the upper side of the Transwell chamber at 37 °C and 5% CO2. The cells were observed under an inverted microscope until astrocyte cells and BCECs were co-cultured to a high density. The co-culture medium was changed three times a week. Transepithelial electrical resistance (TEER) was detected by millicell ERS (Millipore, USA) in the co-culture model at 2, 4, 6, 8, 10, 12, 14 d after BCECs seeding. Only the co-culture model with TEER value constant above 200 Ω cm2 was selected for experiment.At the beginning of the transport experiments, the medium in the donor chamber was replaced with 1.5 mL of FBS-free DMEM containing AA–NPs/DOX, ANG–NPs/DOX, Apt–NPs/DOX, NPs/DOX or free DOX at a concentration of 50 μg mL−1 of DOX. Then cells were incubated on a platform, shaking at 50 rpm at 37 °C. At 1, 6, 12, and 24 h, 400 μL of sample medium was taken from the acceptor chamber and an equal volume of FBS-free DMEM was added to the acceptor chamber for compensation. The samples were freeze dried and the total amount of DOX was analyzed by HPLC after methanol treatment to release DOX in NPs.
2.7 In vitro cytotoxicity assay
In vitro cytotoxicity of NPs and free DOX against C6 glioma cells were evaluated by the MTT assay. Cells were seeded at a density of 5 × 104 cells per mL in a 96-well plate and incubated for 24 h. After pre-incubation, cells were treated with AA–NPs/DOX, ANG–NPs/DOX, Apt–NPs/DOX, NPs/DOX and free DOX with concentrations of 0.01, 0.05, 0.1, 0.5, 1, 5 and 10 μM for 48 h in quadruplicate. Untreated cells were used as controls. Then, 20 μL of MTT solution (5 mg mL−1) was added followed by incubation for another 4 h. Then, the supernatant of each well was removed and 100 μL of DMSO was added to dissolve the metabolized product MTT formazan. Finally, the plate was shaken for 10 minutes, and optical density (OD) of the formazan product was measured by a microplate reader (Infinite M200, Tecan, Switzerland) at 570 nm. The cell viability and the half-maximal inhibitory concentration (IC50) were calculated.2.8 Statistical analysis
Analysis was performed using SPSS19.0 software. One-way ANOVA analysis of variance was used to determine the statistical significance of differences among groups. P < 0.05 was considered statistically significant in all cases.3 Results and discussion
3.1 Preparation and characterizations of AA–NPs/DOX
Fig. 1A showed the scheme of the preparation and structure of our drug delivery system. The nanoparticles contain a biocompatible and biodegradable PLGA core for DOX loading. It is known that PLGA core could significantly enhance hydrophobic drugs encapsulation and retard their diffusion, leading to high drug loading efficiency and sustained drug release.52 The particle surface was sealed by a polyethylene glycol (PEG) functionalized lipid monolayer, which mainly serves two roles: first, the PEG chain not only provides steric hindrance to increase colloidal stability of NPs and reduce protein adsorption,53–55 but also protects the internal PLGA from directly contacting with water thus impeding hydrolysis; in addition, the terminal carboxyl group PEG offers an anchor for further conjugation of Apt and ANG-2. | ||
| Fig. 1 (A) A scheme showing the preparation and structure of the AA–NPs. Inset: the appearance of the final AA–NPs. (B) DLS measurement of the AA–NPs. Inset: TEM image of AA–NPs; the bar represents 100 nm. (C) 1H-NMR spectrum of PLGA NPs and AA–NPs. The structure of DSPE–PEG2000–ANG/Apt and PLGA were also shown. | ||
The nanoparticles were prepared using a standard emulsion/solvent evaporation method as we described previously.45,56 Briefly, the hydrophobic PLGA and DOX were dissolved in organic phase of methanol/chloroform, and then added to the aqueous phase of lipids solution in a dropwise manner. Since PLGA is insoluble in water, it self-assembled into NPs for DOX encapsulation, and the particle surface was capped with the lipids. To conjugate the active ligands on the surface of NPs, two different methods can be used. We first try to directly attach the ligands on above prepared NPs. However, this method resulted in poor NP stability during the reaction, which is likely due to the compromised lipid layer integrity during the conjugation. In addition, we observed significant particle size increase and drug leakage after ligand functionalization (data not shown). We therefore referred to the pre-conjugation method as shown in Fig. 1A, where the ligands were conjugated to the free lipid first prior to the NPs preparation. The conjugate efficiency for both Apt and ANG-2 were determined to be ∼50% (Fig. S1 and S2†). After purification, these functionalized lipids can be used individually to prepare single targeting Apt–NPs (with AS1411) and ANG–NPs (with ANG-2) respectively, or a combination of these two lipids resulted in dual targeting NPs of AA–NPs.
