
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Orthorhombic SnO2 phase observed composite (Sn1−xCex)O2 synthesized by sol–gel route†
M. H. Carvalho *a,
E. C. Pereirab and
A. J. A. de Oliveira*a
*a,
E. C. Pereirab and
A. J. A. de Oliveira*a
aPhysics Department, Federal University of São Carlos, UFSCar, P. O. Box 676, 13565-905, São Carlos, SP, Brazil. E-mail: adilson@df.ufscar.br; helenacarvalho@df.ufscar.br
bChemistry Department, LIEC, Federal University of São Carlos, UFSCar, P. O. Box 676, 13565-905, São Carlos, SP, Brazil
First published on 22nd January 2018
Abstract
Semiconductor oxides are a class of materials used in many different applications, such as catalysis, solar cells and magnetic devices. In particular, composites with different semiconducting oxides follow synergetic effects with changes of electrical conductivity or magnetic properties. In this case, the synthesis conditions are very important for controlling their properties due to structural defects and non-stable phases at room temperature. In this paper, we analyzed the influence of the synthesis temperature and different concentrations of cerium in the formation of the orthorhombic SnO2 phase. The structural and morphological characterization was performed using experimental techniques such as X-ray diffraction (XRD), allied with Rietveld refinement, field emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDS). The reflections in the XRD pattern and clear lattice fringes observed by HRTEM confirm the formation of the orthorhombic SnO2 phase in the (Sn1−xCex)O2 with x = 0, 0.05, 0.1 and 0.3 synthesized at 750 °C. The reducing atmosphere generated by the heat treatment process of the samples and the strain originated due to the difference of ionic radii between the tin and cerium ion can play a crucial role in the formation of the orthorhombic phase of SnO2.
Introduction
Wide band gap semiconductor oxides, such as tin dioxide (SnO2), are the subject of intensive research due to the their applications in different areas, such as gas sensors,1,2 solar cells,3 catalysts4 and optoelectronic devices.5 This oxide can be synthesized in three different crystalline phases: tetragonal, orthorhombic and cubic. The most thermodynamically stable phase, at room temperature, is cassiterite, a rutile crystalline structure with tetragonal symmetry (space group P42/mnm). Orthorhombic and cubic phases are metastable and can be obtained using synthesis under high pressure and temperature, or from a phase transition of the rutile structure at high pressure.6,7 From a different point of view, the orthorhombic SnO2 phase (O-SnO2) exhibits distinct physical and chemical properties compared to the stable phase (tetragonal), motivating the development of synthesis routes to obtain it. With this purpose, the optimization of the O-SnO2 synthesis under atmospheric pressure allows exploring the new magnetic and optical properties, thus, opening new perspectives for different applications.The formation of the orthorhombic phase is directly related to the synthesis method used, as well as to other experimental conditions, such as pressure, and temperature. Although originally this phase was found only in experiments carried out at high pressure and temperature, new synthesis strategies have been proposed to prepare O-SnO2 under atmospheric pressure. In this sense, different physical and chemical routes, such as pulsed laser deposition (PLD)8,9 oxidizing epitaxial tin monoxide films,10 metalorganic chemical vapor deposition (MOCVD)11 and co-precipitation methods12 were used to synthesize this phase. For example, Chen et al. prepared pure O-SnO2 thin films at a pressure of 3 × 10−2 Pa and substrate temperature of 320 °C using pulsed laser deposition (PLD).8 Lamelas et al. investigated the formation of nanoparticles with orthorhombic phase produced by the oxidation of mechanically milled tin monoxide powders, followed by a heating to 575 °C.13 Using a different approach, Zhang et al., proposed to dope the SnO2 with Mn to obtain the orthorhombic phase. The authors reported a mixture of O-SnO2 and T-SnO2 for samples processed at ambient pressure doped with 5 mol% of Mn.12 Using a similar strategy, we investigated the doping process of SnO2 with a rare earth metal (RT). Among the RT, we chose cerium doping, since it has two oxidation states and ionic radius with the coordination number equal to 6, Ce3+ (1.01 Å) and Ce4+ (0.87 Å), respectively, much larger than the Sn4+ (0.69 Å), with the same coordinate number.14 This could induce stress in the SnO2 lattice, possibly leading to the formation of the orthorhombic phase.
From a different point of view, sol–gel routes, such as, the Pechini method,15 have not been used to prepare this oxide phase. Then, considering the importance of orthorhombic SnO2 and the lack of papers using sol-routes to synthesize it, the aim of this work is the synthesis and structural characterization orthorhombic phase of SnO2 in the nanoparticle system (Sn1−xCex)O2, with x = 0, 0.05, 0.1, 0.3, 0.7 and 1.0 using the Pechini method.