The as-prepared AA–NPs were semi-transparent with red opalescence (Fig. 1A), and the color is attributable to DOX loading. The particle size of NPs, Apt–NPs, ANG–NPs and AA–NPs were approximately 160 nm with a narrow distribution (PDI < 0.1) (Table 1), and a representative measurement of AA–NPs was shown in Fig. 1B. The zeta potential of the NPs was approximately −12 mV. It is known that, to rely on the electric charge for NPs stabilization, the zeta potential needs to be kept >25 mV (positive or negative). Therefore, the major force to stabilize the NPs is likely due to the steric hinderance derived from PEG, rather than electrostatic repulsion. Note that the Apt and ANG conjugation have little effect on particle size and zeta potential. It is reasonable as the DSPE–PEG2000–COOH component accounts only for a small fraction of nanoparticle structure (∼4% by mass), and the negative charge is mainly originated from the terminal carboxyl group on PLGA.57 Thus, the colloidal stability of NPs was not affected by active ligands functionalization. The EE and DL were also unaffected by the ligands conjugation to remain at ∼44% and ∼1.7% respectively, which is reasonable as the drug was mainly loaded at the PLGA core. The particle morphology of AA–NPs was further determined by transmission electron micrographs (TEM), indicating the NPs spherical with homogenous size (inset of Fig. 1B). We notice that the particle size measured by TEM was slightly smaller (∼100 nm) than that of DLS detection, which is due to the particle dehydration before TEM measurement. The 1H-NMR was used to confirm that the ligands functionalized DSPE–PEG2000–COOH was attached to the surface of NPs (Fig. 1C). Compared with PLGA NPs (NPs prepared without the addition of DSPE–PEG2000–ANG/Apt), AA–NPs showed a characteristic peak at 3.5 ppm, which is attributed to the methylene protons in DSPE–PEG2000–ANG/Apt. Therefore, the DSPE–PEG2000–ANG/Apt fragment was successful inserted into the NPs structure and the dual targeting delivery system was developed.
| Formulations | Size (nm) | PDI | ζ-Potential (mV) | EE (%) | DL (%) |
|---|---|---|---|---|---|
| NPs | 161.3 ± 2.5 | 0.052 ± 0.03 | −11.4 ± 0.7 | 44.07 ± 0.5 | 1.69 ± 0.04 |
| Apt–NPs | 168.5 ± 1.8 | 0.069 ± 0.024 | −12.8 ± 0.6 | 44.02 ± 1.83 | 1.67 ± 0.19 |
| ANG–NPs | 163.5 ± 1.3 | 0.072 ± 0.031 | −12.4 ± 0.6 | 44.71 ± 3.20 | 1.68 ± 0.14 |
| AA–NPs | 167.2 ± 1.9 | 0.085 ± 0.035 | −13.7 ± 1.2 | 43.42 ± 1.67 | 1.66 ± 0.12 |
In vitro drug release of DOX from different NPs was carried out in the medium of phosphate buffer solution (PBS, pH 7.4) to simulate the physiological environment. Over 80% of the free DOX diffusively cross the dialysis membrane after 4 h, and the process was completed within 8 h (>95% release) (Fig. 2, black trace). By contrast, after encapsulation into nanoparticles, a remarkable sustained drug release was observed. The drug release performance of different nanoparticle formations was almost identical (Fig. 2), which can be explained by that the modifications occurred at the particle surface, while DOX was mainly trapped in the PLGA core. An initial drug release from NPs was observed in the first 24 h (∼40%), followed by a much slower rate thereafter. The cumulative drug release after 140 h was about 50% for different NPs. This drug release performance follows a typical profile of polymer-based NPs,58 suggesting the successful encapsulation of DOX in PLGA NPs core. This sustained drug release property allows for decreased administrating frequency and thus enhances the patient compliance.
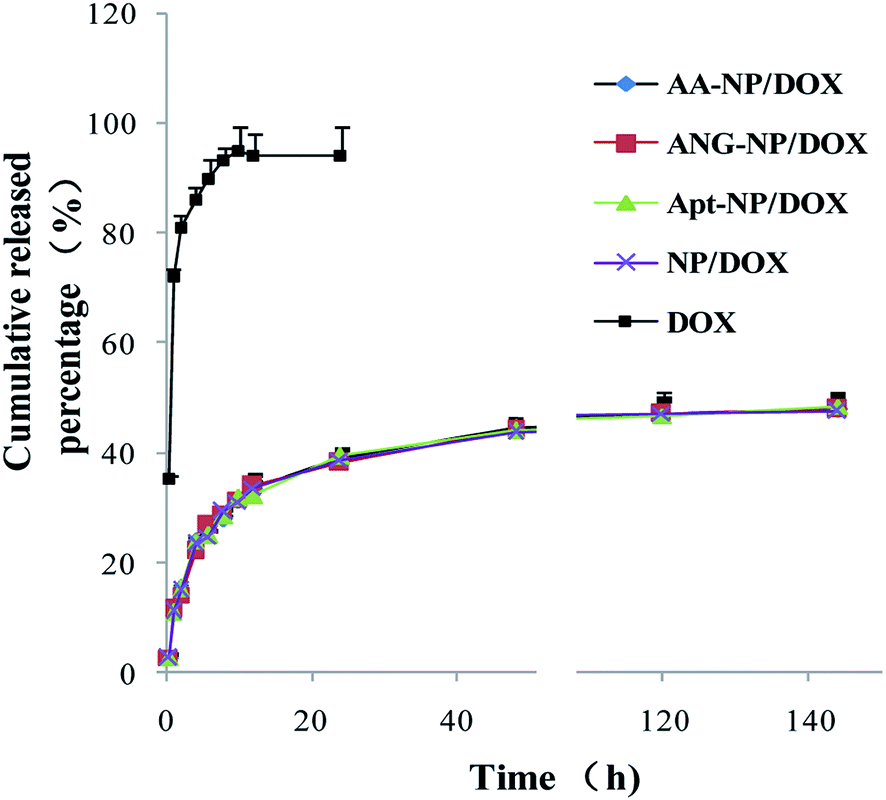 | ||
| Fig. 2 In vitro drug release of free DOX and different nanoparticle formulations as a function of time. Data were shown as mean ± SD (n = 3). | ||
3.2 Cell uptake studies
So far, we have constructed nanoparticles with surface dual functionalized with ANG-2 and AS1411 using a simple yet controllable method. The cell uptake experiments were then designed to evaluate the targeting efficiency of these active ligands through fluorescent microscopy and flow cytometry in vitro. It has been reported that BCECs have a relatively high level of LRP expression on cell surface,38,59 and ANG-2 can specifically bind to LRP for transcytosis of the brain delivery.15,36 Therefore, BCECs was used as a model to study the targeting ability of ANG-2 on nanoparticle surface (Fig. 3A).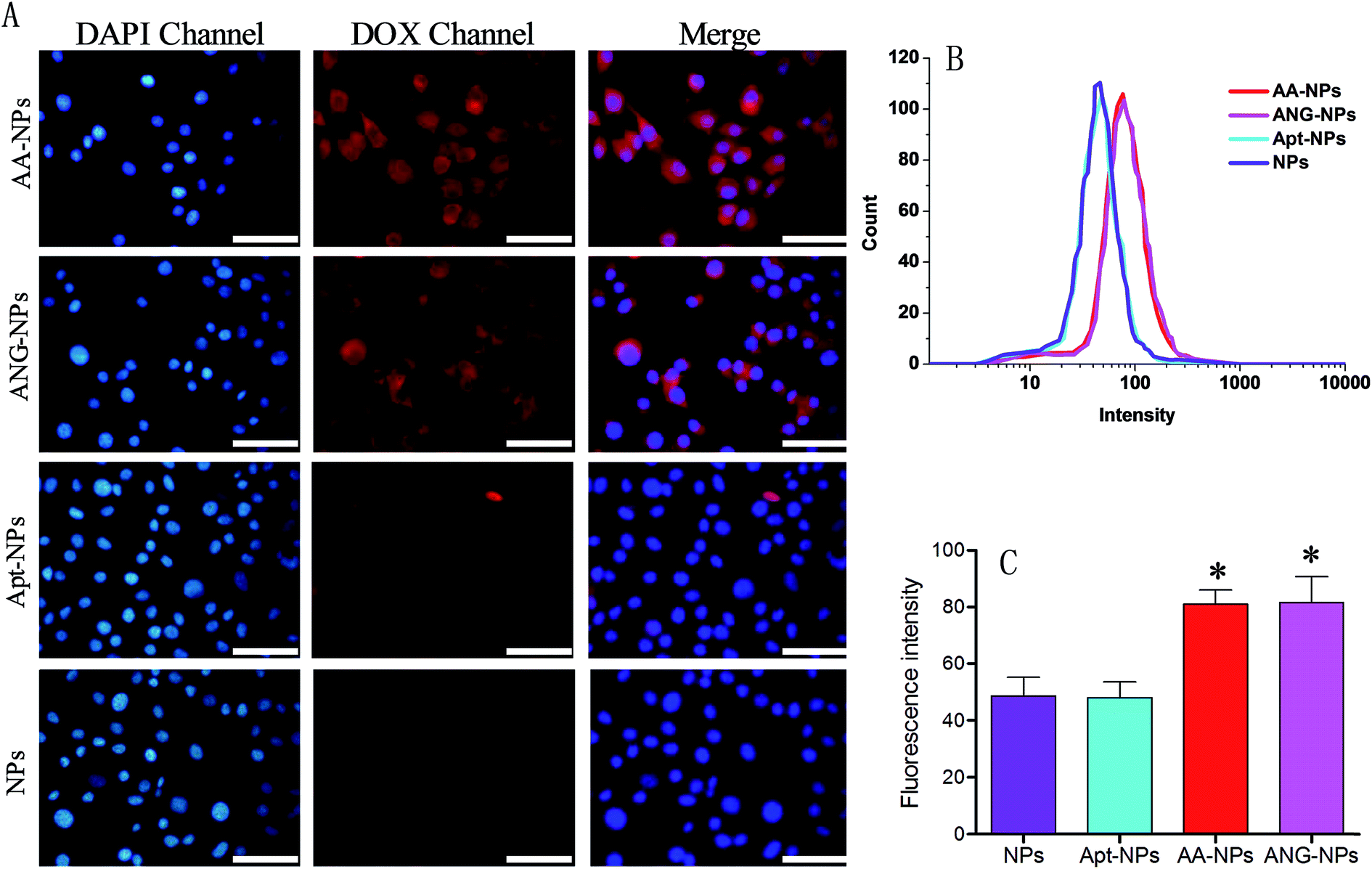 | ||
| Fig. 3 (A) Fluorescence microscopy showing the uptake of AA–NPs, ANG–NPs, Apt–NPs and NPs by BCECs. (B and C) Quantitative flow cytometry test of cell uptake for AA–NPs, ANG–NPs, Apt–NPs and NPs by BCECs (n = 3, *P < 0.05, compared with NPs). Bar, 50 μm. | ||
To localize cells, cell nuclei were stained blue by DAPI (Fig. 3A, lane 1). The NPs were internalized by endocytosis, and were expected to be collapsed and digested by endosomes and lysosomes to release DOX. Therefore, the relative extent of cell uptake of NPs can be indicated by the intrinsic red fluorescence of DOX. The red fluorescence was observed in the cytoplasm for all test groups (Fig. 3A, lane 2), signifying the successful internalization of the NPs, otherwise the fluorescence would be observed mostly on the contour of the cells. Specifically, after incubating with unmodified NPs, only moderate red fluorescence was observed inside cells. A similar uptake was observed for Apt modified NPs. With ANG-2 modification, on the other hand, the ANG–NPs/DOX exhibited obviously stronger red fluorescence signal inside BCECs cells, suggesting the role of ANG-2 in assisting cellular uptake. This result was consistent with previous work that the surface ANG-2 conjugation could enhance cell uptake in intro via LRP-mediated endocytosis.26
For the dual modified NPs (i.e. AA–NPs), we also observed significantly brighter red fluorescence compared with ANG-2 lacking NPs. Therefore, the presence of Apt on NPs surface does not hamper the target binding ability of ANG-2. It should be noted that the AA–NPs was constructed using a mixture of DSPE–PEG2000–ANG-2 and DSPE–PEG2000–Apt at 1:1 molar ratio. Therefore, the surface density of ANG-2 on AA–NPs was only half that of ANG–NPs, while their cell uptake efficiencies were comparable. We rationalized this by that the amount of ANG-2 on AA–NPs is already saturate for cell binding, and after that no further enhancement can be achieved by increasing ANG-2 density. Indeed, similar phenomenon was also observed in our previous work by using another binding ligand for tumor targeting delivery.50 To have a better comparison, we merged the fluorescence of DAPI and DOX channels for different groups, and the results were presented in the third lane of Fig. 3A. It is clearly showed that ANG-2 modified NPs (i.e. AA–NP and ANG–NPs) have relatively higher uptake in each cell compared with ANG-2 free NPs (i.e. Apt–NPs and NPs).