Experiment details
Nanoparticles of (Sn1−xCex)O2 were produced using the Pechini method15 with different Ce molar concentrations (x = 0, 0.05, 0.1, 0.3, 0.7, 1) and synthesis temperatures. Initially, in an 0.5 L of an aqueous solution of 0.25 mol L−1 citric acid (P A Synth), 28.20 g of tin chloride dihydrate (Sigma-Aldrich) was added. The final solution pH was controlled using 1 M/L ammonia solution (Synth) added drop wise until its value is 3. Then, a white powder of tin citrate was obtained.16 The precipitate was washed with 0.7 L of distilled water and dried in the oven at 70 °C for 24 h, resulting in the tin citrate monohydrate (C6H4O7Sn2·H2O).16 Next, the precursor solution were prepared by dissolving 2 × 10−3 mol tin citrate monohydrate (C6H4O7Sn2·H2O) in 4.46 mL ethylene glycol, EG, (Sigma-Aldrich) under stirring at 70 °C, leading to a solution of 0.45 mol L−1. It was necessary to add 80 μL of concentrated nitric acid to completely dissolve the salt. Separately, 2 × 10−3 mol ammonium cerium(IV) nitrate (NH4)2Ce(NO3)6 (Vetec) was dissolved in 4.46 mL of EG, under stirring at 70 °C. Afterward, separately, 2 × 10−2 mol citric acid (CA – P A Synth) was added to these solution, keeping it at constant temperature and stirring. All samples were prepared with the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:40 of total metal salt:citric acid:ethylene glycol, respectively. Then, the proper molar amount of cerium salt solution was added to the Sn salt solution in the following oxide molar composition: 0, 0.05, 0.10, 0.30, 0.70, 1.00. Then, the solutions were polymerized at 110 °C for 1 h and immediately thermal treated using two steps: (i) at 300 °C for 2 hours using a heating rate of 10 °C min−1, to inhibit aggregate formation and, (ii) different final temperatures (500, 580, 650 or 750 °C) for 2 hours to obtain the doped oxides. The second thermal treatment used also 10 °C min−1 as heating rate. The color of the pure SnO2 is white powder, and, for those samples doped with cerium, their color become yellow.
:10:40 of total metal salt:citric acid:ethylene glycol, respectively. Then, the proper molar amount of cerium salt solution was added to the Sn salt solution in the following oxide molar composition: 0, 0.05, 0.10, 0.30, 0.70, 1.00. Then, the solutions were polymerized at 110 °C for 1 h and immediately thermal treated using two steps: (i) at 300 °C for 2 hours using a heating rate of 10 °C min−1, to inhibit aggregate formation and, (ii) different final temperatures (500, 580, 650 or 750 °C) for 2 hours to obtain the doped oxides. The second thermal treatment used also 10 °C min−1 as heating rate. The color of the pure SnO2 is white powder, and, for those samples doped with cerium, their color become yellow.
The crystalline structures of the samples were investigated by X-ray diffraction (XRD) using a Rigaku powder diffractometer (Model D/Max-2500PC) with CuKα radiation with Bragg–Brentano geometry mode θ–2θ in the range 20–100° at a scan speed of 0.5° min−1, with a step of 0.02°. Rietveld refinements17 were performed using the free GSAS-EXPGUI software.18,19 The quality of the fitting procedure can be followed using the goodness of fit (S parameter). It is described in the literature if S is close to 1 indicate an excellent fitting process.20
The morphology and size of the nanoparticles were examined by Field Emission Scanning Electron Microscopy (FE-SEM) using Supra 35-VP Carl Zeiss, operated at 15 kV and by high-resolution Transmission Electron Microscopy (HRTEM) technique, using an FEI microscope (model Tecnai G2 F20, FEI), 300 kV. The chemical analyses of the samples were performed by EDS using a Thermo-Noran EDS.
Results and discussion
Fig. 1 shows X-ray diffraction patterns measured at room temperature for SnO2 samples, synthesized at 500 (a), 580 (b), 650 (c) and 750 °C (d). The orange and violet vertical bars correspond to the indexation to the Inorganic Crystal Structure Database, (ICSD no. 39173 and 157450) for the tetragonal and orthorhombic phase SnO2, respectively.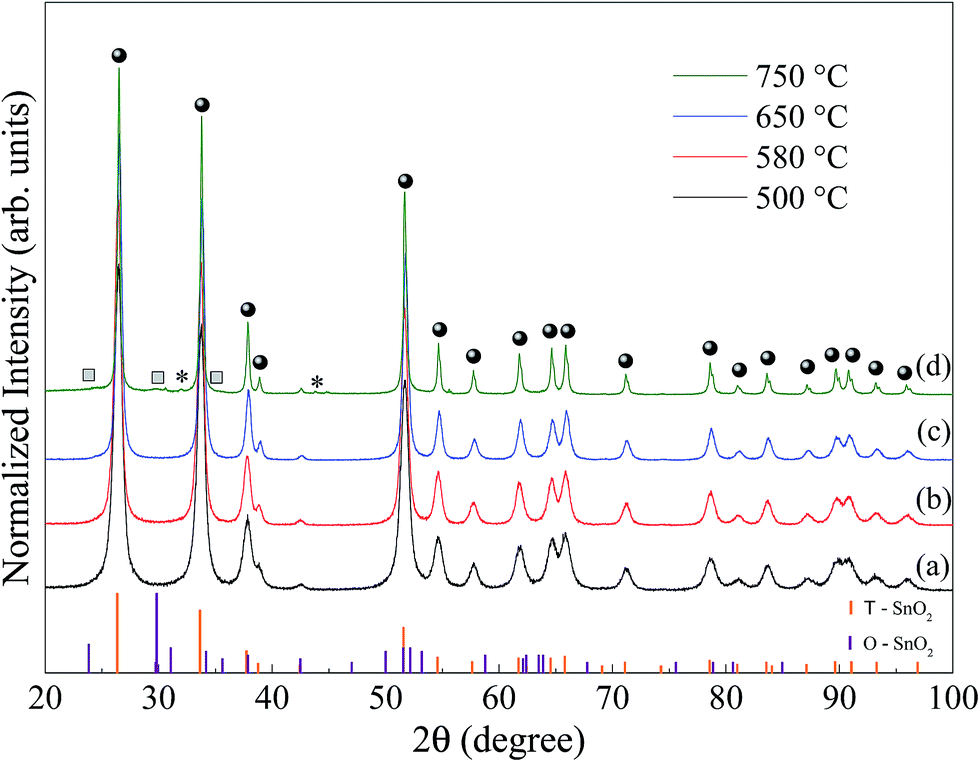 | ||