Quantitative results were obtained from flow cytometry (Fig. 3B and C). In consistent with the above observations, the NPs decorated with ANG-2 displayed fluorescence significantly stronger than those treated with unmodified NPs (∼1.7-fold higher). In addition, Apt and ANG-2 dual decorated NPs (AA–NPs) also showed higher fluorescence, as the case of ANG–NPs.
The target efficiency of AS1411 functionalized NPs was similarly studied by using nucleolin over-expressed C6 glioma cells (Fig. 4A).60 In this case, the Apt modified NPs (i.e., AA–NPs and Apt–NPs) displayed higher level of red fluorescence than those of Apt lacking NPs inside cells, demonstrating that the cellular association of NPs was facilitated by the specific interaction between AS1411 and nucleolin. We also quantified the relative cell uptake by using flow cytometry (Fig. 4B and C). Based on the quantification result, ANG-2 modified NPs also showed a marginally increased fluorescence compared with unmodified NPs (1.5-fold higher). This is because LRP1 is also expressed on glioma cells.15,37 Therefore, ANG-2 modification also gives rise to enhanced NPs uptake by glioma cells. Because of its dual targeting ability (both to BBB and glioma cells), ANG-2 has been widely employed as ligand in brain tumor drug delivery.33,34,61 With Apt decoration, the Apt–NPs displayed even stronger intensity in C6 glioma cells, with 2.4-fold higher than that of unmodified NPs. Therefore, compared with ANG-2, AS1411 has better binding affinity to glioma cells. Interestingly, dual modifying NPs with ANG-2 and AS1411 further enhances the fluorescence slightly, suggesting a synergistic effect of these two ligands for interacting with C6 glioma cells. Collectively, the dual targeting property of the AA–NPs has been demonstrated for both BCECs and C6 glioma cells.
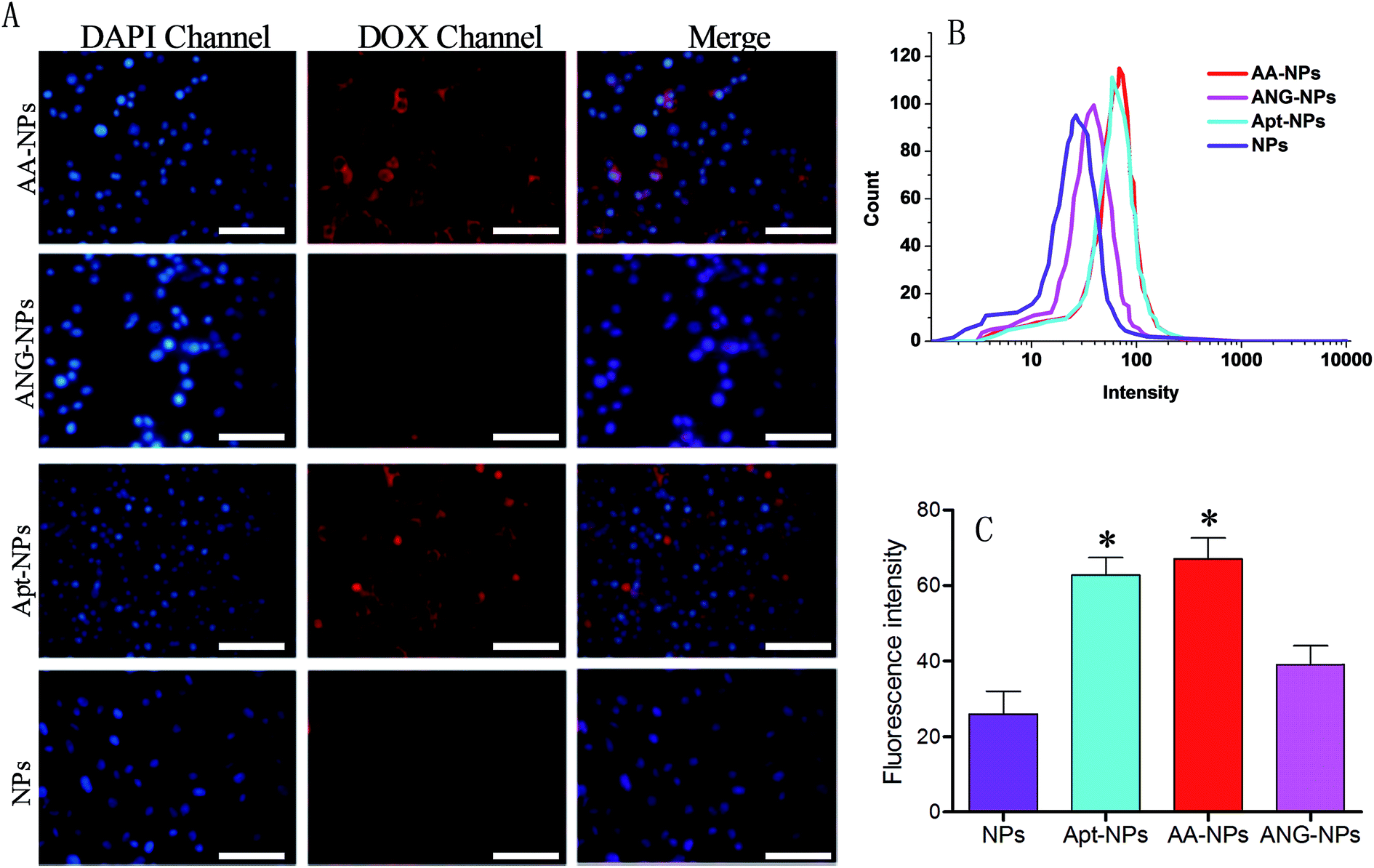 | ||
| Fig. 4 (A) Fluorescence microscopy showing the uptake of AA–NPs NPs, ANG–NPs, Apt–NPs and NPs by C6 glioma cells. (B and C) Quantitative flow cytometry test of cell uptake for AA–NPs, ANG–NPs, Apt–NPs and NPs by C6 glioma cells (n = 3, *P < 0.05, compared with NPs). Bar, 50 μm. | ||
3.3 In vitro evaluation of BBB penetration