| Fig. 1 X-ray diffraction patterns for the SnO2 calcined, (a) 500, (b) 580, (c) 650 and (d) 750 °C for 2 hours. The circles and squares denote the tetragonal and orthorhombic SnO2 peaks, respectively; the asterisk denotes the peaks of metallic Sn; the orange and violet bars, respectively, correspond to the indexation of the diffraction peaks for SnO2 tetragonal (ICSD 39173) and orthorhombic (ICSD 157450). | ||
The XRD patterns shown in Fig. 1a to c are consistent with the tetragonal SnO2 phase (space group: P42/mnm) with lattice parameters, a = b = 4.7358 Å and c = 3.1851 Å.21 Within the limit of resolution in XRD the detection of additional reflections were observed, which excludes the presence of a crystalline secondary phase. In Fig. 1d, samples prepared at 750 °C, in addition to the clear presence of SnO2 tetragonal (marked circles), a second phase, O-SnO2 peaks was identified. In fact, Bragg peaks (11l) and (02l) (see squares labels in Fig. 1d) correspond to the orthorhombic SnO2 (space group: Pbcn) with lattice parameters, a = 4.707 Å, b = 5.710 Å and c = 5.246 Å.22 Moreover, the weak diffraction peak at 2θ = 30.6° and 44.9°, labeled by an asterisk in Fig. 1d, can be indexed as the plane (200) and (211) of the metallic tin phase (Sn), in agreement with that reported in the literature23 (ICSD no. 40038).
The XRD patterns were analyzed using Rietveld refinement.17 A pseudo-Voigt function of Thompson-Cox-Hasting modified by Young and Desai (pV-TCHZ) as the profile function24 used in the fitting process. The full width at half maximum (FWHM) obtained from this procedure was used to estimate the crystallite size 〈DX-ray〉 and is presented in Table 1. In addition, lattice parameters, unit cell volume, lattice strain and goodness of fit, (S), obtained by the Rietveld refinement are also listed in Table 1.
| Temp (°C)/phase | Lattice parameter (Å) | Cell volume V (Å3) | 〈DX-ray〉 (nm) | Strain (%) | S | ||
|---|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | |||||
| 500/T | 4.735 | 4.735 | 3.185 | 71.40 | 19.3 | 0.3411 | 1.34 |
| 580/T | 4.735 | 4.735 | 3.185 | 71.41 | 29.8 | 0.5127 | 1.48 |
| 650/T | 4.742 | 4.742 | 3.188 | 71.67 | 29.9 | 0.6261 | 1.52 |
| 750/T | 4.740 | 4.740 | 3.188 | 71.62 | 50.3 | 0.1260 | 1.82 |
| 750/O | 4.727 | 5.650 | 5.271 | 140.10 | 4.8 | 3.6819 | |
| 750/M | 5.828 | 5.828 | 3.178 | 107.90 | 40.4 | 1.0480 | |
The goodness of fit indicator S shows the good quality of structural refinement data. A change was observed in the lattice parameters and consequently in the volume of the unit cell as a function of the increase in the synthesis temperature.
As shown in Fig. 1 there is a narrowing in the FWHM of the Bragg peak with increasing synthesis temperature. Part of this narrowing is due to the increase in the crystallite size, as calculated by Rietveld refinement (Table 1), which is in good agreement with the literature.25 For the sample calcined at 750 °C, Table 1 shows that the size of the crystallite of the majority phase, tetragonal SnO2, is much larger compared to the size of the crystallite of the orthorhombic SnO2. In contrast, we obtained the highest strain value for the orthorhombic SnO2 and a lower value for tetragonal SnO2.
Fig. 2a shows SEM image for the SnO2 sample calcined at 750 °C. In the inset the particle size histogram was obtained from several SEM images fitted by a log-normal distribution. It clearly observed that pure SnO2 particles are larger (48.0 ± 0.2) and have non-uniform sizes compared to the pure sample calcined at 650 °C (29.4 ± 0.3). These data are shown in Fig. SI.1 (ESI†). High-resolution TEM images (Fig. 2b and c) also show that this sample is highly crystalline, which is clearly observed from the Fourier transform (Fig. 2d) of the image. It is possible to observe two sets of planes with lattice spacing of 3.64 Å and 2.98 Å, respectively, corresponding to planes (110) and (111) of the orthorhombic of SnO2 phase. It was also possible to observe a plane at 3.34 Å, which corresponds to the plane (110) of the tetragonal SnO2 phase (Fig. 2c). The TEM results confirm that both phases of tetragonal SnO2 and orthorhombic coexist in the sample even in the case of non-doped material, x = 0, synthesized at 750 °C, which is consistent with XRD data (see Fig. 1).
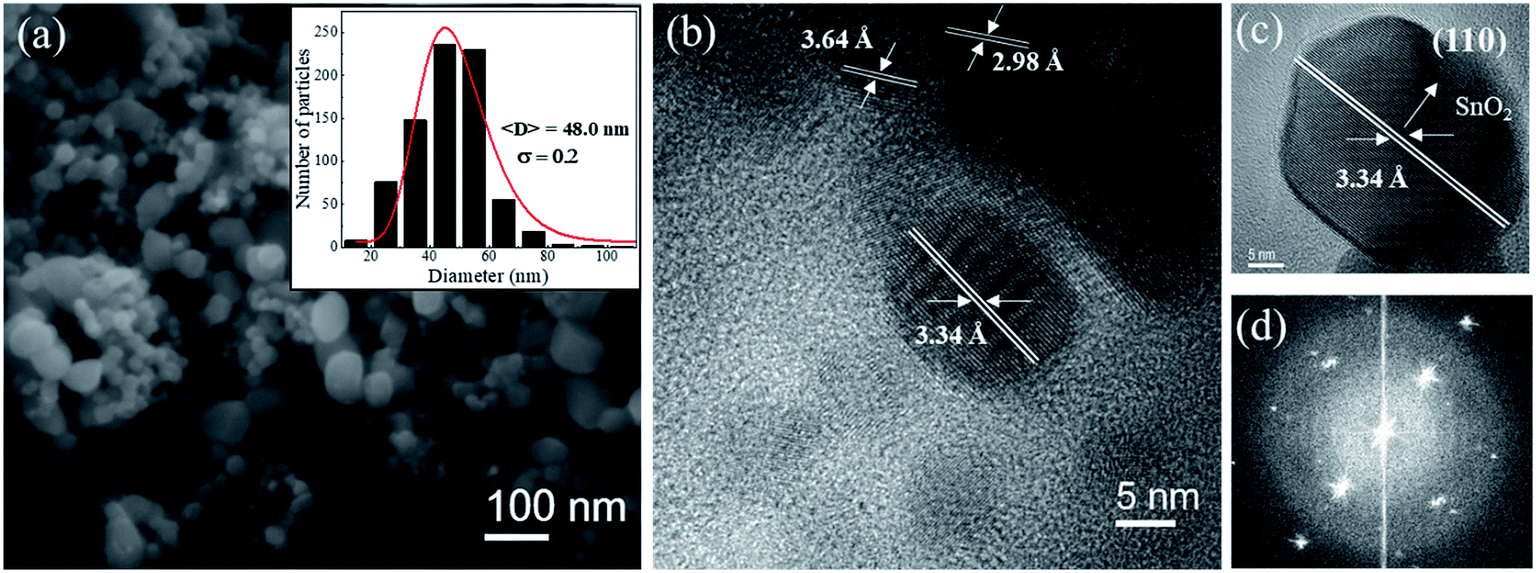 | ||
| Fig. 2 (a). FE-SEM image of the sample SnO2 calcined at 750 °C. (b) HRTEM image of a set of nanoparticles. (c) HRTEM image of an individual SnO2 nanoparticle indicating the interplanar distances characteristic of tetragonal tin dioxide and the corresponding Fourier transform in (d). The insert: particle size histogram fitted with a log-normal size distribution. | ||
Recent studies developed by Zhang et al.12 reported the formation of the orthorhombic phase in SnO2 at atmospheric pressure, for samples doped with 5 mol% of Mn, synthesized by co-precipitation. The authors proposed the formation of this phase with distortion of the lattice generated by the doping process of tetragonal SnO2.12 Based on this paper, we decided to study the solubility limit of cerium in SnO2. It is known that the Ce4+ ion has an ionic radius equal to 0.87 Å with coordination number equal to 6 and Sn4+ is smaller (ionic radius = 0.69 Å), with the same coordination number.14 Therefore, the doping of SnO2 using Ce could induce distortions in the lattice, increasing the concentration of the orthorhombic SnO2 phase.
Fig. 3a shows TEM images for the sample x = 0.3 calcined at 750 °C. A spherical morphology and a reduction in particle size are observed, compared to the undoped sample (Fig. 2a). Fig. 3b shows the HRTEM image which confirms both SnO2 phases, with the d-spacing at 3.34 Å and 2.98 Å corresponding to the tetragonal (110) and orthorhombic (111) planes, respectively. We also observed the d-spacing at 3.12 Å, which corresponds to the plane (111), confirming the segregation of the CeO2 cubic phase. The energy-dispersion X-ray spectroscopy (EDS) characterization (Fig. 3c) confirms that the synthesized sample is composed only of Sn, Ce, and O and the quantitative analysis of both Sn and Ce molar ratios of the experimentally and nominally obtained composition is in good agreement, which is presented in Table SI.2 (ESI†). Fig. 3d shows the reconstructed HRTEM image of one region corresponding to CeO2 particle. It is clearly observed that the sample has structural defects, observed in the Figure by the lack of linear behavior of the crystalline planes. It is possible this occurs due to the distortions in the atomic planes of the CeO2 phase. In the Fig. 3e–g, respectively, the FFT pattern obtained from each region of Fig. 3b is shown, i.e., orthorhombic and tetragonal SnO2 phase and the cubic CeO2 phase.