Having demonstrated the dual targeting efficiency of the AA–NP/DOX, we next studied in vitro BBB penetration ability. BCECs and astrocyte cells were chosen to construct the in vitro BBB model in this study. The permeability of the co-culture model was monitored by transepithelial electrical resistance (TEER), which reflects the structural integrity of the in vitro BBB model. We measured the TEER as a function of time after BCECs seeding. The TEER value gradually increased over time until reaching a plateau of ∼260 Ω cm2 (Fig. 5A), indicating a successful construction of in vitro BBB model. Once the TEER value was constant, different NPs formulations were added to the interior of the Transwell chamber and the transportation of total DOX was measured by HPLC after culturing various hours at 37 °C and 5% CO2.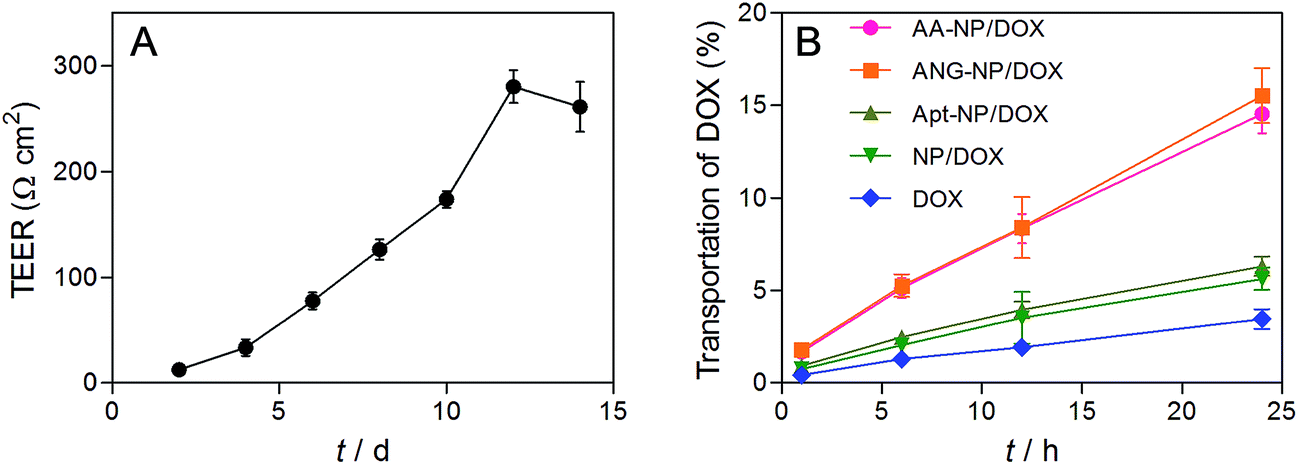 | ||
| Fig. 5 (A) Evaluation of in vitro BBB model by the TEER value. (B) BBB transport efficiency of different formulations. Data were shown as mean ± SD (n = 3). | ||
The transport percentage of DOX across the BBB model over a period of 24 h were shown in Fig. 5B. For all groups, the amount of DOX transportation was in a time-dependent fashion. Longer incubation time results in higher amount of DOX transportation. After 24 h incubation, the transport percentages were calculated to be 14.6%, 15.5%, 6.3%, 5.8% and 3.6% for AA–NPs, ANG–NPs, Apt–NPs, and free DOX, respectively. The transportation of free DOX is the lowest, which is because it is passively diffused into BBB cells and can be easily effluxed by active transporters.10 After formulation of NPs, the accumulative delivery of DOX is moderately higher, which is likely due to the endocytosis mechanism of NPs by cells. Notably, with ANG-2 modification on NPs surface, we observed remarkably stronger transportation, which is ∼2.7- and 4.3-fold higher than that of ANG-2 lacking NPs and free DOX, respectively. This result is consistent with the above cell update study, showing that the transport of DOX across the BBB model was significantly increased by ANG-2 modification. In addition, the AA–NPs achieved a comparable DOX transportation efficiency to that of ANG–NPs, further confirming the largely retained functionality of ANG together with AS1411 modification.