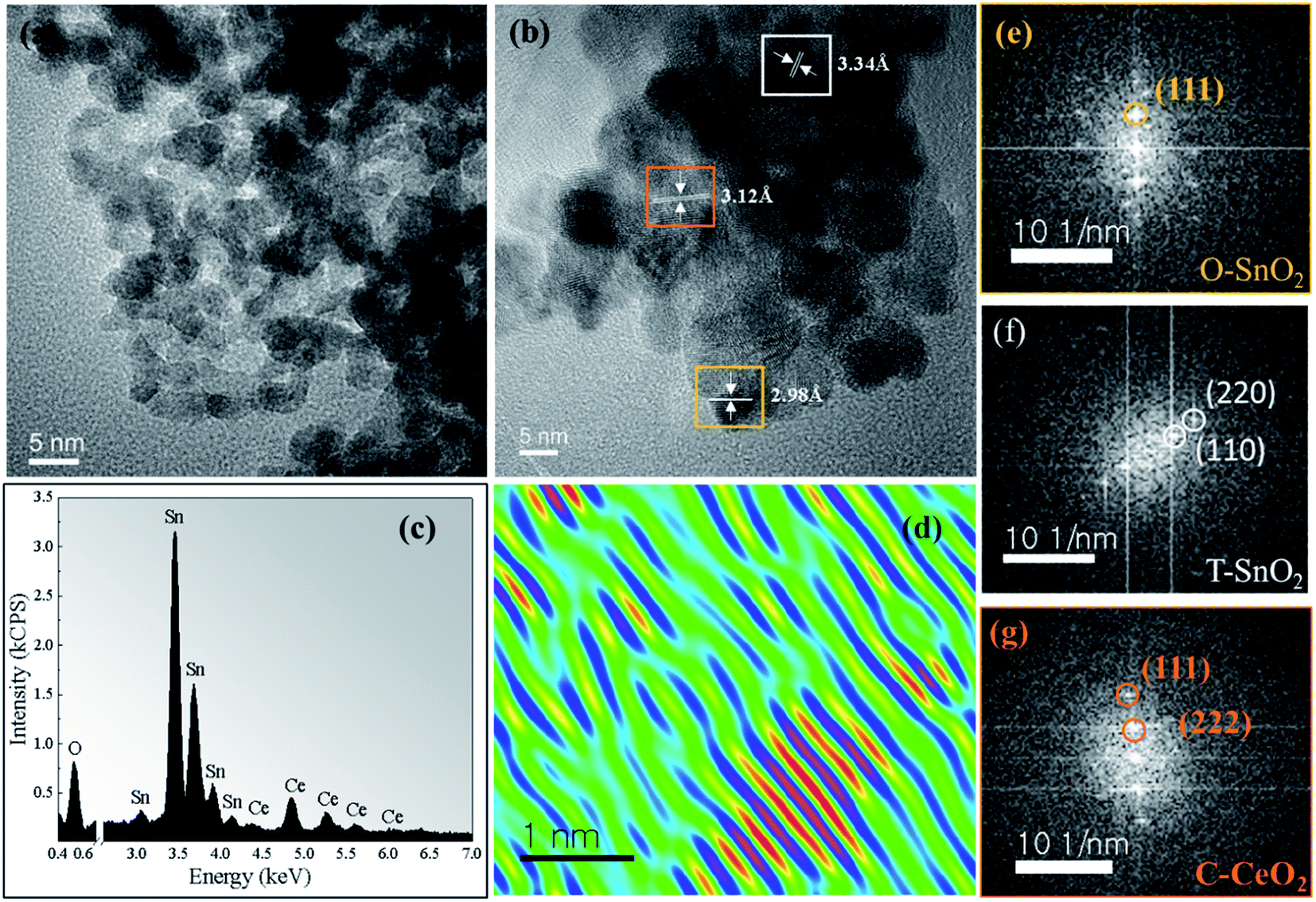 | ||
| Fig. 3 (a) TEM image of the sample x = 0.3 calcined at 750 °C. (b) HRTEM image (c) EDS spectrum of the sample. (d) Reconstructed HRTEM image of a CeO2 particle, which shows the lattice mismatch between CeO2 plans. (e–g) FFT pattern referring to the orthorhombic (O-SnO2), tetragonal (T-SnO2) phase and the cubic (C-CeO2) phase. | ||
The XRD patterns for the samples (Sn1−xCex)O2 changing the molar concentration of Ce (x = 0, 0.05, 0.1, 0.3, 0.7, 1) calcined at 750 °C for 2 hours are shown in Fig. 4. We also show the difference (blue line) between the experimental and calculated patterns, which were evaluated by the Rietveld method. All patterns shown in Fig. 4 indicate that both structures, tetragonal and orthorhombic of SnO2 coexist within the samples in the range from 0 to x = 0.3. No diffraction peak associated to the formation of cerium oxide (CeO2) was detected up to the concentration x = 0.1. However, by increasing the concentration to higher values, x = 0.3, 0.7 and 1, the Bragg peaks of CeO2 fluorite structure can be observed space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and lattice parameter, a = b = c = 5.4124 Å (ICSD no. 72155).
m and lattice parameter, a = b = c = 5.4124 Å (ICSD no. 72155).