3.4 In vitro cytotoxicity assay
Finally, the in vitro cytotoxicity of free DOX and different DOX loaded NPs were studied by MTT assay with C6 glioma cells (Fig. 6). In general, the cytotoxicity progressively increased with higher DOX concentration after 48 h incubation, based on which the IC50 values of each test group were calculated. The IC50 values of AA–NPs, ANG–NPs, Apt–NPs, NPs and free DOX were 0.91, 3.26, 0.78, 6.18, 1.37 μM, respectively. Note that, free DOX showed even stronger inhibitory effect than non-Apt modified NPs, with IC50 value 4.5- and 2.4-fold lower than those of NPs and ANG–NPs, respectively. Indeed, this result was consistent with previous reports.62,63 We rationalized this phenomenon by that free DOX could rapidly diffuse into cancer cells to exert its cytotoxic function, while NPs need a time-consuming step to release DOX for activity. However, after Apt modification, the cytostatic activity of NPs was strongly enhanced. For example, the Apt–NPs and AA–NPs achieved IC50 1.76- and 1.5-fold lower than that of free DOX, respectively. This improved cytotoxicity can be ascribed to the specific interaction between AS1411 aptamer and nucleolin C6 glioma cell membrane, which tremendously enhances the cell uptake of the NPs (P < 0.05).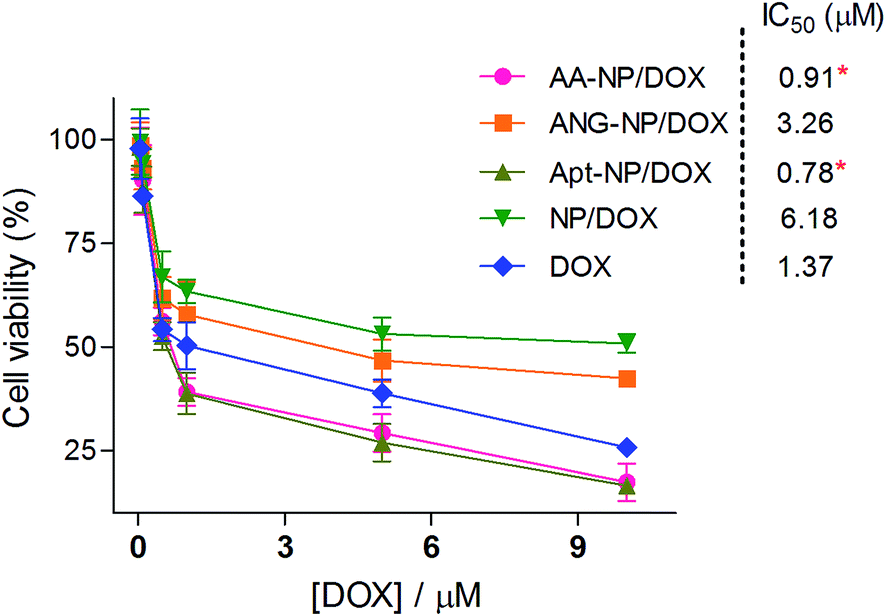 | ||
| Fig. 6 In vitro anti-proliferation effect of different formulations on C6 cells after 48 h incubations. Data were shown as mean ± SD (n = 4, *P < 0.05, as compared with free DOX). | ||
4 Conclusion
In summary, we have developed a robust strategy to construct dual targeting NPs for anti-glioma therapy. Two active ligands of ANG-2 and AS1411 Apt were successfully conjugated to NPs surface through a simple chemical technique with high efficiency and little perturbation on NPs quality. Compared with the unmodified NPs, AA–NPs exhibited significantly higher cellular uptake in both BCECs and C6 glioma cells, improved ability to penetrate the BBB, and better inhibitory effect toward C6 glioma cells. All these results indicated that such lipid capped polymer NPs with active ligands functionalization are promising anticancer drug delivery system for glioma treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Scientific Research Starting Foundation from Central South University and Hunan Scientific Research Plan for Traditional Chinese Medicine (201834).References
- M. L. Goodenberger and R. B. Jenkins, Cancer Genet., 2012, 205, 613–621 CrossRef CAS PubMed.
- Q. T. Ostrom, L. Bauchet, F. G. Davis, I. Deltour, J. L. Fisher, C. E. Langer, M. Pekmezci, J. A. Schwartzbaum, M. C. Turner, K. M. Walsh, M. R. Wrensch and J. S. Barnholtz-Sloan, Neuro-Oncology, 2014, 16, 896–913 CrossRef CAS PubMed.
- M. P. Pinto, M. Arce, B. Yameen and C. Vilos, Nanomedicine, 2017, 12, 59–72 CrossRef CAS PubMed.
- C. Adamson, O. O. Kanu, A. I. Mehta, C. Di, N. Lin, A. K. Mattox and D. D. Bigner, Expert Opin. Invest. Drugs, 2009, 18, 1061–1083 CrossRef CAS PubMed.
- B. Y. S. Ong, S. H. Ranganath, L. Y. Lee, F. Lu, H. S. Lee, N. V. Sahinidis and C. H. Wang, Biomaterials, 2009, 30, 3189–3196 CrossRef CAS PubMed.
- M. G. Hart, R. Grant, R. Garside, G. Rogers, M. Somerville and K. Stein, Cochrane Database Syst. Rev., 2011, 25(3), CD007294 Search PubMed.
- J. M. Pipas, L. P. Meyer, C. H. Rhodes, L. D. Cromwell, C. E. McDonnell, L. S. Kingman, J. R. Rigas and C. E. Fadul, J. Neuro-Oncol., 2005, 71, 301–305 CrossRef CAS PubMed.
- W. M. Pardridge, Mol. Interventions, 2003, 3, 90–105 CrossRef CAS PubMed.