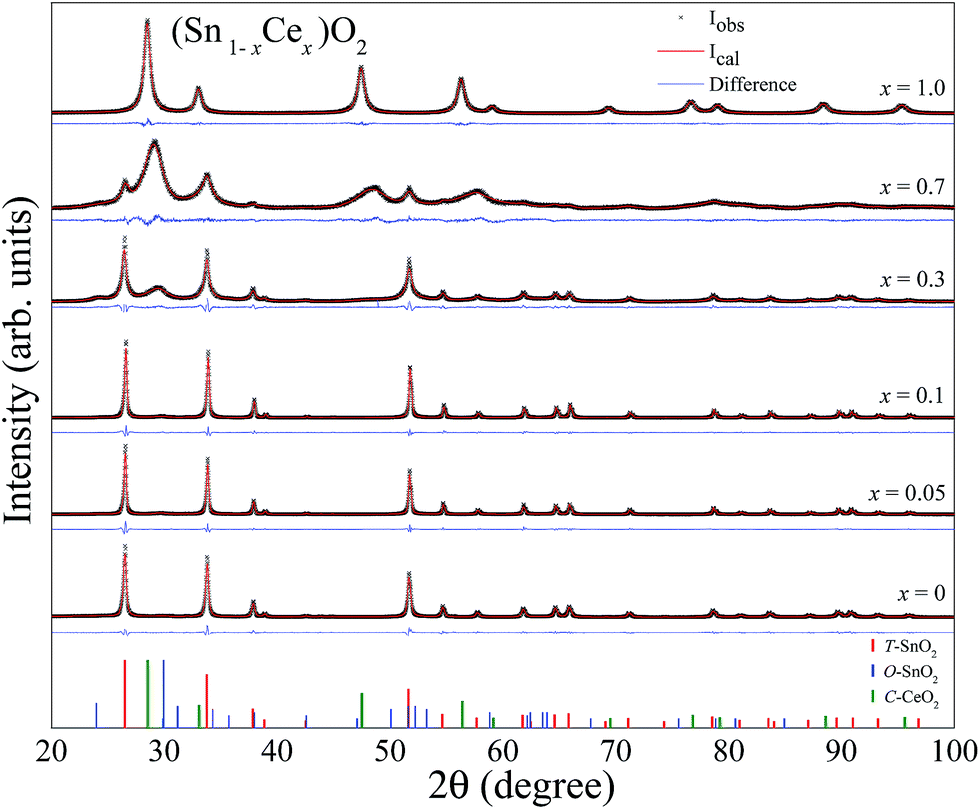 | ||
| Fig. 4 X-ray diffraction patterns for the (Sn1−xCex)O2 (x = 0, 0.05, 0.1, 0.3, 0.7, 1) nanoparticles synthesized at 750 °C. The solid lines are the fittings using the Rietveld method (red lines) and difference between the experimental and calculated patterns (blue lines). | ||
The structural parameters calculated by the Rietveld refinement are listed in Table 2, which show a molar percentage for each phase, lattice parameters, unit cell volume, crystallite size, lattice strain and goodness of fit values, which is an indicator that the refinement quality is suitable. Table 2 shows that while the unit cell volume of the tetragonal SnO2 did not present significant changes as the Ce concentration increases 10 mol%, the orthorhombic of SnO2 phase exhibits an expansion in the structure, i.e., there is an increase in the volume of the unit cell, suggesting the formation of a substitutional solid solution. Since, Ce4+ ions have a larger ionic radius than Sn4+, with the same coordination number, its incorporation into the orthorhombic SnO2 structure justify the increase of the volume of the unit cell. However, the solubility of Ce ions in SnO2 without phase segregation is limited to 10 mol%. Above this concentration, the segregation of cerium oxide phase can be observed. Table 2 shows that the crystallite size of T-SnO2 is much larger than that of O-SnO2 and presents lower strain values. This probably occurs due to the decrease in internal stresses when crystallite size increases. Another important characteristic due to the incorporation of cerium in SnO2 is the disorder in the crystalline structure created by the stacking faults in the crystal due to the difference of ionic radius, resulting in increased strain.
| (Sn1−xCex)O2 | Phases | (% molar) | Lattice parameter (Å) | Cell volume V (Å3) | 〈DX-ray〉 (nm) | Strain (%) | S | ||
|---|---|---|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | |||||||
| x = 0 | T-SnO2 | 87.58 | 4.740 | 4.740 | 3.188 | 71.62 | 50.3 | 0.1260 | 1.82 |
| O-SnO2 | 11.27 | 4.727 | 5.650 | 5.271 | 140.10 | 4.8 | 3.6819 | ||
| M-Sn | 1.15 | 5.828 | 5.828 | 3.178 | 107.90 | 40.4 | 1.0480 | ||
| x = 0.05 | T-SnO2 | 88.97 | 4.740 | 4.740 | 3.188 | 71.62 | 74.3 | 0.1087 | 1.90 |
| O-SnO2 | 10.46 | 4.768 | 5.633 | 5.304 | 142.40 | 12.6 | 3.6086 | ||
| M-Sn | 0.57 | 5.827 | 5.827 | 3.183 | 108.13 | 9.7 | 0.9295 | ||
| x = 0.1 | T-SnO2 | 89.65 | 4.740 | 4.740 | 3.188 | 71.62 | 74.9 | 0.1087 | 1.78 |
| O-SnO2 | 9.44 | 4.701 | 5.719 | 5.305 | 142.60 | 12.2 | 4.0458 | ||
| M-Sn | 0.91 | 5.828 | 5.828 | 3.171 | 107.70 | 27.6 | 1.0337 | ||
| x = 0.3 | T-SnO2 | 50.87 | 4.743 | 4.743 | 3.187 | 71.70 | 25.0 | 0.1125 | 1.76 |
| O-SnO2 | 40.90 | 4.798 | 5.658 | 5.282 | 143.40 | 6.5 | 3.3376 | ||
| CeO2 | 8.23 | 5.323 | 5.323 | 5.323 | 150.70 | 3.6 | 1.1425 | ||
| x = 0.7 | T-SnO2 | 26.43 | 4.748 | 4.748 | 3.196 | 72.05 | 13.7 | 1.4380 | 1.33 |
| CeO2 | 73.57 | 5.325 | 5.325 | 5.325 | 151.06 | 18.8 | 8.3392 | ||
| x = 1 | CeO2 | 100 | 5.416 | 5.416 | 5.416 | 158.89 | 14.1 | 0.0829 | 1.17 |
Fig. 5 shows the summarization of these results plotting the molar concentration obtained by the Rietveld refinement for each oxide as a function of the nominal Ce concentration. The results show that the introduction of different Ce concentrations produces different conditions to stabilize the orthorhombic phase of SnO2. This Figure corroborates the data presented in Fig. 4, in which the amount of O-SnO2 phase increases as the Ce nominal concentration increases to 30 mol%. The maximum amount of O-SnO2 obtained is 41% for the sample synthesized at 750 °C with 30 mol% of Ce dopant.
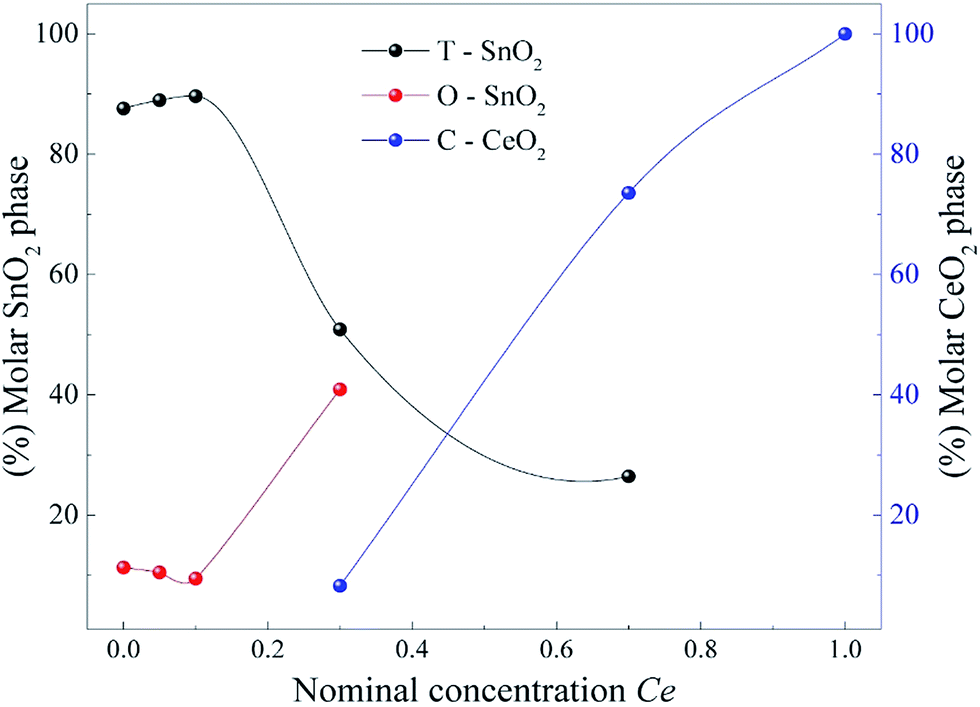 | ||
| Fig. 5 Molar concentration obtained by the Rietveld refinement for each oxide as a function of the nominal Ce concentration. | ||
It is well established in the literature that the orthorhombic SnO2 phase was originally observed only in experiments with high pressures and temperatures.8,9 However, recently Zhang et al. using the chemical co-precipitation method, reported the formation of the orthorhombic phase of SnO2 at atmospheric pressure, when the samples were doped with 5 mol% of Mn.12 Our results show that the orthorhombic phase already appears in the pure sample, although with a small molar fraction, reaching a molar fraction of approximately 41% when we increase the concentration of Ce to x = 0.3 (see Fig. 5).