- R. Karim, C. Palazzo, B. Evrard and G. Piel, J. Controlled Release, 2016, 227, 23–37 CrossRef CAS PubMed.
- W. M. Pardridge, J. Cereb. Blood Flow Metab., 2012, 32, 1959–1972 CrossRef CAS PubMed.
- C. Saraiva, C. Praca, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 34–47 CrossRef CAS PubMed.
- N. J. Abbott, J. Inherited Metab. Dis., 2013, 36, 437–449 CrossRef CAS PubMed.
- W. M. Pardridge, Y. S. Kang, J. L. Buciak and J. Yang, Pharm. Res., 1995, 12, 807–816 CrossRef CAS.
- L. Descamps, M. P. Dehouck, G. Torpier and R. Cecchelli, Am. J. Physiol., 1996, 270, H1149–H1158 CAS.
- M. Demeule, J. C. Currie, Y. Bertrand, C. Che, T. Nguyen, A. Regina, R. Gabathuler, J. P. Castaigne and R. Beliveau, J. Neurochem., 2008, 106, 1534–1544 CrossRef CAS PubMed.
- F. Zhang, C. L. Xu and C. M. Liu, Drug Des., Dev. Ther., 2015, 9, 2089–2100 CrossRef PubMed.
- O. van Tellingen, B. Yetkin-Arik, M. C. de Gooijer, P. Wesseling, T. Wurdinger and H. E. de Vries, Drug Resist. Updates, 2015, 19, 1–12 CrossRef CAS PubMed.
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS PubMed.
- A. Miranda, M. J. Blanco-Prieto, J. Sousa, A. Pais and C. Vitorino, Int. J. Pharm., 2017, 531, 389–410 CrossRef CAS PubMed.
- H. L. Gao, J. Neuroimmune Pharmacol., 2017, 12, 6–16 CrossRef PubMed.
- H. L. Gao, Acta Pharm. Sin. B, 2016, 6, 268–286 CrossRef PubMed.
- H. L. Gao, Z. Q. Pang and X. G. Jiang, Pharm. Res., 2013, 30, 2485–2498 CrossRef CAS PubMed.
- H. Gao, S. Zhang, S. Cao, Z. Yang, Z. Pang and X. Jiang, Mol. Pharm., 2014, 11, 2755–2763 CrossRef CAS PubMed.
- D. Miao, M. Jiang, Z. Liu, G. Gu, Q. Hu, T. Kang, Q. Song, L. Yao, W. Li, X. Gao, M. Sun and J. Chen, Mol. Pharm., 2014, 11, 90–101 CrossRef CAS PubMed.
- Q. Hu, G. Gu, Z. Liu, M. Jiang, T. Kang, D. Miao, Y. Tu, Z. Pang, Q. Song, L. Yao, H. Xia, H. Chen, X. Jiang, X. Gao and J. Chen, Biomaterials, 2013, 34, 1135–1145 CrossRef CAS PubMed.
- H. Xin, X. Jiang, J. Gu, X. Sha, L. Chen, K. Law, Y. Chen, X. Wang, Y. Jiang and X. Fang, Biomaterials, 2011, 32, 4293–4305 CrossRef CAS PubMed.
- P. Figueiredo, V. Balasubramanian, M. A. Shahbazi, A. Correia, D. Wu, C. G. Palivan, J. T. Hirvonen and H. A. Santos, Int. J. Pharm., 2016, 511, 794–803 CrossRef CAS PubMed.
- H. L. Gao, J. Qian, S. J. Cao, Z. Yang, Z. Q. Pang, S. Q. Pan, L. Fan, Z. J. Xi, X. G. Jiang and Q. Z. Zhang, Biomaterials, 2012, 33, 5115–5123 CrossRef CAS PubMed.
- H. L. Gao, Z. Yang, S. J. Cao, Y. Xiong, S. Zhang, Z. Q. Pang and X. G. Jiang, Biomaterials, 2014, 35, 2374–2382 CrossRef CAS PubMed.
- H. L. Gao, Z. Yang, S. Zhang, S. J. Cao, Z. Q. Pang, X. Yang and X. G. Jiang, J. Controlled Release, 2013, 172, 921–928 CrossRef CAS PubMed.
- H. L. Gao, Y. Xiong, S. Zhang, Z. Yang, S. J. Cao and X. G. Jiang, Mol. Pharm., 2014, 11, 1042–1052 CrossRef CAS PubMed.
- G. Huile, P. Shuaiqi, Y. Zhi, C. Shijie, C. Chen, J. Xinguo, S. Shun, P. Zhiqing and H. Yu, Biomaterials, 2011, 32, 8669–8675 CrossRef PubMed.
- X. Y. Sun, Z. Q. Pang, H. X. Ye, B. Qiu, L. R. Guo, J. W. Li, J. F. Ren, Y. Qian, Q. Z. Zhang, J. Chen and X. G. Jiang, Biomaterials, 2012, 33, 916–924 CAS.
- D. L. Ni, J. W. Zhang, W. B. Bu, H. Y. Xing, F. Han, Q. F. Xiao, Z. W. Yao, F. Chen, Q. J. He, J. N. Liu, S. J. Zhang, W. P. Fan, L. P. Zhou, W. J. Peng and J. L. Shi, ACS Nano, 2014, 8, 1231–1242 CrossRef CAS PubMed.
- Y. C. Chen, C. F. Chiang, L. F. Chen, P. C. Liang, W. Y. Hsieh and W. L. Lin, Biomaterials, 2014, 35, 4066–4081 CrossRef CAS PubMed.