The Pechini method proved to be an effective alternative for the synthesis of orthorhombic SnO2, possibly due to the number of defects generated in the thermal treatment process. However, the process of incorporating Ce in the SnO2 phase led to an increase in the concentration of the orthorhombic phase, possibly due to the ionic radius difference of Ce4+ (0.87 Å) and Sn4+ (0.69 Å) leading to the disorder of the crystal structure created by stacking faults in the crystal.
Conclusions
In this work, we have successfully obtained nanoparticles of (Sn1−xCex)O2 using the Pechini method. The reflections on the XRD pattern and clear lattice fringes observed in the HRTEM images confirm the formation of the orthorhombic of SnO2 phase with x = 0, 0.05, 0.1 and 0.3, synthesized at 750 °C. Through the Rietveld refinement analyzes it can be observed that the sample with 0.3 moles of cerium reaches a significant molar fraction of the orthorhombic phase, approximately 41%. The reducing atmosphere generated by the heat treatment of the samples and the strain originated due to the difference of ionic rays between the tin and cerium ion can play a crucial role in the formation of orthorhombic phase SnO2. The synthesis method used proved to be a promising alternative route for the preparation of orthorhombic SnO2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the FAPESP (2013/07296-2 and 2017/24995-2), CAPES and CNPq Brazilian agencies.References
- L. Renard, O. Babot, H. Saadaoui, H. Fuess, J. Brötz, A. Gurlo, E. Arveux, A. Klein and T. Toupance, Nanoscale, 2012, 4, 6806–6813 RSC.
- J. Arbiol, E. Comini, G. Faglia, G. Sberveglieri and J. R. Morante, J. Cryst. Growth, 2008, 310, 253–260 CrossRef CAS.
- C. C. Sno, S. Cells, A. Hossain, J. R. Jennings, Z. Y. Koh and Q. Wang, ACS Nano, 2011, 5, 3172–3181 CrossRef PubMed.
- A. Dodd, A. McKinley, M. Saunders and T. Tsuzuki, Nanotechnology, 2006, 17, 692–698 CrossRef CAS.
- H.-S. Woo, I.-S. Hwang, C. W. Na, S.-J. Kim, J.-K. Choi, J.-S. Lee, J. Choi, G.-T. Kim and J.-H. Lee, Mater. Lett., 2012, 68, 60–63 CrossRef CAS.
- J. Haines and J. M. Le, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 144–154 CrossRef.
- L. G. Liu, Science, 1978, 199, 422–425 CAS.
- Z. Chen, J. K. L. Lai and C.-H. Shek, Appl. Phys. Lett., 2006, 89, 231902 CrossRef.
- F. J. Lamelas and S. A. Reid, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 9347–9352 CrossRef CAS.
- A. Prodan, N. Vene, F. Sevgek and M. Hudomalj, Thin Solid Films, 1987, 147, 313–319 CrossRef CAS.
- L. Kong, J. Ma, Z. Zhu, C. Luan, X. Yu and Q. Yu, Mater. Lett., 2010, 64, 1350–1353 CrossRef CAS.
- Q. Zhang, P. Liu, C. Miao, Z. Chen, C. M. Lawrence Wu and C.-H. Shek, RSC Adv., 2015, 5, 39285–39290 RSC.
- F. J. Lamelas, J. Appl. Phys., 2004, 96, 6195 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- M. P. Pechini, Method of preparing lead and alkaline earth titanates and niobates and coating method using the same to form a capacitor, US3330697 A, Patent, 1967.
- M. M. Besso, Tin salts of citric acid and method of preparation, US3213120, Patent, 1965.
- R. A. Young, The Rietveld Method. I.U.C, Oxford University Press Inc., New York, 1995 Search PubMed.
- A. C. Larson and R. B. Von Dreele, Los Alamos Natl. Lab. Rep., 2004, pp. 86–748 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- L. B. McCusker, R. B. Von Dreele, D. E. Cox, D. Louër and P. Scardi, J. Appl. Crystallogr., 1999, 32, 36–50 CrossRef CAS.
- H. Seki, N. Ishizawa, N. Mizutani and M. Kato, Powder Diffr., 1989, 4, p156–p160 CrossRef.
- L. Gracia, A. Beltrán and J. Andrés, J. Phys. Chem. B, 2007, 111, 6479–6485 CrossRef CAS PubMed.
- J. A. Lee and G. V. Raynor, Proc. Phys. Soc., London, Sect. B, 1954, 67, p737–p747 CrossRef.
- R. Young and P. Desai, Arch. Nauki Mater., 1989, 10, 71–90 Search PubMed.
- A. Maciel, P. Lisboa-Filho, E. Leite, C. Paiva-Santos, W. Schreiner, Y. Maniette and E. Longo, J. Eur. Ceram. Soc., 2003, 23, 707–713 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra12727h |
| This journal is © The Royal Society of Chemistry 2018 |