- M. Demeule, A. Regina, C. Che, J. Poirier, T. Nguyen, R. Gabathuler, J. P. Castaigne and R. Beliveau, J. Pharmacol. Exp. Ther., 2008, 324, 1064–1072 CrossRef CAS PubMed.
- L. Maletinska, E. A. Blakely, K. A. Bjornstad, D. F. Deen, L. J. Knoff and T. M. Forte, Cancer Res., 2000, 60, 2300–2303 CAS.
- S. Ito, S. Ohtsuki and T. Terasaki, Neurosci. Res., 2006, 56, 246–252 CrossRef CAS PubMed.
- W. Han, G. Yin, X. Pu, X. Chen, X. Liao and Z. Huang, J. Biomater. Sci., Polym. Ed., 2017, 28, 1695–1712 CrossRef CAS PubMed.
- C. Chen, Z. Duan, Y. Yuan, R. Li, L. Pang, J. Liang, X. Xu and J. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 5864–5873 CAS.
- Y. Li, X. Zheng, M. Gong and J. Zhang, Oncotarget, 2016, 7, 79401–79407 Search PubMed.
- W. Zhou, P. J. Huang, J. Ding and J. Liu, Analyst, 2014, 139, 2627–2640 RSC.
- A. B. Iliuk, L. Hu and W. A. Tao, Anal. Chem., 2011, 83, 4440–4452 CrossRef CAS PubMed.
- H. M. Meng, H. Liu, H. L. Kuai, R. Z. Peng, L. T. Mo and X. B. Zhang, Chem. Soc. Rev., 2016, 45, 2583–2602 RSC.
- W. Zhou, Y. Zhou, J. Wu, Z. Liu, H. Zhao, J. Liu and J. Ding, J. Drug Targeting, 2014, 22, 57–66 CrossRef CAS PubMed.
- J. Guo, X. Gao, L. Su, H. Xia, G. Gu, Z. Pang, X. Jiang, L. Yao, J. Chen and H. Chen, Biomaterials, 2011, 32, 8010–8020 CrossRef CAS PubMed.
- P. J. Bates, D. A. Laber, D. M. Miller, S. D. Thomas and J. O. Trent, Exp. Mol. Pathol., 2009, 86, 151–164 CrossRef CAS PubMed.
- F. Mongelard and P. Bouvet, Curr. Opin. Mol. Ther., 2010, 12, 107–114 CAS.
- S. P. Jiang, S. N. He, Y. L. Li, D. L. Feng, X. Y. Lu, Y. Z. Du, H. Y. Yu, F. Q. Hu and H. Yuan, Int. J. Nanomed., 2013, 8, 3141–3150 Search PubMed.
- J. Li, J. Mao, J. Tang, G. Li, F. Fang, Y. Tang and J. Ding, RSC Adv., 2017, 7, 22954–22963 RSC.
- Y. Wang, N. Wang, B. Cai, G. Y. Wang, J. Li and X. X. Piao, Neural Regener. Res., 2015, 10, 2011–2017 CrossRef PubMed.
- C. Yu, Y. Hu, J. Duan, W. Yuan, C. Wang, H. Xu and X. D. Yang, PLoS One, 2011, 6, e24077 CAS.
- J. Wang, X. Xing, X. Fang, C. Zhou, F. Huang, Z. Wu, J. Lou and W. Liang, Philos. Trans. R. Soc., A, 2013, 371, 20120309 CrossRef PubMed.
- D. C. Drummond, C. O. Noble, Z. Guo, M. E. Hayes, J. W. Park, C. J. Ou, Y. L. Tseng, K. Hong and D. B. Kirpotin, J. Pharmacol. Exp. Ther., 2009, 328, 321–330 CrossRef CAS PubMed.
- E. Kajiwara, K. Kawano, Y. Hattori, M. Fukushima, K. Hayashi and Y. Maitani, J. Controlled Release, 2007, 120, 104–110 CrossRef CAS PubMed.
- Z. Liu, H. Zhao, L. He, Y. Yao, Y. Zhou, J. Wu, J. Liu and J. Ding, RSC Adv., 2015, 5, 16931–16939 RSC.
- J. Q. Xi, L. Y. Da, C. S. Yang, R. Chen, L. Z. Gao, L. Fan and J. Han, Int. J. Nanomed., 2017, 12, 3331–3345 CrossRef PubMed.
- A. Mukerjee and J. K. Vishwanatha, Anticancer Res., 2009, 29, 3867–3875 CAS.
- R. D. Bell, A. P. Sagare, A. E. Friedman, G. S. Bedi, D. M. Holtzman, R. Deane and B. V. Zlokovic, J. Cereb. Blood Flow Metab., 2007, 27, 909–918 CrossRef CAS PubMed.
- J. Mosafer, M. Teymouri, K. Abnous, M. Tafaghodi and M. Ramezani, Mater. Sci. Eng., C, 2017, 72, 123–133 CrossRef CAS PubMed.
- J. Bruun, T. B. Larsen, R. I. Jolck, R. Eliasen, R. Holm, T. Gjetting and T. L. Andresen, Int. J. Nanomed., 2015, 10, 5995–6008 CAS.
- Y. Guo, Y. Zhang, J. Li, Y. Zhang, Y. Lu, X. Jiang, X. He, H. Ma, S. An and C. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 5444–5453 CAS.
- X. Ying, H. Wen, W. L. Lu, J. Du, J. Guo, W. Tian, Y. Men, Y. Zhang, R. J. Li, T. Y. Yang, D. W. Shang, J. N. Lou, L. R. Zhang and Q. Zhang, J. Controlled Release, 2010, 141, 183–192 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12376k |
| This journal is © The Royal Society of Chemistry 2018 |